

INTRODUCTION
Bacteria and archaea have developed many systems to defend against potentially harmful mobile genetic elements (MGEs), such as viruses and plasmids (1, 2). Clustered regularly interspaced short palindromic repeats (CRISPR) together with their accompanying cas (CRISPR-associated) genes are one of the most prevalent of these systems, occurring in approximately 50% of bacteria and 90% of archaea (3). CRISPR-Cas systems present a particularly formidable defence because they are adaptive, acquiring specific immunity to segments of foreign DNA after exposure to these elements (for reviews see (4, 5)). To combat these potent systems, MGEs have acquired genes encoding inhibitors of CRISPR-Cas systems, known as anti-CRISPRs. First discovered in 2013 (6), knowledge of these proteins has rapidly expanded with 45 distinct families currently recognized (7). In this review, we describe the discovery, functional mechanisms, evolution, and applications of anti-CRISPRs.
CRISPR-Cas SYSTEMS
CRISPR loci are composed of two parts: the first, is an array comprising variable spacers that are primarily derived from foreign genetic elements separated by a palindrome that is repeated throughout the array; the second, is a group of cas genes encoding proteins associated with the array (for review see (8)). The array is transcribed, processed, and complexed with Cas proteins (Figure 1). The resulting CRISPR-Cas complex contains one CRISPR RNA molecule (crRNA) bound to one or more Cas proteins that can specifically recognize invading foreign genetic elements/nucleic acids through complementarity with the crRNA. The site on the invading DNA that is targeted by the spacer, known as the protospacer, is flanked by a protospacer adjacent motif (PAM), which is generally composed of 2 to 4 nucleotides and is specific for a given CRISPR-Cas system. PAMs are crucial in allowing CRISPR-Cas systems to distinguish self from non-self, and prevent the binding of CRISPR complexes to the CRISPR arrays from where they originated. Once foreign nucleic acids are bound, the CRISPR-Cas complex mediates their cleavage and subsequent destruction (Figure 1). A unique power of CRISPR-Cas systems is their ability to adapt to invading MGEs that have not been previously encountered. This is accomplished by excising short sequences from these elements and integrating them into the CRISPR array as new spacers. Thus, immunity to these MGEs is acquired. Despite these commonalities, CRISPR-Cas systems are highly variable – genetically, architecturally, and functionally (9).
Figure 1.
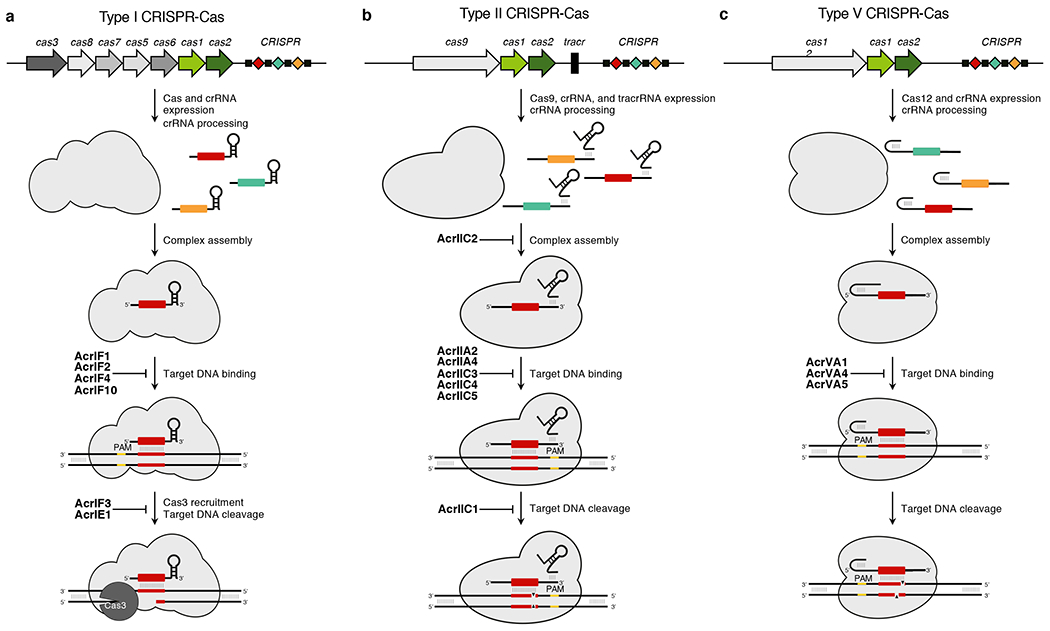
Anti-CRISPR proteins inhibit CRISPR-Cas systems at distinct stages. (a) A generalized type I CRISPR-Cas locus is depicted. Cas genes and the CRISPR array are expressed. The array transcript is subsequently processed into mature crRNA. Cas proteins assemble around the mature crRNA to form the a CRISPR-Cas complex, which identifies foreign DNA targets through complementary base pairing with its crRNA and an appropriate PAM sequence. AcrIF1, AcrIF2, AcrIF4, and AcrIF10 prevent Cascade from interacting with DNA. Annealing of the crRNA triggers R-loop formation, the recruitment of the Cas3 helicase/nuclease, and the destruction of the DNA target. AcrIF3 and AcrIE1 disable Cas3 to prevent target cleavage. (b) A generalized type II CRISPR-Cas locus is depicted. Cas9 and processed crRNA and tracrRNA assemble in a complex that can recognize PAM flanked sequences with complementarity to its crRNA. AcrIIC2 inhibits crRNA loading into Cas9 preventing proper complex assembly. AcrIIA2, AcrIIA4, AcrIIC3, AcrIIC4, and AcrIIC5 prevent the complex from recognizing target DNA. Following target recognition, Cas9 creates a double stranded DNA break target, leading to its destruction. AcrIIC1 inhibits the nuclease activity of Cas9 to prevent target cleavage. (c) A generalized type V CRISPR-Cas locus is depicted. Cas12 and the crRNA are expressed, processed, and assembled into a surveillance complex. The complex binds target DNA with a crRNA complementary, PAM flanked target. AcrVA1, AcrVA4, and AcrVA5 prevent DNA recognition. Target binding triggers the nuclease activity of Cas12, generating a staggered double-stranded DNA break to destroy the target. Cas genes = coloured arrows. CRISPR array: repeats = black boxes; spacers = coloured diamonds. PAM = yellow. cas1 and cas2 encode proteins involved in CRISPR adaptation
The diversity of CRISPR-Cas systems has been recently categorized into two classes, six types, and more than thirty subtypes, which differ in their palindromic repeats, spacer lengths, and complement of unique Cas proteins (9). In Class 1 systems, nucleic acid recognition and cleavage are carried out by a complex of crRNA and at least three different Cas proteins. In Class 2 systems these functions are mediated by a single Cas protein bound to crRNA. The Class 2 systems include the CRISPR-Cas9 systems that are widely used for genome editing applications (for review see (10)). Anti-CRISPRs have been found that block CRISPR-Cas types of both classes.
DISCOVERY OF ANTI-CRISPRS
Initial discovery of anti-CRISPRs
Anti-CRISPRs were first discovered as part of an investigation of the effects of prophages in the bacterial species, Pseudomonas aeruginosa (Pae). Prophages are formed when a bacterial virus (also known as a bacteriophage or phage) integrates its complete genome into the host genome. Prophage formation is a frequently-occurring feature of phages. Although most genes within prophages are silenced, genes that remain expressed often elicit profound physiological effects (11, 12). Bondy-Denomy et al. constructed collection of strains that each contained a different prophage in an effort to systematically investigate their phenotypic effects (13). Using this collection, three prophages were discovered that mediated inhibition of the type I-F CRISPR-Cas system present in the Pae strain under study (6). By comparing the genomes of these phages to those of related phages, a region potentially containing anti-CRISPR (acr) genes was discovered in eight different phages. Ultimately, genes encoding 5 completely distinct Acr protein families (AcrIF1-AcrIF5) that inhibit the type I-F system were identified. In a subsequent study, genes located within the same phage acr region were found to encode four additional Acr families (AcrIE1-AcrIE4). These Acrs inhibit the type I-E CRISPR-Cas system encoded by some Pae strains (14). Remarkably, among these nine newly discovered families, all proteins were fewer than 140 amino acids in length and displayed no sequence similarity to other putative or known proteins.
Protein sequence database searches using Acr proteins from the initial nine families as PSI-BLAST (15) queries identified only closely related homologues encoded within the Pseudomonas genus, making the identification of additional families of anti-CRISPRs challenging. This problem was circumvented by the observation that one gene, referred to as acr associated gene 1 (aca1), was conserved at the 3’-end of every acr region (Figure 1a). The Aca1 protein contains a predicted Helix-Turn-Helix (HTH) DNA-binding domain, suggesting that it might be a regulator of the acr operon (see below). By performing PSI-BLAST searches with the Aca1 protein, regions within prophages and other types of MGEs in which Aca1 homologues were encoded downstream of genes encoding proteins with small sizes were identified. In vivo, activity assays performed with some of these candidates revealed two additional families of Acrs that inhibited the type I-F system of Pae (AcrIF6 and AcrIF7) (16). Fortuitously, the AcrIF6 family emerged as the first to include diverse homologues in a wide variety of bacterial species. Unexpectedly, one AcrIF6 homologue was encoded in a prophage region that did not include an aca1 gene. However, the protein encoded directly downstream of the gene encoding this Acr, contained a predicted HTH DNA-binding domain. Homologues of this new putative DNA-binding protein, named Aca2, were found to be encoded downstream of genes encoding other small proteins that turned out to possess Acr activity (16). This iterative “guilt by association” approach yielded three additional families of Acrs with inhibitory activity against the Pae type I-F system (AcrIF8-AcrIF10) (Figure 2b). PSI-BLAST searches with members of these new families detected homologues in bacterial species as broadly distributed phylogenetically as the type I-F system itself, indicating the potential for Acrs to inhibit most of the known I-F systems (16).
Figure 2.

The commonly used approaches for acr gene discovery. (a) Representative acr regions are shown from the Pae phages in which these genes were first identified. Gray arrows depict predicted open reading frames flanking the acr regions. Dark green arrows correspond to genes encoding the highly conserved anti-CRISPR-associated gene 1 (aca1) predicted to encode a protein with a helix-turn-helix (HTH) motif. The remaining colored arrows represent the acr genes. (b) The “guilt-by-association” method is a homology-based computational search approach that uses the proteins encoded by the acr and aca genes as queries. For example, a search with AcrIF6 (pink arrow) of Pae identified a homologue in Oceanimonas smirnovii. The gene encoding this protein was found adjacent to aca2, which is a distinct HTH protein. Searches with Aca2, resulted in homologues being detected in the genomes of Vibrio parahaemolyticus and Brackiella oedipodis, which led to the discovery of acrIF9 (purple arrow) and acrIIC1 (yellow arrow) positioned upstream of aca2 in these bacteria. Finally, homology-based searches using the protein encoded by acrIIC1 resulted in the discovery of aca3 (green arrow). This back and forth approach has facilitated the discovery of many families of Acrs. (c) The self-targeting approach is also commonly used for acr gene discovery. If a prokaryotic strain has a functional CRISPR-Cas system (e.g., type II-A depicted on the left) and contains a spacer (checkered box in the CRISPR-array) that matches a protospacer (checkered box within the dark gray arrow on the right) located adjacent to a known PAM (red box within the dark gray arrow on the right) within an MGE, then that organism must also have an acr gene—otherwise, its CRISPR-Cas system would degrade its own genome. Black diamonds represent the palindromic repeats in the CRISPR-array. Candidate acr genes are unique genes (patterned blue arrows) present in the targeted MGE (patterned blue arrows; top) and absent from a related MGE that elicits no Acr activity (patterned blue arrows; bottom).
Further PSI-BLAST searches using Aca2 as a query identified putative Acrs encoded within MGEs in strains of Brackiella oedipodis and Neisseria meningitidis (Nme) (17). Since these strains encoded no Class 1 systems but encode type II-C Cas9 systems, the activity of these putative Acrs was tested against the Nme CRISPR-Cas9 system. These experiments revealed three families of Acrs (AcrIIC1-AcrIIC3) that inhibited NmeCas9. Excitingly, these Acrs were also shown to inhibit genome editing in cultured human cells (17).
Further anti-CRISPR discovery through bioinformatics
In addition to the guilt-by-association approach described above, another computational method, the “self-targeting” approach, has been fruitful in identifying new Acrs (Figure 2c). This approach involves searching for bacterial genomes encoding both a functional CRISPR-Cas system and CRISPR spacers that target sites within the same genome. Such strains would not be expected to survive unless their genomes also encoded anti-CRISPRs to inhibit their CRISPR-Cas systems. Rauch et al. identified strains of Listeria monocytogenes (Lmo) bearing self-targeting spacers (18). Within one of these strains, they also identified a targeted prophage, and showed that this prophage conferred anti-CRISPR activity. Subsequent comparison of this phage genome with a closely related phage genome that did not possess anti-CRISPR activity facilitated the identification of an acr region encoding two proteins (AcrIIA1 and AcrIIA2) that efficiently inhibited the type II-A Lmo CRISPR-Cas system. BLAST searches with these Acrs led to additional prophage acr regions, and the identification of two more Acr families, AcrIIA3 and AcrIIA4. Importantly, AcrIIA2 and AcrIIA4 were able to inhibit the Streptococcus pyogenes (Spy) CRISPR-Cas9 system, which is by far the most widely used system for genome editing applications. This inhibition was demonstrated both in a bacterial assay, and in human tissue culture cells (18).
In a subsequent study, a systematic search of 150,000 prokaryotic genomes carried out by Watters et al. identified more than 9000 genomes (~6% of the 150,000) with self-targeting spacers (19). By focusing on self-targeting strains of Moraxella bovoculi, which possesses a type V-A CRISPR-Cas12 system, anti-CRISPRs inhibiting this system (AcrVA1, AcrVA4, AcrVA5) were discovered. A guilt-by-association approach carried out at the same time also uncovered type V-A Acrs in strains of Moraxella catarrhalis (AcrVA1-AcrVA3) (20). These discoveries were notable because type V CRISPR-Cas12 systems are increasingly being used for genome editing applications (21). In the Marino et al. study, AcrIC1, an Acr inhibiting the type I-C CRISPR-Cas system was also discovered (20).
Functional screens and selections to identify anti-CRISPRs
In a purely functional approach to Acr identification, Hynes et al. used a collection of hundreds of virulent phages infecting Streptococcus thermophilus (Sth), a bacterial strain critical in the production of yogurt, to identify phages that did not elicit CRISPR-Cas based immunity (22). One of these phages was confirmed to be resistant to CRISPR-Cas attack even when infecting cells producing spacers designed to target the phage genome. By cloning and expressing all of the genes from this phage, one was found, called acrIIA5, that inhibited the type II-A systems of the Sth and Spy CRISPR-Cas9 systems. A subsequent study using the same functional approach combined with gene neighborhood analysis identified anti-CRISPR AcrIIA6 (23). In a conceptually related approach, a virus infecting the archaeal species, Sulfolobus islandicus, that displayed resistance to CRISPR-Cas, was characterized. By isolating viral mutants that became sensitive to CRISPR-Cas, He et al. discovered an Acr that inhibits the type I-D system of this species. This Acr, called AcrID1, is the only type I-D Acr identified so far (24).
In a recent study, a functional selection was used to isolate genes with Acr activity from human gut and soil metagenomic libraries (25). For the selection, SpyCas9 was co-expressed in E. coli with a single-guide RNA (sgRNA) directing the CRISPR-Cas complex to cleave a second plasmid conferring chloramphenicol resistance (Cmr). Introduction of the Cmr-conferring plasmid into the strain expressing the SpyCas9:sgRNA complex resulted in very few colonies being resistant to Cm due to cleavage of the plasmid by Cas9. However, expression of AcrIIA2 from the Cmr-conferring plasmid resulted in thousands of colonies growing in the presence of Cm because the plasmid is protected from SpyCas9-mediated destruction by the Acr. Random fragments of DNA derived from various metagenomic samples were then cloned into the Cmr-conferring plasmid; thus, allowing for the selection of plasmids encoding proteins inhibiting SpyCas9 based on the bacterium’s ability to grow in the presence of Cm. Using this approach, four genes that encoded proteins (AcrIIA7-AcrIIA10) that could inhibit SpyCas9 DNA cleavage activity in vitro were identified. Three of these four putative Acrs, also bound robustly to SpyCas9 in vitro (25). The homologues of AcrIIA7 and AcrIIA9 were much more broadly distributed among diverse bacterial phyla compared to other previously identified Acrs. Surprisingly, these homologues were not enriched for occurrence in species encoding type II-A CRISPR-Cas systems, which has been a consistent feature of other Acr families. AcrIIA8 displayed sequence similarity and predicted structural similarity to a phage structural protein involved in joining the phage head to the phage tail (26, 27). Our own synteny analysis of the closest homologue of AcrIIA8 in the NCBI database (refseq ID WP_009270720, 80% identical to AcrIIA8), which lies in a prophage of Clostridium butyricum, indicates that it must function as a head-tail joining protein in this phage. In future studies, it will be of great interest to characterize the Acr activity of homologues of these functionally selected Acrs to determine whether these are families of Acrs or whether the detected Acr activity of the proteins isolated may be adventitious, and not reflect their biologically relevant function.
Nomenclature for Acr Proteins
A standard naming convention for Acrs was recently published (7). Acr familes are named for their type (e.g. IF) and are numbered sequentially as they are discovered, so that, for example, the third Acr family blocking a type I-F system is named AcrIF3. To avoid redundancy in names, investigators submit the names for new Acr families as soon as the paper describing them has been accepted. All Acr names are listed in a spreadsheet (https://tinyurl.com/anti-CRISPR), which also lists other useful information about the Acr family, such as publication information and amino acid sequence.
Small Molecule Inhibitors of CRISPR-Cas Function
Due to the many practical applications for Cas9 inhibitors in genome editing applications, some investigators have focused on developing small molecule Cas9 inhibitors as these would have some advantages over proteins. Maji et al. generated a platform to screen large libraries of small-molecules to identify inhibitors of SpyCas9 (28). This high-throughput platform screened small molecule libraries with a fluorescence-polarization-based assay for DNA binding. One molecule, BRD0539, was identified as a SpyCas9 inhibitor that is stable in human plasma. This molecule demonstrated dose and temporal control of SpyCas9 in vitro, in both bacterial and mammalian cells. This approach will likely be useful for identifying small molecule inhibitors for other existing and emerging CRISPR-associated nucleases.
MECHANISMS OF ACR FUNCTION
The tremendous significance of CRISPR-Cas systems for genome editing applications has driven a correspondingly large effort directed at understanding the mechanisms by which these systems are inhibited by Acrs. The ability of these small proteins to potently block the activity of large ribonucleoprotein complexes is remarkable and elucidating the many different means by which this can be achieved is fascinating, providing new insights into the functioning of CRISPR-Cas systems. Below we describe the best characterized mechanisms by which Acrs inhibit type I, type II, and type V CRISPR-Cas systems. A gallery of all of the solved Acr structures is shown in Figure 3.
Figure 3.

All of the solved structures of Acrs are shown. Some of these were solved on their own and some were solved bound to CRISPR-Cas complexes. No significant structural changes have been observed between the bound and unbound forms of Acrs when both have been solved. The PDB identification codes for each structure are shown in parentheses.
MECHANISMS OF TYPE I ACR ACTIVITY
Being the first discovered, Acrs inhibiting the Pae type I-F system were also the first to be characterized in vitro (29). The 350 kDa DNA recognition complex of the type I-F system, known as the Csy complex, is composed of four Cas proteins arrayed along a single 60 nucleotide crRNA (Figure 4). Cas6f, which is the nuclease responsible for processing the CRISPR array transcript into single crRNA molecules, remains bound to a stem-loop at the 3’-end of the crRNA. Six copies of Cas7f form the backbone of the complex, binding non-specifically to the spacer region of the crRNA. A complex of Cas8f and Cas5f binds to the 5’-end of the crRNA, known as the handle (30, 31). The Csy complex searches for a canonical PAM site specific for the type I-F system (3’-GG-5’) and, once found, unwinds the DNA duplex to form a stable R-loop where the target strand hybridizes with the crRNA and the nontarget strand is stabilized by the Cas8f subunit. Cryo-electron microscopy (cryo-EM) structural analysis of the DNA bound Csy complex shows a large conformational change in the complex involving the elongation of the Cas7f backbone and rotation of the Cas8f subunit relative to the non-DNA-bound form (31, 32). The elongation of the Cas7f backbone is required to form stable interactions between the crRNA and the target strand, while the Cas8f rotation is required to expose the R-loop binding channel for nontarget strand stabilization as well as to expose the docking site for Cas3, the nuclease of type I systems. Cas3 is recruited to the Csy complex and binds to Cas8f following formation of the R-loop (32).
Figure 4.

Structures of type I-F Acrs bound to the Pae Csy complex determined by cryoEM. (a) The cryo-EM structure of AcrIF1 bound to the Csy complex in the presence of a crRNA (yellow) is shown. Two copies of AcrIF1 (cyan) bind to the Cas7f backbone (gray) of the complex. Cas8f and Cas5f are not shown here for simplification. On the right is shown a close-up where residues 8-15 and 33-35 of AcrIF1 (blue spheres) reach into the DNA binding groove to sterically clash and prevent target DNA hybridization. These residues are indicated with red arrows. (b) The left image shows AcrIF2 (red) bound to the Cas8f hook domain (cyan) of the Csy complex. The middle image shows the structure of AcrIF10 (magenta) bound to the Cas8f hook domain (cyan). The right image shows that the two Acrs bind in different locations and cause the hook to move in different directions. (c) The left image shows the complete Csy complex. The helical bundle of Cas8f that binds to Cas3 is in cyan in the center of the structure. In the middle image, this bundle is overlaid with AcrIF3. The two helices that overlay well and are in bolder colors are both critical for binding to Cas3. On the right is the structure of Cas3 bound to AcrIF3.
Inhibition of DNA Binding
In vitro studies using purified components showed that AcrIF1, AcrIF2, and AcrIF4 bind directly to the Csy complex (29). AcrIF3 did not bind to the Csy complex, but bound Cas3, thereby preventing the nuclease component of the system from being recruited to the Csy:DNA complex. The binding of either AcrIF1 or AcrIF2 to the Csy complex abrogated its DNA binding activity. However, these Acrs blocked DNA binding in different ways, with AcrIF1 binding to Cas7f subunits and AcrIF2 binding to the Cas8f subunit. Furthermore, AcrIF1 was found to completely occlude the DNA binding site of the Csy complex, while complementary ssDNA molecules could still bind to the 3’-end of the spacer RNA in the presence of AcrF2 even though this Acr blocked binding of dsDNA and ssDNA complementary to the 5’-end of the spacer. AcrF2 was found to compete with dsDNA for binding of the Csy complex, while AcrF1 was still able to bind to Csy complex pre-saturated with DNA.
Structures determined by Cryo-EM confirmed that AcrIF1 binds to Cas7f, showing two molecules per Csy complex bound at the interfaces formed by the pairs of Cas7f subunits closest to the 5’-end of the crRNA (Figure 4a)(30, 31, 33). Binding of AcrIF1 prevents target DNA binding by sterically hindering access to the DNA binding groove. Prior to the cryo-EM studies, the structure of AcrIF1 alone was determined by NMR (34). Structure-guided mutagenesis experiments identified three residues (Y6, Y20, and E31) that are critical for binding to the Csy complex and for inhibiting CRISPR-Cas activity (34). In the cryo-EM structures, these residues were found to be deeply buried in interface between AcrF1 and Cas7f, interacting with K85, a conserved residue in Cas7f. A K85A substitution markedly weakened the interaction between AcrIF1 and the Csy complex (30).
As was shown biochemically (29), cryo-EM structures revealed a single molecule of AcrIF2 bound to the Cas8f subunit of the Csy complex (Figure 4b). The surface of AcrIF2 is negatively charged, facilitating its interaction with positively charged residues in a lysine-rich vise formed at the interface between Cas8f and the closest Cas7f subunit (Cas7.6f). This region on the Csy complex is crucial for DNA binding (30). AcrIF2 was originally thought to act as a DNA mimic by competing with DNA for binding at the lysine-rich vise (30). However, further cryo-EM analysis revealed that though AcrIF2 binds at the interface between Cas8f and Cas7.6f, it only partially overlaps with the region occupied by the DNA. AcrIF2 most likely inhibits the Csy complex by swinging a flexible domain of Cas8f, known as the hook, away from Cas7.6f, resulting in a regional conformational change that is imcompatible with DNA binding (31).
The structure of the Csy complex bound to AcrIF10 (35), an Acr which had not been previously characterized in vitro, revealed that it occupies the same site as the DNA duplex and likely acts as a DNA mimic (Figure 4b). The presence of AcrIF10 induces a local conformational change in the Cas8f hook domain, causing it to swing towards Cas7.6f, locking Cas8f in a similar conformation to that seen upon DNA binding. While AcrIF10 and AcrIF2 both bind to the interface between Cas8f and Cas7.6f, the regions that they occupy overlap minimally, and they cause the Cas8f hook to swing in opposite directions (Figure 4b) (31).
The structure of AcrID1 has also been solved (Figure 3) and it has been shown to bind Cas10d, the large subunit of the I-D CRISPR-Cas complex (24). Since Cas10d has nuclease activity and is involved in DNA-binding, it cannot be predicted at this time which step of CRISPR-Cas function is blocked by this Acr.
Inhibition of DNA Cleavage
In contrast to the Acrs discussed above, AcrIF3 binds to the Cas2-3 nuclease. The fusion of Cas2 (a protein involved in adaptation) and Cas3 (a nuclease critical for interference) is a unique feature of type I-F and type I-F variant CRISPR-Cas systems (9). The structure of the complex of this Acr with PaeCas2-3 has been solved by X-ray crystallography (36) and cryo-EM (37) (Figure 4c). Cas2-3 consists of an N-terminal histidine-aspartate (HD) type nuclease domain, two tandem RecA-like subdomains (RecA1 and RecA1) that together make up the SF2 helicase domain, a linker region, and a C-terminal domain (CTD). AcrIF3, an all-helical protein, binds as a dimer to Cas2-3, covering the HD domain, linker region, RecA2, and the CTD, blocking the DNA-binding tunnel formed by RecA2 and the CTD, and preventing Cas2-3 from accessing substrate DNA. Furthermore, AcrIF3 masks most of the exposed surface between the CTD and linker region that is required for Cas2-3 binding to the Cas8f subunit of the Csy complex after DNA target binding.
Cryo-EM characterization of the Csy complex bound to a dsDNA target sequence showed that DNA binding is accompanied by a large conformational change in a helical bundle domain of Cas8f, which results in a dramatic re-positioning of this region (32). This domain forms the interface for binding to Cas2-3 (Figure 4c). Remarkably, this Cas8f helical domain resembles the structure of AcrIF3 with one central helix displaying five identical residues (Figure 4c). Substituting some of these residues at structurally comparable positions in Cas8f or AcrIF3 abrogated binding of both proteins to Cas2-3. Thus, AcrIF3 appears to be a structural mimic of the Cas8f domain that recruits Cas2-3 to the DNA-bound Csy complex.
Detailed mechanisms for Acrs inhibiting other Type I systems have not been elucidated. However, the mode of action of AcrIE1 (14), which blocks the Pae I-E system, has been shown to be similar to AcrIF3. This Acr also binds to the nuclease (Cas3) and likely prevents its recruitment to the DNA bound I-E CRISPR-Cas complex. The structure of this Acr, determined by crystallography (35), is comprised of a dimer with each subunit containing three α-helices and a small C-terminal β-sheet (Figure 3). Apart from being all helical, the AcrIE1 structure does not resemble that of AcrIF3, suggesting that they inhibit Cas3 and Cas2-3, respectively, in different ways.
A notable feature of AcrIF3 and AcrIE1 is that these Acrs do not prevent specific binding of the CRISPR-Cas complex to its DNA target. Thus, these Acrs convert the CRISPR-Cas system into a specific DNA-binding complex that could perform other functions within the cell, such as repressing transcription. The ability of the Csy complex to specifically repress transcription in the presence of AcrIF3 has been demonstrated (6).
MECHANISMS OF TYPE II ACR ACTIVITY
Type II CRISPR-Cas systems use a single effector protein, denoted Cas9, and are thus designated as Class 2. These systems are by far the most widely studied due to their proven utility in diverse genome editing applications (10, 38). For this reason, mechanistic studies of anti-CRISPRs specific for these systems have been pursued with great vigour.
There are three subtypes of the type II system: II-A, II-B and II-C. These subtypes are distinguished based on CRISPR-Cas locus architecture and Cas9 phylogeny. Cas9 is a multi-domain protein that includes both REC and NUC lobes (Figure 5a) (for reviews see (39–42). In its natural context within the cell, Cas9 binds to two separate RNA molecules, the crRNA, which is processed from the CRISPR array and includes the spacer region, and a constant tracrRNA, which sits adjacent to the CRISPR array in the CRISPR-Cas locus and is required for crRNA processing. In most Cas9-based applications the crRNA and tracrRNA are joined into one molecule called the single-guide RNA (sgRNA). The REC lobe is primarily responsible for recognition of sgRNA with an arginine-rich bridge helix (BH) domain playing a particularly important role. The NUC lobe contains the two domains responsible for Cas9 cleavage of target DNA: the HNH domain that cleaves the crRNA-complementary DNA strand and the three-part RuvC domain (RuvC-I, RuvC-II and RuvC-III) that cleaves the non-complementary DNA strand. At the C-terminal end of Cas9, also within the NUC lobe, are the wedge domain (WED) and PAM-interaction domain (PID), which mediate initial DNA-binding and PAM recognition. Cas9 functions by first binding to sgRNA, which triggers a large conformational change, resulting in formation of the DNA surveillance complex. This complex scans for its cognate PAM sequence and, upon detection, surveys adjacent DNA for potential target sequences. Once Cas9 identifies target DNA with the correct PAM, an R-loop forms and the HNH domain is activated for cleavage of the target strand. Structural change of the HNH domain indirectly induces activation of the RuvC domain, which then cleaves the nontarget strand.
Figure 5.

Structures of Cas9 bound to Acrs. (a) The structure on the left shows SpyCas9 bound to AcrIIA2 (cyan) primarily interacting with the PAM-interacting domain (PID) of Cas9 (pale green), as well as the HNH (light orange) and REC domains (grey). The structure on the right shows SpyCas9 bound to AcrIIA4 (violet) primarily interacting with the PID (pale green), as well as the RuvC domain (pale yellow). Both Acrs bind in the same region, which is also where DNA binds. (b) AcrIIC1 (salmon) and AcrIIC3 (bluepurple) interact with different surfaces of the HNH domain (light orange) from NmeCas9.
Inhibition of DNA Binding
AcrIIA4 was the first type II Acr to be characterized in detail. Highlighting the intense interest in this type of Acr, four papers describing the AcrIIA4 structure were published in less than a year, with the first emerging only four months after its discovery (43–46). Structures of AcrIIA4 in complex with sgRNA-bound SpyCas9 show that that this highly acidic protein binds the PAM-interacting domain of Cas9, mimicking the PAM region of the dsDNA target and preventing DNA binding (Figure 5a) (43, 45, 46). AcrIIA4 also interacts with residues within the RuvC active site, so that nuclease activity would likely also be abrogated if DNA were able to bind. The effectiveness of AcrIIA4 as an inhibitor is emphasized by its strong binding affinity for Cas9:sgRNA (Kd = 4 nM), which is 15-fold higher than the Cas9:sgRNA affinity for target DNA (43). AcrIIA4 binds at least 1000-fold more weakly to Cas9 alone than to Cas9:sgRNA (44), confirming that the conformational changes occurring upon sgRNA binding are required to form the binding site for AcrIIA4, as was deduced from structural comparisons (43).
The mechanism of AcrIIA2 has also been characterized in detail both structurally and biochemically (47, 48). Although AcrIIA2 is entirely unrelated to AcrIIA4 both in sequence and structure, it interacts with the PAM-interacting domain of Cas9 through almost identical local contacts as AcrIIA4 (Figure 5a). These Acrs compete with DNA for binding to Cas9, and also compete with each other. They present a striking example of convergent evolution where different proteins have been selected to bind to the same sites in a similar manner. While displaying overlap with AcrIIA4 in binding the PAM-interacting domain, AcrIIA2 also interacts with the HNH and REC2 domains, which are not contacted by AcrIIA4. A distinguishing feature of AcrIIA2 is its temperature-sensitivity, displaying a substantially lower ability to inhibit in vitro Cas9 activity at 37 °C compared to 22 °C (47). A homologue of AcrIIA2, designated AcrIIA2b, bearing approximately 35% sequence identity to AcrIIA2, is not temperature-sensitive, and is a much more potent inhibitor of Cas9 than AcrIIA2. Structural characterization of this homologue revealed that two key aromatic residues found only in AcrIIA2b lie close to PAM-interacting residues of Cas9 and are responsible for the increased activity of this Acr.
Type II-C inhibitors AcrIIC4 and AcrIIC5 have been shown in vitro and in vivo to block DNA-binding activity, but not the binding of Cas9 to its corresponding sgRNA (49). Further mechanistic details about these Acrs have yet to be published.
Inhibition of DNA Cleavage
AcrIIC1, originally identified as an inhibitor of the type II-C NmeCas9, was found to inhibit the diverse type II-C Cas9 orthologues from Campylobacter jejuni (Cje) and Geobacillus stearothermophilus (Geo), which are 42% and 36% identical to NmeCas9, respectively. Protein interaction studies with domains of GeoCas9 showed that AcrIIC1 binds to the HNH domain. A crystal structure of this Acr bound the HNH domain of NmeCas9 revealed that it binds to the active site surface of the HNH domain by interacting with several highly conserved residues required for catalysis (Figure 5b). Consistent with its binding to a domain responsible for nuclease activity, Cas9:sgRNA bound by AcrIIC1 is still able to bind to target DNA with normal affinity but is unable to cleave the DNA. In this way, AcrIIC1 creates a catalytically dead Cas9 (dCas9) that still binds its DNA target. The ability of AcrIIC1 to inhibit diverse Cas9 orthologues is explicable as the HNH domain is one of the most conserved regions of Cas9.
Inhibition through Dimer Formation
AcrIIC3 inhibits DNA binding by NmeCas9 and induces it to dimerize forming a 2:2 complex of Cas9 and the Acr (50, 51). While a structure of this dimeric complex has not yet been determined, AcrIIC3 has been shown to bind the HNH domain of Cas9 and structures of this complex have been solved (51, 52). Strikingly, AcrIIC3 binds on the opposite side of the HNH domain from AcrIIC1 (Figure 5b). The ability of AcrIIC3 to inhibit only NmeCas9, as compared to the broad specificity of AcrIIC1 is likely explained by the much higher sequence variability observed on the surface bound by AcrIIC3. Supporting this conclusion, substitution of amino acids in the NmeCas9 HNH domain with those found in Cas9 orthologues that are not inhibited by AcrIIC3 resulted in large decreases in binding affinity for this Acr (52). Further interaction studies have indicated that AcrIIC3 also binds the REC lobe. This lobe, which is highly variable in sequence among Cas9 homologues, is required for AcrIIC3-mediated dimerization (51). Since the Cas9 surface bound by AcrIIC3 is not critical for function, it is probably the dimerization mediated by AcrIIC3 that leads to inhibition of DNA binding activity. Important functional surfaces may be buried in the Cas9 dimer interface and/or critical conformational changes may be impeded.
Inhibition of sgRNA Binding
Protein interaction studies with truncated forms of Cas9 showed that AcrIIC2 binds to the Arg-rich BH in the REC lobe of NmeCas9, which is involved in sgRNA interaction (51, 53). Consistent with this binding site, an AcrIIC2:Cas9 complex is blocked from interaction with sgRNA. Additionally, AcrIIC2 binds weakly to Cas9 once the latter is bound to sgRNA. The structure of AcrIIC2 shows a dimer with a large negatively charged surface that serves as the interface for binding the positively charged BH (Figure 5a). Co-crystal structures of AcrIIC2 bound to the BH identified key interacting negative residues on the Acr and key positive residues on Cas9 that were verified by mutagenesis (51, 53). Structural superposition of the BH with that of the NmeCas9:sgRNA binary complex shows that the AcrIIC2 dimer sterically clashes with stem-loops 1 and 2, and slightly overlaps the seed region of the sgRNA, altogether blocking the binding sites of the sgRNA to the bridge helix. When AcrIIC2 is co-expressed with Cas9 in E. coli, the total accumulation of Cas9 is markedly decreased, likely because Cas9 is locked in its apo-form lacking sgRNA, which is more susceptible to digestion by intracellular proteases. Thus, AcrIIC2 may hamper Cas9 activity in the cell both by blocking sgRNA binding and by decreasing the steady-state levels of Cas9.
MECHANISMS OF TYPE V ACR ACTIVITY
Belonging to Class 2, type V CRISPR-Cas systems target dsDNA using the effector protein, Cas12. Similar to Cas9, Cas12 forms a structure comprised of two lobes called REC and NUC (for reviews see (39, 41)). Cas9 and Cas12 both utilize a RuvC-like endonuclease domain to mediate DNA cleavage. However, unlike Cas9, Cas12 generates staggered cuts and lacks a second nuclease (HNH) domain. Outside of the RuvC domain, Cas9 and Cas12 bear little or no similarity either in sequence or structure, even though analogous nomenclature is used in naming the domains of these two enzymes (e.g., WED, PAM-interacting, and BH).
Inhibition of DNA Binding with Enzymatic Activity
AcrVA1 is a broad-spectrum inhibitor of type V systems with in vitro and in vivo activities against four diverse Cas12a orthologues, which range in pairwise percent identities between 30% and 40% (19, 20). The mechanism of Cas12a inhibition by AcrVA1 has been explored in biochemical and structural detail (54, 55). AcrVA1 binds Cas12a:crRNA in the cleft between the REC and NUC lobes, interacting mostly with domains involved in PAM-interaction, thereby acting as a DNA mimic. Strikingly, AcrVA1 also cleaves the crRNA and permanently prevents DNA binding. AcrVA1-mediated crRNA cleavage is specific to Cas12a-bound crRNA and is independent of spacer sequence or length. Further, crRNA cleavage occurs in the presence of a catalytically dead version of Cas12a, implying that AcrVA1 is not altering the nuclease behavior of Cas12a, but rather has nuclease activity itself. Consistent with this hypothesis, AcrVA1 can perform multiple rounds of crRNA cleavage in the presence of a Cas12a:crRNA complex, making AcrVA1 the first anti-CRISPR protein demonstrated to have an enzymatic mechanism (54).
AcrVA5 also inhibits the DNA binding activity of Cas12a, but functions against a more limited set of orthologues as compared to AcrVA1. Surprisingly, this Acr does not form a stable complex with the Moraxella bovoculi (Mb) Cas12a orthologue that it inhibits (56). This observation led to a search for covalent modifications of MbCas12a mediated by AcrVA5, and it was discovered that this Acr mediates acetylation of K635. This residue is crucial for PAM interaction, so that modification at this position completely abolishes DNA binding (Figure 6). An MbCas12a with an Arg residue substituted position 635 is resistant to the inhibitory activity of AcrVA5, as are Cas12a orthologues that naturally possess Arg at this position. Determining the crystal structure of AcrVA5 revealed that it is similar to other acetyltransferases in structure and it is bound to acetyl-CoA (56). Acetylation represents a unique mechanism of Acr function. However, the means by which AcrVA5 recognizes specific Cas12a homologues before acetylating them remains to be discovered.
Figure 6.

AcrVA5 acetylates K635 and prevents MbCas12a from recognizing the PAM. On the left is a superposition of PAM-bound LbCas12a and MbCas12a acetylated at K635. In the close-up on the right, it can be seen that the acetylated K635 (red) sits within the PID and can sterically block recognition of the PAM. Thus, MbCas12a is unable to interact with target DNA when K635 is methylated.
Inhibition of DNA Binding Accompanied by Cas12a Dimerization
AcrVA4 induces dimerization of Cas12a and blocks its DNA binding activity (54, 55, 57). Analysis by cryo-EM shows that AcrVA4 binds primarily to portions of the REC domain that are involved in crRNA binding and pre-crRNA processing, which is also carried out by Cas12a (55, 57). AcrVA4 appears to mimic pre-crRNA (57). The binding of AcrVA4 is far from the DNA binding site, but it is able to allosterically block the conformational changes that are required for Cas12a to bind target DNA. AcrVA4 is a dimer, and structural data show that Cas12a dimerization mediated by AcrVA4 occurs wholly through interactions of the N-terminal domain of AcrVA4. A version of AcrVA4 consisting of only its C-terminal domain, which accounts for all of the binding interactions with Cas12a, was able to efficiently inhibit Cas12a activity without inducing dimerization (57). These data show that the formation of Cas12a dimers is not required for the inhibitory activity of AcrVA4.
SUMMARY OF ACR MECHANISMS
Over the past four years, studies describing detailed biochemical and/or structural characterization of 16 different Acrs have been published. Reflecting the excitement and competition in the CRISPR-Cas field, more than half of these structures have been solved at least twice, and some up to four times. On the bright side, these repeated studies on the same proteins have consistently been in agreement, so we can be confident in our conclusions about Acr mechanisms, which are summarized in Figure 1. Remarkably, among the structures of 16 different Acrs (Figure 3), none show any similarity to any others, reflecting a unique evolutionary origin for each Acr family. Despite this structural diversity, there is some mechanistic overlap. Most common is the binding of Acrs at or close to the PAM-interaction region, a property shared by AcrIF2, AcrIF10, AcrIIA2, AcrIIA4, and AcrVA1. These Acrs are all acidic and, to varying extents, mimic DNA. DNA mimicry is a common theme among many inhibitors of DNA binding proteins including phage-encoded inhibitors of restriction enzymes (58, 59). The second most common activity for Acrs to block is DNA cleavage with AcrIF3, AcrIE1, and AcrIIC1 possessing this activity. Besides AcrIIC2, no Acr blocks biogenesis of the CRISPR-Cas complex. It appears that, in general, Acrs have the most utility if they can directly block the activity of already formed CRISPR-Cas complexes, which would usually be present when foreign DNA is introduced into a cell. The recent findings of Acrs with enzymatic activity is very exciting and we expect that more such examples will arise.
IN VIVO FUNCTION OF ACR AND ACA PROTEINS
As described above, Aca proteins are frequently encoded at the 3’-end of acr gene regions and are highly conserved in MGEs, including prophages. As such, they have served as a key signpost for new acr gene identification. There are currently seven families of Aca proteins (20), whose only commonality is a N-terminal HTH DNA-binding domain. Interestingly, the structures of AcrIIA1 and AcrIIA6 (Figure 3) also contain HTH domains indicating that these Acrs may play a dual role as Acr and Aca proteins (18, 23, 60). Consistent with this idea, AcrIIA1 is extremely widespread and is generally encoded at the 3’-end of acr regions. Until recently, the function of Aca proteins was unknown though it seemed likely that they were involved in regulating acr genes as they appeared to be DNA binding proteins. With respect to regulation of acr genes, it was also not known how Acr proteins accumulate quickly enough after phage DNA entry into the cell to protect against CRISPR-Cas complexes that are already present and poised to destroy the phage genome.
Recently, a study elucidated the function of an Aca protein, denoted Aca1, from Pae phage JBD30 – one of the phages in which Acr proteins were first discovered (61). This study also measured the expression of acr genes early in the infection process. It was shown that acr genes are quickly expressed to very high levels at the onset of phage infection. This expression, driven by a powerful promoter located immediately upstream of the acr genes, is critical for anti-CRISPR deployment during infection as mutant phages lacking this promoter fail to replicate in the presence of CRISPR-Cas. Unexpectedly, Aca proteins were shown to be repressors of acr transcription, and, in the case of JBD30, this repression is essential for phage survival. In the absence of Aca activity, uncontrolled expression from the acr promoter dysregulates the expression of essential genes downstream of the acr operon causing loss of phage viability. This critical function of Aca proteins likely explains their high conservation in acr operons. In addition, the positioning of aca genes within acr operons has likely facilitated the spread of these operons by allowing them to insert into diverse genomic locations without disrupting the surrounding genetic circuitry.
Also related to the question of how Acr proteins are able to accumulate quickly enough after phage DNA entry, two recent studies showed that initial infections by phages producing Acr proteins are unable to completely inactivate CRISPR-Cas systems, and that these phages are inhibited by CRISPR-Cas (62, 63). Although these infections fail, they nonetheless produce Acr protein that progressively accumulates with each unsuccessful infection. Once a critical concentration of Acr protein is reached, the host exists in an immunosuppressed state that is sensitive to subsequent infection. This cooperative behaviour of Acr-producing phages was demonstrated to be a major determinant of replicative success in the presence of CRISPR-Cas; without a sufficient amount Acr protein donated by ‘sacrificial’ phages, the infection dies out (62, 63). The density of phages needed to mount a successful defence against CRISPR-Cas largely depends on the strength of the Acr protein, with weaker Acr proteins requiring higher phage densities, and the potency of the CRISPR-Cas system. Although not explored, the number of acr genes targeting the same CRISPR-Cas system likely affects the phage population densities needed to inactivate CRISPR-Cas. Many acr loci encode multiple distinct acr genes targeting the same CRISPR-Cas subtype. Inhibiting the same system in different ways would likely lower the phage densities necessary to neutralize CRISPR-Cas or perhaps even circumvent the need for cooperation to mount a successful infection. Multiple turnover enzymatic Acrs could also have a similar effect. While cooperation provides one means by which phages can outpace CRISPR-Cas, it is possible that other strategies exist. Additionally, as phages are thought to exist in heterogenous populations, it remains to be determined if other members of the community can exploit the immunosuppressed host generated by the Acr-producing phages.
ACR EVOLUTION AND EVOLUTIONARY IMPACT
To date, over 40 families of Acr proteins have been discovered. There is little similarity between the various families aside from their small size (typically between 50 to 150 amino acids) and a stereotypic association with aca genes, making it difficult to trace their evolutionary origin(s). The paucity of structural or sequence similarity to known proteins suggests that Acrs may be the product of de novo evolution. An intriguing feature of Acrs is that some families have homologues that are very diverse and appear in widespread species with respect to phylogeny, while other families have few homologues that may occur in only one species. Families with limited distribution may have evolved more recently and not had time to spread by horizontal gene transfer, or these families may have narrow specificity, and can only inhibiting the CRISPR-Cas system in one species. Thus, there would be no adaptive drive for their retention in other species.
CRISPR-Cas has been shown, in most cases, to drive phages to extinction when host populations target phages with multiple and diverse spacer sequences. However, under similar conditions, a single acr gene is sufficient to overcome CRISPR-Cas (64), thereby imposing a strong selective pressure on CRISPR-Cas to mutate and diversify to bypass Acr inhibition. Therefore, it would be expected that key residues involved in Acr interaction would vary among Cas proteins. Indeed, the critical residue of MbCas12a targeted by AcrVA5 is not universally conserved, allowing some MbCas12a homologs to escape from AcrVA5-mediated inhibition (56). In N. meningitidis, distinct orthologues of Cas9 with different Acr susceptibilities have been identified (65). Additionally, variation in select residues of Cas8f in strains of P. aeruginosa has been noted, perhaps reflecting hot spots of Acr binding in these systems. On the other hand, many Acrs target essential residues or block multiple functions of CRISPR-Cas (30, 36, 37, 50), limiting the possibility of mutational escape. This may act as a driving force for the diversification of CRISPR-Cas systems. We expect that, in general, broad specificity Acrs, like AcrIIC1, would target highly conserved residues in Cas proteins. Substitutions of these residues that might allow for evasion of Acr inhibition would likely also cause loss of CRISPR-Cas function. Thus, it is more difficult for systems to arise that are not inhibited by these Acrs. While it remains unknown how Acrs may be shaping CRISPR-Cas diversity, these examples suggest that Acrs likely have a profound influence. Additionally, limited mutational escape has the potential to result in the accumulation of multiple CRISPR-Cas systems in a single genome. In a survey, 6% of bacteria and 14% of archaea were found to carry multiple types and subtypes of CRISPR-Cas systems (3).
Since many subtypes of CRISPR-cas systems are composed of distinct Cas proteins, it might be expected that each individual Acr would inhibit only a single CRISPR-Cas system. In most cases this is true, but there are exceptions. For example, the AcrIF6 homologue of Pae strongly inhibits both the I-E and I-F systems of Pae (16). Whereas, other AcrIF6 orthologs, encoded in diverse Gammaproteobacteria MGEs, lacked the cross-subtype dual specificity of AcrIF6Pae. Amino acid substitutions were found that abolished only one of this Acrs’ activities, suggesting that it possesses two separate functional interfaces (16). Additional examples include one AcrVA3 homologue, which inhibits both I-C and V-A systems (20), and AcrIIA5, which inhibits both II-A and II-C type systems (23, 66). These cases present intriguing targets for future mechanistic studies.
In light of the impact of Acrs on CRISPR-Cas systems, it may be expected that new elements participating in the arms race between these systems will be discovered. For example, some strains may possess so called “anti-anti-CRISPR” mechanisms as this type of back and forth escalation phenomenon has been well documented in the evolution of restriction-modification systems (67). Taking advantage of direct anti-CRISPR/CRISPR-Cas interactions, bacteria could upregulate intracellular concentrations of Cas protein to titrate away anti-CRISPRs thereby inactivating their actions. In P. aeruginosa, over expression of target Cas protein was shown to inhibit phages relying on Acr for infection (29), providing proof of concept for this mechanism. Bacteria could also repress anti-CRISPR expression using Aca-like proteins. Heterologous expression of Aca1 from a plasmid has been shown to inhibit Acr-dependent phages (61). Lastly, bacteria could possess dedicated anti-anti-CRISPRs that interact directly with Acr proteins. These types of inhibitor could function by preventing Acrs from binding their Cas protein target or by mediating their degradation. Further exploration of these potential anti-anti-CRISPR mechanisms will contribute to our understanding of the coevolutionary dynamics between bacteria and phages as well as the evolution of CRISPR-Cas systems.
APPLICATIONS OF ANTI-CRISPR PROTEINS
The rapidly expanding palette of CRISPR-Cas technologies has led to a corresponding motivation to develop tools to control and modulate their activities. Acr proteins targeting type II (Cas9) and type V (Cas12a) effectors have drawn particular interest as they may provide temporal, spatial, or conditional control over established genome editing systems. One major application of Acr proteins is their use as “off-switches” for genome editing. Minimizing, if not abolishing, undesired off-target activity is important for CRISPR-Cas9 technology, especially for therapeutic use. Although extensive efforts have led to Cas9 variants with enhanced specificity (68), excessive or prolonged Cas9 activity may increase the likelihood of off-target editing or cytotoxicity, necessitating a means to shut down Cas9 upon achieving a desired outcome. In combination with engineered Cas9 variants and means to regulate Cas9 expression, Acr proteins can act as an additional safeguard to reduce potential adverse effects of Cas9. For instance, timed delivery of AcrIIA4 following SpyCas9-sgRNA editing of a desired target reduces the extent of off-target editing in cells. Limiting the window of SpyCas9 activity likely reduces off-target effects due to differential kinetics of Cas9 on- versus off-target editing (45). Furthermore, as multiplexed CRISPR-Cas gene therapies are developed in an effort to offer treatments for previously untreatable, complex, genetic diseases (Campa et al. 2019), Acrs will become even more important for mitigating the potentially exponential increase in off-target effects as the number of crRNAs rises.
CRISPR technology has also been applied to the development of gene drives, which are genetic elements that force super-Mendelian inheritance to disseminate desired traits in a population. A prominent example is the ongoing development of female-sterility-inducing gene drives in mosquitoes to eradicate vector-borne diseases such as malaria (69, 70). A means of effective control over the spread of a gene drive after its initial release is highly desirable. Acr proteins could be deployed to put a brake on the propagation of a CRISPR-based gene drive after the parental driver organisms are released into the relevant ecosystem. As a proof of concept, temporal control of AcrIIA2 and AcrIIA4 using an inducible promoter has been demonstrated to halt or titrate the efficiency of a SpyCas9-based gene drive in yeasts (71).
Nuclease-inactive dCas9 can be used to tether or recruit various effector proteins to genomic sites of interest. For example, chromatin visualization and targeted gene regulation can be achieved via fusion of fluorescent proteins (FPs) and transcriptional activators or repressors to dCas9, respectively. Technologies based on dCas9 not only allow genome manipulation, but also alteration of the epigenome via fusion of DNA demethylation enzymes (e.g., TET) or histone-modifying effectors (e.g., LSD1 or p300) (72). Acrs that limit DNA binding may also be used to regulate the activities of these functional domains. For example, type II Acr proteins were used to control chromosome labeling by dCas9-FP fusions during live-cell imaging (17), as well as demethylation by dCas9-Tet1 fusions in induced pluripotent stem cells (73). Moreover, Acr proteins have enabled programmable and dynamic gene regulation in CRISPR-based synthetic circuits by regulating CRISPRi (CRISPR interference) and CRISPRa (CRISPR activation) (74).
Acr proteins can often tolerate fusion to epitope tags and fluorescent proteins without compromising their inhibitory potency (71, 75). This offers opportunities to engineer Acr proteins through domain fusions. For instance, an AcrIIA4 hybrid with a light-inducible LOV2 domain has been shown to control SpyCas9- and dSpyCas9-mediated genome and epigenome editing in optogenetics (75). Posttranslational control of Acr proteins was achieved by fusing an inducible destabilization domain (DD) that degrades the protein in the absence of an external ligand known as Shield1 (74). Furthermore, posttranscriptional regulation of Acrs by microRNAs that are expressed in certain cell types enables cell type-specific inhibition of Cas9 activities (76, 77). This strategy was validated for enforcing the tissue specificity of genome editing not only in cultured cells but also in adult mice (78), demonstrating that anti-CRISPRs can function in mammalian tissues in vivo. Acr proteins also have applications in the development adenoviral vectors for Cas9 delivery in mammalian cells. Recently, a helper-dependent adenoviruses (HDAd) vector for transient Cas9 expression in target cells was created (79). By design, these HDAds encode SpyCas9 and a guide that directs the cleavage of the vectors’ own genome after transduction of target cells, thereby allowing transient SpyCas9 expression and function. However, self-cleavage during viral production also occurs, leading to genomic rearrangements that make virus production impossible. AcrIIA2 and AcrIIA4 were used to inhibit SpyCas9 from initiating vector self-cleavage during the viral production; thus greatly improving yield (79). A final potential use of Acr proteins is in development of phage therapies as an alternative to antibiotics to treat bacterial infections (80). Phage therapies, however, may be compromised in pathogenic hosts with active CRISPR-Cas systems such as Pseudomonas aeruginosa (81) and Neisseria meningitidis (82). Since Acr proteins have been found in these and other pathogens, acr genes could be included in the engineering of therapeutic bacteriophages that circumvent multidrug resistance in pathogenic bacteria.
FUTURE PERSPECTIVES
Starting from the first paper in January, 2013, there are now (August, 2019) 99 papers in Pubmed mentioning “anti-CRISPR”. Remarkably, 63 of these have been published in 2018 and 2019, emphasizing the accelerating interest in this field. Despite the rapid accumulation in knowledge about Acrs, we are surely only seeing the “tip of the iceberg” so far. Given the rapidity with which Acrs that inhibit many different CRISPR-Cas systems have been discovered, we anticipate that many more Acr families will be identified in the near future, and that Acrs will eventually be found to inhibit every type of system. With the widespread occurrence of Acrs, it will become crucial to address the question of whether CRISPR-Cas systems are frequently participating in functions outside of phage defence, and whether Acrs participate in these functions by modulating CRISPR-Cas activity rather than complete inhibition. There are already several examples of CRISPR-Cas systems that fulfil non-canonical roles in gene regulation and virulence (83–86). Another area of future interest will be in elucidating more Acr mechanisms. These mechanisms have already proven to be remarkably diverse, and continued focus in this area will provide many more new insights into CRISPR-Cas function and likely many surprises. Finally, it is important to note that CRISPR-Cas systems represent only one of many bacterial anti-phage defence mechanisms, and the number of these systems known has greatly expanded in past 4 years (1, 87–89). While Acrs have been rapidly discovered and characterized due to the excitement surrounding CRISPR-Cas systems, no inhibitors have yet been discovered for these fascinating new anti-phage systems. The discovery approaches applied to Acr systems should be applicable for finding new “anti-anti-phage” systems, and we expect that exciting new fields of study will emerge from these efforts.
ACKNOWLEDGMENTS
This work was supported by grants from the Canadian Institutes for Health Research to A.R.D. (FDN-15427), and by a grant from the U.S. National Institutes of Health (GM125797) to A.R.D. and E.J.S.
REFERENCES
- 1.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, et al. 2018. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359: 1008–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dy RL, Richter C, Salmond GP, Fineran PC. 2014. Remarkable Mechanisms in Microbes to Resist Phage Infections. Annu Rev Virol 1: 307–31 [DOI] [PubMed] [Google Scholar]
- 3.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, et al. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13: 722–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amitai G, Sorek R. 2016. CRISPR-Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol 14: 67–76 [DOI] [PubMed] [Google Scholar]
- 5.Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82: 237–66 [DOI] [PubMed] [Google Scholar]
- 6.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493: 429–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bondy-Denomy J, Davidson AR, Doudna JA, Fineran PC, Maxwell KL, et al. 2018. A Unified Resource for Tracking Anti-CRISPR Names. CRISPR J 1: 304–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrangou R, Marraffini LA. 2014. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol Cell 54: 234–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koonin EV, Makarova KS, Zhang F. 2017. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37: 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright AV, Nunez JK, Doudna JA. 2016. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 164: 29–44 [DOI] [PubMed] [Google Scholar]
- 11.Bondy-Denomy J, Davidson AR. 2014. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol 52: 235–42 [DOI] [PubMed] [Google Scholar]
- 12.Feiner R, Argov T, Rabinovich L, Sigal N, Borovok I, Herskovits AA. 2015. A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat Rev Microbiol 13: 641–50 [DOI] [PubMed] [Google Scholar]
- 13.Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, et al. 2016. Prophages mediate defense against phage infection through diverse mechanisms. ISME J 10: 2854–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawluk A, Bondy-Denomy J, Cheung VH, Maxwell KL, Davidson AR. 2014. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 5: e00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, et al. 2016. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol 1: 16085. [DOI] [PubMed] [Google Scholar]
- 17.Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, et al. 2016. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167: 1829–38 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, et al. 2017. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell 168: 150–8 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watters KE, Fellmann C, Bai HB, Ren SM, Doudna JA. 2018. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 362: 236–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, et al. 2018. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362: 240–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rusk N 2019. Spotlight on Cas12. Nat Methods 16: 215. [DOI] [PubMed] [Google Scholar]
- 22.Hynes AP, Rousseau GM, Lemay ML, Horvath P, Romero DA, et al. 2017. An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat Microbiol 2: 1374–80 [DOI] [PubMed] [Google Scholar]
- 23.Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, et al. 2018. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat Commun 9: 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He F, Bhoobalan-Chitty Y, Van LB, Kjeldsen AL, Dedola M, et al. 2018. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype I-D immunity. Nat Microbiol 3: 461–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uribe RV, van der Helm E, Misiakou MA, Lee SW, Kol S, Sommer MOA. 2019. Discovery and Characterization of Cas9 Inhibitors Disseminated across Seven Bacterial Phyla. Cell Host Microbe 25: 233–41 e5 [DOI] [PubMed] [Google Scholar]
- 26.Lhuillier S, Gallopin M, Gilquin B, Brasiles S, Lancelot N, et al. 2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc Natl Acad Sci U S A 106: 8507–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxwell KL, Yee AA, Arrowsmith CH, Gold M, Davidson AR. 2002. The solution structure of the bacteriophage lambda head-tail joining protein, gpFII. J Mol Biol 318: 1395–404 [DOI] [PubMed] [Google Scholar]
- 28.Maji B, Gangopadhyay SA, Lee M, Shi M, Wu P, et al. 2019. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 177: 1067–79 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, et al. 2015. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526: 136–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, et al. 2017. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169: 47–57 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, et al. 2017. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell 171: 414–26 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, et al. 2019. Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell 74: 132–42 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng R, Xu Y, Zhu T, Li N, Qi J, et al. 2017. Alternate binding modes of anti-CRISPR viral suppressors AcrF1/2 to Csy surveillance complex revealed by cryo-EM structures. Cell Res 27: 853–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maxwell KL, Garcia B, Bondy-Denomy J, Bona D, Hidalgo-Reyes Y, Davidson AR. 2016. The solution structure of an anti-CRISPR protein. Nat Commun 7: 13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pawluk A, Shah M, Mejdani M, Calmettes C, Moraes TF, et al. 2017. Disabling a Type I-E CRISPR-Cas Nuclease with a Bacteriophage-Encoded Anti-CRISPR Protein. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Yao D, Xu JG, Li AR, Xu J, et al. 2016. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat Struct Mol Biol 23: 868–70 [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Ma J, Cheng Z, Meng X, You L, et al. 2016. A CRISPR evolutionary arms race: structural insights into viral anti-CRISPR/Cas responses. Cell Res 26: 1165–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrangou R, Doudna JA. 2016. Applications of CRISPR technologies in research and beyond. Nat Biotechnol 34: 933–41 [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Doval C, Jinek M. 2017. Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Curr Opin Struct Biol 47: 157–66 [DOI] [PubMed] [Google Scholar]
- 40.Jiang F, Doudna JA. 2017. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys 46: 505–29 [DOI] [PubMed] [Google Scholar]
- 41.Stella S, Alcon P, Montoya G. 2017. Class 2 CRISPR-Cas RNA-guided endonucleases: Swiss Army knives of genome editing. Nat Struct Mol Biol 24: 882–92 [DOI] [PubMed] [Google Scholar]
- 42.Mir A, Edraki A, Lee J, Sontheimer EJ. 2018. Type II-C CRISPR-Cas9 Biology, Mechanism, and Application. ACS Chem Biol 13: 357–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong, Guo M, Wang S, Zhu Y, Wang S, et al. 2017. Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546: 436–9 [DOI] [PubMed] [Google Scholar]
- 44.Kim I, Jeong M, Ka D, Han M, Kim NK, et al. 2018. Solution structure and dynamics of anti-CRISPR AcrIIA4, the Cas9 inhibitor. Sci Rep 8: 3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, et al. 2017. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv 3: e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang H, Patel DJ. 2017. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol Cell 67: 117–27 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang F, Liu JJ, Osuna BA, Xu M, Berry JD, et al. 2019. Temperature-Responsive Competitive Inhibition of CRISPR-Cas9. Mol Cell 73: 601–10 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L, Yin M, Wang M, Wang Y. 2019. Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol Cell 73: 611–20 e3 [DOI] [PubMed] [Google Scholar]
- 49.Lee J, Mir A, Edraki A, Garcia B, Amrani N, et al. 2018. Potent Cas9 Inhibition in Bacterial and Human Cells by AcrIIC4 and AcrIIC5 Anti-CRISPR Proteins. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, et al. 2017. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170: 1224–33 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, et al. 2019. Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol Cell 74: 296–309 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim Y, Lee SJ, Yoon HJ, Kim NK, Lee BJ, Suh JY. 2019. Anti-CRISPR AcrIIC3 discriminates between Cas9 orthologs via targeting the variable surface of the HNH nuclease domain. FEBS J [DOI] [PubMed] [Google Scholar]
- 53.Thavalingam A, Cheng Z, Garcia B, Huang X, Shah M, et al. 2019. Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat Commun 10: 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, et al. 2019. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat Struct Mol Biol 26: 315–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Li Z, Daczkowski CM, Gabel C, Mesecar AD, Chang L. 2019. Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe 25: 815–26 e4 [DOI] [PubMed] [Google Scholar]
- 56.Dong L, Guan X, Li N, Zhang F, Zhu Y, et al. 2019. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat Struct Mol Biol 26: 308–14 [DOI] [PubMed] [Google Scholar]
- 57.Knott GJ, Cress BF, Liu JJ, Thornton BW, Lew RJ, et al. 2019. Structural basis for AcrVA4 inhibition of specific CRISPR-Cas12a. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang HC, Chou CC, Hsu KC, Lee CH, Wang AH. 2019. New paradigm of functional regulation by DNA mimic proteins: Recent updates. IUBMB Life 71: 539–48 [DOI] [PubMed] [Google Scholar]
- 59.Wang HC, Ho CH, Hsu KC, Yang JM, Wang AH. 2014. DNA mimic proteins: functions, structures, and bioinformatic analysis. Biochemistry 53: 2865–74 [DOI] [PubMed] [Google Scholar]
- 60.Ka D, An SY, Suh JY, Bae E. 2018. Crystal structure of an anti-CRISPR protein, AcrIIA1. Nucleic Acids Res 46: 485–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stanley SY, Borges AL, Chen K-H, Swaney DL, Krogan NJ, et al. 2019. Anti-CRISPR Associated Proteins are Crucial Repressors of Anti-CRISPR Transcription. Cell: in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Borges AL, Zhang JY, Rollins MF, Osuna BA, Wiedenheft B, Bondy-Denomy J. 2018. Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity. Cell 174: 917–25 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landsberger M, Gandon S, Meaden S, Rollie C, Chevallereau A, et al. 2018. Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity. Cell 174: 908–16 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Houte S, Ekroth AK, Broniewski JM, Chabas H, Ashby B, et al. 2016. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532: 385–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, et al. 2019. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol Cell 73: 714–26 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marshall R, Maxwell CS, Collins SP, Jacobsen T, Luo ML, et al. 2018. Rapid and Scalable Characterization of CRISPR Technologies Using an E. coli Cell-Free Transcription-Translation System. Mol Cell 69: 146–57 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8: 317–27 [DOI] [PubMed] [Google Scholar]
- 68.Kim D, Luk K, Wolfe SA, Kim JS. 2019. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu Rev Biochem 88: 191–220 [DOI] [PubMed] [Google Scholar]
- 69.Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, et al. 2016. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34: 78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kyrou K, Hammond AM, Galizi R, Kranjc N, Burt A, et al. 2018. A CRISPR-Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat Biotechnol 36: 1062–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Basgall EM, Goetting SC, Goeckel ME, Giersch RM, Roggenkamp E, et al. 2018. Gene drive inhibition by the anti-CRISPR proteins AcrIIA2 and AcrIIA4 in Saccharomyces cerevisiae. Microbiology 164: 464–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adli M 2018. The CRISPR tool kit for genome editing and beyond. Nat Commun 9: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu XS, Wu H, Krzisch M, Wu X, Graef J, et al. 2018. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172: 979–92 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakamura M, Srinivasan P, Chavez M, Carter MA, Dominguez AA, et al. 2019. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat Commun 10: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bubeck F, Hoffmann MD, Harteveld Z, Aschenbrenner S, Bietz A, et al. 2018. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat Methods 15: 924–7 [DOI] [PubMed] [Google Scholar]
- 76.Hirosawa M, Fujita Y, Saito H. 2019. Cell-Type-Specific CRISPR Activation with MicroRNA-Responsive AcrllA4 Switch. ACS Synth Biol 8: 1575–82 [DOI] [PubMed] [Google Scholar]
- 77.Hoffmann MD, Aschenbrenner S, Grosse S, Rapti K, Domenger C, et al. 2019. Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res 47: e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee J, Mou H, Ibraheim R, Liang S-Q, Liu P, et al. 2019. Tissue-restricted genome editing in vivo specified by microRNA-repressible anti-CRISPR proteins. RNA: in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palmer DJ, Turner DL, Ng P. 2019. Production of CRISPR/Cas9-Mediated Self-Cleaving Helper-Dependent Adenoviruses. Mol Ther Methods Clin Dev 13: 432–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nobrega FL, Costa AR, Kluskens LD, Azeredo J. 2015. Revisiting phage therapy: new applications for old resources. Trends Microbiol 23: 185–91 [DOI] [PubMed] [Google Scholar]
- 81.van Belkum A, Soriaga LB, LaFave MC, Akella S, Veyrieras JB, et al. 2015. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. MBio 6: e01796–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y 2017. The CRISPR-Cas9 system in Neisseria spp. Pathog Dis 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li R, Fang L, Tan S, Yu M, Li X, et al. 2016. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian host immunity. Cell Res 26: 1273–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Louwen R, Horst-Kreft D, de Boer AG, van der Graaf L, de Knegt G, et al. 2013. A novel link between Campylobacter jejuni bacteriophage defence, virulence and Guillain-Barre syndrome. Eur J Clin Microbiol Infect Dis 32: 207–26 [DOI] [PubMed] [Google Scholar]
- 85.Sampson TR, Napier BA, Schroeder MR, Louwen R, Zhao J, et al. 2014. A CRISPR-Cas system enhances envelope integrity mediating antibiotic resistance and inflammasome evasion. Proc Natl Acad Sci U S A 111: 11163–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS. 2013. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497: 254–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, et al. 2015. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J 34: 169–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ofir G, Melamed S, Sberro H, Mukamel Z, Silverman S, et al. 2018. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol 3: 90–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH, et al. 2014. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507: 258–61 [DOI] [PMC free article] [PubMed] [Google Scholar]