

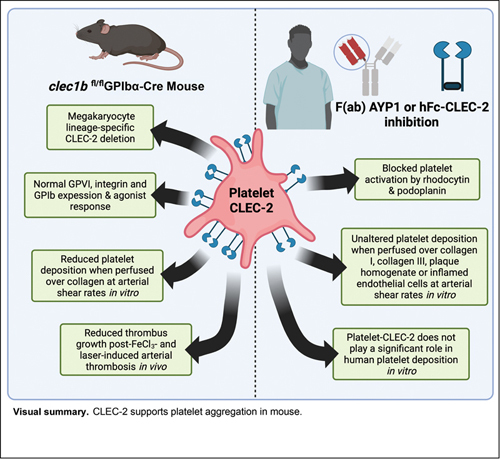
Abstract
C-type lectin-like receptor 2 (CLEC-2) is highly expressed on platelets and a subpopulation of myeloid cells, and is critical in lymphatic development. CLEC-2 has been shown to support thrombus formation at sites of inflammation, but to have a minor/negligible role in hemostasis. This identifies CLEC-2 as a promising therapeutic target in thromboinflammatory disorders, without hemostatic detriment. We utilized a GPIbα-Cre recombinase mouse for more restricted deletion of platelet-CLEC-2 than the previously used PF4-Cre mouse. clec1b fl/fl GPIbα-Cre + mice are born at a Mendelian ratio, with a mild reduction in platelet count, and present with reduced thrombus size post-FeCl 3 -induced thrombosis, compared to littermates. Antibody-mediated depletion of platelet count in C57BL/6 mice, to match clec1b fl/fl GPIbα-Cre + mice, revealed that the reduced thrombus size post-FeCl 3 -injury was due to the loss of CLEC-2, and not mild thrombocytopenia. Similarly, clec1b fl/fl GPIbα-Cre + mouse blood replenished with CLEC-2-deficient platelets ex vivo to match littermates had reduced aggregate formation when perfused over collagen at arterial flow rates. In contrast, platelet-rich thrombi formed following perfusion of human blood under flow conditions over collagen types I or III, atherosclerotic plaque, or inflammatory endothelial cells were unaltered in the presence of CLEC-2-blocking antibody, AYP1, or recombinant CLEC-2-Fc. The reduction in platelet aggregation observed in clec1b fl/fl GPIbα-Cre + mice during arterial thrombosis is mediated by the loss of CLEC-2 on mouse platelets. In contrast, CLEC-2 does not support thrombus generation on collagen, atherosclerotic plaque, or inflamed endothelial cells in human at arterial shear.
Keywords: CLEC-2, thrombosis, platelets, AYP1, thromboinflammation
Introduction
Platelets critically limit blood loss post-injury. Conversely, pathological vascular occlusion due to thrombosis in the arterial or venous systems can cause disorders such as acute coronary syndrome, ischemic stroke, and deep vein thrombosis (DVT). Current antiplatelet therapeutics, such as aspirin and platelet P2Y 12 inhibitors, are effective in limiting arterial thrombosis, but are detrimental to hemostasis. Platelet immune receptors are now recognized to drive thrombosis in the arterial and venous systems, notably at sites of inflammation (thromboinflammation), 1 although increased bleeding upon inhibition remains a major concern.
Platelets contain two immune receptors which signal through an immunoreceptor tyrosine-based activation motif (ITAM), glycoprotein VI (GPVI), and FcγRIIA, characterized by two YxxL sequences. 2 3 In addition, they express C-type lectin-like receptor (CLEC)-2, which has a hemITAM domain, containing a single YxxL sequence. 4 CLEC-2 is a type II transmembrane receptor, composed of a short cytoplasmic tail, a stalk region, and an extracellular, ligand-binding C-type lectin-like domain. 5 It is expressed at high levels in platelets, and at a low level in a subpopulation of myeloid cells. 6 Platelet functions of CLEC-2 have been shown in wound healing, 7 sepsis, 8 inflammation-driven venous thrombosis, 9 10 and inflammatory macrophage migration. 11 CLEC-2 has been demonstrated to contribute to arterial and venous thromboses in mouse models of disease, 9 10 12 13 14 15 with its role in arterial thrombosis being independent of its ITAM signaling domain. 15
Podoplanin is recognized as the major endogenous ligand of CLEC-2, although hemin has more recently been shown as a ligand. 16 Podoplanin is widely expressed outside of vasculature, including on alveolar epithelial cells, fibroblasts, lymphatic endothelial cells, and kidney podocytes, 17 and is upregulated during inflammation on macrophages, T helper (TH) 17 cells, and in stromal cells. 11 18 Hemin is released post-hemolysis, but is ordinarily quenched by scavenging mechanisms. During severe hemolytic disease, such as sickle cell disease, or rhabdomyolysis, hemin induces platelet agglutination and aggregation by ferroptosis, and platelet-CLEC-2 in an ITAM-dependent manner, respectively. 16 19 There is also speculation on the existence of a ligand to CLEC-2 in healthy vasculature to account for its role in arterial thrombosis models in mice. 12 As yet, the identity of this ligand is not known.
Previous studies investigating the contribution of CLEC-2 to arterial thrombosis utilized antibody-induced depletion of CLEC-2 (using the monoclonal antibody, INU1), or by genetic deletion, crossing the clec1b fl/fl and platelet factor (PF)4-Cre mouse models 20 ; both approaches have limitations. Antibody depletion is associated with a marked thrombocytopenia and PF4 is expressed in several cell types beyond platelets, including microglia, subpopulations of monocytes, and T cells. 21 22 23 This is of significance, as several cell types beyond platelets have been shown to support arterial and venous thrombosis, including neutrophils (through NETosis), monocytes (during atheroma formation), and mast cells (during venous thrombosis). 1 24
In this study, we have investigated the contribution of CLEC-2 in mice in vivo and in vitro models of thrombosis using a GPIbα-Cre mouse for genetic deletion of CLEC-2. 25 The clec1b fl/fl GPIbα-Cre + mouse exhibits a 98.7% deletion of platelet-CLEC-2, without deletion in myeloid cells. 25 26 We have investigated the role of CLEC-2 in human blood using F(ab) fragments of a CLEC-2-podoplanin-blocking antibody, AYP1, 27 and recombinant CLEC-2 in in vitro models of platelet adhesion and aggregation at arterial shear rates.
Materials and Methods
Mice
Wild type (WT) C57BL/6 mice (9–12 weeks; males and females) were purchased from Jackson Laboratories (United Kingdom). CLEC-2-deficient mice (clec1b fl/fl GPIbα-Cre) have been described. 26 GPVI-deficient (GP6 −/− clec1b fl/fl GPIbα-Cre − ) mice were also used, but not their double-deficient (CLEC-2 and GPVI) GPIbα-Cre + littermates. All experiments were performed in accordance with UK law (Animal Scientific Procedures Act 1986) with approval of the local ethics committee and UK Home Office approval under PP9677279 and P0E98D513 granted to the University of Birmingham.
Human Material
Venous blood was drawn from healthy, consenting, drug-free volunteers into 3.2% trisodium citrate BD Vacutainers (Becton Dickinson, United Kingdom). Ethical approval was granted by the University of Birmingham Research Ethics Committee (Ref: UHSP/22/BTVR/07). Washed platelets were isolated as previously described, 16 and aggregation was monitored at a concentration of 2 × 10 8 /mL. The use of pooled plaque homogenate material from atherosclerotic patients was approved by the North-West Haydock Research Ethics Committee (Ref: 20/NW/0001). The pooled plaque homogenate was prepared as described. 28
Materials
Further details on materials are found in the Supplementary Material ( Supplementary Tables S1–S5 , available in the online version).
Cell Culture
Human umbilical vein endothelial cells (HUVECs; Promocell, Germany) were cultured in a humidified incubator at 5% CO 2 and 37 ° C in Endothelial Cell Growth Media (Promocell, Germany). For flow adhesions assays, cells were cultured in a 2% gelatin-coated, µ-Slide VI 0.4 perfusion chamber (Ibidi, Germany). Cells were tumor necrosis factor (TNF)-α-activated (10 ng/mL) for 24 hours, blocked with 2% BSA (bovine serum albumin), and citrate-anticoagulated blood was perfused for 3 minutes.
Mouse Platelet Isolation
Blood was taken from the inferior vena cava of terminally CO 2 -narcosed mice, post-isoflurane-anesthesia. Washed platelets were prepared as described. 16 Light transmission aggregometry was performed as described. 16
Platelet Depletion In Vivo
Mice were challenged with an anti-GPIbα antibody (Emfret, Germany) at 0.1 mg/kg by intraperitoneal injection; platelet count was analyzed by saphenous bleed daily and quantified by an ABX Pentra 80 automated hematology analyzer (Horiba, Japan).
In Vivo Thrombosis Assays
Mice (20–25 g) were anaesthetized intraperitoneally by avertin ( 2,2,2-tribromoethanol ; Sigma, United States), before tracheotomy and administration of anti-GP1bβ X488 (Emfret, Germany) at 0.1 µg/kg by cannulation of the jugular vein.
Ferric chloride (FeCl 3 ) injury : the carotid artery was exposed and thrombi were generated by administration of FeCl 3 -soaked filter paper (2 × 1 mm, 10%, 3 minutes). 29
Laser injury : the cremaster muscle was exposed and thrombi were induced by laser injuries of the arterioles. Images were acquired using an Olympus, upright spinning disk confocal microscope (4X Nikon air lens or 40X Nikon water-immersed lens, respectively). Images were analyzed using Slidebook6 software (Intelligent Imaging Innovations, United States), as described. 29
Flow Adhesion Assays
Maastricht flow chamber : citrated whole blood was perfused over Horm collagen (surfaces coated with 100 µg/mL; Nycomed, Switzerland), human collagen III (100 µg/mL; Sigma, United States), or pooled plaque homogenate (500 µg/mL) through a Maastricht parallel flow chamber at arterial shear rate (1,000 s −1 ), as described. 30 Blood was treated with 40 µM PPACK and re-calcified (2 mM). P-Selectin (anti-CD62P, BioLegend, United States), αIIbβ3 (anti-fibrinogen, Dako, United States), and phosphatidylserine (PS)-exposure (Annexin V, ThermoFisher, United States) were measured at 3.5 minutes as described. 30 31 Images were acquired using an EVOS M5000 Imaging system (Invitrogen, United States) by transmitted light imaging (40X lens).
µ-Slide VI 0.1 and 0.4 : whole blood was perfused over Horm collagen (surfaces coated with 100 µg/mL) or HUVECs through a µ-Slide VI 0.1 or 0.4, respectively. Blood was perfused at arterial shear rate (1,000 s −1 ) and shear stress (10 dyn.s/cm 2 ) unless specified. Blood was treated with 2 µM DiOC6 32 33 (ThermoFisher, United States) and 40 µM PPACK. Images were acquired simultaneously of two fields with Z-stacks using an EVOS M5000 Imaging system (ThermoFisher, United States) by fluorescence and transmitted light imaging (20X lens).
Analysis : images were analyzed using parameter specific semi-automated ImageJ scripts, as described. 30 31
Receptor Expression
Receptor surface expression was assessed by Accuri C6 Plus flow cytometry (BD Bioscience, United Kingdom). Antibodies were incubated alongside the agonist for 20 minutes, before fixing in 4% paraformaldehyde (PFA). Data were quantified by FlowJo. Receptor surface expression was visualized by epifluorescent microscopy. Cells were fixed in 4% PFA before blocking with 5% BSA-PBS and incubating with antibodies overnight. Fluorescence intensity was quantified using FIJI v2.1.
Statistics
All data are presented as mean ± standard deviation, unless otherwise stated. The statistical comparison between two groups was analyzed using a t -test and the statistical difference between multiple groups in vitro using one-way ANOVA (analysis of variance) with Tukey's multiple comparisons test. The statistical comparison for in vivo experiments was determined by a Kruskal–Wallis test using Prism 8 (GraphPad Software Inc., United States). Statistical significance was represented as shown: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Results
Thrombus Formation Is Diminished in clec1b fl/fl GPIbα-Cre + Mice Post-FeCl 3 - and Laser-Induced Arterial Injury
Previous studies investigating the role of CLEC-2 on platelets in arterial thrombosis in mice have used a clec1b fl/fl PF4-Cre model to selectively delete the hemITAM receptor on platelets. 14 15 However, several groups have shown that the PF4-Cre + transgene also deletes CLEC-2 on a subpopulation of myeloid cells and lymphocytes, 21 22 23 thereby making it unclear if the observed phenotype is solely due to deletion on platelets. In view of this, we have utilized a GPIbα-Cre transgenic mouse strain, to selectively delete CLEC-2 in the megakaryocyte/platelet lineage 25 34 and compared the result to both littermate control mice and to GPVI-deficient mice. Platelets from GPIbα-Cre + mice (clec1b flox-negative) have been shown to be similar to genetically unaltered platelets. 25 We have previously reported that clec1b fl/fl GPIbα-Cre + mice are born at a Mendelian ratio with a mild reduction in platelet count, possibly as a result of a defect in blood–lymphatic separation, as the GPIbα-Cre itself does not alter platelet count. 25 26 Here, we describe a 20.7 ± 4.6% reduction in circulating platelet count ( Supplementary Fig. S1A , available in the online version), but unchanged platelet volume and erythrocyte and white blood cell (monocyte, neutrophil, and lymphocyte) counts in CLEC-2 fl/fl GPIbα-Cre + mice, compared to GPIbα-Cre − littermate controls ( Supplementary Fig. S1B–G , available in the online version). Furthermore, we show that deletion of platelet-CLEC-2 does not impact the expression of GPVI, GPIb, or CD41 ( Supplementary Fig. S2 , available in the online version), nor washed platelet response to thrombin, collagen-related peptide, or thromboxane A 2 receptor agonist, U46619; CLEC-2-deficient platelets do not respond to rhodocytin ( Supplementary Fig. S3 , available in the online version).
Using the FeCl 3 -induced thrombosis model of the carotid artery, we show that thrombus growth post-injury is mildly impaired in clec1b fl/fl GPIbα-Cre + mice, and more substantially in the absence of GPVI. This could be explained by a delay in thrombus growth, or platelet activation, in GPVI-deficient, but not clec1b fl/fl GPIbα-Cre + mice compared to littermate controls ( Fig. 1A ). The contribution of CLEC-2 to thrombus formation was also assessed using a laser-induced thrombus injury in cremaster muscle arterioles. The time to peak thrombus formation was not delayed in clec1b fl/fl GPIbα-Cre + mice compared to littermate controls, but the peak thrombus size was significantly reduced ( Fig. 1B ). These results show a mild defect in arterial thrombus formation at arterial shear in clec1b fl/fl GPIbα-Cre + mice, in line with previous results in clec1b fl/fl PF4-Cre + mice. 14 15
Fig. 1.
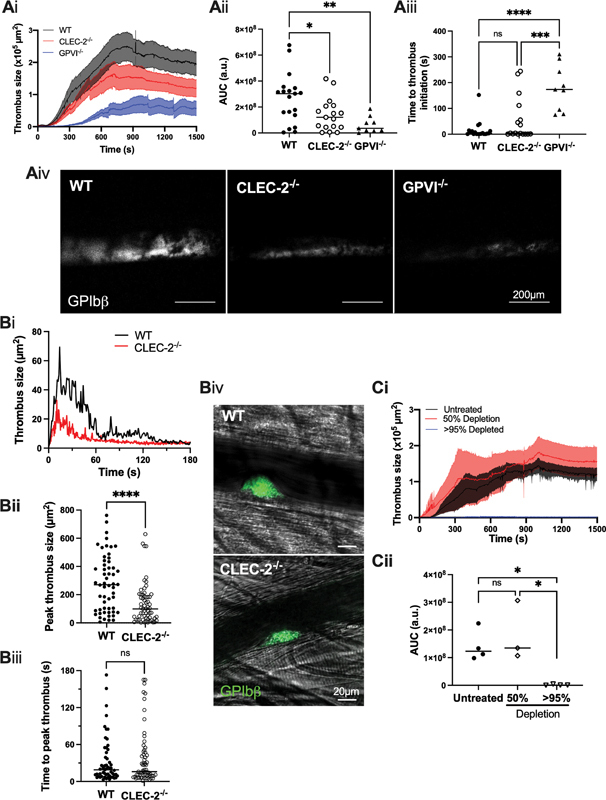
Platelet-CLEC-2-deficiency results in smaller, unstable arterial thrombus formation in mice. ( A ) Wild type (WT; clec1b fl/fl GPIbα-Cre − ), platelet-CLEC-2-deficient (CLEC-2 −/− ; clec1b fl/fl GPIbα-Cre + ), or GPVI-deficient (GPVI −/− ; GP6 −/− clec1b fl/fl GPIbα-Cre − ) mice were challenged with FeCl 3 -soaked filter paper (10%, 3 minutes) on the carotid artery. ( A i) Thrombus size was measured over 25 minutes using an anti-GPIbβ antibody fluorescence using confocal microscopy and ( Aii ) the area under the curve (AUC) was calculated (a.u.= arbitrary units). ( Aiii ) The time taken for a thrombus to reach 1 × 10 4 µm 2 was calculated to quantify formation initiation. ( Aiv ) Representative images are shown at 25 minutes postinjury (WT n = 19 mice; CLEC-2 −/− n = 17 mice; GPVI −/− n = 9 mice). ( B ) WT or CLEC-2 −/− mice were challenged with a laser injury in arterioles of the cremaster muscle. ( Bi ) Thrombus size was observed over 3 minutes by GPIbβ-antibody fluorescence using confocal microscopy, and the median thrombus size of all injuries is shown. ( Bii ) The peak thrombus size and ( Biii ) time to reach peak thrombus sizes were calculated, each dot representing an injury. ( Biv ) Representative images are shown of the peak thrombus (12–15 seconds) postmedian injury of the median mouse/genotype (WT n = 56 injuries, 6 mice; CLEC-2 −/− n = 62 injuries, 6 mice). ( C ) C57BL/6 mice were treated intraperitoneally with a platelet-depletion antibody (0.1 mg/kg), and challenged 24 or 72 hours posttreatment (>95 and 50% depletion, respectively) with FeCl 3 -soaked filter paper (10%, 3 minutes) on the carotid artery. ( Ci ) Thrombus size was observed over 25 minutes by GPIbβ-antibody fluorescence using confocal microscopy and ( Cii ) the AUC was calculated (a.u.= arbitrary units). Differences in WT thrombus sizes post-FeCl 3 injury in (Ai) and (Ci) may reflect differences in lens chip sizes used; the same chip was used during in the relevant data sets, and values are relative to controls. The statistical significance between two groups was analyzed using an unpaired t-test, and the statistical difference between multiple groups was analyzed using one-way ANOVA with Tukey's multiple comparisons test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ANOVA, analysis of variance.
The decrease in thrombus formation in the clec1b fl/fl GPIbα-Cre + mouse could be due to the loss of CLEC-2 and/or the mild thrombocytopenia. To determine the effect of platelet count on thrombus formation, C57BL/6 mice were treated with a platelet-depleting antibody (GPIbα), 8 and the recovery in platelet count was monitored. The FeCl 3 -injury model was then performed in mice when the platelet count had been lowered by >95% or approximately 50% at 24- and 72-hour postdepletion, respectively. Of note, the approximately 50% platelet depletion is greater than the 20.7 ± 4.6% depletion observed in the clec1b fl/fl GPIbα-Cre + mice. Thrombus formation was abolished in mice with a >95% reduction in platelet count but unaltered in mice with an approximately 50% platelet depletion, relative to controls ( Fig. 1C ). This shows that the decrease in platelet count is not the cause of the reduction in thrombus formation.
Together, these data confirm that platelet-CLEC-2 plays an important role in the formation of thrombi in the arterial circulation in mice, and that this is independent of the reduction in platelet count.
CLEC-2 Contributes to Thrombus Formation in Mouse Blood Perfused over Collagen at Arterial Shear Rates ex vivo
The contribution of CLEC-2 to thrombus formation was further investigated ex vivo , using an Ibidi µ-Slide VI 0.1 flow chamber coated with Horm collagen. The platelet count of clec1b fl/fl GPIbα-Cre + mice was normalized to match the level of WT littermate control using washed platelets from a second clec1b fl/fl GPIbα-Cre + mouse ( Fig. 2A ). Specific labeling of the donor platelets showed that both the donor and host platelets contribute to thrombus formation when perfused over collagen at arterial shear ( Supplementary Fig. S4 , available in the online version). Blood from clec1b fl/fl GPIbα-Cre + mice perfused at 1,000 s −1 showed a significant decrease in thrombus surface coverage (38 ± 11%) after 4 minutes compared to WT controls ( Fig. 2B ). Interestingly, there was no change in platelet deposition after 2 minutes of perfusion, suggesting a role for CLEC-2 in thrombus growth, but not in thrombus initiation. These data show that CLEC-2 contributes to the growth of thrombi at arterial shear rates in mouse blood on a collagen surface.
Fig. 2.
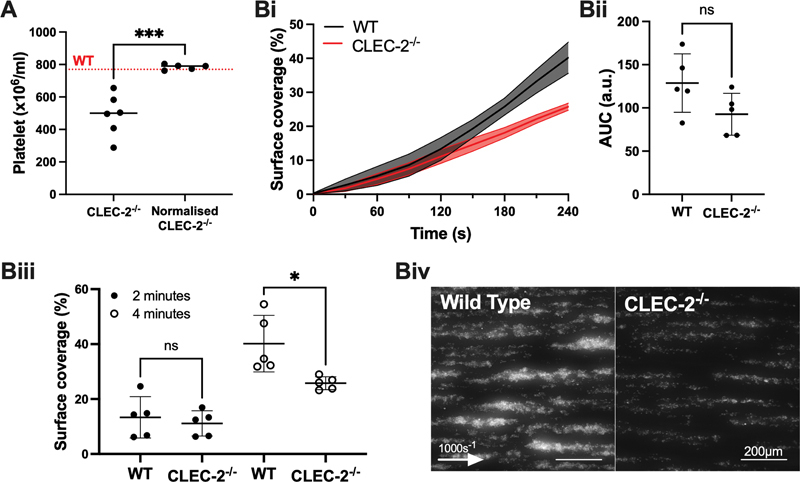
Platelet-CLEC-2 promotes thrombus growth at arterial shear in mice ex vivo . ( A ) Washed platelets from a CLEC-2 −/− (clec1b fl/fl GPIbα-Cre + ) mouse was donated to the whole blood of a secondary CLEC-2 −/− mouse to match WT (clec1b fl/fl GPIbα-Cre − ) platelet counts. The red-dotted line represents median platelet count of littermate WT controls. ( B i) Thrombus surface coverage was measured over 4 minutes using DiOC6 fluorescence (2 µM). ( Bii ) Subsequent area under the curve (AUC) was calculated (a.u. = arbitrary units) and ( Biii ) thrombus formation after 2 and 4 minutes is shown. ( Biv ) Representative images shown after 4 minutes ( n = 5 mice). The statistical significance between two groups was analyzed using an unpaired t-test. * p < 0.05, *** p < 0.001.
CLEC-2 Does Not Impact Thrombus Formation on Collagen or Plaque at Arterial Shear in Human Blood in vitro
Studies were designed to investigate the contribution of CLEC-2 to thrombus formation in human blood using fragments of the human CLEC-2-blocking antibody, AYP1. 27 AYP1 F(ab) and F(ab) 2 (10 µg/mL) inhibited rhodocytin- and podoplanin-expressing HEK-293T cell-induced aggregation of washed platelets ( Fig. 3A, B ; Supplementary Fig. S5A, B , available in the online version). Furthermore, F(ab) and F(ab) 2 (10 µg/mL) inhibited rhodocytin-induced platelet-P-selectin expression in whole blood ( Fig. 3C ). This demonstrates that AYP1 can be used to block platelet activation by the endogenous CLEC-2 ligand, podoplanin, in whole blood bearing in mind that rhodocytin and podoplanin bind to a shared epitope on CLEC-2. 5
Fig. 3.
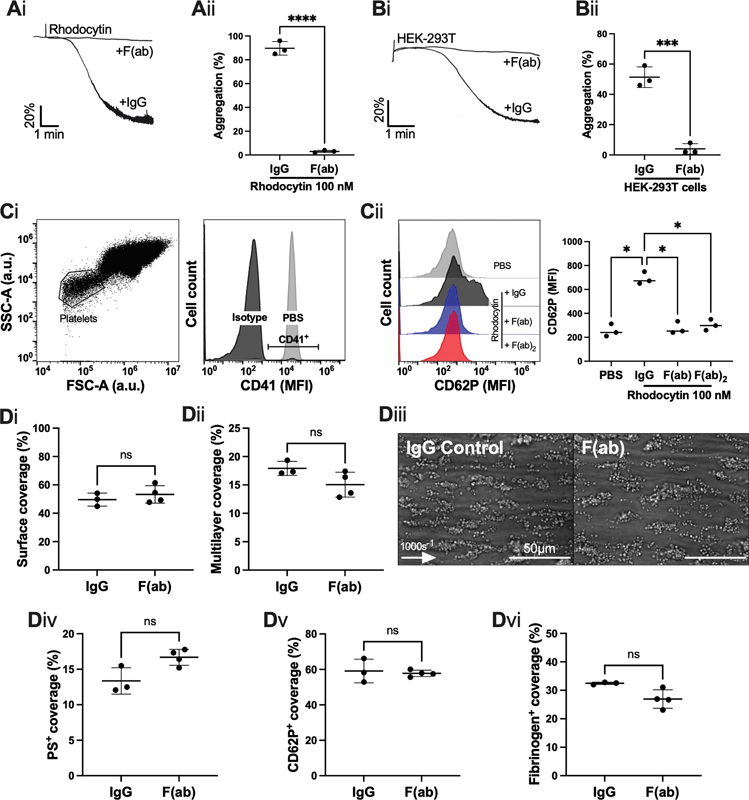
The effect of inhibition of CLEC-2 inhibition by AYP1 fragments on platelet aggregation and thrombus formation on collagen under flow. ( A, B ) Washed platelet aggregation was assessed by light transmission aggregometry, in the presence or absence of AYP1 fragment, F(ab) (10 µg/mL). ( Ai ) Representative platelet aggregation trace induced by rhodocytin (100 nM) and ( Aii ) maximum aggregation ( n = 3 donors). ( Bi ) Representative platelet aggregation trace induced by HEK-293T cells and ( Bii ) final aggregation ( n = 3 donors). ( C ) Inhibition of CLEC-2 by F(ab) or F(ab) in whole blood was assessed by flow cytometry in the presence of rhodocytin (100 nM) for 20 minutes. ( Ci ) Platelets were identified by size, and their expression of CD41 in whole blood, and ( Cii ) their activation was measured by CD62P mean fluorescence intensity (MFI) by an Accuri C6 flow cytometer ( n = 3 donors). ( D ) Whole blood from healthy volunteer donors was perfused through a Maastricht flow chamber at 1000 s −1 , in the presence or absence of F(ab) (10 µg/mL) 10 minutes prior to perfusion. The flow chamber was coated with Horm collagen (100 µg/mL). ( Di ) Thrombus surface coverage and ( Dii ) and multi-layered thrombus size shown after perfusion for 3.5 minutes. ( Diii ) Representative images shown after 3.5 minutes ( n = 3 donors; arrow indicates direction of flow). ( Div ) Phosphatidylserine (PS) exposure, ( Dv ) P-selectin expression (CD62P), and ( Dvi ) fibrinogen coverage were assessed with a semi-automated ImageJ script from fluorescent images, shown in Supplementary Fig. S5 . The statistical significance between two groups was analyzed using a paired t -test and the statistical difference between multiple groups was analyzed using one-way ANOVA with Tukey's multiple comparisons test. * p < 0.05, *** p < 0.001. ANOVA, analysis of variance.
Platelet thrombus generation at arterial shear was assessed using both the Maastricht flow chamber, facilitating small volumes of blood and agonists, and an Ibidi µ-Slide chamber, allowing for cell culture-grade perfusions. Interestingly, neither F(ab) nor F(ab) 2 fragments of AYP1 (10 µg/mL) significantly altered thrombus generation over Horm collagen in the Ibidi µ-Slide VI 0.1flow chamber at 1,000 s −1 ( Supplementary Fig. S5C , available in the online version). Furthermore, AYP1 F(ab) (10 µg/mL) did not alter thrombus formation (surface area and multilayer thrombus size), nor expression of platelet activation markers (PS exposure, P-selectin expression, and αIIbβ3 activation), when perfused over Horm collagen at arterial shear rate (1,000 s −1 ) in the Maastricht flow chamber ( Fig. 3D , Supplementary Fig. S6 , available in the online version).
Horm collagen is 95% type I collagen, whilst the sub-endothelium is composed of a mixture of both collagen type I and collagen type III. For this reason, we extended the studies in the Maastricht flow model to human collagen type III preparation. AYP1 F(ab) (10 µg/mL) also did not alter platelet deposition, surface coverage, or activation on this surface ( Fig. 4A ). Atherosclerotic plaque contains a variety of extracellular matrix proteins and debris of various cells including podoplanin-positive macrophages. 18 To replicate plaque-driven thrombosis in a large-artery intima, blood was perfused over pooled plaque homogenate at arterial shear rate (1,000 s −1 ). Platelet coverage, multilayer thrombus size, and expression of platelet activation markers were not altered in the presence of AYP1 F(ab) (10 µg/mL), compared to controls ( Fig. 4B ).
Fig. 4.
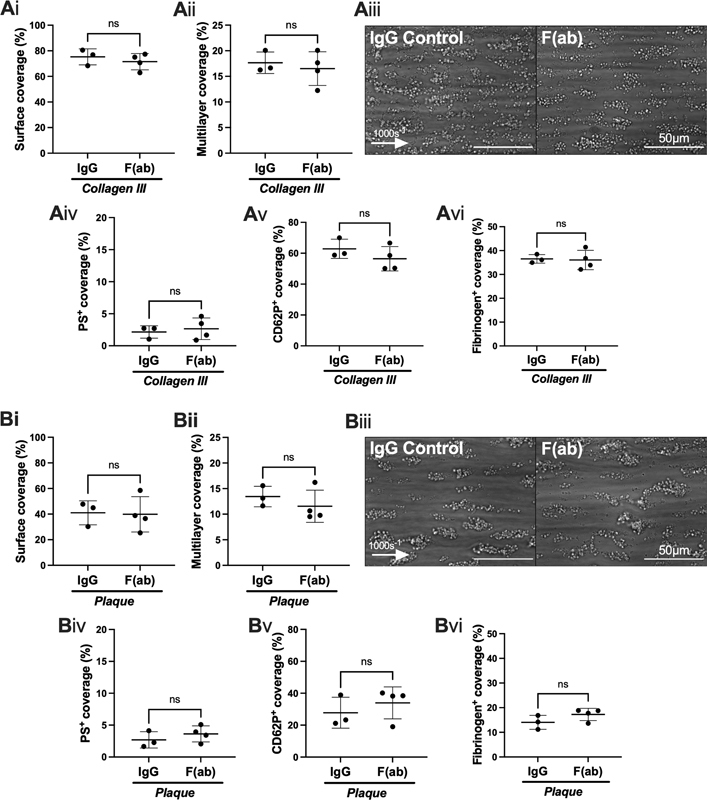
CLEC-2 inhibition by AYP1 fragments does not alter thrombus formation, nor platelet activation in human blood perfused over collagen type III or plaque homogenate. Whole blood from healthy volunteer donors was perfused through a Maastricht flow chamber at 1000 s −1 , in the presence or absence of AYP1 F(ab) (10 µg/mL) for 10 minutes prior to perfusion. ( A ) The flow chamber was coated with collagen III (100 µg/mL). ( Ai ) Thrombus surface coverage and ( Aii ) multi-layered thrombi after 3.5 minutes. ( Aiii ) Representative images shown after 3.5 minutes ( n = 3 donors; arrow indicates direction of flow). ( Aiv ) Phosphatidylserine (PS) exposure, ( Av ) P-selectin expression (CD62P), and ( Avi ) integrin αIIbβ3 activation were calculated from fluorescent images. ( B ) The flow chamber was coated with pooled plaque homogenate (500 µg/mL). ( Bi ) Thrombus surface coverage and ( Bii ) and multi-layered thrombus size after 3.5 minutes. ( Biii ) Representative images shown after 3.5 minutes ( n = 3 donors; arrow indicates direction of flow). ( Biv ) Phosphatidylserine (PS) exposure, ( Bv ) P-selectin expression (CD62P), and ( Bvi ) integrin αIIbβ3 activation were calculated from fluorescent images. The statistical significance between two groups was analyzed using a paired t -test.
These results demonstrate that blockade of CLEC-2 by AYP1 has no effect on thrombus formation when human blood is perfused over collagen types I and III, as well as atherosclerotic plaque at arterial shear in two widely used flow models.
CLEC-2-Ligand Inhibition by Recombinant Human CLEC-2-Fc Does Not Alter Thrombus Surface Coverage
The ligand for CLEC-2 that supports thrombus formation in mice, and potentially in human, is not known. To date, the only known ligand found in the blood is hemin, which is formed from lysed red blood cells, but is rapidly quenched by hemopexin. 16 Importantly, AYP1 does not block hemin-induced aggregation, 16 and indeed may not block the unidentified ligand, proposed in studies of mouse thrombosis models, if present. Therefore, we utilized recombinant human CLEC-2-Fc (hFc-CLEC-2) to block a potential role of hemin or a CLEC-2-ligand that binds to a site distinct to AYP1. In addition, hFc-CLEC-2 (10 µg/mL) blocked rhodocytin- and podoplanin-expressing HEK-293T cell-induced platelet aggregation ( Fig. 5A, B ); we have previously demonstrated that hFc-CLEC-2 inhibits hemin-induced platelet aggregation. 16 Furthermore, hFc-CLEC-2 inhibited rhodocytin-induced platelet activation in whole blood, as shown by P-selectin (CD62P) surface expression ( Fig. 5C ). hFc-CLEC-2 did not alter thrombus formation when perfused over Horm collagen in an Ibidi µ-Slide VI 0.1 flow chamber, or the Maastricht flow chamber ( Fig. 5D , Supplementary Fig. S7 , available in the online version).
Fig. 5.
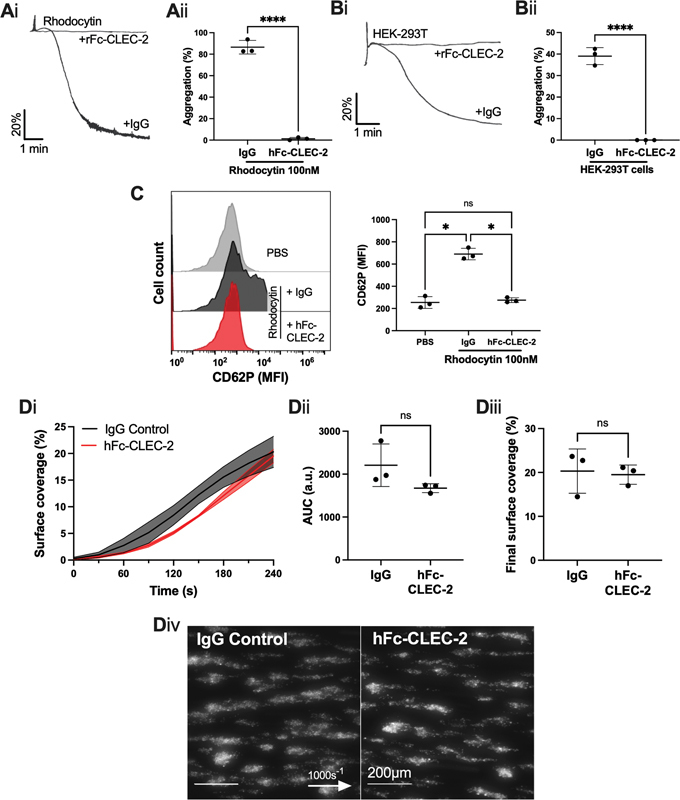
The effect of recombinant human CLEC-2-Fc on platelet aggregation and thrombus formation. ( A, B ) Washed platelet aggregation was assessed by light transmission aggregometry, in the presence or absence of recombinant human CLEC-2-Fc (hFc-CLEC-2) (10 µg/mL). ( Ai ) Representative platelet aggregation trace induced by rhodocytin (100 nM) and ( Aii ) maximum aggregation ( n = 3 donors). ( Bi ) Representative platelet aggregation trace induced by HEK-293T cells and ( Bii ) final aggregation ( n = 3 donors). ( C ) Inhibition of CLEC-2 by hFc-CLEC-2 in whole blood was assessed by flow cytometry in the presence of rhodocytin (100 nM) for 20 minutes. Platelets were identified as shown in Fig. 3(Ci) and their activation was measured by P-selectin (CD62P) median fluorescence intensity (MFI) using an Accuri C6 flow cytometer ( n = 3 donors). ( D ) Whole blood was perfused over Horm collagen-coated (100 µg/mL) µ-Slide VI 0.1 flow chamber at 1000 s −1 , in the presence or absence of hFc-CLEC-2 for 10 minutes prior to perfusion. ( Di ) Thrombus surface coverage over 4 minutes, measured using DiOC6 fluorescence (2 µM). ( Dii ) Subsequent area under the curve (AUC) was calculated (a.u. = arbitrary units). ( Diii ) Thrombus formation after 4 minutes. ( Div ) Representative images shown after 4 minutes ( n = 3 donors; arrow indicates direction of flow). The statistical significance between two groups was analyzed using a paired t -test with Mann–Whitney correction and the statistical difference between multiple groups was analyzed using one-way ANOVA with Tukey's multiple comparisons test. * p < 0.05, **** p < 0.0001. ANOVA, analysis of variance.
These results strengthen the evidence against a role for CLEC-2 in thrombus formation on collagen in human platelets at arterial shear.
CLEC-2 Does Not Alter Platelet Adherence or Leukocyte Recruitment in Blood Perfused over Endothelial Cells
The endothelium plays a critical role in driving thrombus formation at sites of inflammation in the venous system (thromboinflammation) through recruitment of platelets and leukocytes via binding von Willebrand factor- and Mac-1-GPIb, respectively, 35 with additional binding of platelet and endothelial P-selectin to leukocyte-PSGL1. We have therefore investigated whether CLEC-2 also plays a role in the recruitment of platelets and leukocytes by perfusion over a monolayer of resting and activated endothelial cells.
To model human vasculature, a monolayer of primary HUVECs was cultured in an Ibidi µ-Slide VI 0.4 and activated by TNF-α to significantly upregulate the expression of intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule (VCAM-1); VE-cadherin expression was not significantly altered ( Supplementary Fig. S8 , available in the online version). Surface coverage of platelets and leukocytes was significantly increased in TNF-α-activated HUVECs versus unstimulated controls ( Fig. 6A ). CLEC-2 inhibition at venous shear stress by AYP1 F(ab) 2 did not alter leukocyte recruitment, nor platelet surface coverage ( Fig. 6B ). Surface coverage was also unchanged in the presence of AYP1 F(ab) 2 at arterial shear ( Fig. 6C ).
Fig. 6.
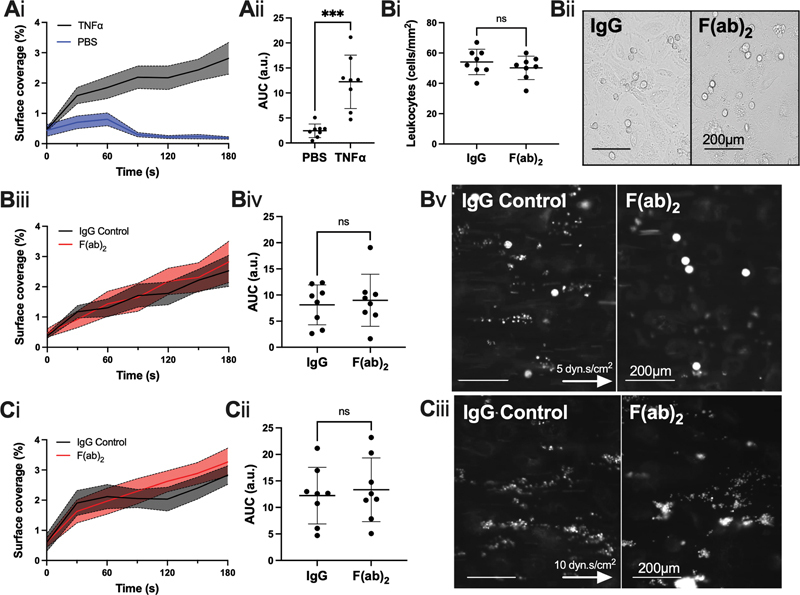
CLEC-2 inhibition by F(ab) 2 does not alter thrombus formation or leukocyte recruitment over activated endothelial cells. ( A ) Whole blood in the presence of an IgG control to F(ab) 2 was perfused in a µ-Slide VI 0.4 flow chamber over 24 h-TNF-α activated (10 ng/mL) or unstimulated, human umbilical vein endothelial cells (HUVEC) for 3 minutes. ( Ai ) Thrombus surface coverage over 3 minutes, measured using DiOC6 fluorescence (2 µM); ( Aii ) subsequent area under the curve (AUC) was calculated (a.u. = arbitrary units). ( B ) Whole blood was perfused at venous shear stress (5 dyn.s/cm 2 ) over 24 h-TNF-α activated (10 ng/mL) HUVEC for 3 minutes in the presence or absence of F(ab) 2 (10 µg/mL) for 10 minutes prior to perfusion. ( Bi ) Leukocyte recruitment was analyzed by manual identification from five fields of view per donor and ( Bii ) representative images were taken postperfusion. ( Biii ) Thrombus surface coverage over 3 minutes, measured using DiOC6 fluorescence (2 µM) and ( Biv ) subsequent area under the curve (AUC) was calculated (a.u. = arbitrary units). ( Bv ) Representative images presented after 4 minutes ( n = 8 donors; arrow indicates direction of flow). ( C ) Whole blood was perfused at arterial shear stress (10 dyn.s/cm 2 ) over 24 h-TNF-α activated (10 ng/mL) HUVEC for 3 minutes in the presence or absence of F(ab) 2 (10 µg/mL) for 10 minutes prior to perfusion. ( Ci ) Thrombus surface coverage over 3 minutes, measured using DiOC6 fluorescence (2 µM) and ( Cii ) subsequent AUC was calculated. ( Ciii ) Representative images shown after 4 minutes ( n = 8 donors; arrow indicates direction of flow). The statistical significance between two groups was analyzed using a paired t-test. *** p < 0.001. IgG, immunoglobulin G.
Our data suggest that CLEC-2 does not contribute to leukocyte recruitment, nor platelet adhesion at arterial or venous shear rates over TNF-α-activated HUVECs.
Discussion
In this study, we have investigated the role of CLEC-2 in platelet aggregate formation under arterial shear rates using both mouse and human blood, as well as in mouse models of arterial thrombosis. We used a recently generated clec1b fl/fl GPIbα-Cre mouse which, in contrast to the clec1b fl/fl PF4-Cre mouse, specifically deletes CLEC-2 on megakaryocytes and platelets. Results confirm a role for mouse CLEC-2 in supporting thrombus growth, but not initiation, both in in vivo and ex vivo studies. 12 36 In contrast, we did not observe a role for CLEC-2 in thrombus formation using human blood perfused over collagen types I and III, plaque, or inflammatory endothelial cells at arterial shear rates using F(ab) fragments of the CLEC-2-podoplanin blocking antibody, AYP1, or recombinant CLEC-2. This suggests that CLEC-2 may not contribute to platelet activation in arterial thrombosis in humans, consistent with the absence of an identified ligand for CLEC-2 in human blood.
The critical role of CLEC-2 in supporting arterial thrombosis in vivo in mice was shown using FeCl 3 and laser injury models, with the former mediated by oxidative stress- and free-radical-dependent lipid peroxidation-induced endothelial damage. 37 38 In addition, FeCl 3 induces direct hemolysis to release free hemin, 39 40 which is rapidly scavenged by hemopexin, 41 42 but may also activate CLEC-2 in this model. On the other hand, speculations of erythrocyte lysis post-laser injury 43 have been countered by more recent studies. 37 Similarly, there is no evidence for significant hemolysis in in vitro flow models. The absence of podoplanin in healthy vasculature and the lack of evidence for a role of hemin suggest the presence of a novel ligand for CLEC-2 in mouse blood to support thrombus formation. The identification of the CLEC-2 ligand is required to further determine its role in the regulation of thrombosis and to establish if this pathway is conserved in human. The observation of thrombus formation in a CLEC-2 signaling-dead transgenic mouse 15 suggests that the ligand supports thrombus formation as an adhesion molecule.
The present study provides evidence against a role for CLEC-2 in platelet aggregation under arterial shear rates in human blood on multiple surfaces, including atherosclerotic plaque. CLEC-2 was inhibited using the CLEC-2-podoplanin blocking monoclonal antibody, AYP1, and recombinant CLEC-2. Whilst a ligand to CLEC-2 in mouse, but not human blood, may explain this trans-species variation, this also may be explained by the copy number of CLEC-2 on mouse platelets, which is 10-fold higher than human platelets. 27 44 45 46 The level of expression may be a key determinant of the role of CLEC-2 as an adhesion receptor. 15
The observation that CLEC-2 does not play a role in platelet aggregation on atherosclerotic plaque material is of particular interest, given reports of expression of podoplanin on advanced human plaque. 18 The plaque material used in this study was pooled from 10 donors and therefore is representative of material from multiple patients. In contrast, the adhesion of platelets on plaque under flow is abrogated when the GPVI–collagen interaction is blocked by an anti-GPVI antibody, suggesting platelet activation by plaque is almost entirely mediated by GPVI. 47 48 More recently, it has been shown that deletion of CLEC-2 (using the PF4-Cre) decreases GPIbα-mediated integrin αIIbβ3 activation, which may also contribute to the loss of platelet activation perfused at arterial shear in the GPIbα-Cre + mice. 49
There are a number of limitations in this study. Whilst in vivo studies were blinded throughout procedures, presentation of blood-filled lymph nodes was evident in platelet-CLEC-2-deficient mice 26 ; to combat this, the analysis of thrombus formation was analyzed by an independent, blinded researcher. The studies in humans are limited to in vitro flow adhesion models and these do not represent all human vascular beds. Haining et al demonstrated that CLEC-2 did not contribute to thrombus occlusion in a mechanical injury of the aorta, which was recently confirmed using a humanized CLEC-2 mouse model. 15 50 This illustrates a varied contribution of vascular beds to thrombus formation in mice.
To conclude, we have strengthened the evidence for the role of platelet CLEC-2 in mouse arterial thrombosis using a clec1b fl/fl GPIbα-Cre + , which has a higher specificity of megakaryocytes/platelet-deletion, relative to clec1b fl/fl PF4-Cre + mouse. In contrast, we did not observe a role of human CLEC-2 in platelet aggregation under flow conditions on a variety of surfaces using CLEC-2-blocking antibody, AYP1, or hFc-CLEC-2. Podoplanin has been shown to be markedly upregulated in the sub-endothelium during DVT and liver inflammation in humans. 51 52 The present results suggest that blocking the CLEC-2–podoplanin interaction in patients at risk of venous thrombosis will not interfere with hemostasis, reinforcing CLEC-2 as a target for a new class of antiplatelet agent in thromboinflammatory disorders.
Acknowledgments
The authors would like to thank Dr. Beata Grygielska for genotyping mice, Dr. Dean Kavanagh for assistance with intravital microscopy optimization, Professor Johannes Eble (Münster University) for gifting rhodocytin and Professor Yotis Senis (University of Strasbourg) for the GPIbα-Cre mice. Visual abstract was created using BioRender.com.
Funding Statement
Funding This work was supported by a college-funded PhD studentship (University of Birmingham), N.J.J. and H.C.B. are funded by a European Union's Horizon 2020 research and innovation program under Marie Sklodowska-Curie grant agreement (766118). S.J.M. is supported by the BHF Accelerator Award (AA/18/2/34218). J.R. is supported by a BHF Intermediate Basic Science Fellowship Application (FS/IBSRF/20/25039). S.P.W. holds a BHF Chair (CH/03/003).
Conflict of Interest None declared.
Author Contributions
J.H.B. designed and performed research, collected, analyzed and interpreted data, and wrote the manuscript; C.W.S.., N.J.J., and H.C.B. performed experiments and analyzed data; Y.D. and M.R.T. produced reagents; S.J.M., N.S.P., and J.R. interpreted data; S.P.W. designed research and interpreted data. All authors edited and approved the final version of the manuscript.
What is known about this topic?
In murine models of venous thrombosis, CLEC-2 has been shown to support thrombus growth during thromboinflammation. 9 10
CLEC-2 has previously been shown to support thrombus growth during models of arterial thrombosis in mice using radiation chimeric mice, a Clec1b fl/fl PF4-Cre mouse, and a CLEC-2-depleting antibody, INU1. 12 13 14
The role of CLEC-2 during arterial thrombosis in mice has been shown to be independent of classical CLEC-2 hemITAM signaling. 15
What does this paper add?
We utilize a GPIbα-Cre to delete CLEC-2 on platelets 25 26 and confirm a role for platelet CLEC-2 in thrombus growth in two models of arterial thrombosis in mice, as well as in ex vivo flow models.
For the first time, we use fragments of human CLEC-2-blocking antibody, AYP1, as well as recombinant CLEC-2, to block CLEC-2-induced platelet activation by its ligands.
We observe no role for CLEC-2 in human blood in thrombus growth at arterial shear rates when perfused over multiple surfaces, including inflamed endothelial cells ex vivo .
Supplementary Material
References
- 1.Rayes J, Watson S P, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. 2019;129(01):12–23. doi: 10.1172/JCI122955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rayes J, Bourne J H, Brill A, Watson S P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res Pract Thromb Haemost. 2019;4(01):23–35. doi: 10.1002/rth2.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rayes J, Jadoui S, Lax S. The contribution of platelet glycoprotein receptors to inflammatory bleeding prevention is stimulus and organ dependent. Haematologica. 2018;103(06):e256–e258. doi: 10.3324/haematol.2017.182162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee R H, Bergmeier W. Platelet immunoreceptor tyrosine-based activation motif (ITAM) and hemITAM signaling and vascular integrity in inflammation and development. J Thromb Haemost. 2016;14(04):645–654. doi: 10.1111/jth.13250. [DOI] [PubMed] [Google Scholar]
- 5.Martin E M, Zuidscherwoude M, Morán L A, Di Y, García A, Watson S P. The structure of CLEC-2: mechanisms of dimerization and higher-order clustering. Platelets. 2021;32(06):733–743. doi: 10.1080/09537104.2021.1906407. [DOI] [PubMed] [Google Scholar]
- 6.Lowe K L, Navarro-Núñez L, Bénézech C. The expression of mouse CLEC-2 on leucocyte subsets varies according to their anatomical location and inflammatory state. Eur J Immunol. 2015;45(09):2484–2493. doi: 10.1002/eji.201445314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wichaiyo S, Lax S, Montague S J. Platelet glycoprotein VI and C-type lectin-like receptor 2 deficiency accelerates wound healing by impairing vascular integrity in mice. Haematologica. 2019;104(08):1648–1660. doi: 10.3324/haematol.2018.208363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rayes J, Lax S, Wichaiyo S. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. 2017;8(01):2239. doi: 10.1038/s41467-017-02402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hitchcock J R, Cook C N, Bobat S. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest. 2015;125(12):4429–4446. doi: 10.1172/JCI79070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Payne H, Ponomaryov T, Watson S P, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. 2017;129(14):2013–2020. doi: 10.1182/blood-2016-09-742999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourne J H, Beristain-Covarrubias N, Zuidscherwoude M. CLEC-2 prevents accumulation and retention of inflammatory macrophages during murine peritonitis. Front Immunol. 2021;12:693974. doi: 10.3389/fimmu.2021.693974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.May F, Hagedorn I, Pleines I. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. 2009;114(16):3464–3472. doi: 10.1182/blood-2009-05-222273. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki-Inoue K. Essential in vivo roles of the platelet activation receptor CLEC-2 in tumour metastasis, lymphangiogenesis and thrombus formation. J Biochem. 2011;150(02):127–132. doi: 10.1093/jb/mvr079. [DOI] [PubMed] [Google Scholar]
- 14.Bender M, May F, Lorenz V. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013;33(05):926–934. doi: 10.1161/ATVBAHA.112.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haining E J, Cherpokova D, Wolf K. CLEC-2 contributes to hemostasis independently of classical hemITAM signaling in mice. Blood. 2017;130(20):2224–2228. doi: 10.1182/blood-2017-03-771907. [DOI] [PubMed] [Google Scholar]
- 16.Bourne J H, Colicchia M, Di Y. Heme induces human and mouse platelet activation through C-type-lectin-like receptor-2. Haematologica. 2021;106(02):626–629. doi: 10.3324/haematol.2020.246488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quintanilla M, Montero-Montero L, Renart J, Martín-Villar E. Podoplanin in inflammation and cancer. Int J Mol Sci. 2019;20(03):E707. doi: 10.3390/ijms20030707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatakeyama K, Kaneko M K, Kato Y. Podoplanin expression in advanced atherosclerotic lesions of human aortas. Thromb Res. 2012;129(04):e70–e76. doi: 10.1016/j.thromres.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Oishi S, Tsukiji N, Otake S. Heme activates platelets and exacerbates rhabdomyolysis-induced acute kidney injury via CLEC-2 and GPVI/FcRγ. Blood Adv. 2021;5(07):2017–2026. doi: 10.1182/bloodadvances.2020001698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tiedt R, Schomber T, Hao-Shen H, Skoda R C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 2007;109(04):1503–1506. doi: 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 21.Pertuy F, Aguilar A, Strassel C. Broader expression of the mouse platelet factor 4-cre transgene beyond the megakaryocyte lineage. J Thromb Haemost. 2015;13(01):115–125. doi: 10.1111/jth.12784. [DOI] [PubMed] [Google Scholar]
- 22.Calaminus S D, Guitart A V, Sinclair A. Lineage tracing of Pf4-Cre marks hematopoietic stem cells and their progeny. PLoS One. 2012;7(12):e51361. doi: 10.1371/journal.pone.0051361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abram C L, Roberge G L, Hu Y, Lowell C A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponomaryov T, Payne H, Fabritz L, Wagner D D, Brill A. Mast cells granular contents are crucial for deep vein thrombosis in mice. Circ Res. 2017;121(08):941–950. doi: 10.1161/CIRCRESAHA.117.311185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagy Z, Vögtle T, Geer M J. The Gp1ba-Cre transgenic mouse: a new model to delineate platelet and leukocyte functions . Blood. 2019;133(04):331–343. doi: 10.1182/blood-2018-09-877787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haining E J, Lowe K L, Wichaiyo S. Lymphatic blood filling in CLEC-2-deficient mouse models. Platelets. 2021;32(03):352–367. doi: 10.1080/09537104.2020.1734784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gitz E, Pollitt A Y, Gitz-Francois J J. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood. 2014;124(14):2262–2270. doi: 10.1182/blood-2014-05-572818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reininger A J, Bernlochner I, Penz S M. A 2-step mechanism of arterial thrombus formation induced by human atherosclerotic plaques. J Am Coll Cardiol. 2010;55(11):1147–1158. doi: 10.1016/j.jacc.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 29.Smith C W, Raslan Z, Parfitt L. TREM-like transcript 1: a more sensitive marker of platelet activation than P-selectin in humans and mice. Blood Adv. 2018;2(16):2072–2078. doi: 10.1182/bloodadvances.2018017756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Witt S M, Swieringa F, Cavill R. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. 2014;5:4257. doi: 10.1038/ncomms5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Geffen J P, Brouns S LN, Batista J. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica. 2019;104(06):1256–1267. doi: 10.3324/haematol.2018.198853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarty O J, Larson M K, Auger J M. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280(47):39474–39484. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vardon Bounes F, Mémier V, Marcaud M. Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci Rep. 2018;8(01):13536. doi: 10.1038/s41598-018-31910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita H, Hashimoto Y, Russell S, Zieger B, Ware J. In vivo expression of murine platelet glycoprotein Ibalpha. Blood. 1998;92(02):488–495. [PubMed] [Google Scholar]
- 35.Kroll M H, Harris T S, Moake J L, Handin R I, Schafer A I. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. J Clin Invest. 1991;88(05):1568–1573. doi: 10.1172/JCI115468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hughes C E, Navarro-Núñez L, Finney B A, Mourão-Sá D, Pollitt A Y, Watson S P. CLEC-2 is not required for platelet aggregation at arteriolar shear. J Thromb Haemost. 2010;8(10):2328–2332. doi: 10.1111/j.1538-7836.2010.04006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stalker T J. Mouse laser injury models: variations on a theme. Platelets. 2020;31(04):423–431. doi: 10.1080/09537104.2020.1748589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grover S P, Mackman N. How useful are ferric chloride models of arterial thrombosis? Platelets. 2020;31(04):432–438. doi: 10.1080/09537104.2019.1678119. [DOI] [PubMed] [Google Scholar]
- 39.Woollard K J, Sturgeon S, Chin-Dusting J P, Salem H H, Jackson S P. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J Biol Chem. 2009;284(19):13110–13118. doi: 10.1074/jbc.M809095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tseng M T, Dozier A, Haribabu B, Graham U M. Transendothelial migration of ferric ion in FeCl3 injured murine common carotid artery. Thromb Res. 2006;118(02):275–280. doi: 10.1016/j.thromres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Hahl P, Davis T, Washburn C, Rogers J T, Smith A. Mechanisms of neuroprotection by hemopexin: modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J Neurochem. 2013;125(01):89–101. doi: 10.1111/jnc.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poillerat V, Gentinetta T, Leon J. Hemopexin as an inhibitor of hemolysis-induced complement activation. Front Immunol. 2020;11:1684. doi: 10.3389/fimmu.2020.01684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hovig T, McKenzie F N, Arfors K E.Measruement of the platelet response to laserinduced microvascular injury. Ultrastructural studies Thromb Diath Haemorrh 197432(2-3):695–703. [PubMed] [Google Scholar]
- 44.Burkhart J M, Vaudel M, Gambaryan S. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–e82. doi: 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 45.Dunster J L, Unsworth A J, Bye A P. Interspecies differences in protein expression do not impact the spatiotemporal regulation of glycoprotein VI mediated activation. J Thromb Haemost. 2020;18(02):485–496. doi: 10.1111/jth.14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435–3445. doi: 10.1074/mcp.M114.038513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jamasbi J, Megens R T, Bianchini M. Cross-linking GPVI-Fc by anti-Fc antibodies potentiates its inhibition of atherosclerotic plaque- and collagen-induced platelet activation. JACC Basic Transl Sci. 2016;1(03):131–142. doi: 10.1016/j.jacbts.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulz C, Penz S, Hoffmann C. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res Cardiol. 2008;103(04):356–367. doi: 10.1007/s00395-008-0722-3. [DOI] [PubMed] [Google Scholar]
- 49.Shao B, Hoover C M, Shi H. Deletion of platelet CLEC-2 decreases GPIbα-mediated integrin aIIbb3 activation and decreases thrombosis in TTP. Blood. 2022;139(16):2523–2533. doi: 10.1182/blood.2021012896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown H C, Beck S, Navarro S.Antibody-mediated depletion of human CLEC-2 in a novel humanised mouse model. bioRxiv 2021. Doi: 2021.10.03.462933 [DOI] [PMC free article] [PubMed]
- 51.Chauhan A, Sheriff L, Hussain M T. The platelet receptor CLEC-2 blocks neutrophil mediated hepatic recovery in acetaminophen induced acute liver failure. Nat Commun. 2020;11(01):1939. doi: 10.1038/s41467-020-15584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolson P LR, Nock S H, Hinds J. Low-dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica. 2021;106(01):208–219. doi: 10.3324/haematol.2019.218545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.