

SUMMARY
Deleterious somatic mutations in DNA methyltransferase 3 alpha (DNMT3A) and TET mehtylcytosine dioxygenase 2 (TET2) are associated with clonal expansion of hematopoietic cells and higher risk of cardiovascular disease (CVD). Here, we investigated roles of DNMT3A and TET2 in normal human monocyte-derived macrophages (MDM), in MDM isolated from individuals with DNMT3A or TET2 mutations, and in macrophages isolated from human atherosclerotic plaques. We found that loss of function of DNMT3A or TET2 resulted in a type I interferon response due to impaired mitochondrial DNA integrity and activation of cGAS signaling. DNMT3A and TET2 normally maintained mitochondrial DNA integrity by regulating the expression of transcription factor A mitochondria (TFAM) dependent on their interactions with RBPJ and ZNF143 at regulatory regions of the TFAM gene. These findings suggest that targeting the cGAS-type I IFN pathway may have therapeutic value in reducing risk of CVD in patients with DNMT3A or TET2 mutations.
Graphical Abstract
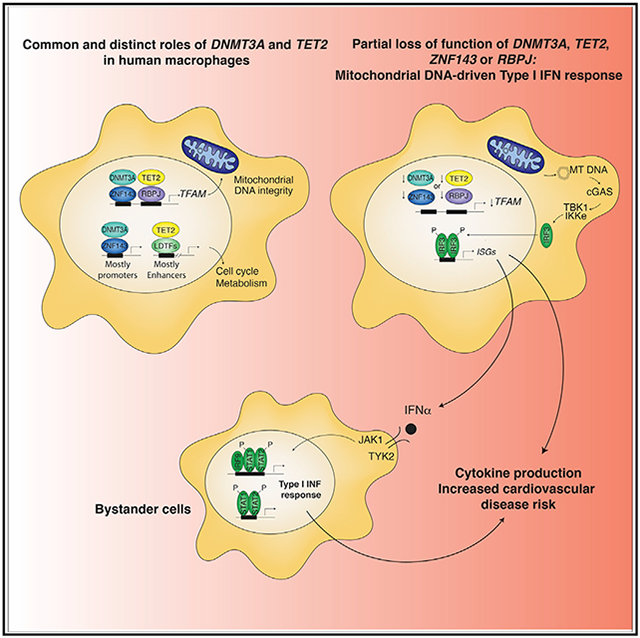
In brief
Clonal hematopoiesis due to mutations in DNMT3A or TET2 increases risk of cardiovascular disease, but mechanisms are presently unclear. Cobo et al. reveal that loss of function of DNMT3A and TET2 results in a type 1 interferon response in macrophages due to impaired mitochondrial DNA integrity.
INTRODUCTION
Cardiovascular disease (CVD) remains the leading cause of mortality in the elderly in western societies (Yazdanyar and Newman, 2009; Rodgers et al., 2019; Ferrucci and Fabbri, 2018; North and Sinclair, 2012). Age is the strongest risk factor for CVD, in part as a surrogate for the cumulative exposure to classical risk factors, although emerging risk factors acquired de novo later in life represent opportunities for modifying age-related CVD risk (Sniderman and Furberg, 2008; D’Agostino et al., 2008; Kannel and Vasan, 2009). Deep sequencing of various tissues and specific cell types has revealed that mutation-driven somatic mosaicism is a hallmark of aging in all tissues. The frequency of somatic mutations is higher in proliferative tissues such as the hematological system, esophagus, and skin (Martincorena et al., 2015; Welch et al., 2012; Fernandez et al., 2016; Forsberg et al., 2017; Vijg, 2014). In the hematopoietic system, mutations can confer a competitive growth advantage to the progenitor cells in which they occur. These somatic mutations can be detected in blood as clones because the mutated stem cells maintain the ability to differentiate into circulating granulocytes, monocytes, and lymphocytes (Cole et al., 2017; Buscarlet et al., 2017; Ko et al., 2011; Moran-Crusio et al., 2011; Li et al., 2011). This process can be associated with the development of hematological malignancies but is called clonal hematopoiesis of indeterminate potential (CHIP) when it is not associated with altered blood-cell counts. It has been estimated that 10%–20% of individuals older than 65 years old display a fraction of white blood cells that carry a mutation in a driver gene (Zink et al., 2017; McKerrell et al., 2015). Of note, these mutations are associated with a >2-fold increase in the risk of developing CVD, with the degree of risk correlated with mutant allele frequency (Jaiswal et al., 2017). CHIP is disproportionately driven by mutations in two cancer-related genes, DNMT3A or TET2, which together represent more than 50% of reported carriers (Jaiswal et al., 2014; 2017bib_Jaiswal_et_al_2017; Xie et al., 2014; Buscarlet et al., 2017; Jaiswal et al., 2014; Genovese et al., 2014; Acuna-Hidalgo et al., 2017). DNMT3A encodes an enzyme that catalyzes the de novo conversion of cytosine into 5-hydroxy-methylcytosine residues at CpG sites, a process that is mechanistically linked to transcriptional repression. Conversely, TET2 encodes an enzyme that catalyzes the conversion of 5-methylcytosine to 5-hydroxy-methylcytosine, a process that initiates a sequence of reactions ultimately resulting in DNA demethylation and reversal of methyl CpG-mediated transcriptional repression. Thus, the observation that loss of function of DNMT3A and TET2 has a similar phenotypic consequence with respect to CVD risk raises the question of whether this phenotype is related to their distinct enzymatic functions (Cobo et al., 2021).
Monocytes and macrophages are key mediators of CVD processes (Cobo et al., 2021; Ghattas et al., 2013; Tabas and Lichtman, 2017). Subsets of lesion-associated macrophages secrete various inflammatory cytokines that activate neighboring cells and help to propagate arterial inflammation and contribute to the pathophysiology of CVD (Ghattas et al.; Yu et al., 2013; Moore et al., 2013; Bobryshev et al., 2016). Experimental evidence supports a role for DNTM3A and TET2 in regulating macrophage function in the context of atherosclerosis. Competitive bone marrow transplantation of DNMT3A- or TET2-deficient cells into a wild-type mice mimic the scenario of clonal hematopoiesis and accelerated atherosclerosis in a process that requires inflammasome activation and overproduction of interleukin-1b (I1b) and other inflammatory cytokines by macrophages (Jaiswal et al., 2017; Fuster et al., 2017, 2020). In addition, reduced expression of Dnmt3a enhances the response of mouse macrophages to lipopolysaccharide (LPS) and IFNg (Pham et al., 2013). Mechanistically, clonal expansion upon Dnmt3a loss leads to osteoporosis in a process that requires IL-20 expression from Dnmt3a mutant macrophages through interferon regulatory factor-3 (IRF3)-NFκB signaling (Kim et al., 2021), consistent with an anti-inflammatory role of DNTM3A. Moreover, reduced expression of DNMT3A in macrophages using small interference RNA (siRNA) leads to increased production of the anti-inflammatory cytokine IL10 in human macrophages exposed to S. aureus (Mba Medie et al., 2019) raising the possibility that mouse macrophages may not fully represent the functions of DNMT3A and TET2 in human macrophages. More broadly, the biological roles of DNMT3A and TET2 in human macrophages and the mechanisms underlying their functions remain poorly understood.
To investigate these roles, we used specific antisense oligonucleotides (ASOs) that reduced the expression of DNMT3A or TET2 between 50% and 75%, modeling the partial loss of function resulting from heterozygous mutations. The consequences of reduced expression of each factor were determined at the RNA by RNA sequencing. In parallel, we correlated transcriptomic changes with the sites of DNMT3A and TET2 binding determined by chromatin immunoprecipitation (ChIP)-sequencing. These studies revealed both distinct and overlapping roles of DNMT3A and TET2, with upregulation of genes involved in the type I interferon (IFN) response being the most significant shared consequence of reduced expression of each protein. Notably, the promoters of these genes were not direct binding sites of either DNMT3A or TET2. Instead, we traced this shared program of dysregulated gene expression to alterations in mitochondrial DNA (mtDNA). Specifically, we provide evidence that reduced expression or somatic loss of function mutations in either DNMT3A or TET2 results in reduced expression of transcription factor A mitochondria (TFAM), a mtDNA-binding protein involved in transcription, replication, and packaging of mtDNA (Scarpulla, 2008, 2011; West and Shadel, 2017). Similar to direct TFAM depletion (West et al., 2015), decreased DNMT3A or TET2 leads to cytosolic release of mtDNA that activates cGAS signaling and induces a cell-autonomous upregulation of interferon-stimulated genes (ISGs) that is then propagated in a non-cell autonomous manner through the production of IFNα.
RESULTS
DNMT3A and TET2 restrain interferon signaling in human monocyte-derived macrophages and atherosclerotic macrophages
Characterization of the expression patterns of DNMT3A and TET2 in monocytes as they differentiated into monocyte-derived macrophages (MDM) showed that DNMT3A and TET2 protein expression are strongly upregulated in a time-dependent manner (Figures 1A and 1B). As MDM are major contributors to the development and clinical complications of atherosclerotic lesions, we focused our attention on these cells. To investigate the roles of DNMT3A or TET2 in MDM, we screened a series of ASOs (Crooke, 2007) for efficacy in reducing DNMT3A and TET2 expression at both mRNA and protein. We used the two most effective DNMT3A ASOs (DNMT3A ASO 1, DNMT3A ASO 2) and TET2 ASOs (TET2 ASO 1, TET2 ASO 2) (Figures 1C and Figure S1A). While both DNMT3A ASOs 1 and 2 selectively reduced expression of DNMT3A, we unexpectedly found that the TET2 ASOs 1 and 2 also reduced DNMT3A expression (Figure 1C).
Figure 1. Transcriptional effect of DNMT3A and TET2 ablation in normal and atherosclerotic macrophages.

(A) Intracellular staining analysis of DNTM3A and TET2 by flow cytometry at different stages of differentiation of monocyte-derived macrophages versus circulating monocytes (day 0) (n = 3 donors/experiment, 1 representative donor is shown). Scale bars: 5 μm.
(B) IF analysis of DNMT3A and TET2 in monocyte-derived macrophages 6 days after differentiation versus 1.5 days (n = 3 donors/experiment, 1 representative donor is shown). Scale bars: 5 μm.
(C) Protein quantification of DNMT3A and TET2 by IF in monocyte-derived macrophages transfected with the indicated ASOs against DNMT3A or TET2. (n = 3 donors).
(D) Bar plots showing the number of differentially expressed genes (DEG) in monocyte-derived macrophages transfected with the indicated ASOs targeting DNMT3A or TET2 (n = 3 donors).
(E) Venn diagram showing overlap between upregulated genes in both DNMT3A ASO and TET2 ASO and associated gene ontology term enrichment.
(F) Log2 fold change of the indicated interferon stimulated genes (ISGs) and other inflammatory genes in MDM treated with DNTM3A ASOs or TET2 ASOs. Adjusted p < 0.05 for all versus control ASO.
(G) Macrophages differentiated from monocytes isolated from individuals with no mutations (left panel) or macrophages isolated from atherosclerotic plaque (right panel) were stratified by DNMT3A or TET2 expression according to single-cell RNA-seq. Data corresponds to normalized expression values. (n = 3/group). Boxes denote upper (DNMT3AHigh and TET2High) and lower (DNMT3ALow and TET2Low) quartiles of expression, excluding cells with 0 values.
(H) Bar graphs of the average expression values of DNMT3A or TET2 in DNMT3ALow versus DNMT3AHigh or TET2Low versus TET2High in healthy macrophages or atherosclerotic macrophages, respectively, as defined in (G).
(I) Boxplot of expression of interferon stimulated genes determined by scRNA-seq data of MDM from healthy donors (left) or from macrophages isolated from atherosclerotic plaques (right) in cells with low DNMT3A or TET2 compared to high DNMT3A or TET2 as defined in (G), respectively. Data corresponds to normalized expression values. Mann-Whitney U test was used to calculate statistical significance. *p < 0.05; **p < 0.01. Also see Figure S1.
To globally assess the impact of reduced TET2 or DNMT3A on gene expression, we performed RNA sequencing of MDM treated with the scrambled control ASO or each of the gene-specific ASOs. This analysis revealed global alterations in mRNA that were proportional to the reduction in DNMT3A or TET2 protein (Figure 1D). Importantly, the genes upregulated through ASOs of different efficacies in reducing TET2 and DNMT3A protein nearly overlapped, producing a consensus gene set of high confidence used for downstream analysis (Figures S1B and S1C left panel). This consensus gene list was enriched for functional annotations associated with type I IFN and cytokine signaling (Figures S1B and S1C right panel). Similar enrichment is observed in the 143 upregulated genes that were shared between the two DNMT3A ASOs and the two TET2 ASOs, including transcripts of inflammatory cytokines and mediators of interferon signaling such as IFIT2, IFIT3, CXCL10, CCL2, CCL3, CCL8, CCL13, and IL1B mRNA (Rusinova et al., 2013; Schoggins and Rice, 2011) (Figures 1E and 1F).
To further establish the specificity of human DNMT3A and TET2 ASOs 1 and 2, we transfected them into mouse bone marrow derived macrophages (BMDM). Transfection of these ASOs did not induce expression a type I IFN response, whereas transfection of recombinant DNA (rDNA) containing immunostimulatory sequences did, indicating specificity of DNMT3A and TET2 ASOs (Figure S1D). As an additional approach, we designed siRNAs targeting DNMT3A and TET2 and transfected them into MDM. The siRNAs directed against TET2 selectively reduced TET2 expression, while the siRNAs directed against DNMT3A reduced DNMT3A and TET2 expression, as was observed for the corresponding DNMT3A ASOs (Figures 1C and S1E). Importantly, all four siRNAs targeting DNMT3A or TET2 induced a type I IFN response (Figure S1E). Lastly, we found upregulation of ISGs in Dnmt3a−/− BMDM (Kim et al., 2021) (Figure S1F) and in DNMT3A−/− human iPSC-derived macrophages (Lim et al., 2021) (Figure S1G), confirming that reduced expression of DNMT3A leads to upregulation of ISGs in the context of genetically driven loss of function. Collectively, these findings strongly support the hypothesis that loss of function of either DNMT3A or TET2 results in a type I IFN response in human monocyte derived macrophages in addition to other molecular consequences that are specific for each factor.
DNMT3A and TET2 are proteins with methyltransferase and methylcytosine dioxygenase activity, respectively. Therefore, we investigated whether activation of ISGs is accompanied by changes in DNA hydroxymethylcytosine (5hmC) and found a similar amount of 5hmC in MDM treated with DNMT3A ASO or TET2 ASO (Figure S1H). Moreover, treatment with the methyltransferase inhibitor 5′-azacytidine or an inhibitor of TET2 catalytic domain (Bobcat339) did not alter the expression of ISGs (Figure S1I), yet 5-azacytidine reduced global DNA methylation (Figure S1J). These data indicate that alterations in DNA methylation are not a major driver of transcriptional changes induced by acute reductions of DNTM3A or TET2 expression in MDM, suggesting an alternative mechanism is at play. In addition, we did not observe changes in three-dimensional conformation of the chromatin around ISG, including CXCL10 or MX1 (Figure S1K, arrowheads).
We next investigated whether reduced DNMT3A or TET2 expression would lead to changes in proliferation of MDM. We found that most of the downregulated genes in DNMT3A ASO or TET2 ASO treated MDM were related to cell cycle and mitosis, including DEPTOR, HELLS, KIFs, STMN1, and MKI67 (Figures S2A-S2C). However, analysis of proliferation, senescence, or apoptosis after treatment with DNMT3A ASO or TET2 ASO revealed no differences compared to control ASO (Figures S2D and S2E). These data indicate that reduced DNMT3A or TET2 expression does not alter cell proliferation, apoptosis, or senescence of MDM under these in vitro culture conditions.
To investigate variation in DNMT3A and TET2 expression in human MDM and its potential relationship to ISGs, we performed single cell RNA-seq sequencing in MDM prepared from three healthy donors and ranked the MDM according to DNMT3A or TET2 mRNA expression (Figures 1G and 1H). We found that MDM with reduced DNMT3A or TET2 as defined by quartiles of expression (Q1 versus Q4 for TET2 and Q1-2 versus Q3-4 for DNMT3A) had a statistically significant upregulation of genes involved in IFN signaling (Figure 1I, left panel). Similar upregulation was observed in macrophages isolated from the atherosclerotic plaque (Depuydt et al., 2020) with lower DNMT3A or TET2 (Figure 1I right panel) when compared to those expressing high DNMT3A or TET2, respectively. These findings suggest that low amounts of wild-type DNMT3A or TET2 expression could predispose human macrophages to a type I IFN responses.
Reduced expression of DNMT3A or TET2 or heterozygous loss of function mutations cause activation of ISGs in bystander cells
Activated macrophages can remodel the local environment non-cell autonomously through the secretion of inflammatory mediators (Honda et al., 2006). The upregulation of ISGs was accompanied by increased IFNA1, IFNA4, and IFNA5 mRNA expression and higher secretion of IFNα (Figure 2A), but not upregulation of IFNB or higher IFNβ secretion (Figure S3A). To investigate whether this was sufficient to activate bystander cells, we treated normal MDM with conditioned media (CM) from MDM treated with DNMT3A ASO or TET2 ASO for 2 h and performed RNA sequencing. We found 116 upregulated genes in MDM treated with CM from DNMT3A ASO and 102 upregulated genes in MDM treated with CM from TET2 ASO (log2 fold change >1 and FDR <0.05). Of these, 32 genes were common between CM DNMT3A ASO and CM TET2 ASO and these genes were enriched in functional pathways related to IFN signaling, including IFITs, MX1, OASL, and IRF7 mRNA (Figures 2B and 2C). Treatment with conditioned media did not alter the expression of DNMT3A or TET2 mRNA or protein, indicating that the non-cell-autonomous activation of IFN signaling was not due to reduced expression of DNMT3A or TET2 (Figure S3B). An increase in IRF7 protein was confirmed by immunofluorescence (IF) microscopy (Figure S3C). We investigated whether signaling by IFNα is required for the non-cell-autonomous activation of ISGs by CM from DNMT3A ASO 1 or TET2 ASO 2 treated MDM. Treatment of CM from DNTM3A ASO or TET2 ASO with blocking antibodies for IFN alpha, beta, and omega receptor chain 2 (anti-IFNαR2 antibody) prevented the upregulation of IFN-related genes (Figure 2D). These results suggest that reduced DNMT3A or TET2 expression leads to non-cell-autonomous activation of IFN signaling through IFNαR2. Moreover, reduced DNMT3A or TET2 expression sensitized MDM to an exacerbated response to IFNα as assessed by upregulation of ISG (Figure 2E, note difference in scale for relative expression). These data indicate that reduced expression of DNMT3A or TET2 primes macrophages towards a more pro-inflammatory state.
Figure 2. Effect of deleterious mutations or ablation of DNTM3A or TET2 in bystander cells.

(A) RT-qPCR (left) of IFNA1, IFNA4, and IFNA5 and protein secretion analysis by ELISA (right) of IFNα in MDM transfected with DNMT3A ASO or TET2 ASO (n ≥ 3 donors).
(B) Venn diagram and pathway analysis of genes differentially expressed in normal MDM incubated with conditioned media from macrophages transfected with DNMT3A ASO 1 or TET2 ASO 2(n = 4 donors).
(C) Representative examples of ISG expression in macrophages treated with conditioned media from macrophages transfected with DNMT3A ASO 1 or TET2 ASO 2.
(D) Analysis by RNA-seq of expression of ISG in macrophages incubated with conditioned media from macrophages transfected with DNMT3A ASO 1 or TET2 ASO 2 with and without IFNαR2 antibody (n = 2 donors).
(E) RT-qPCR of ISGs in MDM transfected with DNMT3A ASO or TET2 ASO with and without treatment with IFNα (n = 4 donors).
(F) Relative concentrations of IFNα in culture supernatant of macrophages differentiated from monocytes from two individuals with DNMT3A mutations or TET2 mutations compared to two individuals without detectable mutation (control 1 and control 2).
(G) RT-qPCR analysis of ISG expression in MDM treated with conditioned media of macrophages with DNMT3A or TET2 mutations versus conditioned media from control individuals. Mann-Whitney U test was used to calculate statistical significance in all panels except in (E) where Student’s t test was used. *p < 0.05; **p < 0.01. Also see Figures S1 and S3.
Next, we investigated whether deleterious mutations in DNMT3A or TET2 in myeloid cells would recapitulate effects found in MDM upon DNMT3A or TET2 depletion. We used MDM from two individuals without detectable DNMT3A or TET2 mutations; from two individuals with DNMT3A mutations DNMT3A c.2638A>G (VAF 21%); DNMT3A c.1717C>T; (VAF 24%); and two individuals with TET2 mutations TET2 c.2176C>T, (VAF 17%) and TET2 c.519delG (VAF 36%);TET2 c.5551_5552insT (VAF 29%) (Table S1). Similar to CM from DNMT3A or TET2 ASO, CM from MDM with DNMT3A or TET2 mutations showed higher abundance of IFNα (Figure 2F) but not IFNβ (Figure S3E). In addition, we found upregulation of ISGs such as IFIT1, IFIT3, IRF7, RSAD2, and MX1 when MDM from healthy donors was cultured with CM from MDM from individuals with DNMT3A or TET2 mutations (Figure 2G). These data indicate that deleterious DNMT3A or TET2 mutations leads to secretion of IFNα and non-cell-autonomous activation of ISGs.
We then investigated whether any molecular signature would be shared among the promoters of genes upregulated by DNMT3A ASO and TET2 ASO. De novo motif enrichment analysis of these promoters revealed over-representation of motifs for IFN regulatory factors (IRFs) and, to a lower extent, NFKB and HOXA1 motifs (Figure 3A). Consistent with this, we found upregulation of IRF1 and IRF7 protein in MDM treated with DNMT3A ASO or TET2 ASO (Figures 3B and S3F). This was accompanied by increased binding of IRF1, IRF7, and the active, phosphorylated form of IRF3 (p-IRF3) to the IFNA1 promoter (Figure 3C) and to the promoters of classical ISGs such as CXCL10, IFIT2, OASL, MX1, and ISG15 (Figures 3D and S3G). These regions also showed increased histone H3K4me3 and H3K27ac deposition (Figures 3E and S3H), consistent with increased mRNA being due to increases in gene transcription. These results suggest that DNMT3A and TET2 restrain an IFN program in MDM mediated by activation of IRFs.
Figure 3. Analysis of the molecular signature and signaling pathway altered by DNTM3A or TET2 ablation in macrophages.

(A) Motif enrichment analyses of the promoters of upregulated genes in MDMs treated with DNMT3A ASOs and TET2 ASOs.
(B) Protein analysis by IF of IRF1 and IRF7 in MDM incubated with reduced DNTM3A or TET2 expression (n = 3 donors).
(C) ChIP-qPCR of IRF1, IRF7, or p-IRF3 at the IFNA1 promoter in MDM incubated with DNTM3A ASO or TET2 ASO (n = 3 donors).
(D) ChIP-qPCR of p-IRF3 at the promoter of CXCL10, IFIT2, OASL, MX1, and ISG15 in mm treated with DNMT3A ASO or TET2 ASO as compared to scramble ASO (n = 4 donors).
(E and F) H3K27ac and RNApolII ChIP signal at the promoters of CXCL10, IFIT2, OASL, MX1, ISG15 in macrophages with reduced DNMT3A or TET2 expression (n = 4 donors). Mann-Whitney U test was used to calculate statistical significance. *p < 0.05; **p < 0.01.
Activation of ISGs is not directly regulated by DNMT3A or TET2 binding
To investigate whether transcriptomic changes in macrophages with reduced DNMT3A or TET2 expression were associated with direct binding of DNMT3A or TET2 to gene regulatory regions, we performed ChIP-sequencing assays for each protein in MDM derived from four healthy donors. Examples of DNMT3A-specific, TET2-specific, and DNMT3A/TET2 co-bound peaks are illustrated in Figure 4A. Notably, approximately 85% of the DNMT3A peaks were co-occupied by TET2 (Figure 4B). Approximately 20% of DNM3TA or TET2 ChIP-seq peaks were in proximal promoter regions and roughly 2% in CpG islands (Figure 4C). Genomic regions bound by DNM3TA with higher occupancy were more highly enriched in promoter regions, while regions with higher TET2 affinity were primarily located within distal genomic regions (Figure S4A).
Figure 4. Genome-wide occupancy of DNMT3A and TET2.

(A) Composite of genomic regions bound by DNMT3A only (left), by DNTM3A and TET2 (center), or by TET2 only (right).
(B) Venn plot showing the overlap of all irreproducible discovery rate (IDR) peaks of DNMT3A and TET2 ChIP-seq (n = 4 donors).
(C) Analysis of genomic distribution of DNMT3A or TET2 peaks (promoters defined by 1,000 bp upstream of TSS).
(D) De novo motif analysis of DNMT3A (left) or TET2 (right) ChIP-seq peaks.
(E) Pathway analysis of genes associated with DNTM3A unique peaks (left), or 25% of top best DNTM3A-TET2 cobound peaks (center), or 25% of top best TET2 unique peaks (right).
(F) Venn plots of integrative analysis of upregulated genes in DNMT3A ASOs and TET2 ASOs with DNTM3A peak (left), TET2 peak (center), or DNMT3A-TET2 peak (right) as defined by 5,000 bp upstream of the TSS. Also see Figure S4.
Motif-enrichment analysis identified binding sites for macrophage lineage determining factors in both DNMT3A and TET2 ChIP-seq peaks, including PU.1, CEBP-NFIL3, AP-1, and MITF-TFE transcription factors (Figure S4B). The most highly enriched motifs in DNMT3A-specific peaks correspond to the promoter associated GFY motif and a STAF motif (Figure 4D). This is consistent with DNMT3A unique peaks being enriched in promoter regions compared to co-bound DNMT3A and TET2 peaks or TET2 unique peaks (34% versus 20% or 20%, respectively) (Figure 4C). DNMT3A-TET2 cobound peaks and TET2 unique peaks were proximal to genes enriched for functional annotations associated with inflammation. In contrast, DNTM3A unique peaks were proximal to genes enriched for functional annotations associated with RNA Pol II transcription, post translational protein modification and cell cycle (Figures 4D and 4E). In addition, we found higher number of tags per position in DNMT3A-TET2 co-bound peaks versus DNMT3A unique peaks or TET2 unique peaks, suggesting cooperative binding (Figure S4C).
Notably, terms related to type I IFN signaling were not strongly enriched among genes associated with specific or shared DNMT3A or TET2 binding sites, suggesting that the activation of ISGs in MDM with reduced DNTM3A or TET2 was indirectly regulated. To investigate this further, we integrated the list of differentially expressed genes in MDM treated with DNMT3A ASOs or TET2 ASOs with DNMT3A or TET2 peaks. Of the mutually upregulated genes, only 25/143 (17.5%) have peaks co-bound by DNMT3A or TET2, 5 of which correspond to ISGs (Figure 4F). Therefore, while genome-wide location analysis of DNMT3A and TET2 provide evidence for both overlapping and distinct roles in MDM, they do not appear to directly regulate most of the differentially regulated genes related to IFN signaling.
Activation of IFN signaling by reduced DNMT3A or TET2 expressing MDM is initiated by cGAS and propagated by IFNαR2
To gain further insight into mechanisms underlying the upregulation of IFNα and ISGs we performed a time-course analysis of gene expression in MDM following DNTM3A or TET2 depletion in the presence or absence of an IFN receptor blocking antibody. These experiments demonstrated that reduction of DNMT3A or TET2 expression resulted in up-regulation of ISGs including MX1, IFIT1, IRF1, IRF7, IFIT2, IFITM1, CMPK2, CXCL10, ISG15, and IFIT3 as early as 12 h, whereas upregulation of other inflammatory transcript genes such as CCL8, IL1B, CCL2, CCL4, CCL3, CXCL2, CXCL1, CXCL5, IL6 and TNFA primarily occurred by 24 h. Notably, the upregulation of genes activated at 24 h was almost completely blocked by treatment with the anti-IFNαR2 antibody, while genes induced at 12 h were largely unaffected (Figure 5A). ASO-mediated reduction of DNMT3A and TET2 resulted in phosphorylation of IRF3 as detected by Immuno blotting (Figure 5B), consistent with our finding of increased phospho-IRF3 at promoters containing IRF recognition motifs (Figures 2D and 2E). Phosphorylation of IRF3 is mediated by phosphorylated TANK binding kinase (TBK1), which was increased by ASO-mediated reduction of both DNMT3A and TET2 (Figure 4B). These results suggested a common role of DNMT3A and TET2 in preventing the phosphorylation and activation of IRFs and downstream gene expression in human MDM.
Figure 5. Role of DNTM3A and TET2 in mitochondrial DNA integrity and activation of cGAS signaling.

(A) RT-qPCR analysis of ISG or other inflammatory genes at 8, 12, or 14 h after incubation with DNMT3A ASO 1 or TET2 ASO 2 in the presence or absence of anti IFNAR2 (n = 4 donors).
(B) Western blot analysis of p-TBK1, p-IRF3, and IFIT2 in lysates of MDM treated with DNMT3A ASO 1 or TET2 ASO 2 (n = 1 pool of 3 donors).
(C) RT-qPCR analysis of ISGs or other inflammatory genes at 8, 12, or 14 h after incubation with DNMT3A ASO 1 or TET2 ASO 2 in the presence or absence of the cGAS inhibitor G-140 (n = 4 donors).
(D) Bar graphs of mitochondrial copy number (top) and increased cytosolic mitochondrial DNA in extracts of MDM treated with DNMT3A ASO 1 or TET2 ASO 2 (n = 3 donors).
(E) Immunofluorescence analysis of mitochondria (HSP60) and DNA in regions associated with mitochondria in MDM treated with DNMT3A ASO 1 or TET2 ASO 2 in the presence or absence of treatment with ddC (arrowheads) (n = 3 donors). Scale bars: 2 μm.
(F) Quantification of cytosolic DNA (nucleoids) in MDM treated with DNMT3A ASO 1 or TET2 ASO 2 in the presence or absence of treatment with ddC (n = 3 donors).
(G) RT-qPCR analysis of ISG or other inflammatory genes at 8, 12, or 14 h after incubation with DNMT3A ASO 1 or TET2 ASO 2 in the presence or absence of treatment with ddC (n = 4 donors).
(H) Representative images by IF analysis of HSP60 and DNA showing larger nucleoids (arrowheads) in macrophages with DNMT3A or TET2 mutations. Scale bars: 2 μm.
(I) Quantification of cytosolic DNA in macrophages with DNMT3A or TET2 mutations. Mann-Whitney U test was used to calculate statistical significance. *p < 0.05; **p < 0.01. Also see Figure S5.
In macrophages, TBK1 is primarily activated by Toll-like receptors (TLRs) that couple to the TRIF signaling pathway or by the cGAS/STING pathway that senses cytoplasmic nucleic acids of viral or host origin (Pandey et al., 2014; Wan et al., 2020; Abe and Barber, 2014). The early upregulation of ISGs without activation of other inflammatory genes is most consistent with activation of cGAS signaling. We therefore investigated whether cGAS activation preceded signaling by IFNAR2. Notably, treatment of MDM with the cGAS inhibitor G-140 (Abe and Barber, 2014; Lama et al., 2019) prevented the upregulation of ISGs and inflammatory genes both at 12 h and 24 h following ASO-mediated depletion of DNMT3A and TET2, consistent with an essential role of cGAS in the activation of the type I IFN response (Figure 5C).
Heterozygosity of the mitochondrial DNA transcription factor TFAM leads to mtDNA depletion (i.e., reduced mtDNA copy number), enlarged nucleoids, release of mtDNA into the cytoplasm, and cGAS-STING activation in fibroblasts and BMDM (West et al., 2015). Similar to the direct effect of reduced Tfam, we found that reduced DNTM3A or TET2 expression led to a lower mtDNA copy number, increased cytosolic mtDNA, and enlarged DNA nucleoids adjacent to mitochondria (HSP60). DNA nucleoids were reduced after treatment of cells with dideoxycytosine (ddC) to deplete mtDNA (Figures 5D, 5E, and 5F). ASO-mediated reduction of DNMT3A or TET2 was also accompanied by more elongated mitochondria (Figures S5A and S5B), but did not affect mitochondrial membrane potential or increase reactive oxygen species (ROS) (Figures S5C and S5D), phenotypes also shared with Tfam heterozygous cells. Importantly, depletion of mtDNA with ddC also almost completely prevented the upregulation of ISGs and inflammatory genes at both 12 and 24 h (Figure 5G). These data suggest that reduced expression of DNMT3A or TET2 results in mitochondria and mtDNA alterations that closely resemble those observed in Tfam heterozygous cells (West et al., 2015).
These findings led us to investigate whether mutations in DNTM3A or TET2 associated with CHIP led to mtDNA accumulation in the cytoplasm. Although it is not possible to distinguish wild-type from mutant cells in these experiments, we found evidence for cytoplasmic DNA nucleoids adjacent to mitochondria in a percentage of cells that correlate with the variant allele frequency of TET2 or DNMT3A mutations of the patient cells (Figures 5H and 5I). These findings are consistent with reduced DNMT3A and TET2 expression resulting in mitochondrial alterations that lead to release of mtDNA and activation of a type I IFN response.
The above results led us to interrogate the status of TFAM in DNTM3A or TET2 ASO-depleted cells. We found downregulation of TFAM mRNA (Figure 6A) and protein assessed by immunofluorescence (IF) (Figures 6B and 6C) in MDM treated with either DNMT3A or TET2 ASOs or with siRNAs for DNTM3A or TET2. Furthermore, around 1,400 differentially expressed genes were detected when comparing MDM treated with small interfering RNAs (siRNAs) against TFAM and control-treated MDM, but no changes in DNMT3A or TET2 expression were detected (Figures 6D and 6E). Moreover, genes upregulated by both DNMT3A ASOs and TET2 ASOs and siTFAM treatment in MDM belonged to interferon signaling (e.g., ISGs) (Figures 6F-6H). These data support the hypothesis that the interferon program activated in MDM with reduced DNMT3A or TET2 is at least in part regulated through TFAM expression. In line with this, in MDM treated with DNMT3A ASO 1 or TET2 ASO 2 we find downregulation of DNMT3A and TET2 (at 6–8 h) precedes downregulation of TFAM at 10 h and accumulation of cytosolic mtDNA at 12 h (Figures 6I, 6J, and S6A). In contrast, incubation of MDM with recombinant DNA or IFNα for 2 h induced both the direct and secondary ISG response within 2 h without affecting TFAM, DNMT3A, or TET2 expression (Figure S6B, left and right, respectively). These data support a mechanism of action in which reduction of DNMT3A or TET2 leads to downregulation of TFAM and subsequent accumulation of cytosolic mtDNA.
Figure 6. DNMT3A and TET2 regulate the TFAM gene to restrain activation of ISGs.

(A) RT-qPCR analysis of TFAM expression in MDM treated with DNMT3A ASOs or TET2 ASOs or siRNAs for DNMT3A or TET2 (n = 5 donors for ASOs and n = 3 donors for siRNA).
(B) Protein expression by IF in MDM treated with DNMT3A ASOs or TET2 ASOs (n = 2 donors). Scale bars: 10 μm.
(C) Quantification of protein expression shown in (B), downregulation in MDM treated with DNMT3A ASOs or TET2 ASOs (n = 2 donors).
(D) Bar plots showing the number of differentially expressed genes in MDM treated with siTFAM 1 or siTFAM 2 versus siControl.
(E) Bar plot showing reduced expression of TFAM (but not DNMT3A or TET2) in MDM treated with siRNAs for TFAM.
(F) Venn plot showing the overlap between genes upregulated by DNTM3A ASOs and TET2 ASOs in MDM incubated with siRNAs for TFAM.
(G) Pathway analysis of genes upregulated by DNMT3A ASOs, TET2 ASOs, and by TFAM siRNA showing enrichment in interferon signaling genes.
(H) Bar plot showing the upregulation of IFN-stimulated genes in cells treated with siRNAs for TFAM.
(I) Protein expression by IF of DNMT3A (left), TET2 (center), and TFAM (right) showing DNMT3A and TET2 reduction at 8 h, whereas TFAM reduction occurs at 10 h in MDM treated with DNMT3A ASOs or TET2 ASOs (n = 5 donors, one representative donor is shown).
(J) Quantification of cytosolic nucleoids in MDM treated with DNMT3A ASOs or TET2 ASOs (n = 3 donors). Mann-Whitney U test was used to calculate statistical significance. *p < 0.05; **p < 0.01 except panel in (I) where Student’s t test was used to calculate statistical significance. Also see Figure S6.
DNMT3A and TET2 directly regulate TFAM expression via RBPJ and ZNF143 to restrain IFN signaling
To investigate the mechanism by which DNMT3A and TET2 regulate TFAM expression, we examined the distribution of their binding sites in the vicinity of the TFAM locus. We observed strong binding of both factors to the TFAM promoter (Figure 7A). From the motif enrichment analysis of DNMT3A and TET2 ChIP-seq peaks, our attention was drawn to the GFY and ZNF143 motifs. These were the top 2 motifs for DNMT3A peaks and were also enriched in DNMT3A and TET2 co-bound peaks (Figure 4D). The last 5 base pairs of the GFY motif are identical to a motif that is recognized by RBPJ at a subset of its genomic binding sites in other cell types (Sakai et al., 2019; Castel et al., 2013). ZNF143 is ubiquitously expressed, and functions in various cancers (Ye et al., 2020; Izumi et al., 2010)(Kawatsu et al., 2014) and regulates promoter-enhancer loops in murine hematopoietic stem cells (Zhou et al., 2021). We therefore performed ChIP-seq analysis of RBPJ and ZNF143 to investigate the possibility that they serve to target DNMT3A and/or TET2 to their genomic binding sites in human MDM.
Figure 7. Combinatorial binding of RBPJ, ZNF143, DNMT3A, and TET2 to coordinate TFAM expression and prevent type-I IFN signaling.

(A) Composites of DNTM3A, TET2, RBPJ ,and ZNF143 ChIP-seq showing binding to TFAM promoter.
(B) Chow-Ruskey plot showing the overlap between DNTM3a, TET2, ZNF143, and RBPJ ChIP-seqs (n = 4 donors for DNMT3A and TET2 ChIP-seqs and n = 2 donors for ZNF143 and RBPJ ChIP-seq).
(C) HOMER analysis of motifs found in the 1,604 peaks co-bound by DNMT3A, TET2, RBPJ, and ZNF143 showing motifs for ZNF143, ELF4, PU.1, and CEBPD.
(D) HOMER analysis of motifs found in ZNF143 peaks showing enrichment for ZNF143, ELF4, RUNX, PU.1, CEBPE, and MITF/TFE.
(E) HOMER analysis of motifs found in RBPJ peaks showing enrichment in PU.1, BORIS, AP-1, CEBPE, PU.1-IRF8, and MITF/TFE.
(F) RT-qPCR of RBPJ, ZNF143, DNTM3A, TET2, or TFAM in MDM treated with siRNAs for RBPJ or ZNF143 (n = 3 donors).
(G) Composites of DNTM3A, TET2, RBPJ, and ZNF143 ChIP-seq showing binding to ZNF143 promoter.
(H) RT-qPCR showing upregulation of CXCL10, RSAD2, ISG15, and IFIT1 in MDM treated with siRNAs for RBPJ or ZNF143 (n = 3 donors).
(I) ChIP-qPCR over a control negative region (negative), proximal or distal region of TFAM promoter showing reduced binding of DNMT3A and TET2 in MDM treated with siRNAs for RBPJ or ZNF143 (n = 3 donors).
(J) Representative images by IF analysis of HSP60 and DNA showing larger nucleoids (arrowheads) in macrophages with DNMT3A or TET2 mutations. Scale bars, 2 μm.
(K) Quantification of cytosolic DNA showing increased number of macrophages treated with siRNA for RBPJ or ZNF143 or TFAM. Mann-Whitney U test was used to calculate statistical significance. *p < 0.05; **p < 0.01. Also see Figure S7.
We identified 43,382 high-confidence binding sites for RBPJ and 3,156 high-confidence ZNF143 binding sites, enabling integration with the genome-wide locations of DNMT3A and TET2 (Figure 7B). Motif enrichment of ZNF143 peaks exhibited substantial overlap with motifs enriched at DNMT3A only peaks, with the GFY and ZNF143 motifs ranked first and second (Figure 7C). Consistent with this, 80% (2,523/3,156) of ZNF143 peaks were co-bound by DNMT3A (Figures 7B, S7A, and S7B), suggesting an important role of ZNF143 in directing genomic binding of DNMT3A. Conversely, motif enrichment of RBPJ peaks exhibited large overlap with motifs enriched at TET2-only sites, including motifs for macrophage lineage determining factors PU.1, AP-1, CEBP, RUNX, and TFE family members (Figures 7D, S7A, and S7B). Corresponding to this, 78% (13,748/17,657) of TET2 only peaks co-localized with RBPJ binding sites (Figure 7B), mostly at distal genomic locations, suggesting a role of RBPJ in directing the DNA binding pattern of TET2. Notably, we identified 1,604 genomic locations at which DNMT3A, TET2, ZNF143, and RBPJ were all co-bound (Figure 7B). This binding pattern was clearly represented at the TFAM promoter (Figure 7A). In contrast, the second upstream binding site of DNMT3A and TET2 was strongly bound by RBPJ, but not ZNF143. Motif enrichment analysis of sites at which all four factors were bound identified the GFY and ZNF143 motifs as the most highly enriched, but at even higher frequencies than observed at DNMT3A-only peaks, (Figures 7E, S7A, and S7B).
The co-occurrence of DNMT3A, TET2, ZNF143 and RBPJ at the TFAM promoter suggested the possibility that ZNF143 and RBPJ function to coordinately recruit DNMT3A and TET2 to this location to induce its expression. To test this hypothesis, we reduced RBPJ and ZNF143 expression using independent pairs of siRNAs. While the ZNF143 siRNAs selectively reduced ZNF143, both siRNAs targeting RBPJ resulted in reduced expression of not only of RBPJ, but also of ZNF143 (Figure 7F). Although the downregulation of ZNF143 by RBPJ siRNAs was unexpected, this was consistent with the observation that both RBPJ and ZNF143 were strongly bound to the ZNF143 promoter (Figure 7G) and provides evidence that RBPJ regulates ZNF143 expression. siRNA ablation of RBPJ or ZNF143 did not alter DNMT3A expression and resulted in increased expression of TET2 (Figure 7G), indicating that reduced expression of TFAM was not due to reduced expression of either of these factors. Importantly, reduced expression of RBPJ or ZNF143 expression alone or in combination led to upregulation of ISGs including CXCL10, RSAD2, ISG15 and IFIT1 (Figure 7H).
We then investigated whether RBPJ and ZNF143 were required for the binding of DNMT3A and TET2 to the TFAM promoter by performing locus-specific ChIP assays for each factor at the promoter and upstream binding site. We found that whereas reduction of both RBPJ and ZNF143 had strongly deleterious effects on binding of DNMT3A and TET2 at the proximal promoter of TFAM, knockdown of RBPJ had a greater effect on their binding to the distal region of TFAM promoter (Figure 7I). This observation correlated with the absence of ZNF143 binding to this location and implies that other factors (e.g., Elf4, SP1, C/EBP etc., Figure 7E) collaborate with RBPJ to mediate DNMT3A and TET2 recruitment to this site. Collectively, these results provide evidence that DNMT3A and TET2 directly regulate the expression of TFAM through interactions with RBPJ and ZNF143. The effects of ablating ZNF143 and RBPJ on the expression of TFAM further predicted that there should also be an activation of the type I IFN response. Consistent with this expectation, knockdown of RBPJ or ZNF143 led to increased expression of ISGs and increased numbers of nucleoids (Figures 7J and 7K), mimicking the effect of reduced DNMT3A, TET2, or TFAM expression.
DISCUSSION
Mutations in the regulators of DNA methylation DNMT3A and TET2 represent more than 70% of all mutations in hematological disorders and in CHIP (Jaiswal et al., 2014, 2017; Fuster et al., 2017). While these mutations contribute to the clonal expansion of hematopoietic progenitor cells, the functions of DNMT3A and TET2 in differentiated human macrophages and the effects of loss-of-function mutations are largely unknown. In this work, we show that in addition to distinct biological roles, DNMT3A and TET2 function in a coordinated manner to maintain mtDNA integrity by regulating TFAM expression. Reduced expression of DNMT3A or TET2 led to upregulation of ISGs that was recapitulated in MDM from individuals with DNMT3A or TET2 mutations. Prior studies in mice demonstrated that TET2 or DNMT3A deficiency can result in exacerbated pro-inflammatory responses in BMDM (Cull et al., 2017) or mast cells (Leoni et al., 2017), respectively, consistent with the findings presented here. However, DNTM3A inhibition has also been shown to reduce the production of type I IFNs in mouse peritoneal macrophages (Li et al., 2016). This apparent discrepancy suggests species or context-specific differences that will require additional studies to resolve.
We also observed an IFN signature in both normal MDM and atherosclerotic lesion macrophages (Depuydt et al., 2020) exhibiting reduced DNTM3A or TET2 expression as assessed by single cell RNA-seq experiments. This finding suggests that low expression of DNMT3A or TET2 in macrophages might promote type 1 IFN responses even in individuals without loss of function mutations. This finding is of potential clinical relevance considering previous studies reporting that IFN signaling is associated with enhanced atherosclerosis and plaque instability (Goossens et al., 2010). It will therefore be important to determine whether low amounts of DNMT3A or TET2 are causally related to increased expression of interferon stimulated genes observed in vivo.
We find production of IFNαs—but not IFNβ—by cells with reduced DNMT3A or TET2 or with DNMT3A or TET2 mutations leads to activation of ISG in neighboring cells through signalling by IFNAR2. We speculate that IFNB is not induced in cells with reduced DNTM3A or TET2 mutations because of the specific requirement of the IFNβ enhanceosome for NFκB (Panne, 2008) and the modest activation of NFκB in MDM in which DNMT3A or TET2 expression is reduced. These findings support the concept that a few DNTM3A or TET2 mutant cells might act in a paracrine manner, accelerating and perpetuating tissue inflammation. Accordingly, this “few bad apples spoil the barrel” scenario may explain how CHIP with a VAF of as little as 10% (affecting 20% of nucleated blood cells) was sufficiently associated with atherosclerotic CVD risk (Jaiswal et al., 2017). Furthermore, CHIP due to mutations in DNMT3A or TET2 with a VAF 2% (affecting 4% of cells) or less was associated with incident heart failure and worse prognosis with heart failure in a dose dependent manner (Dorsheimer et al., 2019; Assmus et al., 2021; Yu et al., 2021).
Despite displaying antagonistic enzymatic activities, loss of function of DNMT3A or TET2 in murine models show overlapping phenotypes in terms of increased HSC fitness (Ostrander et al., 2020), suggesting a common program regulated by DNMT3A and TET2. In our studies, although we cannot rule out an undetectable contribution of changes in DNA methylation, it is unlikely that reduction of the enzymatic functions of DNMT3A or TET2 account for activation of the IFN program. The lack of correlation between DNA methylation and differential gene expression in MDM is consistent with other studies including Dnmt3a-null HSC (Challen et al., 2011) and human samples of acute myeloid leukemia (Ley et al., 2010). The lack of ISG upregulation in MDM treated with the DNA methyltransferase inhibitor azacytidine differs from ovarian cancer cells where it triggers cytosolic sensing of double-stranded RNA causing a type I IFN response and apoptosis (Chiappinelli et al., 2015), indicating a cell-dependent role of DNA methylation by DNMT3A in ISG activation. A role of TET2 outside DNA methylation is also concordant with previous work showing that non-catalytic roles of TET2 are crucial to regulate HSC and progenitor cell homeostasis (Ito et al., 2019). Our finding of direct regulation of gene expression by DNMT3A and TET2, as with TFAM in MDM, may partly explain the convergent cardiovascular phenotypes associated with both DNMT3A and TET2 mutations.
Analyses of the genome-wide binding patterns of DNMT3A and TET2 in human MDMs led to the discovery of their co-recruitment to a specific set of DNA regulatory elements by ZNF143-STAF and RBPJ. This pattern contrasts with a previous finding that RBPJ and ZNF143 peaks are mutually exclusive in leukemia cells (Wang et al., 2011), indicating that additional factors are required to establish their cell-specific DNA binding patterns. Mechanistically, we provided evidence that RBPJ and ZNF143 were required for binding of DNTM3A and TET2 and transcriptional activation of the TFAM promoter. The role of RBPJ in restraining IFN signaling in MDM contrasts with the reported role of RBPJ in M1-like polarization of BMDM treated with TLR agonists (Xu et al., 2012; Foldi et al., 2010). However, the findings here that siRNA mediated reduction of RBPJ or ZNF143 led to loss of TFAM expression and a robust type I IFN response in MDMs suggests a species or context-dependent role of RBPJ in activating inflammatory programs of gene expression.
Despite substantial advances in the treatment of conventional risk factors, such as hypercholesterolemia, CVD remains a major cause of morbidity and mortality. CHIP represents a recently recognized non-conventional risk factor for which there are currently no specific therapeutic approaches. The present findings suggest that possibility that pharmacological inhibition of cGAS or downstream type I IFN signaling could be of therapeutic value. Our findings may also open roads to identifying other scenarios of disease with IFN activation associated to reduced DNMT3A or TET2 expression such as lupus (Piotrowski et al., 2015), endometriosis (Szczepanska et al., 2013), or pancreatic cancer (Vitale et al., 2007). Finally, our results add to the ever-growing list of pathological circumstances that involve mitochondria and mtDNA-mediated inflammation (West and Shadel, 2017; Newman and Shadel, 2018).
Limitations of the study
As these studies were performed in human MDMs in vitro, they likely do not fully model the consequences of loss of function of DNMT3A or TET2 in macrophages that reside in pathological tissue environments. In addition, due to the short duration of in vitro studies and lack of cell proliferation, the potential to record changes in gene expression due to alterations in DNA methylation was limited. Thus, we cannot exclude important consequences of haploinsufficiency of the enzymatic activities of DNMT3A or TET2 in macrophages in tissue environments. Second, we studied a limited number of individuals with DNMT3A or TET2 mutations and were not able to definitively distinguish cells containing mutant DNMT3A or TET2 from non-mutant cells. Although our studies clearly demonstrated activation of IFN signaling at the population level and an increased frequency of DNA nucleoids in MDMs from individuals with DNMT3A or TET2 mutations that scaled with variant allele frequency, this is not the same as defining these phenotypes as a function of each individual cell’s genotype.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher K. Glass (cglass@health.ucsd.edu).
Material availability
This study did not generate new unique reagents.
Data and code availability
The datasets supporting the current study have been deposited in GEO (GSE206030). This study did not generate any unique code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Isolation of monocytes0
Human PBMC were isolated from whole blood by Ficoll density gradient using Ficoll Plaque Premium (Sigma GE Healthcare, #17-544-02) or BD Vacutainer CPT Tubes (BD, 362753) as described somewhere else. PBMC were washed twice with HBSS (Gibco ThermoFisher, #14175095) containing 2% BSA, 1 mm EDTA. Monocytes were obtained by negative selection using a kit (StemCell, #19359).
Experiments using mouse bone marrow derived macrophages (BMDM)
Experiments involving BMDM were carried out under the protocol number S01015. Bone marrow cells were flushed and culture for six days in 15 ng/uL of MCSF. Eight-ten weeks male and females C57/Bl6 mice were used.
METHOD DETAILS
Collection of monocytes from healthy donors and patients with DNTM3A or TET2 mutations
Monocytes from healthy donors were isolated from blood collected using the normal blood donor progam of Scripps Research (https://www.scripps.edu/science-and-medicine/cores-and-services/normal-blood-donor-service/index.html) under the IRB 207533. All patient samples were collected at the University of California San Diego (UCSD) Moores Cancer Center with patient consent under protocols approved by Institutional Review Boards and in accordance with the Declaration of Helsinki. Genomic DNA samples were isolated from peripheral blood mononuclear cells and were genetically characterized by targeted capture of DNA and sequenced on the Illumina platform. Alignment and variant calling were performed using a custom pipeline and variant calls were annotated using validated software utilizing databases of known germline and somatic variants, followed by confirmation of these variants by manual review. A summary of the patient’s samples used in this study is shown in Table S1.
Differentiation of monocytes to macrophages (MDM)
Monocytes were incubated with RPMI 1640 (Sigma Aldrich, # 10-040) medium containing 10% de-complemented FBS (OmegaScientific, #FB-02), 1 mM Sodium Pyruvate (Gibco Thermofisher), Penicillin-Streptomycin (15140122, 1000 U/mL) and 25ng/mL-50 ng/mL of recombinant human M-CSF (StemCell #78057-2) for six days at 37°C and 5% CO2. Fresh media was added 48 h and four days after seeding.
Transfection of MDM or BMDM with antisense oligonucleotides and transfection of MDM with siRNA
Monocyte-derived macrophages or bone marrow derived macrophages (BMDM) were transfected with 250 nM of Scramble ASO or siRNA control, DNMT3A or TET2 ASO (kindly provided by IONIS Pharmaceuticals) or DNMT3A siRNA or TET2 siRNA using DharmaFECT reagents (Horizon, #T-2004-02) for 24 h in medium without antibiotics. The two DNMT3A ASO or TET2 ASO that displayed greater DNMT3A or TET2 protein knockdown were used for the experiments. DNMT3A siRNA #1 (hs.Ri.DNMT3A.13.1), DNMT3A siRNA #2 (hs.Ri.DNMT3A.13.2), TET2 siRNA #1 (hs.Ri.TET2.13.1), TET2 siRNA #2 (hs.Ri.TET2.13.2). After 24 h, transfection media then used to condition untreated MDM for 2 h.
Treatment of MDM with chemicals
MDM were incubated for four days with 3 mg of IgG control (R&D systems, #MAB003) or anti-Interferon Alpha/Beta/Omega Receptor Chain 2 (IFNαR2) blocking antibody (Pbl assay science, #21385-1, Lot 7035), 2′,3′-dideoxycytidine (ddC) or 10 μM of human specific c-GAS inhibitor G-140 (Invivogen, inh-g140). Effect in DNMT3A or TET2 ASO was calculated as relative to that measured in cells transfected with Scramble ASO.
Stimulation of MDM with KLA, R848, IFNα
MDM were incubated for two or five with, 2 pg/mL of IFNα2 (StemCell, #78076) in RPMI 1640 medium containing 10% de-complemented FBS, 1 mM Sodium Pyruvate, Penicillin-Streptomycin and 25–50 ng/ml of recombinant human M-CSF. MDM previously incubated with ASO were incubated with 2 pg/mL IFNα for 5 h.
Crystal violet staining
For staining in MDM, 100K monocytes were initially seeded in 96-well plates and ASO-mediated and three technical replicates were used in all conditions. Transfected cells were washed with PBS and incubated with crystal violet 0.2% (w:v) solution for 10 min at room temperature. Excess of crystal violet solution was removed using water and air dry. Once the plates were dried, 1% SDS solution was added to each well to solubilise the staining and absorbance was read using 570 nm wavelength in a luminometer. The average of two measurements was used for final analysis.
Incubation with siRNA
Monocyte-derived macrophages were transfected with 250 nM of control siRNA or specific siRNA against TFAM (IDT technologies, kindly provided by Gerald Shadel’s laboratory) using DharmaFECT reagents (Horizon, #T-2004-02) for 24 h in medium without antibiotics. Next day, transfection media was replaced with normal media to let the cells recover for 24 h before harvesting.
Immunofluorescence
Briefly, peripheral blood monocytes were differentiated to MDM in chambered slides (Millipore, #C86024) and fixed with BD Cytofix/Cytoperm Buffer (BD, BD554714) for 10 min at room temperature. Cytofix/Cytoperm buffer was removed and MDM were washed twice with HBSS containing 2% BSA and 1 mm EDTA. Cells were kept in wash/permeabilization buffer (BD, BD554714) for 1 h at 4°C or until the experiment was performed. Fixed MDM were blocked using 3% BSA, 0.1% Triton-PBS for 30 min at room temperature and then with the primary antibody overnight at 4°C. For double or triple IF, the corresponding antibodies were added simultaneously and incubated overnight at 4°C. Next day, MDM were washed with 0.1% Triton-PBS, incubated with the appropriate fluorochrome-conjugated secondary antibody and fluorescent probes and nuclei were counter-stained with DAPI. After washing with 0.1% Triton-PBS, slides were mounted with Prolong Gold Antifade Reagent (Life Technology, #10144). Images were taken using a Leica SP8 with light deconvolution microscope or SP5 confocal microscope (Leica). ImageJ was used to quantify signal intensity of IF images. Briefly, three-colored images were split into single-colored images, nuclei were delineated using the freehand selection tool of ImageJ and intensity was calculated as the value of mean/area. Experiments were performed in triplicates. One representative experiment is shown. A list of antibodies and fluorescent probes used for IF with their working concentration is shown below. Scale bars are provided in the figure corresponding figure legend.
Apotracker. Biolegend, #427401. 1/1000.
Bodipy. ThermoFisher, #D3922. 1/1000.
CellEvent. ThermoFisher, #C10723. 1/1000.
CTCF. Cell Signaling, #2899 S. 1/300.
DNA. Abcam, #27156. 1/400-1/1000.
DNMT3A. Abcam, #2850. 1/100-1/200.
HSP60. Cell Signaling, #12165 S. 1/200-1/400.
IRF1. Abcam, #191032. 1/100.
IRF7. Abcam, #115352. 1/100.
KI67. Invitrogen, #53-5698-82. 1/100-1/200.
Phalloidin. Abcam, #176759. 1/1000.
TET2. Abcam, #94580. 1/100-1/200.
TFAM. Cell Signaling, #8076 S. 1/100.
Donkey anti-mouse 488. ThermoFisher, #A21202. 1/200.
Donkey anti-rabbit 555. ThermoFisher, #A31572. 1/200.
Detection of mitocondrial DNA in cytosolic extracts
Digitonin extracts from MDM were generated as described somewhere else (Holden and Horton, 2009); (West et al., 2015). Briefly, 3 million MDM incubated with Scramble ASO, DNMT3A ASO #1 or TET2 ASO #2 were each divided into two equal aliquots, and one aliquot was resuspended in 500 μL of 50 μM NaOH and boiled for 25 min to solubilize DNA and 50mL 1M Tris-HCl pH 8 was added to neutralize the pH. These extracts served normalization controls for total mtDNA. The second equal aliquots were resuspended in 400 mL buffer containing 150 mM NaCl, 50 mM HEPES, pH 7.5, and 15–25 μg/mL digitonin (Promega, #G9441) and incubated for 10 min under mild shaking to allow selective plasma membrane permeabilization, then centrifuged at 1000 g for 4 min three times to pellet intact cells. The cytosolic supernatants were transferred to fresh tubes and spun at 16,000 g for 10 min to pellet any remaining cellular debris, yielding cytosolic preps free of nuclear or contamination from other organelles. DNA was then isolated from these pure cytosolic fractions using DNA ChIP & Celan concentrator (Zymo Research). Quantitative PCR (qPCR) was performed using the KAPA SYBR Fast PCR master mix (Sigma Aldrich, #KK4605) and an ABI-StepOne 96 well plate instrument (Applied Biosystems) on both whole cell extracts and cytosolic fractions using nuclear DNA primers and mtDNA primers (see list below). Ct values obtained for mtDNA abundance for whole cell extracts served as normalizer for the mtDNA values obtained from the cytosolic fractions. Abundance of mtDNA was calculated as average of signal obtained for each mtDNA primer set. The sequence of the primers used and their location in the genomic or mitochondrial DNA is provided below.
| Coordinates | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| chr1:103552993-103553062 | ttcctctgtaaactaaaagaac | acccatatttggcagttaaaca |
| chrM:404-524 | cttttggcggtatgcacttt | gtgtgtgtgtgctgggtagg |
| chrM:2333-2436 | gcataagcctgcgtcagatt | cctgtgttgggttgacagtg |
| chrM:3116-3217 | cctccctgtacgaaaggac | tgggtgggtgtgggtataat |
| chrM:5198-5325 | aattccatccaccctcctct | tgatggtggctatgatggtg |
| chrM:6377-6514 | cttaggggccatcaatttca | gcagctaggactgggagaga |
| chrM:7820-7927 | ccatccctacgcatccttta | gccgtagtcggtgtactcgt |
| chrM:9420-9527 | acaccacctgtccaaaaagg | ggctaggctggagtggtaaa |
| chrM:10831-10931 | aatcaacacaaccacccaca | aggaaaaggttggggaacag |
Gene expression analysis by qRT-PCR
Total RNA was isolated using Direct-zol RNA MicroPrep kit (Zymoresearch, #11-33MB) and treated with DNase I on column. 500 ng-1 μg of RNA solution was enriched in poly-A tailed RNA by incubation with Oligo d(T) Magnetic Beads (NEB, #S1419 S). cDNA was prepared according to the manufacturer’s specifications, using the Superscript III Reverse Transcriptase (ThermoFisher, # 18080093). qRT-PCR analysis was performed using the KAPA SYBR Fast PCR master mix (Sigma Aldrich, #KK4605) and an ABI-StepOne 96 well plate instrument (Applied Biosystems). The sequence of the primers used is provided below. Expression of each gene were normalised to HPRT mRNA using the ΔΔCt method.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| CCL2 | tctgtgcctgctgctcatag | gcactgagatcttcctattggtg |
| CCL3 | gcccttgctgtcctcctct | gggaggtgtagctgaagcag |
| CCL4 | cctcatgctagtagctgccttc | aagcttcctcgcggtgtaa |
| CCL5 | gatccagaagccccttttct | tcagttttccttgtttccactg |
| CCL8 | ttctgcagcgcttctgtg | cacgttaaagcagcaggtga |
| CMPK2 | agttccaggttgttgccatc | atgcaagagggtggtgactt |
| CXCL1 | gaaagcttgcctcaatcctg | caccagtgagcttcctcctc |
| CXCL10 | ctccagtctcagcaccatga | tgatgcaggtacagcgtacag |
| CXCL2 | cacactcaagaatgggcaga | agcttcctccttccttctgg |
| HPRT | tgaggatttggaaagggtgt | gcacacagagggctacaatg |
| IFIT1 | aacaggttttcgcaatcagg | tgacatctcaattgctccaga |
| IFIT3 | agaacaaatcagcctggtca | atggcatttcagctgtgga |
| IFITM1 | agagaggcctgcgacaagt | atgttgatcacggtggacct |
| IFNA1 | acccacagcctggataacag | actggttgccatcaaactcc |
| IL1B | agctgatggccctaaacaga | aagcccttgctgtagtggtg |
| IL6 | ctcagccctgagaaaggaga | tttcagccatctttggaagg |
| IRF7 | cgacccgcacaaggtgta | ctgtggtggtgggacagc |
| ISG15 | ctgagaggcagcgaactcat | agcatcttcaccgtcaggtc |
| MX1 | ccttaggcctcggtcttctc | caccgtgacactgggattc |
| RSAD2 | gtgagcaatggaagcctgat | acggccaataaggacattga |
| TNFA | ccccagggacctctctctaa | tctcagctccacgccatt |
| DNMT3A | aggtggagccatcgaagc | ggaggcgagagttaaaacttaaacata |
| TET2 | agatggctgccctttaggat | ggtgtgctgctgaatgtttg |
| ZNF143 | ggctgtttcctgtcctggt | ggaaactctgtcattccctga |
| RBPJ | ggagttttggcgagagtttg | tgagatcataatggttccgaag |
| TFAM | aacactgcttggaaaaccaaa | cagcttttcctgcggtgaat |
| Hprt | cattaaagcactgaatagaaatagtga | tatgtcccccgttgactgat |
| Oasl1 | actggaccaagcactacacg | tgtagccttctgccacattg |
| Ifit2 | aagaggttgcctggagagtg | ttagctgtcgcagattgctc |
| Cmpk2 | tgctgccagatctgcttaac | aagtactgcccgggcttc |
| Mx1 | agcatggctcaggaggtg | acatccaagaccttgccttc |
| Cxcl10 | cgtcattttctgcctcatcc | tatggccctcattctcactg |
| Rsad2 | acagccaagacatccttcgt | aggaagggttctcctccaga |
Enzyme-linked immunosorbent assay (ELISA)
IFNα (Pbl assay science, #41135-1) was measured by ELISA in conditioned media from ASO-depleted MDM or cultured media used for MDM with DNMT3A or TET2 mutations following manufacturer’s specifications. Measurements of all samples were carried out in duplicates.
Intracellular staining and cytometry
For detection of intracellular proteins by cytometry cells were fixed using Cytofix/Cytoperm Buffer for 10 min at room temperature, washed twice with HBSS containing 2%BSA and 1 mM EDTA, and with Wash/Perm buffer (BD, #BD554714). Cells were incubated with Fc receptors blocking agent TruStain FcX (Biolegend, #422302) for 10 min on ice followed by incubation with desired antibody for 30 min also on ice. Stained cells were pelleted to remove excess of antibody, washed twice, filtered using 35 μm strainer and resuspended in 250–500 μL in HBSS-2%BSA-1 mM EDTA. Unstained cells were used as negative control. Cytometry was performed using a Sony MA900 sorter. A list of fluorophore-conjugated antibodies used for intracellular staining with their working concentration is shown below.
DNMT3A. sc373905-AF488. 1/100-1/200
TET2. sc398535-AF647. 1/100-1/200
KI67. Invitrogen, #53-5698-82. 1/100.
Western blot
It was performed as described somewhere else (Cobo et al., 2018). Briefly, proteins were extracted from MDM using a lysis buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 5 mM EDTA and 0.5% NP-40) supplemented with protease inhibitor and phosphatase inhibitor cocktails. Protein concentration was measured by using DC™ Protein Assay Kit (Bio-Rad Laboratories) according to the manufacturer’s instructions. Protein solutions were boiled at 95°C for 5 min in NuPAGE™ LDS Sample Buffer with NuPAGE™ Sample Reducing Agent for SDS-PAGE. Proteins were resolved using NuPAGE™ 4–12% Bis-Tris Gels and transferred to PVDF membranes (Immobilon, #IPVH00010) previously activated with methanol. Once proteins were transferred, membranes were blocked in a solution of 4% milk in PBS:0.1%Triton for 45 min at room temperature and incubated with specific antibodies overnight at 4°C in mild rotation. Membranes were then washed using PBS:0.1%Triton and incubated with anti-rabbit or anti-mouse antibodies conjugated with HRP (Dako, #P0448 or #P0447, respectively) and developed using Chemidoc molecular imager (BioRad). A list of the antibodies used and working concentration is provided below.
DNMT3A. Abcam, ab2850. 1/1000.
TET2.Abcam, ab94580. 1/1000.
Phopho-TBK1. Cell Signaling, #D52C2. 1/1000.
TBK1. Cell Signaling, #D1B4. 1/1000.
Phospho-IRF3. Cell Signalling, #D6O1M. 1/500.
IRF3. Cell Signaling, #D83B9. 1/1000.
IFIT2. Boster, #A04428. 1/1000.
Actin. Sigma, #A5441. 1/1000.
Transmission electron microscopy (TEM)
Sample preparation and imaging was performed at the Electron Microcopy Unit of the UCSD School of Medicine. Briefly, MDM were pelleted and fixed by immbersion in 2% glutaraldehyde (EMS) diluted in 0.15 M sodium cacodylate buffer at pH 7.4 for at least 4 h. Cells were then postfixed in 1% osmium tetroxide in 0.15 M cacodylate buffer for 60 min and stained in bloc in 2% uranyl acetate (LADD Research Industries, #23620) for 60 min. Samples were dehydrated in ethanol, embedded in Durcupan epoxy resin (Sigma-Aldrich, #D0291), sectioned at 50–60 nm on a Leica UCT ultramicrotome (Leica Microsystems), and picked up on 300 mesh copper grids. Sections were stained with 2% uranyl acetate for 5 min and followed by Sato’s lead stain for 1 min. Grids were viewed and imaged using a JEOL 1400Plus (JEOL) transmission electron microscope coupled with a Gatan OneView digital camera (Gatan).
Analysis of methylcytosine
Analysis of hydroymethylcytosine was performed using ELISA-based assay (https://www.activemotif.com/catalog/1043/global-dna-methylation-assay-line-1?gclid=Cj0KCQjws-OEBhCkARIsAPhOkIbL2w0_w4WfU782biga1nD8eLOTRBGXPDa6h0W0hZQLOEfIQCtGB-kaArX6EALw_wcB#order) using 100 ng of DNA, following manufacture’s specifications. Treatment of MDM with 5-azacytidine was used as positive control.
Single-cell RNA-seq library preparation and analysis of MDM and atherosclerotic macrophages
For normal MDM, MDM from three healthy donors were washed twice with PBS, pelleted, and resuspended in PBS containing 0.04% BSA. Single cells were linked to beads using the 10X Chromium system (10X Genomics). Single-cell sequencing libraries were prepared following manufacturer’s instructions using v3 chemistry. The demultiplexed fastq files produced by cellranger mkfastq were processed by cellranger count (10X genomics) using the human genome reference ‘refdata-cellranger-GRCh38-3.0.0’. For each sample, the filtered_feature_bc_matrix output from cellranger count was analyzed by the Seurat R package, by adapting the code from the Guided Clustering Tutorial (https://satijalab.org/seurat/archive/v3.1/pbmc3k_tutorial.html). Single cell RNA-seq data from atherosclerotic macrophages was provided by KHMP and is available in (Depuydt et al., 2020). The single cell expression data were filtered to include only the genes of interest: DNMT3A, TET2 and genes identified in the REACTOME Interferon alpha and beta signalling gene set. As surrogate of reduced DNMT3A or TET2, we compared cells in the top and bottom quartile of DNMT3A or TET2 expression for MDM or top two quartiles versus bottom two quartiles for macrophages isolated from the atherosclerotic plaque, for the expression of genes in the Reactome Interferon alpha and beta signalling gene set.
RNA-seq library preparation
It was performed as described elsewhere (Gosselin et al., 2017); (Seidman et al., 2020)]. Briefly, MDM were lysed in TRIzol (ThermoFisher, #15596026). RNA and DNase treatment was carried out using Direct-zol RNA MicroPrep kit (Zymoresearch, #11-33MB). 500ng-1 μg total RNA was enriched in poly-A tailed RNA transcripts by double incubation with Oligo d(T) Magnetic Beads (NEB, S1419 S) and fragmented for 9 min at 94°C in 2X Superscript III first-strand buffer containing 10 mM DTT (Invitrogen, #P2325). The 10 μL of fragmented RNA was added to 0.5 μL of Random primers (Invitrogen, #48190011), 0.5 μL of Oligo d(T) primer (Invitrogen, #18418020), 0.5 μL of SUPERase inhibitor (Ambion, #AM2696), 1 μL of 10 mM dNTPs and incubated at 50°C for 3 min. Then, 5.8 μL of water, 1 μL of 10 mM DTT, 0.1 μL of 2 μg/μL Actinomycin D (Sigma, #A1410), 0.2 μL of 1% Tween-20 (Sigma) and 0.5 μL of SuperScript III (Invitrogen, #ThermoFisher 18080044) was added to the mix. Reverse-transcription (RT) reaction was performed at 25°C for 10 min followed by 50°C for 50 min. RT product was purified with RNAClean XP (Beckman Coulter, #A63987) and eluted in 10 μL in 0.01% Tween-20. The RNA-cDNA complex was then added to 1.5 μL of 10X Blue Buffer (Enzymatics, #B0110-L), 1.1 μL of dUTP mix (10 mM dATP, dCTP, dGTP and 20 mM dUTP), 0.2 μL of 5 U/μl RNAseH (Enzymatics, #Y9220L), 1 μL of 10 U/μl DNA polymerase I (Enzymatics, #P7050L), 0.15 μL of 1% Tween 20 and 1.05 μL of nuclease free water; and incubated at 16°C for 2.5 h or overnight. The resulting dsDNA product was purified using 28 μL of SpeedbBead Magnetic Carboxylate (GE Healthcare, #651521050 50250) diluted in 20%PEG8000:2.5M NaCl to a final 13% PEG concentration, washed twice with 80% etOH, air dry and eluted in 40 μL of 0.05% Tween-20. The purified 40 μL of dsDNA was end-repaired by blunting followed by A-tailing and adapter ligation as described elsewhere (Heinz et al., 2010) using BIOO Barcodes (BIOO Scientific, #514104), IDT TruSeq Unique Dual Indexes or Kapa Unique Dual-Indexed Adapters using 15 μL Rapid Ligation Buffer (Enzymatics, #L603-LC-L), 0.33 μL 1% Tween 20 and 0.5 μL T4 DNA ligase HC (Enzymatics, #L6030-HC-L). Libraries were amplified by PCR for 11–15 cycles using Solexa IGA and Solexa IGB primers (AATGATACGGCGACCACCGA and CAAGCAGAAGACGGCATACGA, respectively), purified using 1 μL of SpeedbBead Magnetic Carboxylate in 15.2 μL of 20%PEG8000:2.5M NaCl, washed with 80% etOH and eluted in 0.05% Tween 20. Eluted libraries were quantified using a Qubit dsDNA HS Assay Kit and sequenced on a NextSeq 500 or Hi-Seq 4000 (Illumina, San Diego, California).
Analysis of RNA-seq
FASTQ sequencing files were mapped to the mm10 or hg38 reference genomes for mouse samples or human samples, respectively using STAR with default parameters. Biological and technical replicates were used in all experiments. Quantification of transcripts was performed analyzeRepeats.pl (HOMER) with parameters -condenseGenes -count exons -noadj. Principal Component Analysis (PCA) was obtained based on the Transcripts Per kilobase Million (TPM) on all genes of all samples. Expression value for each transcript was calculated using the analyzeRepeats.pl tool of HOMER with the following parameters -condenseGenes -count exons -tpm. Differential expression analysis was calculated using getDiffExpression.pl tool of HOMER using default parameters (FDR <0.05 and log2fold change >1 or < −1)). For the experiment shown in Figure 5A, Log2 fold change was calculated for CM DNMT3A ASO or CM TET2 ASO as compared to CM Scramble ASO for each individual and the average for the 4 samples was calculated. Pathway analyses were performed using the Molecular Signature Database of GSEA (Subramanian et al., 2005), (Mootha et al., 2003). A spreadsheet containing the expression (TPM) values as well as the differentally expressed genes can be found in GEO (GSE206030).
Motif analysis at the promoter of differentially expressed genes
In silico promoter analysis of differentially expressed genes was performed using the findMotifs.pl tool of Homer searching for motifs of length 8 and 10 and from −2000 to +500 bp relative to the TSS, using 4 threads.
ChIP-seq libraries preparation
For DNMT3A and TET2 ChIP was performed as described previously (Eichenfield et al., 2016; Seidman et al., 2020). Briefly, MDM were fixed with 3 mM Disuccinimidyl-glutarate, DSG, (Proteochem, #C1104) in PBS for 30 min at room temperature followed by 10 min incubation with 1% formaldehyde (ThermoFisher, #28906) at room temperature. Next, 2.625 M Glycine was added to a final concentration of 125 mM to quench fixation. Cells were washed with 0.01%Triton X-100:PBS, scraped and centrifuged for 10 min at 3,000 rpm at 4°C. Cells were washed once again with 0.01%Triton X-100 in PBS, pelleted, snap frozen and stored at −80°C. For ChIP experiments, cells were thawed and permeabilised in 1 mL of ice-cold buffer containing 10 mM HEPES/KOH pH7.9, 85 mM KCl, 1 mM EDTA, 0.2% IGEPAL CA-630 (Sigma Aldrich, #I8896), 1X protease inhibitor cocktail (Sigma, #11836145001) and 1 mM PMSF for 10 min on ice. Cells were then spun down and lysed in 130 μL of lysis buffer containing 20 mM Tris/HCl pH7.5, 1 mM EDTA, 0.5 mM EGTA, 0.1% SDS, 0.4% Sodium Deoxycholate, 1% NP-40, 0.5 mM DTT, 1x protease inhibitor cocktail and 1 mM PMSF and chromatin was sheared by sonication. In all buffers, DTT, protease inhibitor and PMSF were added freshly. Cell lysates were sonicated in a 96 microTUBE Rack (Covaris, #500282) using a Covaris E220 for 25 cycles with the following settings: time, 60 s; duty, 5.0; PIP, 140; cycles, 200; amplitude, 0.0; velocity, 0.0. Sonicated lysates were recovered and spun at 10,000 rpm for 10 min at 4°C to remove cell debris. One percent of sonicated lysate was kept as ChIP input for analysis. Immunoprecipitation mix consisting of Protein G Dynabeads (Invitrogen, #10003D) and DNMT3A antibody cocktail (2 μg of DNMT3A, #ab2850 plus 2 μg of DNMT3A, #C15410085), 5 μL of TET2 antibody (kindly provided by Ali Shilatifard, (Wang et al., 2018), 1.5 μL of RBPJ (H00007702-M01) or 2 μL of RBPJ (#ab25949) was added to sonicated chromatin solution and incubated overnight on rotator at 4°C. Next day, immunocomplexes were placed on a magnet and bead complexes were washed for 1 min with 150 μL of cold buffers as indicated: 3 times lysis buffer, 6 times with wash buffer containing 10 mM Tris/HCl pH7.5, 250 mM LiCl, 1 mM EDTA, 0.7% Na-Deoxycholate and 1% NP-40 alternative; 3 times with TET buffer containing 10 mM Tris/HCl pH 8.0, 1 mM EDTA, 0.2% Tween 20; and 1 time with IDTE buffer containing 10 mM Tris/HCl pH 8.0 and 0.1 mM EDTA. Bead complexes were resuspended in 25 μL of TT buffer containing 10 mM Tris/HCl pH 8.0, 0.05% Tween 20. All wash buffer contained 1X Protease Inhibitor cocktail. ChIP libraries for sequencing were prepared while remained on beads using NEBNext Ultra II Library kit (NEB, #E7645L) by reducing 50% the reaction volume as previously described (Seidman et al., 2020; Heinz et al., 2018). Crosslinks were revered by adding 33.5 μL of mix containing 18.4 μL nuclease free water, 4 μL 10% SDS, 3 μL 0.5 M EDTA, 1.6 μL 0.2M EGTA, 1 μL 10 mg/ml Proteinase K (Biolabs, #P8107 S), 1 μL 10 mg/ml RNase A, 4.5 μL 5M NaCl by incubating at 55°C for 1 h followed by 75°C for 30 min. Dynabeads were removed and libraries were cleaned by adding 2 μL of SpeadBeads in 124 μL of 20% PEG 8000/1.5M NaCl, washed by adding 150 μL 80% etOH, air dry and eluted in 12.5 μL of buffer containing 10 mM Tris/HCl pH 8.0 and 0.05% Tween 20. DNA was PCR-amplified for 14 cycles using NEBNext Ultra II PCR master mix using Solexa 1GA and Solexa 1GB primers. Libraries were size selected 200–500 bp by running in 10% TBE acrylamide gels (ThermoFisher, #EC62752BOX) and sequenced using using either a HiSeq 4000 or a NextSeq 500.
ChIP-seq analysis
ChIP-seq analysis as described somewhere else (Seidman et al., 2020). Briefly, peaks for replicates with corresponding input experiments were obtained using HOMER with the parameters –L 0 –C 0 –fdr 0.9 –minDist 200 –size 200. Only peaks with IDR <0.05 were used for Downstream analysis (Li et al., 2011). The pooled tag directories from all replicates were used for visualization. HOMER mergePeaks to identify overlapping peaks and peaks were marked as DNMT3A unique, DNMT3A and TET2 co-binding, and TET2 unique groups.
HOMER annotatePeaks.pl tool was used to count the number of DNMT3A or TET2 tags in DNMT3ATET2 co-bound peaks. A list of the IDR peaks for DNTM3A, TET2, RBPJ and ZNF143 ChIP-seq can be found in GEO (GSE206030).
Locus-specific ChIP (ChIP-qPCR)
IRF7, IRF1 and phospho-IRF3 (p-IRF3) ChIP was performed as described for DNMT3A or TET2 ChIP. Briefly, cells were permeabilised, lysed chromatin was sheared by sonication using. For H3K4me3, H3K27ac or RNApolII, MDM were fixed with 1% formaldehyde for 15 min at room temperature and fixation was quenched using glycine to a final concentration of 125 mM. MDM were collected by centrifugation after adding Tween 20 to a final concentration of 0.01%. After washing in 0.01%Tween 20 in PBS, MDM pellets were snap frozen or lysed in LB3 buffer containing (10 mM Tris/HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% deoxcycholate, 0.5% sarkosyl, 1 3 protease inhibitor cocktail, and 1 mM sodium butyrate). Lysates were sonicated using a Covaris E-220 for 12 cycles with the following setting: time, 60 s; duty, 5.0; PIP, 140; cycles, 200; amplitude, 0.0; velocity, 0.0; dwell, 0.0. Sonicated chromatin was incubated with XX. ChIP was performed at 4°C with 1 μg of desired antibodies: IRF1 (Abcam, #191032), IRF7 (IRF7. Abcam, #115352), p-IRF3 (Abcam, #76493), H3K27ac (Active Motifs, #39133), H3K4me3 (Millipore, #04-745) or RNApolII (GeneTex, #GTX102535). One percent of sonicated chromatin was kept as ChIP input. Next day, beads were washed, crosslink were reversed, and DNA was purified by adding 2 μL of SpeadBeads in 124 μL of 20% PEG 8000/1.5M NaCl, washed by adding 150 μL 80% etOH, air dry and eluted in 20 μL of buffer containing 10 mM Tris/HCl pH 8.0 and 0.05% Tween-20. Enrichment for IRF1, IRF7, p-IRF3, H3K4me3, H3K27ac or RNApolIIwas normalised to input and to the negative control region (AMY2B). The sequence of the primers used, and their genomic location is provided below.
| Gene promoter | Forward (5′- 3′) | Reverse (5′- 3′) |
|---|---|---|
| AMY2B #1 | tcagcactggattgtagaacttg | aagccacatgtactaaagactgaaa |
| AMY2B #2 | aacctcaaggccaacagaga | tgcaaccacaggtgtagagg |
| CXCL10 #1 | tggattgcaacctttgttttt | tttccctctgctcctcttttt |
| CXCL10 #2 | gccacgattcatcatccagt | gggttcagctccaagacact |
| IFIT2 #1 | ctccggaggaaaaagagtcc | tcccttcagctgacgttaca |
| IFIT2 #2 | aagagcattttggggtgaaa | accccacttcctgctaaggt |
| ISG15 #1 | gtttcttccgctcactctgg | ataagcctgaggcacacacg |
| ISG15 #2 | catggggatgttttccaagt | taaatatcgcgcattccaga |
| MX1 #1 | ccgagaacctgcgtctcc | actcacagaccctgtgctga |
| MX1 #2 | ggcctggcctgacaactat | aacgggtgtgtggaagga |
| OASL #1 | ctgggcaacagagcaagac | cctgtgccagactctaacctc |
| OASL #2 | ctgggcgagacagtgagact | gtgatttcagccagcgttta |
| TFAM proximal | cgaccggatgttagcagatt | ggcaatacacaactccagca |
| TFAM distal | tggaaggtccctttcaacc | tgaaaacattggctctctgg |
In situ Hi-C Library preparation
In situ Hi-C was performed on 1 million formaldehyde-fixed MDM per each condition (Scramble ASO, DNMT3A ASO #1 and TET2 ASO #2) as described previously (Heinz et al., 2018). Briefly, nuclei were isolated by resuspending the fixed, snap frozen cell pellet in 200 μL Wash Buffer (50 mM Tris/HCl pH 7.5, 10 mM NaCl, 1 mM EDTA, 0.5% SDS, 1x protease inhibitor cocktail (SIGMA)). Nuclei were incubated at 37°C for 60 min and then spun down at 1000xg for 5 min at room temperature. Most of the supernatant was discarded, except for 10 μL of liquid with the nuclei that was resuspended in DpnII buffer (25 μL 10% TrixonX-100, 25 μL of 10x DpnII buffer (NEB), 188 μL water) and rotated for 15 min at 37°C. Chromatin was then digested overnight with 2 μL (100 U) DpnII (NEB) at 37°C, rotating end over end at 8 RPM.The next day, nuclei were spun down for 5 min, 1000xg and then 225 μL of the supernatant was discarded, leaving ~25 μL of liquid remaining with the nuclei pellet. Overhangs were filled in with Biotin-14-dATP (Thermo) by adding 75 μL of Klenow master mix (54.45 μL water, 7.5 μL NEBuffer 2 (NEB), 0.35 μL dCTP, 0.35 μL dTTP, 0.35 μL dGTP, 7.5 μL 0.4 mM Biotin-14-dATP, 2 μL 10% Triton X-100, 2.4 μL (12.5 U) Klenow Fragment (Enzymatics, #7060L)) and rotating end over end at room temperature for 40 min. Proximity ligation was performed by transferring the reaction into an Eppendorf tube and adding ligation master mix (322.75 μL water, 40 μL 10x T4 ligase buffer (New England, #N0202), 36 μL 10% TritonX-100, 20% 1000x BSA, and HC T4 DNA ligase (Enzymatics)). Samples were incubated at 16°C overnight rotating end over end. The following day, reactions were stopped by adding 20 μL 0.5M EDTA plus 1 μL 10 μg/μL RNAse A at 42 °C for 15 min. To reverse crosslinks and digest proteins, 31 μL L 5M NaCl, 29 μL of 10% SDS, and 5 μL 20 mg/mL proteinase K were added to each sample. Samples were incubated at 55°C for 1 h, then at 65°C overnight. The following day, DNA was extracted using 600 μL pH 8-buffered phenol/chloroform/isoamyl alcohol (Invitrogen), followed by 550 μL chloroform. DNA was then precipitated with 1.5 μL (15 mg/mL) Glycoblue (Thermo) and 1400 μL 100% ethanol overnight at −20°C, pelleted for 20 min at 160000xg, 4°C and washed 2x with 1 mL 80% EtOH. The pellet was air-dried and dissolved in 131 μL TT buffer (0.05% Tween 20/10 mM Tris pH 8). DNA was then sheared to ~300 bp average size in 130 μL TT on a Covaris E-220 for 120 s, duty cycle 5, PIP 175, and cycles per burst 200. Biotinylated DNA was incubated with 20 μL DynaBeads MyOne Streptavidin T1 beads that had been washed 1x with B&W buffer (2x B&W: 10 mM Tris HCl pH 7.5, 1 mM EDTA, 2M NaCl) and resuspended in 130 μL 2x B&W buffer with 0.2% Tween 20. The binding reaction was incubated for 45 min at room temperature, rotating end over end. The beads were washed 2x with 150 μL 1x B&W plus 0.1% Triton X-100, 1x with 180 μL TET (TE + 0.05% Tween 20) and resuspended in 30 μL ice-cold NebNext Ultra II end prep mix (1.5 μL NebNext Ultra II EndPrep Enzyme, 3.5 μL EndPrep Buffer, 25 μL TT buffer) and incubated 20°C for 30 min followed by 65°C for 30 min. Beads were resuspended in ligation master mix (15 μL NebNext Ultra II ligation master mix, 0.5 μL ligation enhancer) and 1 μL of BIOO Nextflex DNA sequencing adapters were added. The mixture was incubated at 20°C for 20 min and the reaction was stopped using 5 μL 0.5M EDTA. Following this, the beads were washed twice in 1x B&W with 0.1% Triton X-100, twice with TET, and resuspended in 20 μL TT buffer. Libraries were amplified by PCR for 10 cycles (98°C, 30 s; [98°C, 10 s; 63°C, 25 s; 72°C, 30 s]; 72°C 5 min, 4°C hold) using 10 μL of the bead resuspension in a 50 μL reaction with NEBNext Ultra II Q5 mastermix (NEB), 0.5 uM each Solexa 1GA/1GB primers (Solexa 1GA: AATGATACGGCGACCACCGA, Solexa 1GB: CAAGCAGAAGACGGCATACGA). Libraries were precipitated onto magnetic beads by adding 2 μL of Speedbeads, 40 μL 20% PEG/2.5M NaCl and incubating for 15 min at room temperature. The beads were washed 2x with 180 μL 80% EtOH and air dried. Samples were eluted by adding 20 μL TT buffer per sample. Libraries were sequenced to a depth of approximately 100 million reads per experiment on Illumina NovaSeq S4 300. One pool of three donors were used for Hi-C experiment.
Hi-C data mapping, analysis, and visualization
Hi-C fastq files were trimmed at DpnII recognition sites (GATC) and aligned to the human genome (hg38) separately using bowtie 2 (Langmead and Salzberg, 2012). Following trimming, R1 and R2 for each sample were mapped separately to the hg38 reference genome using Bowtie2. After mapping, HOMER Hi-C tag directories were created using the HOMER command makeTagDirectory (Heinz et al., 2010). Hi-C interaction matrices were generated using juicertools (Durand et al., 2016) and were visualized using juicebox (Durand et al., 2016). PC1 values for each sample were calculated using HOMER’s runHiCpca.pl with -res 50000 and were visualized using the UCSC genome browser (Kent et al., 2002). TADs and loops were called using HOMER’s findTADsAndLoops.pl find with parameters -res 3000 and -window 15000. To compare TADs and loops between groups, TADs and loops were merged using merge2Dbed.pl -tad and -loop, respectively. Differential enrichment of these features was then calculated using Homer’s getDiffExpression.pl.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons of quantitative data between groups was carried out using two-tailed Mann-Whitney U test in all cases unless otherwise indicated. Box pots illustrate the median, Q1 and Q3 quartile of the data. Error bars in box plots represent the lowest and highest data point within 1.5x Q1-Q3 range. Bar graphs represent the media. Error bars in bar graphs represent the Standard error of the mean. All plots were generated using Numbers (iWORK’09) or R studio (version 2.15.2 [2012-10-26]). Mann Whitney U-test was used to calculate statistical significance. *p < 0.05; **p < 0.01. Bar graphs represent the mean and error bars represents the standar error of the mean. Box plots represent the median and first and third quartiles of the data; error bars are generated by R software and represent the highest and lowest data within 1.5× interquartile range.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Active Motif | 39133; RRID: AB_2561016 |
| H3K4me3 | Millipore-Sigma | 04-745; RRID:AB_1163444 |
| DNMT3A | Santa Cruz | sc-373905; RRID:AB_10920404 |
| DNTM3A | Abcam | ab2850; RRID:AB_303355 |
| DNMT3A | Diagenode | C15410085; RRID:AB_2916119 |
| CTCF | CST | 2899S; RRID:AB_2086794 |
| DNA | Abcam | ab27156; RRID:AB_470907 |
| HSP60 | CST | 12165S; RRID:AB_2636980 |
| IRF1 | Abcam | ab191032; RRID:AB_2904575 |
| IRF7 | Abcam | ab115352; RRID:AB_10862356 |
| KI-67 | Invitrogen | 53-5698-82; RRID:AB_2802330 |
| TET2 | Abcam | ab94580; RRID:AB_10887588 |
| Phalloidin | Abcam | ab176759; RRID:N/A |
| TFAM | CST | 8076S; RRID:AB_10949110 |
| Donkey anti mouse | ThermoFisher | A21202; RRID:AB_141607 |
| Donkey anti rabbit | ThermoFisher | A31572; RRID:AB_162543 |
| Apotracker | Biolegend | 427401; RRID:N/A |
| CellEvent | Thermo Fisher | C10723; RRID:N/A |
| p-TBK1 | CST | D52C2; RRID:AB_10693472 |
| TBK1 | CST | D1B4; RRID:AB_2255663 |
| p-IRF3 | CST | D6O1M; RRID:AB_2773013 |
| IRF3 | CST | D83B9; RRID:AB_1904036 |
| IFIT2 | Boster | A04428; RRID:AB_2916120 |
| Actin | Sigma | A5441; RRID:AB_476744 |
| TET2 | Provided by Ali Shilatifard | (Wang et al., 2018) |
| Anti rabbit HRP | Dako | P0448; RRID:AB_2617138 |
| Anti mouse HRP | Dako | P0447; RRID:AB_2617137 |
| TrueStain FcX | Biolegend | 422302; RRID:AB_2818986 |
| RNApol II | Gene Tex | GTX102535; RRID:AB_1951313 |
| ZNF143 | Novus Bio | H00007702-M01; RRID:AB_425746 |
| RBPJ | Abcam | ab25949; RRID:AB_778155 |
| Chemicals, peptides, and recombinant proteins | ||
| KAPA SYBR FAST qPCR Master mix (2X) | Kapa Biosystems | Cat#07959427001 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat#10002D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat#10004D |
| SpeedBeads magnetic carboxylate modified particles | GE Healthcare | Cat#65152105050250 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596018 |
| Formaldehyde | Thermo Fisher Scientific | Cat#BP531-500 |
| Disuccinimidyl glutarate | ProteoChem | Cat#c1104-100mg |
| Oligo d(T)25 Magnetic Beads | NEB | Cat#S1419S |
| DTT | Thermo Fisher Scientific | Cat#P2325 |
| SUPERase-In | Ambion | Cat#AM2696 |
| Oligo dT primer | Thermo Fisher Scientific | Cat#18418020 |
| Agencourt RNA Clean XP Beads | Beckman Coulter | Cat#A63987 |
| 10 X Blue Buffer | Enzymatics | Cat#P7050L |
| DNA polymerase I | Enzymatics | Cat#P7050L |
| Random primers | Thermo Fisher Scientific | Cat#48190011 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080044 |
| 5 X first-strand buffer | Thermo Fisher Scientific | Cat#18080044 |
| Actinomycin D | Sigma | Cat#A1410 |
| Proteinase inhibitor cocktail | Sigma-Aldrich | Cat#P8340 |
| PVDF membranes | Immobilon | Cat#IPVH00010 |
| ProLong anti fade | Life Technologies | Cat#10144 |
| DharmaFECT reagents | Horizon | Cat#T-2004-02 |
| IgG control | R&D systems | Cat#MAB003 |
| anti-Interferon Alpha/Beta Receptor Chain 2 (IFNαR2) | Pbl assay science | Cat#21385-1, Lot 7035 |
| IFNα2 | Stem Cell Technology | Cat#78076 |
| 2′,3′-dideoxycytidine (ddC) | Provided by Prof. Gerald Shadel | |
| G-140 | Invivogen | Cat#inh-g140 |
| Chambered slides | Millipore | Cat#C86024 |
| Digitonin | Promega | Cat#G9441 |
| Durcupan epoxy resin | Sigma | Cat# D0291 |
| Sodium pyruvate | Gibco | Cat#11360070 |
| Pen/Strep | Gibco | Cat#15140122 |
| Human MCSF | Stem Cell Technology | Cat#78057-2 |
| Ficoll plaque premium | GE Healthcare | Cat#17-544-02 |
| BD Vacutainer CPT tubes | BD | Cat#362753 |
| Critical commercial assays | ||
| Direct-zol RNA MicroPrep Kit | Zymo Research | Cat#R2062 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#554714 |
| NEBNext Ultra II Library Preparation Kit | NEB | Cat#E7645L |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat#Q32851 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18080051 |
| Chromium Single Cell 3′ Library Kit v2 | 10X Genomics | Cat#120234 |
| Chromium Single Cell 3′ Gel Bead Kit v2 | 10X Genomics | Cat#120235 |
| Chromium Single Cell A Chip Kit v2 | 10X Genomics | Cat#120236 |
| Chromium i7 Multiplex Kit | 10X Genomics | Cat#120262 |
| Global DNA Methylation Assay–LINE-1 | Active Motifs | Cat#55017 |
| ChIP DNA clean & concentrator | Zymogen | Cat #D5205 |
| IFNα ELISA | Pbl assay science | Cat#41135-1 |
| Human monocyte isolation kit | Stem Cell Technology | Cat#19359 |
| Deposited data | ||
| Raw and analysed data | GEO | GEO: GSE206030 |
| Oligonucleotides | ||
| Scamble ASO | IONIS Pharmaceuticals | N/A |
| DNMT3A ASO#1 | IONIS Pharmaceuticals | N/A |
| DNMT3A ASO#2 | IONIS Pharmaceuticals | N/A |
| DNMT3A ASO#3 | IONIS Pharmaceuticals | N/A |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| DNMT3A ASO#4 | IONIS Pharmaceuticals | N/A |
| TET2 ASO#1 | IONIS Pharmaceuticals | N/A |
| TET2 ASO#2 | IONIS Pharmaceuticals | N/A |
| TET2 ASO#3 | IONIS Pharmaceuticals | N/A |
| TET2 ASO#4 | IONIS Pharmaceuticals | N/A |
| siControl | IDT | Cat#51-01-19-09 |
| siDNMT3A#1 | IDT | hs.Ri.DNMT3A.13.1 |
| siDNMT3A#2 | IDT | hs.Ri.DNMT3A.13.2 |
| siTET2#1 | IDT | hs.Ri.TET2.13.1 |
| siTET2#2 | IDT | hs.Ri.TET2.13.2 |
| siTFAM#1 | IDT | Provided by Gerald Shadel |
| siTFAM#2 | IDT | Provided by Gerald Shadel |
| siRBPJ#1 | IDT | hs.Ri.RBPJ.13.1 |
| siRBPJ#2 | IDT | hs.Ri.RBPJ.13.2 |
| siZNF143#1 | IDT | hs.Ri.ZNF143.13.1 |
| siZNF143#2 | IDT | hs.Ri.ZNF143.13.2 |
| Software and algorithms | ||
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| Cell Ranger | https://github.com/10XGenomics/cellranger | |
| FlowJo | https://www.flowjo.com/ | |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| Irreproducibility Discovery Rate (IDR) | (Li et al., 2011) | https://www.encodeproject.org/software/idr/ |
| R package: DeSeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Rstudio | Rstudio | https://www.rstudio.com/ |
| Seurat | https://satijalab.org/seurat/archive/v3.1/pbmc3k_tutorial.html | |
Highlights.
Loss of function of DNMT3A or TET2 in human macrophages induces type I interferons
The type I interferon response is due to mitochondrial DNA release that activates cGAS
DNMT3A and TET2 maintain mitochondrial DNA integrity by regulating TFAM expression
DNMT3A and TET2 regulate TFAM expression by interacting with RBPJ and ZNF143
ACKNOWLEDGMENTS
This work was supported by a Leducq Transatlantic Network Grant 16CVD01(C.K.G., M.d.W.), NIH P01 HL147835 (C.K.G.), EMBO ALTF 960-2018 (IC), R0 AR069876, and the Audrey Geisel Chair in Biomedical Sciences (G.S.S.), Salkexcellerators Postdoctoral Fellowship (K.C.M.), ZonMW 09120011910025 (M.d.W.), the National Headache Foundation (GENIUSII and 2019B016 to M.d.W.), NS047101 (Confocal Microscopy Core), 1KL2TR001444 (T.N.T.).We acknowledge P.M., B.F., C.N., M.H., S.D., Y.A., G.S., J.S., T.P., H.B., J.S., donors from the San Diego Blood Bank, and from the Normal Blood Program of Scripps for having provided blood. We also acknowledge Jana Collier and Martina Pasillas for technical assistance; Ali Shilatifard for providing the TET2 antibody used for ChIP-seq; the electron microscopy core at the School of Medicine at the Univeristy of California, San Diego; the Microscopy Unit Core at the School of Medicine; Laura Antonucci, Elsa Lopez (Michael Karin’s Laboratory, UCSD) for help in doing confocal imaging of MDM; and to LVA for essential drafting of figures.
Footnotes
DECLARATION OF INTERESTS
C.K.G. is a cofounder and member of the scientific advisory board of Asteroid Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2022.06.022.
REFERENCES
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol 88, 5328–5341. 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney L, Veltman JA, Gilissen C, and Hoischen A (2017). Ultra-sensitive sequencing identifies high prevalence of clonal hematopoiesis-associated mutations throughout adult life. Am. J. Hum. Genet 101, 50–64. 10.1016/j.ajhg.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, Rasper T, Abou-El-Ardat K, Herrmann E, Berkowitsch A, et al. (2021). Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur. Heart J 42, 257–265. 10.1093/eurheartj/ehaa845. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, and Orekhov AN (2016). Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. BioMed Res. Int 2016, 9582430. 10.1155/2016/9582430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lépine G, Mollica L, Szuber N, Dubé MP, and Busque L (2017). DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762. 10.1182/blood-2017-04-777029. [DOI] [PubMed] [Google Scholar]
- Castel D, Mourikis P, Bartels SJJ, Brinkman AB, Tajbakhsh S, and Stunnenberg HG (2013). Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071. 10.1101/gad.211912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44, 23–31. 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986. 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo I, Martinelli P, Flández M, Bakiri L, Zhang M, Carrillo-de-Santa-Pau E, Jia J, Sánchez-Arévalo Lobo VJ, Megías D, Felipe I, et al. (2018). Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 554, 533–537. 10.1038/nature25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo I, Tanaka T, Glass CK, and Yeang C (2021). Clonal hematopoiesis driven by DNMT3A and TET2 mutations: role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr. Opin. Hematol 29, 1–7. 10.1097/MOH.0000000000000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole CB, Russler-Germain DA, Ketkar S, Verdoni AM, Smith AM, Bangert CV, Helton NM, Guo M, Klco JM, O’Laughlin S, et al. (2017). Haploinsufficiency for DNA methyltransferase 3A predisposes hematopoietic cells to myeloid malignancies. J. Clin. Invest 127, 3657–3674. 10.1172/JCI93041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST (2007). Antisense Drug Technology: Principles, Strategies and Applications, 2nd Edition (CRC press; ). [Google Scholar]
- Cull AH, Snetsinger B, Buckstein R, Wells RA, and Rauh MJ (2017). Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol 55, 56–70.e13. 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- D’Agostino RB Sr., Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, and Kannel WB (2008). General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation 117, 743–753. 10.1161/CIRCULATI0NAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- Depuydt MAC, Prange KHM, Slenders L, Örd T, Elbersen D, Boltjes A, de Jager SCA, Asselbergs FW, de Borst GJ, Aavik E, et al. (2020). Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res 127, 1437–1455. 10.1161/CIRCRESAHA.120.316770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brune B, Wagner S, Serve H, et al. (2019). Association of mutations contributing to clonal hematopoiesis with prognosis in chronic Ischemic heart failure. JAMA Cardiol. 4, 25–33. 10.1001/jamacardio.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenfield DZ, Troutman TD, Link VM, Lam MT, Cho H, Gosselin D, Spann NJ, Lesch HP, Tao J, Muto J, Gallo RL, Evans RM, Glass CK. Tissue damage drives co-localization of NF-κB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. Elife. 2016. Jul 27;5:e13024. doi: 10.7554/eLife.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández LC, Torres M, and Real FX (2016). Somatic mosaicism: on the road to cancer. Nat. Rev. Cancer 16, 43–55. 10.1038/nrc.2015.1. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, and Fabbri E (2018). Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol 15, 505–522. 10.1038/s41569-018-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldi J, Chung AY, Xu H, Zhu J, Outtz HH, Kitajewski J, Li Y, Hu X, and Ivashkiv LB (2010). Autoamplification of Notch signaling in macrophages by TLR-induced and RBP-J-dependent induction of Jagged1. J. Immunol 185, 5023–5031. 10.4049/jimmunol.1001544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg LA, Gisselsson D, and Dumanski JP (2017). Mosaicism in health and disease - clones picking up speed. Nat. Rev. Genet 18,128–142. 10.1038/nrg.2016.145. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, Ferrer-Pérez A, Matesanz N, Herrero-Cervera A, Sano S, et al. (2020). TET2-Loss-of-Function-Driven clonal hematopoiesis exacerbates experimental insulin resistance in aging and obesity. Cell Rep. 33. 10.1016/j.celrep.2020.108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghattas A, Griffiths H Devitt A, Lip GYH, and Shantsila E (2013). Monocytes in coronary artery disease and atherosclerosis: where are we now? J. Am. Coll. Cardiol 62, 1541–1551. [DOI] [PubMed] [Google Scholar]
- Goossens P, Gijbels MJJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, Vanderlocht J, Beckers L, Buurman WA, Daemen MJAP, et al. (2010). Myeloid Type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 12, 142–153. 10.1016/j.cmet.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222. 10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage--determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, Marazzi I, García-Sastre A, Shaw ML, Benner C. Transcription Elongation Can Affect Genome 3D Structure. Cell. 2018. Sep 6;174(6):1522–1536.e22. doi: 10.1016/j.cell.2018.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden P, and Horton WA (2009). Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2, 243. 10.1186/1756-0500-2-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Takaoka A, and Taniguchi T (2006). Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Ito K, Lee J, Chrysanthou S, Zhao Y, Josephs K, Sato H, Teruya-Feldstein J, Zheng D, Dawlaty MM, and Ito K (2019). Non-catalytic roles of Tet2 are essential to regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep. 28, 2480–2490.e4. 10.1016/j.celrep.2019.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi H, Wakasugi T, Shimajiri S, Tanimoto A, Sasaguri Y, Kashiwagi E, Yasuniwa Y, Akiyama M, Han B, Wu Y, et al. (2010). Role of ZNF143 in tumor growth through transcriptional regulation of DNA replication and cell-cycle-associated genes. Cancer Sci. 707, 2538–2545. 10.1111/j.1349-7006.2010.01725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498. 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med 377, 111–121. 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB, and Vasan RS (2009). Is age really a non-modifiable cardiovascular risk factor? Am. J. Cardiol 104, 1307–1310. 10.1016/j.amjcard.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawatsu Y, Kitada S, Uramoto H, Zhi L, Takeda T, Kimura T, Horie S, Tanaka F, Sasaguri Y, Izumi H, et al. (2014). The combination of strong expression of ZNF143 and high MIB-1 labelling index independently predicts shorter disease-specific survival in lung adenocarcinoma. Br. J. Cancer 110, 2583–2592. 10.1038/bjc.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Slabicki M, Kemp JP, Bick A, Gibson CJ, Griffin G, et al. (2021). Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J. Exp. Med 218, e20211872. 10.1084/jem.20211872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, and Rao A (2011). Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 108, 14566–14571. 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, Gogakos T, Steinberg JI, Miller M, Ramos-Espiritu L, et al. (2019). Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun 10, 2261. 10.1038/s41467-019-08620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C, and Monticelli S (2017). Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 114, E1490–E1499. 10.1073/pnas.1616420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. (2010). DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med 363, 2424–2433. 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, Shen Q, Liu X, Zhu X, Li N, et al. (2016). Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol 17, 806–815. 10.1038/ni.3464. [DOI] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, and Xu M (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518. 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JY, Duttke SH, Baker TS, Lee J, Gambino KJ, Venturini NJ, Ho JSY, Zheng S, Fstkchyan YS, Pillai V, et al. (2021). DNMT3A haploinsufficiency causes dichotomous DNA methylation defects at enhancers in mature human immune cells. J. Exp. Med 218, e20202733. 10.1084/jem.20202733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15 (12), 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mba Medie F, Sharma-Kuinkel BK, Ruffin F, Chan LC, Rossetti M, Chang YL, Park LP, Bayer AS, Filler SG, Ahn R, et al. (2019). Genetic variation of DNA methyltransferase-3A contributes to protection against persistent MRSA bacteremia in patients. Proc. Natl. Acad. Sci. USA 116, 20087–20096. 10.1073/pnas.1909849116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, Understanding Society Scientific Group, Crawley C, Craig J, Scott MA, et al. (2015). Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 10, 1239–1245. 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, and Fisher EA (2013). Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol 13, 709–721. 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24. 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman LE, and Shadel GS (2018). Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J. Cell Biol 217, 3327–3329. 10.1083/jcb.201808118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North BJ, and Sinclair DA (2012). The intersection between aging and cardiovascular disease. Circ. Res 110, 1097–1108. 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander EL, Kramer AC, Mallaney C, Celik H, Koh WK, Fairchild J, Haussler E, Zhang CRC, and Challen GA (2020). Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Rep. 14, 551–560. 10.1016/j.stemcr.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Kawai T, and Akira S (2014). Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol 7, a016246. 10.1101/cshperspect.a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D (2008). The enhanceosome. Curr. Opin. Struct. Biol 18, 236–242. 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Pham D, Yu Q, Walline CC, Muthukrishnan R, Blum JS, and Kaplan MH (2013). Opposing roles of STAT4 and Dnmt3a in Th1 gene regulation. J. Immunol 191, 902–911. 10.4049/jimmunol.1203229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski P, Grobelna MK, Wudarski M, Olesrńska M, and Jagodziński PP (2015). Genetic variants of DNMT3A and systemic lupus erythematosus susceptibility. Mod. Rheumatol 25, 96–99. 10.3109/14397595.2014.902296. [DOI] [PubMed] [Google Scholar]
- Rodgers JL, Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, Karia K, and Panguluri SK (2019). Cardiovascular risks associated with gender and aging. J. Cardiovasc. Dev. Dis 6, E19. 10.3390/jcdd6020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, and Hertzog PJ (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res (Jan;41(Database issue):D1040–6.). 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, Ego KM, Bruni CM, Deng Z, Schlachetzki JCM, et al. (2019). Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity 51, 655–670.e8. 10.1016/j.immuni.2019.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2008).Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev 88, 611–638. 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, and Rice CM (2011. Dec). Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol 1 (6), 519–525. 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. (2020). Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 52, 1057–1074.e7. 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniderman AD, and Furberg CD (2008). Age as a modifiable risk factor for cardiovascular disease. Lancet 371, 1547–1549. 10.1016/S0140-6736(08)60313-X. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepańska M, Mostowska A, Wirstlein P, Malejczyk J, Płoski R, Skrzypczak J, and Jagodziński PP (2013). Polymorphic variants of DNMT3A and the risk of endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol 166, 81–85. 10.1016/j.ejogrb.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Tabas I, and Lichtman AH (2017). Monocyte-Macrophages and T Cells in atherosclerosis. Immunity 47, 621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J (2014). Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev 26, 141–149. 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, van Eijck CHJ, van Koetsveld Ing PM, Erdmann JI, Speel EJM, van der Wansem Ing K, Mooij DM, Colao A, Lombardi G, Croze E, et al. (2007). Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann. Surg 246, 259–268. 10.1097/01.sla.0000261460.07110.f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan D, Jiang W, and Hao J (2020). Research advances in howthe cGAS-STING pathway controls the cellular inflammatory response. Front. Immunol 11, 615. 10.3389/fimmu.2020.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, et al. (2011). Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 108, 14908–14913. 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ozark PA, Smith ER, Zhao Z, Marshall SA, Rendleman EJ, Piunti A, Ryan C, Whelan AL, Helmin KA, et al. (2018). TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv 4, eaau6986. 10.1126/sciadv.aau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278. 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, and Shadel GS (2017). Mitochondrial DNAin innate immune responses and inflammatory pathology. Nat. Rev. Immunol 17, 363–375. 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478. 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhu J, Smith S, Foldi J, Zhao B, Chung AY, Outtz H, Kitajewski J, Shi C, Weber S, et al. (2012). Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol 13, 642–650. 10.1038/ni.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanyar A, and Newman AB (2009). The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin. Geriatr. Med 25, 563–577. vii. 10.1016/j.cger.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B, Yang G, Li Y, Zhang C, Wang Q, and Yu G (2020). ZNF143 in chromatin looping and gene regulation. Front. Genet 11, 338. 10.3389/fgene.2020.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, Brown MR, Griffin G, Desai P, Correa A, et al. (2021). Supplemental association of clonal hematopoiesis with incident heart failure. J. Am. Coll. Cardiol 78, 42–52. 10.1016/j.jacc.2021.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XH, Fu YC, Zhang DW, Yin K, and Tang CK (2013). Foam cells in atherosclerosis. Clin. Chim. Acta 424, 245–252. 10.1016/j.cca.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Yu M, Tirado-Magallanes R, Li B, Kong L, Guo M, Tan ZH, Lee S, Chai L, Numata A, et al. (2021). ZNF143 mediates CTCF-bound promoter-enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat. Commun 12, 43. 10.1038/s41467-020-20282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752. 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have been deposited in GEO (GSE206030). This study did not generate any unique code.