

Abstract
KRAS G12D is the most frequent KRAS mutation in human cancer with particularly high frequencies in pancreatic and colorectal cancer. Informed by the structure of the KRASG12C inhibitor adagrasib, Hallin et al. have now, through multiple rounds of structure‐based drug design, identified and validated a potent, selective, and noncovalent KRASG12D inhibitor, MRTX1133. This study demonstrated that MRTX1133 inhibited both the inactive and active state of KRASG12D and showed potent antitumor activity in several preclinical models of pancreatic and colorectal cancer, especially when combined with cetuximab, a monoclonal antibody against the EGFR, or BYL‐719, a potent PI3Kα inhibitor.
Keywords: colorectal cancer, combination therapies, KRASG12D , noncovalent binding, pancreatic ductal adenocarcinoma
A new study by Hallin et al. has identified and validated a potent, selective, and noncovalent KRASG12D inhibitor, MRTX1133. This inhibitor shows potent antitumor activity in several preclinical models of pancreatic and colorectal cancer, especially when combined either with cetuximab, a monoclonal antibody against the EGFR, or with BYL‐719, a potent PI3Kα inhibitor.
Abbreviations
- CRC
colorectal carcinoma
- GAP
GTPase‐activating protein
- GDP
guanosine diphosphate
- GMPPCP
β,γ‐Methyleneguanosine 5′‐triphosphate sodium salt
- GTP
guanosine triphosphate
- IC50
half maximal inhibitory concentration
- IP
intraperitoneal
- IV
intravenous
- ORR
objective response rate
- RBD
RAS‐binding domain
1. A historical perspective of KRAS‐targeting drugs
KRAS oncogenes have been identified in a quarter of all human tumors and appear with high prevalence in some of the most lethal types of cancer such as pancreatic ductal adenocarcinoma (PDAC, 95%), colorectal carcinoma (CRC, 50%), and lung adenocarcinoma (LUAD, 30%). Yet, KRAS oncoproteins were considered undruggable for decades and patients with KRAS‐mutant tumors remained excluded from personalized medicine approaches [1]. This notion changed recently thanks to the identification of a previously unrecognized pocket in the switch‐II region (switch‐II pocket) of KRAS. Moreover, by taking advantage of the reactive cysteine residue in one of the mutant KRAS isoforms (KRASG12C), Shokat and colleagues developed the first compound to directly block a KRAS oncoprotein [2]. Since the switch‐II pocket is only accessible when KRASG12C is bound to GDP and therefore inactive, binding of a covalent inhibitor requires a substantial degree of nucleotide cycling to effectively block this oncoprotein. Indeed, KRASG12C retains a significant level of nucleotide cycling despite its insensitivity to classical GTPase‐activating protein (GAP)‐stimulated GTP hydrolysis which in this case is mediated via the noncanonical GAP RGS3 [3].
Further efforts in drug development in the following years led to the development and subsequent approval of sotorasib (AMG 510), a drug developed by Amgen (Thousand Oaks, CA, USA). Sotorasib forms a covalent bond with the KRASG12C oncoprotein blocking it in its inactive state and has demonstrated clinical efficacy for a subset of patients with KRAS G12C‐mutant LUAD as well as CRC [4]. A second covalent KRASG12C inhibitor developed by Mirati Therapeutics, adagrasib (MRTX849), recently received the Breakthrough Therapy designation by the FDA [5]. Although the KRAS G12C mutation is particularly frequent in LUAD (40% of KRAS mutations, Fig. 1A), its overall prevalence only encompasses 13% of all KRAS‐mutant tumors, indicating that there is an urgent requirement to target other mutant isoforms.
Fig. 1.
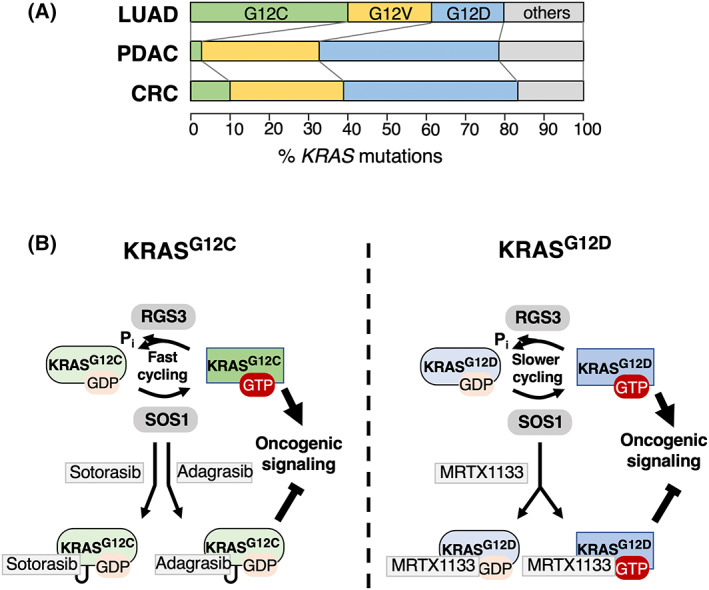
MRTX1133 inhibits KRASG12D. (A) Frequency of mutant KRAS alleles in lung adenocarcinoma (LUAD), pancreatic ductal adenocarcinoma (PDAC), or colorectal carcinoma (CRC). G12C mutations are indicated in green, G12V mutations in yellow and G12D alleles in blue. All other mutations are represented in gray. (B) Left: KRASG12C inhibition by sotorasib and adagrasib. KRASG12C is characterized by fast nucleotide cycling where GTP hydrolysis is catalyzed by the noncanonical GAP RGS3. Sotorasib and adagrasib bind to the switch II pocket in the inactive, GDP‐bound state of KRASG12C and form covalent interactions with the reactive cysteine residue. Right: KRASG12D inhibition by MRTX1133. KRASG12D has two‐ to three‐fold slower cycling rates than KRASG12C. MRTX1133 binds both GDP‐ and GTP‐bound KRASG12D with high affinity without the requirement for covalent interactions.
The most prevalent KRAS mutation in human cancer is KRAS G12D, present in 33% of all cases and the most frequent mutant KRAS allele in PDAC (46%, Fig. 1A). Consequently, the development of inhibitors targeting KRASG12D has always been of particular interest since the identification of the switch‐II pocket. Yet, this mutant isoform lacks the reactive cysteine residue present in KRASG12C, thus imposing a significant challenge to design selective compounds that bind to this mutant isoform in a stable manner. In addition, the GTP hydrolysis rate of KRASG12D is two to three times lower than that of KRASG12C [6]. Nevertheless, informed by the structure of the KRASG12C inhibitor adagrasib, scientists at Mirati Therapeutics were recently able to synthesize, through multiple rounds of structure‐based drug design, a selective, noncovalent KRASG12D inhibitor (MRTX1133) that is active at concentrations in the low nm range [7]. This drug binds to the switch‐II pocket with extraordinary high affinity, thereby obviating the requirement for covalent interactions (Fig. 1B).
2. Validation of the KRASG12D inhibitor MRTX1133
A more recent study has now evaluated the mechanism of action and antitumor activity of MRTX1133 [8]. First, the authors performed a series of assays to validate the binding efficacy of the drug to KRASG12D when compared with wild‐type KRAS. Homogenous time resolved fluorescence (HTRF) as well as surface plasmon resonance (SPR) assays confirmed an IC50 of < 2 nm and a binding KD of 0.2 pm, which are approximately 700‐fold more selective for KRASG12D over wild‐type KRAS. In addition, MRTX1133 was able to prevent binding of a RAF1 RBD peptide to KRASG12D preloaded with the nonhydrolyzable GTP analog GMPPCP with an IC50 of 9 nm. Together with the resolved structure of MRTX1133 associated with KRASG12D bound to GDP or to GMPPCP, the authors observed a conformational change in the switch I and II regions that was incompatible with effector binding. Thus, this indicated that MRTX1133 inhibited both inactive and active KRASG12D states (Fig. 1B). Although this compound inhibited the inactive form with higher potency, its additional activity against the active form is likely to contribute to its higher overall potency.
The authors also tested the cellular activity of MRTX1133 in cell lines carrying the G12D mutation in KRAS [8]. MRTX1133 inhibited downstream signaling pathways in a concentration‐dependent manner with an IC50 of < 3 nm. Moreover, this inhibitory effect was maintained for up to 48–72 h, at least when applied at higher concentrations. The authors also found that MRTX1133 inhibited ERK phosphorylation and cell growth in 2D as well as 3D cultures in 24 out of 25 KRAS G12D‐mutant cell lines tested. In contrast, most non‐KRAS G12D‐mutant cell lines were not inhibited at all and only a few of them responded at higher concentrations. More importantly, MRTX1133 was active in mouse xenograft tumors in the range of 10–30 mg·kg−1. Of note, when tested in a series of 25 human cell line‐ and patient‐derived xenografts (PDX) at 30 mg·kg−1, 11 of them displayed tumor regression rates of > 30%. Interestingly, although 73% of PDAC models responded to the treatment, only 25% of CRC models were affected. Whether this holds true in the clinic remains to be determined, but it could be a consequence of the fact that mutant KRAS acts as a primary driver in PDAC but not in CRC [8]. Finally, MRTX1133 showed a poor bioavailability when applied orally. Nevertheless, it was effective when administered via IP or IV routes. Whether this limitation will affect the clinical utility of this compound remains to be determined. Yet, data recently presented at the NCI RAS Initiative Symposium held in Frederick, MD suggested that formulation strategies to enhance oral absorption and/or increase IV half‐life may increase the probability of augmenting the efficacy in the clinic.
Based on the limited efficacy in some of the preclinical in vivo models, the authors set out to explore factors that constrain the response to MRTX1133 as well as to identify collateral dependencies that could maximize its efficacy. To this end, they conducted a CRISPR/Cas9 sgRNA library screen both in vitro and in vivo. These experiments revealed several tumor suppressor genes such as PTEN, KEAP1, NF1, or RB1 that conferred at least partial resistance to MRTX1133. Interestingly, KEAP1 also scored strongly in one of the in vivo models of PDAC, suggesting that KEAP1 could be a key modifier of antitumor response.
Since KRAS inhibition as a monotherapy does not result in prolonged tumor regression in lung cancer [4, 5], the authors anticipated that combination therapies would likely increase the therapeutic benefit of MRTX1133. The results of the CRISPR/Cas9 sgRNA library screen suggested that, among others, inhibition of EGFR and PTPN11 (SHP2) could synergize with MRTX1133. Informed by these results, the authors selected several compounds that could target these and other proteins and tested whether they could observe a synergistic effect with MRTX1133. Interestingly, combinatorial treatment with MEK, ERK, SHP2, or SOS1 inhibitors did not substantially enhance the activity of MRTX1133 to the same extent previously observed with KRASG12C inhibitors. However, the HER2 family inhibitors, afatinib and cetuximab, as well as the selective PI3Kα inhibitor, BYL‐719, did show a synergistic effect in PDAC and CRC cell lines. Based on these results, the authors combined MRTX1133 with cetuximab, an EGFR inhibitor approved for KRAS WT CRC, and with the PI3Kα inhibitor BYL‐719. These combinations were even efficient in models in which either drug as a monotherapy had no effect. Importantly, both drug combinations did not cause significant body weight loss in mice when used at effective doses, suggesting that there could be therapeutic windows when used in human patients.
The findings described by Hallin et al. may have a significant impact for patients with KRAS G12D‐mutant tumors [8]. Until now, the therapeutic options for patients with KRAS G12D‐positive PDAC were limited to old chemotherapy strategies, such as gemcitabine, 5‐fluorouracil, or taxanes. PDAC is one of the types of cancer with the lowest survival rates and accumulating evidence indicates that mutations in KRAS are an early event that drives this disease. Hence, a drug that could block the initiating event in those PDAC patients with KRAS G12D mutations could lead to substantially improved patient survival. As a proof‐of‐principle, recent clinical data with sotorasib (21% ORR) and adagrasib (50% ORR) in KRAS G12C‐mutant PDAC provide initial evidence, although KRAS G12C mutations comprise less than 2% of KRAS‐mutated PDAC (Fig. 1A) [9, 10].
Several mechanisms have been described that, at least in cell culture and mouse models of PDAC, allow survival of tumor cells in the absence of mutant KRAS [11, 12]. This is not unexpected since lung cancers treated with KRASG12C inhibitors are also capable of rapidly developing resistance. Therefore, combination therapies are likely to be more effective and the current study by Hallin et al. already proposes several potent combinations [8]. In contrast to PDAC, KRAS mutations are usually not considered an initial driving event in CRC, instead being responsible for the progression of adenomas to malignant carcinomas. This property might be one of the reasons for the limited effect of KRASG12C inhibitors in CRC patients [13]. Whether the combinatorial therapies of MRTX1133 outlined in this study will increase the efficacy of this novel KRAS inhibitor remains to be determined.
Finally, it will be of great interest to see how MRTX1133 will perform in clinical trials. Based on prior experience with sotorasib or adagrasib, it is likely that combination therapies will be much more effective. Until then, additional preclinical studies will shed light on potential mechanisms of resistance and guide clinicians to select appropriate combination therapies. Interrogation of resistance mechanisms in patients treated with sotorasib or adagrasib revealed secondary mutations in KRAS that prevented these drugs from binding to the switch‐II pocket, as well as mutations in effectors of related signaling pathways such as EGFR, BRAF or activating mutations in other RAS paralogs [14, 15]. Given the high affinity binding mode of MRTX1133, similar mutations, either preventing high‐affinity binding to the switch‐II pocket or activating other signaling pathways, can be expected. Yet, most tumors acquire resistance without the presence of novel mutations [14, 15]. Understanding the molecular mechanisms responsible for the appearance of resistance will provide new treatment avenues to increase the clinical efficacy of these compounds. Regardless of these potential limitations, the development of MRTX1133 is undoubtedly a major step forward toward the implementation of much‐needed effective therapies for patients with KRAS G12D‐mutant tumors.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by grants from the European Research Council (ERC‐2015‐AdG/695566, THERACAN), the CRIS Cancer Foundation and the CIBERONC to M.B. M.B. is a recipient of an Endowed Chair from the AXA Research Fund.
Comment on: https://doi.org/10.1038/s41591-022-02007-7
Contributor Information
Matthias Drosten, Email: mdrosten@usal.es.
Mariano Barbacid, Email: mbarbacid@cnio.es.
References
- 1. Drosten M, Barbacid M. Targeting KRAS mutant lung cancer: light at the end of the tunnel. Mol Oncol. 2022;16:1057–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K‐Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li C, Vides A, Kim D, Xue JY, Zhao Y, Lito P. The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science. 2021;374:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skoulidis F, Ki BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutations. N Engl J Med. 2021;384:2371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jänne PA, Riely GJ, Gadgel SM, Heist RS, Ou SHI, Pacheco JM, et al. Adagrasib in non‐small‐cell lung cancer harboring a KRASG12C mutation. N Engl J Med. 2022;387:120–31. [DOI] [PubMed] [Google Scholar]
- 6. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer‐associated KRAS mutations. Mol Cancer Res. 2015;13:1325–35. [DOI] [PubMed] [Google Scholar]
- 7. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2022;65:3123–33. [DOI] [PubMed] [Google Scholar]
- 8. Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, et al. Anti‐tumor efficacy of a potent and selective non‐covalent KRASG12D inhibitor. Nat Med. 2022;28:2171–82. [DOI] [PubMed] [Google Scholar]
- 9. Strickler JH, Satake H, Hollebecque A, Sunakawa Y, Tomasini P, Bajor DL, et al. First data for sotorasib in patients with pancreatic cancer with KRAS p.G12C mutation: a phase I/II study evaluating efficacy and safety. J Clin Oncol. 2022;40(36_suppl):360490. [Google Scholar]
- 10. Bekaii‐Saab TS, Spira AI, Yaeger R, Buchschacher GL, McRee AJ, Sabari JK, et al. KRYSTRAL‐1: updated activity and safety of adagrasib (MRTX849) in patients with unresectable or metastatic pancreatic cancer and other gastrointestinal tumors harboring a KRASG12C mutation. J Clin Oncol. 2022;40(4_suppl):519.34878805 [Google Scholar]
- 11. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hou P, Kapoor A, Zhang Q, Li J, Wu C, Li J, et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discovery. 2020;10:1058–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeiffer P, Qvortrup C. KRASG12C inhibition in colorectal cancer. Lancet Oncol. 2022;23:10–1. [DOI] [PubMed] [Google Scholar]
- 14. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibitionin cancer. N Engl J Med. 2021;384:2382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao Y, Murciano‐Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599:679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]