

Abstract
Digenic Alport syndrome refers to the inheritance of pathogenic variants in COL4A5 plus COL4A3 or COL4A4 or in COL4A3 plus COL4A4. Where digenic Alport syndrome includes a pathogenic COL4A5 variant, the consequences depend on the sex of the affected individual, COL4A5 variant “severity,” and the nature of the COL4A3 or COL4A4 change. A man with a pathogenic COL4A5 variant has all his collagen IV α3α4α5-heterotrimers affected, and an additional COL4A3 or COL4A4 variant may not worsen disease. A woman with a pathogenic COL4A5 variant has on average 50% of her heterotrimers affected, which is increased to 75% with a further COL4A3 or COL4A4 variant and associated with a higher risk of proteinuria. In digenic Alport syndrome with pathogenic COL4A3 and COL4A4 variants, 75% of the heterotrimers are affected. The COL4A3 and COL4A4 genes occur head-to-head on chromosome 2, and inheritance is autosomal dominant when both variants affect the same chromosome (in cis) or recessive when they affect different chromosomes (in trans). This form of digenic disease results in increased proteinuria and a median age of kidney failure intermediate between autosomal dominant and autosomal recessive Alport syndrome. Previous guidelines have suggested that all pathogenic or likely pathogenic digenic variants should be identified and reported. Affected family members should be identified, treated, and discouraged from kidney donation. Inheritance within a family is easier to predict if the two variants are considered independently and if COL4A3 and COL4A4 variants are known to be inherited on the same or different chromosomes.
Keywords: Alport syndrome, kidney failure, genetic renal disease, proteinuria, collagen, COL4A3, COL4A4, COL4A5 genes, digenic Alport syndrome
Introduction
Alport syndrome (OMIM #301050) is the second most common cause of genetic kidney failure after polycystic kidney disease (1,2). Inheritance is X linked where there is a single pathogenic variant in the COL4A5 gene and autosomal recessive where there are pathogenic variants in both copies of the COL4A3 or COL4A4 gene. “Autosomal dominant” Alport syndrome is sometimes used where there is a heterozygous pathogenic COL4A3 or COL4A4 variant, but the term is a misnomer because there are no extrarenal or “syndromic” features. This condition is also known as thin basement membrane nephropathy (3). The three COL4A3–COL4A5 genes code for the collagen IV α3- to α5-chains, which form the collagen IV α3α4α5-heterotrimer (4). A network of these trimers forms the backbone of the basement membranes in the kidney, ear, and eye (4).
The distinguishing features of all forms of Alport syndrome are persistent hematuria, a family history of hematuria, and, sometimes, kidney failure together with basement membrane thinning or lamellation (5). Proteinuria and FSGS are recognized increasingly (6). Hearing loss, lenticonus, and fleck retinopathy are also common; however, the hearing loss may be coincidental, and the ocular abnormalities may not be recognized.
Men with X-linked Alport syndrome and men and women with autosomal recessive disease have a similar phenotype with kidney failure, hearing loss, lenticonus, and a fleck retinopathy. Ninety percent of men with X-linked disease have kidney failure by the age of 40 (7); and 15%–30% of women with X-linked disease have kidney failure by the age of 60. Hearing loss and fleck retinopathy are common, but lenticonus does not occur (8). Most individuals with autosomal dominant Alport syndrome have hematuria only, and while a few develop proteinuria and kidney failure, hearing loss and ocular abnormalities are very rare (9,10).
Two additional forms of inheritance are now recognized, and the definitions differ; however, the following interpretations have been adopted for Alport syndrome (3). “Digenic” disease refers to two pathogenic variants in different COL4A3–COL4A5 genes (11). This differs from “modifying” variants, where a heterozygous pathogenic COL4A3–COL4A5 variant occurs together with a pathogenic change in a gene encoding another podocyte or glomerular filtration barrier protein (12,13).
Some geneticists prefer to use “double heterozygotes” rather than digenic variants where the pathogenic changes affect genes that on their own cause disease. However, digenic disease is used here to be consistent with the recent classification of Alport syndrome and previous reports (3,11,14–16). Digenic inheritance refers to variants at two loci that explain the phenotype of some affected individuals better than a variant at a single locus (17). Both loci may be equally important, but one may also increase the disease risk or severity (17,18). The outcome depends on whether the affected individual is a man with a COL4A5 change, in which case all heterotrimers are already affected. The second variant may also determine who develops clinical features or has disease at a younger age. This is because the second variant often increases the proportion of heterotrimers that are defective. Although digenic disease contributes to the variability of clinical features in some families, this is not a common explanation.
Types of Digenic Inheritance
Digenic Alport syndrome is due to a pathogenic variant in COL4A5 plus one in COL4A3 or COL4A4 or due to a pathogenic variant in COL4A3 plus one in COL4A4. These will be considered separately because their population frequencies, modes of inheritance, and molecular and clinical consequences are different. For digenic variants affecting COL4A5 plus COL4A3 or COL4A4, many of these features depend on whether the COL4A5 variant affects a man or woman, and in all patients, variant severity also contributes to the phenotype (19)
Pathogenic Variants in COL4A5 Plus COL4A3 or COL4A4 in Men or Women
The number of published reports of digenic Alport syndrome has increased with the widespread adoption of massively parallel sequencing for routine genetic testing. Massively parallel sequencing has dramatically improved the ability to detect multiple variants in different genes contemporaneously in a single individual. Previous practice was to sequence the COL4A3–COL4A5 genes where there were the characteristic features of X-linked or autosomal recessive Alport syndrome: that is, kidney failure, hearing loss, and ocular abnormalities. After a pathogenic variant was found, the laboratory ceased to look for a further change. This meant that few digenic variants were identified, most included a COL4A5 change, and only some of the digenic variants were reported as such.
Here, we have reviewed all of the published digenic variants found in a literature search and after requests to testing laboratories attending the 2021 International Workshop on Alport Syndrome (20) where at least some clinical data (age, sex, proteinuria, and age at kidney failure) were also available. This strategy identified 43 COL4A5 plus COL4A3/COL4A4 variants and 32 COL4A3 plus COL4A4 variants.
It is, however, possible to deduce the relative population frequencies of digenic variants more accurately from recently published data for pathogenic variants in COL4A5 (of about one in 2000) and for COL4A3 or COL4A4 (of about one in 100) (1). Thus, digenic inheritance of a COL4A5 variant plus a COL4A3 or COL4A4 variant occurs in about 1/2000 × 1/100=1/200,000 if both occur independently and, alternatively, in about 1% of all individuals with X-linked Alport syndrome. This was confirmed in a study of 417 mainly children with suspected Alport syndrome, where six had pathogenic variants in both COL4A5 and COL4A3 or COL4A4 (21).
Inheritance
With COL4A5 plus COL4A3/COL4A4 variants, the changes are inherited independently from one parent or from both. In these patients, inheritance of the individual COL4A5 plus COL4A3/COL4A4 variants each resembles the inheritance pattern of the underlying variant, namely X linked or autosomal dominant. Inheritance in an individual family is easier to understand if the inheritance of each variant is considered separately.
Thus, for a man with a pathogenic COL4A5 variant, none of his sons will inherit the COL4A5 variant, but all of his daughters will; furthermore, half of his sons and half of his daughters will inherit the COL4A3 or COL4A4 variant as an independent event. Thus, none of his sons have digenic disease, but half of his daughters will.
For a woman with a pathogenic COL4A5 variant as well as a pathogenic COL4A3 or COL4A4 variant, half of her sons and half of her daughters will inherit the pathogenic COL4A5 variant, and as an independent event, half of her sons and half of her daughters will inherit the pathogenic COL4A3 or COL4A4 variant. Thus, if she had four children, on average, one has a COL4A5 variant plus a COL4A3 or COL4A4 variant (and digenic disease), one has a COL4A5 variant only, one has a COL4A3 or COL4A4 variant only, and one has neither variant.
The clinical consequences also depend on the effect of the variants on the collagen IV α-chains and whether they affect the interaction of the impaired chains.
Disease Pathogenesis
In order to understand the effects of an additional pathogenic variant, it is important to understand why some pathogenic COL4A5 variants result in more severe disease and more pronounced proteinuria or earlier-onset kidney failure.
Pathogenic COL4A5 variants that result in severe disease include truncating variants, major rearrangements, large deletions, and splicing variants that induce exon skipping or intronic retention (7,19). Premature stop codons typically result in nonsense-mediated decay of the mRNA transcript and absence of the collagen IV α3α4α5-heterotrimer from affected membranes (22). “Severe” COL4A5 variants result in severe disease with early-onset kidney failure, hearing loss, and, often, lenticonus and central fleck retinopathy. In contrast, missense variants produce less disruption of the collagen IV α-chains, which may be partly retained within the endoplasmic reticulum, resulting in increased ER stress. However, the collagen IV α-chains also persist in smaller amounts within the affected membranes (23,24). These variants are associated with milder disease with later-onset kidney failure and hearing loss and are less often associated with lenticonus. Hypomorphic variants also result in milder disease with late-onset proteinuria and kidney failure in the sixth or seventh decade often without hearing loss, lenticonus, or the central fleck retinopathy (25,26). These variants include Gly substitutions that are adjacent to a noncollagenous sequence or a non-Gly substitution. p.Gly624Asp is a common hypomorphic COL4A5 variant in people of European ancestry (26).
On their own, heterozygous pathogenic COL4A3 and COL4A4 variants have a less severe effect than pathogenic heterozygous COL4A5 variants. They usually result in hematuria and sometimes proteinuria, but kidney failure is less likely; additionally, hearing loss, lenticonus, and fleck retinopathy are very rare if they occur at all (9,10). Sometimes, hematuria and proteinuria are not present.
Pathogenic Variants in COL4A5 Plus COL4A3 or COL4A4 in Men
The major consequences of digenic variants where one affects COL4A5 depend on the sex of the individual and the variant severity. In men, a COL4A5 variant affects all of the collagen IV α3α4α5-heterotrimers (Figure 1A). If the COL4A5 variant is already severe (a large rearrangement or a truncating or splice variant), the heterotrimers are often absent, and kidney failure is likely before the age of 30 (7). The addition of even a severe COL4A3 or COL4A4 variant may have no consequences because the collagen IV α3- and α4-chains are destroyed rather than being expressed in basement membranes. However, if the COL4A5 variant is a missense variant resulting in milder disease, then the collagen IV α3α4α5-heterotrimer is abnormal, and the additional COL4A3 or COL4A4 variant may further disrupt half of the already abnormal heterotrimers (Figure 1A).
Figure 1.
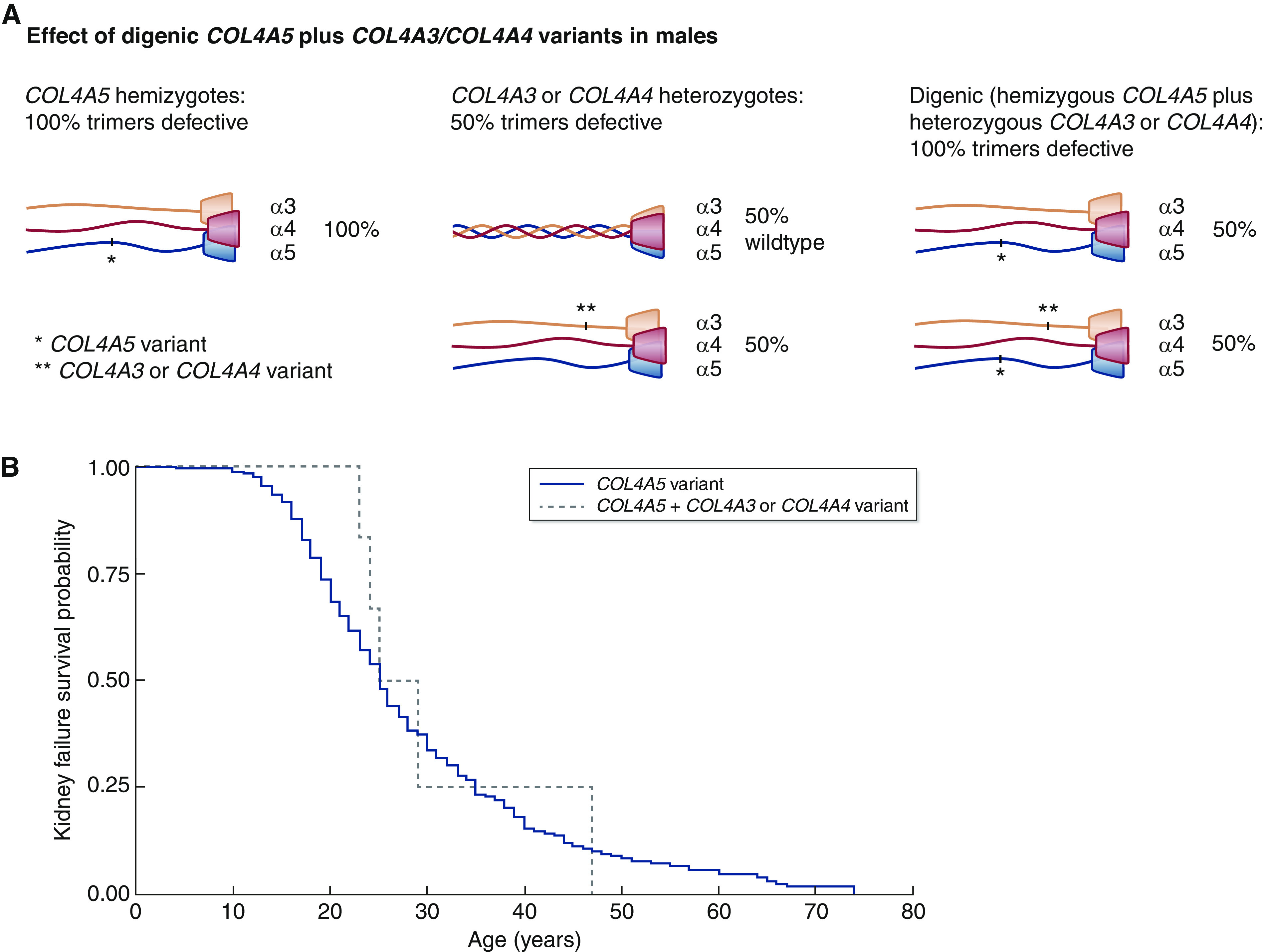
Digenic COL4A5 plus COL4A3 or COL4A4 variants in males are not associated with greater disease severity and earlier-onset kidney failure (P=0.75). (A) Effect of digenic COL4A5 plus COL4A3/COL4A4 variants on heterotrimer formation in men (16). (B) Kidney survival in men with a COL4A5 variant (from LOVD) compared with a COL4A5 plus a COL4A3 or COL4A4 variant. The data and references for the digenic variants are summarized in Table 1, and full data are provided in Supplemental Table 1. The COL4A5 variant (solid line) and the COL4A5 plus a COL4A3 or COL4A4 variant (dashed line) are shown. LOVD, Leiden Open Variation Database.
Although these effects are likely, it is still difficult to confirm that the combination of COL4A5 and COL4A3 or COL4A4 variants results in more severe disease because so few individuals with digenic disease have been described, many affected children are too young to have kidney failure, and glomerular basement membrane (GBM) lamellation and hearing and ocular defects may not be known. Common clinical data are often sparse and rarely include the age at which proteinuria was first detected, which may be more relevant for such studies than the age at kidney failure.
In addition, some individuals reported with a COL4A5 variant may have an undetected pathogenic change in COL4A3 or COL4A4. Other extrarenal factors are also important in determining the risk of kidney failure, such as hypertension control and avoiding diabetes and becoming overweight (27,28). Furthermore, the age at kidney failure has been further delayed with the widespread use of ACE inhibitor treatment (29).
A review of all reported digenic COL4A5 variants in 16 men with a median age of 16 years (5–55) found that the COL4A5 change was severe in five (31%) and that the additional variant was severe in only two (13%) (16,21,30–32) (Table 1). Digenic disease was associated with proteinuria in 12 (12 of 13; 92%) and kidney failure in five (five of 15; 33%), which developed at a median age of 27 (range, 23–47), as well as hearing loss (six of ten; 60%) and ocular abnormalities (three of six; 50%). In one study, there were more affected boys with proteinuria (21), but there was no difference in the median age at kidney failure of 24 years compared with 23 years without the additional COL4A3 or COL4A4 variant (P=0.75, comparison with Leiden Open Variation Database results) (Figure 1B, Supplemental Table 1). Digenic Alport syndrome with a hypomorphic COL4A5 and a further pathogenic COL4A3 or COL4A4 variant may be associated with more severe disease with earlier-onset proteinuria and a younger age at kidney failure, but there is no evidence for this currently.
Table 1.
Clinical features in men with digenic Alport syndrome where one variant affects COL4A5 compared with features with one pathogenic COL4A5 variant only
| Feature | X-Linked Alport Syndrome in Men, n=315 (7) | X-Linked Alport Syndrome in Men after ACE Inhibitor Treatment, n=282 (29) | Digenic Alport Syndrome in Men Including COL4A5 Variant, n=16 |
|---|---|---|---|
| Age range (median), yr | Not provided | 13 (0–73) | 16 (5–55) |
| COL4A5 severe variant | 75/195 families (38%) | 125 (47%) | 5 (31%) |
| p.G624D in COL4A5 | Not specified | 0 | 4 |
| COL4A3 | Not applicable | Not applicable | 11 |
| COL4A4 | Not applicable | Not applicable | 5 |
| Additional variant is severe | Not applicable | Not applicable | 2/16 |
| GBM lamellation, splitting, or basket-weave pattern | 88/98 (90%) at a mean age of 12.5 yr | Not specified | 5/5 |
| GBM thinning | 12/98 (12%) at a mean age of 11 yr | Not specified | Not specified |
| Abnormal collagen IV α3α4α5-immunohistochemistry | 14/16 (88%) | 64/146 (44%) | Not specified |
| Hematuria | 191/193 (99%) | 262/264 (99%) | 14/14 (100%) |
| Proteinuria | 207/218 (95%) | 238 (91%) | 12/13 (92%) |
| No. with kidney failure, n (%) | 282/360 (78) | 61/282 (22) | 5/15 (33) |
| Age at kidney failure (median), yr | 25 (9 to >41) | 35 (32–40) | 25 (23–47) |
| Hearing loss, n (%) | 239/303 (79) | 77 (32) | 6/10 (60) |
| Ocular abnormalities | 57/162 (35%) | 13 (6%) | 3/6 (50%) |
“Severe” COL4A5 variants include major rearrangements, truncating, and splice site variants. Precise numbers were not always provided but have been deduced from percentages or figures in manuscripts. GBM, glomerular basement membrane; No., number.
Pathogenic Variants in COL4A5 Plus COL4A3 or COL4A4 in Women
Twice as many women inherit a pathogenic COL4A5 variant because they have two X chromosomes (27). This means that they are therefore twice as likely to have digenic disease with a pathogenic COL4A5 variant plus a pathogenic COL4A3 or COL4A4 variant.
Overall, in such women, the pathogenic COL4A5 variant affects half of the heterotrimers, and the additional COL4A3 or COL4A4 variant means that 75% of the heterotrimers are defective (Figure 2B) (16). However, the situation is more complicated because inheritance is different for COL4A5 where there is X-chromosome inactivation and for the COL4A3 or COL4A4 variant. A severe COL4A5 variant results in the loss of the collagen IV α3α4α5-heterotrimer from on average half of the cells, whereas a severe COL4A3 or COL4A4 variant results in only half of the total collagen IV α3α4α5-heterotrimers in each cell. For a mild COL4A5 variant, on average half of the cells have a defective heterotrimer, and for a mild COL4A3 or COL4A4 variant, half of the heterotrimers in each cell are defective.
Figure 2.
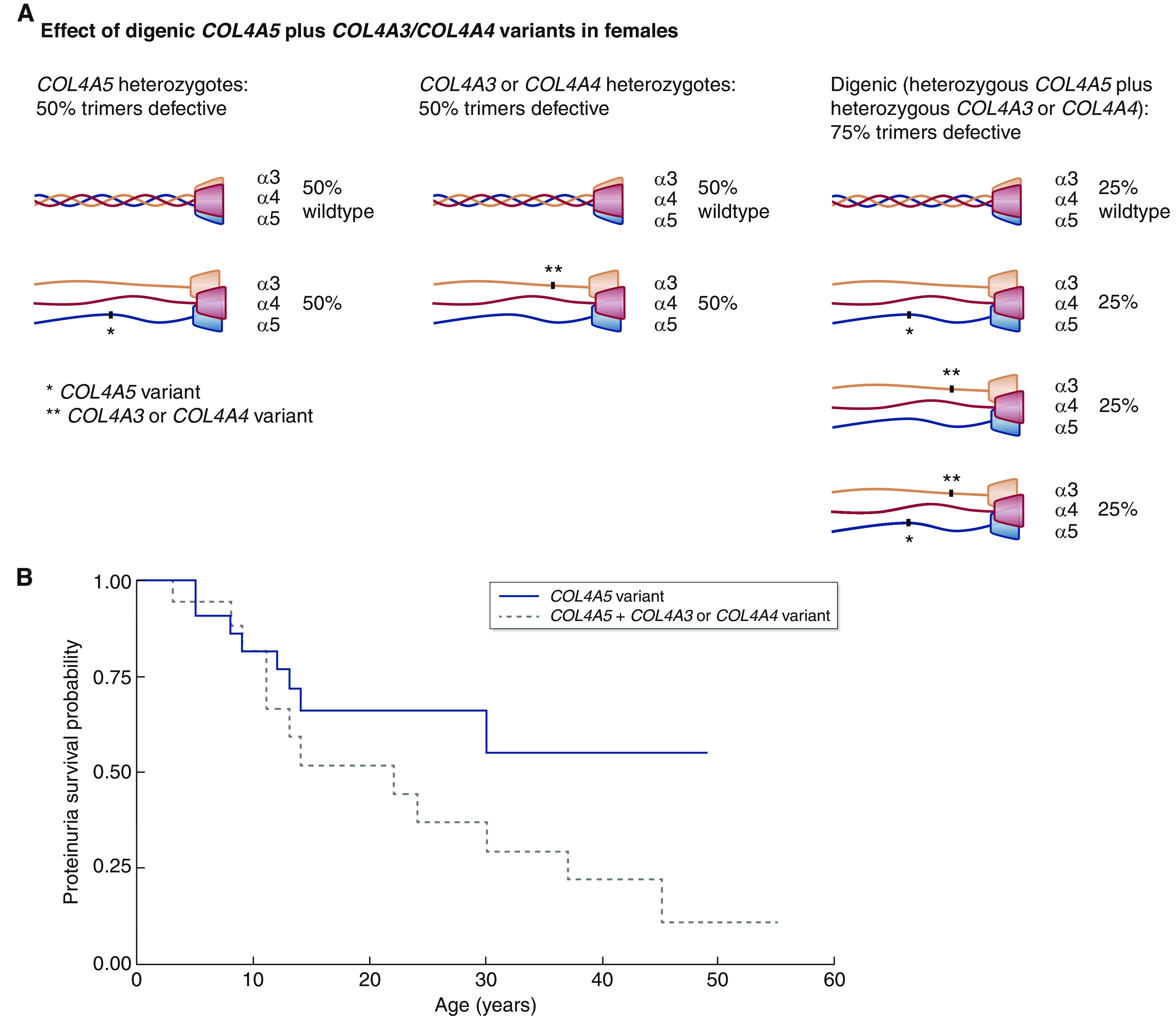
Digenic COL4A5 plus COL4A3 or COL4A4 variants in females are associated with a trend to earlier onset of proteinuria (P=0.09). (A) Effect of digenic COL4A5 plus COL4A3/COL4A4 variants on heterotrimer formation in women (16). (B) Proteinuria in women with a COL4A5 variant (37) compared with a COL4A5 plus a COL4A3 or COL4A4 variant (Table 2). The data and references for the digenic variants are summarized in Table 2, and full data are provided in Supplemental Table 2. The COL4A5 variant (solid line) and the COL4A5 plus a COL4A3 or COL4A4 variant (dashed line) are shown.
Data were available from 26 women with a pathogenic COL4A5 variant and a COL4A3 or COL4A4 variant with a median age of 13 years (range, 3–55) (Table 2) (15,16,21,30,33–37). In seven patients (27%), the COL4A5 variant was severe. Sixteen (62%) had proteinuria, which had developed at a median of 22 years (range, 3–45), and only two had kidney failure, which developed at 40 or 44 years. Three (15%) had hearing loss, and one (7%) had unspecified ocular abnormalities. There was GBM lamellation in five of six examined (83%) and GBM thinning in one of six (17%). At present, too few women have been described with digenic variants that include a COL4A5 change to demonstrate an earlier age at kidney failure. However, when compared with women with severe COL4A5 variants (37), those with pathogenic digenic changes demonstrated a trend to proteinuria at a younger age (P=0.09) (Figure 2B).
Table 2.
Clinical features in women with digenic Alport syndrome where one variant affects COL4A5 compared with features with one pathogenic COL4A5 variant only
| Feature | X-Linked Alport Syndrome in Women, n=349 (8) | X-Linked Alport Syndrome in Women, n=275 (44) | X-Linked Alport Syndrome in Women, n=24 (37) | Digenic Alport Syndrome in Women Including COL4A5 Variant, n=26 |
|---|---|---|---|---|
| Age range (median), yr | Not provided | 24 (0–92) | 8 (1–41) | 13 (3–55) |
| COL4A5 severe variant | 191/315 (61%) | 138 (50%) | 14/24 (58%) | 7 (27%) |
| COL4A3 | Not applicable | Not applicable | Not applicable | 15 |
| COL4A4 | Not applicable | Not applicable | Not applicable | 11 |
| Additional variant is severe | Not applicable | Not applicable | Not applicable | 7/26 (27%) |
| GBM lamellation, splitting, or basket-weave pattern | 20/28 (71%) | Not specified | 16/16 (basket weave in 8/16) | 5/6 (83%) |
| GBM thinning | 6/28 (21%) | Not specified | 16/16 | 1/6 (17%) |
| Abnormal collagen IV α3α4α5-immunohistochemistry | 7/9 (78%) | Not specified | Not specified | Not specified |
| Hematuria | 309 (96%) | 232 (98%) | 23/23 (100%) | |
| Proteinuria | 176/234 (75%) | 175 (73%) | 10/24 (42%) | 16/26 (62%) |
| Age at proteinuria onset (median), yr | Not specified | 7 | 13 (5–30) | 12 (3–45) |
| No. with kidney failure, n (%) | 51/288 (18) | 33 (12) | 1/24 (4) | 2/24 (8) |
| Age at kidney failure (median), yr | 38 (18–70) | 65 | 40 | 40, 44 |
| Hearing loss, n (%) | 239/303 (79) | 15 (6) | 4/24 (17) | 3/20 (15) |
| Ocular abnormalities | 57/162 (35%) | 4 (2%) | 0/24 (0%) | 1/14 (7%) |
GBM, glomerular basement membrane; No., number.
Thus, women with digenic disease that includes a COL4A5 variant have a higher risk of developing proteinuria, which also increases their risk of kidney failure (7). This suggests that women with digenic disease that includes a pathogenic COL4A5 variant have a worse prognosis that those with only a pathogenic variant in COL4A5.
Course
The clinical course depends on the sex of the affected individual, and the severity of both the COL4A5 variant and the COL4A3 or COL4A4 variant, that is whether both variants are individually associated with severe disease.
Management
Previous guidelines for the management of Alport syndrome have addressed digenic forms of the disease (5,14,38,39). These are summarized here.
All individuals who undergo testing for Alport syndrome should be examined for variants in all of the COL4A3, COL4A4, and COL4A5 genes because additional pathogenic variants in these genes may worsen clinical features.
Men with digenic variants that include a pathogenic COL4A5 variant should typically be treated from the time of diagnosis with renin-angiotensin-aldosterone blockade (38). Women with digenic variants that include a pathogenic COL4A5 variant should be treated from the time of diagnosis because of their higher risk of proteinuria and kidney failure (38). These recommendations should be applied less stringently in children because of their increased risks with severe dehydration due to vomiting and diarrhea on top of ACE inhibitor use.
All first-degree family members should undergo genetic testing because they may have both pathogenic variants or only one, and half of the sons of an affected women may themselves develop kidney failure with only the COL4A5 variant (39).
Individuals with digenic disease should be discouraged from kidney donation because of their own risk of kidney failure (14,39).
Inheritance is complicated and easier to understand if the variants are considered separately. Advice about the disease in future generations will be different in men and women with a pathogenic COL4A5 variant.
Pathogenic Heterozygous Variants in COL4A3 Plus COL4A4
Population Frequency
On the basis of population studies, digenic disease with a pathogenic variant in COL4A3 plus a pathogenic variant in COL4A4 is more common than a pathogenic variant in COL4A5 plus one in COL4A3 or COL4A4 because pathogenic COL4A3 and COL4A4 variants are more common (1). This form of digenic disease is predicted to affect about one in 200 (for the approximate frequency of a pathogenic COL4A3 variant) × one in 200 (for the COL4A4 variant) = one in 40,000 of the population or one in 200 of those with suspected familial hematuria (where the index patient has hematuria and is likely to have a pathogenic heterozygous COL4A3 or COL4A4 variant). About 0.5% of all COL4A3 or COL4A4 variants have a second variant because digenic disease must also include a variant in the other gene. This occurs about five times as often as the combination of pathogenic variants in COL4A5 and another in COL4A3 or COL4A4.
Inheritance
Because COL4A3 and COL4A4 occur back-to-back on chromosome 2 with a short intervening sequence (40), the two pathogenic variants in COL4A3 and COL4A4 may occur on the same (in cis) or opposite (in trans) chromosomes. These different patterns affect the mode of inheritance and whether disease occurs in successive generations. Where both variants occur on the same chromosome, they are inherited together in consecutive generations, and the pattern mimics autosomal dominant inheritance. This occurs in about half of the patients, and only one parent is affected with a digenic phenotype and, typically, hematuria.
Thus, for an affected man or woman with pathogenic variants in both COL4A3 and COL4A4 on the same chromosome, half of their children inherit both pathogenic variants and have digenic disease. Successive generations with the digenic variants will pass these on to half of their children in each generation.
In contrast, when the abnormal COL4A3 and COL4A4 genes affect different chromosomes, which also occurs in half of the patients, both parents typically have hematuria, and the inheritance pattern mimics that of autosomal recessive disease.
Thus, for an affected man or woman with pathogenic variants in both COL4A3 and COL4A4 on different chromosomes, on average half of their children have a pathogenic COL4A3 or COL4A4 variant, but none have both variants and digenic disease.
There may, however, be an ascertainment bias in the reporting of families with these two inheritance patterns because individuals with variants on different chromosome are likely to have more severe disease and to undergo genetic testing more frequently.
Pathogenesis
A single pathogenic variant in COL4A3 or COL4A4 in both men and women results in 50% of the collagen IV α3α4α5-heterotrimers being defective, but two pathogenic variants in these genes result in 75% of the heterotrimers being defective (16) (Figure 3A).
Figure 3.
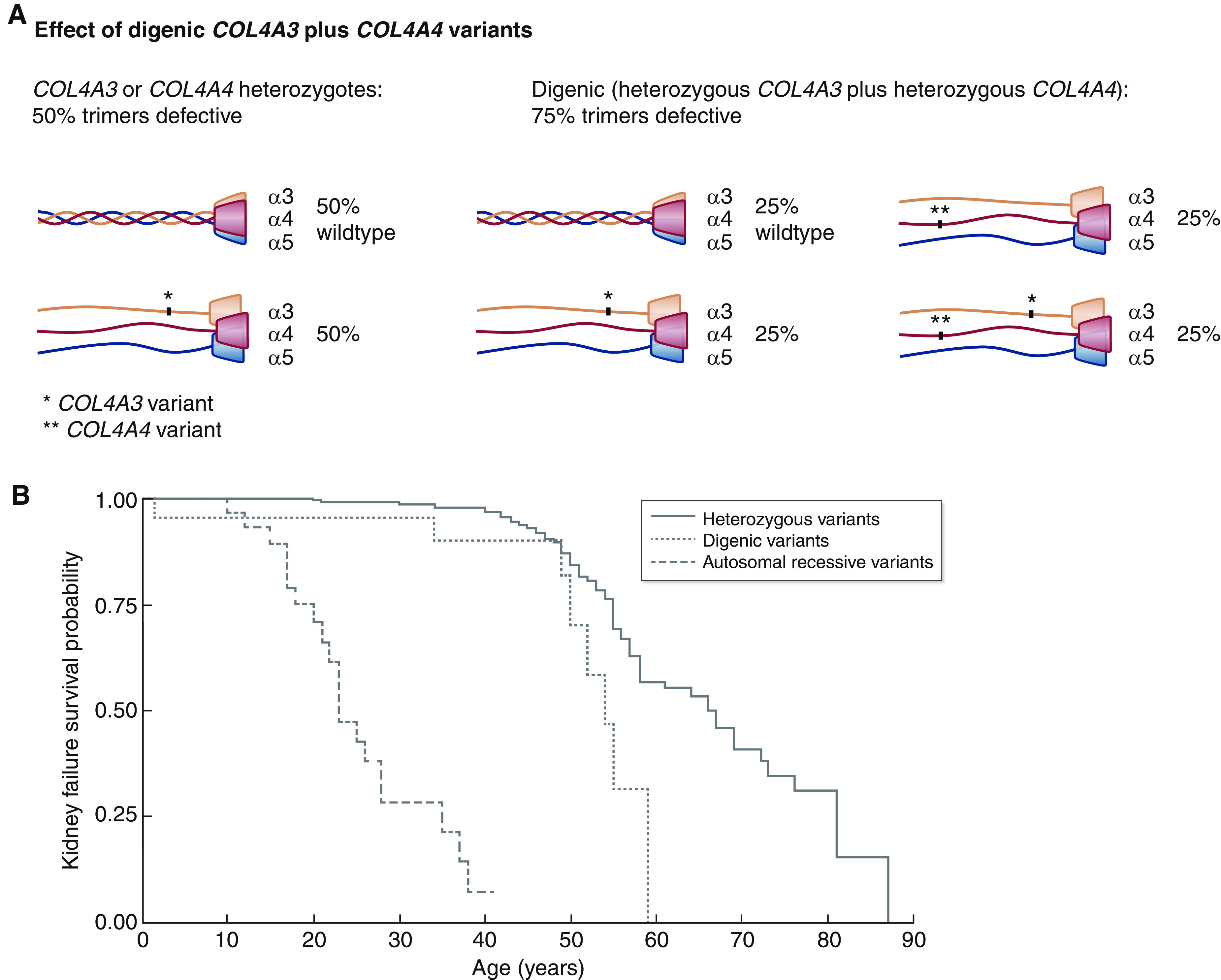
Digenic COL4A3 plus COL4A4 variants are associated with disease severity intermediate between autosomal dominant and autosomal recessive Alport syndrome (P=0.01, P<0.001, respectively). (A) Effect of digenic COL4A3 plus COL4A4 variants on heterotrimer formation (16). (B) Kidney survival in digenic Alport syndrome where pathogenic variants affect COL4A3 and COL4A4 compared with autosomal recessive (43) or autosomal dominant (9) Alport syndrome. The data and references for the digenic variants are summarized in Table 3, and full data are provided in Supplemental Table 3. Heterozygous variants (solid line), digenic variants (dotted line), and autosomal recessive variants (dashed line) are shown.
Clinical Features
The phenotype of a heterozygous or single pathogenic variant is typically hematuria, whereas proteinuria is less common; kidney failure is uncommon, and hearing loss and ocular abnormalities are rare. Hearing loss occurs from many other causes, but any individual with ocular abnormalities and a single pathogenic variant should undergo rigorous examination for a further COL4A3–COL4A5 variant.
The cohort of 32 individuals reported with digenic pathogenic variants in COL4A3 plus COL4A4 (9,15,16,36,41,42) included ten men and 15 women with a median age of 34 years (1–66); there were no severe variants in 15 (47%) individuals, one severe variant in 12 (38%) individuals, and two severe variants in five (16%) individuals (Table 3, Supplemental Table 3). Nineteen (66%) had proteinuria, which was the same likelihood as for autosomal dominant disease of a hospital-based cohort (65%), and the median age at kidney failure was 54 years (n=8), which was later than for autosomal recessive Alport syndrome (43) (23; range, 10–38; P<0.001) and younger than for autosomal dominant Alport syndrome (9) (66; range, 58–73; P=0.01) (Figure 3B). In addition, four (27%) had hearing loss, which was more often than for autosomal dominant Alport syndrome but less often than for autosomal recessive disease (23 of 35; 67%). None had an ocular abnormality. One individual had GBM lamellation, and two had membrane thinning. Thus, the risk of kidney failure for a pathogenic heterozygous COL4A3 or COL4A4 variant on its own is very small, but the addition of a further pathogenic change may increase this risk.
Table 3.
Clinical features in digenic Alport syndrome where pathogenic variants affect COL4A3 and COL4A4 compared with autosomal recessive or autosomal dominant Alport syndrome
| Feature | Digenic Alport Syndrome, n=32 | Autosomal Dominant Alport Syndrome, n=252 (9) | Autosomal Recessive Alport Syndrome, n=40 (43) |
|---|---|---|---|
| Age range (median), yr | 34 (1–66) | 48 (5–87) | 31 (6–54) |
| COL4A3 | 32 | 107 (35 families) | 20/40 (50%) |
| COL4A4 | 32 | 133 (43 families) | 20/40 (50%) |
| No severe variants | 15 (47%) | 136 (57%) | 8 (20%) |
| One severe variant | 12 (38%) | 104 (43%) | 12 (30%) |
| Two severe variants | 5 (16%) | Not applicable | 20 (50%) |
| GBM lamellation or splitting | 1/3 (33%) | Not specified | 34/36 (94%) |
| GBM thinning | 2/3 (67%) | Not specified | 2/36 (6%) but biopsy performed as a child |
| Abnormal collagen IV α3α4α5-immunohistochemistry | Not specified | Not specified | Not specified |
| Hematuria | Not specified | 232/252 (92 %) | Not specified |
| Proteinuria | 19/29 (66%) | 157/241 (65%) | Not specified |
| Age at proteinuria onset (median), yr | 43 (5–59) taken from age at proteinuria recorded | Not specified | Not specified |
| No. with kidney failure, n (%) | 8 (25) | 61 (24) | 20/34 (59) |
| Age at kidney failure (median), yr | 54 (n=8) | 67 (58–73) | 22.5 (10–38) |
| Hearing loss, n (%) | 4/15 (27) | 11/131 (8) | 23/35 (67) |
| Ocular abnormalities | 0/7 | 2/75 (2%) | 10/18 (56%) |
GBM, glomerular basement membrane; No., number.
Course
Again, the clinical course depends largely on the nature of the underlying pathogenic variants, but it appears that the risk of proteinuria is increased with digenic variants in COL4A3 and COL4A4 compared with that of pathogenic heterozygous variants.
Management
Again, these recommendations appear in various expert guidelines (5,14,38,39), and they are consistent with the recommendations for digenic disease due to a pathogenic COL4A5 variant plus a COL4A3 or COL4A4 variant. Individuals with digenic variants should be treated from the time of diagnosis (38). As for other forms of Alport syndrome, it is important to minimize the amount of proteinuria and to control any hypertension (5,14). At-risk first-degree family members should undergo genetic testing (14). First-degree family members should be strongly discouraged from kidney donation (14). Inheritance is complicated and easiest to understand if variants are considered separately.
In conclusion, digenic variants are rare but recognized increasingly; their inheritance is complicated, and except for X-linked Alport syndrome in men where the phenotype is already severe, they are probably associated with increased proteinuria and an earlier age at kidney failure. Individuals with digenic variants should be treated early, their affected family members identified and treated, and those with digenic variants dissuaded from kidney donation.
Many digenic variants are probably overlooked, especially where the second variant is hypomorphic. There are also occasional reports of digenic variants being associated with secondary FSGS and with cystic kidney disease as occurs with other COL4A3–COL4A5 variants.
There are still limited data on GBM lamellation, hearing loss, and ocular abnormalities in digenic disease with pathogenic COL4A3 and COL4A4 variants, but the data on age at proteinuria suggest that the phenotype is intermediate between autosomal dominant and autosomal recessive Alport syndrome.
Disclosures
O. Bielska reports other interests or relationships with the Polish Society for Pediatric Nephrology, a nonprofit organization. S. Daga reports a patent with A. Renieri, Silvestro Giovanni Conticello, A.M. Pinto, Ilaria Meloni, Francesco Donati, Susanna Croci, and Diego Lopergolo titled “CRISP R/Cas9 system for gene therapy” (102018000020230 [I0178645] BRE/RRI-mma). D.P. Gale reports consultancy agreements with Jude Bio and Reata Inc.; research funding from Sanofi and Travere; honoraria from Alexion, Novartis, and Otsuka; and serving as a trustee for AlportUK and Chair of the Renal Association Rare Diseases Committee. B. Lipska-Zietkiewicz has received honoraria from Takeda and Travere for giving lectures; reports serving as Section Editor of Experimental Nephrology and Genetics for Nephron, a scientific journal by Karger; and reports serving as a member of the European Society of Pediatric Nephrology, a Hereditary Glomerulopathies WG Co-Chair, a member of the International Pediatric Nephrology Association, and a Molecular Diagnostics Task-Force Member; and other interests and relationships with the European Reference Network for Rare Kidney Diseases. A. Renieri reports research funding from Travere Therapeutics, Inc. H. Rothe reports employment with Leipzig Dialysis Center, Leipzig, Germany and the Weisswasser Center for Nephrology and Metabolism; honoraria from VIFOR Pharma; and speakers bureau for VIFOR Pharma. J. Savige reports other interests or relationships with the Alport Foundation Australia and the PKD Australia Scientific Board. All remaining authors have nothing to disclose.
Funding
None.
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Author Contributions
J.T. Gibson, B. Lipska-Zietkiewicz, and J. Savige conceptualized the study; M. Aksenova, E. Ars, O. Bielska, A. Cerkauskaite, S. Daga, D.P. Gale, J.T. Gibson, A.M. Pinto, A. Renieri, H. Rothe, and J. Savige were responsible for data curation; J. Savige was responsible for investigation; E. Ars, J.T. Gibson, and J. Savige were responsible for formal analysis; J.T. Gibson and J. Savige were responsible for methodology; J. Savige was responsible for project administration; M. Aksenova, E. Ars, O. Bielska, A. Cerkauskaite, S. Daga, D.P. Gale, J.T. Gibson, B. Lipska-Zietkiewicz, A.M. Pinto, A. Renieri, H. Rothe, and J. Savige were responsible for resources; J.T. Gibson was responsible for visualization; J. Savige provided supervision; D.P. Gale, J.T. Gibson, B. Lipska-Zietkiewicz, and J. Savige wrote the original draft; and M. Aksenova, E. Ars, O. Bielska, A. Cerkauskaite, S. Daga, D.P. Gale, J.T. Gibson, B. Lipska-Zietkiewicz, A.M. Pinto, A. Renieri, H. Rothe, and J. Savige reviewed and edited the manuscript.
Supplemental Material
This article contains the following supplemental material online at https://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.03120322/-/DCSupplemental.
Supplemental Material. Supplemental methods.
Supplemental Table 1. Genotype-phenotype correlation in men with digenic Alport syndrome including a COL4A5 variant.
Supplemental Table 2. Genotype-phenotype correlation in women with digenic Alport syndrome including a COL4A5 variant.
Supplemental Table 3. Genotype-phenotype correlation in men and women with digenic Alport syndrome with a COL4A3 and COL4A4 variant.
References
- 1.Gibson J, Fieldhouse R, Chan MMY, Sadeghi-Alavijeh O, Burnett L, Izzi V, Persikov AV, Gale DP, Storey H, Savige J; Genomics England Research Consortium : Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J Am Soc Nephrol 32: 2273–2290, 2021. 10.1681/ASN.2020071065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, Paterson AD, Pei Y: Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol 29: 2593–2600, 2018. 10.1681/ASN.2018050493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, Nozu K, Renieri A, Rheault M, Wang F, Gross O: Alport syndrome: A unified classification of genetic disorders of collagen IV α345: A position paper of the Alport Syndrome Classification Working Group. Kidney Int 93: 1045–1051, 2018. 10.1016/j.kint.2017.12.018 [DOI] [PubMed] [Google Scholar]
- 4.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG: Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med 348: 2543–2556, 2003. 10.1056/NEJMra022296 [DOI] [PubMed] [Google Scholar]
- 5.Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F: Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 24: 364–375, 2013. 10.1681/ASN.2012020148 [DOI] [PubMed] [Google Scholar]
- 6.Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, Venkat-Raman G, Ennis S: Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 31: 961–970, 2016. 10.1093/ndt/gfv325 [DOI] [PubMed] [Google Scholar]
- 7.Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 11: 649–657, 2000. 10.1681/ASN.V114649 [DOI] [PubMed] [Google Scholar]
- 8.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol 14: 2603–2610, 2003. 10.1097/01.asn.0000090034.71205.74 [DOI] [PubMed] [Google Scholar]
- 9.Furlano M, Martínez V, Pybus M, Arce Y, Crespí J, Venegas MDP, Bullich G, Domingo A, Ayasreh N, Benito S, Lorente L, Ruíz P, Gonzalez VL, Arlandis R, Cabello E, Torres F, Guirado L, Ars E, Torra R: Clinical and genetic features of autosomal dominant Alport syndrome: A cohort study. Am J Kidney Dis 78: 560–570.e1, 2021. 10.1053/j.ajkd.2021.02.326 [DOI] [PubMed] [Google Scholar]
- 10.Matthaiou A, Poulli T, Deltas C: Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clin Kidney J 13: 1025–1036, 2020. 10.1093/ckj/sfz176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deltas C: Digenic inheritance and genetic modifiers. Clin Genet 93: 429–438, 2018. 10.1111/cge.13150 [DOI] [PubMed] [Google Scholar]
- 12.Voskarides K, Papagregoriou G, Hadjipanagi D, Petrou I, Savva I, Elia A, Athanasiou Y, Pastelli A, Kkolou M, Hadjigavriel M, Stavrou C, Pierides A, Deltas C: COL4A5 and LAMA5 variants co-inherited in familial hematuria: Digenic inheritance or genetic modifier effect? BMC Nephrol 19: 114, 2018. 10.1186/s12882-018-0906-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daga S, Fallerini C, Furini S, Pecoraro C, Scolari F, Ariani F, Bruttini M, Mencarelli MA, Mari F, Renieri A, Pinto AM: Non-collagen genes role in digenic Alport syndrome. BMC Nephrol 20: 70, 2019. 10.1186/s12882-019-1258-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savige J, Lipska-Zietkiewicz BS, Watson E, Hertz JM, Deltas C, Mari F, Hilbert P, Plevova P, Byers P, Cerkauskaite A, Gregory M, Cerkauskiene R, Ljubanovic DG, Becherucci F, Errichiello C, Massella L, Aiello V, Lennon R, Hopkinson L, Koziell A, Lungu A, Rothe HM, Hoefele J, Zacchia M, Martic TN, Gupta A, van Eerde A, Gear S, Landini S, Palazzo V, Al-Rabadi L, Claes K, Corveleyn A, Van Hoof E, van Geel M, Williams M, Ashton E, Belge H, Ars E, Bierzynska A, Gangemi C, Renieri A, Storey H, Flinter F: Guidelines for genetic testing and management of Alport syndrome. Clin J Am Soc Nephrol 17: 143–154, 2022. 10.2215/CJN.04230321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, Pasini A, Barbano G, Pinciaroli AR, Garosi G, Frullanti E, Pinto AM, Mencarelli MA, Mari F, Renieri A, Ariani F: Alport syndrome: Impact of digenic inheritance in patients management. Clin Genet 92: 34–44, 2017. 10.1111/cge.12919 [DOI] [PubMed] [Google Scholar]
- 16.Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, Miglietti N, Antonucci MF, Cetta F, Sayer JA, van den Wijngaard A, Yau S, Mari F, Bruttini M, Ariani F, Dahan K, Smeets B, Antignac C, Flinter F, Renieri A: Evidence of digenic inheritance in Alport syndrome. J Med Genet 52: 163–174, 2015. 10.1136/jmedgenet-2014-102822 [DOI] [PubMed] [Google Scholar]
- 17.Schäffer AA: Digenic inheritance in medical genetics. J Med Genet 50: 641–652, 2013. 10.1136/jmedgenet-2013-101713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazzo A, Raimondi D, Daneels D, Moreau Y, Smits G, Van Dooren S, Lenaerts T: Understanding mutational effects in digenic diseases. Nucleic Acids Res 45: e140, 2017. 10.1093/nar/gkx557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savige J, Storey H, Il Cheong H, Gyung Kang H, Park E, Hilbert P, Persikov A, Torres-Fernandez C, Ars E, Torra R, Hertz JM, Thomassen M, Shagam L, Wang D, Wang Y, Flinter F, Nagel M: X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS One 11: e0161802, 2016. 10.1371/journal.pone.0161802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daga S, Ding J, Deltas C, Savige J, Lipska-Ziętkiewicz BS, Hoefele J, Flinter F, Gale DP, Aksenova M, Kai H, Perin L, Barua M, Torra R, Miner JH, Massella L, Ljubanović DG, Lennon R, Weinstock AB, Knebelmann B, Cerkauskaite A, Gear S, Gross O, Turner AN, Baldassarri M, Pinto AM, Renieri A: The 2019 and 2021 International Workshops on Alport Syndrome. Eur J Hum Genet 30: 507–516, 2022. 10.1038/s41431-022-01075-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Ding J, Zhang H, Yao Y, Xiao H, Wang S, Wang F: Effect of heterozygous pathogenic COL4A3 or COL4A4 variants on patients with X-linked Alport syndrome. Mol Genet Genomic Med 7: e647, 2019. 10.1002/mgg3.647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bateman JF, Boot-Handford RP, Lamandé SR: Genetic diseases of connective tissues: Cellular and extracellular effects of ECM mutations. Nat Rev Genet 10: 173–183, 2009. 10.1038/nrg2520 [DOI] [PubMed] [Google Scholar]
- 23.Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, Karaiskos C, Savva I, Paraskeva R, Dweep H, Sticht C, Anastasiadou N, Zouvani I, Goumenos D, Felekkis K, Saleem M, Voskarides K, Gretz N, Deltas C: Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol 25: 260–275, 2014. 10.1681/ASN.2012121217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Mohammad M, Wang Y, Tan R, Murray LS, Ricardo S, Dagher H, van Agtmael T, Savige J: The chemical chaperone, PBA, reduces ER stress and autophagy and increases collagen IV α5 expression in cultured fibroblasts from men with X-linked Alport syndrome and missense mutations. Kidney Int Rep 2: 739–748, 2017. 10.1016/j.ekir.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pierides A, Voskarides K, Kkolou M, Hadjigavriel M, Deltas C: X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia 17: 207–213, 2013 [PMC free article] [PubMed] [Google Scholar]
- 26.Żurowska AM, Bielska O, Daca-Roszak P, Jankowski M, Szczepańska M, Roszkowska-Bjanid D, Kuźma-Mroczkowska E, Pańczyk-Tomaszewska M, Moczulska A, Drożdż D, Hadjipanagi D, Deltas C, Ostalska-Nowicka D, Rabiega A, Taraszkiewicz J, Taranta-Janusz K, Wieczorkiewicz-Plaza A, Jobs K, Mews J, Musiał K, Jakubowska A, Nosek H, Jander AE, Koutsofti C, Stanisławska-Sachadyn A, Kuleszo D, Ziętkiewicz E, Lipska-Ziętkiewicz BS: Mild X-linked Alport syndrome due to the COL4A5 G624D variant originating in the Middle Ages is predominant in Central/East Europe and causes kidney failure in midlife. Kidney Int 99: 1451–1458, 2021. 10.1016/j.kint.2020.10.040 [DOI] [PubMed] [Google Scholar]
- 27.Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, Finlay M, Flinter F: Alport syndrome in women and girls. Clin J Am Soc Nephrol 11: 1713–1720, 2016. 10.2215/CJN.00580116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kashtan CE, Gross O: Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol 36: 711–719, 2021. 10.1007/s00467-020-04819-6 [DOI] [PubMed] [Google Scholar]
- 29.Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, Ishiko S, Nakanishi K, Shima Y, Nagase H, Takeda H, Rossanti R, Ye MJ, Nozu Y, Ishimori S, Ninchoji T, Kaito H, Morisada N, Iijima K, Nozu K: Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int 98: 1605–1614, 2020. 10.1016/j.kint.2020.06.038 [DOI] [PubMed] [Google Scholar]
- 30.Choi M, Anistan YM, Eckardt KU, Gollasch M, Nickel P: Possible digenic disease in a Caucasian family with COL4A3 and COL4A5 mutations. Nephron 141: 213–218, 2019. 10.1159/000495764 [DOI] [PubMed] [Google Scholar]
- 31.Domingo-Gallego A, Pybus M, Bullich G, Furlano M, Ejarque-Vila L, Lorente-Grandoso L, Ruiz P, Fraga G, López González M, Piñero-Fernández JA, Rodríguez-Peña L, Llano-Rivas I, Sáez R, Bujons-Tur A, Ariceta G, Guirado L, Torra R, Ars E: Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol Dial Transplant 37: 687–696, 2022. 10.1093/ndt/gfab019 [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Sun BC, Zhao BG, Ma QS: An overview of the multi-pronged approach in the diagnosis of Alport syndrome for 22 children in Northeast China. BMC Nephrol 21: 294, 2020. 10.1186/s12882-020-01962-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daga S, Baldassarri M, Lo Rizzo C, Fallerini C, Imperatore V, Longo I, Frullanti E, Landucci E, Massella L, Pecoraro C, Garosi G, Ariani F, Mencarelli MA, Mari F, Renieri A, Pinto AM: Urine-derived podocytes-lineage cells: A promising tool for precision medicine in Alport syndrome. Hum Mutat 39: 302–314, 2018. 10.1002/humu.23364 [DOI] [PubMed] [Google Scholar]
- 34.Han KH, Park JE, Ki CS: De novo mutations in COL4A5 identified by whole exome sequencing in 2 girls with Alport syndrome in Korea. Korean J Pediatr 62: 193–197, 2019. 10.3345/kjp.2018.06772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokota K, Nozu K, Minamikawa S, Yamamura T, Nakanishi K, Kaneda H, Hamada R, Nozu Y, Shono A, Ninchoji T, Morisada N, Ishimori S, Fujimura J, Horinouchi T, Kaito H, Nakanishi K, Morioka I, Taniguchi-Ikeda M, Iijima K: Female X-linked Alport syndrome with somatic mosaicism. Clin Exp Nephrol 21: 877–883, 2017. 10.1007/s10157-016-1352-y [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Chen C, Wei Y, Zhao G, Liu L, Wang C, Zhang J, Kong X: Novel mutations of COL4A3, COL4A4, and COL4A5 genes in Chinese patients with Alport Syndrome using next generation sequence technique. Mol Genet Genomic Med 7: e653, 2019. 10.1002/mgg3.653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mastrangelo A, Giani M, Groppali E, Castorina P, Soldà G, Robusto M, Fallerini C, Bruttini M, Renieri A, Montini G: X-linked Alport syndrome in women: Genotype and clinical course in 24 cases. Front Med (Lausanne) 7: 580376, 2020. 10.3389/fmed.2020.580376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savige J, Ariani F, Mari F, Bruttini M, Renieri A, Gross O, Deltas C, Flinter F, Ding J, Gale DP, Nagel M, Yau M, Shagam L, Torra R, Ars E, Hoefele J, Garosi G, Storey H: Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol 34: 1175–1189, 2019. 10.1007/s00467-018-3985-4 [DOI] [PubMed] [Google Scholar]
- 39.Savige J, Storey H, Watson E, Hertz JM, Deltas C, Renieri A, Mari F, Hilbert P, Plevova P, Byers P, Cerkauskaite A, Gregory M, Cerkauskiene R, Ljubanovic DG, Becherucci F, Errichiello C, Massella L, Aiello V, Lennon R, Hopkinson L, Koziell A, Lungu A, Rothe HM, Hoefele J, Zacchia M, Martic TN, Gupta A, van Eerde A, Gear S, Landini S, Palazzo V, Al-Rabadi L, Claes K, Corveleyn A, Van Hoof E, van Geel M, Williams M, Ashton E, Belge H, Ars E, Bierzynska A, Gangemi C, Lipska-Ziętkiewicz BS: Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur J Hum Genet 29: 1186–1197, 2021. 10.1038/s41431-021-00858-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson BG, Reeders ST, Tryggvason K: Type IV collagen: Structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem 268: 26033–26036, 1993 [PubMed] [Google Scholar]
- 41.Rungsung I, Sahay M, Dalal A: Digenic variations of human COL4A3 and COL4A4 genes result in early onset renal failure. Gene Rep 19: 100602, 2020 [Google Scholar]
- 42.Kamiyoshi N, Nozu K, Fu XJ, Morisada N, Nozu Y, Ye MJ, Imafuku A, Miura K, Yamamura T, Minamikawa S, Shono A, Ninchoji T, Morioka I, Nakanishi K, Yoshikawa N, Kaito H, Iijima K: Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin J Am Soc Nephrol 11: 1441–1449, 2016. 10.2215/CJN.01000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA: COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol 24: 1945–1954, 2013. 10.1681/ASN.2012100985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, Yamanouchi S, Minamikawa S, Morisada N, Nakanishi K, Shima Y, Yoshikawa N, Ninchoji T, Morioka I, Kaito H, Iijima K: Natural history and genotype-phenotype correlation in female X-linked alport syndrome. Kidney Int Rep 2: 850–855, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.