

Abstract
Cancer-associated fibroblasts (CAFs) are a heterogeneous population of cells. At one end of the spectrum are alpha-smooth muscle actin expressing myoCAFs (myofibroblast CAFs) and at the other end are the interferon (IFN) and Janus Kinase/Signal Transducer and Activator of Transcription responsive iCAFs (inflammatory CAFs). Both types of CAFs promote tumor growth. While myoCAFs foster immune exclusion and limit tumor spread, iCAFs create a highly immunosuppressive environment and foster the seeding of distant metastases. However, iCAFs also represent a tumor vulnerability. They are competent to undergo necroptosis, a highly immunogenic form of cell death that is triggered when Z-DNA or Z-RNA (collectively called ZNA) is sensed by the IFN-induced ZNA binding protein 1 (ZBP1). The sequestering of ZNA ligands by the p150 isoform of the double-stranded RNA-specific deaminase ADAR1 protects iCAFs from cell death. ZBP1-dependent necroptosis in iCAFs can be triggered by administering an orally available small molecule that generates sufficient amounts of ZNA to bypass ADAR1 inhibition. The therapeutic approach of targeting Z-prone sequences (called flipons) is agnostic to the mutations driving cancer progression. By exploiting the tumor vulnerability posed by expression of ZBP1-dependent immunogenic cell death pathways in iCAFs, flipon therapeutics offer new hope for improved clinical outcomes.
Keywords: Immunity, Innate; Programmed Cell Death 1 Receptor; Alarmins; Inflammation; Tumor Microenvironment
Highlights.
Inflammatory cancer-associated fibroblasts (iCAFs) portend poor patient survival.
iCAFs can be selectively targeted with a small molecule that activates necroptosis.
This orally available compound triggers iCAF death by inducing left-handed Z-DNA.
The drug increases immune checkpoint blockade efficacy in preclinical melanoma models.
The challenge
While the success of immune checkpoint blockers (ICB) has proven the worth of immunotherapies in the clinic, most patients still do not experience lasting remissions. Tumors with a large fibroblast component are especially difficult to treat, and include a subset of breast, pancreatic and lung cancers.1 Myriad roles for cancer-associated fibroblasts (CAFs) in promoting cancer growth and spread have been proposed and specifically targeted by a spectrum of therapeutic modalities.2 3 Our focus here is on a novel therapeutic approach that exploits the highly immunogenic cell death pathway of necroptosis present in a subset of interferon (IFN)-activated inflammatory CAFs (iCAFs). The pathway depends on the sensing of the non-canonical left-handed Z-DNA or Z-RNA (collectively called ZNA) double helix by ZNA binding protein 1 (ZBP1). The subsequent ZBP1-dependent necroptotic response is normally inhibited within tumors by another IFN-induced ZNA binding protein, the p150 isoform of ADAR1, which also binds ZNAs. Both proteins recognize ZNA through related Zα domains.4 5 We describe here how an orally available small molecule can bypass ADAR1 to trigger ZBP1 activation and necroptosis in iCAFs to cause immunogenic cell death. The molecule acts by inducing Z-DNA formation by Z-prone sequences called flipons.6 This novel approach raises the hope that flipon therapeutics can improve the effectiveness of ICB in the clinic as we demonstrated with preclinical models of melanoma.
Cancer-associated fibroblasts
The heterogeneity of CAFs has recently been highlighted by single cell sequencing approaches.2 The studies have built on earlier work identifying two major subsets of tumor resident CAFs: myoCAFs and iCAFs.7 The myoCAFs (cancer-associated myofibroblasts) express alpha-smooth muscle actin (encoded by ACTA2) and often form a palisade around the tumor cells, which excludes beneficial immune cell infiltration but also limits cancer cell intravasation and metastatic spread. Clinically, a predominance of myoCAFs correlates with resistance to immunotherapy.8 The differentiation of myoCAFs from mesenchymal stem cells resident in the tumor microenvironment (TME) is favored by cross-talk between transforming growth factor-β (TGF-β) and the Hedgehog signaling pathway.9 In contrast, depletion of myoCAFs allows the emergence of iCAFs and results in a higher rate of metastases, as shown in models of mouse pancreatic ductal adenocarcinoma (PDAC).10 While the differentiation of iCAFs is prevented by TGF-β produced by myoCAFs, their growth is driven by interleukin 1 (IL-1)-induced leukemia inhibitory factor, IL-6, IFN and potentially other activators of inflammatory Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathways.11 The iCAFs are highly immunosuppressive and cooperate with adhesion G protein-coupled receptor E1 (F4/80+) programmed cell death ligand 1 (PD-L1)+, major histocompatibility complex II (MHCIIlow) tumor-associated macrophages (TAMs) to induce immune resistance, as reported for genetically engineered KRASG12D proto-oncogene mouse models of PDAC.12 While a predominance of myoCAFs in the tumor bed is bad because of immune exclusion, iCAFs are worse as they portend the onset of metastatic disease.
Many early therapeutic strategies were aimed at myoCAFs, primarily because of their role in dampening immune responses. At the time, the part played by myoCAFs in repressing iCAF differentiation was unknown3 and approaches, which included TGF-β and Hedgehog signaling pathway inhibitors, produced results that were contrary to expectations: in experimental models where myoCAFs were eliminated by Sonic Hedgehog deletion, an immunosuppressive environment developed within the tumor mass, with expansion of CD4+CD25+FOXP3+ regulatory T cells and concomitant loss of effector CD8+ T cells,13 while in the 4T1 breast cancer model, blockade of TGF-β resulted in depletion of myoCAFs but resulted in the emergence of immunosuppressive macrophages.14 Targeting myoCAFs by blockade of TGF-β and Hedgehog pathways was also complicated by on-target side effects, reflective of the role these pathways perform in normal tissue maintenance. Overall, the earlier focus on myoCAFs yielded new information, but did not improve clinical outcomes.
ZBP1 and necroptosis
Interestingly, JAK/STAT signaling pathways not only increase iCAF abundance, but also sensitize cells to ZBP1-dependent necroptotic cell death, which is highly immunogenic. For example, components of this cell death pathway, including ZBP1 itself, are induced by IFN via JAK1/STAT1 signaling.15 ZBP1-mediated necroptosis is triggered by left-handed ZNA conformations, where the two strands of a DNA or RNA double-helix twist to the left rather than to the right as occurs with Watson-Crick B-DNA or A-RNA. ZNA prone flipons are present in the genomes of many promoters, retroelements and pathogens.5 6 16–18 Their sequence motifs often have alternating purine, pyrimidine motifs with guanosine preferred over adenosine and cytidine favored over thymidine.19
ZNA activates ZBP1 by engaging its N-terminal Zα domains and triggers binding to receptor interacting protein kinase 3 (RIPK3) through the receptor-interacting protein homology interaction motifs present on both proteins20 21 (figure 1B). The subsequent phosphorylation of mixed lymphocyte kinase like by RIPK3 promotes the lethal assembly of pores in nuclear and cytoplasmic membranes that lead to necroptotic cell death. Alternatively, both ZBP1 and RIPK3 can associate with RIPK1 to initiate apoptosis, a less inflammatory form of cell death.15 22 23 Apoptosis is favored over necroptosis by the association of ZBP1 with RIPK1 with caspase 8 (CASP8) protease further modulating outcomes through cleavage of both RIPK1 and RIPK3.24 25
Figure 1.

ZBP1-initiated cell death signaling and its repression by ADAR1. (A) ADAR1 suppresses immune responses in multiple ways through its different domains. The ADAR1 Zα domain binds ZNA and inhibits ZBP1 activation while the A-RNA binding domains (RBDs1–3) inhibit activation of the double-stranded RNA (dsRNA) sensors melanoma differentiation-associated gene 5 (encoded by IFIH1), RIG-I (encoded by DDX58) and the protein kinase PKR (encoded by EIF2AK2).44 Together the Zα and RBD domains of ADAR1 localize the enzyme to substrates where adenosine to inosine editing (A→I) occurs, a modification that can alter the stability of dsRNA regions and also recode messenger RNAs since inosine is translated as guanosine. Zβ is not known to bind nucleic acids. (B) Binding of ZNA activates ZBP1. ZBP1 activates RIPK1 and RIPK3 by engaging in RHIM-based associations with these proteins. Phosphorylation of MLKL by RIPK3 leads to the formation of membrane pores and necroptosis, while activation of CASP8 by RIPK1 results in apoptosis, a relatively non-inflammatory form of cell death. RIPK1 sequesters ZBP1 In some settings, and the RIPK1/CASP8 complex can also inhibit necroptosis.24 25 In addition, ZBP1 can associate with toll receptor associated with TIR-domain-containing adapter-inducing interferon-β (TRIF, encoded by TICAM1) and activate NF-κB (not shown).45 Stimulatory pathways are indicated with green arrows while inhibitory ones are colored blue with ZNA dependent outcomes labeled yellow. The arrows with blunt ends indicate suppressive interactions. CASP8, caspase 8; MLKL, mixed lymphocyte kinase like; NF-kB, nuclear factor-kB; PKR, protein kinase RNA; pMLKL, phosphorylation of MLKL; RBDs, dsRNA-binding domains; RHIMs, receptor-interacting protein homology interaction motifs; RIPK3, receptor interacting protein kinase 3; ZBP1, ZNA binding protein 1; ZNA, Z-DNA or Z-RNA collectively.
Notably, the p150 isoform of ADAR1 negatively regulates ZBP1 activation by binding ZNA through its Zα domain (figure 1).5 26–28 ADAR1 also engages A-RNA through its double-stranded RNA (dsRNA) binding domains (RBDs) and prevents IFN induction by the helicase melanoma differentiation-associated gene 5 (encoded by IFIH1). The sequestration of dsRNA also prevents inhibition of protein translation by PKR (protein kinase RNA-activated, encoded by EIF2AK2) (figure 1A).29 ADAR1 further inhibits these responses through the adenosine to inosine editing of A-RNA, destabilizing dsRNA while decreasing its potential to form Z-RNA. By arresting both ZBP1 activation and IFN induction, ADAR1 p150 maintains immune silence4 (figure 1).
DAMPs and immunogenic cell death
Tumors exploit a number non-inflammatory mechanisms, such as apoptosis and efferocytosis, to eliminate cells and their debris (figure 1).30 However, when tumor cells die by necroptosis, they leak out various proteins and nucleic acids with strong inflammatory and immunogenic potential. These damage-associated molecular patterns (DAMPs) comprise heat-shock, high-mobility group box 1 (HMGB1), annexin A1, calreticulin and histone proteins, DNA and RNA and many other signaling molecules including ATP.31 Those originating from the nucleus are especially immunogenic, as shown in mouse models of severe influenza.17 Chemokines such as IL-33, IL-1α and C-X-C motif chemokine ligand 1 are also released and attract neutrophils and other immune cells to the site of injury.32 The damage to normal tissue could far exceed that necessary to contain a threat if this response was left unchecked.33 Indeed, there is an immunosuppressive arm to the necroptotic response that leads to resolution and tissue repair. Tumors subject to necroptosis can suppress the immune response against cancer cells by exploiting counter measures that depend on encapsulation of necroptotic cells or on increased levels of prostaglandin E2, oxidized HMGB1 or galectin-9.30 34–36 DAMPs like SAP130 (Sin3A-associated protein 130), acting through macrophage-inducible C-type lectin-initiated pathways, further promote the differentiation of infiltrating myeloid cells into F4/80+PD-L1+ MHCIIlow TAMs that suppress antitumor responses.12 32 The tumor-driven counter-measures that inhibit immune cell activation can be blocked by the use of ICB, allowing DAMP-activated dendritic cells (DCs) to emerge. As these DCs differentiate, they produce cytokines and present cancer cell-derived antigens to CD8+ T cells and stimulate robust immune responses which, when combined with ICB approaches, can produce tumor rejection (figure 2).
Figure 2.
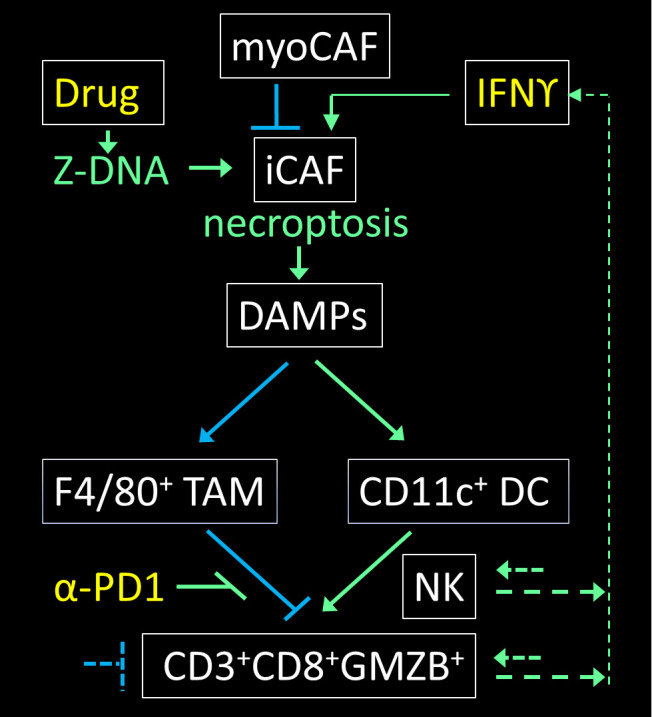
ZNA induced by a small molecule bypasses the need for ADAR1 inhibition and directly activates ZBP1 to induce necroptosis in iCAFs. The DAMPs released induce both immunosuppressive TAMs (tumor associated macrophages) that express the F4/80 adhesion protein and immunostimulatory CD11c+ DCs (dendritic cells). The amplification of anti-cancer cytotoxic CD8+ T cells and natural killer (NK) cells is favored by ICB with antibodies like anti-PD1 (α-PD1) that prevent binding of the PDL1 and PDL2 ligands expressed by TAMs to the immune-inhibitory programmed cell death 1 (PD1) receptor expressed on T cells. The net effect of α-PD1 is stimulatory as it negates a negative interaction. The antitumor immune cells, along with bystander cells, also produce cytokines that signal through the JAK1/STAT1 pathway and, by enhancing ZBP1 expression in iCAFs, increase the vulnerability of these cells to necroptosis.11 15 46 Stimulatory effects are indicated with green arrows while inhibitory pathways are colored blue with therapeutic agents amplifying antitumor responses labeled yellow. The arrows with a blunt end indicate suppressive interactions, while the dotted lines are representative of additional nodes that are potentially targetable but that are not detailed here. DAMPs, damage-associated molecular patterns; F4/80, adhesion G protein-coupled receptor E1 encoded by ADGRE1; iCAF; inflammatory CAF; ICB, immune checkpoint blocker; IFN, interferon; JAK/STAT, Janus Kinase/Signal Transducer and Activator of Transcription; PDL1, programmed cell death ligand 1; myoCAF (α-smooth muscle actin (encoded by ACTA2) positive cancer-associated myofibroblasts);TAMs, tumor-associated macrophages; ZBP1, ZNA binding protein 1; ZNA, Z-DNA or Z-RNA collectively.
Targeting iCAFs
Given these experimental findings, we wondered whether triggering intratumor necroptosis in the presence of ICB would enhance treatment outcomes. We hypothesized that by directly activating ZBP1 in the TME, we could overcome silencing of immunogenic cell death by ADAR1 p150. An analysis of human melanoma samples from The Cancer Genome Atlas revealed that CAFs expressed the entire necroptosis machinery,5 whereas other non-transformed cell types do not.37 Thus, a therapeutic window of opportunity existed for the selective activation of necroptosis in CAFs of the TME without toxicity to major organs. If successful, this strategy would boost DC-induced beneficial antitumor immune responses engendered by ICBs. Further, such an approach would be agnostic to the presence of any cancer cell mutations, including those that drive malignancy and others that inactivate the necroptotic machinery (a frequent occurrence during tumorigenesis).38 The next step was to find a way to implement this strategy.
CBL0137, the first flipon therapeutic
While CAFs express the necroptosis pathway, there is insufficient ZNA ligand in these cells to activate ZBP1. For example, the transcription of ZNA prone flipons within fibroblasts is not dysregulated as it is in cancer cells. Further, any ZNA formed is quenched by ADAR1, either through direct sequestration or by editing of dsRNAs (figure 1).5 The challenge was to find a ZNA-inducing agent that not only bypasses ADAR1 to activate necroptosis in iCAFs, but one that also spares normal tissues. (figure 2). ICBs would then provide a way to overcome the immunosuppression that occurs as part of the normal wound repair response and that is induced by DAMPs.12 32 The combination of a ZNA-inducing drug and ICB would then initiate and amplify antitumor responses through a positive feedback loop where the release of IFN from immune cells would drive the differentiation of additional drug-susceptible, necroptosis-competent iCAFs. (figure 2).
A screen of clinical compounds identified the curaxin CBL0137 as an inducer of Z-DNA in all mammalian cell types tested.5 Further experimentation demonstrated that the molecule only induced necroptosis in cells competent for ZBP1-initiated death signaling. CBL0137 also triggered ZBP1-dependent necroptosis in CAFs in vivo when tested in mouse models of melanoma and drove regression of tumors that are either completely (B16.F10) or partially (YUMMER 1.7) refractory to ICB (α-programmed cell death 1) monotherapy. Further, we were able to show infiltration of tumors with CD11c+ DCs, as well as CD8+ cytotoxic T cells.5 We also demonstrated the CBL0137-induced T cell priming in draining lymph nodes and promoted ICB-driven abscopal responses.
CBL0137 has been trialed in humans as a monotherapy where the primary concern was a manageable combined thrombocytopenia/neutropenia occurring in 2 of the 83 patients treated.39 These findings may represent an off-target effect as we noted that CBL0137 can induce both ZBP1-dependent and non-ZBP1-dependent cell death in bone marrow derived myeloid cells. Further study will reveal if these effects impact outcomes when CBL0137 is combined with ICB in the clinic. Overall, the combination of ZBP1-induced necroptosis and immune checkpoint blockade holds significant promise for the treatment of checkpoint resistant/refractory malignancies.
Future directions
While we have carried out proof in principle studies demonstrating that the combination of ZBP1-dependent necroptosis and ICB can improve cancer treatment outcomes, there is still much to be explored. Most importantly, which tumors are likely to respond? Any solid tumor type with a significant population of iCAFs (eg, PDAC, cutaneous melanomas) will be targets for approaches directly targeting ZBP1 in the TME, as we have outlined here. More broadly, the dependence of tumors on the IFN-induced ADAR1 p150 isoform for survival likely indicates that they are also susceptible to IFN-induced ZBP1 dependent necroptosis. A survey of 324 tumors (30 cancer types) performed at the Sanger Center gives hope that this approach may be quite generally applicable, as dependency on ADAR expression for survival was found in 42% of cancers, with more than 70% of head and neck tumors, 50% of central nervous system tumors, 40% of lung tumors and over 12% of large intestine cancers vulnerable to ADAR gene deletion (see supplementary table 3 in reference 40). Interestingly, the dependence of tumor cells on ADAR1 protein for their survival was unrelated to the mutational load or microsatellite instability status of the tumors.41 Although ICB responsiveness is not predicted by ADAR1 expression, it is encouraging that the poor ADAR1 editing of an empirically determined set of RNAs correlates with better patient survival outcomes42
Other important questions also arise. Are there better molecules than CBL0137 to induce ZNA formation? Do any of the small-molecule epigenetic modifiers currently in the clinic activate ZBP1-dependent necroptosis by derepressing the expression of retroelement Z-prone flipons? Do drugs that inhibit topoisomerases promote ZNA-necroptosis by freezing flipons in the Z-DNA state, preventing their relaxation back to B-DNA? Which other available immunotherapies will work with flipon therapeutics to amplify anticancer immune responses? Can ADAR1 p150 targeted molecules be developed to unleash both ZBP1-dependent necroptosis along and A-RNA-stimulated antitumor responses? Alternatively, are there additional activators of necroptosis, such as the irradiation of tumors or the herpes virus T-Vec, that can subdue a dominant myoCAF population which otherwise would limit the clinical response to a combined flipon/ICB immunotherapy? All these approaches selectively target tumors over normal cells and offer the hope of reversing the bad-to-worse outcomes CAFs generate.
jitc-2022-005704supp001.pdf (1.2MB, pdf)
Acknowledgments
The graphical abstract incorporates elements from43 that are modified under the CC BY-NC 4 license.
Footnotes
Twitter: @insideoutbio
Contributors: The manuscript evolved out of a long-term collaboration with many contributions from both parties. AH wrote the initial draft and prepared the figures with input and editing from SB.
Funding: This study was funded by NIH grants CA168621, CA190542 to SB; InsideOutBio IOBG202209010 to AH.
Competing interests: SB is a co-inventor on a provisional patent application ‘Combination of curaxins and immune checkpoint inhibitors for treating cancer’ filed by the Fox Chase Cancer Center. AH is a founder of InsideOutBio that is focused on developing immunotherapeutics for the treatment of cancers.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Not applicable.
References
- 1.Joshi RS, Kanugula SS, Sudhir S, et al. The role of cancer-associated fibroblasts in tumor progression. Cancers 2021;13:1399. 10.3390/cancers13061399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pereira BA, Vennin C, Papanicolaou M, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends Cancer 2019;5:724–41. 10.1016/j.trecan.2019.09.010 [DOI] [PubMed] [Google Scholar]
- 3.Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020;20:174–86. 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herbert A. ADAR and immune silencing in cancer. Trends Cancer 2019;5:272–82. 10.1016/j.trecan.2019.03.004 [DOI] [PubMed] [Google Scholar]
- 5.Zhang T, Yin C, Fedorov A, et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 2022;606:594–602. 10.1038/s41586-022-04753-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbert A. A genetic instruction code based on DNA conformation. Trends Genet 2019;35:887–90. 10.1016/j.tig.2019.09.007 [DOI] [PubMed] [Google Scholar]
- 7.Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–96. 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakravarthy A, Khan L, Bensler NP, et al. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 2018;9:4692. 10.1038/s41467-018-06654-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Javelaud D, Pierrat M-J, Mauviel A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett 2012;586:2016–25. 10.1016/j.febslet.2012.05.011 [DOI] [PubMed] [Google Scholar]
- 10.Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–34. 10.1016/j.ccr.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spinelli FR, Colbert RA, Gadina M. JAK1: number one in the family; number one in inflammation? Rheumatology 2021;60:ii3–10. 10.1093/rheumatology/keab024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ajina R, Malchiodi ZX, Fitzgerald AA, et al. Antitumor T-cell immunity contributes to pancreatic cancer immune resistance. Cancer Immunol Res 2021;9:386–400. 10.1158/2326-6066.CIR-20-0272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steele NG, Biffi G, Kemp SB, et al. Inhibition of Hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin Cancer Res 2021;27:2023–37. 10.1158/1078-0432.CCR-20-3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grauel AL, Nguyen B, Ruddy D, et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun 2020;11:6315. 10.1038/s41467-020-19920-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingram JP, Thapa RJ, Fisher A, et al. ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J Immunol 2019;203:1348–55. 10.4049/jimmunol.1900216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehdipour P, Marhon SA, Ettayebi I, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020;588:169–73. 10.1038/s41586-020-2844-1 [DOI] [PubMed] [Google Scholar]
- 17.Zhang T, Yin C, Boyd DF, et al. Influenza virus Z-RNAs induce ZBP1-Mediated necroptosis. Cell 2020;180:1115–29. 10.1016/j.cell.2020.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiao H, Wachsmuth L, Kumari S, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 2020;580:391–5. 10.1038/s41586-020-2129-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herbert A. Z-DNA and Z-RNA in human disease. Commun Biol 2019;2:7. 10.1038/s42003-018-0237-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 2008;181:6427–34. 10.4049/jimmunol.181.9.6427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebsamen M, Heinz LX, Meylan E, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 2009;10:916–22. 10.1038/embor.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook WD, Moujalled DM, Ralph TJ, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ 2014;21:1600–12. 10.1038/cdd.2014.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thapa RJ, Ingram JP, Ragan KB, et al. DAI senses influenza A virus genomic RNA and activates RIPK3-Dependent cell death. Cell Host Microbe 2016;20:674–81. 10.1016/j.chom.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oberst A, Dillon CP, Weinlich R, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011;471:363–7. 10.1038/nature09852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton K, Wickliffe KE, Maltzman A, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016;540:129–33. 10.1038/nature20559 [DOI] [PubMed] [Google Scholar]
- 26.Hubbard NW, Ames JM, Maurano M, et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 2022;607:769–75. 10.1038/s41586-022-04896-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao H, Wachsmuth L, Wolf S, et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature 2022;607:776–83. 10.1038/s41586-022-04878-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Reuver R, Verdonck S, Dierick E, et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 2022;607:784–9. 10.1038/s41586-022-04974-w [DOI] [PubMed] [Google Scholar]
- 29.Hartner JC, Walkley CR, Lu J, et al. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 2009;10:109–15. 10.1038/ni.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boada-Romero E, Martinez J, Heckmann BL, et al. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 2020;21:398–414. 10.1038/s41580-020-0232-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature Committee on cell death 2018. Cell Death Differ 2018;25:486–541. 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seifert L, Werba G, Tiwari S, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016;532:245–9. 10.1038/nature17403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balachandran S, Rall GF. Benefits and perils of necroptosis in influenza virus infection. J Virol 2020;94. 10.1128/JVI.01101-19. [Epub ahead of print: 16 04 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hangai S, Ao T, Kimura Y, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U S A 2016;113:3844–9. 10.1073/pnas.1602023113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rapoport BL, Steel HC, Theron AJ, et al. High mobility group box 1 in human cancer. Cells 2020;9. 10.3390/cells9071664. [Epub ahead of print: 10 07 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teo Hansen Selnø A, Schlichtner S, Yasinska IM, et al. High mobility group box 1 (HMGB1) induces Toll-like receptor 4-mediated production of the immunosuppressive protein galectin-9 in human cancer cells. Front Immunol 2021;12:675731. 10.3389/fimmu.2021.675731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herbert A, Fedorov A, Poptsova M. Mono a Mano: ZBP1's love-hate relationship with the kissing virus. Int J Mol Sci 2022;23:3079. 10.3390/ijms23063079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan MJ, Kim Y-S. The serine threonine kinase RIP3: lost and found. BMB Rep 2015;48:303–12. 10.5483/BMBRep.2015.48.6.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarantopoulos J, Mahalingam D, Sharma N, et al. Results of a completed phase I trial of CBL0137 administered intravenously (IV) to patients (PTS) with advanced solid tumors. JCO 2020;38:3583–83. 10.1200/JCO.2020.38.15_suppl.3583 [DOI] [Google Scholar]
- 40.Behan FM, Iorio F, Picco G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019;568:511–6. 10.1038/s41586-019-1103-9 [DOI] [PubMed] [Google Scholar]
- 41.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. 10.1186/s13073-017-0424-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siddiqui J, Miles WO. RNA editing signatures identify melanoma patients who respond to pembrolizumab or nivolumab treatment. Transl Oncol 2021;14:101197. 10.1016/j.tranon.2021.101197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janji B, Hasmim M, Parpal S, et al. Firing up the cold tumors by targeting Vps34. Oncoimmunology 2020;9:1809936. 10.1080/2162402X.2020.1809936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogel OA, Han J, Liang C-Y, et al. The p150 isoform of ADAR1 blocks sustained RLR signaling and apoptosis during influenza virus infection. PLoS Pathog 2020;16:e1008842. 10.1371/journal.ppat.1008842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muendlein HI, Connolly WM, Magri Z, et al. ZBP1 promotes inflammatory responses downstream of TLR3/TLR4 via timely delivery of RIPK1 to TRIF. Proc Natl Acad Sci U S A 2022;119:e2113872119. 10.1073/pnas.2113872119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishizuka JJ, Manguso RT, Cheruiyot CK, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019;565:43–8. 10.1038/s41586-018-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-005704supp001.pdf (1.2MB, pdf)