

Abstract
Non-oncogenic cancer drivers often impinge on complex signals that create new addictions and vulnerabilities. PKCλ/ι suppresses tumorigenesis by blocking metabolic pathways that regulate fuel oxidation and create building blocks for the epigenetic control of cell differentiation. Reduced levels of PKCλ/ι unleash these pathways to promote tumorigenesis, but the simultaneous activation of the STING-driven interferon cascade prevents tumor initiation by triggering immunosurveillance mechanisms. However, depending on the context of other signaling pathways, such as WNT/β-Catenin or PKCζ, and timing, PKCλ/ι deletion can promote or inhibit tumorigenesis. In this review, we discuss in detail the molecular and cellular underpinnings of PKCλ/ι functions in cancer with the perspective of the crosstalk between metabolism and inflammation in the tumor microenvironment.
Keywords: Cancer, interferon, protein kinase C, autophagy, metabolism, tumor microenvironment
Contextual dual oncogene or tumor suppressor role of the atypical PKCs
The atypical Protein Kinase C (aPKC) family of kinases only includes two members: PKCζ and PKCλ/ι. The PRKCZ gene encodes PKC in humans (Prkcz in mice), while PKCλ/ι is encoded by the PRKCI gene in humans and the Prkci gene in mice [1, 2] (Figure 1). Both kinases have been shown to play critical roles in key cellular functions, including controlling cell polarity, inflammation, and tumor transformation (Table 1) [2–5]. Studies in mouse models and more recent analysis of human specimens and data sets have established PKCζ as a tumor suppressor [6–11]. Instead, PKCλ/ι has been shown to have putative cell-autonomous tumor-promoting functions in models of Ras-induced lung adenomas and BCR-ABL-induced leukemia [12–15]. However, it is clear now that PKCλ/ι involvement in cancer is more complex. Thus, PKCλ/ι also shows tumor suppressor activities in other contexts, such as therapy-resistant prostate cancer, mesenchymal colorectal cancer, and hepatocellular carcinomas [11, 16–19]. It should be kept in mind that while the implication of PKCλ/ι as a tumor promoter in lung cancer and leukemia is based on research focused on the tumor epithelial cell compartment, the role of PKCλ/ι as a tumor suppressor becomes even more apparent when considered a critical player in orchestrating transformation and immunological signals in the tumor microenvironment. In this review, we will provide an in-depth discussion integrating the role of PKCλ/ι in epigenetics and metabolism, together with new evidence linking PKCλ/ι to the control of autophagy and lysosomal biology as a critical mechanism of tumor promotion. We will also discuss the crosstalk with the interferon pathway, which is central in immunosurveillance to prevent tumor initiation. Finally, we will discuss these fundamental cell functions in three cancer paradigms: colorectal cancer, prostate cancer, and hepatocellular carcinoma.
Figure 1. aPKCs have a contextual oncogene or tumor suppressor role.
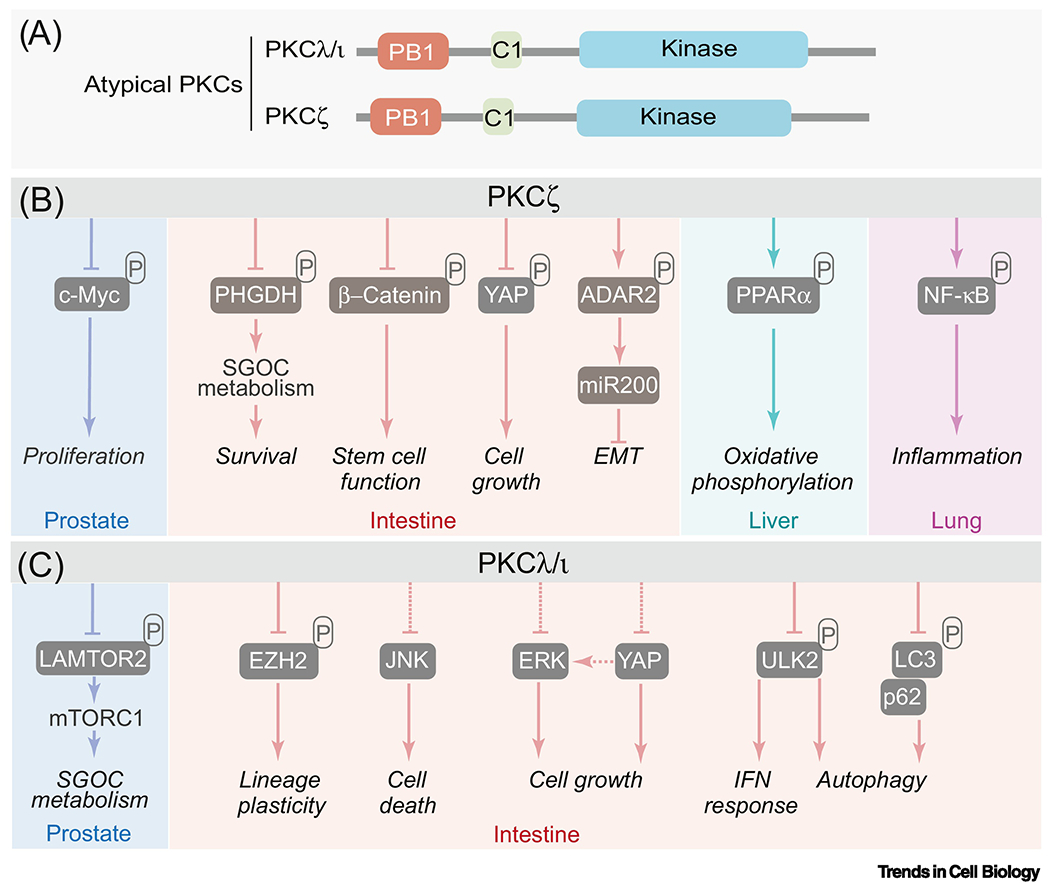
(A) The atypical protein kinase C subfamily is constituted by two members: PKCζ and PKCλ/ι. The aPKCs differ from the other members of the PKC family in that they lack functional C2 and the C1 domain does not bind diacylglycerol. The regulatory domain of the aPKCs also harbor a PB1 domain, a highly conserved protein interaction module that regulates their binding to other PB1-containing proteins such as the autophagy receptors and signaling adaptor p62.
(B-C) PKCζ (B) and PKCλ/ι (C) and their direct phosphorylation substrates and binding partners. The aPKCs have a contextual oncogene or tumor suppressor role in different types of cancer. They have pleiotropic biological functions mediated by phosphorylation of substrates or binding to selective interactors. SGOC: serine glycine one-carbon; EMT: epithelial mesenchymal transition; IFN: interferon.
Table 1.
Contextual-dependent role of the aPKCs and their binding partners in cancer
| PKC | Interactors | Organism/Cancer type | aPKC expression | Proposed role/Mechanism | References |
|---|---|---|---|---|---|
| PKCζ | PHGDH | Colorectal cancer | Reduced PKCζ | Metabolic reprogramming and tumor suppressor/Phosphorylation substrate | [6] |
| PKCζ | ADAR2 | Metastatic colorectal cancer | Reduced PKCζ | Tumor suppressor role, repressor of miR200 and EMT/Phosphorylation substrate | [10] |
| PKCζ | β-Catenin and YAP | Intestinal cells and colorectal cancer | Reduced PKCζ | Tumor suppressor role, repressor of stemness/Phosphorylation substrate | [9] |
| PKCζ | c-Myc | Prostate cancer | Reduced PKCζ | Tumor suppressor role/Phosphorylation substrate | [8] |
| PKCλ/ι | p62 | Hepatocellular carcinoma | Reduced PKCλ/ι | Tumor suppressor role/PB-1 adapter | [54, 55] |
| PKCλ/ι | LAMTOR2 | Neuroendocrine Prostate cancer | Reduced PKCλ/ι | Repressor of mTOR and lineage plasticity/Phosphorylation substrate | [19] |
| PKCλ/ι | EZH2 | Colorectal cancer | Reduced PKCλ/ι | Repressor of Paneth cell differentiation/Phosphorylation substrate | [17] |
| PKCλ/ι | ULK2 | Colorectal cancer | Reduced PKCλ/ι | Inactivation of interferon pathway and autophagy/Phosphorylation substrate | [18] |
| PKCλ/ι | SOX2 | Lung squamous carcinoma | Increased PKCλ/ι | Activation of stemness/Phosphorylation substrate | [12] |
Epigenetic and metabolic control of lineage plasticity
Previously published data had shown that the selective knock-out (KO) of PKCζ alone in intestinal epithelial cells (IECs) resulted in resistance to glucose deprivation by the upregulation of a glutamine-driven serine biosynthetic pathway [6]; it also showed that PKCζ-defective IECs were rendered with increased stemness features due to the activation of YAP/β-Catenin signaling [9]. Furthermore, when PKCζ was KO in intestinal cancer cells, these became more metastatic through a mechanism involving the downregulation of miR-200s [10]. However, none of these effects were sufficient to generate transformation spontaneously and consistently required additional oncogenic drivers, such as the inactivation of the Apc gene [6, 9, 17]. Strong evidence that PKCλ/ι was a tumor suppressor in intestinal tumorigenesis came from the analysis of mice in which it was KO in the intestinal epithelium together with PKCζ [11, 20]. Indeed, the conditional double PKCζ and PKCλ/ι deficiency in that mouse model resulted in spontaneous, aggressive serrated tumors in the small intestine and the colon without any additional oncogenic stimuli [11, 20].
PKCλ/ι modulates epigenetic mechanisms in cell differentiation
The tumor suppressor function of PKCλ/ι in intestinal carcinogenesis is rooted in its role in the regulation of the homeostasis of the cells of the intestinal secretory pathway. Thus, the initial evidence that PKCλ/ι could be involved in modulating cell differentiation and lineage plasticity stands from the observation that PKCλ/ι-deficient IECs showed a defective epithelial secretory pathway characterized by the accumulation of immature Paneth/goblet cell progenitors associated with a reduction in the number of mature Paneth cells, with the few remaining being mislocalized [17]. Paneth cells are normally located at the bottom of the crypt playing a central role in the metabolic homeostasis of intestinal stem cells and creating a sterile environment through the secretion of lysozyme and anti-microbial peptides. This is of paramount importance in an organ, like the intestine, that is in intimate contact with a massive microbiota component [21, 22]. This Paneth/goblet defect, together with possible alterations in other progenitors in the crypt, resulted in chronic intestinal inflammation that, nonetheless, was not sufficient to spontaneously drive tumorigenesis but that cooperated with the inactivation of Apc, or the activation of the β-Catenin pathways in a mutagen/inflammatory-driven mouse model system [17]. Therefore, the inactivation of either PKCζ or PKCλ/ι generated independent and distinct conditions conducive to tumorigenesis when this process was driven by the WNT pathway [6, 17]. The mechanism underlying the defects in the intestinal secretory lineage of PKCλ/ι-deficient IECs involves the upregulation of EZH2, a member of the Polycomb complex essential for the methylation of histones [17], which serves to repress gene expression through the increased content of H3K27-Me3 (Figure 2) [23]. Central transcription factors in this process are Atoh1 and Gfi1, whose levels are unleashed upon the inhibition of the Notch pathway [24–26]. Higher EZH2 in PKCλ/ι-deficient IECs represses the Atoh1 promoter activity by accumulating H3K27-Me3 [17]. EZH2 is a direct substrate of PKCλ/ι [17], which reduces its stability through a still-to-be-identified mechanism. In this regard, recent evidence demonstrates that EZH2 is subjected to a complex regulation by multiple post-translational modifications (PTMs) including phosphorylation, acetylation, methylation, ubiquitylation, O-GlcNAcylation, and sumolylation [27]. These PTMs impact many aspects of EZH2 biology including its stability, enzymatic activity, localization, binding to other subunits of polycomb repressive complex 2 (PRC2) [27]. Furthermore, the loss of PKCλ/ι in IECs results in JNK-induced cell death, which, together with the accumulation of EZH2 that represses the expression of critical transcriptional master regulators of the intestinal secretory pathway, leads to an inflammatory microenvironment conducive to cancer [17].
Figure 2. PKCλ/ι controls lineage cell differentiation through EZH2-dependent epigenetic reprogramming.
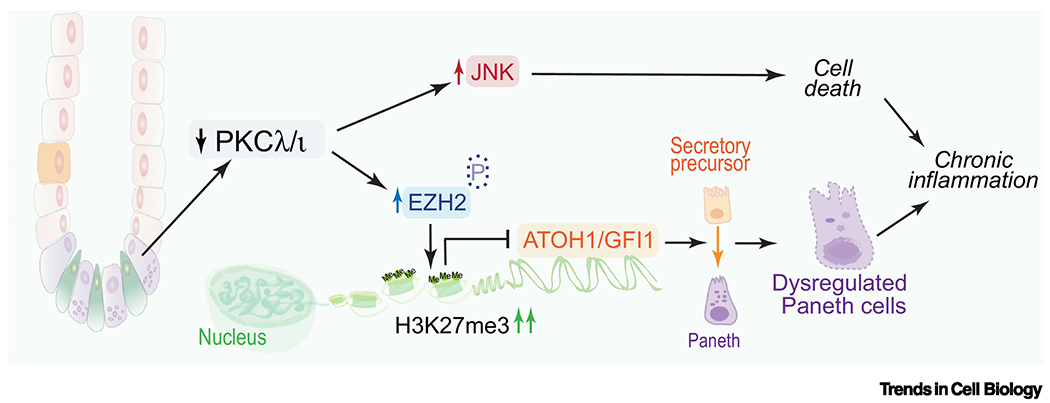
PKCλ/ι regulates lineage plasticity at least in part through the control of the stability of EZH2 by direct phosphorylation. The loss of EZH2 phosphorylation upon PKCλ/ι deficiency increases EZH2 and H3K27me3 levels, which represses the expression of two master regulators of Paneth cell differentiation, ATOH1, and GFI1. The downregulation of these two genes results in a disruption of Paneth cell homeostasis that leads to an inflammatory microenvironment conducive to cancer.
The connection of PKCλ/ι with the epigenetic control of lineage plasticity in the intestine echoes its role in castration-resistant prostate cancer. Thus, the loss of PKCλ/ι in PTEN-deficient prostate epithelial cells promotes tumorigenesis in vivo by inducing the reprogramming of tumor cells towards a partial basal cell lineage with neuroendocrine characteristics [19]. Studies in different cell lines and the bioinformatics and histological analyses of human prostate cancer datasets and tissue specimens established that this effect of PKCλ/ι deficiency is cell-autonomous and of human relevance [19]. Lineage plasticity is a developmental process of cell reprogramming and adaptive resistance employed by cells to evade therapeutic pressures[28]. This process plays a crucial role in tumor progression to metastasis and resistance to targeted therapies, and as such it is becoming a major clinical problem in many types of cancers, including prostate cancer [28–30]. While naive neuroendocrine prostate cancer (NEPC) is a sporadic disease, its incidence is rising due to therapy resistance to more powerful anti-androgen drugs [28, 31, 32]. Thus, although androgen deprivation therapy is very effective for the early management of prostate cancer [33], tumors become resistant, which leads to a lethal outcome known as castration-resistant prostate cancer (CRPC). However, although second-generation androgen receptor (AR) inhibitors have been successfully implemented as a second-line therapy [34, 35], the response to these potent drugs declines with time, and most patients again develop resistance [33, 36]. One of the most aggressive forms of resistant tumors is NEPC, which arises as a consequence of changes in the lineage of cancer cells from luminal (AR-positive) to basal (AR negative). This new stage of tumorigenesis is very aggressive and metastatic and displays markers of neuroendocrine differentiation [32, 37]. These tumors have small cell histological features, suggesting that similar cellular and molecular mechanisms might also be important in other types of cancers, like in lung small cell carcinomas [29, 30]. Future research should provide insights into the potential role of PKCλ/ι in this type of tumors, and in light with prior evidence suggesting a pro-tumorigenic role of this kinase in non-small cell lung carcinomas (NSCLC) [38]. The loss of PKCλ/ι in prostate epithelial cells induces changes in the DNA methylome through a mechanism dependent on DNA methyltransferase, consistent with the epigenetic modifications underlying lineage reprogramming in CRPC (Figure 3) [19]. This evidence suggests that inhibition of DNA methylation might reverse the basal/neuroendocrine state of CRPC cells and promote the transition to a more luminal phenotype, which would restore sensitivity to androgen-deprivation therapy [39]. Collectively, all these studies lead to the conclusion that PKCλ/ι is a novel and critical regulator of the epigenetic control of lineage cell differentiation both in the intestinal and prostate epithelia with dire results when it is lost or inactivated.
Figure 3. PKCλ/ι regulates the crosstalk between epigenetic reprogramming and metabolism.
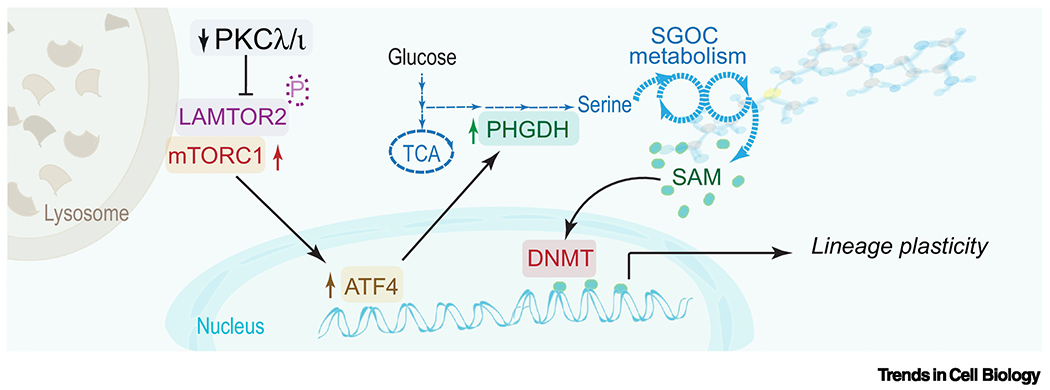
The loss of PKCλ/ι results in the upregulation of serine biosynthesis through a mTORC1/ATF4-driven pathway. PKCλ/ι represses mTORC1 activation by phosphorylation of LAMTOR2, a component of the Ragulator complex that anchors mTORC1 to the lysosome membrane, enhancing its activity. Loss of PKCλ/ι activates mTORC1 to drive an ATF4-dependent gene transcription program resulting in the upregulation of the serine biosynthetic pathway that is key for synthesizing S-adenosyl methionine (SAM), which is the universal obligate methyl donor for DNA methylation by DNA methyltransferases (DNMT).
Lysosomal positioning and mTORC1 activity in ATF4 upregulation and one-carbon metabolism
The control of DNA methylation levels in prostate cancer cells and likely histone methylation in IECs is enabled by activating the one-carbon metabolism through the upregulation of the serine biosynthetic pathway (Figure 3) [19]. This type of metabolic reprogramming is key for the synthesis of S-adenosyl methionine (SAM), the universal and obligate donor of methyl groups in many biochemical reactions [40, 41]. Therefore, PKCλ/ι by generating SAM facilitates the modulation of gene expression through DNA methylation in prostate cancer. This process most likely provides the substrate for the enhanced EZH2-driven methylation of H3K27 that results in the repression of Atoh1/Gfi1 intestinal secretory progenitor cells (Figure 2) [17]. Therefore, PKCλ/ι emerges as a new node in the crosstalk between the epigenetic control of gene expression and the generation of the metabolic building blocks required for this process to proceed (Figure 3).
The precise mechanism whereby PKCλ/ι represses the serine one-carbon metabolism is by inhibiting the expression of ATF4 [19], which is the master transcription factor of many metabolic enzymes in this pathway, including PHGDH, MTHFD2, and PSAT1 (Figure 3) [40, 42]. PKCλ/ι deficiency results in the upregulation of ATF4 expression through the upregulation of mTORC1 [19], which had previously been shown to regulate ATF4 transcriptionally [43, 44]. mTORC1 is central to the modulation of cell growth and metabolism [45]; under basal conditions, it is anchored to the lysosomes through the Ragulator complex, which together with the Rag and Rheb GTPases are required and sufficient for mTORC1 activity [46, 47]. A central component of Ragulator is LAMTOR2, which has been shown to associate with RagA/B and RagC/D also to control lysosomal positioning [47]. PKCλ/ι was found to directly interact with LAMTOR2 in an unbiassed Bio-ID proteomics approach, which revealed the association of PKCλ/ι with several other components of the endosomal/lysosomal system, including LAMP2, KIF2C/B, and the V-ATPases [19]. PKCλ/ι directly phosphorylates LAMTOR2 at serine 30, which is located at the interface with the Rag GTPases, consistent with a model whereby PKCλ/ι-induced phosphorylation of LAMTOR2 could impact complex formation, recruitment to lysosomes, and ultimately mTORC1 activation. Indeed, the loss of PKCλ/ι or the expression of a LAMTOR2 alanine-30 mutant induced the perinuclear aggregation of lysosomes and increased the amount of mTOR and RagC proximal to the nucleus, where Rheb, a key activator of mTORC1, resides [19]. Furthermore, these effects of PKCλ/ι deficiency were rescued by treating these cells with Ciliobrevin D, a dynein inhibitor that blocks the retrograde transport system [19]. Therefore, PKCλ/ι orchestrates a lysosome-regulated mechanism to activate a mTORC1-ATF4 axis to unleash the one-carbon metabolism for SAM synthesis and the subsequent epigenetic regulation of lineage differentiation.
PKCλ/ι is a new player in the regulation of autophagy
All these studies collectively point to a model whereby a critical role of PKCλ/ι is to modulate lysosome topology. However, a new connection between PKCλ/ι and the lysosome has been revealed in recent studies, which is related to autophagy activation and its crosstalk with metabolism, the stress response, and immunomodulatory pathways.
Autophagy control by the p62-PKCλ/ι-LC3 ternary complex regulates oxidative metabolism
The first evidence that PKCλ/ι might be involved in autophagy comes from an unbiased proteomics effort to determine the role of PKCλ/ι in hepatocytes and IECs in hepatocellular carcinoma (HCC) and intestinal cancer, respectively. Those studies revealed that LC3 and ULK2 were two interacting partners of PKCλ/ι (Figures 4A and 4B) [18, 48–50]. LC3 is a central component of the autophagy machinery that also binds the adaptor p62 (encoded by the SQSTM1 gene) through its specific LIR (LC3-interacting region) domain (Figures 1 and 4A) [51]. This is of great interest from a molecular cell biology point of view because p62 was identified several years ago as an aPKC-interacting partner in an unbiassed 2-hybrid screen [52]. Both p62 and the aPKCs harbor in their N-terminal regions respective PB1 domains that account for p62 oligomerization and its interaction with PKCλ/ι (Figure 1) [53–55]. Therefore, the existence of a p62-PKCλ/ι complex was known for a long time, but its mechanistic involvement in autophagy has been recently clarified. Thus, p62, PKCλ/ι and LC3 form a ternary complex whereby p62 helps anchor PKCλ/ι to LC3, which results in the PKCλ/ι-induced phosphorylation of LC3 at serine 12 [48, 49]. It is well accepted that p62 is a positive component of the autophagy machinery by bringing ubiquitinated aggregates to the autophagosome for their degradation [55–57]. However, p62 also favors the homeostatic inhibition of autophagy through the PKCλ/ι-triggered phosphorylation of LC3’s S12 [48, 49]. The negative regulation of LC3 by S12 phosphorylation can be explained by modeling its structure with a p62 peptide. Accordingly, S12 phosphorylation creates a destabilizing clash with D15, leading to structural changes that account for the reduced binding of p62 to LC3 [48]. These biochemical experiments and structural predictions explain why an alanine mutant of LC3 at serine 12, which cannot be phosphorylated by PKCλ/ι, drives increased autophagy vesicles and flux [48]. Therefore, the loss of PKCλ/ι promotes the accumulation of unphosphorylated LC3 that binds more tightly to p62, and likely other components of the autophagic machinery, fostering autophagy [48]. This process has two consequences. On the one hand, it favors the generation of fatty acids for oxidation [48]. On the other, it creates an environment favorable to the detoxification of damaged mitochondria, which accumulate as a consequence of the enhanced oxidative stress created by the activation of oxidative metabolism in PKCλ/ι-deficient hepatocytes (Figure 4A, Key Figure) [48]. However, to fully understand the complexity of the crosstalk between the different metabolic pathways controlled by PKCλ/ι, it should be taken into consideration that PKCλ/ι represses basally oxidative phosphorylation pathways by inhibiting the expression of its master regulator, PPARα (Figure 4A) [48]. In this way, PKCλ/ι deficiency promotes PPARα-mediated oxidative phosphorylation while simultaneously unleashing autophagy that feeds the pathway and helps detoxify excessive ROS production [48]. Therefore, PKCλ/ deficiency provides the mechanisms that allow cancer and normal cells to sustain levels of ROS that are not toxic but sufficient to drive tumorigenesis [48]. An important consequence of increased ROS production by PKCλ/ι-deficient cells is the activation of the transcription factor NRF2 (Figure 4A) [48], which serves two purposes. First, it also contributes to ROS detoxification by transcriptionally upregulating oxidative stress scavengers [58]; this, together with the upregulation of autophagy, maintains oxidative stress within acceptable levels. However, increased ROS-induced NRF2 results in another pro-tumorigenic function. Thus, NRF2 accumulation in hepatocytes leads to their mitogenic activation through still-to-be fully understood pathways [48].
Figure 4. The regulation of autophagy and the interferon pathway by PKCλ/ι determines cancer outcome.
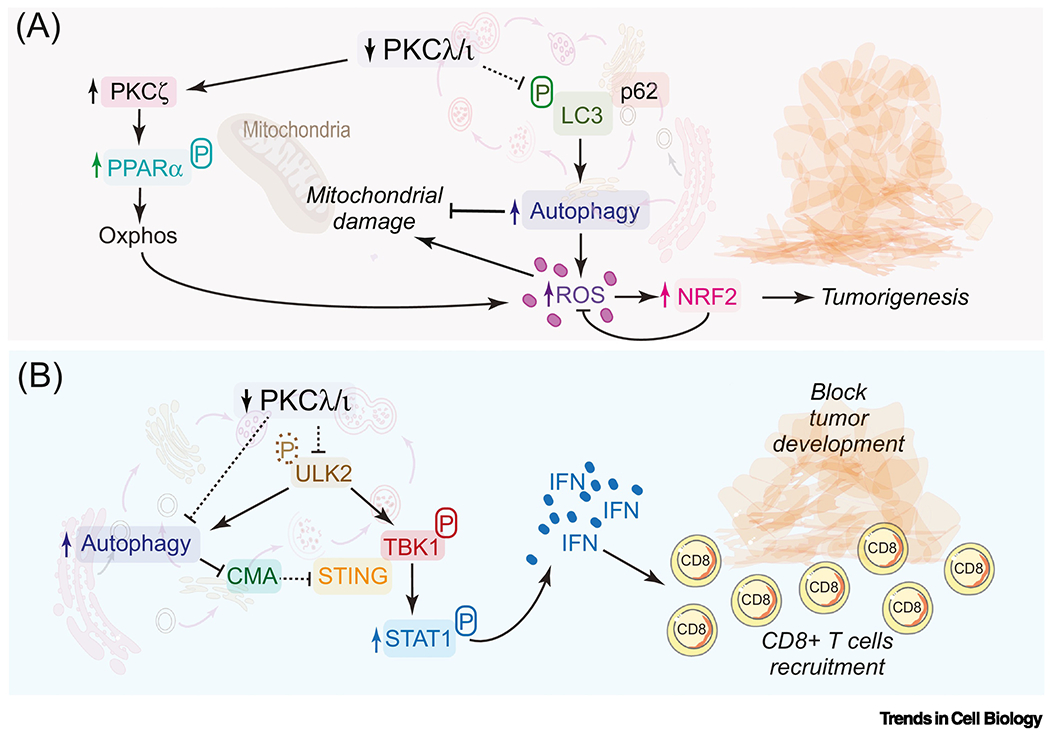
(A) PKCλ/ι represses oxidative phosphorylation pathways by inhibiting the expression of PPARα and autophagy in liver. PKCλ/ι forms a ternary complex with p62 and LC3, whereby PKCλ/ι modulates the interaction of LC3 with p62, which is inhibited by the constitutive phosphorylation of LC3 by PKCλ/ι. Loss of PKCλ/ι enhances autophagy, which increases ROS production that leads to NRF2 activation to promote tumorigenesis. The loss of PKCλ/ι enhances PPARα activity and autophagy, sustaining an oxidative metabolism required for tumor development.
(B) The loss of PKCλ/ι unleashes the ULK2-TBK1-STAT1-IFN pathway to promote an anti-tumorigenic response orchestrated by the recruitment of CD8+ T cells in intestinal epithelial cells. PKCλ/ι regulates the IFN pathway and autophagy through ULK2 by phosphorylation. Autophagy upregulation by loss of PKCλ/ι serves to sustain the activation of the IFN pathway by preventing the degradation of STING by CMA.
PKCλ/ι, autophagy and the interferon response
From all these studies, we can conclude that PKCλ/ι is mechanistically tied to the lysosome’s function through the phosphorylation of LAMTOR2, which regulates mTORC1, and through the phosphorylation of LC3, that contributes to autophagy’s role in oxidative phosphorylation and ROS homeostatic control (Figures 3 and 4A). However, more recent data link PKCλ/ι to autophagy through another player. The unbiassed proteomics Bio-ID strategy described above also revealed that PKCλ/ι interacted with proteins involved in autophagy [47] and with components of the intracellular trafficking from the endoplasmic reticulum to the Golgi [18]. A critical new interactor identified in that study was ULK2 [18] (Figure 4B). This observation reinforced the notion that PKCλ/ι is a fine-tune modulator of autophagy because ULK2, like ULK1, can positively regulate this process [59]. However, these unexpected new findings revealed a novel and unanticipated signaling connection whereby PKCλ/ι regulates interferon (IFN) activation. These recent studies showed that ULK2 phosphorylates and activates another kinase termed TBK1 that plays a critical role during IFN signaling [60]. Thus, by phosphorylating the transcription factor IRF3, which is the master regulator of the synthesis of IFNα/β [61], TBK1 drives the generation of type I IFNs (IFNα/β), whereas ULK1/2 also impacts autophagy [62]. The TBK1-IRF3 pathway is controlled by the adaptor STING transitions from the endoplasmic reticulum to the Golgi to activate this pathway [63, 64]. The fact that PKCλ/ι interacts with components of the ER-Golgi network, together with its ability to bind and phosphorylate ULK2, uncovers the molecular mechanism that underly previous observations that the loss of PKλ/ι in IECs promoted the IFN cascade that recruited CD8+ T cells to increase anti-cancer immunosurveillance (Figure 4B) [18, 50].
The connection of PKCλ/ι with ULK2 also shed new light on mechanistic questions linking the IFN response and different autophagy mechanisms. That is, ULK2 phosphorylation by PKCλ/ι at serine 150 inhibited ULK2 activity and promoted its degradation through a Vps4AB-dependent mechanism, suggestive of microautophagy [18]. Central to this process is the K63-type polyubiquitination of the ULK2’s K146 residue by the NDFIP1-NEDD4L ubiquitin ligase complex [18]. Inactivation of ULK2 by PKCλ/ι maintains S172 phosphorylation of TBK1 on check, which renders cells with low levels of IRF3 phosphorylation and basally reduced IFN signaling [18]. In this way, PKCλ/ι loss unleashes the ULK2-TBK1-IRF3-IFN pathway in parallel with the activation of autophagy (Figure 4B) [18]. Altogether, these observations open a new paradigm whereby autophagy is positively modulated concomitantly to the stimulation of IFN signaling by the simple inhibition of PKCλ/ι [18, 50]. These findings beg the question: what is the role of autophagy in the IFN cascade activated by PKCλ/ι loss? Recent observations showed that inhibiting autophagy severely reduced the enhanced activation of IFN in PKCλ/ι-deficient cells because of reduced STING levels [18]. This is apparently counterintuitive because previous data suggested that STING was a target of autophagy itself [65], although later work challenged that concept [64]. In this regard, the studies on PKCλ/ι revealed a connection between autophagy (also known as macroautophagy) and chaperone-mediated autophagy (CMA). According to this model, STING is basally degraded by CMA to maintain IFN under unstimulated conditions [18]. However, the activation of autophagy by PKCλ/ι deficiency represses CMA through a mechanism that still needs to be identified, rendering cells with high basal levels of STING that synergize with the increased activation of the TBK1-IRF3 axis further to stimulate the IFN response (Figure 4B). These check-and-balance mechanisms are central to preventing the pro-tumorigenic potential of PKCλ/ι-deficient cells through activating immunosurveillance, at least in the intestinal cancer paradigm.
The complex relations between the IFN pathway, autophagy, and immunosurveillance
All the studies described in the preceding section should be considered in the conceptual framework of others addressing the crosstalk between autophagy and the immune response [66, 67], which have generated complex and, on many occasions, contradictory conclusions. This important topic has already been discussed in previous very informative reviews [68, 69]. Here we will examine recent new evidence that sheds light on this seemingly multifaceted question. At a cell-autonomous level, it seems that autophagy’s role in controlling the IFN cascade and immunosurveillance processes is heavily dependent on the context. Thus, if the activation of autophagy is accompanied by a parallel induction of immunogenic cell death (ICD), as is the case in response to certain chemotherapeutics, autophagy is a required event. However, the mechanism still needs to be fully understood [70]. However, if the conditions involve the activation of the cGAS-STING pathway by, for example, the release of mitochondrial DNA in response to radiotherapy, then autophagy will inhibit the pathway by limiting the number of damaged mitochondria [71]. Also, autophagy has recently been shown to degrade major histocompatibility complex (MHC) class I proteins, at least in pancreatic cancer, suggesting that inhibiting autophagy prevents immunoevasion [72]. If we keep in mind that the need for autophagy for the activation of the IFN/immunosurveillance cascade seems to be dictated by the ICD status, then the evidence that the loss of PKCλ/ι in IECs results in the activation of markers of ICD is consistent with the positive role of autophagy in the activation of immunosurveillance in PKCλ/ι-deficient pre-neoplastic lesions [18].
The new finding that PKCλ/ι regulates the IFN pathway has important consequences for our understanding of the dual role of this kinase as a pro and anti-tumor molecule. Thus, recent experiments showed that the inducible genetic inactivation of PKCλ/ι in already established β-Catenin-driven tumors reduces tumor load, associated with triggering IFN signaling and the recruitment of CD8+ T cells [18]. These new findings strongly suggest that inhibitors of PKCλ/ι might be used to make tumors immunologically “hot” and, therefore, potentially susceptible to immunotherapy. However, this again would be context-dependent due to the cell-autonomous and non-autonomous role of PKCλ/ι as a tumor suppressor [2]. In this regard, an important aspect of all these studies is the opposite effects of PKCλ/ι in intestinal cancer, depending on the timing of its inactivation. If PKCλ/ι is lost concomitantly to tumor initiation by APC inactivation, in that case it enhances tumorigenesis because the β-Catenin pathway synergizes with the inflammatory state created by PKCλ/ι deficiency, which is further boosted by the β-Catenin-induced activation of immunoevasion [17]. Therefore, if the ability of PKCλ/ι deficiency to activate the immunosurveillance mechanisms is blunted, then PKCλ/ι loss drives tumorigenesis characterized by tubular adenomas [17].
Similarly, when PKCλ/ι is constitutively inactivated in IECs in which PKCζ has also been genetically ablated, mice develop intestinal tumors, but in this case through the serrated pathway, which is likely driven by the activation of a YAP-MAPK cascade, independent of β-Catenin signaling [11]. In this case, the immunosurveillance state activated by PKCλ/ι loss is ablated by the lack of PKCζ [11], which is required for IFN activation through a mechanism not understood. These PKCλ/ι-PKCζ doubly deficient tumors correspond to the invasive mesenchymal phenotype, which has the worst prognosis in patients and for which better therapies are sorely needed [11, 16, 20]. In this regard, while the single genetic inactivation of PKCλ/ι in IECs is not sufficient to drive tumorigenesis, if the levels of CD8+ T cells are reduced with neutralizing antibodies, then the full carcinogenic potential of PKCλ/ι deficiency is unleashed, and serrated mesenchymal tumors develop without any other oncogenic stimuli [11]. This contrasts with the APC-deficient model that does not progress beyond the adenoma stage even under conditions of PKCλ/ι deficiency [17]. Therefore, the context, timing, and balance of signals accompanying the loss of PKCλ/ι deficiency seem to be critical to determine the outcome of the mechanisms that drive tumor development.
Challenges and opportunities in the therapeutic targeting of PKCλ/ι in cancer
Several studies reporting the direct pharmacologic inhibition of PKCλ/ι have met three important caveats. First, it is extremely difficult to generate PKCλ/ι-specific chemical inhibitors without also inhibiting PKCζ due to the high degree of identity in their catalytic domains. This important problem has not been adequately addressed yet and has been recently discussed in a previous review [2]. The second potential caveat is liver toxicity; as demonstrated recently and discussed above, PKCλ/ι inhibition in hepatocytes creates a cell-autonomous and cell-non-autonomous oxidative state that drives tumorigenesis [48]. However, it should be kept in mind that PKCλ/ι is lost chronically in this hepatocyte-selective model, which is quite different from a situation in which this kinase might be inhibited acutely in a therapeutic setting, and the putative liver toxicity caused by excessive oxidative stress could be managed to mitigate its side effects. Third, inhibiting PKCλ/ι as an anti-cancer strategy must take into consideration that although some data suggest that this kinase might be required for tumor initiation in certain cancer models, it is a tumor suppressor, as demonstrated in more complex systems recapitulating the interplay between the cancer epithelium and the tumor microenvironment [2]. However, while inhibiting PKCλ/ι in the context of cells deficient also in other tumor and immunosuppressors such as PTEN (in the prostate) or APC (in the intestine) drives higher levels of malignancy, the fact that the single inactivation of PKCλ/ι in IECs causes a potent immunosurveillance response in the absence of other mutations that prevents tumor initiation, suggest that putative selective inhibitors of PKCλ/ι might be useful to enable immunotherapy by switching the tumor phenotype from immunologically cold to infiltrated by cytotoxic T cells, as demonstrated in our own studies using inducible IEC-specific single PKCλ/ι KO models [11, 18]. On the other hand, a potential barrier in using immune checkpoint inhibitors in cancer therapy is the severe desmoplastic and immunosuppressive response associated with the mesenchymal phenotype of tumors deficient for PKCλ/ι and PKCζ [11]. However, as shown recently, that hurdle can be overcome by combining anti-stroma with immune checkpoint inhibition, suggesting that although the mesenchymal phenotype of intestinal tumors is a clear promoter of malignancy, their reactive stroma also creates a vulnerability that can be targeted therapeutically [11]. One very attractive possibility is that the simultaneous reduction in the content of both aPKCs might be developed as biomarkers to identify mesenchymal cancer patients susceptible to this type of combination treatment.
Concluding remarks
Considerable effort has been devoted to identifying the mutagenic landscape of different cancers. While some of this knowledge has been leveraged to generate new therapies that have been relatively successful in the clinic, it seems that a better understanding of the complex interactions between different non-oncogenic players, such as epigenetics, metabolism, and the IFN cascades, will generate new therapeutic targets in a mutational-agnostic manner. On many occasions, decreased levels of gene products, like in the case of PKCλ/ι, create new dependencies and tumor vulnerabilities. Since restoring the activity/levels of these new non-oncogenic suppressors is challenging, a complete characterization of the signals from cells deficient in these tumor-suppressive players is critical to identify points of action for new therapeutics. In the case of PKCλ/ι-defective cells, it will be necessary to find ways to inactivate its enzymatic activity in tumors (to activate immunosurveillance) while simultaneously preventing the unleashing of pro-tumoral signals. This, along with several other pending important mechanistic details, remain to be investigated (see Outstanding Questions).
Outstanding questions.
While it is clear that PKCλ/ι inactivation promotes EZH2-mediated repression of key transcription factors involved in lineage differentiation and also promotes the generation of methyl donor groups through the serine-one-carbon pathway, what remains to be determned is the precise mechanisms whereby PKCλ/ι regulates the levels of EZH2, and how that impacts the epigenome of differentiating cells in the intestine and the prostate epithebum.
The fact that PKCλ/ι represses mTORC1 through the regulation of lysosome position with respect to the nucleus, which is instrumental for the activation of ATF4, also begs the question: what other metabolic mechanisms are being impinged by PKCλ/ι in a mTORC1-dependent, but ATF4-independent, manner that contributes to the cell-autonomous antitumorigenic effects of PKCλ/ι?
By maintaining STING levels through the inhibition of CMA, increased levels of macroautophagy by PKCλ/ι loss support the actuation of the IFN pathway triggered via TBK1–IRF3. What remains to be determined is how macroautophagy inhibits this process and whether it also impinges on other IFN-independent cell functions.
While PKCλ/ι loss unleashes the actuation of the IFN cascade, how PKCζ inhibition blocks that effect remains to be determined, which is essential for understanding the immunoevasion mechanisms triggered in colorectal cancer by WNT-independent pathways.
In summary, PKCλ/ι inactivation generates signals that converge into enhanced tumorigenesis by creating a pro-tumoral inflammatory environment, like in the intestinal epithelium, or unleashing the autophagy-OXPHOS-NERF2 cascade, like in hepatocytes. In contrast, in situations in which the tumor is already fully developed and initiated by a mechanism independent of PKCλ/ι deficiency, the activation of the IFN-mediated immunosurveillance cascade prevails. These data should be considered in the context of the dual role of PKCλ/ι loss in different types of cancers. Something that has not been explored, but that would be of great interest, is to determine whether the activation of the IFN pathway contributes to the inhibition of lung tumorigenesis and leukemia reported upon PKCλ/ι loss in mouse models driven by specific oncogene activation [12–15]. This is undoubtedly an important aspect to resolve if PKCλ/ι can ever be considered a viable potential therapeutic target in cancer.
Highlights.
PKCλ/ι regulates epithelial cell lineage differentiation in the intestine and the prostate through epigenetic mechanisms, which are enabled, at least in prostate cancer cells, by the upregulation of ATF4, a critical step in the serine biosynthetic pathway for the generation of S-adenosyl-methionine, the obligate universal donor of methyl groups.
ATF4 is transcriptionally activated by mTORC1 through the PKCλ/ι-induced phosphorylation of LAMTOR2 at serine-30, which governs the aggregation of lysosomes and increases the amount of mTOR and RagC proximal to the nucleus, where RheB activates mTOR.
PKCλ/ι, in complex con p62, inhibits autophagy by directly phosphorylating LC3 at serine-12, while PKCλ/ι inactivation renders cells with increased autophagy activity that promotes oxidative phosphorylation triggered by PPARα and serves to eliminate ROS-damaged mitochondria, maintaining ROS levels below toxic concentrations, which enables the accumulation of NRF2 to activate tumorigenesis.
The loss of PKCλ/ι results in the activation of a new ULK2-TBK1 cascade that further reinforces the activation of autophagy to prevent the degradation of STING that helps stimulate the interferon pathway to promote CD8+ T-cell recruitment and activation that inhibits tumor initiation and progression.
Acknowledgments
Research in the author’s laboratories was supported by grants by NCI and NIDDK of the National Institutes of Health under awards numbers: R01CA207177, R01DK108743, R01CA250025, and R01CA265892 to J.M; R01CA218254 and R01CA246765 to M.T.D.-M; J.M. and M.T.D.-M. are Homer T. Hirst III Professors of Oncology in Pathology.
Footnotes
Declaration of Interests
J.M. and M.T.D-M are co-founders, shareholders, and members of the board of ZelamBio Inc. J.F.L and A.D. have no interests to declare.
References
- 1.Griner EM and Kazanietz MG (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7 (4), 281–94. [DOI] [PubMed] [Google Scholar]
- 2.Reina-Campos M et al. (2019) The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell 36 (3), 218–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moscat J and Diaz-Meco MT (2000) The atypical protein kinase Cs - Functional specificity mediated by specific protein adapters. Embo Reports 1 (5), 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vorhagen S and Niessen CM (2014) Mammalian aPKC/Par polarity complex mediated regulation of epithelial division orientation and cell fate. Exp Cell Res 328 (2), 296–302. [DOI] [PubMed] [Google Scholar]
- 5.Hong Y (2018) aPKC: the Kinase that Phosphorylates Cell Polarity. F1000Res 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma L et al. (2013) Control of Nutrient Stress-Induced Metabolic Reprogramming by PKCzeta in Tumorigenesis. Cell 152 (3), 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galvez AS et al. (2009) Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol 29 (1), 104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JY et al. (2013) c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc Natl Acad Sci U S A 110 (16), 6418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llado V et al. (2015) Repression of Intestinal Stem Cell Function and Tumorigenesis through Direct Phosphorylation of beta-Catenin and Yap by PKCzeta. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shelton PM et al. (2018) The Secretion of miR-200s by a PKCzeta/ADAR2 Signaling Axis Promotes Liver Metastasis in Colorectal Cancer. Cell Rep 23 (4), 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakanishi Y et al. (2018) Simultaneous Loss of Both Atypical Protein Kinase C Genes in the Intestinal Epithelium Drives Serrated Intestinal Cancer by Impairing Immunosurveillance. Immunity 49 (6), 1132–1147 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Justilien V et al. (2014) The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 25 (2), 139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin N et al. (2019) Protein Kinase Ciota and Wnt/beta-Catenin Signaling: Alternative Pathways to Kras/Trp53-Driven Lung Adenocarcinoma. Cancer Cell 36 (2), 156–167 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nayak RC et al. (2019) The signaling axis atypical protein kinase C lambda/iota-Satb2 mediates leukemic transformation of B-cell progenitors. Nat Commun 10 (1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray NR and Fields AP (1997) Atypical protein kinase C iota protects human leukemia cells against drug-induced apoptosis. J Biol Chem 272 (44), 27521–4. [DOI] [PubMed] [Google Scholar]
- 16.Kasashima H et al. (2020) Stromal SOX2 Upregulation Promotes Tumorigenesis through the Generation of a SFRP1/2-Expressing Cancer-Associated Fibroblast Population. Dev Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi Y et al. (2016) Control of Paneth Cell Fate, Intestinal Inflammation, and Tumorigenesis by PKClambda/iota. Cell Rep 16 (12), 3297–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linares JF et al. (2021) PKClambda/iota inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reina-Campos M et al. (2019) Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 35 (3), 385–400 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakanishi Y et al. (2019) Serrated Colorectal Cancer: The Road Less Travelled? Trends Cancer 5 (11), 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adolph TE et al. (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503 (7475), 272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clevers HC and Bevins CL (2013) Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol 75, 289–311. [DOI] [PubMed] [Google Scholar]
- 23.Sauvageau M and Sauvageau G (2010) Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7 (3), 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shroyer NF et al. (2005) Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev 19 (20), 2412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noah TK and Shroyer NF (2013) Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol 75, 263–88. [DOI] [PubMed] [Google Scholar]
- 26.Shroyer NF et al. (2007) Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology 132 (7), 2478–88. [DOI] [PubMed] [Google Scholar]
- 27.Li Z et al. (2020) Post-translational modifications of EZH2 in cancer. Cell Biosci 10 (1), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintanal-Villalonga A et al. (2020) Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol 17 (6), 360–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubin MA et al. (2020) Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol Cell 80 (4), 562–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies AH et al. (2018) Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol 15 (5), 271–286. [DOI] [PubMed] [Google Scholar]
- 31.Beltran H et al. (2019) The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res 25 (23), 6916–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aggarwal R et al. (2018) Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 36 (24), 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson PA et al. (2015) Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15 (12), 701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Bono JS et al. (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364 (21), 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scher HI et al. (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367 (13), 1187–97. [DOI] [PubMed] [Google Scholar]
- 36.Sandhu S et al. (2021) Prostate cancer. Lancet 398 (10305), 1075–1090. [DOI] [PubMed] [Google Scholar]
- 37.Beltran H et al. (2016) Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22 (3), 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Regala RP et al. (2005) Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res 65 (19), 8905–11. [DOI] [PubMed] [Google Scholar]
- 39.Conteduca V et al. (2021) Epigenetics in prostate cancer: clinical implications. Transl Androl Urol 10 (7), 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reina-Campos M et al. (2020) The complexity of the serine glycine one-carbon pathway in cancer. J Cell Biol 219 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reid MA et al. (2017) The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19 (11), 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeNicola GM et al. (2015) NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47 (12), 1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben-Sahra I et al. (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339 (6125), 1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park Y et al. (2017) mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep 19 (6), 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saxton RA and Sabatini DM (2017) mTOR Signaling in Growth, Metabolism, and Disease. Cell 168 (6), 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bar-Peled L et al. (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150 (6), 1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sancak Y et al. (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320 (5882), 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudo Y et al. (2020) PKClambda/iota Loss Induces Autophagy, Oxidative Phosphorylation, and NRF2 to Promote Liver Cancer Progression. Cancer Cell 38 (2), 247–262 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moscat J and Diaz-Meco MT (2020) The interplay between PRKCI/PKClambda/iota, SQSTM1/p62, and autophagy orchestrates the oxidative metabolic response that drives liver cancer. Autophagy 16 (10), 1915–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moscat J et al. (2022) Immunosurveillance, interferon, and autophagic networking in cancer: the PRKCI-ULK2 paradigm. Autophagy 18 (1), 226–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pankiv S et al. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282 (33), 24131–45. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez P et al. (1998) Localization of atypical protein kinase C isoforms into lysosome- targeted endosomes through interaction with p62. Mol Cell Biol 18 (5), 3069–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moscat J et al. (2006) Cell Signaling and Function Organized by PB1 Domain Interactions. Mol Cell 23 (5), 631–640. [DOI] [PubMed] [Google Scholar]
- 54.Moscat J and Diaz-Meco MT (2009) To aggregate or not to aggregate? A new role for p62. EMBO Rep 10 (8), 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moscat J et al. (2016) p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 167 (3), 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moscat J and Diaz-Meco MT (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137 (6), 1001–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirkin V and Rogov VV (2019) A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol Cell 76 (2), 268–285. [DOI] [PubMed] [Google Scholar]
- 58.Hayes JD et al. (2020) Oxidative Stress in Cancer. Cancer Cell 38 (2), 167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee EJ and Tournier C (2011) The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 7 (7), 689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao P et al. (2018) TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 172 (4), 731–743 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saleiro D et al. (2015) Central role of ULK1 in type I interferon signaling. Cell Rep 11 (4), 605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vargas JNS et al. (2019) Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell 74 (2), 347–362 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka Y and Chen ZJ (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5 (214), ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gui X et al. (2019) Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567 (7747), 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prabakaran T et al. (2018) Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J 37 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X et al. (2020) Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 53 (1), 43–53. [DOI] [PubMed] [Google Scholar]
- 67.Deretic V (2021) Autophagy in inflammation, infection, and immunometabolism. Immunity 54 (3), 437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia H et al. (2021) Autophagy in tumour immunity and therapy. Nat Rev Cancer 21 (5), 281–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarke AJ and Simon AK (2019) Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 19 (3), 170–183. [DOI] [PubMed] [Google Scholar]
- 70.Galluzzi L et al. (2017) Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol 14 (4), 247–258. [DOI] [PubMed] [Google Scholar]
- 71.Yamazaki T et al. (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21 (10), 1160–1171. [DOI] [PubMed] [Google Scholar]
- 72.Yamamoto K et al. (2020) Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581 (7806), 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]