

Abstract
Rationale
Although persistent fibroblast activation is a hallmark of idiopathic pulmonary fibrosis (IPF), mechanisms regulating persistent fibroblast activation in the lungs have not been fully elucidated.
Objectives
On the basis of our observation that lung fibroblasts express TBXA2R (thromboxane–prostanoid receptor) during fibrosis, we investigated the role of TBXA2R signaling in fibrotic remodeling.
Methods
We identified TBXA2R expression in lungs of patients with IPF and mice and studied primary mouse and human lung fibroblasts to determine the impact of TBXA2R signaling on fibroblast activation. We used TBXA2R-deficient mice and small-molecule inhibitors to investigate TBXA2R signaling in preclinical lung fibrosis models.
Measurements and Main Results
TBXA2R expression was upregulated in fibroblasts in the lungs of patients with IPF and in mouse lungs during experimental lung fibrosis. Genetic deletion of TBXA2R, but not inhibition of thromboxane synthase, protected mice from bleomycin-induced lung fibrosis, thereby suggesting that an alternative ligand activates profibrotic TBXA2R signaling. In contrast to thromboxane, F2-isoprostanes, which are nonenzymatic products of arachidonic acid induced by reactive oxygen species, were persistently elevated during fibrosis. F2-isoprostanes induced TBXA2R signaling in fibroblasts and mediated a myofibroblast activation profile due, at least in part, to potentiation of TGF-β (transforming growth factor-β) signaling. In vivo treatment with the TBXA2R antagonist ifetroban reduced profibrotic signaling in the lungs, protected mice from lung fibrosis in three preclinical models (bleomycin, Hermansky-Pudlak mice, and radiation-induced fibrosis), and markedly enhanced fibrotic resolution after bleomycin treatment.
Conclusions
TBXA2R links oxidative stress to fibroblast activation during lung fibrosis. TBXA2R antagonists could have utility in treating pulmonary fibrosis.
Keywords: IPF, Hermansky-Pudlak syndrome, oxidative stress, transforming growth factor-β, thromboxane–prostanoid receptor
At a Glance Commentary
Scientific Knowledge on The Subject
Although persistent fibroblast activation and proliferation are hallmarks of idiopathic pulmonary fibrosis, mechanisms regulating this in the lungs have not been fully elucidated. The role of thromboxane–prostanoid signaling and its receptor, TBXA2R (thromboxane–prostanoid receptor), have not been investigated.
What This Study Adds to the Field
TBXA2R links oxidative stress to fibroblast activation during lung fibrosis. Blockade of TBXA2R prevents and treats lung fibrosis due to multiple causes (bleomycin, genetic, and radiation). TBXA2R antagonists could have utility in treating pulmonary fibrosis.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive syndrome characterized by destruction of the gas-exchange units of the lung and pathologic accumulation of extracellular matrix (ECM) (1, 2). Despite decades of work, the mechanisms driving progressive pulmonary fibrosis remain incompletely understood, and most patients with IPF succumb to their disease within 3–5 years of diagnosis (1). Injury to and dysfunction of the lung epithelium is hypothesized to play a prominent role in disease initiation (3, 4), while fibroblasts are the primary effector cell type producing pathologic ECM (5, 6). There is a clear need to better understand the molecular mechanisms responsible for persistent fibroblast activation in IPF to develop new, more effective therapies for this disease.
Prostaglandins and their receptors have been studied extensively in pulmonary fibrosis, with the majority of work focused on antifibrotic effects. Prostaglandin E2 (7), as well as prostacyclin (8, 9), is protective against bleomycin-induced fibrosis, and prostaglandin E2 production is reduced in IPF fibroblasts (10). In addition, prostaglandin D2 reduces bleomycin-induced inflammation and protects against fibrosis (11). Thromboxane A2 (TXA2) is classically produced by platelets and functions to induce platelet aggregation and smooth muscle contraction. Our group previously reported that a decreased prostacyclin:TXA2 ratio is associated with pulmonary fibrosis (12). We have also shown that antagonists of TBXA2R (thromboxane–prostanoid receptor) are protective from pulmonary hypertension-induced right ventricular remodeling (13) and cardiac fibrosis (14), further suggesting that TBXA2R could have a role in fibrotic remodeling in the lungs.
In these studies, we found that TBXA2R expression is increased in IPF lungs, particularly in fibroblasts, and drives persistent fibroblast activation. Using several preclinical models, we demonstrated that inhibition of TBXA2R signaling can ameliorate pulmonary fibrosis. Given the availability of safe and effective TBXA2R antagonists, our studies support the feasibility of TBXA2R-directed therapy for the treatment of pulmonary fibrosis.
Methods
Subjects and Samples
IPF tissue samples were obtained from explanted lungs removed at the time of lung transplantation. Nonfibrotic control tissue samples were obtained from lungs declined for organ donation.
Study Approval
All studies involving human samples were approved by the Vanderbilt Institutional Review Board (060165, 171657, and 192004). Subjects provided informed consent before the collection of samples used in these studies. All animal studies were approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Mouse Models
TBXA2R-floxed mice (15) were obtained from The Jackson Laboratory (#21985). These mice have loxp sites surrounding TBXA2R exon 2, which under the influence of Cre recombinase is excised, eliminating protein production. These mice were crossed to mice with tamoxifen-inducible cre expression under the control of the universally expressed Rosa26 (reverse orientation splice acceptor βgeo line 26) locus (Rosa26-CreER; The Jackson Laboratory #4847) (both on C57Bl/6 background) (16). In combination, these Rosa26-CreER+TBXA2Rf/f mice are referred to as TBXA2RiKO and have global deletion of TBXA2R when induced by tamoxifen. Hermansky-Pudlak syndrome (HPS) mice included “pale ear” mice (The Jackson Laboratory #525), which carry a constitutive mutation in Hps1 (HPS1 biogenesis of lysosomal organelles complex 3 subunit 1) (17), and “pearl” mice (The Jackson Laboratory #3215), which carry a mutation in Hps2, also called Ap3b1 (adaptor-related protein complex 3, beta 1 subunit) (18).
Bleomycin-Induced Fibrosis Models
Bleomycin (Hospira Inc.) was purchased from the Vanderbilt University Medical Center pharmacy. Bleomycin (0.04 U) in 100 μl saline was delivered by direct intratracheal (i.t.) instillation under anesthesia, as described previously (19–21). Tamoxifen-inducible transgenic mice were treated with tamoxifen (400 mg tamoxifen citrate per kilogram chow ad libitum) for 2 weeks before bleomycin instillation (4 wk before bleomycin instillation). Lungs were harvested after euthanasia by pentobarbital at designated time points. Right lungs were tied off and snap frozen for estimation of collagen and extraction of RNA and protein, and left lungs were inflated to 25 cm H2O with 10% neutral buffered formalin for histology.
Mice were randomized to receive either 25 mg/kg/day CPI211 (ifetroban; Cumberland Pharmaceuticals) in drinking water or normal drinking water (vehicle). In a follow-up experiment, 50 mg/kg/day ozagrel HCl hydrate (Combi-Blocks), a TXA2 synthase inhibitor, was given to mice, and several mice were alternatively treated with ethanol vehicle as negative control animals. This ozagrel dose is similar to that used in other studies (22) and produced a roughly fourfold decrease in thromboxane B2 (TXB2) concentrations in a control experiment (see Figure E5 in the online supplement). The final concentration of ethanol in drinking water was approximately 0.04%. All drugs were pretested for palatability to ensure normal consumption of drinking water. Mice were weighed and water was changed once a week.
Radiation-Induced Fibrosis Model
Male/female mice aged 10–12 weeks were randomly assigned to the vehicle- or ifetroban-treated group. Isoflurane-anesthetized mice were placed on a 37°C recirculating water heating pad, and the thorax was administered 17 Gy (300-kVp/10-mA X-rays) at 1.64 Gy/min, as we previously described (23). With the exception of the thorax, the entire animal was shielded by a custom lead block 2.5 cm thick.
Details of assays, measurements, and other analyses are included in the online supplement.
Statistics
Values are expressed as mean ± SEM, and sample size is given for each figure. Two-way ANOVA or one-way ANOVA followed by Tukey’s post hoc test or the Wilcoxon signed rank nonparametric test for experiments with n < 8 were performed in Prism (GraphPad) or JMP Pro 16 (SAS Institute) to determine statistical significance.
Results
TBXA2R Expression in Fibroblasts Is Increased during Lung Fibrosis in Mice and Humans
To determine the role of TBXA2R in pulmonary fibrosis, we first investigated TBXA2R expression in the lungs of patients with IPF. In lung tissue lysates, quantification of TBXA2R by western blot demonstrated significantly higher TBXA2R concentrations in IPF compared with control lungs (Figure 1A). By examining our recently published single-cell RNA sequencing data set generated from explanted lungs with pulmonary fibrosis and lungs from nonfibrotic control subjects (declined donors) (6), we observed that TBXA2R expression was detected in several different subsets of fibroblasts, in addition to endothelial cells and smooth muscle cells (Figure 1B). Consistent with transcriptomic data, dual immunofluorescence staining showed TBXA2R colocalized with a fibroblast marker (S100A4) in areas of fibrotic remodeling in IPF lung tissue sections, but costaining was not observed in lung parenchyma from nonfibrotic control subjects (Figure 1C). In mouse lung tissue, TBXA2R expression increased after bleomycin challenge as measured by western blot (Figure 1D). Similar to our observation in IPF lung tissue, TBXA2R expression was localized to areas of fibrosis and colocalized with the fibroblast marker S100A4 (Figure 1E). The observed upregulation of TBXA2R expression during fibrosis suggests that signaling through this receptor could affect fibrotic remodeling.
Figure 1.
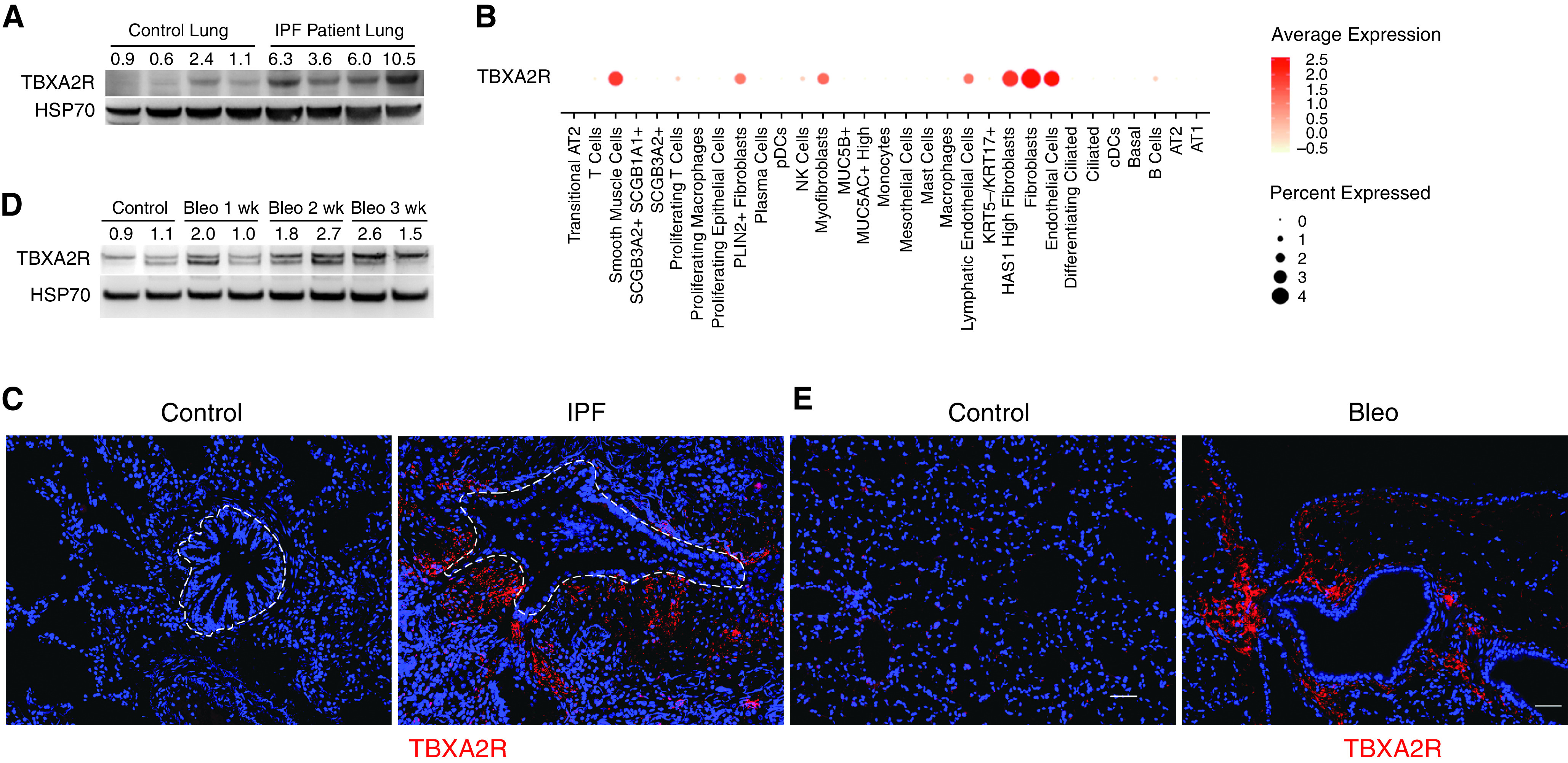
TBXA2R (thromboxane–prostanoid receptor) is upregulated in fibroblasts during lung fibrosis. (A) Western blot nondiseased for TBXA2R in lung tissue from patients with idiopathic pulmonary fibrosis (IPF) compared with nondiseased control subjects. (B) Dot plot depicting TBXA2R gene expression in jointly analyzed single-cell RNA sequencing data from explanted lungs of 20 patients with pulmonary fibrosis and 10 nondiseased control subjects (primary data from Reference 6). (C) Representative immunostaining for TBXA2R in IPF and control lung sections. TBXA2R is red, and vimentin is green. (D) Western blot for TBXA2R expression in lungs of mice at baseline and 1–3 weeks after intratracheal bleomycin. (E) Immunostaining for TBXA2R in mouse lung sections at 3 weeks after treatment with vehicle (control) or bleomycin. TBXA2R is red. Scale bar, 50 μm. AT1 = alveolar type 1; AT2 = alveolar type 2; Bleo = bleomycin; cDC = classical dendritic cell; HAS1 = hyaluronan synthase 1; HSP70 = heat shock protein 70; KRT5 = keratin 5; KRT17 = keratin 17; MUC5AC = mucin 5AC, oligomeric mucus/gel-forming; MUC5B = mucin 5B, oligomeric mucus/gel-forming; NK = natural killer; pDC = plasmacytoid dendritic cell; PLIN2 = perilipin 2; SCGB1A1 = secretoglobin family 1A member 1; SCGB3A2 = secretoglobin family 3A member 2.
To test the effects on TBXA2R signaling on lung fibrosis, we generated inducible TBXA2R-deficient mice by crossing TBXA2R-floxed mice (TBXA2Rf/f) with a universal tamoxifen-inducible Cre recombinase line (see Methods) (TBXA2RiKO hereafter). These mice develop normally and have no spontaneous phenotype except for a mildly increased bleeding time (15). Tamoxifen treatment (400 mg/kg chow ad libitum for 14 d) led to ∼75% reduction in TBXA2R protein in lung tissue (see Figures E1A and E1B). The resulting TBXA2RiKO and age-matched adult wild-type (WT) control animals were treated with tamoxifen and then challenged with i.t. bleomycin (0.04 U). At Day 21 after bleomycin, TBXA2RiKO mice were protected from lung fibrosis as determined by morphometric analysis (Figures 2A and 2B) and measurement of hydroxyproline content (Figure 2C) compared with control animals. To determine whether TXA2-mediated signaling through TBXA2R was responsible for protection from fibrosis in TBXA2RiKO mice, we used ozagrel, which is a highly efficacious small-molecule inhibitor of the enzyme responsible for generation of TXA2 (24). In these studies, treatment with ozagrel using a standard dosing strategy in drinking water from Days 7–21 after bleomycin did not cause a significant difference in fibrosis (Figures 2D–2F). Although the nonsignificant difference might become statistically significant with a larger number of animals, the difference in effect size between TBXA2RiKO (Figures 2B and 2C) and ozagrel (Figures 2E and 2F) suggests that an alternative ligand could be responsible for profibrotic TBXA2R signaling.
Figure 2.
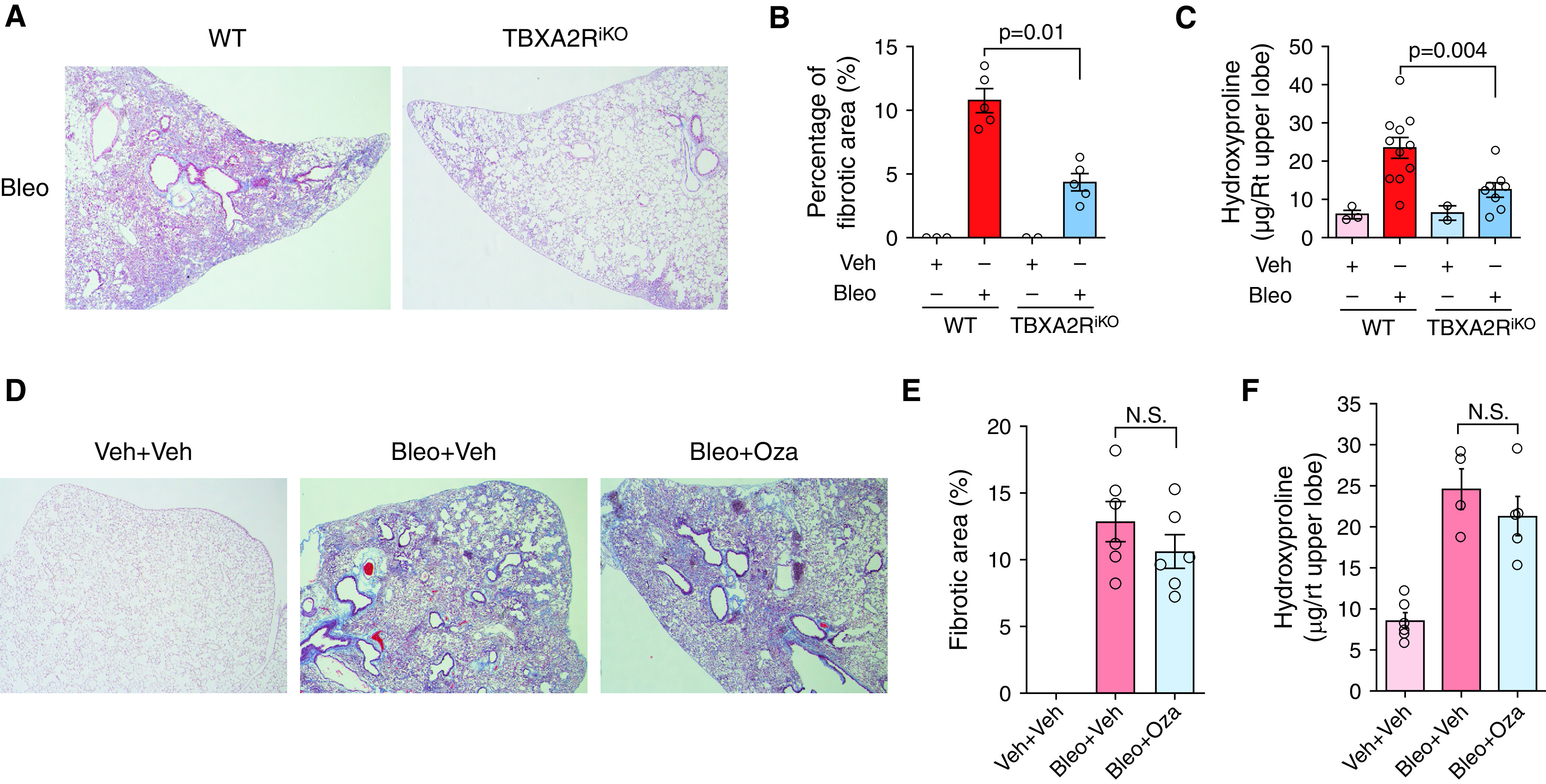
Deleting TBXA2R (thromboxane–prostanoid receptor) protects against experimental lung fibrosis. (A) Representative Masson’s trichrome–stained lung sections from wild-type (WT) and TBXA2RiKO (Rosa26-CreER+TBXA2Rf/f) mice at 21 days after intratracheal bleomycin (Bleo). (B) Morphometric evaluation of lung fibrosis on lung sections. n = 3 for control WT and TBXA2RiKO mice, and n = 5 for the Bleo treatment groups. (C) Hydroxyproline content in the right upper lobe. n = 3 for control WT mice, n = 2 for control TBXA2RiKO mice, n = 11 for the Bleo-treated WT mice, and n = 8 for the Bleo-treated TBXA2RiKO mice. Statistical comparison was performed using the Wilcoxon rank sum test (also P < 0.05 by two-way ANOVA with Tukey’s honestly significant difference test). (D) Representative Masson’s trichrome–stained lung sections from WT mice at 21 days after treatment with intratracheal Bleo or vehicle (Veh), with or without thromboxane synthesis inhibitor (Oza) beginning on Day 7. (E) Morphometric evaluation of lung fibrosis on lung sections. n = 6 for each group. (F) Hydroxyproline content in right upper lobe. n = 6 for each group. Error bars in all panels denote SEM. N.S. = not significant by Wilcoxon rank sum test; Oza = ozagrel; Rt = right.
F2-Isoprostanes Induce TBXA2R Signaling
F2-isoprostanes (F2-IsoPs) are nonenzymatic products of free radical–induced peroxidation of arachidonic acid (25) that have been reported to be increased in the lungs of patients with IPF (26). F2-IsoPs have structural similarities to TXA2 and can activate TBXA2R signaling (27). We measured F2-IsoPs and TXB2, the major stable metabolite of TXA2, in the lungs of mice at baseline and after i.t. bleomycin. TXB2 was increased in lung tissue by Day 1 after bleomycin and subsequently returned toward baseline by Day 7 (Figure 3A). In contrast, F2-IsoPs were increased in the lungs throughout the 21-day time course (Figure 3B).
Figure 3.
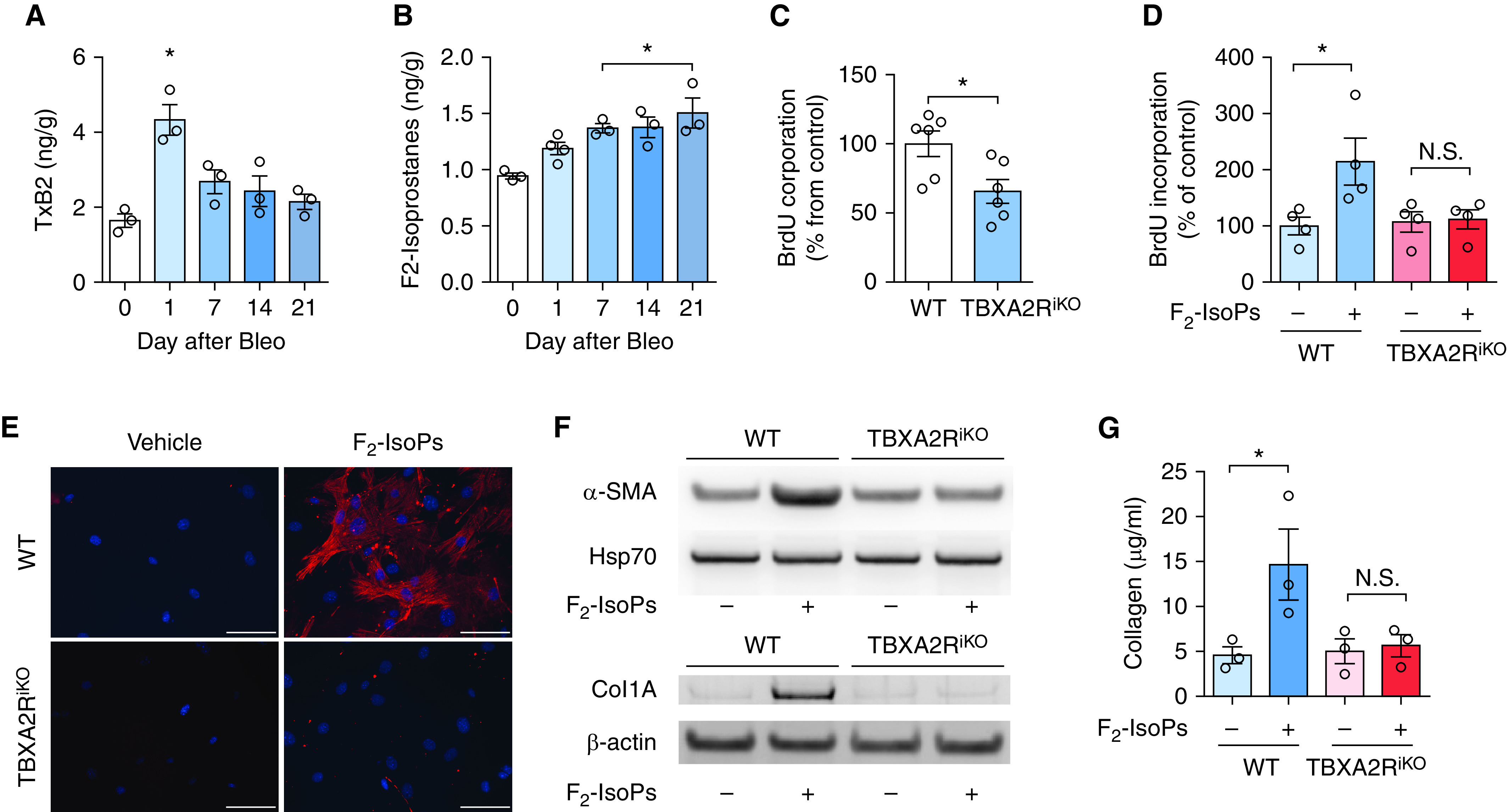
F2-isoprostanes (F2-IsoPs) activate TBXA2R signaling in fibrosis. (A) TxB2 was measured in the lungs of mice at baseline and 1–3 weeks after bleomycin (Bleo). n = 3 per group. *P < 0.05 compared with baseline. (B) F2-IsoPs measured in the lungs of mice at baseline and 1–3 weeks after intratracheal Bleo. n = 3 or 4 per group. *P < 0.05 compared with baseline. (C) Primary lung fibroblasts from Rosa-creER/Tbxa2rf/f mice were treated ex vivo with 4OH-tamoxifen to obtain TBXA2R-deleted fibroblasts (TBXA2RiKO) and BrdU incorporation was measured at 48 hours under full-serum (10%) conditions. n = 6 mice per group. *P < 0.05 compared with wild-type (WT) fibroblasts. (D) F2-IsoP (100 nM) stimulation of WT and Tbxa2riKO fibroblasts under reduced serum conditions (2.5%) measured at 48 hours. n = 4 per group. *P < 0.05. (E) Immunocytochemistry evaluating for α-SMA (α-smooth muscle actin) expression 48 hours after F2-IsoP treatment. α-SMA, red; DAPI, blue. Scale bars, 40× image with 100 μm. (F) Western blots for α-SMA and collagen 1 expression at 48 hours after F2-IsoP treatment. (G) Densitometry for collagen 1. n = 3 lungs per condition. All comparisons were significant both by nonparametric (Wilcoxon rank sum) and parametric (ANOVA with post hoc test) measures. Error bars denote SEM. BrdU = bromodeoxyuridine; Hsp70 = heat shock protein 70; N.S. = not significant; TxB2 = thromboxane B2; TBXA2R = thromboxane–prostanoid receptor.
To determine whether F2-IsoPs were responsible for TBXA2R-driven profibrotic phenotypes in lung fibroblasts, we isolated mouse lung fibroblasts (MLFs) from WT and tamoxifen-treated TBXA2RiKO mice and cultured these cells in serum-containing media, which contains isoprostanes (28) as well as latent TGF-β (transforming growth factor-β) (29). When grown on tissue culture plates in 10% serum-containing media, TBXA2RiKO MLFs had ∼50% less proliferation compared with WT MLFs (Figure 3C). Under low-serum conditions (2.5%), however, TBXA2RiKO MLFs had similar bromodeoxyuridine incorporation compared with WT MLFs. In low-serum conditions, addition of F2-IsoPs (100 nM; Cayman Chemicals) to the culture medium enhanced proliferation of WT but not TBXA2R-deficient MLFs (Figure 3D). In addition, treatment with F2-IsoPs induced α-SMA (α-smooth muscle actin) expression in WT MLFs but not TBXA2RiKO MLFs (Figures 3E and 3F). We also measured collagen 1 expression by western blot (Figure 3G) and collagen accumulation in media (Figure 3H) and found that F2-IsoPs upregulated collagen synthesis and secretion by WT MLFs but not TBXA2RiKO MLFs. Together, these studies indicate that F2-IsoPs can activate fibroblast proliferation and induce myofibroblast differentiation via TBXA2R signaling.
TBXA2R Activation Potentiates TGF-β Signaling
As myofibroblast differentiation is classically induced by TGF-β, we wondered whether TBXA2R could regulate TGF-β signaling (30). We transfected WT and TBXA2RiKO MLFs with a Smad2/3-binding element–driven luciferase construct and added F2-IsoPs to activate TBXA2R signaling at 48 hours after transfection. In addition to stimulating increased luciferase activity (Figure 4A), western blots showed canonical and noncanonical TGF-β pathway activation in WT MLFs but not TBXA2RiKO MLFs (Figures 4B and 4C). Similarly, stimulation of fibroblasts with a specific TBXA2R agonist, U-46619 (100 nM; Cayman Chemicals), induced TGF-β pathway activation (see Figure E2). To test whether TGF-β receptors were involved in induction of the TGF-β pathway by F2-IsoPs, we treated WT MLFs with F2-IsoPs with or without addition of the TGF-β receptor inhibitor LY210976. As shown in Figure 4D, F2-IsoP treatment resulted in robust induction of TGF-β targets SerpinE1 (serpin family E member 1) and Timp1 (tissue inhibitor of metalloproteinase-1), which was prevented by LY2109761 (Figure 4D). These studies indicate that F2-IsoPs promote fibroblast activation through TBXA2R-mediated potentiation of TGF-β signaling.
Figure 4.

F2-isoprostane (F2-IsoP) induction of the TGF-β pathway requires TBXA2R. (A) Luciferase assay was performed to quantify Smad2/3 transcriptional activity after transfection with Smad2/3-binding element–driven luciferase reporter construct using wild-type (WT) and Tbxa2riKO (Rosa26-CreER+TBXA2Rf/f) lung fibroblasts at 8 hours after F2-IsoP or vehicle treatment. n = 3 for each group. *P < 0.05. (B) Western blot for T-Smad2, P-Smad2, and Timp1 (tissue inhibitor of metalloproteinase-1) using WT and Tbxa2riKO lung fibroblasts at 24 hours after F2-IsoP or vehicle treatment. (C) Western blot for T-Akt, P-Akt, T-p44/42, and P-p44/42 using WT and Tbxa2riKO lung fibroblasts at 24 hours after F2-IsoP or vehicle treatment. Western blots were performed in triplicate samples and repeated at least twice. (D) WT mouse lung fibroblasts were treated with or without F2-IsoPs (100 nM) and LY2109761, an inhibitor of TGF-β receptor type 1/type II kinases, for 24 hours. Quantitative PCR analysis for Serpine1 and Timp1 gene expression was performed. n = 3 for each group. *P < 0.05. Significance was determined using the Wilcoxon rank sum test. Error bars denote SEM. Ctrl = control; Hsp70 = heat shock protein 70; N.S. = not significant; P-Akt = phospho-Akt; P-p44/42 = phospho-p44/42; P-Smad2 = phospho-Smad2; SERPINE1 = serpin family E member 1; T-Akt = total Akt; TBXA2R = thromboxane–prostanoid receptor; TGF-β = transforming growth factor-β; T-p44/42 = total p44/42; T-Smad2 = total Smad2.
A Small-Molecule Inhibitor of TBXA2R Signaling Blocks Fibroblast Activation
For these studies, we isolated MLFs from WT mice and stimulated them with F2-IsoPs in the presence of a small-molecule TBXA2R antagonist (ifetroban 0.3 μM) or vehicle. Ifetroban (originally known as BMS-180,291) is a highly selective, potent (half maximal inhibitory concentration ∼ 1,020 nM), orally available TBXA2R antagonist (31). Ifetroban treatment reduced Smad2/3, p44/42, and Akt phosphorylation, as well as Timp1 and α-SMA protein expression (Figure 5A).
Figure 5.

TBXA2R antagonism reduces proliferation, migration, and activation of fibroblasts from lungs of mice and patients with IPF. (A) Lung fibroblasts were isolated from lungs of mice and treated with F2-isoprostanes, ifetroban (Ifet), or both. Western blots are shown for evaluation of α-SMA, total Smad2/3 (T-Smad2/3), phospho-Smad2/3 (P-Smad2/3), Timp1 (tissue inhibitor of metalloproteinase-1), total p44/42 (T-p44/42), phospho-p44/42 (P-p44/42), total Akt (T-AKT), and phospho-Akt (P-AKT). (B–E) Lung fibroblasts were isolated from explanted lungs of six patients with IPF and treated with Ifet (300 nM) or vehicle. (B) Scratch wound closure assay measured as wound area closed at 24 hours. *P < 0.05 by Wilcoxon signed rank test. Error bars denote SEM. (C) Cell proliferation measured by BrdU incorporation. *P < 0.05 by Wilcoxon signed rank test. Error bars denote SEM. (D) Quantitative PCR for evaluation of profibrotic gene expression. Lines connect cells from the same patient with and without Ifet. Average of vehicle controls was set to 1. n = 6 per group. *P < 0.05 compared with vehicle-treated group by paired t test (each treated culture was paired to the same culture with no treatment). (E) Western blot for evaluation of T-Smad2/3, P-Smad2/3, Timp1, T-p44/42, P-p44/42, T-AKT, and P-AKT. ACTA2 = actin alpha 2, smooth muscle; α-SMA = α-smooth muscle actin; BrdU = bromodeoxyuridine; COL1A1 = collagen type I alpha 1 chain; COL1A2 = collagen type I alpha 2 chain; Hsp70 = heat shock protein 70; IPF = idiopathic pulmonary fibrosis; SERPINE1 = serpin family E member 1; TBXA2R = thromboxane–prostanoid receptor; Veh = vehicle.
Next, we examined the impact of TBXA2R antagonism on the activated fibroblast phenotype of human lung fibroblasts isolated from explanted lungs of patients with IPF. In these studies, ifetroban treatment (0.3 μM) significantly inhibited migration (scratch wound closure) (Figure 5B); proliferation (Figure 5C); expression of profibrotic genes, including α-SMA, collagen type 1, and serpin E1; and activation of profibrotic signaling via Smad2/3, extracellular-regulated kinase (p42/44), and AKT pathways (Figures 5D and 5E). Together, these studies further support the conclusion that signaling through TBXA2R regulates important profibrotic functions of activated lung fibroblasts from mice and humans.
Ifetroban Treatment Attenuates Fibrosis in Mouse Models of Lung Fibrosis
To test whether pharmacological inhibition of TBXA2R could be used therapeutically to prevent or treat lung fibrosis, we treated WT mice (C57Bl/6 background) with ifetroban (25 mg/kg/day in drinking water) or standard drinking water (vehicle) beginning 3 days before a single i.t. injection of bleomycin (0.04 U) or beginning on Day 7 after bleomycin and continuing until harvest at Day 21 (Figure 6A, top two lines). Ifetroban treatment blocked bleomycin-induced fibrosis when administered for the entire 21-day course or administered only during the fibrotic phase (Days 7–21), as assessed by morphometric analysis (Figures 6B and 6C) and hydroxyproline content (a major component of collagen) (Figure 6D). Although epithelial apoptosis and inflammation have been correlated with subsequent fibrosis (20, 21, 32, 33) in the bleomycin model, ifetroban did not affect epithelial apoptosis (Figures E3A and E3B) or inflammatory cell recruitment (Figure E4).
Figure 6.
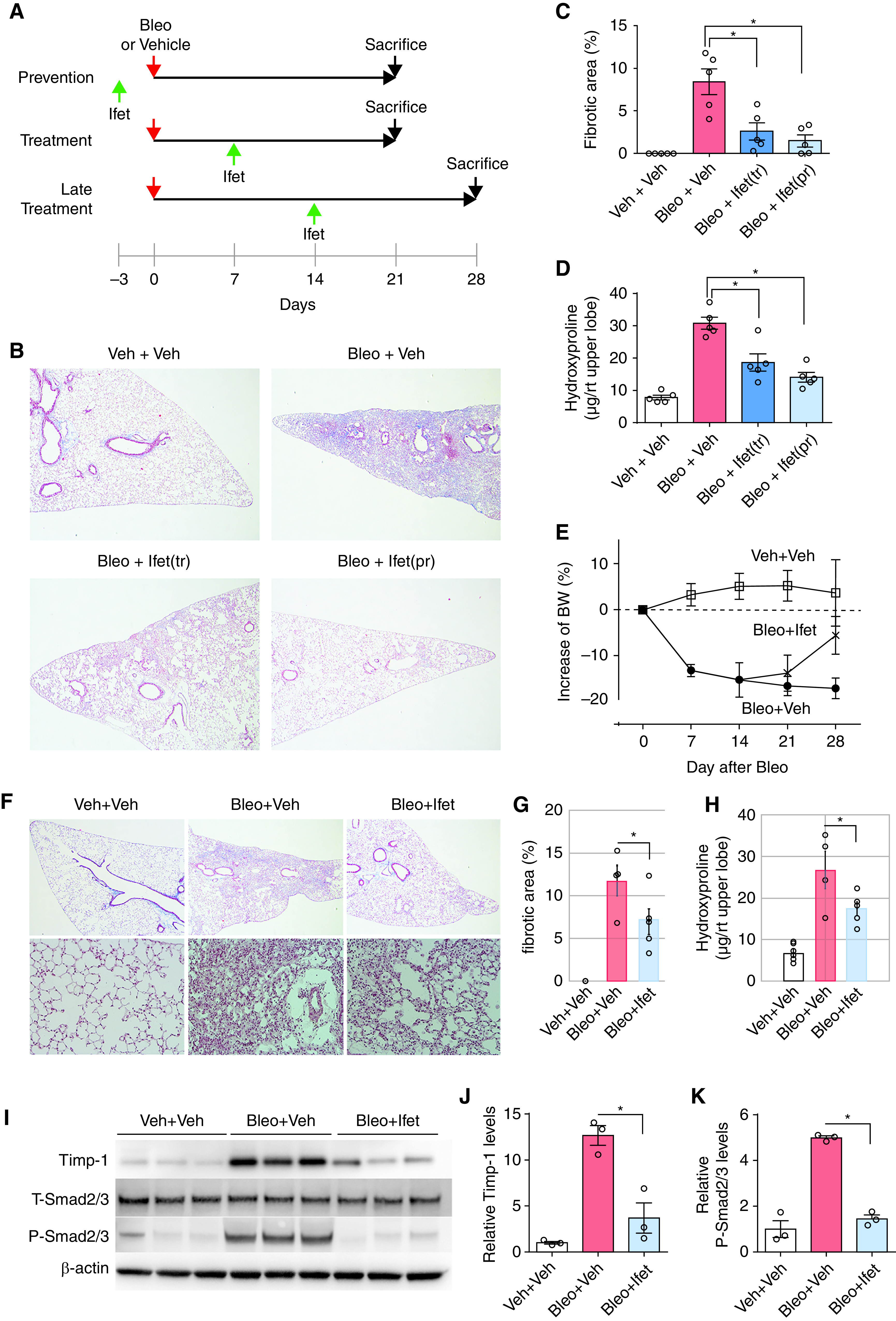
TBXA2R signaling regulates bleomycin (Bleo)–induced fibrosis, even when started 7 and 14 days after injury. (A) Experimental schema showing the timing of ifetroban (Ifet) start and date of sacrifice for panels. The top two lines correspond to B–D in this figure and the bottom line to E–K. (B–D) Wild-type mice were treated with intratracheal Bleo (0.04 U) or vehicle (Veh), with or without Ifet (25 mg/kg/day). Ifet treatment (Ifet[tr]) was delivered from Day 7 to Day 21 after Bleo. Ifet prevention (Ifet[pr]) was started 3 days before Bleo treatment and continued to Day 21. (B) Representative Masson’s trichrome–stained lung sections. (C) Morphometric evaluation of lung fibrosis using these lung sections. (D) Hydroxyproline content in right upper lobe. *P < 0.05 by Wilcoxon signed rank test. (E) Change in weight after Bleo treatment. Data are presented as mean ± SEM. Significance was determined using ANOVA with repeated measures. (F) Representative Masson’s trichrome–stained lung sections from wild-type mice at 28 days after treatment with intratracheal Bleo or Veh with or without Ifet treatment begun at Day 14. (G) Morphometric evaluation of lung fibrosis on lung sections. Data are presented as mean ± SEM. (H) Hydroxyproline content quantified from the right upper lobe. Data are presented as mean ± SEM. *P = 0.05 by Wilcoxon signed rank test. (I) Western blot for Timp-1 (tissue inhibitor of metalloproteinase-1), phospho-Smad2/3, and total Smad2/3 in lung tissue. (J) Densitometry for Timp-1. *P < 0.05 by Wilcoxon signed rank test. (K) Densitometry for total and phospho-Smad2/3 (n = 5 per group). *P < 0.05 by Wilcoxon signed rank test. Error bars denote SEM. BW = body weight; P-Smad2/3 = phospho-Smad2/3; rt = right; TBXA2R = thromboxane–prostanoid receptor; T-Smad2/3 = total Smad2/3.
To determine whether ifetroban treatment could hasten the resolution of fibrosis, we performed additional experiments in which drug or vehicle was started on Day 14 and continued until Day 28 (Figure 6a, bottom line). In these studies, ifetroban attenuated weight loss (Figure 6E) and accelerated the resolution of fibrosis (Figures 6F–6H). Consistent with an antifibrotic effect, proteins associated with fibrosis, including Timp1 and Smad2 phosphorylation downstream of canonical TGF-β signaling, were both strongly induced by bleomycin and blocked by ifetroban treatment (Figures 6I–6K). Cumulatively, studies in the bleomycin model showed that ifetroban reduces fibrosis and enhances resolution when given during the fibrotic phase of bleomycin-induced fibrosis without affecting bleomycin-induced inflammation or epithelial apoptosis, thereby suggesting direct effects of TBXA2R signaling on fibrogenesis.
We next tested whether ifetroban could prevent fibrosis in a model of genetic susceptibility to lung fibrosis, HPS. HPS is an autosomal recessive disease in which several of the genetic subtypes (HPS1, HPS2, and HPS4) have highly penetrant pulmonary fibrosis with onset in early adulthood (34). Naturally occurring mutations in HPS mice reliably model important features of the human disease, including susceptibility to profibrotic stimuli (35–37). To induce fibrosis in HPS mice, low doses of bleomycin (0.025 U) delivered by i.t. injection result in a rapidly progressive pulmonary fibrosis phenotype (35–37). We treated HPS1 mice and HPS2 mice (with loss-of-function mutations in the HPS1 gene and the HPS2 gene, respectively) with ifetroban beginning on the day before i.t. bleomycin and continuing until Day 7 after bleomycin, when lungs were harvested. Ifetroban treatment significantly attenuated lung fibrosis in HPS1 (Figure 7A) and HPS2 mice (Figures 7B and 7C) as determined by measurement of lung collagen.
Figure 7.
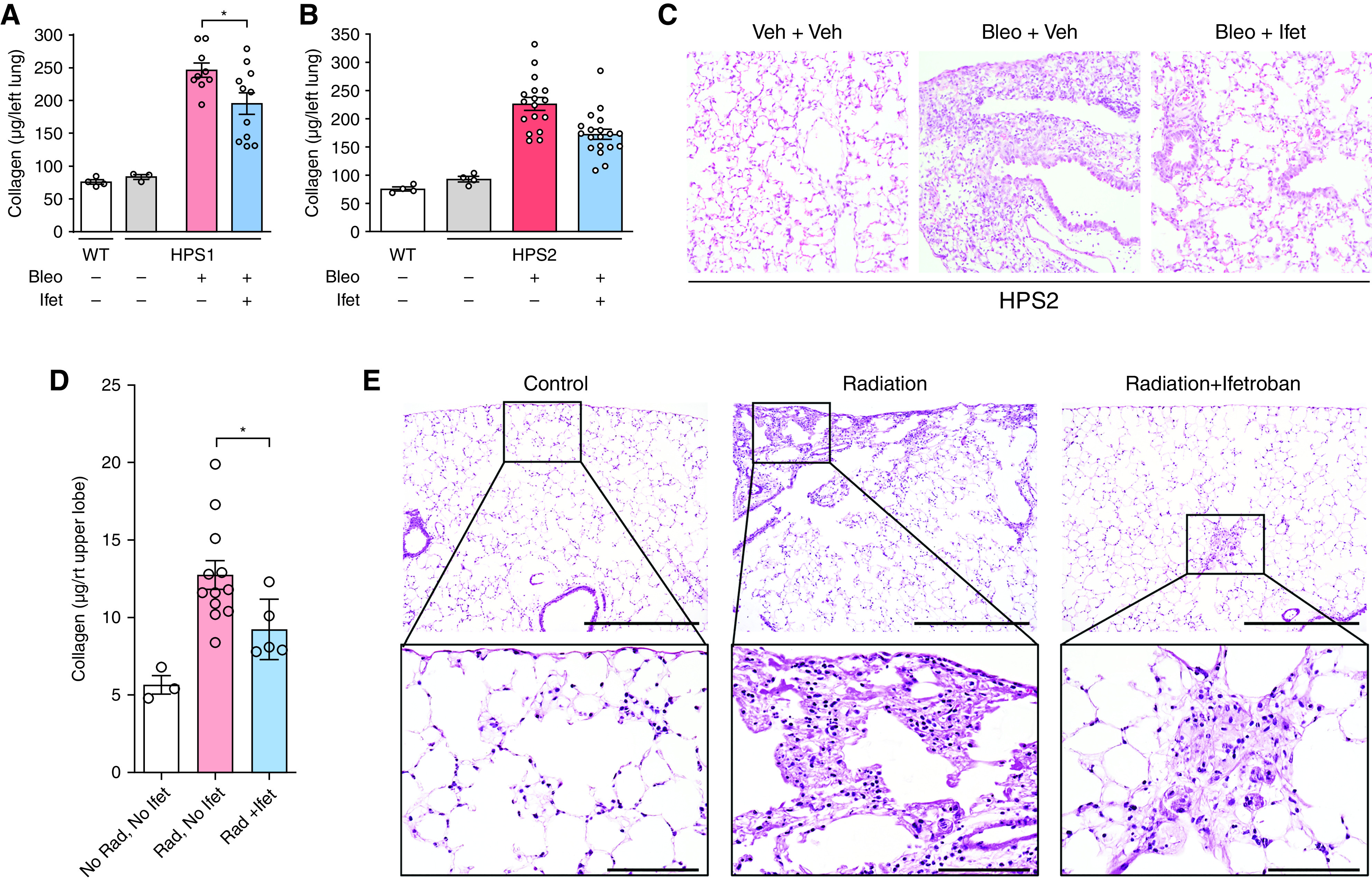
TBXA2R antagonism attenuates Hermansky-Pudlak syndrome (HPS)-associated fibrosis and radiation-induced fibrosis. (A and B) HPS1, HPS2, or wild-type control mice were treated with ifetroban (Ifet) or vehicle, followed by intratracheal bleomycin (0.025 U), and lungs were harvested on Day 7. Sircol assay was conducted to quantify collagen content in lungs of HPS1 mice or HPS2 mice. n = 3 or 4 per group unchallenged mice and 9–17 per group HPS mice. (C) Representative histology for HPS2 mice with or without Ifet pretreatment. (D and E) Sircol assay quantifying collagen content (D) and representative histology from lungs of mice exposed to thoracic irradiation (17 Gy) together with Ifet (n = 5) or vehicle (n = 12) and harvested at 4 months after irradiation (E). Top row, scale bars, 4× image with 1000 μm; bottom row, scale bars, 40× with 100 μm. *P < 0.05. For all quantitative figure elements, data are presented as mean ± SEM, with data from each individual mouse overlaid as circles. Statistical analysis was performed using ANOVA, followed by Tukey’s honestly significant difference test for comparisons between individual groups (*P < 0.05). Bleo = bleomycin; Rad = radiation; TBXA2R = thromboxane–prostanoid receptor; Veh = vehicle; WT = wild-type.
We also used a model of radiation-induced fibrosis to investigate the antifibrotic effects of ifetroban. Immediately after thoracic irradiation (17 Gy), mice were administered ifetroban in drinking water or vehicle and continued treatment until lungs were harvested 4 months after irradiation. Compared with placebo, ifetroban-treated mice had significantly reduced lung collagen content after exposure to ionizing radiation (Figures 7D and 7E). In summary, treatment with the small-molecule TBXA2R inhibitor ifetroban was beneficial in three preclinical models of lung fibrosis, thereby underscoring the promise of TBXA2R as a therapeutic target for lung fibrosis.
Discussion
Transient fibroblast activation is an adaptive process that is crucial for wound healing and other forms of injury repair. Although considerable progress has been made in understanding the signaling and molecular processes that activate fibroblasts in the lung and other organs during injury repair, the mechanisms that lead to persistent and pathologic fibroblast activation remain less well defined. In these studies, we demonstrate a novel paradigm linking oxidative stress to persistent fibroblast activation through F2-IsoP–induced TBXA2R signaling, which is a previously unrecognized regulator of the TGF-β pathway.
In the context of lung fibrosis, reactive oxygen species (ROS) are produced by a variety of cells, including epithelial cells (38, 39) and fibroblasts (40, 41). Reduced NADP oxidase 4 has been shown to be an important source of ROS in lung fibrosis (42, 43). ROS have been shown to enhance fibroblast proliferation, induce collagen formation (41), and promote apoptosis resistance (43–45). F2-IsoPs are produced by nonenzymatic conversion of arachidonic acid in the presence of ROS (25, 26) and have been shown to function as an alternative ligand for TBXA2R (46, 47). F2-IsoPs are increased in serum, BAL, and exhaled breath condensate from patients with IPF (48–50) and have previously been suggested to contribute to bleomycin-induced fibrosis in rats (51). Our findings indicate that F2-IsoPs can mediate the effects of ROS on fibroblasts and that F2-IsoP–induced fibroblast activation can be prevented by TBXA2R antagonism or genetic deletion.
Although TBXA2R performs diverse functions beyond platelet biology, its role in lung fibrosis has not been previously investigated. In our studies, we found that TBXA2R signaling in fibroblasts modulates TGF-β signaling. Although TGF-β pathway activation in fibroblasts is typically mediated by the serine/threonine kinase activity of TβRI (TGF-β receptor I), receptor activation independent of ligand (TGF-β)–mediated interactions between TβRII (TGF-β receptor II) and TβRI has been reported (52, 53). In this regard, G protein–coupled receptors other than TBXA2R, including G protein–coupled receptors for endothelin-1 and thrombin, have been reported to induce Smad2/3 phosphorylation in smooth muscle and endothelial cells, respectively (54, 55). We also saw an increase in both MAPK and AKT phosphorylation with isoprostanes, inhibited by TBXA2R blockade. This could be a downstream effect of TGF-β activation; mitogen-activated protein kinase and AKT have both been reported to be nontranscriptionally activated by TGF-β (56–58). However, we also cannot rule out direct regulation of these pathways by TBXA2R signaling. Elucidation of molecular mechanisms linking TBXA2R activation to these pathways will require additional studies.
Although these data provide compelling evidence that TBXA2R signaling is involved in pulmonary fibrosis, there are important limitations of these studies. First, there are likely cell type–specific effects of TBXA2R. Both our studies and other recent single-cell RNA sequencing studies (5, 6, 59) found TBXA2R in a variety of other pulmonary cell types, with the exact concentration varying by study. Many of these cell types, including endothelial cells, smooth muscle cells, and platelets, may also affect fibrogenesis in the lungs. Further studies will be required to discern the effects of TBXA2R antagonism in these cell types of fibrogenesis. Second, we have not conclusively shown that F2-IsoPs are the major ligand for activation of TBXA2R signaling during in vivo lung fibrosis. This would require a strategy to reduce ROS, which would likely have additional effects on lung fibrosis not mediated directly through TBXA2R. Third, additional work is required to determine whether TBXA2R is an important therapeutic target in fibrotic lung diseases other than IPF and fibrotic conditions in other organs.
Together, these studies have demonstrated a key role of TBXA2R signaling in pulmonary fibrosis, and TBXA2R represents an attractive target with translational potential. In these experiments, we demonstrate that the small molecule ifetroban effectively inhibits TBXA2R signaling and pulmonary fibrosis in multiple experimental models. Ifetroban and other TBXA2R antagonists have been or are currently in human studies for the secondary prevention of coronary artery disease (60), allergic asthma (61), and other conditions (62–65), and appear to be safe and well-tolerated. Therapies aimed at interrupting TBXA2R signaling have the potential for rapid translation into clinical trials with the goal of improving outcomes for patients with IPF.
Footnotes
Supported by NIH grants R01HL151016 (T.S.B.), R01HL135011 (E.J.C.), R01HL095797 (J.D.W.), P01HL108800 (J.D.W.), P01HL092870 (T.S.B.), R01HL145372 (J.A.K. and N.E.B.), R01HL119503 (L.R.Y.), K24HL143281 (L.R.Y.), and T32HL094296 (N.I.W.); NHLBI grant K08HL130595 (J.A.K.); and the Doris Duke Charitable Foundation (J.A.K.). Ifetroban was provided by Cumberland Pharmaceuticals.
Author Contributions: T.S. performed experiments, drafted the manuscript, and had intellectual input into experimental design. J.C., X.C., T.P.S., V.V.P., S.G., W.H., A.R., N.I.W., J.E.C., and H.T. assisted with specific experiments and assays. J.A.K. and N.E.B. provided and analyzed single-cell RNA sequencing data. M.L.F. assisted with radiation experiments. L.R.Y. and P.G. performed the Hermansky-Pudlak syndrome mouse experiments. Y.T., E.J.C., C.S., J.J.G., J.A.K., T.S.B., and J.D.W. conceived of the project, designed experiments, and drafted and revised the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202106-1503OC on June 28, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med . 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 2. Hewlett JC, Kropski JA, Blackwell TS. Idiopathic pulmonary fibrosis: epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol . 2018;71-72:112–127. doi: 10.1016/j.matbio.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pardo A, Selman M. Idiopathic pulmonary fibrosis: new insights in its pathogenesis. Int J Biochem Cell Biol . 2002;34:1534–1538. doi: 10.1016/s1357-2725(02)00091-2. [DOI] [PubMed] [Google Scholar]
- 4. Winters NI, Burman A, Kropski JA, Blackwell TS. Epithelial injury and dysfunction in the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci . 2019;357:374–378. doi: 10.1016/j.amjms.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, et al. Single cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv . 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, et al. Single-cell RNA-sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv . 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moore BB, Ballinger MN, White ES, Green ME, Herrygers AB, Wilke CA, et al. Bleomycin-induced E prostanoid receptor changes alter fibroblast responses to prostaglandin E2. J Immunol . 2005;174:5644–5649. doi: 10.4049/jimmunol.174.9.5644. [DOI] [PubMed] [Google Scholar]
- 8. Zhu Y, Liu Y, Zhou W, Xiang R, Jiang L, Huang K, et al. A prostacyclin analogue, iloprost, protects from bleomycin-induced pulmonary fibrosis in mice. Respir Res . 2010;11:34. doi: 10.1186/1465-9921-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, et al. COX-2-derived prostacyclin protects against bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2006;291:L144–L156. doi: 10.1152/ajplung.00492.2005. [DOI] [PubMed] [Google Scholar]
- 10. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest . 1995;95:1861–1868. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kida T, Ayabe S, Omori K, Nakamura T, Maehara T, Aritake K, et al. Prostaglandin D2 attenuates bleomycin-induced lung inflammation and pulmonary fibrosis. PLoS ONE . 2016;11:e0167729. doi: 10.1371/journal.pone.0167729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cruz-Gervis R, Stecenko AA, Dworski R, Lane KB, Loyd JE, Pierson R, et al. Altered prostanoid production by fibroblasts cultured from the lungs of human subjects with idiopathic pulmonary fibrosis. Respir Res . 2002;3:17. doi: 10.1186/rr166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. West JD, Voss BM, Pavliv L, de Caestecker M, Hemnes AR, Carrier EJ. Antagonism of the thromboxane-prostanoid receptor is cardioprotective against right ventricular pressure overload. Pulm Circ . 2016;6:211–223. doi: 10.1086/686140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. West JD, Galindo CL, Kim K, Shin JJ, Atkinson JB, Macias-Perez I, et al. Antagonism of the thromboxane-prostanoid receptor as a potential therapy for cardiomyopathy of muscular dystrophy. J Am Heart Assoc . 2019;8:e011902. doi: 10.1161/JAHA.118.011902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cyphert JM, Allen IC, Church RJ, Latour AM, Snouwaert JN, Coffman TM, et al. Allergic inflammation induces a persistent mechanistic switch in thromboxane-mediated airway constriction in the mouse. Am J Physiol Lung Cell Mol Physiol . 2012;302:L140–L151. doi: 10.1152/ajplung.00152.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci . 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, et al. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol . 2003;285:L643–L653. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- 18. Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT. Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics . 2000;69:370–379. doi: 10.1006/geno.2000.6350. [DOI] [PubMed] [Google Scholar]
- 19. Burman A, Kropski JA, Calvi CL, Serezani AP, Pascoalino BD, Han W, et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight . 2018;3:e99543. doi: 10.1172/jci.insight.99543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanjore H, Cheng D-S, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem . 2011;286:30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A . 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carter PR, McElhatten RM, Zhang S, Wright WS, Harris NR. Thromboxane-prostanoid receptor expression and antagonism in dextran-sodium sulfate-induced colitis. Inflamm Res . 2011;60:87–92. doi: 10.1007/s00011-010-0240-2. [DOI] [PubMed] [Google Scholar]
- 23. Traver G, Mont S, Gius D, Lawson WE, Ding GX, Sekhar KR, et al. Loss of Nrf2 promotes alveolar type 2 cell loss in irradiated, fibrotic lung. Free Radic Biol Med . 2017;112:578–586. doi: 10.1016/j.freeradbiomed.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dogné J-M, de Leval X, Benoit P, Delarge J, Masereel B. Thromboxane A2 inhibition: therapeutic potential in bronchial asthma. Am J Respir Med . 2002;1:11–17. doi: 10.1007/BF03257158. [DOI] [PubMed] [Google Scholar]
- 25. Montuschi P, Barnes PJ, Roberts LJ., II Isoprostanes: markers and mediators of oxidative stress. FASEB J . 2004;18:1791–1800. doi: 10.1096/fj.04-2330rev. [DOI] [PubMed] [Google Scholar]
- 26. Montuschi P, Ciabattoni G, Paredi P, Pantelidis P, du Bois RM, Kharitonov SA, et al. 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am J Respir Crit Care Med . 1998;158:1524–1527. doi: 10.1164/ajrccm.158.5.9803102. [DOI] [PubMed] [Google Scholar]
- 27. Gardi C, Arezzini B, Monaco B, De Montis MG, Vecchio D, Comporti M. F2-isoprostane receptors on hepatic stellate cells. Lab Invest . 2008;88:124–131. doi: 10.1038/labinvest.3700712. [DOI] [PubMed] [Google Scholar]
- 28. Schmitt D, Shen Z, Zhang R, Colles SM, Wu W, Salomon RG, et al. Leukocytes utilize myeloperoxidase-generated nitrating intermediates as physiological catalysts for the generation of biologically active oxidized lipids and sterols in serum. Biochemistry . 1999;38:16904–16915. doi: 10.1021/bi991623w. [DOI] [PubMed] [Google Scholar]
- 29. Oida T, Weiner HL. Depletion of TGF-β from fetal bovine serum. J Immunol Methods . 2010;362:195–198. doi: 10.1016/j.jim.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai J, Liu B, Guo T, Zhang Y, Wu X, Leng J, et al. Effects of thromboxane prostanoid receptor deficiency on diabetic nephropathy induced by high fat diet and streptozotocin in mice. Eur J Pharmacol . 2020;882:173254. doi: 10.1016/j.ejphar.2020.173254. [DOI] [PubMed] [Google Scholar]
- 31. Ogletree ML, Harris DN, Schumacher WA, Webb ML, Misra RN. Pharmacological profile of BMS 180,291: a potent, long-acting, orally active thromboxane A2/prostaglandin endoperoxide receptor antagonist. J Pharmacol Exp Ther . 1993;264:570–578. [PubMed] [Google Scholar]
- 32. Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, Boomershine CS, et al. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol . 2011;300:L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med . 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vicary GW, Vergne Y, Santiago-Cornier A, Young LR, Roman J. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Ann Am Thorac Soc . 2016;13:1839–1846. doi: 10.1513/AnnalsATS.201603-186FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Young LR, Gulleman PM, Bridges JP, Weaver TE, Deutsch GH, Blackwell TS, et al. The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am J Respir Crit Care Med . 2012;186:1014–1024. doi: 10.1164/rccm.201207-1206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Young LR, Gulleman PM, Short CW, Tanjore H, Sherrill T, Qi A, et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight . 2016;1:e88947. doi: 10.1172/jci.insight.88947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young LR, Pasula R, Gulleman PM, Deutsch GH, McCormack FX. Susceptibility of Hermansky-Pudlak mice to bleomycin-induced type II cell apoptosis and fibrosis. Am J Respir Cell Mol Biol . 2007;37:67–74. doi: 10.1165/rcmb.2006-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheresh P, Kim S-J, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta . 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anathy V, Lahue KG, Chapman DG, Chia SB, Casey DT, Aboushousha R, et al. Reducing protein oxidation reverses lung fibrosis. Nat Med . 2018;24:1128–1135. doi: 10.1038/s41591-018-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bocchino M, Agnese S, Fagone E, Svegliati S, Grieco D, Vancheri C, et al. Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS ONE . 2010;5:e14003. doi: 10.1371/journal.pone.0014003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, et al. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum . 2001;44:2653–2664. doi: 10.1002/1529-0131(200111)44:11<2653::aid-art445>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 42. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med . 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med . 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J . 2014;43:1448–1458. doi: 10.1183/09031936.00095113. [DOI] [PubMed] [Google Scholar]
- 45. Sanders YY, Liu H, Zhang X, Hecker L, Bernard K, Desai L, et al. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol . 2013;1:8–16. doi: 10.1016/j.redox.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bauer J, Ripperger A, Frantz S, Ergün S, Schwedhelm E, Benndorf RA. Pathophysiology of isoprostanes in the cardiovascular system: implications of isoprostane-mediated thromboxane A2 receptor activation. Br J Pharmacol . 2014;171:3115–3131. doi: 10.1111/bph.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Acquaviva A, Vecchio D, Arezzini B, Comporti M, Gardi C. Signaling pathways involved in isoprostane-mediated fibrogenic effects in rat hepatic stellate cells. Free Radic Biol Med . 2013;65:201–207. doi: 10.1016/j.freeradbiomed.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 48. Fois AG, Paliogiannis P, Sotgia S, Mangoni AA, Zinellu E, Pirina P, et al. Evaluation of oxidative stress biomarkers in idiopathic pulmonary fibrosis and therapeutic applications: a systematic review. Respir Res . 2018;19:51. doi: 10.1186/s12931-018-0754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Psathakis K, Mermigkis D, Papatheodorou G, Loukides S, Panagou P, Polychronopoulos V, et al. Exhaled markers of oxidative stress in idiopathic pulmonary fibrosis. Eur J Clin Invest . 2006;36:362–367. doi: 10.1111/j.1365-2362.2006.01636.x. [DOI] [PubMed] [Google Scholar]
- 50. Malli F, Bardaka F, Tsilioni I, Karetsi E, Gourgoulianis KI, Daniil Z. 8-isoprostane levels in serum and bronchoalveolar lavage in idiopathic pulmonary fibrosis and sarcoidosis. Food Chem Toxicol . 2013;61:160–163. doi: 10.1016/j.fct.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 51. Arezzini B, Vecchio D, Signorini C, Stringa B, Gardi C. F2-isoprostanes can mediate bleomycin-induced lung fibrosis. Free Radic Biol Med . 2018;115:1–9. doi: 10.1016/j.freeradbiomed.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 52. Wojciech S, Ahmad R, Belaid-Choucair Z, Journé AS, Gallet S, Dam J, et al. The orphan GPR50 receptor promotes constitutive TGFβ receptor signaling and protects against cancer development. Nat Commun . 2018;9:1216. doi: 10.1038/s41467-018-03609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dayati P, Rezaei HB, Sharifat N, Kamato D, Little PJ. G protein coupled receptors can transduce signals through carboxy terminal and linker region phosphorylation of Smad transcription factors. Life Sci . 2018;199:10–15. doi: 10.1016/j.lfs.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 54. Kamato D, Burch ML, Osman N, Zheng W, Little PJ. Therapeutic implications of endothelin and thrombin G-protein-coupled receptor transactivation of tyrosine and serine/threonine kinase cell surface receptors. J Pharm Pharmacol . 2013;65:465–473. doi: 10.1111/j.2042-7158.2012.01577.x. [DOI] [PubMed] [Google Scholar]
- 55. Burch ML, Ballinger ML, Yang SNY, Getachew R, Itman C, Loveland K, et al. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor. J Biol Chem . 2010;285:26798–26805. doi: 10.1074/jbc.M109.092767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guo H, Jian Z, Liu H, Cui H, Deng H, Fang J, et al. TGF-β1-induced EMT activation via both Smad-dependent and MAPK signaling pathways in Cu-induced pulmonary fibrosis. Toxicol Appl Pharmacol . 2021;418:115500. doi: 10.1016/j.taap.2021.115500. [DOI] [PubMed] [Google Scholar]
- 57. Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene . 2005;24:5742–5750. doi: 10.1038/sj.onc.1208928. [DOI] [PubMed] [Google Scholar]
- 58. Hamidi A, Song J, Thakur N, Itoh S, Marcusson A, Bergh A, et al. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Sci Signal . 2017;10:eaal4186. doi: 10.1126/scisignal.aal4186. [DOI] [PubMed] [Google Scholar]
- 59. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med . 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bots ML, Ford I, Lloyd SM, Laurent S, Touboul PJ, Hennerici MG, Prevention of Cerebrovascular and Cardiovascular Events of Ischemic Origin with Terutroban in Patients with a History of Ischemic Stroke or Transient Ischemic Attack Vascular Ultrasound Study Investigators Thromboxane prostaglandin receptor antagonist and carotid atherosclerosis progression in patients with cerebrovascular disease of ischemic origin: a randomized controlled trial. Stroke . 2014;45:2348–2353. doi: 10.1161/STROKEAHA.114.004775. [DOI] [PubMed] [Google Scholar]
- 61. Dogné J-M, de Leval X, Benoit P, Rolin S, Pirotte B, Masereel B. Therapeutic potential of thromboxane inhibitors in asthma. Expert Opin Investig Drugs . 2002;11:275–281. doi: 10.1517/13543784.11.2.275. [DOI] [PubMed] [Google Scholar]
- 62. Gaussem P, Reny J-L, Thalamas C, Chatelain N, Kroumova M, Jude B, et al. The specific thromboxane receptor antagonist S18886: pharmacokinetic and pharmacodynamic studies. J Thromb Haemost . 2005;3:1437–1445. doi: 10.1111/j.1538-7836.2005.01468.x. [DOI] [PubMed] [Google Scholar]
- 63. Malini PL, Strocchi E, Zanardi M, Milani M, Ambrosioni E. Thromboxane antagonism and cough induced by angiotensin-converting-enzyme inhibitor. Lancet . 1997;350:15–18. doi: 10.1016/S0140-6736(96)12045-6. [DOI] [PubMed] [Google Scholar]
- 64. Guth BD, Narjes H, Schubert H-D, Tanswell P, Riedel A, Nehmiz G. Pharmacokinetics and pharmacodynamics of terbogrel, a combined thromboxane A2 receptor and synthase inhibitor, in healthy subjects. Br J Clin Pharmacol . 2004;58:40–51. doi: 10.1111/j.1365-2125.2004.02083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hussein Z, Samara E, Locke CS, Orchard MA, Ringham GL, Granneman GR. Characterization of the pharmacokinetics and pharmacodynamics of a new oral thromboxane A2-receptor antagonist AA-2414 in normal subjects: population analysis. Clin Pharmacol Ther . 1994;55:441–450. doi: 10.1038/clpt.1994.54. [DOI] [PubMed] [Google Scholar]
- 66. West JD, Austin ED, Gaskill C, Marriott S, Baskir R, Bilousova G, et al. Identification of a common Wnt-associated genetic signature across multiple cell types in pulmonary arterial hypertension. Am J Physiol Cell Physiol . 2014;307:C415–C430. doi: 10.1152/ajpcell.00057.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hemnes AR, Zhao M, West J, Newman JH, Rich S, Archer SL, et al. Critical genomic networks and vasoreactive variants in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med . 2016;194:464–475. doi: 10.1164/rccm.201508-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PDM, Milne GL, Potter CMF, et al. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A . 2012;109:17597–17602. doi: 10.1073/pnas.1209192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc . 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- 70. Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, et al. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol . 2013;49:180–189. doi: 10.1165/rcmb.2012-0366MA. [DOI] [PMC free article] [PubMed] [Google Scholar]