

Abstract
Cancer stem cells (CSCs) and cells in a cancer stem cell-like (CSCL) state have proven to be responsible for tumor initiation, growth, and relapse in Prostate Cancer (PCa) and other cancers; therefore, new strategies are being developed to target such cellular populations. TLR3 activation-based immunotherapy using Polyinosinic:Polycytidylic acid (PIC) has been proposed to be used as a concomitant strategy to first-line treatment. This strategy is based on the induction of apoptosis and an inflammatory response in tumor cells. In combination with retinoids like 9cRA, this treatment can induce CSCs differentiation and apoptosis. A limitation in the use of this combination is the common decreased expression of TLR3 and its main positive regulator p53. observed in many patients suffering of different cancer types such as PCa. Importantly, human exposure to certain toxicants, such as iAs, not only has proven to enrich CSCs population in an in vitro model of human epithelial prostate cells, but additionally, it can also lead to a decreased p53, TLR3 and RA receptor (RARβ), expression/activation and thus hinder this treatment efficacy. Therefore, here we point out the relevance of evaluating the TLR3 and P53 status in PCa patients before starting an immunotherapy based on the use of PIC +9cRA to determine whether they will be responsive to treatment. Additionally, the use of strategies to overcome the lower TLR3, RARβ or p53 expression in PCa patients, like the inclusion of drugs that increase p53 expression, is encouraged, to potentiate the use of PIC+RA based immunotherapy in these patients.
Keywords: Prostate cancer, Immunotherapy, TLR3, p53, RARβ, Poly (I:C), PIC, 9-cis retinoic acid, Arsenic, Cellular rewiring
Prostate cancer (PCa) and Staging System
Prostate cancer (PCa) is the second most common type of cancer in men worldwide. In 2020, 1,414,259 new cases were identified globally [1] and the International Agency for Research on Cancer (IARC) indicate that the number of PCa deaths was 6.8% of total global cancer deaths such year [2]. By July 2022, PCa has been the neoplasm with the highest estimated prevalence, corresponding to 27% of all cancer cases in men [3]. It is estimated that by 2030 there will be 7 million new cases and 499,000 PCa deaths worldwide [4].
Early diagnosis of this neoplasm is possible with routine checkups, in which the physician requests laboratory tests for the determination of prostate specific antigen (PSA) in the blood and through digital rectal examination; if necessary, prostate biopsy to perform pathological analysis. No PSA analysis is needed for individuals under the age of 50 who show no risk factors. Prostate biopsy is suggested for PSA values above the upper limit (4 ng/ml) since it suggests the presence of prostate epithelium abnormalities [5].
The PCa staging procedure developed in 2002 by the American Joint Committee on Cancer (AJCC), is used to establish stages of prostatic neoplasm development, as well as to differentiate between adenocarcinoma and prostate epidermoid cancer, based on the neoplastic tissue extent (T), lymphatic invasion (N) and metastasis (M) [6].
Another useful tool for PCa staging is the Gleason score, which is established through the pathological observation of at least 3 microscopic fields of prostate tissue from the patient’s biopsies. In Gleason score, cells observed are graded on scale of 1 to 5 and the results in each field are summed. Grades less than 6 correspond to low-grade cancer; a value of 7 is a mean grade and greater than 8 establishes a high-grade neoplasm [7, 8].
TNM stage I is established when no abnormality is detected in the prostate, the Gleason score yields a value less than 6 and PSA values are up to 4 ng/mL. No pain is present at this stage and no pharmacological treatment is required. Continuous medical check-ups are scheduled, and it is recommended to modify habits such as improving the diet and avoiding exposure to heavy metals [8].
In stage II, the physician identifies a tumor located only in the prostate gland during examination. Symptoms such as back pain and difficulty urinating may occur. In this state there are high PSA titles (>4 ng/mL) [9].
Stage III of PCa can be defined based on the tumor’s invasion of other tissues, which includes extraprostatic invasion of surrounding tissue and seminal duct invasion. In stage IV there is invasion of surrounding tissues such as the rectum, urethral ducts and pelvic muscles [10].
The determination of such PCa stages allows an estimate of patient prognosis, as well as decisions on the therapeutic strategy to be used. Depending on the stage, strategies such as androgen deprivation or surgical removal of prostate and testicles, chemotherapy, radiation therapy and/or immunotherapy will be employed [11].
The treatment effectiveness will depend on the diagnosis timing and tumor stage. Stage III typically has a survival rate is less than 30%. For stage IV, the response to treatment and therefore survival, is significantly decreased [12].
The above issues are components that will determine life expectancy, which decreases with more advanced tumor development. This determination can also depend on cellular factors, which will be addressed in the next section.
Stem Cells and their Role in cancer
Stem cells (SC) are a type of cells with pluripotency and self-renewal capabilities. Embryonic stem cells (ESCs) are present during embryonic development and are responsible for the formation of an individual’s different tissues. On the other hand, SCs present in fully developed tissues are called somatic stem cells (SSCs) and can give rise to new cells of specialized tissues. These cells generally can be recognized by a decreased expression of ESC markers, such as Cluster of Differentiation (CD) markers, such as CD14, CD45 and CD133 and the human leukocyte antigen HLA-DR of Major Histocompatibility Complex II (MHC-II), among others [13, 14].
In the prostate and other tissues, SCs exist as a subpopulation of proliferative basal cells. In addition, there are two other cell subpopulations corresponding to excretory luminal cells and neuroendocrine cells, which have an intermediate state of differentiation [15]. The SCs in general can be recognized by the expression of different biomarkers, including extracellular matrix receptor III (CD44) and prominin-1 (CD133), which are expressed in early progenitor cells [16].
Because of their survival abilities, SCs can accumulate modifications that lead to a promotion towards a malignant phenotype [17] When a malignant neoplastic transformation occurs, SSCs over-express some markers typical of ESCs, as well as genes that encode signal-transducing proteins, such as Kristen Rat Sarcoma virus (KRAS), B cell lymphoma 2 (Bcl-2), or Sonic Hedgehog (SHH). On the other hand, other genes decrease their expression, like those that encode for tumor suppressors such as phosphatidyllinositol-3,4,5-triphosphate 3-phosphatase (PTEN), the transcription factor P53, or the anti-apoptotic protein BAX (apoptosis regulator) [18, 19].
As malignant transformation occurs, the expression patterns of other transcription factors, such as Octamer binding 4 (Oct4), Sex determining region Y-box 2 (SOX2), Nanog homeobox (Nanog), and SHH that play a relevant role in maintaining the phenotype of ESCs (pluripotency, survival and self-renewal) are also increased or re-expressed [20, 21]. In this transformation, SCs develop an aggressive phenotype that implies the loss the proliferation regulation. SCs that undergo this transformation are known as cancer stem cells (CSCs) [17] CSCs show increased expression of characteristic ESCs genes, such as CD44 or CD133; transcription factors mentioned above; genes related to drug resistant such as ATP-binding cassette super-family G member 2 (ABCG2) and aldehyde dehydrogenase (ALDHA1); genes involved in DNA repair mechanisms such as ataxia-telangiectasia-mutated-and-Rad3-related kinase (ATR-Chk1) and ataxia telangiectasia mutated protein kinase 1 and 2 (ATM-Chk2); surface molecules such as integrin α2β1 or epithelial cell adhesion molecule (EpCAM) [22–26]. These observations have led to the hypothesis that CSCs are responsible for tumor initiation and progression, resistance to different therapies such as radiation therapy, radical prostatectomy and androgen deprivation, as well as for cancer recurrence [17].
A phenomenon observed in PCa is that there is a certain cell hierarchy, where a small group of CSCs are responsible for leading tumor initiation and development, while cancer cells that are in more external tissue layers correspond to cancerous cells with different levels of differentiation [18].
In addition to CSCs present in prostate tumors, there is a cell type that also possesses self-renewal and multipotent capabilities know as cancer stem cell-like (CSCL) state. Since CSCL cells share some CSC markers associated with cell survival and expansion, therapies directed against CSCs must also contemplate CSCLs, which at any given time may be also responsible for giving rise to a new tumor [27]. A strategy to treat CSCLs was proposed by Gupta et al., 2019 in a study in human mammary epithelial cell line (HMLE) and its neoplastic transformed derivate (HMLER) in which they developed a screening method to identify compounds targeting CSCs, and highlighted salinomycin. Despite this, they theorize that non-CSCs can experience a regression (de-differentiation) acquiring CSCL properties, for which they recommend that CSC-focused treatments be used in combination with non-CSC targeted drugs [28]. When CSCLs are grown in vitro in suspension, they can organize into structures called spheroids which can generate tumors in vivo. Moreover, in some cell types, CD44 and CD133 surface markers are overexpressed in these spheroid cells, but they also express tissue-specific differentiation markers [29].
Clearly, the SCs in the three types of prostate tissue (basal, luminal and neuroendocrine cells) [30], are not the only ones capable of developing cancerous characteristics. The differentiated somatic cells are also capable of this. A hypothesis suggests that differentiated cells in the prostate gland undergo a differentiation regression towards an CSCL phenotype. Some studies suggest that the genesis of PCa begins with basal prostate cells that over-express CD133 [31]. Other works carried out in PCa patients from Israel have shown that tumor protein 63 (p63), a prostate basal cell biomarker, is absent in prostate carcinomas, for which they hypothesize that prostate tumoral cells have a secretory origin, since all basal cells, even basal SCs, express p63 [32]. Furthermore, other studies on neuroendocrine prostate cancer (NEPC) models, developed from prostatectomy samples, suggest that NEPC could originate from prostatic epithelial cells that overexpress n-myc proto-oncogene protein (N-Myc) and myristoylated Serine Threonine Kinase (myr-Akt) [33]. Together, these studies that evaluated prostatic basal, luminal or neuroendocrine cell participation in tumoral development, suggest that somatic cells can undergo a cell dedifferentiation process to form CSC and CSCL involved in the tumor initiation. [14, 34].
Prostate CSCs express CD44, CD133, epidermal growth factor receptor 2 (HER2), ALDH and integrin α2β1 [13, 26], as well as markers such as platelet surface Glycoprotein (CD151), NK cell receptor (CD160) and podocalyxin (TRA-60–1). These markers are associated with a phenotype characterized by giving rise to tumors with a hierarchical organization in prostate cell malignant development [35]. There is also evidence of increased expression of genes encoding for transcription factors such as SOX2, Nanog, Translocation-Associated Notch Protein TAN-1(Notch) and Oct4 [36]. The expression of these genes is associated with the epithelium-mesenchymal transition (EMT), which is also seen in PCa, as it promotes migration and the generation of tumors outside the prostate gland [37]. During EMT, the receptors associated with the Wingless-Type MMTV (Wnt) pathway, transforming growth factor β (TGF-β), extracellular signal receiver (Notch), SHH (organogenesis and cell division) and tyrosine kinase, are also activated [38].
On the other hand, deceased expression of androgen receptor (AR) is also related to tumor growth, since CSCs can grow without relying on interaction with androgens. Some PCa cell models, such as DU145 (responsive to AR), show an increased auto-renovation when the gene codifying AR is muted [31]. Interestingly, it has been described that CSCs can re-express AR, and in some cases, they produce their own androgens, allowing them to survive hormonal deprivation therapies [39]. This implies that tumor initiation can occur at low androgen concentrations and subsequently increase in size due to the expansion of CSCs which produce their own androgens.
Besides androgens, estrogens are also involved in PCa, in some cases as negative regulators. That is why estrogen receptors (ER) become relevant in the treatment of this neoplasm. In a study by Hussain and colleagues in 2011, tissues from PCa patients expressing CD49 were analyzed. The addition of 6 μM ER-Agonist (8b-VE2) for 72 hours, led to the induction of apoptosis in CD49+ cells with CSC properties [40]. These observations suggest that ERs can be considered as therapeutic targets for strategies focused on the tumor microenvironment.
Another interesting observation is that SSCs are able to carry out periods of inactivity, which include a period of cell cycle arrest, leading the cells to survive against antiproliferative therapies when a cancerous transformation occurs [17].
On the other hand, within the tumor, CSC reside in a microenvironment that promotes hypoxia (tumoral characteristic), and this induces the expression of genes such as hypoxia inducible factor 1 (HIF-1) that occurs in the early stages of tumor development, as well as the expression of drug resistance molecules in SCCs such as ABCG2 [26]. It has also been described that decreased activity of DNA methyltransferases (DNMTs) can lead to global hypomethylation of DNA, thus establishing an early event in stages before complete cancerous transformation occurs [41].
In this regard, in a study carried out in mouse induced pluripotent stem cells (miPSCs) that were cultivated with mouse Lewis carcinoma cell lines (LLC) conditioned media to induce their transformation into CSCL (CD44+/ALDH+), an increased expression of hypomethylated genes associated with Phosphatidylinositol 3-kinase (PI3K) -Akt pathway was observed [42].
CSCs in PCa also express genes associated with survival against radiation therapy. For example, the Chk1 gene (Checkpoint kinase1) phosphorylation protects against cytotoxic damage from ionizing radiation and BRCA1/BRCA2-Containing Complex, Subunit 5 (RAD51), together with Chk1, is responsible for events associated with DNA repair [26].
Due to their survival, self-renewal, and drug resistance characteristics, CSCs present in prostate tumors are considered as an aggressive subpopulation that can initiate tumoral foci. For this reason, the study of mechanisms that can induce SCs to acquire a malignant phenotype and CSC markers, can help to improve therapeutic strategies in PCa. Implications in cancerous phenotype development will be addressed in the next section.
Risk Factors and Exposure to iAs in Prostate cancer
Several factors increase the risk for PCa development, such as age, hereditary traits, race or obesity, and environmental factors, such as exposure to toxicants [43]. Other factors associated with an increased risk of PCa include breast cancer susceptibility markers, such as protein 1 and 2 (BRCA1 and BRCA2) genes mutation, which, in their non-mutated form, participate in DNA repair mechanisms [44]. On the other hand, certain genetic conditions, such as Klinefelter syndrome (XXY), and other conditions such as acromegaly and testicular hypofunction have been associated with decreased probability of prostate cancer [44, 45]. Moreover, patients with diabetes show a decreased probability of developing PCa. Metformin treatment appears to represent a protective factor for the development of PCa [46].
In regard to environmental factors, human exposure to inorganic arsenic (iAs) is now well-recognized as an important risk factor for the development of PCa [47]. iAs is common contaminant in aquifers and represents the most important source of human exposure to this metalloid [48]. Recently iAs has been detected in crops for human use, such as rice, whose cultivation frequently uses iAs contaminated water; in the Americas it has been reported that the amount of iAs in rice is above the global average (66 g/kg of rice grain) [49]. Importantly, exposure to iAs is not limited only to natural sources. Anthropogenic activities that increase human exposure to iAs include mining, the use of paints (arsenic is a component found in some brands), wood preservatives, electronic circuits, insecticides and pesticides [50, 51]. Thus, exposure can also occur through inhalation of particles suspended in the air, reaching 1 to 10 g per day [52].
IAs-Induced Malignant Transformation and the Role of Stem Cells
The role of arsenic in the development of PCa appears to be mainly due to the fact that its elimination is mostly renal, exposing the entire genitourinary system to the harmful effects of iAs and its highly toxic methylated metabolites [48, 51].
In studies conducted to help understand and to define the mechanisms associated with the malignant transformation of human prostate cells by iAs, Tokar et al. (2010) demonstrated that iAs induces malignant transformation of non-tumorigenic RWPE-1 cells following chronic exposure to sodium arsenite (5 μM) for 30 weeks, resulting in a new cell line (CAsE-PE) [18]. Importantly, CAsE-PE cells show an increased expression of SC markers such as CD133 suggesting that iAs induces overaccumulation of CSCs in this cell line. The study also suggested that SCs have an advantage over differentiated cells in terms of ability to survive the toxic effects of iAs [20, 53].
To support the above assertions, Tokar and colleagues also demonstrated that a subpopulation of SCs isolated from RWPE-1, called WPE-stem, show an innate resistance to iAs-induced lethality and apoptosis. This was related to higher expression of anti-apoptotic proteins (Bcl-2 and MT) and lower expression of proapoptotic proteins (BAX, caspases 3, 8, 7 and 9) in WPE-stem cells compared with parental RWPE-1 cells. WPE-stem cells also showed higher expression of stress-associated genes (SOD1 and PRODH) compared to the differentiated RWPE-1 cells [20, 54].
Interestingly, exposure of WPE-stem to 5 μm sodium arsenite also led to a malignant transformation of these cells to a CSC phenotype (named As-CSC cells). However, unlike the RWPE-1 malignant transformation which took 30 weeks exposure, WPE-stem transformation occurred in only 18 weeks. According to this work, SCs appear to be particularly sensitive to sodium arsenite-induced transformation compared with differentiated counterparts [20].
As-CSCs were more invasive and proliferative than differentiated and malignantly transformed CAsE-PE cells, and also showed a higher activity of matrix metalloproteinase 9 (MMP-9) [55], which was associated with the development of aggressive tumors when they were heterotransplanted in immunodeficient mice. Other characteristics of As-CSCs included increased expression self-renewal associated genes (p63, ABCG2, BMI-1, SHH, OCT-4, NOTCH-1) and decreased expression of tumor suppressor PTEN [19]. This implies that SCs acquiring the cancerous phenotype due to their exposure to iAs tend to develop characteristics that improve survival and resistance to xenobiotic compounds, thus promoting their resistance to multiple treatments, and suggesting that strategies that target CSCs will be required in iAs-associated PCa.
Therapeutic Strategies against PCa
General Chemotherapy and Immunotherapy
Prostate tumor development is dependent on androgenic hormones, which is why early-stage PCa treatment is based on androgen deprivation which, in some cases, may require a prostatectomy. Pharmacological alternatives for blocking androgen synthesis include gonadotropin-releasing hormone agonists (e.g. leuprolide, goserelin or trytoreline); antiandrogens (e.g. flutamide and bicalutamide); and testosterone synthesis inhibitors such as abiraterone [56].
Androgen deprivation therapy is the prelude to treatment with radiation therapy, chemotherapy, and immunotherapy in later stages of PCa. Radiation therapy applies to patients at low or moderate risk according to the treating physician’s prognosis and is always applied in conjunction with androgen deprivation therapy. Radiation therapy aims to induce irreparable damage to tumor cell DNA [57].
In patients whose cancer has evolved into a castration-resistant state (CRPC), it is recommended to continue with antiandrogenic therapy, as well as to add dexamethasone as a second-line hormone treatment, given its corticosteroid ability to decrease inflammation around the neoplastic region [58]. As part of chemotherapy, drugs such as docetaxel are often used due to their anti-tumor mechanism involving the induction of tubulin assembly into stable microtubules, inhibiting their re-polymerization, and thus affecting events associated with tumor cell mitosis [59].
As part of stage III antineoplastic therapy, some strategies have a biological approach, also known as immunotherapy. This tool involves the stimulation of a patient’s immune system to induce an anti-tumor response. There are two types of approaches: 1) passive immunotherapy, where short-term components of innate immunity or restoration of T-helper cell-mediated immunity (Th-1) are stimulated by the administration of exogenous cytokines and monoclonal antibodies [60]; or 2) active immunotherapy, in which different strategies are used to stimulate the patient’s own adaptive immune response, resulting in the activation of immune cells, and the production of antibodies targeting tumor-specific antigens [61].
The proposed active immunotherapy strategies include vaccines such as Sipuleucel-T, a drug that constitutes an autologous vaccine, where the patient’s dendritic cells are activated in vitro with a specific antigen of prostate cancer and subsequently reintroduced into the patient to induce an immune response against the tumor [62]. Specifically, antigen-presenting cells (ATCs) are extracted and subsequently exposed to two antigens, prostate acid phosphatase (PAP) and a macrophage colony stimulation factor granulocyte (GM-CSF) recombinant antigen. These components (PAP + GM-CSF) are fused into a recombinant protein known as PA2024 [63]. ATCs are cultivated in the presence of PA2024 resulting in their maturation to APC8015s, which are re-implanted in the patient [64].
Other strategies also include the use of an immune system component agonist which is able to induce cellular defense responses or the programmed cell death in cancer cells. This strategy is called non-specific immunotherapy [61]. More specifically, this strategy consists of using cytokines, chemokines, or other receptor agonists whose activity does not depend on the response against a specific antigen. Examples of this strategy involve the exogenous administration of interferon gamma (IFN), IL-2, IL15, agonists of a tumor necrosis factor receptor superfamily member (CD40 agonist), OX-2 membrane glycoprotein (CD200 agonist), among others [65]. The induction of apoptosis and an anti-tumoral pathway through the activation of toll like receptors (TLRs)-has been shown to be promising to treat cancer [66].
Therapeutic Agents Targeting CSCs
Given their increased survival capacity and based on the evidence supporting that CSCs are responsible for the initiation, progress and recurrence of cancer, several proposed therapeutic strategies are primarily focused on the elimination of CSCs. The effectiveness of such strategies relies on targeting different markers characterizing SCs and CSCs to achieve effective tumor eradication.
Some CSC markers that are targeted by anti-tumor therapies include drug efflux pumps such as the ABCG2 protein. For example, Gefinitib has been demonstrated in preclinical studies to improve Irinotecan bioavailability by blocking ABCG2 [67]. Other methods target proteins associated with proliferation, like Epidermal Growth Factor Receptor (EGFR) that is targeted by monoclonal antibodies such as cetuximab (blocking EGFR ligand binding) and erlotinib (blocking EGFR intracellular tyrosine kinase activity) [68].
It is worth mentioning that, for the most part, CSC-focused strategies are in the clinical trials phase or are just being suggested as a treatment. Among these are the use of signaling pathway inhibitors such as the compound ETC-159 (Wnt inhibitor) which is in phase I clinical studies [69]. Another example is LY3039478 (Notch inhibitor) which is still being evaluated in phase II clinical studies [69]. Another strategy focuses on the use of nanoparticles as a SC marker-based delivery system [70]. Another proposed strategy includes the use of dichloroetyl acetate (DCA) which can induce a mitochondrial metabolism change from glycolytic to oxidative, by inhibition of pyruvate dehydrogenase kinase (PDK) phosphorylation of pyruvate dehydrogenase. In alveolar basal (A549) cancer cells DCA can induce inhibition of tumor growth by the above mechanism [71]. Other studies propose the use of chloroquine to inhibit the formation of autophagosome and inhibit autophagy. This strategy was tested in CD44+/CD24− cells from an MDA-MB-231 cell line [72].
The above-described strategies are generally proposed to be used in conjunction with general antineoplastic drugs as part of a comprehensive approach to prostate cancer therapy that has resisted conventional drug treatment. Drugs such as salinomycin promote events associated with apoptosis in CSCs, such as increased Bax expression. In addition, this strategy was shown to increase the activity of caspase 3 in an independent prostate cancer model of androgens using the PC-3 prostate cell line [73]. Another alternative uses Rottlerin, a compound that inhibits the protein-kinase C-Delta (PKC-δ) pathway and induces activation of caspase −3, −8, or − 9 [74]. These effects result in inhibition of cell proliferation. And alternate effect is the induction of apoptosis through the mitochondrial membrane depolarization, in CSC of a model of pancreatic cancer [75].
Still other strategies involve using molecules to block the signaling pathways that are overexpressed in CSCs [69]. An example of such molecules is Sulforaphan, which works by blocking nuclear enhancer of light kappa chains of activated B cells (NF-κB) inhibiting some pathway associated with Akt intermediaries [76].
Another important strategy involves the induction of cellular differentiation. Although different compounds have been proposed, only two have been shown to affect the degree of differentiation of CSCs. These are drugs that reverse epigenetic changes in tumor cells, such as histone deacetylase enzyme inhibitors (HDACs) that remove acetyl groups of histones, reducing genetic expression. Since HDACs are responsible for regulating the acetylation of histones that make up the nucleosome, they catalyze the removal of acetyl groups from the NH2 end of lysine residues. One of these HDAC inhibitory drugs is called suberoylanilide hydroxamic acid (HSA), which, along with other HDAC inhibitors are in phase I studies of clinical trials [77]. Another useful compound is retinoic acid (RA), or vitamin A, whose function is based on the induction of gene expression associated with cell differentiation through its binding to heterodimeric receptors RAR (α or β) and RXR [78]. Alitretinoin (from Ligand Pharmaceuticals) is a commercial presentation of 9-cis-retinoic acid that has been successfully used for the treatment of promyelocytic leukemia [79].
CSC Differentiation by Retinoids
Vitamin A (retinol or retinoic acid) and the retinoids are compounds capable of inducing the differentiation of SCs [80]. Among the three retinoids, the first retinoids generated include All Trans Retinoic Acid (ATRA, also called Tretinoin), 9-cis Retinoic acid (9cRA, Alitretinoin) and 13-cis retinoic acid (13cRA, isotretinoin). All of these retinoids are used as a treatment for different neoplasms such as acute promyelocytic leukemia [81]; generations II and III of retinoids are used as therapies for skin diseases [80].
ATRA, 9cRA and 13cRNA modulate the growth and differentiation of a variety of normal and malignant cells in vitro and in vivo [82]. 13cRA has been shown to be effective in suppressing premalignant oral injury and in preventing the development of second primary cancers in the upper aerodigestive tract in patients with head and neck cancer [83]. The retinoids induce SC differentiation through a mechanism involving the cytoplasm-to-nucleus transport of CBRP, a protein that carries retinoic acid (CRBP). This is followed by binding to retinoic acid receptors (RAR α or β) that are heterodimerized with RXR, and then binding to retinoid acid response elements (RAREs) in DNA where they activate the transcription of genes associated with SC differentiation, such as cytokeratin 18 (K18), Homeobox Protein Hox-A1 (Hoxa1), SRY-Box Transcription Factor 9 (Sox9), Forkhead Box Protein O3 (FOX03A), among other genes [77]. Among the alpha, beta and gamma RARs variants, the beta variant (β) has the greatest affinity for RAREs [84] (Fig. 1).
Fig. 1.
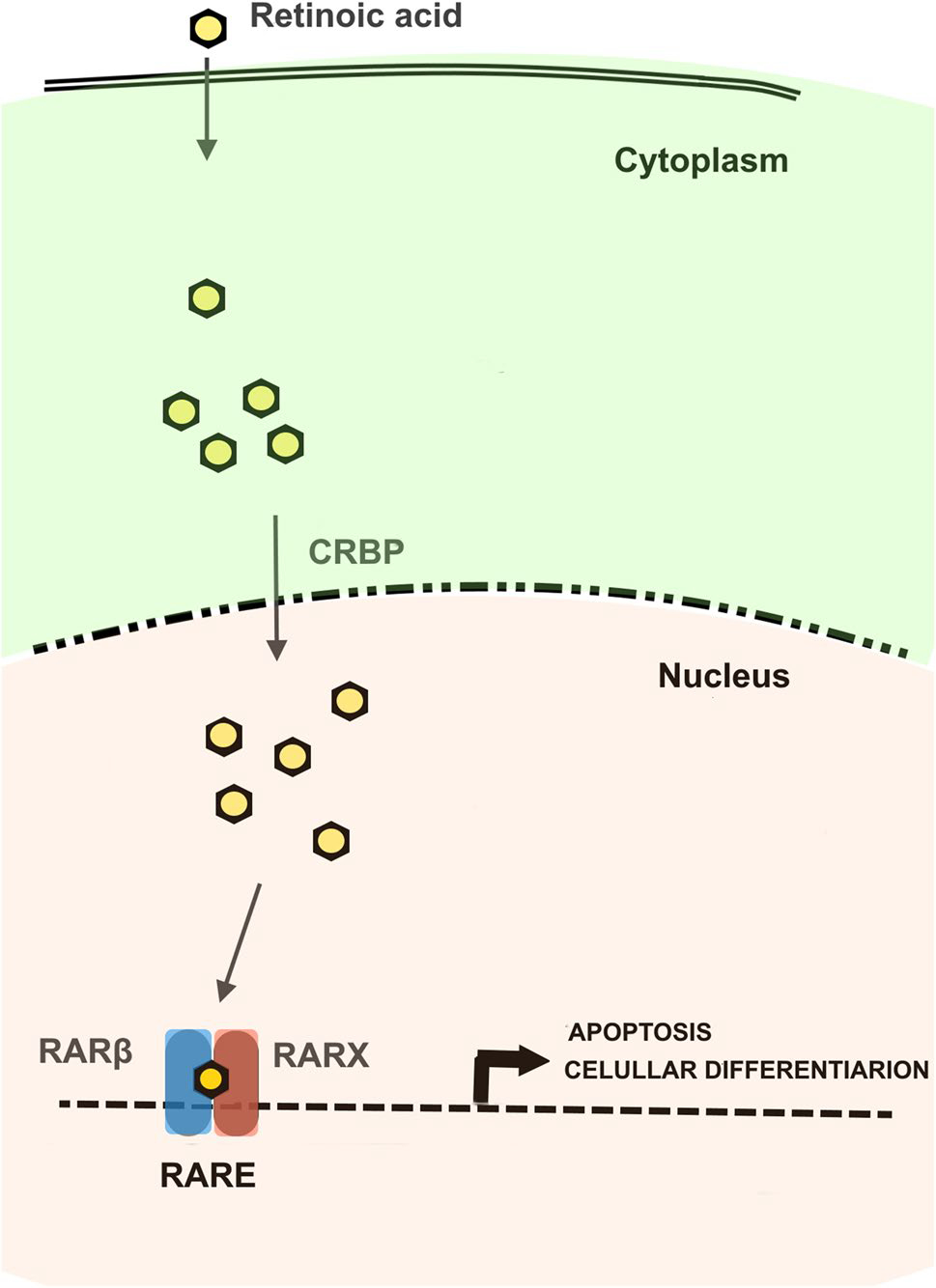
Retinoic acid and retinoids mechanism of action. Modified from Bushue et al., 2010
Although ATRA is one of the most commonly used retinoids as an antineoplastic differentiation strategy, 9cRA (Alitretinoin) has been shown to interact with RARβ with higher affinity [79, 85]. This implies that 9cRA may show better results in the induction of genes involved with CSC differentiation [79].
However, a deficiency in RARβ mRNA expression is commonly found in different cancers, therefore turning tumor cells non-responsive to the action of retinoids. This may be the case for CSCLs cells which are often resistant to treatment with retinoids. In fact, in CSCs RARβ would not be transcribed due inactivation by RAR promoter region methylation during tumorigenesis and therefore its interaction with RA would not occur [86].
Activation of TLRs Receptors in PCa Therapy
Toll-like receptors (TLRs) are components of the innate immune system and are expressed by dendritic cells, macrophages, natural killer cells, adaptive immunity T cells and B cells, and also by non-immune cells like epithelial, endothelial, and fibroblasts [87]. To date, ten different TLRs have been identified in humans, with TLRs 1, 2, 4, 5, 6 and 10 being located in the cellular cytoplastic membrane, while TLRs 3, 7, 8 and 9 are located in endosomal membranes [88].
TLRs are glycoproteins with ecto, transmembrane, and cytoplasmatic domains. The ectodomain consists of leucine-rich repetitions (LRRs) that mediate the recognition of pathogen-associated molecular patterns (PAMPs). The cytoplasmic toll/IL-1 receptor (TIR) domain initiates the signaling cascade after the corresponding PAMP is recognized. The PAMPs recognized by the different TLRs may be lipopolysaccharides, lipoproteins, proteins and nucleic acids from infectious agents (fungus, bacteria, parasites and viruses, respectively) [89].
The canonical anti-microbial response mechanism of TLRs family involves the activation of signaling pathways that induce an inflammatory response. Once the ligand binds to its corresponding TLR, the activated pathway can be divided as dependent or independent of the activation of myeloid differentiation primary response 88 (Myd88). The Myd88-dependent pathway consists of PAMPs recognition through lysine residues present in the TLR recognition domain. Subsequently, the interaction of TIR domain containing adaptor protein (TIRAP)/ mal T cell differentiation protein (MAL) and translocation associated membrane protein (TRAM) is required to collectively activate Myd88 [87].
Following ligand stimulation, Myd88 recruits Interleukin 1 receptor associated kinase 4 (IRAK4) and TNF receptor associated factor 6 (TRAF6). TRAF6 activates a TGF-beta-activated kinase 1 (TAK1)/ TAK1-Binding Protein 1 (TAB1) / TAK1-binding protein 2 (TAB2)/ TAK1-binding protein 3 (TAB3) complex through ubiquitination. In this way, the TAK1 complex activates the IkappaB Kinase (IKK) complex that catalyzes I-Kappa-B (IκBs), which is destroyed by the proteosome pathway. This allows NF-kB to be translocated into the nucleus [90]. TAK1 simultaneously activates the MAPK pathway promoting activation of Activator Protein 1 (AP-1). Both NF-kB and AP-1 induce inflammatory responses by activating transcription of genes such as interferon I (IFN I), cytokines such as interleukin 2, 6 and 12, as well as growth factors, including granulocyte colony and macrophage stimulator factor (GM-CSF) [87].
The alternative Myd88-independent pathway involves the TIR-domain-containing adapter-inducing interferon-β (TRIF) activation [87]. This pathway is activated after TLR3 recognizes double stranded RNA (dsRNA) (TLR4 ligand activates both routes) [91]. The cytoplasmic domain interacts with TRIF which in turn does so with TRAF6 and TRAF3. This induces recruitment with Receptor-Interacting Protein-1 (RIP-1), which in turn activates the TAK1 complex finally leading to the activation of NF-κB and MAPK, thus inducing the expression of inflammatory cytokines (Fig. 2) [87].
Fig. 2.
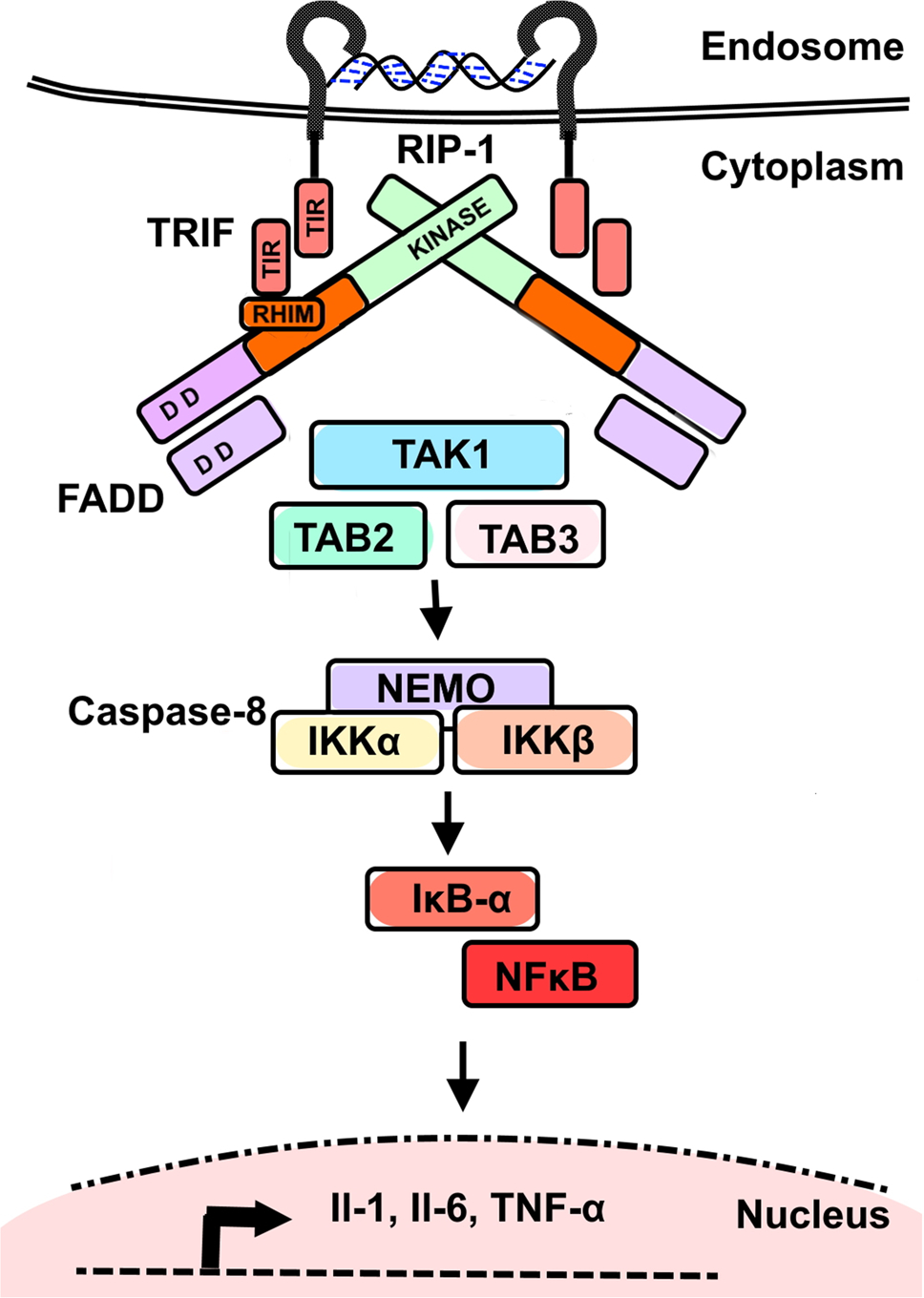
Endosomal TLR3-activation by dsRNA and TRIF-associated inflammatory pathway activation. Modified from Estornes et al., 2013
Interestingly, TLR3 activation is also capable of inducing an apoptotic response by means of an alternate route, in which, after activation with dsRNA, the cytoplasmic portion of TLR3, having a TIR domain, binds the TRIF adapter which has an RIP Homotypic Interaction Motif (RHIM) domain, allowing RHIM interaction with RIP1. RIP1 then recruits Fas Associated Via Death Domain (FADD) through homotypic interaction between its domain of death (DD), while FADD recruits caspasa-8 through its respective Death Effect Domain (DED) (Fig. 3). These molecules form a Caspase 8/FADD/FADD-like IL-1-converting enzyme-inhibitory protein (c-FLIP)/ RIP-1/ TRIF complex which is similar to the Death Induction Signaling Complex (DISC) that is capable of activating Caspase 3 and 9. This DISC-like complex is able to activate mechanisms such as DNA fragmentation by Caspase-activated Deoxyribonucleases (CAD), degradation of cytoskeleton by Rho 1 kinase (ROCK-1), and protein degradation [90, 92].
Fig. 3.
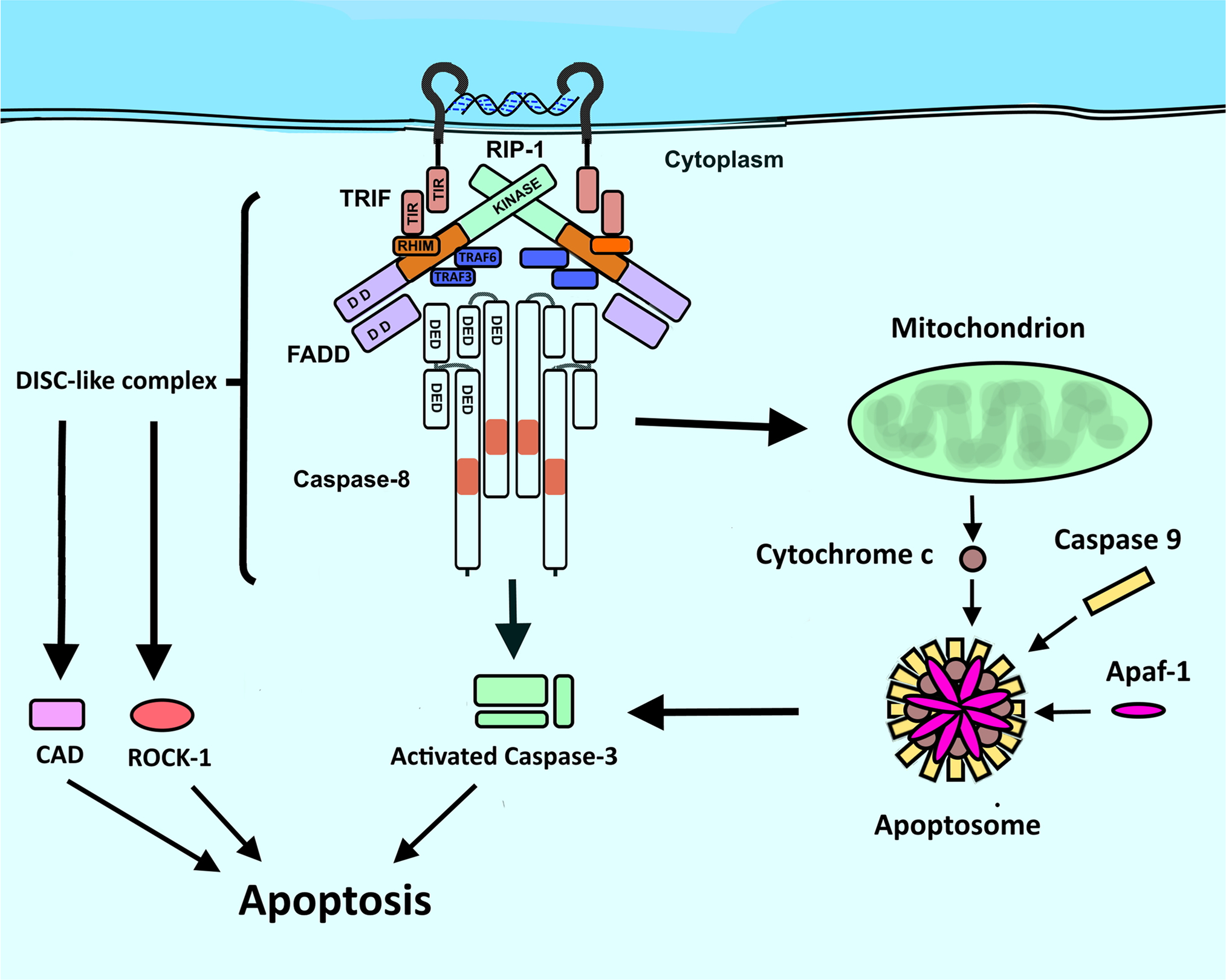
TRIF-associated apoptosis induction via Death Induction Signaling-like complex formation in TLR3 activation. Modified from Estornes et al., 2013
In this context, PCa immunotherapy strategies have been developed consisting of the stimulation of an immune component which induces a pro-apoptotic response based on TLR activation with their synthetic ligands [93].
Unlike the stimulation of TLR4, TLR6 TLR7, TLR8 and TLR9 in cancer cells, which induces a pro-tumor effect, TLR3 and TLR5 stimulation with synthetic ligands has been shown to have anti-tumor effects on different cancer models. Therefore, these latter receptors have been proposed as targets for the treatment of different types of tumors [90, 94].
The TLR5 agonist, Entolimod (also known as CBLB502), corresponds to a synthetic analogue of its natural ligand (flagellin), which activates a Myd88-dependent signaling pathway that induces antitumor effects mediated by the synthesis of pro-inflammatory cytokines. Entolimod has been used in phase I clinical studies [95]. Synthetic TLR3 ligands, such as Polyinosinic:polycytidylic acid (PIC) and Polyadenylic:polyuridylic acid (PAU) are analogues of dsRNA and activate inflammatory and anti-tumor apoptotic responses through the TRIF-pathway. PIC has pharmaceutical names such as Rintatolimod and Hiltonol, which have a formulation including cabroxymethylcellulose and poly-L-lysine as stabilizing agents [96]. Rintatolimod and Hiltonol are in phase II clinical studies, while PAU is still in preclinical phase I studies [97].
TLR3 Agonist, Polyinosinic:Polycytidylic Acid (PIC)
Drugs used as TLR3 agonists (dsRNA) have been proposed for cancer therapy in breast, prostate, bladder, and other tissues, since they induce pro-apoptotic and pro-inflammatory effects through an Myd88-independent signaling pathway. TLR3 stimulation activates the TRIF adaptor protein, which allows IKK protein to be released and activate NF-KB transcription factor triggering, in turn, other transcription factors activation such as Interferon Regulation Factor 3 and 7 (IRF3 and IRF7), as well as the activation of Signal And Transducer Activator Of Transcription 1 (STAT1) [98].
It is important to point out that independently of TLR3, other cytoplasmic receptors may recognize dsRNA in the form of a PIC agonist, such as: dsRNA-dependent kinase protein (PKR), retinoic acid-inducible gene product (RIG-I), or gene 5 product associated with melanoma differentiation (MDA 5). These intermediaries can induce pro-apoptotic states by activating the same protein kinases and transcription factors that are activated through the activation of TLR3 [99]. Importantly, among the proposals to improve the interaction of PIC in cells with sufficient expression levels of TLR3, the administration of PIC through vehicles such as liposomes seems to be promising [100].
PIC +9cRA to Treat PCa
In 2014, Galli and colleagues described an interesting synergic anti-tumor pathway based on activation of TLR3 by PIC combined with the use of 9cRA, which induces activation of both an apoptotic cellular response and SC differentiation. According to this study, the anti-tumor effect of this combination is achieved by the activation of a pathway through which PIC increases the expression of microRNAs −29b, −29c, −148b and − 152 that target DNMTs (DNMT1, DNMT3a and DNMT3b) mRNAs, thus inhibiting their translation. This event is indirectly responsible for the demethylation of RARβ promoter and therefore induces its reexpression in DU-145 human prostate cancer cells (Fig. 4) [101]. These same results were described for prostatic mouse cells (TRAMP) and metastatic pleural effusion breast cancer cells (MDA-MB-231). What is more relevant in this antitumor pathway, is the synergistic action of PIC and 9cRA by inducing the expression of RARβ thus making CSCs and CSCLs susceptible to the retinoid-induced differentiation effect, while PIC mainly induces apoptosis in differentiated tumor cells.
Fig. 4.

PIC +9cRA TLR3-associated anti-cancer pathway. The synergistic pathway begins with the recognition of PIC by its receptor Toll-Like receptor 3 (TLR3) within and endosome, where it triggers responses associated with inflammation, apoptosis and indirect induction of RARβ expression. The other part of the pathway implies that 9cRA in turn interacts with retinoic acid receptors alpha, beta and gamma (RAR α, β, γ) and retinoid x receptors (RXR) to induce the expression of genes associated with cell differentiation, apoptosis and TLR3 expression
Interestingly, 9cRA was also shown to indirectly increase the expression of TLR3, whose promoter contains a sequence of interferon binding/stimulation response element (ISRE/IRF-E) and a binding site for the signal transducer protein and transcription activator 1 (STAT1) [102]. This effect was described in breast cancer mammary gland cell lines Hs578T, H3396, T47D and ZR-75-1 (BT-474 mammary gland control cell line) in which the exposure to 9cRA (1 μM) for 48 hours resulted in the expression and activity of transcription factors such as IRF1 and STAT1 in cancer cells, thus leading 9cRA to activate the TLR3-promoting region for transcription [102].
Further evidence of the effectiveness of 9cRA + PIC as anti-tumoral therapy was shown in prostate cancer cell line (LNCaP) and involved its ability to inhibit the cell cycle and induce apoptosis [103] through the interaction of TIR domain with Protein Kinase C alfa (PKC-α) and the activation of C-Jun N-terminal kinases (JNK) and mitogen-activated protein kinases (p38), leading to formation of a complex with caspase 8 [92].
Therefore, it is strongly suggested that the 9cRA + PIC combination promotes cell differentiation and induces apoptosis in CSCs and CSCLs, making this a promising strategy in cancers which are resistant to conventional cancer chemotherapy. In addition, administration of PIC may be directed to a specific site, to some extent, by subcutaneous administration, and thus decreases possible collateral damage to healthy tissues [104].
Therefore, the use of TLR3 ligands alone or in combination with retinoids is promising to treat PCa and other tumors. However, it is important to note that TLR3 expression in some PCa patients may be significantly decreased [105]. This observation implies that immunotherapy based on TLR3 activation by PIC could be ineffective in this group of patients [15] In this context, it would be relevant to identify those individuals with PCa who show a decreased expression of TLR3 in order to predict the treatment effectiveness, as well as to identify the status of those factors that may modify TLR3 expression or function.
Modification of TLR3 gene and protein expression as a result of environmental factors seems to occur more often than expected. For example, in a study conducted by Chen et al., alveolar macrophages exposed to Cobalt II metal-organic frameworks (Co(II)-MOF) for 72 h lead to a decrease in the relative expression of TLR3 [106].
In an in vivo study, male mice C57BL/6 J exposed to sodium arsenite at concentrations ranging from 10 to 100 ppb for 5 weeks, showed altered expression of TRIF6 and, thus, a modified TLR3 response [107]. Studies comparing lung biopsies from smokers to those of non-smokers found that TLR3 expression was significantly lower in the smoker group [108].
Our group recently demonstrated that TLR3 expression is significantly decreased in prostate stem and epithelial cells malignantly transformed in vitro by 5 uM sodium arsenite. Interestingly, non-exposed (control) SCs express TLR3 at a significantly lower level that non-exposed (control) differentiated epithelial cells. Additionally, arsenite-transformed SCs were non-responsive to PIC alone or in combination with 9-cRA in terms of RARβ and SC differentiation markers expression compared with non-exposed prostate SCs. These transformed SCs showed decreased miR-29c, −148b and − 152 expressión which correlated with a significantly increased DNMT1 and DNMT3A expression. [109]
On the basis of such observations, besides the TLR3 determination, the identification and evaluation of cellular factors which may positively or negatively regulate TLR3 becomes relevant to determine the effectiveness of a proposed anti-cancer therapy. In this regard, it is known that interleukin 12 and IFN-α both have the ability to positively regulate TLR3 expression. However, the main regulator of TLR3 and other TLRs is the tumor suppressor protein p53 [110]. This protein regulates the expression of TLRs through binding to specific p53 response elements (P53RE) located at the promoter region of TLR encoding genes [111]. Therefore, p53 status is currently used as a predictor of a successful PIC-based anti-tumor therapy [112].
However, p53 expression or function is frequently altered in PCa, which can be the result of mutations, gene deletions, or gene silencing through promoter methylation [112]. Additionally, the protein Murine Double Minute (MDM2) expression, the main p53 negative regulator, is also significantly increased in PCa, thus decreasing the cell cycle regulatory activities of p53 [113]. These observations could suggest that use of PIC+9cRA as antineoplastic therapy might not be effective in individuals with decreased p53 expression since such condition will, in turn, lead to a decreased TLR3 expression. In this sense, the administration of MDM2 antagonists like Nutilin [114], which inhibits p53/MDM2 interaction leading to p53 stabilization, has been shown to work as a cell cycle regulator and inducer of p53-associated genes expression, including those encoding TLRs. The Nutilin mechanism of action involves the occupancy of MDM2 binding pocket in p53 protein [115].
Therefore, the use of p53 positive regulators arises as an alternative to improve the response to the combination of PIC and 9cRA through activation of TLR3 [116]. This would allow TLR3 to induce its pro-inflammatory, pro-apoptotic and cellular differentiation effects. It is worth mentioning that pro-inflammatory and proapoptotic responses in turn, leads to the increased expression of p53 [117].
In the above scenario, administration of Doxorubicin (Dox), another p53 inducer, could also indirectly improve the TLR3 expression or function through increased p53 expression. In this way the combination of doxorubicin with PIC could help synergistically to increase the pro-apoptotic response associated with TLR3 [111]. Administration of RA has also been observed to induce the expression of p53 [116]. Therefore, Dox, PIC and RA could be used in combination to modify p53 expression, to increase TLR3 expression and to induce CSCs and CSCL differentiation as an antineoplastic therapy in PCa patients.
As previously stated, it is relevant to predict an adequate response to anti-tumor treatment based on the use of PIC and 9cRA combination. In regard to p53 and its potential antitumor effects, Indian subjects chronically exposed to iAs showed significantly high methylation of the p53 gene suggesting possible gene silencing. These p53 methylation values were associated with the level of iAs in the exposed population [118]. Other studies have shown decreased p53 and Bax transcript expression in RWPE-1 prostate epithelial cells that were acutely exposed to low-level iAs (100 pg/mL) for 72 hours and chronically for 90 days, contributing to malignant transformation [47].
Unfortunately, however, it is estimated that approximately 20% of patients with PCa and 53% of patients with castration-resistant metastatic PCa, have mutations in TP53 [119].
In cases where p53 deletion or mutations are present, it becomes practically inadequate to use inducers of p53 like Dox, Nutilin PIC+RA. In such cases, autologous vaccine strategies have been proposed, where peripheral blood mononuclear cells (PBMC) are isolated and cultured with IL-4 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to generate dendritic cells (DC). In turn, those DCs may be transduced with a recombinant viral vector containing a wild type p53 sequence (rAd-p53). These DC-rAdp53 can activate cytotoxic T lymphocytes (CTL) to recognize and attack LNCaP prostate cancer cells (also infected with rAdp53). This strategy could be used for scenarios where p53 expression is compromised or has loss of function mutations [120].
In summary, the combination of PIC+9cRA is an effective alternative for PCa treatment since it can induce cellular differentiation of CSCs and CSCLs and a proapoptotic response in malignant cells. However, the effectiveness of this strategy will depend on the level of TLR3 expression and RARβ in the tumor, which in turn depends on p53 protein status, as the main TLR3 regulator. In patients who have a decreased expression of p53 or increased activity of their negative regulator MDM2, the antitumor pathway based on the activation of TLR3 could be reactivated through a p53 agonist(s) or an inhibition of MDM2 in a combination therapy. However, in those cases of p53 deletion or mutation, other innovative strategies such as autologous DC-rAdp53 vaccine to improve p53 expression and reactivate TLR3 antitumor pathway may be considered to treat PCa.
Note: Most of the information in the literature regarding the efficacy of TLR3 and RAR combination to treat PCa comes from in vitro studies with PCa cell lines expressing TLR3; to know how useful this combination may be to treat PCa, changes in the expression of TLR3 and RARβ in prostate adenocarcinoma cases (PACa) –the most common PCa type– were analyzed using OncoDB (http://oncodb.org/) [121] which provides differential gene expression data between tumor samples of the 33 cancer types contained in The Cancer Genome Atlas (TCGA) and matched healthy tissue. Comparison between 505 tumor and 52 healthy tissue samples showed that both TLR3 and RARβ are downregulated in PACa (TLR3, Log2 Fold Change: −1.16, FDR: 1.30*10−9; RARβ, Log2 Fold Change: −1.01, FDR: 3.80*10−10). To further analyze how TLR3 and RARβ contribute when differentiating between PACa and healthy groups, principal component analysis (PCA) was used. For this purpose, preprocessed TCGA RNA-seq data of 502 PACa tumor and 52 matched healthy tissue samples was retrieved from Rahman et al., 2015 [122] (GEO accession: GSE62944). Normalization of raw gene counts and PCA were conducted using DESeq2 [123]. To isolate the contribution of TLR3 and RARβ to sample grouping, 3 recently reported reference genes for cancer transcriptomic analyses (HNRNPL, PCBP1 and RER1; [124]) were used as background for the PCA together with TLR3 or RARβ. This simplified PCA approach revealed that 81% and 71% of the data variability can be explained by TLR3 and RARβ signal, respectively, when separating PACa tumor samples from healthy tissue, which is higher than the reference gene background alone (63%) and resembles the observed behavior when using other well-known PCa markers, such as PCA3 (96%) or MMP9 (86%).
Despite this situation, the use of PIC to activate the TLR3-anti-tumoral pathway is currently in phase II clinical trials, which suggests that even with a low TLR3 expression, the use of PIC alone or in combination with RA may be effective to treat PCa. However, as previously indicated, these observations support the need to perform analysis of TLR3 status in individual prostate tumor tissues before starting an immunotherapy based on TLR3 activation in PCa patients.
Cellular Rewiring, Phenotype Plasticity and Chemotherapy Resistance
It is relevant to consider that another important challenge for PCa therapy, which also may also be a source of new CSCs, is the phenomenon called cellular rewiring, commonly observed in cancer cells, which implies the acquisition of new properties of intracellular proteins leading to the reengineering of intracellular signaling pathways. The origins of this phenomenon are variated and include oncogenic fusions, mutations or amplifications of polypeptides promoting in this way malignancy, cellular phenotype plasticity and PCa therapy resistance [125].
Specifically, in PCa, high throughput methods have demonstrated that most patients with CRPC have alterations in the gene codifying for androgen receptor (AR) or in its signaling pathways, commonly in association with PTEN loss [126]. Such alterations in AR, include AR reactivation or increased expression, local steroid synthesis, ligand binding domain (LBD) mutation, modified intracellular signaling and alternative splicing of AR mRNA [127]. Classical studies carried out on LNCaP cells show that a single mutation at codon 868 (Thr to Ala) can lead to an AR that is able to bind different steroids and therefore activate different signaling pathways [128].
As known, the change from androgen-dependent (ADPC) to androgen independent (AIPC) state is a marker of a bad prognosis in PCa patients. Although AR is essential for tumor growth and cell survival in both ADPC and AIPC, its role in androgen independent cancer was not clear some years ago. In 2010 Wang and colleagues, using and androgen independent cell line LNCaP-abl obtained from long-term androgen deprivation culture of AIPC clinical samples, demonstrated that AR-regulated genes in AIPC were not the same as those regulated by androgen in ADPC. For example, in AIPC, AR directly increased the expression of M-phase cell cycle genes, mainly the gene UBE2C encoding the Ubiquitin-conjugating enzyme E2 C, which is required for the destruction of mitotic cyclins CDC20, and CDK1 in the cell cycle progression. They also analyzed epigenetic marks around M-phase AR-bound gene enhancers and found H3K4 methyl marks (transcription activation) and the recruitment of transcription factors such as FOXA1 at the AR target enhancer sequence, mainly in those sequences that positively regulate UBE2c gene expression [129]. This is a highly illustrative example of how an altered AR receptor produced after long-term androgen deprivation, leads to a new activated set of AR targets that induce cellular malignant transformation or increases the aggressiveness of cancer cells.
Loss of PTEN is associated with castration resistance through a cross-talking between PI3K and AR signaling, two apparently independent pathways. Pharmacological inhibition of PI3K or AKT (which are normally regulated by PTEN) increases AR expression through a mechanism involving the receptor of human tyrosine kinase EGFR/HER, a regulator of cell growth and proliferation [130, 131]. This was first described in a retrospective study conducted with PCa tissues from patients who had only radical prostatectomy, and patients who received the anti-androgen bicalutamide for 120 days before surgery. Authors indicated that treatment with the drug was associated with a significant reduction of PTEN levels which correlated with increased levels of p-AKT, EGFR and the protein HER2 [131]. Later, Carver et al., (2011), demonstrated by transcriptome analysis in mouse and human PCa, that the inhibition of PI3K pathway in PTEN (−) PCa, leads to the activation of AR through feedback signaling to the receptor tyrosine kinase HER2/HER3. Conversely, AR inhibition led to the AKT activation due to reduced levels of protein FKBP5, a molecular co-chaperone of the glucocorticoid receptor (GR complex), which has been demonstrated to have an inhibitory effect on this receptor signaling pathway. Therefore, these observations support the use of therapy targeting both pathways simultaneously.
As mentioned above, the AR constitutive activity in CRPC may also be the result of an aberrant alternative splicing which causes the production of an AR variant lacking the ligand binding domain (LBD) but conserving the DNA binding and amino-terminal transactivation domains. These variants create a great challenge for an effective anti-AR therapy. The more frequently expressed variant in prostate tumors is the AR-V7 which mRNA contains only the 1,2,3 AR exons, but lacks 4 to 8 exons. The produced protein has sixteen new amino acids at the carboxyterminal domain. Importantly, CRPC expressing AR-V7 are typically refractory to the second line ADT [129].
Although it is well known that AR and AR-V7 regulate many common genes, such as androgen signaling, it has also been demonstrated that AR-V7 and other AR variants are able to regulate the expression of unique target genes [127, 129, 132].
Recently Xia L et al., (2022) described that the mechanism of producing the AR-V7 involves the largest protein of the mRNAs polyadenylation complex, named cleavage and polyadenylation specific factor (CPSF1), and its negative regulators, the ubiquitin ligases complex SIAH1. Authors describe that an increased methylation of the adenosine 6 (m6A) of SIAH1 mRNA decreases its translation and leads to CPSF1 increased activity, which interacts with the spliceosome system. CPSF1 causes the production of AR-V7 due to the alternative use of the 3′ splice site next to cryptic exon 3 (CE3) causing the loss of exons 4 to 8, generating a truncated AR that lacks ligand binding domain, but retains its transactivation domain [133].
In a worse scenario of CRPC, other spliced variants of different proteins may also increase the transcriptional activity of AR gene targets (normal or aberrant). This is the case with the transmembrane protein called neuropilin-2, which specific spliced variant form a complex and stabilizes AR and nuclear pore proteins inducing the characteristic CPRC gene expression. Importantly, the inhibition of this protein leads to a sensitization to anti-AR therapies as enzalutamide [134].
On the other hand, an interesting relationship between AR and DNA repair has also been described. Polkinghorn et (2013), using a clinically validated xenograft model of CRPC expressing AR (LNCaP-AR) and human primary prostate cancer tumors (which are AR signaling-dependent), demonstrated that the expression of DNA repair genes is significantly enriched. Authors described a signature of 144 DNA repair genes that were significantly associated with canonical AR output in CR cells. With an in vitro model, they showed that among these 144 genes, AR directly modulates the expression of 32 of them, by binding to enhancers of gene promoters in classical consensus DNA sequences, and their expression is increased after exposure to androgens. These results suggest that androgens induce DNA repair through the regulatory effect of AR as transcription factor. Authors further demonstrated that, by accelerating radiation-induced damaged DNA repair, AR can promote PCa radio resistance in androgen sensitive cancer. Such observations are the base of ADT and radiation combined therapy [135].
Importantly, AR-V7 is also responsible for epithelial-mesenchymal transition (EMT) and the expression of SC signature genes a property observed after ADT, named cellular plasticity which is proposed as a critical event for cancer progression, metastasis, and therapy resistance in this type of neoplasia [136] (Fig 5).
Fig. 5.
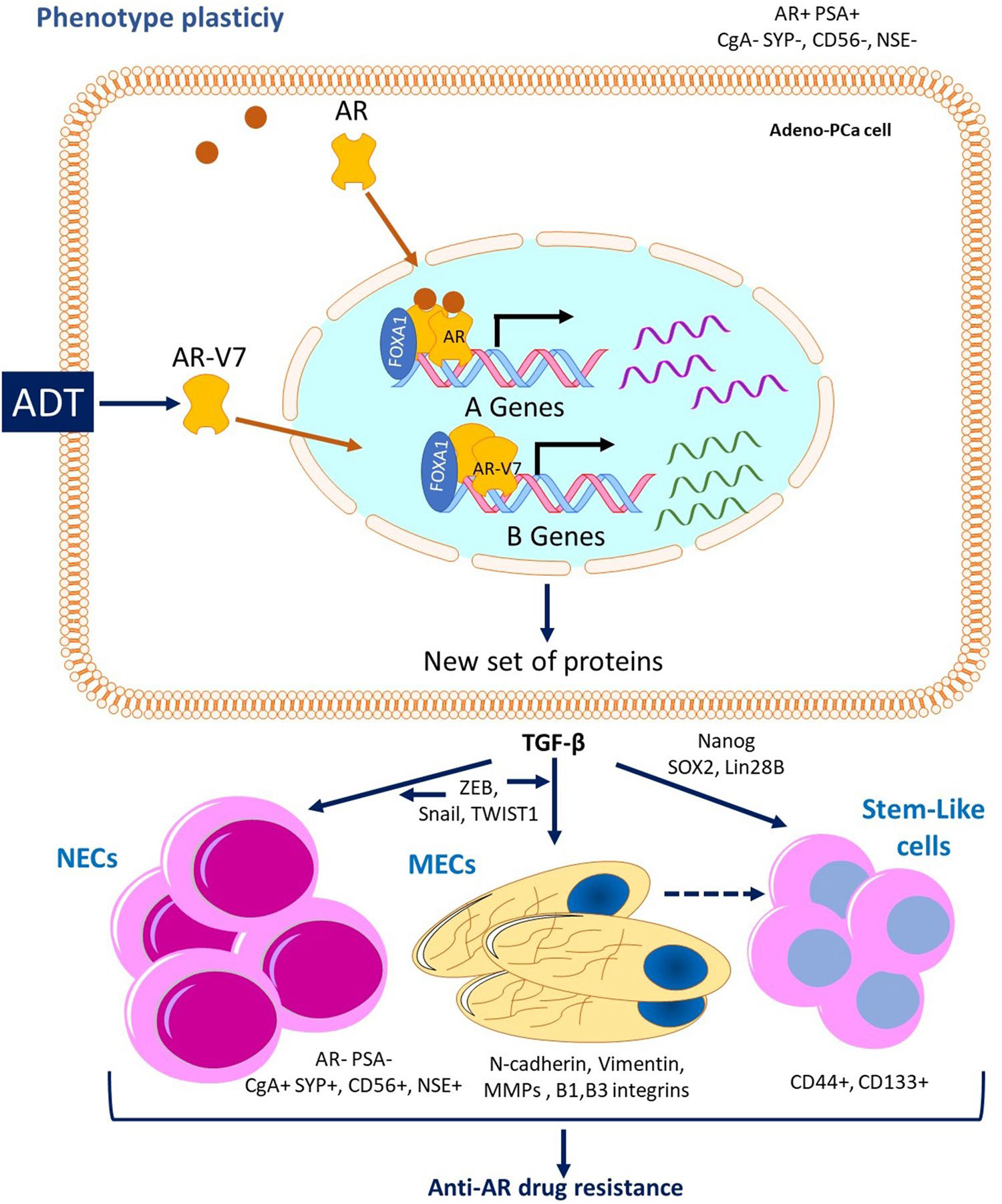
Anti-AR drug resistance caused by phenotype plasticity. Because of ADT, AR-V7 is produced by mRNA abnormal alternative splicing, leading to a protein that lacks ligand binding domain, but conserves DNA binding and transactivation domains. Androgen-independent AR-V7 activates the transcription of a new set of genes producing epithelial plasticity, were TGF-β is a critical factor inducing the production of neuroendocrine, mesenchymal and stem cells showing resistance to anti-AR therapy
Kong et al., (2015) showed that the AR-v7 transfection of different cell lines, lead not only to the expression of EMT genes such as fibronectin, but also to the expression of SC markers such as Nanog and Lin28B. In vitro androgen deprivation also led to the increased expression of SC factors Lin28B, Nanog and SOX2, and the increased expression of EMT markers ZEB1, N-cadherin and Vimentin. Moreover, the over-expression of AR-V7, but not the full-length AR, lead to the promotion of cell migration and clonogenicity and to an enhanced prostasphere formation. These observations suggest that AR variants contribute to PCa progression through the induction of EMT and the acquisition of SC characteristics [137].
Indeed, over-expression or reactivation of stemness factors due to cell pathways rewiring may induce cellular plasticity from one cell type to SCs. Studies from Tang and colleagues, have identified increased Nanog expression as a key factor contributing to cell dedifferentiation and CRPC since induction of Nanog expression in LNCaP cells gradually reprogrammed the cells into androgen insensitive CRPC cells with SC characteristics through four steps: 1) Competitive Nanog binding to AR/FOXA1 DNA sites, inhibiting AR-mediated differentiation; 2) activating transcription of stemness genes as ABCG2 and WNT5A in association with AR/FOXA1; 3) Nanog mediated increased transcription of genes involved in migration, invasion and metastasis; and 4) transactivation of genes associated with proliferation and cell cycle progression [136].
The high relevance of Nanog in this process is supported by the fact that its inhibition, or the inhibition SOX2 that regulates Nanog, could lead to depletion of prostate SCs [138, 139]. Therefore, in principle, compounds or strategies that regulate the stemness-associated pathways such as Nanog, Sox2, and Wnt/β-catenin could be used together with anti-AR therapies to target SCs.
On the other hand, prostate tumors may also switch lineage from adenocarcinomas to neuroendocrine-like tumors. Typical neuroendocrine markers are chromogranin A (CgA), synaptophysin (SYP), neuron specific enolase (NSE), and CD56 (Parimi et al., 2014), and are found mainly after starting the treatment with AR-inhibitors such as abiraterone/enzalutamide. This transition through one cell phenotype to another, is associated with abnormal gene expression pathways and drug targets, and the ability to invade other tissues and evade anti-tumor immune responses. Importantly, phenotype plasticity can induce AR splice variants production, and as previously mentioned, AR splice variants may reciprocally activate factors promoting epithelial plasticity. Among them, TGF-β is a critical factor, since it in turn activates a series of transcription factors that regulate epithelial plasticity. As was reviewed by Somarelli and colleagues (2020), the master regulators of EMT and NEPC are the Snail/ZEB family members as well as TWIST1 and the WNT/β catenin pathway, all of which are also implicated in therapy resistance [132].
Therefore, there is a big spectrum of cell phenotypes residing in PCa tumors, some expressing AR and/or AR variants, and others expressing neuroendocrine mesenchymal and/or stem cells phenotype. These different phenotypes can have different sensitivities to antitumor drugs, thus presenting a huge challenge for therapy; however, all these three cell types do express TLR3 [140], therefore they also are a possible target for PIC + RA immunotherapy.
Together, all this knowledge supports the individual characterization of tumor cells to find the correct combination of drugs and strategies to successfully treat PCa.
Final Thoughts
The study of CSCs as a central cell type in carcinogenesis has allowed for the identification of these cells as responsible for generating aggressive cancer phenotypes, tissue invasion and could alter the effectiveness of the current pharmacologic therapies. This has also led to the identification of characteristics permitted to improve therapeutic strategies based on biological markers of the CSC subpopulations. Consequently, there are great opportunities for development of new strategies focused on eliminating these cells.
Other factors that could be used to enrich antineoplastic treatment are the concomitant administration of specific immunotherapy of markers such as MDR. In this regard, new approaches have been shown to be efficient by inducing CSC differentiation, thus making them responsive for conventional therapies, such as the use of combination of TLR3 agonist and retinoids. Despite this, new challenges arise in the optimization of these concomitant therapies.
Different factors, cellular, genetic, and environmental, can influence the efficiency of these therapies, so determining the status of these molecules, their genes and the factors that modulate them, becomes critical before starting a chemotherapy and immunotherapy and this aspect represents another new area of opportunity for science.
Acknowledgements
Authors acknowledge the contribution of Jesús Gómez Montalvo, member of the RNA Metabolism and Extracellular Vesicles Consortium, National Institute of Genomic Medicine, (INMEGEN), Mexico City, for the rigorous analysis done from various databases to determine the expression of TLR3 and RARβ in clinical samples from patients with PCa.
Footnotes
Conflict of Interest The authors declare that they have no conflicts of interest.
Declarations
Ethics Approval Not applicable.
Consent to Participate The authors declare that they have given their consent to participate in the preparation of this article.
Consent for Publication The authors declare that they have given their consent for this article to be published.
Code Availability Not applicable.
Data Availability
Not applicable.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, & Bray F (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians, 71(3), 209–249. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. (2018). Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. International Journal of Cancer, 144(9), 1941–1953. [DOI] [PubMed] [Google Scholar]
- 3.American Cancer Society (2022). 2022-cancer-facts-and-figures. Atlanta: American Cancer Society. Retrieved from https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2022.html [Google Scholar]
- 4.Ferlay J, Shin HR, Bray F, et al. (2010). Cancer incidence and mortality worldwide: IARC CancerBase; No. 10. [Google Scholar]
- 5.P A-H. (2009). Diagnóstico y Tratamiento del Cáncer de Próstata en el Segundo y Tercer Nivel de Atención. Guía de Práctica Clínica GPC, 1–102. [Google Scholar]
- 6.Heidenreich A, Bellmunt J, Bolla M, Joniau S, Mason M, Matveev V, … Zattoni F (2011). EAU guidelines on prostate cancer. Part I: Screening, diagnosis, and treatment of clinically localised disease. Actas Urológicas Españolas (English Edition), 35(9), 501–514. 10.1016/j.acuroe.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 7.Heidenreich A, Aus G, Bolla M, Joniau S, Matveev VB, Schmid HP, & Zattoni F (2008). European Association of Urology EAU guidelines on prostate cancer. European Urology, 53(1), 68–80. 10.1016/j.eururo.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 8.Bluethmann SM, Wang M, Wasserman E, Chen C, Zaorsky NG, Hohl RJ, & McDonald AC (2020). Prostate cancer in Pennsylvania: The role of older age at diagnosis, aggressiveness, and environmental risk factors on treatment and mortality using data from the Pennsylvania Cancer Registry. Cancer Med, 9(10), 3623–3633. 10.1002/cam4.3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis CW, Qazi J, Hippe DS, et al. (2019). Patterns of distant metastases in 215 Merkelcell carcinoma patients: Implications for prognosisand surveillance. Cancer Medicine, 00, 1–9. 10.1002/cam4.2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butler SS, Muralidhar V, Zhao SG, Sanford NN, Franco I, Fullerton ZH, ... & Nguyen PL. (2020). Prostate cancer incidence across stage, NCCN risk groups, and age before and after USPSTF grade D recommendations against prostate-specific antigen screening in 2012. Cancer, 126(4), 717–724. [DOI] [PubMed] [Google Scholar]
- 11.DeSantis CE, Miller KD, Dale W, Mohile SG, Cohen HJ, Leach CR, ... & Siegel RL. (2019). Cancer statistics for adults aged 85 years and older, 2019. CA: a Cancer Journal for Clinicians, 69(6), 452–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyslop J, Paul A, Seddon J, & Hopper J (2008). Last updated: 15 December 2021. Prostate cancer: diagnosis and treatment, full guideline. National Institute for Health and Clinical Excellence. London. https://www.nice.org.uk/guidance/ng131 [Google Scholar]
- 13.Macías-Abraham C, del Valle-Pérez LO, Hernández-Ramírez P, & Ballester-Santovenia JM (2010). Características fenotípicas y funcionales de las células madre mesenquimales y endoteliales. Revista Cubana de Hematología, Inmunología y Hemoterapia, 26(4), 256–275. [Google Scholar]
- 14.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, … Stewart R (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318(5858), 1917–1920. [DOI] [PubMed] [Google Scholar]
- 15.Gu G, Yuan J, Wills M, & Kasper S (2007). Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Research, 67(10), 4807–4815. [DOI] [PubMed] [Google Scholar]
- 16.Hou Y, Chao Y, Tung H, Wang H, & Shan Y (2014). Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer, 120(17), 2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tokar EJ, Ancrile BB, Cunha GR, & Webber MM (2005). Stem/progenitor and intermediate cell types and the origin of human prostate cancer. Differentiation, 73(9–10), 463–473. [DOI] [PubMed] [Google Scholar]
- 18.Tokar EJ, Qu W, Liu J, Liu W, Webber MM, Phang JM, & Waalkes MP (2010). Arsenic-specific stem cell selection during malignant transformation. Journal of the National Cancer Institute, 102(9), 638–649. 10.1093/jnci/djq093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira S-M, García-Echeverría C, … Reddy VA (2009). The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proceedings of the National Academy of Sciences, 106(1), 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokar EJ, Diwan BA, & Waalkes MP (2010). Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environmental Health Perspectives, 118(1), 108–115. 10.1289/ehp.0901059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsao T, Beretov J, Ni J, Bai X, Bucci J, Graham P, & Li Y (2019). Cancer stem cells in prostate cancer radioresistance. Cancer Letters, 465(August), 94–104. 10.1016/j.canlet.2019.08.020 [DOI] [PubMed] [Google Scholar]
- 22.Kalantari E, Saadi FH, Asgari M, Shariftabrizi A, Roudi R, & Madjd Z (2017). Increased expression of ALDH1A1 in prostate Cancer is correlated with tumor aggressiveness: A tissue microarray study of Iranian patients. Applied Immunohistochemistry and Molecular Morphology, 25(8), 592–598. 10.1097/PAI.0000000000000343 [DOI] [PubMed] [Google Scholar]
- 23.Kalantari E, Asgari M, Nikpanah S, Salarieh N, Asadi Lari MH, & Madjd Z (2017). Co-expression of putative Cancer stem cell markers CD44 and CD133 in prostate carcinomas. Pathology and Oncology Research, 23(4), 793–802. 10.1007/s12253-016-0169-z [DOI] [PubMed] [Google Scholar]
- 24.Tu SM, & Lin SH (2012). Prostate cancer stem cells. Clinical Genitourinary Cancer, 10(2), 69–76. 10.1016/j.clgc.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez NE, Zhang Z, Madamanchi A, Boyd KL, O’Rear LD, Nashabi A, … Zutter MM (2011). The α2β1 integrin is a metastasis suppressor in mouse models and human cancer. Journal of Clinical Investigation, 121(1), 226–237. 10.1172/JCI42328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cojoc M, Mäbert K, Muders MH, & Dubrovska A (2015). A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Seminars in Cancer Biology, 31, 16–27. 10.1016/j.semcancer.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 27.Lee E, Yang J, Ku M, Kim NH, Park Y, Park CB, … Cheong JH (2015). Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death and Disease, 6, 1–10. 10.1038/cddis.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, & Lander ES (2009). Identification of selective inhibitors of Cancer stem cells by high-throughput screening. Cell, 138(4), 645–659. 10.1016/j.cell.2009.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu T, Xu F, Du X, Lai D, Liu T, Zhao Y, … Liu Z (2010). Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Molecular and Cellular Biochemistry, 340(1–2), 265–273. 10.1007/s11010-010-0426-5. [DOI] [PubMed] [Google Scholar]
- 30.Di Zazzo E, Galasso G, Giovannelli P, Di Donato M, Di Santi A, Cernera G, … Sinisi AA (2016). Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget, 7(1), 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins AT, Berry PA, Hyde C, Stower MJ, & Maitland NJ (2005). Prospective identification of tumorigenic prostate cancer stem cells. Cancer Research, 65(23), 10946–10951. 10.1158/0008-5472.CAN-05-2018 [DOI] [PubMed] [Google Scholar]
- 32.Signoretti S, Waltregny D, Dilks J, Isaac B, Lin D, Garraway L, … Loda M (2000). P63 is a prostate basal cell marker and is required for prostate development. American Journal of Pathology, 157(6), 1769–1775. 10.1016/S0002-9440(10)64814-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, … Witte ON (2016). N-Myc drives neuroendocrine prostate Cancer initiated from human prostate epithelial cells. Cancer Cell, 29(4), 536–547. 10.1016/j.ccell.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, … Daidone MG (2005). Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Research, 65(13), 5506–5511. [DOI] [PubMed] [Google Scholar]
- 35.Rajasekhar VK, Studer L, Gerald W, Socci ND, & Scher HI (2011). Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-κB signalling. Nature Communications, 2(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampayo RG, & Bissell MJ (2019). Cancer stem cells in breast and prostate: Fact or fiction? In Advances in cancer research (Vol. 144, pp. 315–341). Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borovski T, Felipe De Sousa EM, Vermeulen L, & Medema JP (2011). Cancer stem cell niche: The place to be. Cancer Research, 71(3), 634–639. [DOI] [PubMed] [Google Scholar]
- 38.Takebe N, Harris PJ, Warren RQ, & Ivy SP (2011). Targeting cancer stem cells by inhibiting Wnt, Notch, and hedgehog pathways. Nature Reviews Clinical oncology, 8(2), 97–106. [DOI] [PubMed] [Google Scholar]
- 39.Leao R, Domingos C, Figueiredo A, Hamilton R, Tabori U, & Castelo-Branco P (2017). Cancer stem cells in prostate cancer: Implications for targeted therapy. Urologia Internationalis, 99(2), 125–136. [DOI] [PubMed] [Google Scholar]
- 40.Hussain S, Lawrence MG, Taylor RA, Lo CYW, BioResource APC, Frydenberg M, … Risbridger GP (2012). Estrogen receptor β activation impairs prostatic regeneration by inducing apoptosis in murine and human stem/progenitor enriched cell populations. PLoS One, 7(7). 10.1371/journal.pone.0040732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patra SK, Patra A, Zhao H, & Dahiya R (2002). DNA methyltransferase and demethylase in human prostate cancer. Molecular Carcinogenesis, 33(3), 163–171. 10.1002/mc.10033 [DOI] [PubMed] [Google Scholar]
- 42.Oo AKK, Calle AS, Nair N, Mahmud H, Vaidyanath A, Yamauchi J, et al. (2018). Up-regulation of PI 3-kinases and the activation of PI3K-Akt signaling pathway in Cancer stem-like cells through DNA Hypomethylation mediated by the Cancer microenvironment. Translational Oncology, 11(3), 653–663. 10.1016/j.tranon.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain AL, Sidana A, Maruf M, Sugano D, Calio B, Wood BJ, & Pinto PA (2019). Analyzing the current practice patterns and views among urologists regarding focal therapy for prostate cancer. In Urologic Oncology: Seminars and Original Investigations (Vol. 37, pp. 182–1e1). Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nyberg T, Frost D, Barrowdale D, Evans DG, Bancroft E, Adlard J, … Brewer C (2020). Prostate cancer risks for male BRCA1 and BRCA2 mutation carriers: A prospective cohort study. European Urology, 77(1), 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watts EL, Goldacre R, Key TJ, Allen NE, Travis RC, & Perez-Cornago A (2020). Hormone-related diseases and prostate cancer: An English national record linkage study. International Journal of Cancer, 147(3), 803–810. 10.1002/ijc.32808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng X, Song M, Preston MA, Ma W, Hu Y, Pernar CH, … Zhang Y (2020). The association of diabetes with risk of prostate cancer defined by clinical and molecular features. British Journal of Cancer, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh KP, Kumari R, Treas J, & Dumond JW (2011). Chronic exposure to arsenic causes increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor a (mtTFA) in human prostate epithelial cells. Chemical Research in Toxicology, 24(3), 340–349. 10.1021/tx1003112 [DOI] [PubMed] [Google Scholar]
- 48.Roh T, Lynch CF, Weyer P, Wang K, Kelly KM, & Ludewig G (2017). Low-level arsenic exposure from drinking water is associated with prostate cancer in Iowa. Environmental Research, 159, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carey M, Meharg C, Williams P, Marwa E, Jiujin X, Farias JG, … Meharg AA (2019). Global sourcing of low-inorganic arsenic Rice grain. Exposure and Health. 10.1007/s12403-019-00330-y. [DOI] [Google Scholar]
- 50.Benbrahim-Tallaa L, & Waalkes MP (2008). Inorganic arsenic and human prostate cancer. Environmental Health Perspectives, 116(2), 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen C-J, & Wang C-J (1990). Ecological correlation between arsenic level in well water and age-adjusted mortality from malignant neoplasms. Cancer Research, 50(17), 5470–5474. [PubMed] [Google Scholar]
- 52.Ahn J, Boroje IJ, Ferdosi H, Kramer ZJ, & Lamm SH (2020). Prostate Cancer incidence in US counties and low levels of arsenic in drinking water. International Journal of Environmental Research and Public Health, 17(3), 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tokar EJ, Qu W, & Waalkes MP (2011). Arsenic, stem cells, and the developmental basis of adult cancer. Toxicological Sciences, 120(SUPPL.1), 192–203. 10.1093/toxsci/kfq342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Achanzar WE, Brambila EM, Diwan BA, Webber MM, & Waalkes MP (2002). Inorganic arsenite-induced malignant transformation of human prostate epithelial cells. Journal of the National Cancer Institute, 94(24), 1888–1891. [DOI] [PubMed] [Google Scholar]
- 55.Ngalame NNO, Tokar EJ, Person RJ, & Waalkes MP (2014). Silencing KRAS overexpression in arsenic-transformed prostate epithelial and stem cells partially mitigates malignant phenotype. Toxicological Sciences, 142(2), 489–496. 10.1093/toxsci/kfu201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomsen FB, Sandin F, Garmo H, Lissbrant IF, Ahlgren G, Van Hemelrijck M, … Stattin P (2017). Gonadotropin-releasing hormone agonists, orchiectomy, and risk of cardiovascular disease: Semi-ecologic, nationwide, population-based study. European Urology, 72(6), 920–928. [DOI] [PubMed] [Google Scholar]
- 57.Pajonk F, Vlashi E, & McBride WH (2010). Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells, 28(4), 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Venkitaraman R, Lorente D, Murthy V, Thomas K, Parker L, Ahiabor R, … Parker C (2015). A randomised phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. European Urology, 67(4), 673–679. [DOI] [PubMed] [Google Scholar]
- 59.Lu X, Yang F, Chen D, Zhao Q, Chen D, Ping H, & Xing N (2020). Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. International Journal of Biological Sciences, 16(7), 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cha H-R, Lee JH, & Ponnazhagan S (2020). Revisiting immunotherapy: A focus on prostate cancer. Cancer Research, 80(8), 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monjazeb AM, Hsiao H-H, Sckisel GD, & Murphy WJ (2012). The role of antigen-specific and non-specific immunotherapy in the treatment of cancer. Journal of Immunotoxicology, 9(3), 248–258. [DOI] [PubMed] [Google Scholar]
- 62.Graff JN, & Chamberlain ED (2015). Sipuleucel-T in the treatment of prostate cancer: An evidence-based review of its place in therapy. Core Evidence, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bilusic M, Heery C, & Madan RA (2011). Immunotherapy in prostate cancer: Emerging strategies against a formidable foe. Vaccine, 29(38), 6485–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel PH, & Kockler DR (2008). Sipuleucel-T: A vaccine for metastatic, asymptomatic, androgen-independent prostate cancer. Annals of Pharmacotherapy, 42(1), 91–98. [DOI] [PubMed] [Google Scholar]
- 65.Kawasaki BT, Mistree T, Hurt EM, Kalathur M, & Farrar WL (2007). Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochemical and Biophysical Research Communications, 364(4), 778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harashima N, Inao T, Imamura R, Okano S, Suda T, & Harada M (2012). Roles of the PI3K/Akt pathway and autophagy in TLR3 signaling-induced apoptosis and growth arrest of human prostate cancer cells. Cancer Immunology, Immunotherapy, 61(5), 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stewart CF, Leggas M, Schuetz JD, Panetta JC, Cheshire PJ, Peterson J, … Germain GS (2004). Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Research, 64(20), 7491–7499. [DOI] [PubMed] [Google Scholar]
- 68.Shukla S, Ohnuma S, Ambudkar V, & S. (2011). Improving Cancer chemotherapy with modulators of ABC drug transporters. Current Drug Targets, 12(5), 621–630. 10.2174/138945011795378540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du F-Y, Zhou Q-F, Sun W-J, & Chen G-L (2019). Targeting cancer stem cells in drug discovery: Current state and future perspectives. World Journal of Stem Cells, 11(7), 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Corato R, Gazeau F, Le Visage C, Fayol D, Levitz P, Lux F, … Wilhelm C (2013). High-resolution cellular MRI: Gadolinium and iron oxide nanoparticles for in-depth dual-cell imaging of engineered tissue constructs. ACS Nano, 7(9), 7500–7512. 10.1021/nn401095p. [DOI] [PubMed] [Google Scholar]
- 71.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, … Michelakis ED (2007). A mitochondria-K+ channel Axis is suppressed in Cancer and its normalization promotes apoptosis and inhibits Cancer growth. Cancer Cell, 11(1), 37–51. 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 72.Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, & Menendez JA (2011). Autophagy positively regulates the CD44+CD24−/low breast cancer stem-like phenotype. Cell Cycle, 10(22), 3871–3885. 10.4161/cc.10.22.17976 [DOI] [PubMed] [Google Scholar]
- 73.Kim K-Y, Yu S-N, Lee S-Y, Chun S-S, Choi Y-L, Park Y-M, … Ahn S-C (2011). Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochemical and Biophysical Research Communications, 413(1), 80–86. [DOI] [PubMed] [Google Scholar]
- 74.Song KS, Kim JS, Yun EJ, Kim YR, Seo KS, Park JH, … Hwang BD (2008). Rottlerin induces autophagy and apoptotic cell death through a PKC-δ-independent pathway in HT1080 human fibrosarcoma cells: The protective role of autophagy in apoptosis. Autophagy, 4(5), 650–658. 10.4161/auto.6057. [DOI] [PubMed] [Google Scholar]
- 75.Singh BN, Kumar D, Shankar S, & Srivastava RK (2012). Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochemical Pharmacology, 84(9), 1154–1163. [DOI] [PubMed] [Google Scholar]
- 76.Kim HN, Kim DH, Kim EH, Lee MH, Kundu JK, Na HK, … Surh YJ (2014). Sulforaphane inhibits phorbol ester-stimulated IKK-NF-κB signaling and COX-2 expression in human mammary epithelial cells by targeting NF-κB activating kinase and ERK. Cancer Letters, 351(1), 41–49. 10.1016/j.canlet.2014.03.037. [DOI] [PubMed] [Google Scholar]
- 77.Urvalek AM, & Gudas LJ (2014). Retinoic acid and histone deacetylases regulate epigenetic changes in embryonic stem cells. Journal of Biological Chemistry, 289(28), 19519–19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taddei A, Roche D, Bickmore WA, & Almouzni G (2005). The effects of histone deacetylase inhibitors on heterochromatin: Implications for anticancer therapy? EMBO Reports, 6(6), 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fritz K, Tiplica GS, Salavastru C, & Onder M (2013). Alitretinoin und off-label-use. Hautarzt, 64(10), 748–751. 10.1007/s00105-013-2593-2 [DOI] [PubMed] [Google Scholar]
- 80.Vahlquist A, & Rollman O (1987). Clinical pharmacology of 3 generations of retinoids. Dermatology, 175(Suppl. 1), 20–27. [DOI] [PubMed] [Google Scholar]
- 81.Yoham AL, & Casadesus D (2020). Tretinoin. In StatPearls [Internet]. StatPearls Publishing. [PubMed] [Google Scholar]
- 82.Xiao J-H, Durand B, Chambon P, & Voorhees JJ (1995). Endogenous retinoic acid receptor (RAR)-retinoid X receptor (RXR) Heterodimers are the major functional forms regulating retinoid-responsive elements in adult human keratinocytes binding of ligands to rar only is sufficient for rar• rxr heterodimers to co. Journal of Biological Chemistry, 270(7), 3001–3011. [DOI] [PubMed] [Google Scholar]
- 83.Tomita A, Kiyoi H, & Naoe T (2013). Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (as 2 O 3) in acute promyelocytic leukemia. International Journal of Hematology, 97(6), 717–725. [DOI] [PubMed] [Google Scholar]
- 84.Allenby G, Janocha R, Kazmer S, Speck J, Grippo JF, & Levin AA (1994). Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors alpha, beta, and gamma. Retinoic acid receptor gamma binds all-trans-retinoic acid preferentially over 9-cis-retinoic acid. Journal of Biological Chemistry, 269(24), 16689–16695. [PubMed] [Google Scholar]
- 85.Bushue N, & Wan YJY (2010). Retinoid pathway and cancer therapeutics. Advanced Drug Delivery Reviews, 62(13), 1285–1298. 10.1016/j.addr.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu X, Chai S, Wang P, Zhang C, Yang Y, Yang Y, & Wang K (2015). Aldehyde dehydrogenases and cancer stem cells. Cancer Letters, 369(1), 50–57. 10.1016/j.canlet.2015.08.018 [DOI] [PubMed] [Google Scholar]
- 87.Kawasaki T, & Kawai T (2014). Toll-like receptor signaling pathways. Frontiers in Immunology, 5(SEP), 1–8. 10.3389/fimmu.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawai T, & Akira S (2006). TLR signaling. Cell Death & Differentiation, 13(5), 816–825. [DOI] [PubMed] [Google Scholar]
- 89.Heidarzadeh M, Roodbari F, Hassanpour M, Ahmadi M, Saberianpour S, & Rahbarghazi R (2020). Toll-like receptor bioactivity in endothelial progenitor cells. Cell and tissue Research, 1–8. [DOI] [PubMed] [Google Scholar]
- 90.Barton GM, & Medzhitov R (2003). Toll-like receptor signaling pathways. Science, 300(5625), 1524–1525. [DOI] [PubMed] [Google Scholar]
- 91.Premkumar V, Dey M, Dorn R, & Raskin I (2010). MyD88-dependent and independent pathways of toll-like receptors are engaged in biological activity of triptolide in ligand-stimulated macrophages. BMC Chemical Biology, 10. 10.1186/1472-6769-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Estornes Y, Micheau O, Renno T, & Lebecque S (2013). Dual role of TLR3 in inflammation and cancer cell apoptosis. World’s largest Science, Technology & Medicine Open Access book publisher, 247–270. [Google Scholar]
- 93.Adams S (2009). Toll-like receptor agonists in cancer therapy. Immunotherapy, 1(6), 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma S, Zhu L, Davoodi M, Harris-White M, Lee JM, St John M, … Dubinett S (2013). TLR3 agonists and proinflammatory antitumor activities. Expert Opinion on Therapeutic Targets, 17(5), 481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brackett CM, Kojouharov B, Veith J, Greene KF, Burdelya LG, Gollnick SO, … Gudkov AV (2016). Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8+ T-cell axis. Proceedings of the National Academy of Sciences, 113(7), E874–E883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheng Y, & Xu F (2010). Anticancer function of polyinosinic-polycytidylic acid. Cancer Biology & Therapy, 10(12), 1219–1223. [DOI] [PubMed] [Google Scholar]
- 97.Zhao S, Zhang Y, Zhang Q, Wang F, & Zhang D (2014). Toll-like receptors and prostate cancer. Frontiers in Immunology, 5(JUL), 1–6. 10.3389/fimmu.2014.00352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kolla V, Lindner DJ, Weihua X, Borden EC, & Kalvakolanu DV (1996). Modulation of interferon (IFN)-inducible gene expression by retinoic acid up-regulation of STAT1 protein in IFN-unresponsive cells. Journal of Biological Chemistry, 271(18), 10508–10514. [DOI] [PubMed] [Google Scholar]
- 99.Liu J, Guo YM, Hirokawa M, Iwamoto K, Ubukawa K, Michishita Y, … Sawada K (2012). A synthetic double-stranded RNA, poly I: C, induces a rapid apoptosis of human CD34+ cells. Experimental Hematology, 40(4), 330–341. 10.1016/j.exphem.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 100.Colapicchioni V, Palchetti S, Pozzi D, Marini ES, Riccioli A, Ziparo E, … Caracciolo G (2015). Killing cancer cells using nanotechnology: Novel poly (I: C) loaded liposome–silica hybrid nanoparticles. Journal of Materials Chemistry B, 3(37), 7408–7416. [DOI] [PubMed] [Google Scholar]
- 101.Galli R, Paone A, Fabbri M, Zanesi N, Calore F, Cascione L, … Lovat F (2013). Toll-like receptor 3 (TLR3) activation induces microRNA-dependent reexpression of functional RARβ and tumor regression. Proceedings of the National Academy of Sciences, 110(24), 9812–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bernardo AR, Cosgaya JM, Aranda A, & Jiménez-Lara AM (2013). Synergy between RA and TLR3 promotes type I IFN- dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells. Cell Death and Disease, 4(1), 1–10. 10.1038/cddis.2013.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, … Riccioli A (2008). Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-α-dependent mechanism. Carcinogenesis, 29(7), 1334–1342. [DOI] [PubMed] [Google Scholar]
- 104.Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, & Vacchelli E (2020). Trial watch: TLR3 agonists in cancer therapy. OncoImmunology, 9(1), 1771143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gonza LO, & Gonza M (2011). Study of TLR3 , TLR4 , and TLR9 in prostate carcinomas and their association with biochemical recurrence, 217–226. 10.1007/s00262-010-0931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Y, Luo L, Zhang SG, Ding R, Zhou J, & Yang C (2020). A porous co(II)–MOF for selective C2H2/CO2 separation and treatment activity on virus-induced COPD via reducing tlr3 gene expression. Journal of Coordination Chemistry, 73(9), 1450–1463. 10.1080/00958972.2020.1786886 [DOI] [Google Scholar]
- 107.Kozul CD, Hampton TH, Davey JC, Gosse JA, Nomikos AP, Eisenhauer PL, … Hamilton JW (2009). Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung. Environmental Health Perspectives, 117(7), 1108–1115. 10.1289/ehp.0800199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Todt JC, Freeman CM, Brown JP, Sonstein J, Ames TM, McCubbrey AL, … Curtis JL (2013). Smoking decreases the response of human lung macrophages to double-stranded RNA by reducing TLR3 expression. Respiratory Research, 14(1), 1–15. 10.1186/1465-9921-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alvarado-Morales I, Olivares-Illana V, Arenas-Huertero C, Reynaga-Hernández E, Layseca-Espinosa E, Tokar EJ, & Escudero-Lourdes C (2021). Human prostate epithelial cells and prostate-derived stem cells malignantly transformed in vitro with sodium arsenite show impaired toll like receptor-3 (TLR3)-associated anti-tumor pathway. Toxicology Letters, 350, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K, … Li J-D (2008). p53 regulates toll-like receptor 3 expression and function in human epithelial cell lines. Molecular and Cellular Biology, 28(21), 6557–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, & Resnick MA (2011). The toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genetics, 7(3). 10.1371/journal.pgen.1001360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Strohmeyer D, Rössing C, Bauerfeind A, Kaufmann O, Schlechte H, Bartsch G, & Loening S (2000). Vascular endothelial growth factor and its correlation with angiogenesis and p53 expression in prostate cancer. The Prostate, 45(3), 216–224. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Z, Li M, Wang H, Agrawal S, & Zhang R (2003). Antisense therapy targeting MDM2 oncogene in prostate cancer: Effects on proliferation, apoptosis, multiple gene expression, and chemotherapy. Proceedings of the National Academy of Sciences of the United States of America, 100(20), 11636–11641. 10.1073/pnas.1934692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Logan IR, McNeill HV, Cook S, Lu X, Lunec J, & Robson CN (2007). Analysis of the MDM2 antagonist nutlin-3 in human prostate cancer cells. The Prostate, 67(8), 900–906. [DOI] [PubMed] [Google Scholar]
- 115.Menendez D, Shatz M, & Resnick MA (2013). Interactions between the tumor suppressor p53 and immune responses. Current Opinion in Oncology, 25(1), 85–92. [DOI] [PubMed] [Google Scholar]
- 116.Mrass P, Rendl M, Mildner M, Gruber F, Lengauer B, Ballaun C, … Tschachler E (2004). Retinoic acid increases the expression of p53 and proapoptotic caspases and sensitizes keratinocytes to apoptosis: A possible explanation for tumor preventive action of retinoids. Cancer Research, 64(18), 6542–6548. [DOI] [PubMed] [Google Scholar]
- 117.MacPherson CW, Shastri P, Mathieu O, Tompkins TA, & Burguière P (2017). Genome-wide immune modulation of TLR3-mediated inflammation in intestinal epithelial cells differs between single and multi-strain probiotic combination. PLoS One, 12(1), 1–18. 10.1371/journal.pone.0169847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chanda S, Dasgupta UB, GuhaMazumder D, Gupta M, Chaudhuri U, Lahiri S, … Chatterjee D (2006). DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicological Sciences, 89(2), 431–437. 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- 119.Quinn DI, Stricker PD, Kench JG, Grogan J, Haynes AM, Henshall SM, … Mahon KL (2019). P53 nuclear accumulation as an early Indicator of lethal prostate Cancer. British Journal of Cancer, 121(7), 578–583. 10.1038/s41416-019-0549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Saito H, Kitagawa K, Yoneda T, Fukui Y, Fujsawa M, Bautista D, & Shirakawa T (2017). Combination of p53-DC vaccine and rAd-p53 gene therapy induced CTLs cytotoxic against p53-deleted human prostate cancer cells in vitro. Cancer Gene Therapy, 24(7), 289–296. 10.1038/cgt.2017.21 [DOI] [PubMed] [Google Scholar]
- 121.Tang G, Cho M, & Wang X (2022). OncoDB: An interactive online database for analysis of gene expression and viral infection in cancer. Nucleic Acids Research, 50(D1), D1334–D1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rahman M, Jackson LK, Johnson WE, Li DY, Bild AH, & Piccolo SR (2015). Alternative preprocessing of RNA-sequencing data in the Cancer genome atlas leads to improved analysis results. Bioinformatics, 31(22), 3666–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Love MI, Huber W, & Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jo J, Choi S, Oh J, Lee S-G, Choi SY, Kim KK, & Park C (2019). Conventionally used reference genes are not outstanding for normalization of gene expression in human cancer research. BMC Bioinformatics, 20(10), 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carceles-Cordon M, Kelly WK, Gomella L, Knudsen KE, Rodriguez-Bravo V, & Domingo-Domenech J (2020). Cellular rewiring in lethal prostate cancer: The architect of drug resistance. Nature Reviews Urology. 10.1038/s41585-020-0298-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shen MM, & Abate-Shen C (2010). Molecular genetics of prostate cancer: New prospects for old challenges. Genes and Development. Cold Spring Harbor Laboratory Press. 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Basil P, Robertson MJ, Bingman WE, Dash AK, Krause WC, Shafi AA, … Weigel NL (2022). Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Scientific Reports, 12(1). 10.1038/s41598-022-09371-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Veldscholte J, Ris-Stalpers C, Kuiper G, Jenster G, Berrevoets C, Claassen E, … Mulder E (1990). A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochemical and Biophysical Research Communications, 173(2), 534–540. [DOI] [PubMed] [Google Scholar]
- 129.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, … Brown M (2009). Androgen receptor regulates a distinct transcription program in androgen-independent prostate Cancer. Cell, 138(2), 245–256. 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, … Scher H (2011). Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell, 19(5), 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Festuccia C, Gravina GL, Muzi P, Pomante R, Ventura L, Vessella RL, … Bologna M (2007). Bicalutamide increases phospho-Akt levels through Her2 in patients with prostate cancer. Endocrine-Related Cancer, 14(3), 601–611. 10.1677/ERC-07-0118. [DOI] [PubMed] [Google Scholar]
- 132.Somarelli JA, Armstrong AJ, Sheth MU, Ware KE, & Jolly MK (2020). Phenotypic plasticity and lineage switching in prostate cancer. In Phenotypic Switching: Implications in Biology and Medicine (pp. 591–615). Elsevier. 10.1016/B978-0-12-817996-3.00021-9. [DOI] [Google Scholar]
- 133.Xia L, Han Q, Duan X, Zhu Y, Pan J, Dong B, … Sha J (2022). m6A-induced repression of SIAH1 facilitates alternative splicing of androgen receptor variant 7 by regulating CPSF1. Molecular Therapy - Nucleic Acids, 28, 219–230. 10.1016/j.omtn.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dutta S, Polavaram NS, Islam R, Bhattacharya S, Bodas S, Mayr T, … Darehshouri A (2022). Neuropilin-2 regulates androgen-receptor transcriptional activity in advanced prostate cancer. Oncogene, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, … Sawyers CL (2013). Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discovery, 3(11), 1245–1253. 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tang DG (2022). Understanding and targeting prostate cancer cell heterogeneity and plasticity. Seminars in Cancer Biology. Academic Press. 10.1016/j.semcancer.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, & Sarkar FH (2015). Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate, 75(2), 161–174. 10.1002/pros.22901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang J, Chen M, Zhu Y, Dai X, Dang F, Ren J, … Gan W (2019). SPOP promotes nanog destruction to suppress stem cell traits and prostate cancer progression. Developmental Cell, 48(3), 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang X, Jin J, Wan F, Zhao L, Chu H, Chen C, … Teng H (2019). AMPK promotes SPOP-mediated NANOG degradation to regulate prostate cancer cell stemness. Developmental Cell, 48(3), 345–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bianchi F, Milione M, Casalini P, Centonze G, le Noci VM, Storti C, … Pastorino U (2019). Toll-like receptor 3 as a new marker to detect high risk early stage non-small-cell lung Cancer patients. Scientific Reports, 9(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.