

Summary
Tumors with BRCA1 mutations have poor prognoses due to genomic instability. Yet this genomic instability has risks and BRCA1-deficient (def) cancer cells must develop pathways to mitigate these risks. One such risk is the accumulation of unfolded proteins in BRCA1-def cancers from increased mutations due to their loss of genomic integrity. Little is known about how BRCA1-def cancers survive their genomic instability. Here we show that BRCA1 is an E3 ligase in the endoplasmic reticulum (ER) that targets the unfolded protein response (UPR) stress sensors, Eukaryotic Translation Initiation Factor 2-alpha Kinase 3 (PERK) and Serine/Threonine-Protein Kinase/Endoribonuclease Inositol-Requiring Enzyme 1 (IRE1) for ubiquitination and subsequent proteasome-mediated degradation. When BRCA1 is mutated or depleted, both PERK and IRE1 protein levels are increased, resulting in a constitutively activated UPR. Furthermore, the inhibition of protein folding or UPR signaling markedly decreases the overall survival of BRCA1-def cancer cells. Our findings define a mechanism used by the BRCA1-def cancer cells to survive their increased unfolded protein burden which can be used to develop new therapeutic strategies to treat these cancers.
Subject areas: Cell biology, Functional aspects of cell biology, Cancer
Graphical abstract
Highlights
-
•
High level of unfolded proteins is detected in BRCA1-deficient cancer cells
-
•
BRCA1 is an ERAD E3 ligase that targets PERK and IRE1 for proteasomal degradation
-
•
Under-ubiquitination of PERK and IRE1 results in a constitutively activated UPR
-
•
Further stressing UPR is lethal to BRCA1-def cancer cells
Cell biology; Functional aspects of cell biology; Cancer.
Introduction
Breast cancer is the most prevalent cancer in women worldwide1 and breast cancer with mutations in the BRCA1 gene will accumulate more genetic mutations over time than most other cancers.2 BRCA1 is a tumor-suppressor protein that promotes genome integrity through pleiotropic functions, including DNA double-strand break repair by homologous recombination (HR).3,4,5,6 Mutations in the BRCA1 gene will render a cell susceptible to genomic instability through defective DNA repair, leading to an increased risk of breast cancer.7 Mice deficient in BRCA1 have been shown to develop basal-like mammary carcinomas.8 With increased genetic mutations in BRCA1-def cells, it is reasonable to hypothesize that the cells would also generate more mutant proteins. However, while there have been significant advances in understanding how mutations in BRCA1 affect genomic integrity, the effect of mutations in BRCA1 on protein homeostasis is less known. Therefore, a deeper understanding of protein homeostasis in BRCA1-def cancer cells will allow us to better understand the downstream impact of BRCA1 deficiency.
BRCA1 protein was initially thought to be primarily nuclear in location but recent studies have demonstrated that it is also found in other subcellular locations, including the endoplasmic reticulum (ER)9,10,11,12 The ER is a cellular organelle where about one-third of all proteins are synthesized. Proteins secreted to extracellular environment, membrane proteins, and proteins that reside inside the ER lumen are all synthesized and folded into their native conformation in this organelle.13,14,15,16 To resolve ER stress and restore protein homeostasis, cells have evolved conserved quality-control mechanisms in the ER termed the Unfolded Protein Response (UPR), endoplasmic reticulum-associated degradation (ERAD) and autophagy.16 ERAD is a highly conserved protein quality control machinery used by the ER to eliminate misfolded or unassembled proteins via the cytosolic ubiquitin-proteasome system (UPS).17,18,19 During the retrotranslocation and ubiquitination steps of the ERAD process, an E3 ubiquitin ligase will form a complex with other substrate-recognizing proteins such as SEL1L and DERLIN-1 to assemble a retrotranslocon and initiate the polyubiquitination of the substrate that targets it for proteasomal degradation.20,21,22,23,24,25,26,27,28
Although BRCA1 was identified more than two decades ago,29 it was not until recently that the role of the RING domain began to emerge. Studies have now revealed multiple mechanisms by which this domain confers the tumor suppressor functions of BRCA1. Sequences encompassing the RING domain of BRCA1 mediate its stable association with the BRCA1-associated RING domain protein 1 (BARD1) which is important for their mutual stability,30,31,32,33 nuclear localization, recruitment to DNA damage sites,34,35 and ubiquitin E3 ligase activity.36,37,38
In this present work, we have investigated the effect of BRCA1 deficiency on protein homeostasis by exploring its E3 ligase activity targeting proteins for degradation outside the nucleus. We have established a connection between BRCA1, the ERAD, and the UPR and identified the dependency of BRCA1-def cancer cells on the UPR for survival. Furthermore, we show that we can selectively target BRCA1-def cancer cells by inhibiting the UPR chaperone protein, cyclophilin B.
Results
BRCA1-def breast cancer cells have a high level of unfolded proteins
To address whether increased genomic instability would lead to increased unfolded protein production, we labeled BRCA1-deficient (def) and BRCA1-replete (+) breast cancer cells with tetraphenylethene maleimide (TPE-MI), a fluorescent dye that targets unfolded proteins.39 TPE-MI fluorescence is activated upon binding to free cysteine thiols, normally buried in the core of globular proteins but exposed upon unfolding. In Figure 1, MDA-MB-436 BRCA1+ or BRCA1-def breast cancer cells were labeled with TPE-MI before fixation for confocal immunofluorescence microscopy (IF) or harvested for flow cytometry. In IF analysis, fixed cells were further labeled with concanavalin-A to outline the ER subcellular compartment. Our results show that the TPE-MI signal intensity is significantly higher (1.4-fold increase) in BRCA1-def cancer cells than in BRCA1+ cancer cells (Figures 1A and 1C). In flow cytometric analysis, a 4-fold increase of TPE-MI fluorescence signal was detected in BRCA1-def cells when compared to the BRCA1+ cells (Figures 1B and 1D).
Figure 1.

Increased accumulation of unfolded protein in BRCA1-def cells, localization of BRCA1 protein to the ER, and interaction with ERAD E3 ligase complex components
(A) Photomicrographs of MDA-MB-436(+ or def) cells labeled with TPE-MI (light/blue) and concanavalin-A (red). Scale = 20 μm. TPE = TPE-MI. + = BRCA1-replete. def = BRCA1-deficient.
(B) Representative flow cytometry dot plots of TPE-high population in BRCA1-def and BRCA1+ cells.
(C and D) Quantitative analysis of TPE signal intensity of each cell preparation.
(E) Confocal microscopy images of BRCA1-proficient MDA-MB-231 breast cancer cells. Top, cells were immunostained with BRCA1 (red) and DERL1 (DERLIN-1) (yellow) antibodies. Bottom, cells were immunostained with secondary antibodies conjugated with fluorophores as control. All cells were counterstained with DAPI (blue). Scale = 5 μM. Green = GFP-KDEL.
(F and G) Immunoprecipitation (IP) of BRCA1 protein pull-downs of DERL1 and SEL1L from MCF7 (F) or MDA-MB-231 (G) cytoplasmic protein extract. IB = immunoblotting. IN = Input. Ig = non-specific immunoglobulin fragments. IgG(H) & IgG(L) = heavy and light chains. BRCA1 IB: ∗∗ = truncated BRCA1. ∗∗∗ = delta11q isoform. Data are represented as mean ± SD. P value was calculated by Student’s two-tailed, unpaired t-test. ∗∗∗ <0.001. See also Figures S1–S3.
We repeated the experiment with HCC1937 BRCA1+ and HCC1937 BRCA1-def breast cancer cells. In IF analysis, we observed a 2.9-fold higher TPE-MI signal intensity in the BRCA1-def cancer cells (Figures S1A and S1C). When analyzed by flow cytometry, a 1.4-fold increase of TPE-MI fluorescence was detected from the BRCA1-def cells when compared to the BRCA1+ cells (Figures S1B and S1D).
BRCA1 interacts with SEL1L and DERLIN-1 without mediating their ubiquitination
Confocal photomicrographs of BRCA1-proficient MDA-MB-231 breast cancer cells transduced with baculoviruses that carried a GFP transgene with a KDEL ER retention signal sequence are shown in Figure 1E. BRCA1 protein (red) localized to the nucleus (blue), cytoplasm, and ER (green). DERLIN-1 (yellow) co-localized with BRCA1 protein in the ER and cytoplasmic compartments (Figure 1E). Single-color panels (white background) and the z-stack images (black background) are shown in Figure S2. These results indicate that the BRCA1 protein is present in both nucleus and cytosol and can colocalize with the ERAD component DERLIN-1 in the ER. BRCA1/DERLIN-1 colocalization immunostaining was also performed in MCF7 cancer cells with similar results (Figure S3).
After confirming the cytoplasmic and ER localization of wildtype BRCA1 protein, we investigated whether BRCA1 also played a role in the ERAD machinery through its E3 ligase activity. We found that BRCA1 can form protein complex with the ERAD retrotranslocon components, SEL1L and DERLIN-1 (Figures 1F, 1G, S4A, and S4B). However, our analyses shows that there was no difference in protein or ubiquitination levels of SEL1L or DERLIN-1 in BRCA1-depleted MCF7 or MDA-MB-231 cells compared to controls (Figures S4C and S4D). Therefore, BRCA1 does not appear to mediate the ubiquitination or degradation of SEL1L or DERLIN-1. However, the interaction between these three proteins does imply that BRCA1 is a component of the ERAD E3 ligase complex.
BRCA1 depletion results PERK and IRE1 overexpression
We hypothesized that since BRCA1 could be acting as an E3 ligase in the ERAD machinery, its deficiency might have an impact on the UPR signaling. To test this hypothesis, we transiently depleted BRCA1 in MCF7 and MDA-MD-231 cells and analyzed the expression of the UPR stress sensor proteins PERK, IRE1, and ATF6 (Figures 2A and 2B). While the effect of BRCA1 deficiency on ATF6 was less clear, the expression of both PERK and IRE1 proteins was significantly increased when BRCA1 was depleted (Figures 2A and 2B). BRCA1-def cancer cells also demonstrated much higher PERK and IRE1 protein expression compared to cognate BRCA1+ cells (Figure 2C). Quantitative analyses of PERK and IRE1 protein signal intensities detected from BRCA1+ and BRCA1-def cancer cells are summarized in Figures S5A and S5C. We also over-expressed BRCA1 or BARD1 protein in BRCA1-def cells to determine whether it would have an opposite effect on the protein levels of PERK and IRE1. Figure 2D show both PERK and IRE1 protein levels were significantly reduced when BRCA1 or BARD1 was overexpressed.
Figure 2.

BRCA1 protein interacts with and ubiquitinates PERK and IRE1
(A–D) Total protein extracts were isolated from (A) control or BRCA1-depleted MCF7, (B) control or BRCA1-depleted MDA-MB-231cells, (C) MDA-MB-436 cells (+ or def), and (D) control, BRCA1 or BARD1 over-expressing MDA-MB-436 BRCA1-def cells.
(E and F) Immunoprecipitation (IP) of BRCA1 protein pull-downs of PERK and IRE1 from MCF7 (E) or MDA-MB-231 (F) cytoplasmic protein extract. BRCA1 IB: ∗ = hyperphosphorylated BRCA1, ∗∗ = truncated BRCA1. ∗∗∗ = delta11q isoform. IRE1 IB: ∗ = possible ubiquitinated IRE1.
(G and H) Ubiquitination analysis of PERK and IRE1 in (G) MCF7 cells or (H) MDA-MB-231 cells. Cells were transfected with control or BRCA1 siRNA. BRCA1 siRNA transfected cells were either untreated or treated with DMSO or Bortezomib overnight before harvesting for protein analysis.
(I and J) In vitro ubiquitination analysis of (I) PERK or (J) IRE1 with E1, UBE2J1, BRCA1, BARD1, and/or ubiquitin. Ubiquitinated PERK or IRE1 was detected with an anti-Ub antibody. Ub = Ubiquitin. Bortz = Bortezomib. Data are represented as mean ± SD. P value was calculated by Student’s two-tailed, unpaired t-test. ∗ <0.05. See also Figures S4–S6.
These data suggested that BRCA1 may regulate the UPR stress sensors PERK and IRE1 at the protein level. To confirm this, we performed qPCR analysis on BRCA1-depleted MDA-MB-231 and MCF7 cells (Figure S5D) and BRCA1- or BARD1-overexpressing MDA-MB-436 BRCA1-def cells (Figure S5E) and demonstrated that neither PERK nor IRE1 mRNA levels was significantly affected by BRCA1 depletion or BRCA1/BARD1 overexpression.
BRCA1 mediates the ubiquitination of PERK and IRE1
Since there was an increase in protein levels of PERK and IRE1 in the BRCA1-depleted MCF7 or MDA-MB-231 cells that were independent of their mRNA levels, we postulated that BRCA1 may be targeting PERK and IRE1 for ubiquitination. We performed co-immunoprecipitation assays with cytoplasmic protein extracts from BRCA1-proficient MCF7 (Figure 2E) and MDA-MB-231 (Figure 2F) cells. Our results show that BRCA1 can form protein complexes with PERK and IRE1. Reverse co-immunoprecipitation using PERK or IRE1 antibodies also pulled down BRCA1 protein (Figure S6A). We repeated the co-immunoprecipitation experiments with proteins isolated from MCF7 ER microsomes and our results show that BRCA1 can form complexes with PERK and IRE1 from proteins isolated from this cellular subfraction (Figure S6B). We then examined the ubiquitination levels of PERK and IRE1 in control and BRCA1-depleted MDA-MB-231 and MCF7 cells. Additional BRCA1-depleted MDA-MB-231 and MCF7 cells were also prepared for DMSO or bortezomib treatment. Our ubiquitination analyses show that there were significant decreases in the ubiquitination of PERK and IRE1 in BRCA1-depleted cells compared to the controls (Figures 2G, 2H, and S6C). Bortezomib treatment resulted in a significant increase of ubiquitinated PERK and IRE1 in the BRCA1-depleted cells, suggesting that ubiquitinated PERK and IRE1 are subjected to proteasomal degradation (Figures 2G, 2H, and S6C). Protein levels of BRCA1, PERK, IRE1, ubiquitin, and GAPDH in control or BRCA1-depleted cells and with or without bortezomib treatment confirmed the proteasome-mediated degradation of IRE1 and PERK (Figure S6D).
BRCA1 ubiquitinates PERK and IRE1
To determine whether BRCA1 can directly ubiquitinate PERK and IRE1, we performed in vitro ubiquitination assays using full-length BRCA1, BARD1, PERK, and IRE1 proteins. Figures 2I and 2J demonstrate that BRCA1 in the presence of BARD1 can ubiquitinate both PERK and IRE1 efficiently. However, while strong PERK and strong IRE1 ubiquitination signals could be seen when both BRCA1 and BARD1 were present (Figures 2I and 2J, lanes 5–7), a mild IRE1 ubiquitination signal was detected when only BRCA1 or BARD1 was used (Figure 2J, lanes 1–2) suggesting that a less efficient ubiquitination process can also be carried out on some protein substrates by either BRCA1 or BARD1 protein alone. In addition, IRE1 was ubiquitinated more rapidly by the BRCA1/BARD1 E3 ligase heterodimer than PERK. Maximal ubiquitination signal of IRE1 could be reached at 10 min or less whereas the maximal ubiquitination signal of PERK required at least 30 min to achieve (Figures 2I and 2J). We repeated the assay using an unrelated protein, NSP15,40 as a negative control for the specificity of the BRCA1 E3 ligase function (Figure S6E).
We also examined BARD1 protein levels in BRCA1+ and BRCA1-def cancer cells. We speculated that if the heterodimer formation is important for the mutual stability of BRCA1 and BARD1, BARD1 should be less stable in the BRCA1-def cells since there will be fewer stable BRCA1 proteins available to form heterodimers. Our protein analysis shows that the level of BARD1 protein was indeed lower in BRCA1-def cells than in BRCA1+ cells (Figure S7).
Depletion of unfolded protein response components decreases the clonal survival of the BRCA1-def cancer cells
Our data imply that BRCA1 deficiency itself results in a survival pathway for the protein mutational burden caused by the inherent genomic instability in these cells. BRCA1 deficiency promotes UPR signaling by constitutively under-ubiquitinating and thereby increasing the expression and activation of PERK and IRE1 (Figure 2). To test whether these UPR signaling pathways support the survival of BRCA1-def cancer cells, we depleted key ERAD and UPR components in BRCA1-def cancer cells and examined their clonal survival in colony formation assays. While all ERAD/UPR components depleted had some impact on the clonal survival of at least one of the BRCA1-def cancer cell lines, only the inhibition of IRE1, BiP or cyclophilin B (CypB) showed severe lethality in all three of the BRCA1-def cancer cell lines (Figure 3A, red asterisks). Furthermore, despite the profound impact of CypB inhibition on the viability of BRCA1-def cells, inhibition of CypB in BRCA1+ cells had little effect on their survival (Figure 3A, blue asterisk & Figure 3B). To further support this selective targeting of BRCA1-def cells, we analyzed additional BRCA1-def breast/ovarian cancer cell lines and found that they are also susceptible to CypB depletion (Figure S8A). These results suggest a specific targeting of BRCA1-def cancer cells can be achieved by depleting CypB. The efficiency of all siRNA transfection was confirmed by Western blot (Figure S8B). We repeated the BRCA1-def cancer cell survival assays using two distinct single CypB siRNAs respectively. Figure 3C shows that while both siCypB-A and siCypB-B can target BRCA1-def cancer cells effectively, siCypB-B did not induce synthetic lethality in BRCA1+ cancer cells.
Figure 3.

Depleting UPR components is lethal to the BRCA1-def cancer cells
(A) MDA-MB-436 (BRCA1-def or +), HCC1937 BRCA1-def and SUMPT149 BRCA1-def breast cancer cells were transfected with control, CypB, IRE1, GRP94, HSP70, BiP, PERK or DERL1 siRNA and assessed for cell survival.
(B) HCC1937 BRCA1+, MDA-MB-231, MCF7, and MCF10A cells were transfected with CypB siRNA and assessed for cell survival.
(C) MDA-MB-436 (+ or def) and HCC1937 (+ or def) breast cancer cells were transfected with control, CypB-A or CypB-B siRNA. Top, western blot analysis. Bottom, clonal cell survival analysis.
(D) MDA-MB-436 BRCA1-def cells were transfected with either (i) pCMV vector and siRNA control, (ii) PPIB/pCMV and siRNA control, (iii) pCMV control and CypB 3′UTR siRNA or (iv) PPIB/pCMV and CypB 3′UTR siRNA for cell survival and Western blot analysis. N.S. = not significant. F-CypB = Flag-tagged cyclophilin B. Bar charts are the summary of the quantitative analysis of the colony forming units (CFU) from each siRNA transfection experiment. Data are displayed as mean ± SD. P values were calculated by Student’s two-tailed, unpaired t-test. See also Figures S7 and S8.
To further validate that the severe lethality induced in the BRCA1-def cancer cells was specifically caused by CypB depletion, we performed a rescue assay in MDA-MB-436 BRCA1-def cancer cells. Figure 3D shows that in CypB (PPIB) over-expressing/siCypB BRCA1-def cells, cell viability was rescued back to ∼80% from <10% when cells were transfected with parental vector and CypB 3′UTR siRNAs (pCMV/siCypB) compared to pCMV/siControl.
Cyclophilin inhibitors increase unfolded proteins and protein disulfide isomerase aggregation in BRCA1-def cancer cells
There are multiple cyclophilin B inhibitors, and three have been clinically tested, with one, cyclosporin A (CsA) achieving FDA approval for the prevention of rejection of transplanted solid organs.41,42,43 To determine whether CypB is critical to alleviating the unfolded protein burden in BRCA1-def cancer cells, we treated the MDA-MB-436 BRCA1+ and BRCA1-def cells with CsA for 2 days and assessed the amount of unfolded protein in the CsA-treated cells. In Figures 4A–4D, DMSO- or CsA-treated MDA-MB-436 BRCA1+ or BRCA1-def breast cancer cells were labeled with TPE-MI before fixation for immunofluorescence staining (IF) or harvested for flow cytometry. Our results show that the TPE-MI signal intensity is significantly higher in both CsA-treated BRCA1+ and BRCA1-def cells (∼60% and 20% increase respectively). In flow cytometric analysis, a nine-fold increase of TPE-MI-high population in CsA-treated BRCA1+ cells and an ∼20% increase in the TPE-MI-high population in CsA-treated BRCA1-def cells were detected when compared to their DMSO-treated counterparts. This is consistent with the BRCA1-def cells having a higher baseline of unfolded proteins than the BRCA1+ cells.
Figure 4.

Cyclophilin inhibitor treatment increases unfolded proteins and PDI aggregates formation in BRCA1-def cancer cells
(A) Photomicrographs of DMSO- or CsA-treated MDA-MB-436 (+ or def) cells labeled with TPE-MI (light/blue) and quantitative analysis of TPE signal intensity of each cell preparation. Scale = 20 μm. TPE = TPE-MI. + = BRCA1-replete. def = BRCA1-deficient.
(B) Representative flow dot plots of TPE signals in DMSO- or CsA-treated MDA-MB-436 (+ or def) breast cancer cells and quantitative analysis of TPE-high cell population of each cell preparation.
(C and D) Quantitative analysis of TPE signal intensity of each cell preparation.
(E) Confocal microscopy images of DMSO- or CsA-treated MDA-MB-436 (+ or def) breast cancer cells. Cells were co-immunostained with CypB (red) and PDI (green) antibodies. Top, 2 days post-treatment. Bottom, 3 days post-treatment. All cells were counterstained with DAPI (blue). Scale = 5 μM. Bar charts are quantitative analyses of cell populations with PDI aggregate formation.
(F) Depletion of PDI induced severe synthetic lethality in MDA-MB-436 BRCA1-def cancer cells. Top, Western blot analysis. Middle, representative images of colony forming units (CFUs) from control or CypB knock-down cancer cells after 14 days culturing. Bottom, quantitative analysis of the clonal colony formation assay. Data are displayed as mean ± SD. P values were calculated by Student’s two-tailed, unpaired t-test. See also Figure S8.
It has been reported that protein disulfide isomerase (PDI), a canonical UPR biomarker, interacts with CypB and promotes protein folding,44,45 so we examined whether CypB depletion would affect PDI. In our confocal microscopy analysis, we found that PDI protein levels are normally lower in BRCA1+ cells compared to the BRCA1-def cells (Figure 4E), probably due to a higher demand for protein folding in the latter. After treating with CsA for 2 days, only a moderate level of CypB was seen in BRCA1+ cells which is consistent with previous reports that showed CsA could promote the secretion of CypB outside of the cells.46,47 However, higher expression of PDI is seen in BRCA1-def cells, and aggregates of PDI protein colocalized with CypB appeared in ∼10% of the BRCA1-def cells after 2 days of CsA treatment. At day 3 post-treatment, >30% of the BRCA1-def cells have numerous large PDI protein aggregates (Figure 4E), implying that CypB function is especially important to PDI folding when PDI is highly expressed, such as when the high level of unfolded proteins are present in BRCA1-def cells. These PDI protein aggregates were rarely detected in CsA-treated BRCA1+ cells (Figure 4E).
We also performed cell survival assay on PDI-depleted MDA-MB-436 BRCA1+ and BRCA1-def cells. Figure 4F shows that the survival of BRCA1+ cells was not significantly affected by PDI depletion whereas more than 60% of BRCA1-def cells were killed when PDI was depleted (Figure 4F). These results are similar to CypB inhibition in BRCA1+ and BRCA1-def cancer cells (Figures 3 and S8A).
Cyclophilin inhibitor treatment is detrimental to the survival of BRCA1-def cancer cells
Although CsA is a cyclophilin inhibitor, it is also an immunosuppressant because it also inhibits calcineurin. Two potent CsA derivatives, NIM811 and Alisporivir (Debio-025), inhibit CypB but do not interact with calcineurin. They have both undergone early-phase clinical trials and have been found to be safe. To investigate the therapeutic potential of suppressing CypB in the BRCA1-def breast cancer cells, we treated BRCA1-def and BRCA1+ cell lines with CsA, NIM811, or Alisporivir and examined their viability post-treatment. Figure 5A shows that similar to CypB siRNA depletion, CsA also induces severe synthetic lethality in all BRCA1-def breast cancer cell lines tested. For BRCA1-def HCC1937, SUM149PT, and UWB1.289 cells, the IC50 value of CsA was ≤1 μM whereas the IC50 value for the BRCA1-def MDA-MB-436 cells was 1 μM, which is a clinically achievable level.
Figure 5.

Cyclophilin inhibitors induce severe synthetic lethality in BRCA1-def cancer cells in vitro and in vivo
(A) BRCA1-def cancer cells (MDA-MB-436, HCC1937, SUM149PT or UWB1.289), BRCA1-proficient cancer cells (MDA-MB-436, MDA-MB-231 or MCF7) and non-tumorigenic mammary epithelial cells (MCF10A) were seeded one day prior to drug treatment. A single continuous dose of CsA, NIM811 or Alisporivir was added to the corresponding cell lines. Quantitative analysis of each survival curve is shown in graphs. CsA = Cyclosporin A. CFU = Colony forming units. DMSO = dimethyl sulfoxide vehicle.
(B) CsA treatment of human BRCA1-def breast cancer cells in a murine model. Representative luciferase images of each treated subject were taken by an In Vivo Imaging systems (IVIS). Animals were either treated with vehicle (control) or CsA (treatment) for 6 weeks post-tumor formation.
(C) Quantitative analysis of tumor progression from each group was summarized in graph.
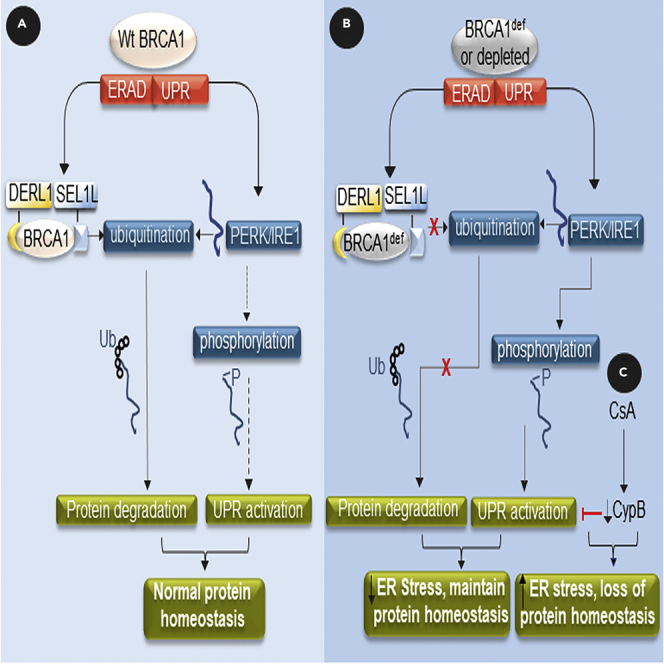
(D) Schematic diagram depicts the model of wildtype and mutant BRCA1 regulation of the ERAD and UPR signaling pathways to maintain protein homeostasis in the ER. Wt = wildtype. def = deficient. Ub = ubiquitin. P = phosphate. Dashed arrow = less favored pathway. Ubiquitination and phosphorylation modifications in the diagram are simplified for clarity. Data are displayed as mean ± SD. P values were calculated by Student’s two-tailed, unpaired t-test.
We also examined some key UPR signaling pathway components in MDA-MB-436 BRCA1+ and BRCA1-def cells with or without CsA treatment. Figure S9A shows that the UPR signaling pathway is more active in the BRCA1-def cells than in the BRCA1-replete cells. Protein levels of PERK and IRE1 (native and phosphorylated), phosphorylated-eIF2a, ATF4, BiP, and GRP94 are all elevated in the BRCA1-def cells. QPCR analysis on the mRNA level of XBP1 also shows a five-fold increase of spliced (sp) XBP1 and total XBP1 in the BRCA1-def cells compared to the BRCA1+ cells (Figure S9B). Up to a 10-fold increase of sp XBP1 and total XBP1 mRNA was detected in CsA-treated BRCA1+ cells (Figure S9B), indicating that CypB inhibition activates the UPR in these cells. Contrarily, a slight decrease of sp XBP1 mRNA level was observed when the BRCA1-def cells were treated with CsA (Figure S9B), perhaps because the cells have already committed to a cell death fate.
The IC50 values of the BRCA1-def cancer cells treated with NIM811 or Alisporivir are similar to the results of the CsA treatment (Figure 5A). The IC50 values of CsA or NIM811 treatment for the BRCA1+ cell lines, MDA-MB-436+, MDA-MB-231, and MCF10A cells, were up to 7-fold higher than the BRCA1-def cancer cells (Figure 5A). Another BRCA1+ breast cancer cell line, MCF7, also has a 3-fold higher IC50 value compared to the BRCA1-def cancer cells. Since all BRCA1-def cancer cell lines show similar sensitivity to CsA, NIM811, and Alisporivir, the mechanism of synthetic lethality in the BRCA1-def cancer cells is likely due to the inhibition of CypB and not the inhibition of calcineurin.
Cyclosporin A treatment in murine xenograft model
After demonstrating that BRCA1-def cancer cells were equally susceptible to CypB inhibition in vitro, we established a murine xenograft model to investigate the efficacy of CsA on BRCA1-def tumors in vivo. Luciferase-expressing SUM149PT BRCA1-def breast cancer cells were used to induce tumor formation subcutaneously in athymic nude mice. Figure 5B shows images of animals from either control (vehicle) or CsA-treated group captured by an In Vivo Imaging System during a six-week study period. Figure 5C shows the summary of the tumor progression of the animals from each group. We found that CsA treatment at 22.5 mg/kg/q2d intraperitoneally was effective in suppressing the growth of the BRCA1-def tumors without any signs of treatment toxicity.
Figure 5D depicts a schematic diagram of our findings: (A) Wildtype BRCA1 protein is a component of ERAD, interacting with SEL1L/DERLIN-1 and regulating the protein levels of PERK/IRE1 via ubiquitination, and (B) BRCA1-deficiency results in higher PERK and IRE1 protein levels and increased activation via auto-phosphorylation. (C) In the presence of cyclophilin inhibitors such as CsA, the UPR survival pathway is compromised, resulting in cell death.
Discussion
BRCA1 protein consists of four major domains: the RING domain, the BRCA1 serine domain, and 2 BRCT domains.48,49 The BRCT domains are phosphopeptide recognition modules, and the RING finger domain has ubiquitin E3 ligase activity.49 The BRCT domains bind to phosphorylated partners such as CtIP that are involved in the homologous recombination DNA damage response.50,51 The RING domain of BRCA1, together with its interacting partner BARD1, constitutes a potent ubiquitin E3 ligase.52,53 Here, we investigated the dynamic between the loss of DNA-repair activity and the loss of E3 ligase activity of BRCA1 protein in BRCA1-def cancer cells. When BRCA1 is mutated or depleted, HR DNA repair becomes less efficient, which drives cells towards error-prone DNA repair pathways, especially to repair replication fork damage.54,55 We hypothesized that the loss of DNA repair and subsequent genomic instability in BRCA1-def cancer cells would increase unfolded/misfolded mutant proteins. We confirmed this hypothesis by demonstrating increased levels of unfolded proteins in BRCA1-def cancer cells compared to BRCA1+ cancer cells (Figures 1 and S1).
In this study, we also uncovered a previously unknown E3 ligase function of BRCA1 in the ER, ubiquitinating PERK and IRE1 for proteasome-mediated degradation. This finding establishes a role for BRCA1 in the UPR56 and identifies new targets for BRCA1-BARD1 ubiquitination for proteasomal destruction. The UPR can initially decrease the unfolded protein burden but continued high levels of unfolded proteins will lead to cell death.57 Since BRCA1-def cancer cells have a high mutational burden,2 resulting in more unfolded/misfolded proteins, the under-ubiquitination of PERK and IRE1 due to BRCA1 deficiency leads to a constitutively activated UPR. This in turn can assist in processing the higher levels of unfolded proteins in these cells and thereby restore protein homeostasis, alleviate ER stress, and promote survival.
Currently, there are at least a dozen of ERAD E3 ligases reported to contribute to the ubiquitination of misfolded proteins in the ERAD. Several are transmembrane proteins such as HRD1.58,59,60,61,62,63 Others are cytoplasmic E3 ligases that have been demonstrated to be involved in ERAD.64,65,66,67,68,69 HRD1 forms ERAD E3 ligase complex with SEL1L and DERLIN-1 and promotes unfolded protein recognition and ubiquitination, recruitment to the retrotranslocon, retrotranslocation, extraction, and transport to the proteasome for degradation.70,71 Several endogenous substrates for SEL1L/HRD1-mediated ERAD, including IRE1 and p53, have been identified.72,73,74,75,76 Although it has been shown that expression of HRD1 is significantly downregulated in breast cancer tissues,77 it is possible that other ERAD E3 ligases beside BRCA1 are functional in the breast cancer cells. Therefore, the impact of BRCA1 deficiency on the ERAD machinery in breast cancer cells is currently unknown.
Our study has uncovered a pro-survival pathway used by BRCA1-def cancer cells to address mutant protein homeostasis by constitutively over-expressing PERK and IRE1 via loss of ubiquitination. The susceptibility of BRCA1-def cancer cells to the depletion of UPR components supports the notion that the UPR signaling pathway is instrumental to the survival of these cells. The same mutation that results in more unfolded proteins creates a pathway to deal with them. This is a crucial component of BRCA1-def oncogenesis, since without this pathway BRCA1-def cells would not survive transformation. This is likely one reason why biallelic BRCA1 loss does not result in cell death but becomes an oncogenic event.
Our data show that CypB depletion or inhibition can specifically kill BRCA1-def cancer cells (Figures 3 and S8). CypB is a peptidyl-prolyl isomerase (PPIase) enzyme that can localize to the ER or the nucleus.78,79 CypB assists in folding newly synthesized proteins and regulates stability, localization, and activity of mature proteins.80,81 As such, CypB can attenuate ER stress via its PPIase activity82 and suppress unfolded protein-induced cell death. We hypothesize that the loss of CypB increases unfolded proteins past a level where the increased levels and activation of PERK/IRE1 are able to maintain protein homeostasis in BRCA1-def cells (Figures 4A and 4B). The appearance of large PDI protein aggregates in CsA-treated BRCA1-def cancer cells also resembles the PDI aggregate formation in Brefeldin-A-treated cells which suggests compromised vesicle formation and protein transport network in the Golgi apparatus,83 and may be a mechanism by which CypB loss causes BRCA1-def cell death.
Taken together, our study shows that disrupting the protein homeostasis via down-regulating CypB is a new and important target in BRCA1-def cancers with a potentially wide therapeutic index since its depletion or inhibition does not affect BRCA1-replete/BRCA1-proficient cells. There is already an enormous clinical experience with the safety of CsA and some with Alisporivir and NIM811, therefore, this study has provided evidence for a new therapeutic strategy to treat BRCA1-def breast and ovarian cancers.
Limitations of the study
The behavior of tumors in situ is not necessarily replicated by immortalized cell lines. Thus, future animal studies will be necessary to validate the role of CypB inhibitors for the treatment of in vivo BRCA1-def cancers in order to provide a solid rationale for advancement to clinical trials.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ATF4 | Cell Signaling | Cat# 11815; RRID: AB_2616025 |
| Anti-ATF6 | Cell Signaling | Cat# 65880; RRID: AB_2799696 |
| Anti-BARD1 | Thermo Fisher | Cat# 22964-1-AP; RRID: AB_2879190 |
| Anti-BRCA1 | Novus Biologicals | Cat# AF2210; RRID: AB_2067618 |
| Anti-BRCA1 | Santa Cruz Biotech | Cat# sc-6954; RRID: AB_626761 |
| Anti-Cyclophilin B | Abcam | Cat# ab 178397 |
| Anti-Cyclophilin B | ThermoFisher | Cat# MA5-31392; RRID: AB_2787028 |
| Anti-DERLIN 1 | Santa Cruz Biotech | Cat# sc-293385 |
| Anti-DERLIN 1 | Origene Technologies | Cat# TA301554 |
| Anti-GAPDH | Cell Signaling | Cat# 2118; RRID: AB_561053 |
| Anti-IRE1 | Cell Signaling | Cat# 3294; RRID: AB_823545 |
| Anti-IRE1 (phospho) | Abcam | Cat# ab124945; RRID: AB_11001365 |
| Anti-IRE1 | Santa Cruz Biotech | Cat# sc-100772; RRID: AB_1124115 |
| Anti-PERK | Cell Signaling | Cat# 5683; RRID: AB_10841299 |
| Anti-PERK (phospho) | Abcam | Cat# ab192591; RRID: AB_2728666 |
| Anti-PERK | Santa Cruz Biotech | Cat# sc-377400; RRID: AB_2762850 |
| Anti-SEL1L | Santa Cruz Biotech | Cat# sc-377350 |
| Anti-Ubiquitin agarose beads conjugated | Santa Cruz Biotech | Cat# sc-8017; RRID: AB_628423 |
| Anti-Ubiquitin | Cell Signaling | Cat# 43124; RRID: AB_2799235 |
| Anti-Flag M2 affinity gel | Millipore Sigma | Cat# A2220; RRID: AB_10063035 |
| Anti-Strep-tactin | Neuromics | Cat# 2-1502-001 |
| Anti-GRP94 | Cell Signaling | Cat# 2104; RRID: AB_823506 |
| Anti-BiP | Cell Signaling | Cat# 3177; RRID: AB_2119845 |
| Anti-HSP70 | Cell Signaling | Cat# 46477; RRID: AB_2687559 |
| Anti-eIF2a | Cell Signaling | Cat# 5324; RRID:AB_10692650 |
| Anti-eIF2a (phospho) | Cell Signaling | Cat# 3398; RRID: AB_2096481 |
| Bacterial and virus strains | ||
| CellLight ER GFP, BacMam 2.0 | Thermo Fisher | Cat# C105090 |
| Chemicals, peptides, and recombinant proteins | ||
| Tetraphenylethene maleimide | AIEgen Biotech | Cat# AIETM TPE-MI |
| Concanavalin-A TRITC conjugated | Thermo Fisher | Cat# C860 |
| Bortezomib | SelleckChem | Cat# S1013 |
| UBE1 | R&D Systems | Cat#E−305-025 |
| UBE2J1 | R&D Systems | Cat# E2-750 |
| BRCA1 | Active Motif | Cat# 31113 |
| Ubiquitin | Boston Biochem | Cat# K-200B |
| Cyclosporin A | SelleckChem | Cat# S2286 |
| NIM811 | MedChem Express | Cat# HY-P0025 |
| Alisporivir (Debio-025) | MedChem Express | Cat# HY-12559 |
| Critical commercial assays | ||
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Fisher | Cat# 78835 |
| Quick-RNA Miniprep kit | Zymo Research | Cat# R1054 |
| SuperScriptTM III First-strand Synthesis SuperMix | Fisher | Cat# 11-752-050 |
| SYBR Green Supermix | Bio-Rad | Cat# 1725121 |
| Endoplasmic Reticulum Isolation Kit | Sigma | Cat# ER0100 |
| Deposited data | ||
| https://data.mendeley.com/datasets/72gjb45yjv/1 | ||
| Experimental models: Cell lines | ||
| MDA-MB-436 BRCA1-def | ATCC | Cat# HTB-130; RRID: CVCL_0623 |
| MDA-MB-436 BRCA1-replete (BRCA1+) | Johnson et al., 2013 | NA |
| HCC-1937 BRCA1-def | ATCC | Cat# CRL-2336; RRID: CVCL_0290 |
| HCC-1937 BRCA1-replete (BRCA1+) | Zhang et al., 2004 | NA |
| SUM140PT BRCA1-def | Asterand Biosciences | Cat# SUM149PT; RRID: CVCL_3422 |
| UWB1.289 BRCA1-def | ATCC | Cat# CRL-2945; RRID: CVCL_B079 |
| UWB1.289 BRCA1 replete ∗BRCA1+) | ATCC | Cat# CRL-2946; RRID: CVCL_B078 |
| MDA-MB-231 | ATCC | Cat# HTB-26; RRID: CVCL_0062 |
| MCF7 | ATCC | Cat# HTB-22; RRID: CVCL_0031 |
| HEK293 | ATCC | Cat# CRL-1573; RRID: CVCL_0045 |
| MCF10A | ATCC | Cat# CRL-10317; RRID: CVCL_0598 |
| Experimental models: Organisms/strains | ||
| Athymic nude mice | Jackson Laboratories | Cat# 002019; RRID: IMSR_JAX:002019 |
| Oligonucleotides | ||
| EIF1AK3 | OriGene | Cat# HP208096 |
| ERN1 | OriGene | Cat# HP205316 |
| BRCA1 | OriGene | Cat# HP210038 |
| BARD1 | OriGene | Cat# HP200444 |
| GAPDH | OriGene | Cat# HP205798 |
| sp XBP1 forward primer, 5′- TGCTGAGTCCGCAGCAGGTG -3′; reverse primer, 5′- GCTGGCAGGCTCTGGGGAAG -3′ | NM_001079539.1 | Fung et al., 2014 |
| Total XBP1 forward primer, TTGTCACCCCTCCAGAACATC. reverse primer, TCCAGAATGCCCAACAGGAT | ||
| Control siRNA | Dhamarcon | Cat# D-001810-10-20 |
| BRCA1 siRNA | Dhamarcon | Cat# sL0034610000 |
| CypB siRNA (pooled) | Santa Cruz Biotech | Cat# sc-35145 |
| CypB-A siRNA | Santa Cruz Biotech | Cat# sc-35145a |
| CypB-B siRNA | Santa Cruz Biotech | Cat# sc-35145b |
| CypB 3′UTR siRNA | Horizon Discovery | Cat# CTM-710441 |
| DERLIN-1 siRNA | Santa Cruz Biotech | Cat# sc-60519 |
| IRE1 siRNA | Santa Cruz Biotech | Cat# sc-40705 |
| BiP siRNA | Santa Cruz Biotech | Cat# sc-29338 |
| GRP94 siRNA | Santa Cruz Biotech | Cat# sc-35523 |
| HSP70 siRNA | Santa Cruz Biotech | Cat# sc-29352 |
| PERK siRNA | Santa Cruz Biotech | Cat# sc-36213 |
| PDI siRNA | Santa Cruz Biotech | Cat# sc-36201 |
| Recombinant DNA | ||
| pLenti PGK V5-LUC Neo plasmid | Addgene | Cat# w623-2; RRID: Addgene_21471 |
| BRCA1/pcDNA3.1 | Yeung et al., 2008 | NA |
| BARD1 | Sino Biologicals | Cat# HG15850-CF |
| PERK | OriGene | Cat# RC214993 |
| IRE1 | OriGene | Cat# RC215023 |
| PPIB | OriGene | Cat# RC203180 |
| NSP15 | OriGene | Cat# VC100587 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad v.7.04 | Prism software | https://www.graphpad.com/ |
| Living Image v.4.7.3 | PerkinElmer | https://www.perkinelmer.com/lab-products-and-services/resources/in-vivo-imaging-software-downloads.html#LivingImage |
| FlowJo-Win64–10.8.0 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Kimi Y Kong (kongk@uthscsa.edu).
Materials availability
All newly created reagents made during this study are available upon request to the lead contact after signature of a University of Texas standard materials transfer agreement.
Experimental model and subject details
Cell lines
MDA-MB-436 (ATCC), MCF7 (ATCC) and MDA-MB-231 (ATCC) human breast cancer cells were cultured in D-MEM medium (SH30022.01, Life Technologies) supplemented with 10% fetal bovine serum (FBS) (S12450, Atlanta Biologicals) and 1% penicillin and streptomycin (pen/strep) (SV30010, Life Technologies). HCC1937 human breast cancer cells (ATCC) were cultured in RPMI1640 medium (SH3002701, Life Technologies) supplemented with 10% fetal bovine serum and 1% pen/strep. SUM149PT human BRCA1mut breast cancer cells (Asterand Bioscience) were cultured in Ham’s F-12 medium (11765054, Invitrogen) supplemented with 5% heat inactivated FBS (16140063, Hyclone), 10 mM HEPES (15630080, Invitrogen), 1 μg/mL hydrocortisone (07925, StemCell Technologies) and 5 μg/mL insulin (19278, Sigma-Aldrich). MCF10A human mammary epithelial cells (ATCC) were cultured in MEGM kit medium (CC-3150, Lonza) supplemented with 100 ng/mL cholera toxin (C8052, Sigma). UWB1.289 human ovarian cancer cells (ATCC) were cultured in 50% RPMI1640 medium and 50% MEGM (CC-3150, Lonza) without antibiotics. MDA-MB-436 BRCA1+ (BRCA1-replete) and HCC1937 BRCA1+ (BRCA1-replete) cells were created by stably re-introducing wildtype BRCA1 cDNA back into their BRCA1-def parental cells.84,85
Plasmids
BARD1 pCMV3, BRCA1/pcDNA3, PERK, IRE1, NSP15, PPIB/pCMV plasmid, pCMV parental vector were used in this study.
Animals
8 weeks old female athymic nude mice were purchased one week prior to cell implantation. All the animals were housed with 12 h light-dark cycle. Access to chow and water was ad libitum to the animals. All animal care, maintenance and procedures complied with the regulations of IACUC and University of Florida Animal Housing Facility (IACUC#201710042).
Method details
Transfection, drug treatment and survival assays
BRCA1, Cyclophilin B (CypB), DERL1, IRE1, GRP94, HSP70, BiP (GRP78), or PERK were selectively depleted using siRNA transient transfection (Lipofectamine RNAiMAX transfection Reagent, Life Technologies). In this study, siRNA control, BRCA1, CypB, CypB-A, CypB-B, CypB 3′UTR, DERLIN-1, IRE1, GRP94, HSP70, BiP, PDI and PERK were used. Briefly, the day prior to transfection, cells (MDA-MB-436, HCC1937, SUM149PT, MCF7, MDA-MB-231 or MCF10A) were plated at a density of 1.4 × 105 per well in a 6-well plate. Transfection reagents were prepared by mixing 6 μL of RNAiMAX/250 μL Opti-MEM (Life Technologies) to 50 nM of siRNA/250 μL Opti-MEM at RT for 20 min before adding to cells. 0.5 mL of fresh medium was added to each well between 4-6 h after transfection for all cell types. Cells were harvested at 2d post-transfection for clonal colony formation analysis of survival, Western analysis, immunofluorescence or other assays. All survival assays were performed at least three times in triplicate (n ≥ 9).
Clonal cell survival was determined by seeding transfected cells (1,000 MDA-MB-436 [+ or def], HCC1937 [+ or def], SUM149PT, MCF7 or MCF10A) per well of a 6-well plate. Cells were cultured for 7–10 days (SUM149PT, MCF10A) or 14–18 days (MDA-MB-436 BRCA1 [+ or def], HCC1937 [+ or def]) to allow formation of clonal colonies. Cells were then rinsed with 1X PBS, fixed with 1% formaldehyde for 10 min and stained with 0.1% crystal violet before counting. Colonies ≥50 cells were counted as a surviving clone. Unpaired Student t tests were used for all statistical analysis, unless otherwise indicated. For drug treatment, 1,000 breast or ovarian cancer cells (MBA-MD-436, HCC1937, SUM149PT, UWB1.289, MBA-MD-231 and MCF7) or non-tumorigenic mammary epithelial cells (MCF10A) were seeded per well in a 6-well plate one day prior to drug treatment. Different dosages (1–10 or 1–20 μM) of the selected drug were added to each cell type one day after cell seeding and proceeded to clonal cell survival assay as described above. Drugs used in this assay were Cyclosporin A (CsA), NIM811 and Alisporivir.
CypB rescue assay
One day after the cells were seeded, 1.5 μg of pCMV parental vector or PPIB/pCMV plasmid were transfected into MDA-MB-436 BRCA1-def cells using Fugene 6 transfection reagent (3 μL per 1 μg of DNA) (E2692, Promega) for overnight at 37°C. Culture medium was replaced next day and cells were proceeded for siRNA transfection within an hour. Cells were harvested two days post-transfection of siRNA for survival assay and Western blot analysis. The assay was performed at least 3 times.
Immunofluorescence and microscopy
To label unfolded proteins, we followed a protocol from a previous study.39 Briefly, cells were rinsed with PBS prior to the staining and then treated with freshly diluted tetraphenylethene maleimide (TPE-MI) (50 μM in culture medium) for 30 min at 37°C. The TPE-MI solution was removed and cells were fixed for microscopy (Zeiss Axiovert 200M) or harvested for flow cytometric analysis (Cytek Aurora). Results were collected from three separate experiments. For microscopic analysis, cells were labelled with concanavalin-A. We used ImageJ software to measure the signal intensity of the grey area from each cell in the micrographs, the data summary is the average of the signal intensities from a total of a hundred cells per cell preparation.
Confocal immunofluorescent analysis was performed as following: MCF7 or MDA-MB-231 cells were cultured in a 35 mm FluoroDish cell culture dish (FD35-100, World Precision Instruments) followed by baculoviral transduction of the ER-GFP construct (GFP-KDEL). Cells were fixed the next day with 4% paraformaldehyde (28906, Thermo Fisher Scientific) for 10 min at ambient temperature, rinsed with 1X PBS thrice, incubated with primary antibodies in 5% FBS, 5% BSA (A9647, Sigma), 0.2% saponin (84510, Sigma-Aldrich) and 0.01% sodium azide (S2002, Sigma-Aldrich) overnight at 4°C. BRCA1 (Abcam) and DERLIN-1 antibodies were used at 5 μg/mL concentration. The cells were then washed with 1X PBS thrice. Secondary antibodies (A11004, A-21235, 1:400, Thermo Fisher Scientific) were added to the cells at ambient temperature and protected from light for 1 h. After washing thrice with 1X PBS, cell culture dishes were mounted in an anti-fade solution containing DAPI (P-36931, Thermo Fisher Scientific).
MDA-MB-436 (BRCA1+ or BRCA1-def) cells were prepared similarly for confocal imaging. Cells were treated with either DMSO (control) or 1 μM CsA one day after cell seeding and fixed at 48 h post-treatment. CypB mouse antibodies and PDI rabbit antibodies were used at 1:300 and 1:100 respectively. PDI protein aggregate cell population was calculated by counting cells with large PDI aggregate formation divided by total number of the cells in the same field. Each set of data was gathered from five random fields of image from each cell preparation.
All images were taken by a Zeiss LSM 710 confocal either via a 63X or 100X objective (UTHSA Optical Imaging Facility) and processed by Zen Black edition software (Carl Zeiss Microscopy). Photomicrographs of distinct cell populations were taken at equal magnifications and equal fluorescence intensities. Each immunofluorescence assay was performed at least 3 times.
Western blot and immunoprecipitation assays
Protein expression of BARD1, BRCA1, CypB, PERK, IRE1, ATF6, SEL1L, DERLIN-1, and the constitutively expressed GAPDH was monitored by standard western blotting. Antibodies were obtained as follows: PERK, IRE1, ATF6, BRCA1, DERLIN-1, SEL1L, GAPDH, CypB, Rabbit IgG HRP-conjugated (NA934-1ML, Thermo Fisher Scientific), Mouse IgG HRP-conjugated (NA931-1ML, Thermo Fisher Scientific) and Goat IgG HRP-conjugated (sc-2020, Santa Cruz Biotech). SuperSignal West Pico Chemiluminescent Substrate (ECL) (34078, Thermo Fisher Scientific) and High Performance Chemiluminescence film (45001508, Thermo Fisher Scientific) were used for the ECL signal detection. Total protein was extracted from all transfected cells using Pierce IP lysis buffer (87788, Fisher Scientific).
For immunoprecipitation, cytoplasmic protein was extracted from MCF7 or MDA-MB-231 cells using NE-PER Nuclear and Cytoplasmic Extraction Reagents. Briefly, 3.5 μg of antibody (control or target) was added to 20 μL of Protein G Plus/Protein A agarose beads in 250 μL of 1X IP lysis buffer for each preparation. The antibody-agarose bead suspension was incubated for 45 min at ambient temp with shaking and then washed and spun down thrice with 0.5 mL 1X IP lysis buffer for each wash. Beads were subsequently resuspended with 250 μL of 1X IP lysis buffer. 25–60 μg of cytoplasmic protein, depends on the targeted pull-down protein, was added to each antibody-beads preparation for incubation overnight at 4°C with shaking. Beads were washed in PBS and spun down thrice next day. Antigens that bound to control or target antibody was eluted by adding 10 μL of H2O and then boiling the beads for 5 min. The eluants were subjected for Western blot analysis. All protein lysis buffers were supplemented with cocktail of protease (PI 78403, ThermoFisher Scientific) and phosphatase inhibitors (PI 78428, ThermoFisher Scientific). Crude protein extract was used as Input for each reaction.
Protein isolation from endoplasmic reticulum microsomes was performed using the Endoplasmic Reticulum Isolation kit. Briefly, cells were washed with 10 volumes of PBS and centrifuged at 600Xg for 5 min. The packed cell volume (PCV) was estimated and the cells resuspended in a volume of 1X hypotonic extraction buffer (10mM HEPES, 25mM KCL, 1mM EGTA and 1X protease inhibitor cocktail) equivalent to 3 times the PCV and then incubated on ice for 20 min. Cells were centrifuged at 600xg for 5 min and the supernatant was removed by aspiration. The new PCV was measured. A volume of 1x Isotonic Extraction Buffer (10 mM HEPES, pH 7.8, with 0.25 M sucrose, 1 mM EGTA and 25 mM potassium chloride) equivalent to 2 times the new PCV was added and the resuspended cells transferred to a 7 mL Dounce homogenizer. The cells were lysed with 10 strokes of the Dounce homogenizer and then the homogenate was centrifuged at 1000xg for 10 min at 4°C. The thin floating lipid layer was carefully removed by aspiration without aspirating the post nuclear supernatant. The remaining supernatant was transferred to a new centrifuge tube and the pellet discarded. The supernatant was centrifuged at 12,000xg for 15 min at 4°C. The floating lipid layer was carefully removed without aspirating the post mitochondrial supernatant. The remaining supernatant was transferred to a new tube and the pellet discarded. This supernatant fraction, which is the post mitochondrial fraction (PMF), is the source for microsomes.
To isolate proteins from the microsomes, we measure the volume of the PMF (V mL) and prepare a volume of 8mM Calcium Chloride Solution equivalent to 7.5 times of the PMF (V mL). The PMF was transferred to a beaker containing a magnetic stir bar. A volume of 8mM Calcium Chloride Solution equivalent to 7.5 times of the PMF was added dropwise to the beaker with constant stirring. The Calcium Chloride Solution is mixed with the PMF and stirred for additional 15 min at 4°C. The sample was centrifuged at 8000xg for 10 min at 4°C. The enriched rough endoplasmic reticulum (RER) microsomes will be in the pellet. The supernatant was removed and the pellet was resuspended in 1X Isotonic Retraction Buffer supplemented with fresh protease inhibitor cocktail. The suspension was homogenized using a pellet pestle in a microcentrifuge tube. Protein concentration was determined by using 1X Bradford reagent. 15 μg of nuclear or ER proteins were used for WB. 5μg of ER protein was used for each IP and co-IP reaction.
Each analysis was performed at least 3 times.
Ubiquitination analysis
BRCA1-proficient breast cancer cells MDA-MB-231 and MCF7 were transiently transfected with control or BRCA siRNA for 48 h as described above. Cytoplasmic protein extract was prepared as described under the Immunoprecipitation Assay section. 300 μg of protein and 35 μL of ubiquitin (Ub) antibody conjugated-agarose beads were mixed in 250 μL of 1X IP lysis buffer and incubated overnight at 4°C with shaking. Beads were washed and spun down thrice next day. Antigens that bound to the target antibody were eluted by adding 10 μL of H2O and by boiling the beads for 5 min. The eluants were subjected to Western blot analysis. For Bortezomib treatment, additional BRCA1 siRNA-transfected cells were prepared and treated with 100 nM of Bortezomib or equal amount of DMSO (control) overnight before protein extraction. Each analysis was performed at least 3 times.
In vitro ubiquitination assay
HEK293 cells (ATCC) was transfected with BARD1, PERK, IRE1 or NSP15 plasmid with FuGENE 6 reagent. Full-length BARD1, PERK, IRE1 and NSP15 proteins were purified individually using anti-Flag M2 affinity gel. Purified proteins were confirmed by Western blot. Concentration of the proteins was estimated by Coomassie Blue staining with known concentrations of Bovine Serum Albumin (A9647, Millipore Sigma). The in vitro ubiquitination assay was carried out similar to a previous study with some modifications.86 Briefly, 100 ng of PERK or IRE1 was mixed with 3 μL of the 10X reaction buffer (K-200B, Boston Biochem), 1 μL UBE1, 2.5 μL UBE2J1, 100 ng BRCA1, 60 ng BARD1, 3 μL of the 10X Mg2+-ATP (K-200B, Boston Biochem) and 3 μL of the 10X ubiquitin (K-200B, Boston Biochem) in a 30 μL reaction. Except for the zero time point mixture, all reactions were incubated at 37°C for their respective time points. All reactions were stopped by addition of SDS sample buffer before loading to 10% SDS-PAGE gel (NP03031BOX, Thermo Fisher) for electrophoresis. Ubiquitinated proteins were detected by an anti-ubiquitin antibody. Strep-tactin antibody for NSP15 protein. Each analysis was performed at least 3 times.
RT-qPCR
Total RNA was extracted from control or BRCA1 siRNA transiently transfected breast cancer cells (MCF7 or MDA-MB-231) using Quick-RNA Miniprep Kit (Zymo Research). cDNAs were generated by Invitrogen™ SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR kit following supplier’s instruction (Fisher Scientific). For each qPCR reaction, 2 μL of cDNA from each sample was used with SYBR Green Supermix. Each reaction was prepared in triplicates in each experiment. Primer pairs targeted EIF1AK3 (PERK), ERN1 (IRE1), BRCA1, BARD1 and GAPDH were used in qPCR reactions. All reactions were run in 96-well plate and analyzed by Applied Biosystem 7500 Real-Time PCR System (ThermoFisher Scientific). Each analysis was performed at least 3 times.
Murine xenograft studies
Luciferase-expressing SUM149PT BRCA1-mutant breast cancer cells were created by transfecting the cancer cells with pLenti PGK V5-LUC Neo plasmid and luciferase expressing clones were subsequently selected by G418 drug treatment. To induce tumor formation, two million cancer cells were subcutaneously injected into NU/J female mice >10 weeks old. Drug treatment (vehicle or CsA) begun once the tumors became detectable in the IVIS In Vivo Imaging system (PerkinElmer) using the Living Image v4.5.5 software. DMSO vehicle (control) or CsA (treatment) was administered intra-peritoneally to the animals every other day and animals were scanned once a week to monitor tumor progression. Tumor-derived from SUM149PT BRCA1-def breast cancer cells appeared by 2 weeks post-injection. Three doses of CsA (5.5, 11.25 or 22.5 mg/kg) were tested in this study and same amount of vehicle was used for the control group. 5 mg/mL CsA stock was prepared in 2% DMSO (D128-500, Fisher Scientific), 30% PEG300 (NC0630366, Fisher Scientific) and 5% Tween 80 (P1754, Sigma-Aldrich) in sterilized deionized/distilled H2O per supplier’s recommendation and diluted in DMSO. Power analysis of the animal size was based on sample size formula, N = 8(CV)2[1+(1-PC)2]/(PC)2, to reach the error = 0.05, Power = 0.80, percentage change in mean (PC) = 20%, co-efficient of variation (CV) = 10–15% (varies between the experiments). The power analysis found that 5 mice per group was the minimal number of mice to obtain statistical significance. We have included 6 animals for each group in our study. All animal care and maintenance complied with the regulations of IACUC and University of Florida Animal Housing Facility (IACUC#201710042). Each analysis was performed at least 3 times.
Quantitation and statistical analysis
Statistical details of each experiments can be found in the Figure Legends. The numbers of experimental replicates are indicated in the Figure Legends or STAR Methods. Statistical comparison included two-tailed Student’s t test as specified in the Figure Legends using Prism software (GraphPad v.7.04). Data presented in graphs are means ± SEMs from three or more replicas as shown unless stated otherwise.
Additional resources
Supplementary information is available for this paper.
Acknowledgments
This research was supported by NIH R01 GM109645, CA139429, and CA205224 to RH; CPRIT Individual Investigator grant RP220269 to RH; NIH R01 GM084020 to EW, RH; Gatorade Trust Start-Up Fund 00123001 and Gatorade Pilot Project Award 00127907 to KYK. UT Health Start-Up Fund 163254-22100-M1522 to RH, KYK. Lori Zabar Women’s Health Research Award of the Foundation for Women’s Wellness to KYK. Confocal images were generated in the Core Optical Imaging Facility which is supported by UTHSCSA and NIH-NCI P30 CA54174 (CTRC at UTHSCSA). We would also like to thank the support from UF IACUC and the animal facility staff for the animal study.
Author contributions
Conceptualization, K.Y.K.; experiments, G.S, M.Y., A.J., T.A.T., L.P., A.K., and N.K.; data analysis, R.H., G.S., A.J., N.K., M.B., E.A.W., and K.Y.K.; writing – initial draft, K.Y.K.; writing – review and editing, R.H., E.A.W., and K.Y.K.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105626.
Supplemental information
Data and code availability
-
•
We have deposited all raw data of the Western blot membrane scans that were used to generate the Figures in this manuscript to the Mendeley Data with https://data.mendeley.com/datasets/72gjb45yjv/1.
-
•
This study did not report original codes.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Clark N.M., Martinez L.M., Murdock S., deLigio J.T., Olex A.L., Effi C., Dozmorov M.G., Bos P.D. Regulatory T cells support breast cancer progression by opposing IFN-gamma-Dependent functional reprogramming of myeloid cells. Cell Rep. 2020;33:108482. doi: 10.1016/j.celrep.2020.108482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kraya A.A., Maxwell K.N., Wubbenhorst B., Wenz B.M., Pluta J., Rech A.J., Dorfman L.M., Lunceford N., Barrett A., Mitra N., et al. Genomic signatures predict the immunogenicity of BRCA-deficient breast cancer. Clin. Cancer Res. 2019;25:4363–4374. doi: 10.1158/1078-0432.CCR-18-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moynahan M.E., Chiu J.W., Koller B.H., Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 4.Scully R., Chen J., Ochs R.L., Keegan K., Hoekstra M., Feunteun J., Livingston D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 5.Scully R., Chen J., Plug A., Xiao Y., Weaver D., Feunteun J., Ashley T., Livingston D.M. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 6.Snouwaert J.N., Gowen L.C., Latour A.M., Mohn A.R., Xiao A., DiBiase L., Koller B.H. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- 7.Tung N., Lin N.U., Kidd J., Allen B.A., Singh N., Wenstrup R.J., Hartman A.R., Winer E.P., Garber J.E. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J. Clin. Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shakya R., Szabolcs M., McCarthy E., Ospina E., Basso K., Nandula S., Murty V., Baer R., Ludwig T. The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc. Natl. Acad. Sci. USA. 2008;105:7040–7045. doi: 10.1073/pnas.0711032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y., Chen C.F., Riley D.J., Allred D.C., Chen P.L., Von Hoff D., Osborne C.K., Lee W.H. Aberrant subcellular localization of BRCA1 in breast cancer. Science. 1995;270:789–791. doi: 10.1126/science.270.5237.789. [DOI] [PubMed] [Google Scholar]
- 10.Coene E., Van Oostveldt P., Willems K., van Emmelo J., De Potter C.R. BRCA1 is localized in cytoplasmic tube-like invaginations in the nucleus. Nat. Genet. 1997;16:122–124. doi: 10.1038/ng0697-122. [DOI] [PubMed] [Google Scholar]
- 11.De Potter C.R., Coene E.D., Schelfhout V.R. Localization of BRCA1 protein at the cellular level. J. Mammary Gland Biol. Neoplasia. 1998;3:423–429. doi: 10.1023/a:1018740216630. [DOI] [PubMed] [Google Scholar]
- 12.Hedgepeth S.C., Garcia M.I., Wagner L.E., 2nd, Rodriguez A.M., Chintapalli S.V., Snyder R.R., Hankins G.D.V., Henderson B.R., Brodie K.M., Yule D.I., et al. The BRCA1 tumor suppressor binds to inositol 1, 4, 5-trisphosphate receptors to stimulate apoptotic calcium release. J. Biol. Chem. 2015;290:7304–7313. doi: 10.1074/jbc.M114.611186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukau B., Weissman J., Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Oakes S.A., Papa F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 16.Moon H.W., Han H.G., Jeon Y.J. Protein quality control in the endoplasmic reticulum and cancer. Int. J. Mol. Sci. 2018;19:E3020. doi: 10.3390/ijms19103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampton R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002;14:476–482. doi: 10.1016/s0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 18.Vembar S.S., Brodsky J.L. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brodsky J.L., Wojcikiewicz R.J.H. Substrate-specific mediators of ER associated degradation (ERAD) Curr. Opin. Cell Biol. 2009;21:516–521. doi: 10.1016/j.ceb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodsky J.L. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho P., Goder V., Rapoport T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 22.Christianson J.C., Shaler T.A., Tyler R.E., Kopito R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christianson J.C., Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenblatt E.J., Olzmann J.A., Kopito R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehnert M., Sommer T., Jarosch E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014;16:77–86. doi: 10.1038/ncb2882. [DOI] [PubMed] [Google Scholar]
- 26.Sun S., Shi G., Han X., Francisco A.B., Ji Y., Mendonça N., Liu X., Locasale J.W., Simpson K.W., Duhamel G.E., et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA. 2014;111:E582–E591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki M., Otsuka T., Ohsaki Y., Cheng J., Taniguchi T., Hashimoto H., Taniguchi H., Fujimoto T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol. Biol. Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Y., Shibata Y., Yun C., Ron D., Rapoport T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 29.Miki Y., Swensen J., Shattuck-Eidens D., Futreal P.A., Harshman K., Tavtigian S., Liu Q., Cochran C., Bennett L.M., Ding W., et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 30.Meza J.E., Brzovic P.S., King M.C., Klevit R.E. Mapping the functional domains of BRCA1. Interaction of the ring finger domains of BRCA1 and BARD1. J. Biol. Chem. 1999;274:5659–5665. doi: 10.1074/jbc.274.9.5659. [DOI] [PubMed] [Google Scholar]
- 31.Hashizume R., Fukuda M., Maeda I., Nishikawa H., Oyake D., Yabuki Y., Ogata H., Ohta T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 32.Joukov V., Chen J., Fox E.A., Green J.B., Livingston D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA. 2001;98:12078–12083. doi: 10.1073/pnas.211427098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minten E.V., Kapoor-Vazirani P., Li C., Zhang H., Balakrishnan K., Yu D.S. SIRT2 promotes BRCA1-BARD1 heterodimerization through deacetylation. Cell Rep. 2021;34:108921. doi: 10.1016/j.celrep.2021.108921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fabbro M., Rodriguez J.A., Baer R., Henderson B.R. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 2002;277:21315–21324. doi: 10.1074/jbc.M200769200. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez J.A., Schüchner S., Au W.W.Y., Fabbro M., Henderson B.R. Nuclear-cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene. 2004;23:1809–1820. doi: 10.1038/sj.onc.1207302. [DOI] [PubMed] [Google Scholar]
- 36.Xia Y., Pao G.M., Chen H.W., Verma I.M., Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003;278:5255–5263. doi: 10.1074/jbc.M204591200. [DOI] [PubMed] [Google Scholar]
- 37.Ruffner H., Joazeiro C.A., Hemmati D., Hunter T., Verma I.M. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. USA. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mallery D.L., Vandenberg C.J., Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002;21:6755–6762. doi: 10.1093/emboj/cdf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen M.Z., Moily N.S., Bridgford J.L., Wood R.J., Radwan M., Smith T.A., Song Z., Tang B.Z., Tilley L., Xu X., et al. A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat. Commun. 2017;8:474. doi: 10.1038/s41467-017-00203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho S., Clipstone N., Timmermann L., Northrop J., Graef I., Fiorentino D., Nourse J., Crabtree G.R. The mechanism of action of cyclosporin A and FK506. Clin. Immunol. Immunopathol. 1996;80:S40–S45. doi: 10.1006/clin.1996.0140. [DOI] [PubMed] [Google Scholar]
- 42.Ciechomska I., Legat M., Golab J., Wesolowska A., Kurzaj Z., Mackiewicz A., Kaminska B. Cyclosporine A and its non-immunosuppressive derivative NIM811 induce apoptosis of malignant melanoma cells in in vitro and in vivo studies. Int. J. Cancer. 2005;117:59–67. doi: 10.1002/ijc.21153. [DOI] [PubMed] [Google Scholar]
- 43.Gallay P.A., Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des. Devel. Ther. 2013;7:105–115. doi: 10.2147/DDDT.S30946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horibe T., Yosho C., Okada S., Tsukamoto M., Nagai H., Hagiwara Y., Tujimoto Y., Kikuchi M. The chaperone activity of protein disulfide isomerase is affected by cyclophilin B and cyclosporin A in vitro. J. Biochem. 2002;132:401–407. doi: 10.1093/oxfordjournals.jbchem.a003236. [DOI] [PubMed] [Google Scholar]
- 45.Meunier L., Usherwood Y.K., Chung K.T., Hendershot L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell. 2002;13:4456–4469. doi: 10.1091/mbc.e02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price E.R., Jin M., Lim D., Pati S., Walsh C.T., McKeon F.D. Cyclophilin B trafficking through the secretory pathway is altered by binding of cyclosporin A. Proc. Natl. Acad. Sci. USA. 1994;91:3931–3935. doi: 10.1073/pnas.91.9.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee J., Choi T.G., Ha J., Kim S.S. Cyclosporine A suppresses immunoglobulin G biosynthesis via inhibition of cyclophilin B in murine hybridomas and B cells. Int. Immunopharmacol. 2012;12:42–49. doi: 10.1016/j.intimp.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 48.Clark S.L., Rodriguez A.M., Snyder R.R., Hankins G.D.V., Boehning D. Structure-function of the tumor suppressor BRCA1. Comput. Struct. Biotechnol. J. 2012;1:e201204005. doi: 10.5936/csbj.201204005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huen M.S.Y., Sy S.M.H., Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010;11:138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manke I.A., Lowery D.M., Nguyen A., Yaffe M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 51.Yu X., Chini C.C.S., He M., Mer G., Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 52.Baer R., Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002;12:86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 53.Wu L.C., Wang Z.W., Tsan J.T., Spillman M.A., Phung A., Xu X.L., Yang M.C., Hwang L.Y., Bowcock A.M., Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 54.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lok B.H., Carley A.C., Tchang B., Powell S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeung B.H.Y., Kwan B.W.Y., He Q.Y., Lee A.S., Liu J., Wong A.S.T. Glucose-regulated protein 78 as a novel effector of BRCA1 for inhibiting stress-induced apoptosis. Oncogene. 2008;27:6782–6789. doi: 10.1038/onc.2008.290. [DOI] [PubMed] [Google Scholar]
- 57.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 58.Fang S., Ferrone M., Yang C., Jensen J.P., Tiwari S., Weissman A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hassink G., Kikkert M., van Voorden S., Lee S.J., Spaapen R., van Laar T., Coleman C.S., Bartee E., Früh K., Chau V., Wiertz E. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005;388:647–655. doi: 10.1042/BJ20041241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeon Y.J., Khelifa S., Ratnikov B., Scott D.A., Feng Y., Parisi F., Ruller C., Lau E., Kim H., Brill L.M., et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;27:354–369. doi: 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kikkert M., Doolman R., Dai M., Avner R., Hassink G., van Voorden S., Thanedar S., Roitelman J., Chau V., Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- 62.Nadav E., Shmueli A., Barr H., Gonen H., Ciechanover A., Reiss Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem. Biophys. Res. Commun. 2003;303:91–97. doi: 10.1016/s0006-291x(03)00279-1. [DOI] [PubMed] [Google Scholar]
- 63.Younger J.M., Chen L., Ren H.Y., Rosser M.F.N., Turnbull E.L., Fan C.Y., Patterson C., Cyr D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 64.Fry W.H.D., Simion C., Sweeney C., Carraway K.L., 3rd Quantity control of the ErbB3 receptor tyrosine kinase at the endoplasmic reticulum. Mol. Cell Biol. 2011;31:3009–3018. doi: 10.1128/MCB.05105-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo X., Shen S., Song S., He S., Cui Y., Xing G., Wang J., Yin Y., Fan L., He F., Zhang L. The E3 ligase Smurf1 regulates Wolfram syndrome protein stability at the endoplasmic reticulum. J. Biol. Chem. 2011;286:18037–18047. doi: 10.1074/jbc.M111.225615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Magadán J.G., Pérez-Victoria F.J., Sougrat R., Ye Y., Strebel K., Bonifacino J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010;6:e1000869. doi: 10.1371/journal.ppat.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meacham G.C., Patterson C., Zhang W., Younger J.M., Cyr D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 68.Yoshida Y., Chiba T., Tokunaga F., Kawasaki H., Iwai K., Suzuki T., Ito Y., Matsuoka K., Yoshida M., Tanaka K., Tai T. E3 ubiquitin ligase that recognizes sugar chains. Nature. 2002;418:438–442. doi: 10.1038/nature00890. [DOI] [PubMed] [Google Scholar]
- 69.Yoshida Y., Tokunaga F., Chiba T., Iwai K., Tanaka K., Tai T. Fbs2 is a new member of the E3 ubiquitin ligase family that recognizes sugar chains. J. Biol. Chem. 2003;278:43877–43884. doi: 10.1074/jbc.M304157200. [DOI] [PubMed] [Google Scholar]
- 70.Araki K., Nagata K. Protein folding and quality control in the ER. Cold Spring Harbor Perspect. Biol. 2011;3:a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kadowaki H., Satrimafitrah P., Takami Y., Nishitoh H. Molecular mechanism of ER stress-induced pre-emptive quality control involving association of the translocon, Derlin-1, and HRD1. Sci. Rep. 2018;8:7317. doi: 10.1038/s41598-018-25724-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujita H., Yagishita N., Aratani S., Saito-Fujita T., Morota S., Yamano Y., Hansson M.J., Inazu M., Kokuba H., Sudo K., et al. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J. 2015;34:1042–1055. doi: 10.15252/embj.201489897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ji Y., Kim H., Yang L., Sha H., Roman C.A., Long Q., Qi L. The Sel1L-Hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep. 2016;16:2630–2640. doi: 10.1016/j.celrep.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong S., Yang Y., Xu Y., Wang Y., Zhang Y., Melo-Cardenas J., Xu X., Gao B., Thorp E.B., Zhang D.D., et al. Endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls B-cell immunity through degradation of the death receptor CD95/Fas. Proc. Natl. Acad. Sci. USA. 2016;113:10394–10399. doi: 10.1073/pnas.1606742113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun S., Shi G., Sha H., Ji Y., Han X., Shu X., Ma H., Inoue T., Gao B., Kim H., et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015;17:1546–1555. doi: 10.1038/ncb3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamasaki S., Yagishita N., Sasaki T., Nakazawa M., Kato Y., Yamadera T., Bae E., Toriyama S., Ikeda R., Zhang L., et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase 'Synoviolin'. EMBO J. 2007;26:113–122. doi: 10.1038/sj.emboj.7601490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Y.M., Wang H.J., Chen F., Guo W.H., Wang Y.Y., Li H.Y., Tang J.H., Ding Y., Shen Y.C., Li M., et al. HRD1 suppresses the growth and metastasis of breast cancer cells by promoting IGF-1R degradation. Oncotarget. 2015;6:42854–42867. doi: 10.18632/oncotarget.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rycyzyn M.A., Clevenger C.V. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. Proc. Natl. Acad. Sci. USA. 2002;99:6790–6795. doi: 10.1073/pnas.092160699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dieriks B., Van Oostveldt P. Spatiotemporal behavior of nuclear cyclophilin B indicates a role in RNA transcription. Int. J. Mol. Med. 2012;29:1031–1038. doi: 10.3892/ijmm.2012.937. [DOI] [PubMed] [Google Scholar]
- 80.Price E.R., Zydowsky L.D., Jin M.J., Baker C.H., McKeon F.D., Walsh C.T. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl. Acad. Sci. USA. 1991;88:1903–1907. doi: 10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan I., Roitenberg N., Cohen E. Vesicle-mediated secretion of misfolded prion protein molecules from cyclosporin A-treated cells. Faseb J. 2018;32:1479–1492. doi: 10.1096/fj.201700598RRR. [DOI] [PubMed] [Google Scholar]
- 82.Kim J., Choi T.G., Ding Y., Kim Y., Ha K.S., Lee K.H., Kang I., Ha J., Kaufman R.J., Lee J., et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J. Cell Sci. 2008;121:3636–3648. doi: 10.1242/jcs.028654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Araujo T.L.S., Fernandes C.G., Laurindo F.R.M. Golgi-independent routes support protein disulfide isomerase externalization in vascular smooth muscle cells. Redox Biol. 2017;12:1004–1010. doi: 10.1016/j.redox.2017.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson N., Johnson S.F., Yao W., Li Y.C., Choi Y.E., Bernhardy A.J., Wang Y., Capelletti M., Sarosiek K.A., Moreau L.A., et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA. 2013;110:17041–17046. doi: 10.1073/pnas.1305170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J., Willers H., Feng Z., Ghosh J.C., Kim S., Weaver D.T., Chung J.H., Powell S.N., Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004;24:708–718. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim B.J., Chan D.W., Jung S.Y., Chen Y., Qin J., Wang Y. The Histone variant MacroH2A1 is a BRCA1 ubiquitin ligase substrate. Cell Rep. 2017;19:1758–1766. doi: 10.1016/j.celrep.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
We have deposited all raw data of the Western blot membrane scans that were used to generate the Figures in this manuscript to the Mendeley Data with https://data.mendeley.com/datasets/72gjb45yjv/1.
-
•
This study did not report original codes.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.