

Abstract
Background
Calcareous outcrops, rocky areas composed of calcium carbonate (CaCO3), often host a diverse, specialized, and threatened biomineralizing fauna. Despite the repeated evolution of physiological and morphological adaptations to colonize these mineral rich substrates, there is a lack of genomic resources for calcareous rock endemic species. This has hampered our ability to understand the genomic mechanisms underlying calcareous rock specialization and manage these threatened species.
Results
Here, we present a new draft genome assembly of the threatened limestone endemic land snail Oreohelix idahoensis and genome skim data for two other Oreohelix species. The O. idahoensis genome assembly (scaffold N50: 404.19 kb; 86.6% BUSCO genes) is the largest (~ 5.4 Gb) and most repetitive mollusc genome assembled to date (85.74% assembly size). The repetitive landscape was unusually dominated by an expansion of long terminal repeat (LTR) transposable elements (57.73% assembly size) which have shaped the evolution genome size, gene composition through retrotransposition of host genes, and ectopic recombination. Genome skims revealed repeat content is more than 2–3 fold higher in limestone endemic O. idahoensis compared to non-calcareous Oreohelix species. Gene family size analysis revealed stress and biomineralization genes have expanded significantly in the O. idahoensis genome.
Conclusions
Hundreds of threatened land snail species are endemic to calcareous rock regions but there are very few genomic resources available to guide their conservation or determine the genomic architecture underlying CaCO3 resource specialization. Our study provides one of the first high quality draft genomes of a calcareous rock endemic land snail which will serve as a foundation for the conservation genomics of this threatened species and for other groups. The high proportion and activity of LTRs in the O. idahoensis genome is unprecedented in molluscan genomics and sheds new light how transposable element content can vary across molluscs. The genomic resources reported here will enable further studies of the genomic mechanisms underlying calcareous rock specialization and the evolution of transposable element content across molluscs.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-022-09000-x.
Keywords: LTR expansion, Oreohelix, Comparative genomics, Genome skim, Gene family evolution, Limestone
Background
Calcareous rocks such as limestone, marble, or dolomite are required ingredients for cement production and thus are necessary ‘building blocks’ for infrastructure. Regions containing these valuable resources often host a diverse and specialized group of species that have adapted to the unique abiotic conditions present in calcareous habitats [1, 2]. A product of this specialization is that many calcareous rock endemics are characterized by narrow ranges (e.g. a single hill, cave, or outcrop) which increases the risk of extinction from disturbance [3]. Balancing the societal needs for carbonate rock and habitat requirements of calcareous rock endemic species can be particularly challenging as there may be little middle ground between economic interests and biodiversity preservation. While there is growing global interest to safeguard calcareous rock endemic diversity [4], government and private quarrying has already caused the extinction of more than 20 calcareous rock endemic species with more likely to follow [5, 6].
Central to this industry-biodiversity conflict is determining what level of conservation priority should be given to species that are edaphically specialized to calcareous habitats. While many calcareous rock endemics are clearly threatened, resources available for conservation actions are limited. Calcareous edaphic specialist species are often, but not always, members of recent radiations that are spread across several calcareous outcrops [2]. A large number of closely related edaphically specialized species spread across numerous isolated outcrops can make targeted species conservation infeasible with the resources and political will available [7]. Instead, policymakers may opt to preserve the most species rich outcrops and allow development to proceed on less diverse areas [5, 8]. However, this conservation approach omits considerations of the evolutionary processes generating calcareous rock endemic diversity. The process of edaphic speciation can be abrupt (i.e. polyploidy, chromosomal rearrangement or inversions) or relatively gradual (i.e. polygenic loci under divergent selection over several hundred generations) (reviewed in [2, 9, 10]). Failure to account for these evolutionary processes in conservation plans of calcareous rock endemics may result in misapplying protective actions to morphologically distinct ecotypes of existing species and/or permitting the destruction of habitat for fully reproductively isolated edaphically specialized species [2].
One of the major groups of calcareous rock endemics that are increasingly being threatened by development are land snails [8, 11]. More than a quarter of all IUCN Red List land snail species with near threatened or higher threat assessments appear to reside primarily or exclusively on calcareous substrates (371 of 1460 species) [12]. Unlike other edaphic specialists that must physiologically and morphologically adapt to tolerate higher concentrations of calcium carbonate (CaCO3) in calcareous habitats, land snails must acquire sufficient CaCO3 from the environment to biomineralize their shell [13] and may exploit greater environmental availability of CaCO3 at calcareous outcrops to increase biomineralization output [14]. Indeed, a number of calcareous rock endemic land snails express thickened or elaborate calcareous ornaments (e.g. prominent calcareous ribs or keels) [15] which may require an abundance of environmental CaCO3 to develop normally. Edaphic specialization by land snails to calcareous habitats may be distinct from classical examples of calcareous edaphic evolution in that land snails are often specializing to an environment favorable to their physiological needs and not to one requiring substantial metabolic adaptations [2]. Given these differences, the genomic mechanisms underlying calcareous edaphic specialization in land snails may be broadly dissimilar from those identified in genomic studies of other calcareous edaphic specialists. Key to unravelling the genomic mechanisms associated with edaphic specialization are genomic assemblies which provide important context for evolutionary and functional inference. Unfortunately, the scarcity of genomic resources available for calcareous rock endemic land snails has hindered our understanding of the origins of edaphic diversity.
An ideal system to study the process of calcareous edaphic specialization in land snails should have repeated transitions to calcareous rock environments to serve as replicates and transitions at different stages of the edaphic specialization process. Oreohelix ‘Mountainsnails’ are suitable candidates as there are numerous transitions from a smooth form which occurs on a variety of geologic substrates to various ornamented forms that predominantly inhabit calcareous substrates [16], have hybrid zones at calcareous outcrop boundaries between forms that indicate recent divergence [17, 18], and phylogenetic studies indicate some ornamented species are of considerable age [16]. Furthermore, many highly ornamented species are declining in population size [18] and are considered to be highly threatened by conservation authorities [19]. Uncovering the processes involved in edaphic specialization in Oreohelix would have considerable bearing on the systematics and conservation status of its members.
Here, we assembled a draft genome assembly of the costate Mountainsnail, Oreohelix idahoensis, a threatened limestone endemic land snail species from the Northwest United States that expresses thickened shell ornaments. In comparison to other gastropod genome assemblies, there were several significantly expanded gene families putatively related to stress response and biomineralization. We also show from analyses of Oreohelix genomes skims and comparisons to other mollusc genome assemblies that repeat content is substantially elevated in the O. idahoensis genome. The genomic resources reported here will serve as a foundation for the conservation genomics of this threatened species and for understanding the genomic basis of resource constrained biomineralization.
Results
Genome assembly and annotation of O. idahoensis
Through utilizing PacBio CLR long reads (324.5 Gb) and 10X genomics linked reads (425.8 Gb), we generated a relatively contiguous and complete genome assembly of O. idahoensis (Table 1). Genomescope 2.0 [20] analysis of 31-mer counts estimated that the O. idahoensis genome was 7.01Gb in size, has a heterozygosity of 0.51% (Supplementary Fig. 1), and a repeat content of 74.9%. Estimates of genome size using raw read mapping rates using ModEst however showed the genome size to be 8.49 Gb [21]. The final pre-polished assembly was produced by merging the hybrid Supernova-DBG2OLC [22] assembly produced from both long and linked reads with the canu v.1.9 [23] assembly generated solely from long reads (Supplementary Table 1). The subsequently polished genome draft was 5.40 Gb in size and was composed of 23,228 scaffolds (scaffold N50 of 404.19 kb). The high repeat content of the genome draft is a likely contributing factor for the difference between the final assembly size (5.40 Gb), the estimated genome size using k-mers (7.01 Gb), and estimated genome size using back mapping rates (8.49 Gb) as repetitive regions are prone to collapse during assembly [23]. The estimated heterozygosity of the O. idahoensis draft assembly using genomescope was lower than other published land snail genomes (e.g. C. nemoralis: 1.42%; C. unifasciata: 1.09%) [24, 25], which likely reflects the isolation and small population size of this threatened species [26]. The draft genome was fairly complete with regard to coding regions as 86.6% of metazoan Benchmarking Universal Single-Copy Orthologs (BUSCO) genes were single-copy or duplicated in the assembly (single copy: 77.3%, duplicated: 9.1%, fragmented: 6.6%) [27]. In terms of contiguity and BUSCO scores, the O. idahoensis genome draft assembly compares favorably to many other large molluscan genomes (Table 1).
Table 1.
Genome assembly statistics of the O. idahoensis genome draft and closely related or large molluscan genomes
| Genome assembly | Size (Gb) | Scaffold N50 (Kb) | BUSCO score | Source |
|---|---|---|---|---|
| Oreohelix idahoensis | 5.40 | 404 | 86.6 | This study |
| Euprymna scolopes | 5.11 | 3724 | 96.9 | Belcaid et al. 2019 |
| Callistoctopus minor | 5.09 | 466 | 76.2 | Kim et al. 2018 |
| Cepaea nemoralis | 3.49 | 330 | 87.2 | Saenko et al. 2021 |
| Candidula fasciata | 1.29 | 246 | 92 | Chueca et al. 2021 |
The final annotation set produced by integrating transcript, protein and ab-initio evidence contained 27,692 predicted protein-coding genes which is within the range of other known gastropod genome assemblies [25, 28]. Mean gene size was 34,594 bp which is roughly three times larger than previous land snail genome assemblies (e.g. C. nemoralis mean gene size: 9629 bp; C. unifasciata median gene size: 11,931 bp) [24, 25], reflecting the influence of repetitive elements on intron size (78.44% of genes had repetitive elements nested in genic regions; Table 2). A total of 92.1% of all genes had a hit to either the NCBI non-redundant nucleotide database, InterProscan [29], or UniProt (Supplementary Table 2). Scanning of scaffolds for contamination with conterminator v.1.0 [30] revealed a single potential 1.2 kb insert of Escheria coli DNA in one scaffold of the assembly. Blasting of the contaminated region against the NCBI nucleotide database did not result in any alignments to E. coli sequences so the putative contaminated region was retained in the assembly.
Table 2.
Gene characteristics of the O. idahoensis draft assembly
| Feature | Value |
|---|---|
| Number of protein codeing genes | 27,692 |
| Number of mRNAs | 46,907 |
| Mean isoforms per gene | 1.69 |
| Mean exon number per mRNA | 5.6 |
| Mean gene length (bp) | 34,594 |
| Mean mRNA length (bp) | 31,244 |
| Mean mRNA exon length (bp) | 265 |
| Mean intron length (bp) | 5523 |
| Number of genes with repetitive element overlap | 21,722 |
Transposable element content and evolution
To evaluate the repeat content of the O. idahoensis genome draft and place the proportion of identified elements in context with other mollusc genome assemblies, we created custom repeat libraries for O. idahoensis and five other previously published mollusc genomes (Cepaea nemorals, Candidula faciata, Achatina immaculata, Radix auricularia, and Euprymna scolopes) using the EDTA pipeline v.1.9.9 [31]. Repeat content of the O. idahoensis genome draft was estimated to be 85.74% which is higher than all currently available mollusc genome assemblies, including those larger than 2.5 Gb [32]. Most of the repeat content in the O. idahoensis draft assembly is classified as transposable elements and a small proportion are predicted to be simple and low-complexity repeats (Supplementary Table 3). The most common repetitive elements were long terminal repeat transposons (LTR) which make up 57.73% of the assembly (Fig. 1; Supplementary Table 3) and are predominantly of the Gypsy/DIRS1 family (32.53% assembly size). This estimate of LTR content is well outside of the known range of LTR content across molluscan genomes (previous estimates are 2–8% LTR content) [33]. DNA transposons compose the remainder of the repeat landscape and are largely terminal invert repeat sequences (Supplementary Table 3).
Fig. 1.

Repetitive element landscapes of O. idahoensis and other large or closely related molluscan genomes. Repeat classes on the bottom right. The bottom axis depicts Kimura-divergence from consensus with greater divergence being indicative of past repeat expansion. The left axis measures the total proportion of the genome draft occupied by the repeat class. Outline drawings were created with BioRender
Our reannotations of five other molluscan genomes revealed significantly greater LTR content than was previously reported in each assembly (Fig. 1). This may be a result of previous studies not utilizing a dedicated LTR detection step in their repeat annotation pipeline, which is now recommended or standard practice for several popular repeat annotation programs including RepeatModeler 2 [34] and EDTA. The depauperate number of long and short interspersed nuclear elements (LINE/SINE) in our repeat annotations of prior assemblies compared to previous estimates may be a product of this difference as LINE and SINE elements can be nested within larger LTR elements [35].
We examined repeat expansion and contraction across the six mollusc genome assemblies by comparing the Kimura sequence divergence of each identified transposable element to its consensus element within the repeat library. The repeat landscape of O. idahoensis indicates that repetitive elements, predominantly LTRs, have undergone expansion recently (Fig. 1). Using a Kimura divergence cut-off of 5%, over 25% of the O. idahoensis genome draft assembly is attributable to recent repeat expansion, primarily LTRs. While some land snail genome assemblies such as C. nemoralis and A. immaculata showed evidence of repeat expansions of DNA and LTR transposons, neither were to the same degree as O. idahoensis. All other mollusc genomes analyzed had older repeat expansions and lower repeat content.
To understand how LTRs may be related to species divergence within Oreohelix, we compared repeat content estimated from genome skims, LTR insertion times, and Oreohelix species divergence estimates from a previous study [16]. Analysis of genome skims using RESPECT v. 1.0.0 [36] revealed O. idahoensis contains roughly 2–3 fold of high copy repetitive k-mers per million bases compared to a closely related smooth, non-limestone endemic O. s. strigosa and a distantly related smooth, non-limestone species O. jugalis (Table 3). We estimated LTR insertion times by comparing divergence between LTR sister pairs using a previously established molluscan substitution rate (see Materials and Methods). Most of the LTR insertions in the O. idahoensis genome preceded the split of O. idahoensis from the O. haydeni complex (Fig. 2). 84.57% of estimated LTRs insertion times in the O. idahoensis genome occurred after the median age of the O. strigosa complex – subrudis split (Fig. 2).
Table 3.
Estimates of repetitive k-mer content and genome size from Oreohelix genome skims
| Sample | Genome Size (Gb) | Coverage | HRCM1 |
|---|---|---|---|
| O. s. strigosa | 6.18 | 7.54 | 497 |
| O. jugalis- A | 6.53 | 5.75 | 997 |
| O. jugalis- B | 6.11 | 5.95 | 811 |
| O. idahoensis- A | 8.99 | 8.53 | 1807 |
| O. idahoensis- B | 9.11 | 5.60 | 1595 |
1HCRM stands for high copy repeats per million and is the average count of the 10 most represented k-mers in a sample of a million base pairs. Higher HCRM values are indicative of greater transposable element content
Fig. 2.

Estimated LTR insertion times in the O. idahoensis genome draft and divergence times between major Oreohelix groups. Upper and lower 95% highest posterior density divergence are depicted at each node in red. Divergence time estimates are taken from Linscott et al. [16]
We then calculated the ratio of solo LTRs to intact LTRs to estimate the frequency of ectopic recombination in the O. idahoensis genome draft. The presence of solo LTRs indicates that ectopic recombination between LTR elements has occurred and can be compared against the total proportion of intact LTR elements to estimate LTR removal rates and rate of ectopic recombination [37]. We found a high ratio of solo to intact LTR elements present in the O. idahoensis genome across LTR families (median ratio: 6.33, Supplementary Table 4), which indicates that there has been a large number of ectopic recombination events that have reduced the number of active LTR elements and facilitated genomic rearrangement.
Phylogenetic analysis and gene family expansion
To evaluate the phylogenetic placement of O. idahoensis, we first aligned single copy orthologs of six gastropod genomes using Orthofinder v.2.4.0 [38], including two other land snail genomes (Achatina fulica, and Candidula unifasciata,), a freshwater snail genome (Biomalpharia glabriata), a sea-slug genome (Aplysia californica), and a Caenogastropod snail (Pomacea canaliculata) to serve as an outgroup. Our approach identified a total of 20,598 orthogroups across the six gastropod species, with 1197 single-copy orthogroups across all species and 300 orthogroups specific to O. idahoensis. We then constructed an ultrametric time-calibrated phylogenetic tree from the multiple sequence alignment of single copy orthologs produced by Orthofinder using BEAST v.1.8.4 [39] (for further details see Materials and Methods). Overall, our phylogeny of gastropod genomes closely matches those from a previous phylogenetic study in terms of topology and branching events (overlap in 95% highest posterior density estimates of divergence events inferred by [40]; Fig. 3A). Our results show that O. idahoensis split from C. unifasciata between 82.3 and 93.7 MYA (95% highest posterior density estimate), which is consistent with the earliest fossil evidence of Oreohelicidae in Cretaceous-Tertiary boundary deposits [41].
Fig. 3.

Gene family evolution and GO enrichment analysis of O. idahoensis genome draft. A BEAST Phylogenetic tree constructed using single-copy orthologs of gastropod species. Upper and lower 95% highest posterior density divergence are depicted at each node in red. Gene family expansion/contraction values are depicted for a given branch with expansion in purple and contraction in orange. Timescale abbreviations: Pz, Permian; Tr, Triassic; J, Jurassic; K, Cretaceous; T, tertiary; Q, Quaternary. B GO enrichment analysis of 119 significantly expanded gene families in O. idahoensis. C GO enrichment analysis of 58 expressed retrocopies with InterProScan GO annotation in the O. idahoensis genome. Photo credit: O. idahoensis, Richard A. Salsbury; C. unifasciata, Sebatsian SANT CC BY-NC 4.0; A. fulica Alexander R. Jenner CC BY-SA 3.0; B. glabriata, Lewis et al., CC BY 2.5; A. californica, Jerry Kirkhart CC BY 2.0; P. canaliculata, Cheng Te Hsu CC BY-SA 4.0. Figure layout inspired by [42]
To better understand gene family evolution within the O. idahoensis genome, we used CAFE v.3.03 [43] to analyze gene family expansion and contraction across Gastropoda. We found 115 significant expansions and 39 significant contractions (P value < 0.01) on the branch leading to Oreohelix (Fig. 3)A). GO enrichment analyses of these expanded orthogroups in clusterProfiler [44] revealed a high proportion of the genes were enriched for GO functions related to nucleotide binding, G-protein coupled peptide receptors, chromatin, and serine-type endopeptidase inhibitors (Fig. 3B). InterProScan identifiers of genes containing significantly enriched GO terms indicated that AIG1, serine protein kinases, histone H2A/H2B, and SERPINs (Fig. 3B) were the primary expanded gene families containing enriched GO terms. The large expansion of AIG1 genes is notable as this gene family is known to rapidly expand and contract across Mollusca and its expression is associated with heat stress and parasitic load [45, 46]. The O. idahoensis genome contains 112 partial or complete copies of this gene which is the largest number of AIG1 elements of any molluscan genome [46]. The expansion of histone H2A/H2B genes is also notable as histone variants are often associated with distinct chromatin regions and suggest a diverse toolset for epigenetically regulating transcription [47]. Of final note is the expansion of serine protein kinases and SERPINs, both proteins have been detected in mollusc shell proteomes and mantle tissue across molluscan species [48].
Retrotransposition of host genes
Given the large proportion of repetitive elements in the O. idahoensis genome draft assembly, we scanned the assembly for retrocopies produced through retrotransposition of host genes using Retroscan v.1.1 [49]. We identified 492 retrocopies of which 22 were intact, 146 were retrogenes, 275 were chimerical retrogenes, and 49 were pseudogenes (see Material and Methods for definitions of retrocopy types). Of these retrocopies, 80 were expressed in the O. idahoensis transcriptome (see Materials and Methods). The distribution of synonymous mutations (Ks) across retrocopies inferred using Retroscan revealed most retrocopies are recent and that expressed retrocopies can be of older age (Fig. 4A). The ratio of synonymous to non-synonymous mutations (Ka/Ks) for expressed retrocopies indicate that they appear to largely be under purifying selection as there was no expressed retrocopy with a Ka/Ks ratio greater than 1 (Fig. 4B).
Fig. 4.
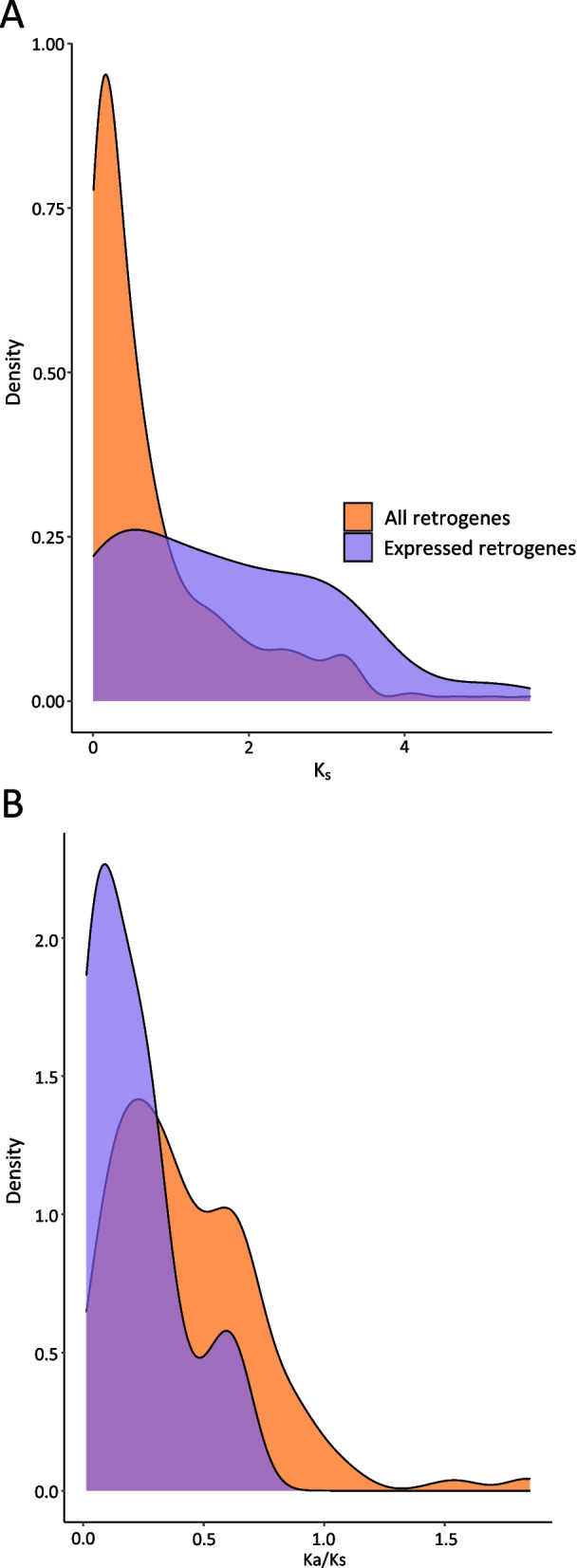
O. idahoensis retrocopy Ks divergence and Ka/Ks ratios. A Expressed and non-expressed retrocopy kernel-density estimates of Ks divergence. B Expressed and non-expressed retrocopy kernel-density estimates of Ka/Ks scores. Ka/Ks values less than 1 indicate purifying selection
GO enrichment analyses of the expressed retrocopies using ClusterProfiler revealed that monooxygenase activity, GTP binding, and microtubule cytoskeleton GO terms were significantly enriched. These GO terms were associated with InterProScan classified gene families of cytochrome P450, Rho GTPases, and α/β tubulin (Fig. 3C). Many of the GO terms within expressed retrocopies overlapped between genes, likely reflecting the fusion of different genes in chimeric retrocopies [49].
Genome size and LTR expansion
The O. idahoensis genome is estimated to be more than twice the size of other previously published land snail genome assemblies [50]. To determine whether the large size of the O. idahoensis genome draft assembly is a product of an ancient whole genome duplication event in the branch leading to O. idahoensis, we estimated ancient whole genome duplication using Ks plots of whole paranome sequences and a molluscan nuclear substitution rate in WGD v.1.0.1 [51]. We detected a single peak in our Ks plot (1.1–1.5; Supplementary Fig. 2) which was dated to be approximately 66–91 MYA. This age estimate is consistent with the established ancient whole genome duplication at the base of Styllomatophoran land snails detected by previous studies [40, 52].
We examined the possibility of recent whole-genome duplication using Gaussian Mixture Models of different ploidy models of heterozygous SNPs distributions using nQuire [53]. The best performing ploidy model was a diploid model as it had the lowest delta likelihood from the free model (diploid delta likelihood: 698275; triploid delta likelihood: 78931; tetraploid delta likelihood 1,375,106). These results suggest the large size of the O. idahoensis genome is likely not attributable to an ancient whole genome duplication specific to Oreohelicidae or to a recent whole genome duplication event.
Finally, we examined genome size estimates of the closely related O. s. strigosa and distantly related O. jugalis by analyzing k-mer counts of genome skims using RESPECT v.1.0.0 [36]. Genome size estimates were roughly 1.5 times greater for O. idahoensis genome skims compared to either smooth form species (~ 9 Gb: ~ 6 Gb; Table 3). While the specific base pair estimates for genome size from genome skims may be off for all Oreohelix due to their relatively high repeat content [36], the relative differences in estimated genome size of O. idahoensis and other Oreohelix species is likely real given their stark differences in repetitive k-mer content.
Historical demography of O. idahoensis
Given the low heterozygosity and threatened status of O. idahoensis, it may be expected that this species has undergone a severe population bottleneck recently. Historical demography trends revealed by PSMC [54] revealed substantial decline in effective population size in the last several million years followed by stasis (Fig. 5). Both PSMC and heterozygosity estimates indicate O. idahoensis has a low effective population size that may be a product of a population bottleneck and/or increased selfing.
Fig. 5.

O. idahoensis historical effective population size (Ne) trajectory obtained from PSMC
Discussion
Elucidating the genomic features associated with adaptive divergence can be challenging without suitable genome assemblies to provide evolutionary and functional context. In this study, we generated a high quality draft-assembly of the limestone endemic land snail O. idahoensis and genome skims from two other Oreohelix species to understand the genomic features underlying edaphic specialization in land snails. Unlike other studies of sessile edaphic specialists (e.g. serpentine or karst plants) [55], in this study we do not find that any evidence of a recent or ancient whole genome duplication event specific to O. idahoensis and instead find the O. idahoensis genome draft assembly is characterized by massive expansion of transposable elements.
Transposable elements are increasingly being studied for their ability to quickly generate novel genetic variation which may facilitate local adaptation [56]. Transposable element activity has been linked to rapid adaptive divergence in ants [57], several mammal species [58], Anolis lizards [59], and plants [60]. While the specific roles of transposable elements in the process of edaphic specialization remain to be elucidated within Oreohelix, our study suggests that LTRs, which compose more than half of the genome, have shaped three major genomic processes in O. idahoensis: genome size expansion, gene composition through retrotransposition of host genes, and ectopic recombination.
Similar to other large genome plant and animal species [60, 61], replication of LTRs and other transposable elements may be the primary driver of the large genome size of O. idahoensis compared to other land snail genera (e.g. C. unifasciata and C. nemoralis; Fig. 1) and other Oreohelix species (Figs. 1, 2 and 3). Our analysis of ancient and recent whole genome duplications lends support to this hypothesis as we detected no recent or ancient whole genome duplication specific to Oreohelicidae. Given the recent expansion of LTRs and other transposable elements in the O. idahoensis genome draft (Fig. 1) and large difference in repetitive k-mer content and genome size between genome skims of O. idahoensis and other Oreohelix species (Table 3), we conclude that the large genome size of O. idahoensis is a product of recent transposable element expansion.
The high proportion and recent activity of LTRs in the O. idahoensis genome suggests that these elements may be engines of genomic novelty which may contribute to the remarkable phenotypic variation present in Oreohelix. Retrotransposition of host genes by transposable elements has generated several retrocopies which have been incorporated as alternative splice variants of expressed host genes. Furthermore, we detected a high ratio of solo LTRs to intact elements (median family ratio: 6.33) which are typically produced through recombination between flanking LTR elements. Compared to plants which often have similar LTR content to O. idahoensis, the ratio of solo to intact elements in O. idahoensis genome draft is generally higher than most plant species [37] and may be indicative of increased genome wide ectopic recombination. Ectopic recombination between transposable elements can cause chromosomal translocations and inversions which can alter linkage relationships between genes and facilitate reproductive isolation between populations [62, 63]. When taken in context with our findings that the closely related non-limestone endemic O. s. strigosa contains much lower repetitive k-mer content than O. idahoensis, our results suggest that LTR mediated ectopic recombination could be a major mechanism underlying edaphic specialization in Oreohelix. However, it is also possible that stress associated with CaCO3 habitat specialization or secondary contact with neighboring species may facilitate decoupling of epigenetic control mechanisms on LTRs which may enable transposable element expansion [62]. Further research leveraging comparative genomic approaches on full genome assemblies of O. idahoensis and closely related smooth form non-limestone endemics are needed to test these hypotheses.
Gene family expansion analysis revealed several expanded gene families putatively related to stress/immunity (AIG1), DNA-packaging (histone H2A/H2B), and biomineralization (serine proteases and SERPINs) which are likely tied to the ecology and genomic architecture of O. idahoensis. The expansion of AIG1 genes may be related to the high frequency of parasites present in many Oreohelix populations [64] and thermal extremes that species experience in semi-arid montane environments [65]. Expansions of histone genes are often associated with greater epigenetic transcriptional control mechanisms [47]. The expansion of histone genes in O. idahoensis may permit a greater diversity of silencing mechanisms to regulate transposable element activity [66]. Serine proteases and SERPINs are involved in a variety of cellular processes (e.g. innate immunity) but are also two halves of a biomineralization system in which serine proteases promote crystal nucleation and SERPINs regulate the process [67]. These gene families are promising targets for future studies examining ecological specialization and genomic architecture in Oreohelix.
Conclusions
We utilized genome skims and a hybrid de novo assembly approach to examine the genomic features associated with edaphic specialization to calcareous habitats in Oreohelix land snails. The limestone endemic O. idahoensis genome is the largest and most repetitive mollusc genome assembly published to-date and has substantial LTR content which has shaped genome size, ectopic recombination, and gene composition. The availability of this genome will facilitate studies into the molecular basis of heightened biomineralization in limestone environments and aids in the conservation genetics of this threatened group of land snails.
Methods
Sample collection and sequencing
Three species were sampled for this study: O. idahoensis, O. jugalis, and smooth O. s. strigosa. Specimens of O. idahoensis (10) and O. jugalis (2) were collected from a limestone outcrop and alluvial deposits at the limestone outcrop border, respectively, at Lucile, Idaho during three collection trips during the spring of 2017–2019. A single specimen of O. s. strigosa was collected from Barret Creek above the north shore of Lake Chelan, Washington in the fall of 2021. A single adult O. idahoensis specimen was selected for genome assembly and a portion of the abdominal region of the foot muscle (1.2 g) was removed and shipped on dry ice to the HudsonAlpha Institute for Biotechnology (Birmingham, AL) for total genomic DNA extraction and library preparation. DNA was extracted using a MagAttract HMW DNA Kit from Qiagen (PN- 67563) and a 10x linked read library was prepared using a Chromium v2 Genome Reagent kit (10X Genomics; PN-120258) with 2.5 ng of DNA as starting material. This same DNA extract was then shipped to the University of Idaho Institute for Bioinformatics and Evolutionary Studies Genomics Resource Core (IBEST-GRC) for continuous long-read (CLR) library prep for the PACBIO Sequel II system. The linked-read library was size selected for 450 bp insert size and sequenced on three Illumina HiSeq X Ten lanes producing 1.42 billion read pairs for a total yield of 425.8 Gb of linked reads. Fragment analyzer traces before and after CLR library prep confirmed the mean fragment length to be > 50 kb, the CLR library was sequenced using three Sequel II SMRT Cell 8 M’s producing 46 million reads totaling 324.5 Gb of unique molecular data.
Two separate RNA extractions were prepared from six pooled mantle-edge tissue and one whole body of O. idahoensis using TRIzol® Reagent (Life Technologies, Gaithersburg, USA) following manufacturer’s instructions. To check the quality of each RNA extract, we opted to assess RNA fragment length (Agilent systems M5310AA) over RNA integrity number (i.e. RIN) as there is no observable 28S peak for Oreohelix and many other gastropod species [68]. An equal proportion from each sample was combined for SMRT Bell sequencing on the PacBio Sequel I platform at the University of Washington PacBio Sequencing Services lab. Library preparation followed the Iso-Seq protocol with cDNA amplification of polyadenylated transcripts [69]. The two libraries were then pooled and sequenced using two Sequel I SMRT Cell 1 M v3’s. Consensus full-length non-chimeric (FLNC) sequences were called using SMRT Link v6.0.0 and output as fasta files for later analysis (https://www.pacb.com/support/softwaredownloads/).
To compare genome repetitiveness and genome size across Oreohelix species, we generated genome-skims for two O. idahoensis, two O. jugalis, and one O. s. strigosa sample. DNA was extracted from abdominal foot tissue for each sample using a Qiagen DNeasy blood and tissue kit and used input for library preparation using a NEBNext® Ultra DNA Library Prep Kit (PN- E7370L). All libraries were pooled and sequenced on a single 150 bp paired-end Novaseq S4 lane resulting in ~ 185 Gb of output.
Genome feature estimation
As many assembly software packages require an estimate of genome size as a starting parameter for assembly (e.g. canu), we used a k-mer counting approach to estimate genome size using our Illumina linked read data. First, we removed the 10x molecule information encoded in the first 22 bp (16 bp barcode, 6 bp nmer-primer) of our Illumina paired-end linked-read data using longranger v.2.2.2 (github.com/10XGenomics/longranger). Scrubbed linked read pairs were then used to estimate genome size, heterozygosity, and repeat content through k-mer analysis. We counted the number of distinct canonical 31-mers using Jellyfish v.2.2.6 [70] and generated a histogram with a max coverage threshold of 1,000,000 for analysis using Genomescope v.2.0 [20]. We chose 31-mers for genome feature estimation using Genomescope v. 2.0 as higher k-mers are more suited to estimating genome size for highly repetitive genomes [20]. We also utilized ModEst on default parameters with our trimmed linked read-data which estimates genome size using mapping rates and coverage data of raw reads to genome assemblies to estimate genome size for O. idahoensis [21]. ModEst was not used for our genome skim data as there was insufficient coverage (< 5) to accurately estimate genome size with this approach when reads were aligned to individual draft assemblies constructed using the pipeline [21]. Instead, genome-skim data for all Oreohelix species were analyzed using RESPECT v.1.0.0 on default settings to estimate genome size and repeat content [36].
Independent and hybrid assembly of linked and long reads
We utilized the 10x Genomics proprietary software Supernova v.2.1.1 [71] to generate a linked-read assembly using 2.1 billion reads, which is the maximum number of input reads that can be passed into Supernova at the time of this study (32 bit cap). All remaining parameters were kept at their default values. The resulting assembly was then output using the ‘pseudohap1’ option and a minimum contig length of 1 kb.
We opted to utilize the natively cluster capable genome assembly software canu v.1.9 [23] over other long-read assemblers given concerns that the high repeat content and large size of the O. idahoensis genome would likely lead to excessive run times. PacBio CLR long-reads were error corrected using canu v.1.9 [23] with the recommended settings for large and repetitive genomes: ‘corMhapFilterThreshold=0.0000000002 corMhapOpti”–=“--threshold 0–0 --num-hashes –2 --num-min-matche–3 --ordered-sketch-size 1–0 --ordered-k-mer-size–4 --min-olaplength 2–0 --repeat-idf-scal” 50” mhapBlockSize=500 ovlMerDistinct=0.975’. The unitgging step was performed using version 2.1.1 with the same assembly parameters as a new version of canu had been released since the initiation of the pipeline [72]. Initial polishing of the raw canu contigs using long reads and Racon v1.4.21 [73] resulted in a modest loss in BUSCO score so we opted to only polish using a single round of NextPolish v.1.3.1 [74] and the barcode-scrubbed linked reads created from longranger. All alignments required for polishing were done using minimap2 v.2.16 [75] unless otherwise stated. The polished canu assembly was then purged of duplicate regions and haplotig sequences using purge_dups on default settings [76].
We created a hybrid assembly following the previously published Supernova-DBG2OLC assembly pipeline [22]. Briefly, Supernova assembled contigs are aligned to uncorrected, compressed long-read contigs using DBG2OLC v1.0.0 [77] which then forms a backbone graph for assembly. This backbone graph is then output and available for polishing using the DBG2OLC consensus module or other user chosen polishing software. The Supernova-DBG2OLC backbone graph was generated using the following parameters in DBG2OLC: ‘k 17 AdaptiveTh 0.0001 K-merCovTh 2 MinOverlap 15 RemoveChimera 1 Contigs supernova_scaftigs.fa f CLR.fasta.’ We then polished the raw Supernova-DBG2OLC contigs using Racon for two rounds of long read polishing followed by one round of short read polishing using NextPolish.
Contigs were merged from both the canu and Supernova-DBG2OLC assemblies to produce a more contiguous assembly using quick-merge v.0.3 [78]. We chose to use the canu assembly as the query sequence and the Supernova-DBG2OLC as the reference to merge contigs as the canu assembly was more fragmented than the Supernova-DBG2OLC assembly. The two assemblies were merged by whole genome alignment using nucmer v.4.0.0rc1 [79] to identify overlaps (parameters: -l 100) and quick-merge using the following settings: ‘-hco 5 -c 1.5 -l 340000 -ml 10000.’ The merged assembly was then polished once with long reads and once using short reads using Racon and NextPolish, respectively. This polished assembly was then corrected for misassemblies using linked read barcode information by tigmint v.1.2.2 [80] on default settings except to set the max molecule length as 70,000 bp, which was slightly above the mean molecule length reported by Supernova (63,810 bp). The tigmint corrected assembly was then scaffolded using ARCS v.1.2.1 with default parameters [81]. Small scaffolds less than 5000 bp were then removed to facilitate faster repeat masking and gene annotation.
Genome assembly quality evaluation
All assemblies were assessed for gene content completeness using the Benchmarking Universal Single Copy Ortholog v4.0.2 (BUSCO) metazoan gene set before and after polishing to ensure assemblies did not become overly polished [82]. The final merged and scaffolded assembly was screened for contaminants using conterminator v.1.0 [30] on the NCBI nonredundant nucleotide database (downloaded on October 26, 2021). Conterminator was run using default parameters except to only consider potential contaminations between O. idahoensis (NCBI:txid 2,584,915) and other taxa in the NCBI database. Identified contaminated regions were then blasted against the NCBI nonredundant nucleotide database to confirm contamination.
Repeat identification and divergence
A de novo library of transposable elements and repeats was generated for O. idahoensis using the EDTA v1.9.9 pipeline on default settings [31]. EDTA is a combined structural and homology repeat detection pipeline that integrates several structural transposable element detection programs with homology searches using RepeatModeler 2.0 [34]. The EDTA transposable element library was then used for genome masking with RepeatMasker v4.1.1 [34] using rmblastn v2.2.28 (http://www.repeatmasker.org/RMBlast.html) as the search engine. A repeat landscape plot was then created using the transposable elements consensus alignments produced by RepeatMaskers and calculating the Kimura divergence from the consensus using the RepeatMasker script ‘calcdivergencefromalign.pl’ and the R-package ggplot2 [83].
To place the identified repeat content and divergence history of the O. idahoensis genome draft in context with other previously published mollusc genome assemblies, we generated individual EDTA repeat libraries for five other previously published mollusc genomes (Cepaea nemorals, Candidula faciata, Achatina immaculata, Radix auricularia, and Euprymna scolopes). These species were selected based on their close phylogenetic relatedness to O. idahoensis or for having a similar genome size (i.e. Euprymna scolopes). Each individual repeat library was used to mask and generate repeat landscape plots following the same procedure as the O. idahoensis genome draft.
LTR insertion times can be estimated by using a known mutation rate and comparing LTR divergence between sister pairs. We estimated the insertion times of intact LTR elements using a molluscan substitution rate of 1.645 × 10− 9 per site per year [84]. Insertion times of LTR elements were then placed in a phylogenetic context by comparing estimated divergence times of Oreohelix species from a previous study [16] with LTR insertion times estimated from the current study (Fig. 2).
We then investigated whether long terminal repeat transposons (LTR) were involved in reshaping structural variation in the O. idahoensis genome draft by estimating the proportion solo LTRs to intact LTR elements. LTRs are mobile transposable elements are composed of two flanking long terminal repeats and an internal sequence which enables replication through a copy-and-paste mechanism. Solo LTRs are formed by ectopic recombination between two LTR regions at non-homologous locations on the same or another chromosome [37]. The ratio of solo LTRs to intact LTRs can be used to estimate the level of ectopic recombination within the genome and deletion of LTR elements [37]. To measure the ratio of solo LTRs to intact LTRs in the genome assembly, we first used the ‘solo_finder.pl’ script from LTR_retriever [85] to identify solo LTR candidates. The ‘solo_finder.pl’ script reports solo LTR candidates have at least 80% coverage of an LTR element in the intact LTR library, possess with an alignment score greater than 300, and are at least 100 bp in length. Then, we filtered solo LTR candidates that were within 25 kb of a scaffold end as these may represent true intact elements that were not fully assembled. Finally, we calculated the ratio of intact vs solo LTR families using the ‘solo_intact_ratio.pl‘script from LTR_retriever.
Genome annotation
We utilized information from Iso-Seq reads, protein homology, and ab initio predictions to annotate the O. idahoensis genome draft. The FLNC reads obtained from Iso-Seq sequencing were mapped to the soft-masked genome draft using minimap2, and transcript models were called using TAMA [86]. The mapped sequences were first collapsed with tama_collapse.py with the parameters ‘-s input.sam -f idaho_scaff_final.fa -p input.collapse -x no_cap -sjt 20 -lde 2 -a 100 -z 100’ for each library (mantle and whole body) and then merged together with the tama_merge.py script. The final set of assembled transcripts were then output as a fasta file use as transcript evidence for genome annotation.
We used MAKER (v.3.01.03) [87] to annotate the O. idahoensis genome in three rounds using transcript evidence from the TAMA processed Iso-Seq libraries, protein evidence from the Cepaea nemoralis, Candidula unifasciata, and Achatina fulicula genome assemblies, and the O. idahoensis EDTA repeat library to soft-mask transposable elements in gene models. In the first round, transcript and protein evidence were aligned to the genome draft using BLASTn and BLASTx [88] and refined for splice site alignment using Exonerate v2.4.0 to generate the initial set of gene models [89]. Given the large LTR content identified in the O. idahoensis genome draft (see below) and the positive correlation between LTR content and intron length across the tree of life [90, 91], we ran the first round of MAKER annotation with three values of the ‘split_hit’ parameter that controls max intron length (the default of 10,000 bp, 50,000 bp, and 100,000 bp). The transcriptome produced with a ‘split_hit’ value of 100,000 bp had the greatest number of complete BUSCO genes and was selected for further annotation refinement. MAKER employs annotation edit distance (AED) scores to assess the quality of generated gene models with respect to prior evidence (i.e. a lower AED score indicates greater agreement between gene model and protein/transcript evidence: 0 complete agreement, 1 no evidence). After the first round, 96.1% of the 22,384 genes had an AED score < 0.5 which indicates almost all gene models were well-supported by transcript or protein homology evidence. The second and third rounds of MAKER utilized the ab-initio predictors SNAP (version 2006-07-28) and Augustus v.3.3.3 [92] to generate new gene models. For each round, SNAP was trained on the prior rounds gene models that had AED scores lower than 0.25 and were of at least 50 amino acids in length. Augustus was retrained using the BUSCO metazoan gene set with the ‘-long’ parameter. The retrained Augustus models were then used to predict gene models during the second and third rounds of MAKER. For the final round of MAKER, we only retained gene models with AED scores < 0.5 resulted in 27,692 gene models. The resulting proteins from the gene models were then aligned against the NCBI nonredundant protein (downloaded on 26 October 2021), InterPro (accessed 26 October 2021) [29], and Uniprot databases (Swissprot and Trembl; accessed 26 October 2021). The longest isoform for each gene was then exported for protein phylogenetic reconstruction.
Phylogenetic analysis and divergence time estimation
We placed the O. idahoensis genome draft in a phylogenetic context by first aligning single copy orthologs of several mollusc genome assemblies (Pomacea canaliculata, Biomalpharia glabrata, Achatina fulica, and Candidula unifasciata,) using Orthofinder v.2.4.0 with the ‘-M msa’ flag [38]. We then constructed an ultrametric time-calibrated tree using BEAST v.2.5.1 [39] with the following settings: WAG amino acid matrix, four gamma rate categories, estimate proportion of invariant sites and substitution rate, uncorrelated relaxed log-normal clock, birth-death model, runtime of 50,000,000 generations, burn-in of 10%, and setting tree height to be calibrated using the fossil date for the Caenogastropoda-Heterobranchia split (390 MYA offset log-normal distribution with a mean of 30) [40, 52]. Convergence was assessed visually using Tracer v.1.7.1 and effective sample size was confirmed to be greater than 200 for all parameters. We chose to omit the C. nemoralis genome assembly in our phylogenetic analysis due to the large number of genes in the C. nemoralis genome which may be a result of incomplete repeat masking [24, 25]. The inclusion of non-masked repetitive elements can affect downstream gene family expansion by altering estimates of ancestral gene family size between species [43].
Gene family evolution
The orthogroup gene family counts generated by Orthofinder and the maximum clade credibility consensus BEAST ultrametic tree were used as input into CAFE v.3.03 [43] to examine gene family expansion across Gastropods. We estimated gene family expansion in CAFE by estimating a single birth-death parameter and a significance level of 0.01. Gene families identified as significantly expanding n O. idahoensis and containing GO annotations by InterProScan were then retained for GO enrichment analysis.
Retrocopy identification
We scanned the genome for retrocopies produced through retrotransposition of host genes using Retroscan v.1.1 on that hard masked O. idahoensis genome assembly using default settings [49]. Retrocopies are partial or complete duplications of host genes but lack the introns of their parent sequence due to the mechanism of retrotransposition. Retrocopies inferred from retroscan correspond to four categories: (1) retrocopies that are entirely intact genes produced through retrotransposition which are labelled as ‘intact;’ (2) retrocopies that retain the open-reading-frame of the parent gene but recruit promoter and enhancer regions which make them functional are labelled ‘retrogenes;’ (3) retrocopies which have fused with other genes and are transcribed as introns or alternative splice variants of exons are labelled as ‘chimeric retrogenes;’ and (4) retrocopies with stop codons or no promoter regions are labelled as ‘pseudogenes.’ Expressed retrocopies were identified by blasting all retrocopies that did not contain a stop codon (retrogenes, chimeric retrogenes, and intact) to the TAMA assembled transcripts. Retrocopies were only considered expressed if they had an exact full-length match to one of the TAMA transcripts. All expressed retrocopies were then functionally annotated by InterProScan and those containing GO terms were retained for GO enrichment analysis.
GO enrichment analysis
We utilized the R package clusterProfiler v3.14 to determine enriched GO terms in the expanded gene families and expressed retrocopies in the O. idahoensis genome [44]. First, full GO term ontologies were built using the ‘buildGOmap’ for foreground gene sets (e.g. expressed retrocopies) and the background gene set consisting of all genes (including retrocopies) within the O. idahoensis genome draft assembly. Then, GO enrichment analysis was performed using the ‘enricher’ function on default settings except to specify a Benjamini-Hochberg correction for p-value estimation (pvalueCutoff = 0.05, pAdjustMethod = “BH”).
Detecting ancestral and recent whole genome duplication
To determine whether ancient whole-genome duplication has led to the large genome size of O. idahoensis, we generated Ks plots of whole paranome sequences using WGD v.1.0.1 [51]. Plots were examined visually for peaks in synonymous substitution values for paralogous genes in the O. idahoensis genome draft which may correspond to a genome duplication event. We estimated the timing of any inferred whole genome duplication events by dividing the estimated Ks peak value by a molluscan substitution rate of 1.645 × 10− 9 per site per year [84].
We also examined the possibility of a recent genome duplication by comparing allelic frequencies of heterozygous SNPs using nQuire v.1.0.0 [53]. In brief, nQuire estimates the best ploidy model (diploid, triploid, tetraploid) that fits the distribution of heterozygous SNPs mapped to the reference assembly by assuming diploids should have a frequency of 0.5/0.5, triploids 0.33/0.66, and tetraploids 0.25/0.75 and 0.5/0.5 [53]. We aligned the linked-reads scrubbed of 10x barcodes to the O. idahoensis genome draft assembly using minimap2 and then removed all alignments with a mapping quality less than 20. The bam file was then input into nQuire and ‘denoised’ of low frequency allele ratios following the recommendation of the authors [53]. The model of ploidy producing the lowest delta likelihood compared to the free Gaussian Mixture Model was then considered to be the ploidy of the O. idahoensis genome draft.
Historical demography
Given the threatened conservation status of O. idahoensis, we also investigated historical demography of this species using PSMC [54]. We first generated input files for PSMC by using alignments of our barcode trimmed linked reads to our genome using minimap2 [75] and samtools mpileup v. 1.9 [93] to estimate heterozygous bases. We utilized a minimum mapping quality of 30 for samtools mpileup and required both pairs to align to the genome. PSMC was run on the input files for 20 generations and Ne was inferred across 74 time intervals (18 + 25*2 + 2 + 4) using default settings. PSMC results were scaled using the same molluscan substitution rate as previous analyses [84] and a generation time estimate taken from unpublished mark-recapture data (5.6 years; Tronstad, personal communication).
Supplementary Information
{kind=link}
{kind=link}
Acknowledgements
We thank Joel Sauder from Idaho Fish and Game for facilitating permits and aiding in sample collection. A special thanks are given to the University of Idaho Genomics Resource Core staff scientists Dan New and Matt Fagnan for their services and input on sequencing strategies. We would also like to thank Katy Munson from University of Washington PacBio Sequencing Services for Iso-Seq sample preparation and sequencing. We are grateful to Amanda Stahlke, Ian Oiler, Bernandette Johnson, Kelly Martin, and John G. Phillips for several helpful discussions related to this work. Additional thanks are given to Selkirk Wildlife Science for permitting early access to their Oreohelix sequencing data. Special thanks are given to Lusha Tronstad of the Wyoming Natural Heritage program for providing generation time estimates from her ongoing mark-recapture study of Oreohelix.
Authors’ contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by T. Mason Linscott, Andrea González-González, and Takahiro Hirano. Genome assembly and analysis was performed by T. Mason Linscott. Christine E. Parent facilitated project management and edited all drafts of the article. The manuscript was written by T. Mason Linscott and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by a National Science Foundation Graduate Research Fellowship program award to T. Mason Linscott (NSF 1842399). Research reported in this publication was also supported by an CAREER grant from the National Science Foundation (NSF 1751157) to Christine E. Parent. Additional sequencing was made possible through the NSF Idaho EPSCoR GEM3 Program under award number OIA-1757324 and the United States Fish and Wildlife Service under grant number F12AP01527 to Selkirk Wildlife Sciences. This work was supported in part by NIH COBRE Phase III grant P30GM103324.
Availability of data and materials
All sequence data used for this publication has been made available through NCBI BioProject accession number PRJNA727022. The genome assembly has been deposited in GenBank under JAMPIV000000000.1. The version used in the present study is 1.0.0. Annotation files, repeat libraries, and coding sequences are available on FigShare (10.6084/m9.figshare.c.6008425.v1).
Declarations
Ethics approval and consent to participate
Animal specimens used in this study were collected under Idaho state permit 160302 and Washington state permit LUCID 21–105.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Clements R, Sodhi NS, Schilthuizen M, Ng PKL. Limestone karsts of Southeast Asia: imperiled arks of biodiversity. BioScience. 2006;56:733–742. doi: 10.1641/0006-3568(2006)56[733:LKOSAI]2.0.CO;2. [DOI] [Google Scholar]
- 2.Rajakaruna N. Lessons on evolution from the study of edaphic specialization. Bot Rev. 2018;84:39–78. doi: 10.1007/s12229-017-9193-2. [DOI] [Google Scholar]
- 3.Staude IR, Navarro LM, Pereira HM. Range size predicts the risk of local extinction from habitat loss. Glob Ecol Biogeogr. 2020;29:16–25. doi: 10.1111/geb.13003. [DOI] [Google Scholar]
- 4.BirdLife/FFI/IUCN/WWF . Extraction and biodiversity in limestone areas. 2014. [Google Scholar]
- 5.Schilthuizen M, Clements R. Tracking extinction from space. Tentacle. 2008;16:8–9. [Google Scholar]
- 6.Kiew R, Rahman RA. Plant diversity assessment of karst limestone, a case study of Malaysia’s Batu caves. NC. 2021;44:21–49. doi: 10.3897/natureconservation.44.60175. [DOI] [Google Scholar]
- 7.Shogren JF, Tschirhart J, Anderson T, Ando AW, Beissinger SR, Brookshire D, et al. Why economics matters for endangered species protection. Conserv Biol. 1999;13:1257–1261. doi: 10.1046/j.1523-1739.1999.98414.x. [DOI] [Google Scholar]
- 8.Liew T-S, Price L, Clements GR. Using Google earth to improve the management of threatened limestone karst ecosystems in peninsular Malaysia. Trop Conserv Sci. 2016;9:903–920. doi: 10.1177/194008291600900219. [DOI] [Google Scholar]
- 9.Lowry DB, Rockwood RC, Willis JH. Ecological reproductive isolation of coast and inland races of Mimulus Guttatus. Evolution. 2008;62:2196–2214. doi: 10.1111/j.1558-5646.2008.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolář F, Fér T, Štech M, Trávníček P, Dušková E, Schönswetter P, et al. Bringing together evolution on serpentine and polyploidy: spatiotemporal history of the diploid-tetraploid complex of Knautia arvensis (Dipsacaceae) PLoS One. 2012;7:e39988. doi: 10.1371/journal.pone.0039988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lydeard C, Cowie RH, Ponder WF, Bogan AE, Bouchet P, Clark SA, et al. The global decline of nonmarine mollusks. BioScience. 2004;54:321–330. doi: 10.1641/0006-3568(2004)054[0321:TGDONM]2.0.CO;2. [DOI] [Google Scholar]
- 12.The IUCN Red List of Threatened Species. Version 2021–3. https://www.iucnredlist.org. Accessed on 23 Mar 2022.
- 13.Barker GM. The biology of terrestrial molluscs. Wallingford, Oxfordshire, UK: CABI; 2001.
- 14.Goodfriend GA. Variation in land-snail shell form and size and its causes: a review. Syst Biol. 1986;35:204–223. doi: 10.1093/sysbio/35.2.204. [DOI] [Google Scholar]
- 15.Alonso MR, López-Alcántara A, Rivas P, Ibáñez M. A biogeographic study of Iberus gualtierianus (L.)(Pulmonata: Helicidae) Soosiana. 1985;13:1–10. [Google Scholar]
- 16.Linscott TM, Weaver K, Morales V, Parent CE. Assessing species number and genetic diversity of the Mountainsnails (Oreohelicidae) Conserv Genet. 2020;21:971–985. doi: 10.1007/s10592-020-01302-5. [DOI] [Google Scholar]
- 17.Pilsbry HA. Land mollusca of North America:(north of Mexico). Acad Nat Sci. 1939;1:412-553.
- 18.Frest TJ, Johannes EJ. Land snail survey of the lower Salmon River drainage, Idaho. 1997. [Google Scholar]
- 19.NatureServe. 2022. NatureServe Explorer [web application]. NatureServe, Arlington, Virginia. Available https://explorer.natureserve.org/. (Accessed 22 Mar 2022).
- 20.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfenninger M, Schönnenbeck P, Schell T. ModEst: accurate estimation of genome size from next generation sequencing data. Mol Ecol Resour. 2022;22:1454–1464. doi: 10.1111/1755-0998.13570. [DOI] [PubMed] [Google Scholar]
- 22.Ma ZS, Li L, Ye C, Peng M, Zhang Y-P. Hybrid assembly of ultra-long Nanopore reads augmented with 10x-genomics contigs: demonstrated with a human genome. Genomics. 2019;111:1896–1901. doi: 10.1016/j.ygeno.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saenko SV, Groenenberg DS, Davison A, Schilthuizen M. The draft genome sequence of the grove snail Cepaea nemoralis. G3. 2021;11:jkaa071. doi: 10.1093/g3journal/jkaa071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chueca LJ, Schell T, Pfenninger M. De novo genome assembly of the land snail Candidula unifasciata (Mollusca: Gastropoda) G3. 2021;11:jkab180. doi: 10.1093/g3journal/jkab180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hohenlohe PA, Funk WC, Rajora OP. Population genomics for wildlife conservation and management. Mol Ecol. 2021;30:62–82. doi: 10.1111/mec.15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seppey M, Manni M, Zdobnov EM. Gene prediction. Springer; 2019. BUSCO: assessing genome assembly and annotation completeness; pp. 227–245. [DOI] [PubMed] [Google Scholar]
- 28.Guo Y, Zhang Y, Liu Q, Huang Y, Mao G, Yue Z, et al. A chromosomal-level genome assembly for the giant African snail Achatina fulica. Gigascience. 2019;8:giz124. doi: 10.1093/gigascience/giz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinegger M, Salzberg SL. Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 2020;21:1–12. doi: 10.1186/s13059-020-02023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ou S, Su W, Liao Y, Chougule K, Agda JR, Hellinga AJ, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20:1–18. doi: 10.1186/s13059-019-1905-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z, Zhang L, Hu J, Wang J, Bao Z, Wang S. The evo-devo of molluscs: insights from a genomic perspective. Evol Dev. 2020;22:409–424. doi: 10.1111/ede.12336. [DOI] [PubMed] [Google Scholar]
- 33.Thomas-Bulle C, Piednoël M, Donnart T, Filée J, Jollivet D, Bonnivard E. Mollusc genomes reveal variability in patterns of LTR-retrotransposons dynamics. BMC Genomics. 2018;19:1–18. doi: 10.1186/s12864-018-5200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020;117:9451–9457. doi: 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su W, Ou S, Hufford MB, Peterson T. A tutorial of EDTA: extensive De novo TE annotator. Plant Transposable Elem. 2021:55–67. [DOI] [PubMed]
- 36.Sarmashghi S, Balaban M, Rachtman E, Touri B, Mirarab S, Bafna V. Estimating repeat spectra and genome length from low-coverage genome skims with RESPECT. PLoS Comput Biol. 2021;17:e1009449. doi: 10.1371/journal.pcbi.1009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jedlicka P, Lexa M, Kejnovsky E. What can long terminal repeats tell us about the age of LTR retrotransposons, gene conversion and ectopic recombination? Front Plant Sci. 2020;11:644. doi: 10.3389/fpls.2020.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:1–14. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Doğan Ö, Guiglielmoni N, Guichard A, Schrödl M. The de novo genome of the “Spanish” slug Arion vulgaris Moquin-Tandon, 1855 (Gastropoda: Panpulmonata): massive expansion of transposable elements in a major pest species. bioRxiv. 2021.
- 41.Pierce HG, Constenius KN. Late Eocene-Oligocene nonmarine mollusks of the northern Kishenehn Basin, Montana and British Columbia. Ann Carnegie Mus Pittsbg. 2001;70:1–112. doi: 10.5962/p.215142. [DOI] [Google Scholar]
- 42.Yu T, Hu Y, Zhang Y, Zhao R, Yan X, Dayananda B, et al. Whole-genome sequencing of Acer catalpifolium reveals evolutionary history of endangered species. Genome Biol Evol. 2021;13:evab271. doi: 10.1093/gbe/evab271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30:1987–1997. doi: 10.1093/molbev/mst100. [DOI] [PubMed] [Google Scholar]
- 44.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guerin MN, Weinstein DJ, Bracht JR. Stress adapted Mollusca and Nematoda exhibit Convergently expanded Hsp70 and AIG1 gene families. J Mol Evol. 2019;87:289–297. doi: 10.1007/s00239-019-09900-9. [DOI] [PubMed] [Google Scholar]
- 46.Lu L, Loker ES, Zhang S-M, Buddenborg SK, Bu L. Genome-wide discovery, and computational and transcriptional characterization of an AIG gene family in the freshwater snail Biomphalaria glabrata, a vector for Schistosoma mansoni. BMC Genomics. 2020;21:190. doi: 10.1186/s12864-020-6534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurumizaka H, Kujirai T, Takizawa Y. Contributions of histone variants in nucleosome structure and function. J Mol Biol. 2021;433:166678. doi: 10.1016/j.jmb.2020.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Marin F. Mollusc shellomes: past, present and future. J Struct Biol. 2020;212:107583. doi: 10.1016/j.jsb.2020.107583. [DOI] [PubMed] [Google Scholar]
- 49.Wei Z, Sun J, Li Q, Yao T, Zeng H, Wang Y. RetroScan: an easy-to-use pipeline for Retrocopy annotation and visualization. Front Genet. 2021;12. [DOI] [PMC free article] [PubMed]
- 50.Gomes-dos-Santos A, Lopes-Lima M, Castro LFC, Froufe E. Molluscan genomics: the road so far and the way forward. Hydrobiologia. 2020;847:1705–1726. doi: 10.1007/s10750-019-04111-1. [DOI] [Google Scholar]
- 51.Zwaenepoel A, Van de Peer Y. Wgd—simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics. 2019;35:2153–2155. doi: 10.1093/bioinformatics/bty915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu C, Ren Y, Li Z, Hu Q, Yin L, Wang H, et al. Giant African snail genomes provide insights into molluscan whole-genome duplication and aquatic–terrestrial transition. Mol Ecol Resour. 2021;21:478–494. doi: 10.1111/1755-0998.13261. [DOI] [PubMed] [Google Scholar]
- 53.Weiß CL, Pais M, Cano LM, Kamoun S, Burbano HA. nQuire: a statistical framework for ploidy estimation using next generation sequencing. BMC Bioinformatics. 2018;19:122. doi: 10.1186/s12859-018-2128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gottlieb LD. Rethinking classic examples of recent speciation in plants. New Phytol. 2004;161:71–82. doi: 10.1046/j.1469-8137.2003.00922.x. [DOI] [Google Scholar]
- 56.Schrader L, Schmitz J. The impact of transposable elements in adaptive evolution. Mol Ecol. 2019;28:1537–1549. doi: 10.1111/mec.14794. [DOI] [PubMed] [Google Scholar]
- 57.Schrader L, Kim JW, Ence D, Zimin A, Klein A, Wyschetzki K, et al. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat Commun. 2014;5:5495. doi: 10.1038/ncomms6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ricci M, Peona V, Guichard E, Taccioli C, Boattini A. Transposable elements activity is positively related to rate of speciation in mammals. J Mol Evol. 2018;86:303–310. doi: 10.1007/s00239-018-9847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feiner N. Accumulation of transposable elements in Hox gene clusters during adaptive radiation of Anolis lizards. Proc R Soc B Biol Sci. 2016;283:20161555. doi: 10.1098/rspb.2016.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Galindo-González L, Mhiri C, Deyholos MK, Grandbastien M-A. LTR-retrotransposons in plants: engines of evolution. Gene. 2017;626:14–25. doi: 10.1016/j.gene.2017.04.051. [DOI] [PubMed] [Google Scholar]
- 61.Sun C, Shepard DB, Chong RA, López Arriaza J, Hall K, Castoe TA, et al. LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Genome Biol Evol. 2012;4:168–183. doi: 10.1093/gbe/evr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serrato-Capuchina A, Matute DR. The role of transposable elements in speciation. Genes. 2018;9:254. doi: 10.3390/genes9050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.González J, Petrov DA. Evolution of genome content: population dynamics of transposable elements in flies and humans. In: Anisimova M, editor. Evolutionary genomics: statistical and computational methods, volume 1. Totowa, NJ: Humana Press; 2012. pp. 361–383. [DOI] [PubMed] [Google Scholar]
- 64.Dempsey ZW, Burg TM, Goater CP. Spatiotemporal patterns of infection for emerging larval liver fluke (Dicrocoelium dendriticum) in three species of land snail in southern Alberta, Canada. J Parasitol. 2019;105:155–161. doi: 10.1645/18-124. [DOI] [PubMed] [Google Scholar]
- 65.Schweizer M, Triebskorn R, Köhler H-R. Snails in the sun: strategies of terrestrial gastropods to cope with hot and dry conditions. Ecol Evol. 2019;9:12940–12960. doi: 10.1002/ece3.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friedli M, Trono D. The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol. 2015;31:429–451. doi: 10.1146/annurev-cellbio-100814-125514. [DOI] [PubMed] [Google Scholar]
- 67.Yarra T, Ramesh K, Blaxter M, Hüning A, Melzner F, Clark MS. Transcriptomic analysis of shell repair and biomineralization in the blue mussel, Mytilus edulis. BMC Genomics. 2021;22:437. doi: 10.1186/s12864-021-07751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Natsidis P, Schiffer PH, Salvador-Martínez I, Telford MJ. Computational discovery of hidden breaks in 28S ribosomal RNAs across eukaryotes and consequences for RNA integrity numbers. Sci Rep. 2019;9:19477. doi: 10.1038/s41598-019-55573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gonzalez-Garay ML. Introduction to isoform sequencing using Pacific biosciences technology (Iso-Seq) In: Wu J, editor. Transcriptomics and gene regulation. Dordrecht: Springer Netherlands; 2016. pp. 141–160. [Google Scholar]
- 70.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol. 2016;34:303–311. doi: 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020;30:1291–1305. doi: 10.1101/gr.263566.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaser R, Sović I, Nagarajan N, Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu J, Fan J, Sun Z, Liu S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–2255. doi: 10.1093/bioinformatics/btz891. [DOI] [PubMed] [Google Scholar]
- 75.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guan D, McCarthy SA, Wood J, Howe K, Wang Y, Durbin R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye C, Hill CM, Wu S, Ruan J, Ma Z(S) DBG2OLC: efficient assembly of large genomes using Long erroneous reads of the third generation sequencing technologies. Sci Rep. 2016;6:31900. doi: 10.1038/srep31900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakraborty M, Baldwin-Brown JG, Long AD, Emerson JJ. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44:e147–e147. doi: 10.1093/nar/gkw654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol. 2018;14:e1005944. doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackman SD, Coombe L, Chu J, Warren RL, Vandervalk BP, Yeo S, et al. Tigmint: correcting assembly errors using linked reads from large molecules. BMC Bioinformatics. 2018;19:393. doi: 10.1186/s12859-018-2425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yeo S, Coombe L, Warren RL, Chu J, Birol I. ARCS: scaffolding genome drafts with linked reads. Bioinformatics. 2018;34:725–731. doi: 10.1093/bioinformatics/btx675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller DE, Staber C, Zeitlinger J, Hawley RS. Highly contiguous genome assemblies of 15 Drosophila species generated using Nanopore sequencing. G3. 2018;8:3131–3141. doi: 10.1534/g3.118.200160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wickham H. ggplot2. WIREs Comput Stat. 2011;3:180–185. doi: 10.1002/wics.147. [DOI] [Google Scholar]
- 84.Allio R, Donega S, Galtier N, Nabholz B. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Molecular biology and evolution. 2017;34(11):2762-2772. [DOI] [PubMed]
- 85.Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of Long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–1422. doi: 10.1104/pp.17.01310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kuo RI, Cheng Y, Zhang R, Brown JWS, Smith J, Archibald AL, et al. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genomics. 2020;21:751. doi: 10.1186/s12864-020-07123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang D, Su Y, Wang X, Lei H, Yu J. Transposon-derived and satellite-derived repetitive sequences play distinct functional roles in mammalian intron size expansion. Evol Bioinformatics Online. 2012;8:EBO.S9758. doi: 10.4137/EBO.S9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Michael TP. Plant genome size variation: bloating and purging DNA. Brief Funct Genomic. 2014;13:308–317. doi: 10.1093/bfgp/elu005. [DOI] [PubMed] [Google Scholar]
- 92.Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(suppl_2):W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data used for this publication has been made available through NCBI BioProject accession number PRJNA727022. The genome assembly has been deposited in GenBank under JAMPIV000000000.1. The version used in the present study is 1.0.0. Annotation files, repeat libraries, and coding sequences are available on FigShare (10.6084/m9.figshare.c.6008425.v1).