

Abstract
Exchange of mobile functional genes within microbiota benefits the microbial community. However, the status of the mobile gene pool in environment is still largely unclear, impeding the understanding on the process of gene transfer in natural microbial communities. The release of extracellular vesicles (EVs) by diverse organisms has been proposed to be a vital way in the complex networks of interactions between microbes and their habitats. In this study, we hypothesized that microbial EVs encapsulating functional DNA are widely distributed in the environmental matrix. The prevalence, source and DNA cargoes of EVs in three types of typical microbial habitats were studied. High abundance of EVs comparable to the bacterial concentration was found in human faeces, wastewater and soil. Metagenomic analysis showed the diverse and differential taxonomy of EVs‐associated DNA compared to source microbiome. An array of efficient EVs producing species was identified. A wide variety of mobile genes including glycoside hydrolase family 25 were enriched. Antibiotic resistance genes co‐localizing with mobile genetic elements were abundant in the EVs. This study provides novel insights into the prevalent EVs as a reservoir for the mobile functional genes in the natural environment.
Keywords: antibiotic resistance genes, extracellular vesicles, functional genes, microbial habitats
1. INTRODUCTION
Intercellular communication via material and information exchange is predominant for bacteria in nature to execute sophisticated tasks, such as biofilm development and response to environmental stimuli (Groussin et al., 2021; Solano et al., 2014). With regard to gene transfer, there is a gene flow across microbial communities. These mobile genes synchronize the behaviour of microbes and benefit the microbial community (Smillie et al., 2011). For example, frequent gene exchange by horizontal gene transfer (HGT) plays a prominent role in the niche expanding of antibiotic resistance genes (ARGs) and thus enhances antibiotic resistance (Arnold et al., 2021; Broaders et al., 2013; Ellabaan et al., 2021; Soucy et al., 2015; Zhu et al., 2017). Transfer of genes encoding mobile glycoside hydrolases across microbiota could spread the ability of polysaccharide metabolism and thus facilitate the growth of microbiota (Brito et al., 2016). All these show crucial roles of mobile gene flow across environmental and gut microbiota. However, the status of this mobile gene pool is still unclear in the field (Brito et al., 2016). This elicits a surprising disconnect on the knowledge between laboratory experiments and natural environments (Brito, 2021).
Almost all living organisms are capable of releasing EVs. As spherical and lipid‐bilayered nanoscale particles, EVs can encapsulate, concentrate and protect their cargoes including DNA, RNA, proteins, and lipids (Toyofuku et al., 2019). The cargoes can stay longer, travel further distance against dilution and degradation in EVs, which facilitate the efficient delivery to the target cells. EVs mediate deliveries of ARGs, virulence determinants, essential nutrients, extracellular electrons, and quorum‐sensing molecules within or across species under laboratory culture conditions (Liu et al., 2019; Tashiro et al., 2017; Toyofuku et al., 2017; Wu et al., 2020). In the field studies, EVs were isolated from seawater using density gradient centrifugation, and these EVs carry varied types of cargoes in marine ecosystems (Biller et al., 2017, 2014). EVs were also found in indoor dust and showed potential promoting effects on lung metastasis (Dinh et al., 2020). In our previous study, we found the prevalence of ARGs and mobile genetic elements (MGEs) in indoor dust EVs and their microbial origins were studied in indoor dust by metagenomic sequencing (Qin et al., 2022). In sewage, EVs were ultraconcentrated by a 100 kDa tangential flow filtration. Metagenomics showed the change of ARGs associated with EVs‐like particles upon antibiotic treatment (Maestre‐Carballa et al., 2019). These studies confirmed the existence of microbial EVs in field, highlighting the necessity to further study the origin, cargoes, and function of EVs in the environmental microbiota. In this study, we found the prevalence of EVs in three common types of microbial habitats including human faeces, wastewater and soil. These EVs population are of varied microbial origins and carried DNA encoding proteins with diverse functions, providing insight into the roles of EVs as reservoirs of functional genes in natural environment.
2. MATERIALS AND METHODS
2.1. Sampling and characteristics
Faecal samples were obtained from a pool of volunteer donors using a randomised list. The collection was approved by the ethical committee of the Institute of Urban Environment, Chinese Academy of Sciences. Stools were collected in a sterile container and immediately returned to researchers within 1 h after defecation. The information on bowel habits, anthropometrics, and medication, as well as diet, were all collected, details were shown in Supplementary Table 1. For wastewater, samples were collected from influents of five wastewater treatment plants in January 2021 in Xiamen city, China. 30 L wastewater was stored in sterile plastic containers and transported to the laboratory within 2 h for immediate processing. Water qualities, including temperature, pH, and conductivity were measured in situ using a portable probe (HachHQ40d, Loveland, CO, USA), and details were shown in Supplementary Table 1. Soils were sampled from ten parks locating in urban and rural areas of Xiamen. Soils from varied habitats including the grassland, shrub‐grassland, forest lawn, and shrubbery were pooled together and 500 g mixed soils were obtained from each park. The soil characteristics, including water content, pH, total organic carbon (TOC), total nitrogen (TN), total carbon (TC), total phosphorus (TP) and carbon nitrogen ratio (TC/TN) were measured (Supplementary Table 1).
For bacterial strains, both Escherichia coli strain DH5α and Staphylococcus aureus RN4220 were purchased from Guangdong Microbial Culture Collection Center, China. The ampicillin‐resistant E. coli strain E4742 was isolated from wastewater following the procedure as shown in the supplementary methods.
2.2. Isolation of EVs from environmental media and bacterial culture
The protocols for extracting EVs from liquid and solid phases are given below. Faecal EVs were isolated as previously described (Tulkens et al., 2020). Briefly, 45 g per specimen were resuspended in endotoxin‐free PBS with the ratio of 1:10 (weight:volume) and mixed at 37°C for 30 min. For soil, 600 g soil per sample was stirred in PBS with the ratio of 1:5 (weight:volume) under 220 rpm/min at 10°C for 8 h. Then both suspensions were centrifuged at 10,000 × g for 15 min at 4°C to remove bacteria and insoluble matter. The supernatant was filtered with 0.45 and 0.22 μm polyether sulfone membrane (Merck Millipore, Ireland) using a vacuum filtration apparatus. For wastewater, samples were filtered sequentially through 3, 0.8, 0.45 and 0.22 μm polyether sulfone membrane. All the above filtrates were concentrated by a tangential flow filter (100 kDa cutoff, Millipore). Filtration with 0.22 μm membrane was then performed again to ensure the depletion of bacteria. Ultracentrifugation (100,000 × g, 4°C, 2 h) was carried out to obtain the EVs pellet using SW41Ti rotor (Beckman Coulter, USA). The pellet was washed one time with PBS using the same centrifugation condition. EVs in bacterial culture were isolated as previously reported with modifications (Manning & Kuehn, 2011). Briefly, the bacteria were cultured in LB broth at 37°C until exponential growth stage (OD600 = 1.2). Then, the bacterial culture was centrifuged at 10,000 × g for 15 min at 4°C, and the supernatant was filtered through 0.45 and 0.22 μm filters. The filtrate was concentrated and ultra‐centrifugated as above.
2.3. Characterization of EVs
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were applied to characterize the EVs as shown in the supplementary materials. Size distribution and particle concentration of EVs were measured using a nanoflow cytometry (N30, NanoFCM, Xiamen, China) following a previous study (Tian et al., 2020). A mixture of silica nanospheres of four different diameters (68 ± 2 nm, 91 ± 3 nm, 113 ± 3 nm, and 155 ± 3 nm) were used as references. The particle concentration of EVs in environmental media was normalized to the weight or volume of samples. Total protein content was determined using the Micro BCA Protein Assay Reagent Kit (Thermo Fisher Scientific, Waltham, USA) following the manufacturer's specifications.
2.4. Extraction of microbial DNA and EVs‐associated DNA
For faeces, 0.2 g fresh sample was applied to DNA extraction using a Fast DNA Stool Mini Kit (Qiagen, Germany). For wastewater, 50 ml sample was filtered through a 0.22 μm polyether sulfone membrane (Merck Millipore). The filters were cut into small pieces and applied for DNA extraction by a FastDNA Spin Kit (MP Bio, USA) according to the manufacturer's guidelines. This kit was also used to extract DNA from 0.5 g soil per sample. All DNA samples were stored at −20°C until further analysis.
To make sure that the detected DNA was associated with EVs, EVs samples were pre‐treated for 30 min with 2U of Turbo DNase (Invitrogen, Carlsbad, CA, USA) in 1X DNase reaction buffer (Invitrogen, Carlsbad, CA, USA). Another round of the above process was performed to digest the DNA residue. Then DNase was deactivated by the addition of 15 mM EDTA and heating at 75°C for 15 min. Genomic DNA extracted from wastewater was applied to the same treatment with EVs, which was used as a positive control to confirm the effectiveness of the DNase treatment. After the elimination of DNA outside of EVs, EVs samples were lysed with 0.125% Triton X‐100 at 37°C for 30 min to release EVs‐associated DNA, which was then extracted using Monarch PCR&DNA Cleanup kit (New ENGLAND BioLabs). The concentration of DNA was quantified using a Qubit high‐sensitivity (HS) assay kit for double‐stranded DNA (Thermo Fisher Scientific, USA) with a Qubit 4.0 Fluorometer (Thermo Fisher Scientific, USA).
2.5. Length of EVs‐associated DNA fragments
Evaluation of DNA fragment length was performed by Agilent 2100 Bioanalyzer using Agilent Bioanalyzer High Sensitivity DNA assay kit according to the manufacturer's instruction. The lower marker (35 bp) and the upper marker (10,380 bp) were used.
2.6. Metagenomics sequencing and bioinformatical analysis
DNA library construction and sequencing were performed by Illumina platform at Majorbio Co., Ltd. (Shanghai, China). A 150 bp paired‐end library was constructed and the actual insert size ranges from 447 to 520 bp. The raw sequencing reads for each sample were independently processed for quality control using the fastp (https://github.com/OpenGene/fastp). The MEGAHIT (Li et al., 2015) was used to assemble different depth sequences with defaulting settings. Assembly statistics (number of contigs, number of bp in contigs, contig N50, contig N90, the longest and the shortest contig) were calculated. The unassembled reads for each sample were pooled and reassembled for further analysis. Genes were predicted by MetaGeneMark (Zhu et al., 2010). The sequences with the best splicing effect were selected, and the open reading frame (ORF) was predicted. Genes with length greater than or equal to 100 bp were selected and translated into amino acid sequences. In all, a total of 46 samples were sequenced (Supplementary Table 4).
2.7. Construction of taxonomic and functional profiling
A non‐redundant gene catalogue combining all of the predicted genes across all samples was generated with CD‐HIT software with a 90% identity as the threshold for genus assignment, and covering more than 90% of the shorter gene were clustered together (Fu et al., 2012), finally, cluster representatives shorter than 300 bp were discarded. To predict taxonomic profiling of the metagenomic samples, BLASTP was run and an e‐value cut‐off of less than 1E−5 was used. The results were obtained through the taxonomic information database corresponding to the NCBI GenBank database (v20200604). In the follow‐up analysis, the virus and unclassified gene was excluded based on taxonomic profiling data (Extended Data Figure 4). Gene abundance was calculated based on the number of aligned reads and gene length in each unigene alignment. We aligned putative amino acid sequences, which were translated from the updated gene catalogue, against the proteins in Kyoto Encyclopaedia of Genes and Genomes database (KEGG, v94.2) and carbohydrate‐active enzymes database (CAZy, v20200408) using BLASTP (default parameter e‐value ≤ 1E−5). The ARGs from each metagenomic assemblies were identified by blasting protein sequences against the Comprehensive Antibiotic Research Database (CARD, v3.0.9) database using stringent cut off (identity > 80% and alignment length > 25). To explore the distribution of mobile genetic elements (MGEs) including integrons, ISs, and plasmids, BLASTP was used to align the clean reads and assembled contigs against the databases downloaded from INTEGRALL (Moura et al., 2009), Isfinder (Siguier et al., 2015), and the NCBI RefSeq database, respectively (default parameter e‐value ≤ 1E−5 and identity > 80%). It is important to note that we reflected the identified fragments onto these MGEs, which inevitably led to overcounting of chromosomal sequences, thus the MGEs in this study actually referred to “MGEs‐like” (Zhou et al., 2019). The co‐occurrence of ARGs and MGEs was identified if they were both located on the same contig.
2.8. Statistical analysis
Sequence‐based gene abundance profiling was performed by standardizing gene abundance by gene lengths in the sample (Qin et al., 2012). We calculated α‐diversity and β‐diversity parameters to assess the different features of microbial composition among individuals. The Shannon diversity index per individual was calculated using the function vegdist in R package vegan (Oksanen et al., 2013) (version 4.0.5). Bray‐Curtis was calculated using the function diversity from the same package, and Permutational multivariate analysis of variance (PERMANOVA) was used to corroborate the differences between different samples.
Functional gene analysis was performed mainly on KEGG Orthologue (KO), CAZy family and ARGs. The total abundance of genes that belong to each KEGG category (the KEGG Class at level 2) and CAZy family was designated as comparison parameter. Wilcoxon paired rank‐sum test was used to calculate the significance level. Differentially abundant ARGs at type level between EVs and their habitats were revealed using non‐parametric factorial Kruskal‐Wallis (KW) sum‐rank test and linear discriminant analysis effect size (LEfSe) (Segata et al., 2011). p < 0.05 and LDA score > 3.0 was considered statistically significant.
2.9. Identification of marker species
To identify the dominant EVs‐producers, firstly, we performed one‐tailed Wilcoxon paired rank‐sum test, and identified 89, 1186 and 1288 species (p < 0.05, False Discovery Rate (FDR) correction) in faeces, wastewater and soil, respectively. Then, we adopted Maximum Relevance Minimum Redundancy (mRMR) to perform a feature selection (Qin et al., 2012). With the “mRMRe” package of the R software to perform the incremental selection (De Jay et al., 2013), we finally selected three sets of top 10 markers as the optimal selection for dominant EVs‐producers.
3. RESULTS
3.1. Prevalence of EVs in natural microbial habitats
Differential centrifugation was used to collect EVs from environmental matrix in our study. Transmission electron microscopy (TEM) revealed that bilayered spherical EVs existed in faeces, wastewater and soil (Figure 1a–c and Extended Data Figure 1), exhibiting similar morphology with EVs released by an E.coli strain E4742 isolated from wastewater, as well as that from S. aureus (Figure 1d and e). SEM showed the EVs on the bacterial cell membrane (Figure 1f). Nano flow cytometry reported the average size around 70 nm for all three EVs populations (Figure 1g). EVs were found in faeces (1010–1011/g, wet weight), soil (107–108/g, wet weight) and wastewater (104–105/ml). To be noted, we cannot rule out the potential co‐existence of viral particles in the EV samples prepared by ultracentrifugation. We further determined the bacterial concentration in each field sample using absolute quantification of 16S rRNA encoding genes. The particle concentration, as well as particle to protein ratio were positively correlated to the field bacterial concentrations (Figure 1h), indicating that bacterial abundance might be a determining factor for the levels of environmental EVs. We did not find other significantly‐related environmental factors (Supplementary Table 2). The prevalence across faeces, wastewater and soil suggests the potential roles of EVs in the microbial communication in the field.
FIGURE 1.
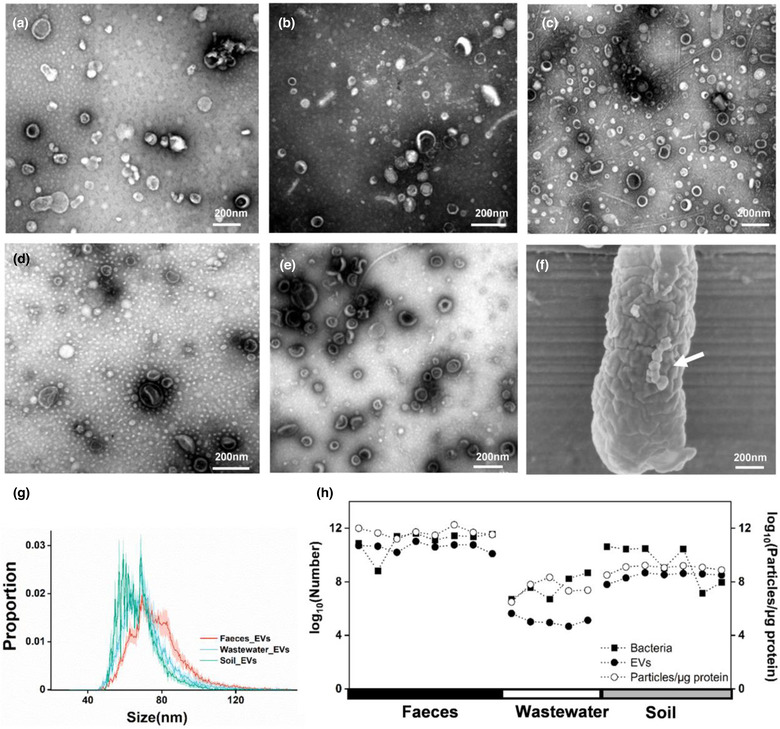
Occurrence of EVs in environment. Transmission electron microscopy of EVs from a, human fecal samples. b, Wastewater samples. c, Soil samples. d, Culture media of Escherichia coli E4742 strain. e, Culture media of S. aureus, scale bar, 200 nm. f, Scanning electron microscopy of EVs on the surface of Escherichia coli E4742, scale bar, 200 nm. g, Size distribution of EVs from faeces, wastewater and soil were analyzed by nano flow cytometry. h, Concentrations of bacteria, EV particles, and the ratio of particles to proteins in corresponding sampling sites across three habitats. Soil samples with negative PCR results are not shown.
3.2. Taxonomic source of EVs‐associated DNA
We next explored the taxonomic host of environmental EVs‐associated DNA using metagenomics. EVs samples were treated with two rounds of DNase to eliminate DNA outside of EVs before subjecting to extraction of EVs‐associated DNA (Extended Data Figure 2a–e). The EVs‐associated DNA fragments across three EVs populations were distributed in the length range of 35 bp to >10 kb and about 53.38% of the fragments were in the range from 200 to 1000 bp (Extended Data Figure 2f, Supplementary Table 3). Metagenomic sequencing predicted high diversity of the taxonomic composition of EVs‐associated DNA ranging from all domains to 5358 species within all three populations of EVs (Figure 2a). Of the species‐level richness assessed in EVs, Shannon's Diversity Indices were significantly higher in wastewater and soil compared with faeces. The microbial taxonomic profiles of EVs‐associated DNA were less diverse than that of the corresponding source habitats but no significant difference in evenness (Figure 2a). As expected, most of the EVs‐associated DNA was predicted to be from bacteria (Figure 2b).
FIGURE 2.
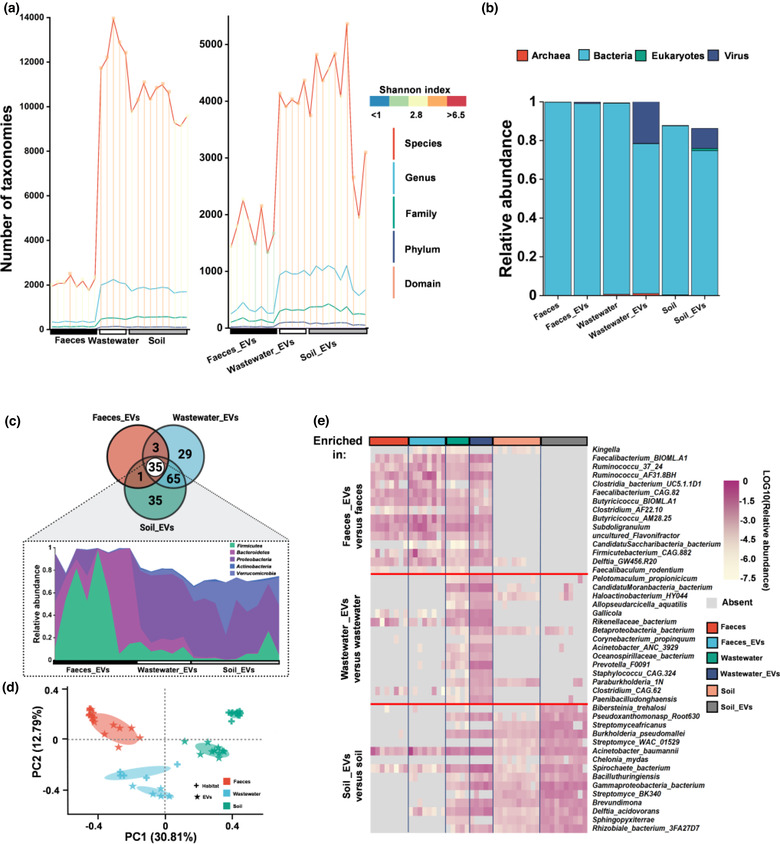
Microbial compositions of EVs‐associated DNA from three habitats. a, The number of microbial taxa predicted by metagenomic sequencing. Different colors of lines indicate different taxonomic levels. The gradient color of vertical lines represents the value of Shannon Index as indicated in the upper right panels. b, Compositional differences at domain levels between microbiota and its derived EVs. The bars indicate the median proportion of different domains. Different colors indicate different domains as presented in the figure. Compositions of unclassified are not shown. c, Venn diagram of the numbers of overlapping and unique phyla. Flow chart showed the relative abundance of five shared dominant microbial phyla among EVs samples (faeces_EVs, n = 8; wastewater_EVs, n = 5; soil_EVs, n = 10). d, PCoA plots of Bray‐Curtis distance of microbial composition at genus level. Each dot represented one sample. Colors were used to distinguish the habitats, and the symbol of plus and asterisk indicated the habitat samples and its derived EVs samples, respectively. Ellipses represented the 95% confidence interval. e, Heat map of the top 15 marker species enriched in the EVs (Maximum Relevance Minimum Redundancy). The complete list was included in Supplementary Table 6
Thirty‐five shared phyla of bacteria, dominated by Gram‐negative bacteria (GNB) including Proteobacteria, Bacteroidetes and Verrucomicrobia, as well as Gram‐positive bacteria (GPB), including Firmicutes and Actinobacteria, contributed the most to all three populations of EVs‐associated DNA (Figure 2c). The median ratio of GPB: GNB varied across EVs populations (Supplementary Table 5). Members of the Firmicutes taxa contributed 47.2% (median, Inter Quartile Range (IQR) 12.8%–71.0%) faecal EVs‐associated DNA, while Proteobacteria produced 55.2% (median, IQR 50.3%–59.5%) of the EVs‐associated DNA in the wastewater and 39.6% (median, IQR 37.1%–48.8%) in the soil, respectively (Figure 2c). This provided field evidences that GPB also released large amount of EVs‐associated DNA. As expected, principal co‐ordinates analysis (PCoA) revealed that the taxonomic composition of EVs were similar to the microbiomes in their source habitats (Figure 2d, Extended Data Fig. 5a and b). Among three habitats, fecal EVs owned greater similarity with faeces microbiome while the EVs from wastewater and soil showed less clustering with respective microbiome (Figure 2d). Species significantly enriched (p < 0.05, FDR correction) in the EVs compared to their source microbiome were defined as efficient producers that were prominent to produce EVs‐associated DNA. In this study, 89, 1186, and 1288 out of 1905, 4357, and 5358 species were identified as efficient producers respectively in faeces, wastewater and soil (Supplementary Table 6). About 60.7% of the efficient producers belong to Firmicutes in faeces habitats, while 50.4% and 34.7% of the efficient producers belong to Proteobacteria in wastewater and soil. Notably, six species of Proteobacteria (Acinetobacter junii, Delftia acidovorans, Delftia lacustris, Delftia tsuruhatensis, Pseudomonas putida, and Sphingobium) were efficient producers in all habitats. Composition of respective top 15 marker producers of EVs‐associated DNA differed across habitats (Figure 2e). Marker EVs producers in the faeces, such as Kingella, possessed very low abundance in wastewater and soil. Pelotomaculum had a high proportion of EVs‐associated DNA in wastewater but was almost absent in the other two habitats. Bibersteinia was a marker producer in the soil but absent in the faeces and wastewater.
3.3. Functional characterization of EVs‐associated DNA
We next tracked the EVs‐associated functional genes produced by the efficient producers. EVs equipped with all categories of functional genes of source microbiota at the level 2 KEGG Class, although there were differences in the quantity (Extended Data Fig. 6a). These suggested that a large proportion of host genome can be encapsulated into EVs. Comparison of the relative levels for each functional gene in EVs versus their source microbiome revealed unique patterns of genes retention. The fecal EVs may be functionally enriched in translation and metabolic pathways, and various types of functional genes were enriched in wastewater and soil, such as cellular processes, information processes, and metabolic pathways (Figure 3a and Extended Data Fig. 6b). As major contributors, Firmicutes contributed primarily metabolic pathway enrichment in faeces EVs and soil EVs. It also contributed mostly genetic information processing pathway in wastewater EVs. Proteobacteria contributed mostly to metabolic pathway enrichment in faeces EVs and soil EVs, human diseases pathway enrichment in wastewater EVs (Supplementary Table 7).
FIGURE 3.
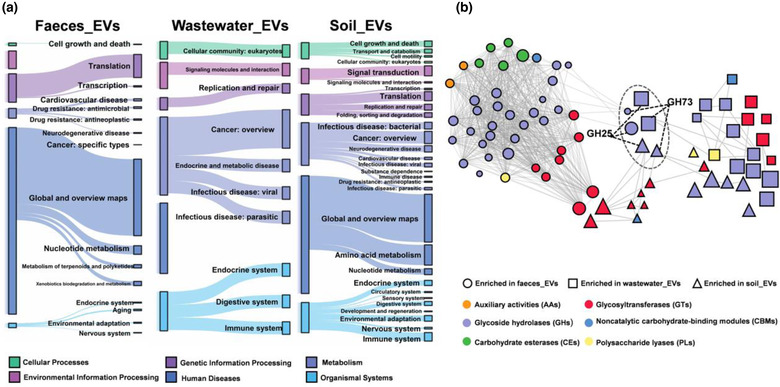
Functional genomic repertoire in EVs compared to source habitat samples. a, KEGG orthologues (the KEGG Class at level 2) of the EVs that were released by efficient producers, tracked using Sankey plots. The heights of the rectangles indicate the relative abundance of KEGG categories, and each function had a distinct color. The lines connected to subcategories (from level 1 to level 2). b, Diversity and distribution of genes encoding CAZymes associated with EVs that were released by efficient producers. A co‐occurrence network of CAZymes that were enriched in EVs compared to habitats. The size of the nodes indicated the relative abundance. Nodes were grouped into six functional classes as shown in the figure. Connecting lines represent that there was a significant spearman correlation between nodes (p < 0.05)
Using carbohydrate‐active enzymes (CAZymes) database, 24,249, 53,830 and 62,572 genes enconding enzymes were annotated in EVs‐associated DNA from faeces, wastewater and soil, respectively. Glycoside hydrolases (GH) were found to be abundant in mobile gene pools across microbiota to facilitate the use of multiple polysaccharides (Brito et al., 2016; Rakoff‐Nahoum et al., 2014). Multiple genes encoding enzymes were enriched in EVs compared with their habitat microbiome (Supplementary Table 8 and Figure 3b). GH and glycosyltransferases (GT) dominated the EVs‐enriched enzymes‐encoding genes, whereas GH73 and GH25 families were enriched in all three EVs populations. As major contributors, Firmicutes contributed mostly GT2 enrichment in faeces EVs, GH25 in wastewater EVs, GH23 and GT8 in soil EVs. Proteobacteria contributed mostly GT4 enrichment in faeces EVs, GH23 in wastewater EVs and GH24 in soil EVs (Supplementary Table 9). A co‐occurrence network predicting potential relationships between these genes revealed closer interrelation in the faecal EVs compared to the other two EVs populations.
3.4. Occurrence of EVs‐associated ARGs
A total of 332 ARGs belonging to 16 antibiotic classes had hits in EVs‐associated DNA across all three types of habitats (Figure 4a). The median relative abundance of ARGs was 1.03 × 10−4 in faecal EVs, 1.42 × 10−4 in wastewater EVs and 3.40 × 10−5 in soil EVs. The relative abundances were lower than those of respective source microbiomes (Figure 4a and Extended Data Fig. 7a, c). ARGs conferring resistance to tetracycline, macrolides‐lincosamids‐streptogramins (MLS) and aminoglycoside, accounted for 77.62% of the total ARGs in faecal EVs. In wastewater EVs, MLS, beta‐lactam, and tetracycline resistance genes accounted for 58.41%. In soil EVs, multidrug and beta‐lactam resistance genes contributed 86.98% (Extended Data Fig. 7a). PCoA showed the cluster of ARGs in the EVs from faeces and wastewater, which also occurred in both habitats (Extended Data Fig. 7d). The relative abundance of 51, 30 and 16 ARGs were significantly different between EVs and their corresponding source microbiome (Extended Data Fig. 8). EVs‐associated ARGs were mostly traced back to 5 phyla, containing 65, 66 and 17 species respectively in faeces, wastewater and soil (Figure 4b). Notably, Firmicutes contributed the largest abundance of faecal EVs‐associated ARGs (49.97%), while Proteobacteria dominated both wastewater and soil EVs‐associated DNA (60.29% and 81.39%, respectively), which were quite different from their relative abundance in the source microbiome.
FIGURE 4.
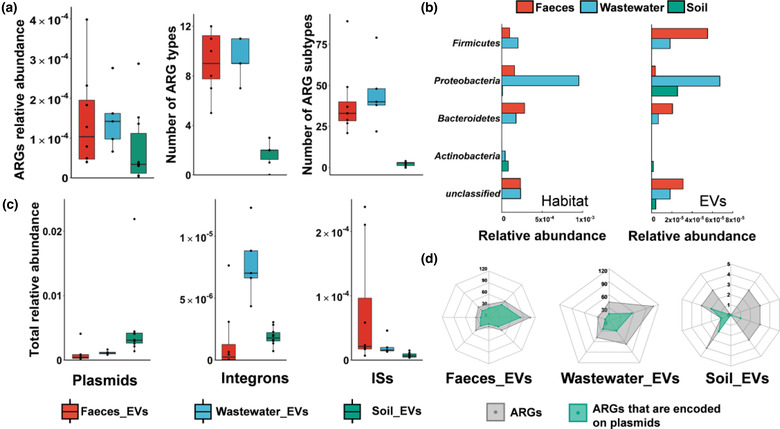
Characterization of ARG and MGE profiles in the EVs samples. a, Relative abundance of EVs‐associated ARGs at the level of total relative abundance, antibiotic types, and drug types. Data were presented as box plots (middle line, median; lower hinge, first quartile; upper hinge, third quartile; lower whisker, the smallest value at most 1.5 × the interquartile range from the hinge; upper whisker, the largest value no further than 1.5 × the interquartile range from the hinge; data beyond the whiskers were outlying points). b, Characterization of the bacteria phyla of ARG profiles in EVs and corresponding sourced habitat. Abundance of ARGs that were found in various bacteria phyla. To facilitate viewing, only those five dominant phyla are shown. Habitat samples and EVs samples are shown separately. c, Total relative abundance of MGEs, including plasmids, integrons and ISs in the EVs samples. d, The number of the total ARGs and the ARGs encoded on the plasmid. The grey plots represented the number of ARGs in each sample, while the green plots indicated the number of ARGs encoded on the plasmid in each sample.
Successful HGT process of ARGs usually requires the assistance of mobile genetic elements (MGEs) (MartãNez et al., 2015; Sentchilo et al., 2013). The abundances of EVs‐associated MGEs were 0.4%, 1.1% and 3.1% in faeces, wastewater, and soil, respectively. The plasmid was the dominant type in both EVs and habitat samples (Figure 4c and Extended Data Fig. 7b). In the EVs from faeces, wastewater, and soil, 70%, 43.6% and 26% ARGs were carried on plasmids, respectively (Figure 4d), showing the differential transfer potential of ARGs in varied EVs populations. It is of great importance to further check whether these transferred ARGs showed antibiotic phenotype in the recipient bacteria.
4. DISCUSSION
4.1. Large quantity of EVs in natural microbial habitats
To date, it is unknown to what extent EVs exist in a variety of natural environments. The way to better isolate EVs from complex environmental milieu still needs extensive study. Differential centrifugation was used in this work to obtain a balance between purity and recovery rate, as further purification like density gradient centrifugation could lead to more loss of EVs within varied density in field samples. With regard to the EVs‐associated DNA in the study, by comparing 16S rDNA copies in matched samples from differential centrifugation and density gradient centrifugation, we found that the relative ratio of bacterial EVs‐associated 16S rDNA protected by DNase did not significantly increase after density gradient centrifugation. This showed that sequenced DNA was largely representative of DNA in bacterial EVs obtained from differential centrifugation following removal of free DNA by DNase treatment (Extended Data Fig. 3). Thus, we believed that differential centrifugation is likely suitable in our study. However, it should be aware that we cannot rule out the potential co‐existence of virus and other nanosized particles.
From TEM and nano FCM, we showed abundant EVs in typical habitats including faeces, wastewater and soil, even at the level of 1011 per gram of faeces. Our finding, together with previous studies on dust and seawater, confirmed the wide existence of EVs in the natural environment. TEM images gave similar size ranges between the study and previous findings. However, the size determined by nano FCM in this study was relatively smaller than that in dust, seawater and human stool determined by nanoparticle tracking analysis (NTA) (Biller et al., 2014; Dinh et al., 2020; Tulkens et al., 2020). This is likely due to that NTA measured the hydrodynamic diameter of nanoparticles, which is relatively larger than naive diameter. The particle number of EVs showed comparable levels with the bacterial number with significantly positive correlation across varied habitat samples. Microbial abundance can be the determining factor for the EVs levels in environmental media. We did not find significant correlations between the particle concentration and other characteristics of the samples; however, the reliability of the correlation relationship was limited due to the small sample size. Data from previous studies all confirmed that lipid‐bilayer nanoparticles can tolerate high temperature, bio‐degradation and other stresses (Schulz et al., 2020; Zhang et al., 2021), which implied the potential function under extreme environment. In a previous study, archaea from hot spring were able to perform intercellular communication through EVs (Liu et al., 2021). Large quantity provides the basis for the potential intercellular communication function of EVs in natural environment.
4.2. Diverse taxonomic source of EVs‐associated DNA
The EVs‐associated DNA were extracted after the removal of free DNA in the samples by two rounds of Turbo DNase digestion. To be noted, similar to previous studies (Biller et al., 2014), there was still DNA that was not accessible to the DNase. One of the possibilities is that they can be tightly bound to the EVs surface. Previous studies have revealed the diverse parent cells of EVs in stool and dust using 16S amplicon sequencing of the EVs‐associated DNA (Choi et al., 2019; Samra et al., 2019). In our study, metagenomic sequencing was used to predict the origin of EVs‐associated DNA which is experted to provide more information compared to16S amplicon sequencing. For all three habitat types, EVs‐associated DNA were released mainly by bacteria and partially by eukaryotes and archaea. To be noted, as eukaryotic EVs were reported to contain relatively few DNA, the proportion of eukaryotic EVs might be underestimated (Jeppesen et al., 2019). In addition, EVs from certain types of bacteria might carry greater DNA content, thus overestimating the proportion of these EVs. Further study on the relative ratio of DNA content to the particle number of EVs should be calculated across bacterial species. Metagenomic analysis showed that the DNA cargoes carried by EVs for each habitat type was clearly distinct from the cellular DNA in the microbiota of origin. DNA enriched in EVs differs by habitat type. For one specific species, it may release different abundance of EVs‐associated DNA across different habitats. This can be attributed to the varied environmental conditions. Temperature, pH, and nutrition can also affect the secretion of EVs from microbes (Baumgarten et al., 2012; Bonnington & Kuehn, 2016; Keenan et al., 2008; Prados‐Rosales et al., 2014). Exposure of bacteria to antibiotics also affected the EVs production (Bos et al., 2021; Maestre‐Carballa et al., 2019; Toyofuku et al., 2019). Importantly, we identified numerous efficient producers in three typical habitats, for example, Pseudomonas putida, with relatively low abundance in the habitat, was found to be an efficient producer in three habitats simultaneously. These species could amply their roles in the microbiota communities via secreting EVs. One reported evidence is that the releasing of EVs is a significant way of exerting the role of catabolism of lignin‐derived aromatic compounds in P. putida (Salvachã°a et al., 2020). Another interesting finding was that EVs‐associated DNA predicted to be from two common GPB phyla Firmicutes and Actinobacteria, possessed relatively higher abundance than its cellular DNA. The relative abundance of Firmicutes reached almost half of the total fecal EVs. This inferred that GPB also released large amounts of EVs in the natural habitats. Given that EVs secretion is a significant way of extending its function, efficient EVs producing species can be specifically endowed with more significance even their relative abundance was small in the habitat.
4.3. EVs can be a mediator of microbial interaction via reservoiring functional genes
In accordance with previous studies that EVs contain almost the whole genome of their original host (Biller et al., 2014), we find that EVs equip with almost all categories of functional genes of source microbiota. Further, a substantial proportion of genes were enriched in efficient producers‐derived EVs, including genes associated with metabolic pathways and signalling pathways. Previous studies have also shown that EVs play a key role in endocrine intercellular communication via exchange of biological information (Paschon et al., 2016; Sherman et al., 2021). We next studied the enzymes abundance in the efficient producers‐derived EVs. Notably, the glycoside hydrolases families of GH73 and GH25 are enriched in EVs in all three types of samples compared with their habitat microbiome. Both families contain lysozymes which are essential for cleaving the peptidoglycan component of the bacterial septum and thus the destruction or reproduction of bacteria (Vollmer et al., 2008). As a peptidoglycan‐degrading lysozyme, GH25 integrated independently in all domains of life through horizontal gene transfer (Metcalf et al., 2014). However, it is still unknown how these genes were transferred horizontally within species so far apart. Enrichment of GH25 family genes in EVs across environment will provide an avenue for the transfer of this family. Moreover, of the correlations within enriched‐functional genes in EVs, closer interrelation was found in faecal EVs compared with wastewater and soil EVs. These results suggested it may be important to determine the different roles these genes have via EVs.
We also found a variety of ARGs associated with EVs from faeces, wastewater and soils, showing that EVs can be a reservoir of ARGs in real environment. The successful HGT process usually requires a certain level of ARGs with the assistance of plasmids and integrons (MartãNez et al., 2015; Sentchilo et al., 2013). We further found that plasmids, and other types of MGEs were also detected in EVs‐associated DNA. High proportion of ARGs (26%–70%) were encoded on plasmids in the EVs from faeces and wastewater. EVs can be a storage vector for the contigs of ARGs and MGEs which are prevalent in environmental media (An et al., 2018; Tennstedt et al., 2003). Previous studies have shown that EVs can mediate inter‐ or intra‐species transfer of plasmids and ARGs based on laboratory studies (Bielaszewska et al., 2020; Rumbo et al., 2011; Yaron et al., 2000). Considering the large amount of EVs production in human stool daily, EVs can be an efficient vector for the transfer of ARGs from human stools to wastewater. EVs can even cross domain barrier (Koeppen et al., 2016). This provides the basis for cargo shuttling of EVs within microbial community in real environment. It is important to further check whether ARGs can be transferred by EVs and help recipient bacteria to obtain antibiotic phenotype in the field.
To be noted, the bioinformatic approach assembled mapped short sequences onto complete gene sequences, so we cannot guarantee that the DNA sequenced belonged to full length functional genes or operons. Here we did find the DNA fragment length can be up to >10 kb and about 53.38% of the fragments were in the range from 200 to 1000 bp. Previous studies reported that 97.0%–99.3% of EVs‐associated reads were successfully mapped to the genome (Langlete et al., 2019). Future work should interrogate EV‐associated DNA samples further with PCR and long reads sequencing.
A new understanding from this study is that large amount of EVs provide an efficient way for microbes to maintain and potentially transfer extracellular mobile functional genes in various habitats. This broadens our understanding on the existing status of extracellular DNA and other public goods in the environment. EVs‐mediating transfer of genes encoding enzymes and ARGs is beneficial to the bacterial community against environmental stress. In this view, gaining a better mechanistic understanding of EVs could lead to developing novel strategies to deal with antibiotic resistance issues.
In all, our results confirm the prevalence of microbial EVs released from diverse organisms in faeces, wastewater, and soil. Metagenomic sequencing confirms the distinct taxonomic composition of EVs‐associated DNA compared to its source microbiome. Bacteria are the dominant resource in all samples. With a wide range of functional genes encapsulated, the prevalence of EVs paves new insight into the status of extracellular DNA in the environment. Considering the well‐known horizontal transfer roles, EVs can be a vehicle for inter‐cellular communication within natural microbiota, especially via the transfer of extracellular mobile functional genes. Further study in broader sampling scale, especially that with environmental pressure is warranted to the generalizability of our findings.
AUTHOR CONTRIBUTIONS
L.T.Z. and H.N.H. conducted the experiment and generated the metagenomic data. R.D.A.L. and Y.F.Q. helped with the extracellular vesicles isolation and characterization experiment. X.L.A and J.Q.S analysed the data on antibiotic resistance genes. Q.S.H. and Y.G.Z. wrote the paper, designed the experiment and got the funding. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
Authors declare that they have no competing interests.
Supporting information
Supplementary Table 1. Sampling information and sample characteristics.
Supplementary Table 2. Correlation between sample characteristics and particle concentrations of EVs.
Supplementary Table 3. The proportion of EVs‐associated DNA fragments from 200 bp to 1000 bp.
Supplementary Table 4. Metagenomic data of e ach sample after quality control.
Supplementary Table 5. Biological proportion and classification of samples at the phylum level.
Supplementary Table 6. Detailed information of species that were enriched in EVs.
Supplementary Table 7. Detailed information of the differential KEGG profiles secreted by major contributors Firmicutes and Proteobacteria between EVs and habitats.
Supplementary Table 8. Detailed information of enzyme encoding genes that were enriched in EVs.
Supplementary Table 9. Detailed information of the differential enzyme encoding genes secreted by major contributors Firmicutes and Proteobacteria between EVs and habitats.
Extended Data Fig. 1| Representative transmission electron microscopy of EVs from wastewater samples ( JM, YD, XL, QP, BZ ) and soil samples (JG, LYZ, HL, NH, ZS).
Extended Data Fig. 2| Quantitation and characterization of EVs‐associated DNA.
Extended Data Fig. 3| Quantitation of 16S rDNA copies of fecal EVs derived from differential centrifugation (DC) and density gradient centrifugation (DGC), respectively.
Extended Data Fig. 4| Summary of a bioinformatic workflow.
Extended Data Fig. 5| Composition of microbiota and EVs in different habitats.
Extended Data Fig. 6| Diversity and distribution of functional categories in the habitat samples and corresponding EVs samples.
Extended Data Fig. 7| Characterization of ARG and MGE profiles in the microbiota from each habitat.
Extended Data Fig. 8| Characterization of ARG profiles.
ACKNOWLEDGEMENTS
We thank Dr. Steven Biller for his valuable advice. We would also thank Zihan Guo for his help in the soil sampling. This work was supported by National Natural Science Foundation of China (32161143016, 42177362, 42250410328), Fujian Provincial Department of Science and Technology (2021J06037, 2022T3063), Chinese Academy of Sciences (CAS‐WX2021SF‐0302), and National Basic Science Data Center “Environment Health DataBase” (NO. NBSDC‐DB‐21).
Zhu, L.‐T. , Huang, H.‐N. , Avellán‐Llaguno, R. D. , Qin, Y. , An, X.‐L. , Su, J.‐Q. , Huang, Q. , & Zhu, Y.‐G. (2022). Diverse functional genes harboured in extracellular vesicles from environmental and human microbiota. Journal of Extracellular Vesicles, 11, e12292. 10.1002/jev2.12292
Li‐Ting Zhu and Hai‐Ning Huang contributed equally to this work.
Contributor Information
Qiansheng Huang, Email: qshuang@iue.ac.cn.
Yong‐Guan Zhu, Email: ygzhu@iue.ac.cn.
DATA AVAILABILITY STATEMENT
Raw sequencing data has been uploaded to Science Data Bank https://datapid.cn/31253.11.sciencedb.01118.
REFERENCES
- An, X.‐L. I. , Chen, Q.‐L. , Zhu, D. , Zhu, Y.‐G. , Gillings, M. R. , & Su, J.‐Q. (2018). Impact of wastewater treatment on the prevalence of integrons and the genetic diversity of integron gene cassettes. Applied and Environmental Microbiology, 84, e02766–17. 10.1128/AEM.02766-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, B. J. , Huang, I.‐T. , & Hanage, W. P. (2021). Horizontal gene transfer and adaptive evolution in bacteria. Nature Reviews Microbiology, 20, 206–218. 10.1038/s41579-021-00650-4 [DOI] [PubMed] [Google Scholar]
- Baumgarten, T. , Sperling, S. , Seifert, J. , Von Bergen, M. , Steiniger, F. , Wick, L. Y. , & Heipieper, H. J. (2012). Membrane vesicle formation as a multiple‐stress response mechanism enhances pseudomonas putida DOT‐T1E cell surface hydrophobicity and biofilm formation. Applied and Environmental Microbiology, 78, 6217–6224. 10.1128/aem.01525-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielaszewska, M. , Daniel, O. E. , Karch, H. , & Mellmann, A. (2020). Dissemination of the blaCTX‐M‐15 gene among enterobacteriaceae via outer membrane vesicles. Journal of Antimicrobial Chemotherapy, 75, 2442–2451. 10.1093/jac/dkaa214 [DOI] [PubMed] [Google Scholar]
- Biller, S. J. , Mcdaniel, L. D. , Breitbart, M. , Rogers, E. , Paul, J. H. , & Chisholm, S. W. (2017). Membrane vesicles in sea water: Heterogeneous DNA content and implications for viral abundance estimates. The ISME Journal, 11, 394–404. 10.1038/ismej.2016.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller, S. J. , Schubotz, F. , Roggensack, S. E. , Thompson, A. W. , Summons, R. E. , & Chisholm, S. W. (2014). Bacterial vesicles in marine ecosystems. Science, 343, 183–186. 10.1126/science.1243457 [DOI] [PubMed] [Google Scholar]
- Bonnington, K. E. , & Kuehn, M. J. (2016). Outer membrane vesicle production facilitates LPS remodeling and outer membrane maintenance in salmonella during environmental transitions. mBio, 7, e01532‐16. 10.1128/mBio.01532-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, J. , Cisneros, L. H. , & Mazel, D. (2021). Real‐time tracking of bacterial membrane vesicles reveals enhanced membrane traffic upon antibiotic exposure. Science Advances, 7, eabd1033. 10.1126/sciadv.abd1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito, I. L. (2021). Examining horizontal gene transfer in microbial communities. Nature Reviews Microbiology, 19, 442–453. 10.1038/s41579-021-00534-7 [DOI] [PubMed] [Google Scholar]
- Brito, I. L. , Yilmaz, S. , Huang, K. , Xu, L. , Jupiter, S. D. , Jenkins, A. P. , Naisilisili, W. , Tamminen, M. , Smillie, C. S. , Wortman, J. R. , Birren, B. W. , Xavier, R. J. , Blainey, P. C. , Singh, A. K. , Gevers, D. , & Alm, E. J. (2016). Mobile genes in the human microbiome are structured from global to individual scales. Nature, 535, 435–439. 10.1038/nature18927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broaders, E. , Gahan, C. G. M. , & Marchesi, J. R. (2013). Mobile genetic elements of the human gastrointestinal tract: Potential for spread of antibiotic resistance genes. Gut Microbes, 4, 271–280. 10.4161/gmic.24627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J.‐P. , Jeon, S. G. , Kim, Y.‐K. , & Cho, Y. S. (2019). Role of house dust mite‐derived extracellular vesicles in a murine model of airway inflammation. Clinical and Experimental Allergy, 49, 227–238. 10.1111/cea.13295 [DOI] [PubMed] [Google Scholar]
- De Jay, N. , Papillon‐Cavanagh, S. , Olsen, C. , El‐Hachem, N. , Bontempi, G. , & Haibe‐Kains, B. (2013). mRMRe: An r package for parallelized mRMR ensemble feature selection. Bioinformatics, 29, 2365–2368. 10.1093/bioinformatics/btt383 [DOI] [PubMed] [Google Scholar]
- Dinh, N. T. H. , Lee, J. , Lee, J. , Kim, S. S. , Go, G. , Bae, S. , Jun, Y. e. I. n. , Yoon, Y. J. , Roh, T. Y. , & Gho, Y. S. (2020). Indoor dust extracellular vesicles promote cancer lung metastasis by inducing tumour necrosis factor‐alpha. Journal of Extracellular Vesicles, 9, 1766821. 10.1080/20013078.2020.1766821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellabaan, M. M. H. , Munck, C. , Porse, A. , Imamovic, L. , & Sommer, M. O. A. (2021). Forecasting the dissemination of antibiotic resistance genes across bacterial genomes. Nature Communications, 12, 2435. 10.1038/s41467-021-22757-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. , Niu, B. , Zhu, Z. , Wu, S. , & Li, W. (2012). CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groussin, M. , Poyet, M. , Sistiaga, A. , Kearney, S. M. , Moniz, K. , Noel, M. , Hooker, J. , Gibbons, S. M. , Segurel, L. , Froment, A. , Mohamed, R. S. , Fezeu, A. , Juimo, V. A. , Lafosse, S. , Tabe, F. E. , Girard, C. , Iqaluk, D. , Nguyen, L. e. T. T. u. , Shapiro, B. J. , … Alm, E. J. (2021). Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell, 184, 2053–2067.e18. 10.1016/j.cell.2021.02.052 [DOI] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Fenix, A. M. , Franklin, J. L. , Higginbotham, J. N. , Zhang, Q. , Zimmerman, L. J. , Liebler, D. C. , Ping, J. , Liu, Q. i. , Evans, R. , Fissell, W. H. , Patton, J. G. , Rome, L. H. , Burnette, D. T. , & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177, 428–445.e18. 10.1016/j.cell.2019.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan, J. I. , Davis, K. A. , Beaugie, C. R. , Mcgovern, J. J. , & Moran, A. P. (2008). Alterations in helicobacter pylori outer membrane and outer membrane vesicle‐associated lipopolysaccharides under iron‐limiting growth conditions. Innate Immunity, 14, 279–290. 10.1177/1753425908096857 [DOI] [PubMed] [Google Scholar]
- Koeppen, K. , Hampton, T. H. , Jarek, M. , Scharfe, M. , Gerber, S. A. , Mielcarz, D. W. , Demers, E. G. , Dolben, E. L. , Hammond, J. H. , Hogan, D. A. , & Stanton, B. A. (2016). A novel mechanism of host‐pathogen interaction through sRNA in bacterial outer membrane vesicles. Plos Pathogens, 12, e1005672. 10.1371/journal.ppat.1005672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlete, P. , Krabberod, A. K. , & Winther‐Larsen, H. C. (2019). Vesicles from vibrio cholerae contain AT‐Rich DNA and shorter mRNAs that do not correlate with their protein products. Frontiers in Cellular and Infection Microbiology, 10, 2708. 10.3389/fmicb.2019.02708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Liu, C.‐M. , Luo, R. , Sadakane, K. , & Lam, T.‐W. (2015). MEGAHIT: An ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics, 31, 1674–1676. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Cvirkaite‐Krupovic, V. , Commere, P.‐H. , Yang, Y. , Zhou, F. , Forterre, P. , Shen, Y. , & Krupovic, M. (2021). Archaeal extracellular vesicles are produced in an ESCRT‐dependent manner and promote gene transfer and nutrient cycling in extreme environments. The ISME Journal, 15, 2892–2905. 10.1038/s41396-021-00984-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Jing, X. , Ye, Y. , Zhan, J. i. , Ye, J. , & Zhou, S. (2019). Bacterial vesicles mediate extracellular electron transfer. Environmental Science & Technology Letters, 7, 27–34. 10.1021/acs.estlett.9b00707 [DOI] [Google Scholar]
- Maestre‐Carballa, L. , Lluesma Gomez, M. , Angla Navarro, A. , Garcia‐Heredia, I. , Martinez‐Hernandez, F. , & Martinez‐Garcia, M. (2019). Insights into the antibiotic resistance dissemination in a wastewater effluent microbiome: Bacteria, viruses and vesicles matter. Environmental Microbiology, 21, 4582–4596. 10.1111/1462-2920.14758 [DOI] [PubMed] [Google Scholar]
- Manning, A. J. , & Kuehn, M. J. (2011). Contribution of bacterial outer membrane vesicles to innate bacterial defense. Bmc Microbiology, 258, 11, 10.1186/1471-2180-11-258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, J. L. , Coque, T. M. , & Baquero, F. (2015). Prioritizing risks of antibiotic resistance genes in all metagenomes. Nature Reviews Microbiology, 13, 396–396. 10.1038/nrmicro3399-c2 [DOI] [PubMed] [Google Scholar]
- Metcalf, J. A. , Funkhouser‐Jones, L. J. , Brileya, K. , Reysenbach, A.‐L. , & Bordenstein, S. R. (2014). Antibacterial gene transfer across the tree of life. Elife, 3, e04266. 10.7554/eLife.04266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura, A. , Soares, M. , Pereira, C. , Leitao, N. , Henriques, I. , & Correia, A. (2009). INTEGRALL: A database and search engine for integrons, integrases and gene cassettes. Bioinformatics, 25, 1096–1098. 10.1093/bioinformatics/btp105 [DOI] [PubMed] [Google Scholar]
- Oksanen, J. . (2007). Multivariate analysis of ecological communities in R: vegan tutorial. University of Oulu, Oulu.
- Paschon, V. , Takada, S. H. , Ikebara, J. M. , Sousa, E. , Raeisossadati, R. , Ulrich, H. , & Kihara, A. H. (2016). Interplay between exosomes, microRNAs and toll‐like receptors in brain disorders. Molecular Neurobiology, 53, 2016–2028. 10.1007/s12035-015-9142-1 [DOI] [PubMed] [Google Scholar]
- Prados‐Rosales, R. , Weinrick, B. C. , Piqué, D. G. , Jacobs, W. R. , Casadevall, A. , & Rodriguez, G. M (2014). Role for mycobacterium tuberculosis membrane vesicles in iron acquisition. Journal of Bacteriology, 196, 1250–1256. 10.1128/JB.01090-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, J. , Li, Y. , Cai, Z. , Li, S. , Zhu, J. , Zhang, F. , Liang, S. , Zhang, W. , Guan, Y. , Shen, D. , Peng, Y. , Zhang, D. , Jie, Z. , Wu, W. , Qin, Y. , Xue, W. , Li, J. , Han, L. , Lu, D. , … Wang, J. (2012). A metagenome‐wide association study of gut microbiota in type 2 diabetes. Nature, 490, 55–60. 10.1038/nature11450 [DOI] [PubMed] [Google Scholar]
- Qin, Y. , Guo, Z. , Huang, H. , Zhu, L. , Dong, S. , Zhu, Y.‐G. , Cui, L. i. , & Huang, Q. (2022). Widespread of potential pathogen‐derived extracellular vesicles carrying antibiotic resistance genes in indoor dust. Environmental Science & Technology, 56, 5653–5663. 10.1021/acs.est.1c08654 [DOI] [PubMed] [Google Scholar]
- Rakoff‐Nahoum, S. , Coyne, M. J. , & Comstock, L. E. (2014). An ecological network of polysaccharide utilization among human intestinal symbionts. Current Biology, 24, 40–49. 10.1016/j.cub.2013.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbo, C. , Fernández‐Moreira, E. , Merino, M. A. , Poza, M. , Mendez, J. A. , Soares, N. C. , Mosquera, A. , Chaves, F. , & Bou, G. (2011). Horizontal transfer of the OXA‐24 carbapenemase gene via outer membrane vesicles: A new mechanism of dissemination of carbapenem resistance genes in acinetobacter baumannii. Antimicrobial Agents and Chemotherapy, 55, 3084–3090. 10.1128/AAC.00929-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvachúa, D. , Werner, A. Z. , Pardo, I. , Michalska, M. , Black, B. A. , Donohoe, B. S. , Haugen, S. J. , Katahira, R. , Notonier, S. , Ramirez, K. J. , Amore, A. , Purvine, S. O. , Zink, E. M. , Abraham, P. E. , Giannone, R. J. , Poudel, S. , Laible, P. D. , Hettich, R. L. , & Beckham, G. T. (2020). Outer membrane vesicles catabolize lignin‐derived aromatic compounds in pseudomonas putida KT2440. PNAS, 117, 9302–9310. 10.1073/pnas.1921073117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samra, M. , Nam, S. K. , Lim, D. H. , Kim, D. H. , Yang, J. , Kim, Y.‐K. , & Kim, J. H. (2019). Urine bacteria‐derived extracellular vesicles and allergic airway diseases in children. International Archives of Allergy and Immunology, 178, 150–158. 10.1159/000492677 [DOI] [PubMed] [Google Scholar]
- Schulz, E. , Karagianni, A. , Koch, M. , & Fuhrmann, G. (2020). Hot EVs ‐ How temperature affects extracellular vesicles. European Journal of Pharmaceutics and Biopharmaceutics, 146, 55–63. 10.1016/j.ejpb.2019.11.010 [DOI] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W. S. , & Huttenhower, C. (2011). Metagenomic biomarker discovery and explanation. Genome Biology, 12, R60. 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentchilo, V. , Mayer, A. P. , Guy, L. , Miyazaki, R. , Green Tringe, S. , Barry, K. , Malfatti, S. , Goessmann, A. , Robinson‐Rechavi, M. , & Van Der Meer, J. R. (2013). Community‐wide plasmid gene mobilization and selection. The ISME journal, 7, 1173–1186. 10.1038/ismej.2013.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, C. D. , Lodha, S. , & Sahoo, S. (2021). Sorting in therapeutic development for cardiovascular disease. Cells, 10, 1500. 10.3390/cells10061500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier, P. , Gourbeyre, E. , Varani, A. , Ton‐Hoang, B. , & Chandler, M. (2015). Everyman's guide to bacterial insertion sequences. Microbiology Spectrum, 3, MDNA3‐0030‐2014. 10.1128/microbiolspec.MDNA3-0030-2014 [DOI] [PubMed] [Google Scholar]
- Smillie, C. S. , Smith, M. B. , Friedman, J. , Cordero, O. X. , David, L. A. , & Alm, E. J. (2011). Ecology drives a global network of gene exchange connecting the human microbiome. Nature, 480, 241–244. 10.1038/nature10571 [DOI] [PubMed] [Google Scholar]
- Solano, C. , Echeverz, M. , & Lasa, I. I. (2014). Biofilm dispersion and quorum sensing. Current Opinion in Microbiology, 18, 96–104. 10.1016/j.mib.2014.02.008 [DOI] [PubMed] [Google Scholar]
- Soucy, S. M. , Huang, J. , & Gogarten, J. P. (2015). Horizontal gene transfer: Building the web of life. Nature Reviews Genetics, 16, 472–482. 10.1038/nrg3962 [DOI] [PubMed] [Google Scholar]
- Tashiro, Y. , Hasegawa, Y. , Shintani, M. , Takaki, K. , Ohkuma, M. , Kimbara, K. , & Futamata, H. (2017). Interaction of bacterial membrane vesicles with specific species and their potential for delivery to target cells. Frontiers in Microbiology, 8, 571. 10.3389/fmicb.2017.00571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennstedt, T. , Szczepanowski, R. , Braun, S. , Puhler, A. , & Schluter, A. (2003). Occurrence of integron‐associated resistance gene cassettes located on antibiotic resistance plasmids isolated from a wastewater treatment plant. Fems Microbiology Ecology, 45, 239–252. 10.1016/S0168-6496(03)00164-8 [DOI] [PubMed] [Google Scholar]
- Tian, Y. e. , Gong, M. , Hu, Y. , Liu, H. , Zhang, W. , Zhang, M. , Hu, X. , Aubert, D. , Zhu, S. , Wu, L. , & Yan, X. (2020). Quality and efficiency assessment of six extracellular vesicle isolation methods by nano‐flow cytometry. Journal of Extracellular Vesicles, 9, 1697028. 10.1080/20013078.2019.1697028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku, M. , Morinaga, K. , Hashimoto, Y. , Uhl, J. , Shimamura, H. , Inaba, H. , Schmitt‐Kopplin, P. , Eberl, L. , & Nomura, N. (2017). Membrane vesicle‐mediated bacterial communication. The ISME Journal, 11, 1504–1509. 10.1038/ismej.2017.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku, M. , Nomura, N. , & Eberl, L. (2019). Types and origins of bacterial membrane vesicles. Nature Reviews Microbiology, 17, 13–24. 10.1038/s41579-018-0112-2 [DOI] [PubMed] [Google Scholar]
- Tulkens, J. , De Wever, O. , & Hendrix, A. n. (2020). Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nature Protocols, 15, 40–67. 10.1038/s41596-019-0236-5 [DOI] [PubMed] [Google Scholar]
- Vollmer, W. , Blanot, D. , & De Pedro, M. A. (2008). Peptidoglycan structure and architecture. Fems Microbiology Review, 32, 149–167. 10.1111/j.1574-6976.2007.00094.x [DOI] [PubMed] [Google Scholar]
- Wu, R. , Tao, Y. e. , Cao, Y. , Zhou, Y. , & Lin, H. (2020). Streptococcus mutans membrane vesicles harboring glucosyltransferases augment candida albicans biofilm development. Frontiers in Microbiology, 11, 581184. 10.3389/fmicb.2020.581184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron, S. , Kolling, G. L. , Simon, L. , & Matthews, K. R. (2000). Vesicle‐mediated transfer of virulence genes from Escherichia coli O157:H7 to other enteric bacteria. Applied and Environmental Microbiology, 66, 4414–4420. 10.1128/aem.66.10.4414-4420.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Ghosh, S. , Kumar, M. , Santiana, M. , Bleck, C. K. E. , Chaimongkol, N. , Altan‐Bonnet, N. , & Shuai, D. (2021). Emerging pathogenic unit of vesicle‐cloaked murine norovirus clusters is resistant to environmental stresses and UV(254) disinfection. Environmental Science & Technology, 55, 6197–6205. 10.1021/acs.est.1c01763 [DOI] [PubMed] [Google Scholar]
- Zhou, S. , Zhu, Y. , Yan, Y. , Wang, W. , & Wang, Y. (2019). Deciphering extracellular antibiotic resistance genes (eARGs) in activated sludge by metagenome. Water Research, 161, 610–620. 10.1016/j.watres.2019.06.048 [DOI] [PubMed] [Google Scholar]
- Zhu, W. , Lomsadze, A. , & Borodovsky, M. (2010). Ab initio gene identification in metagenomic sequences. Nucleic Acids Research, 38, e132. 10.1093/nar/gkq275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y.‐G. , Zhao, Y. I. , Li, B. , Huang, C.‐L. , Zhang, S. i.‐Y. u. , Yu, S. , Chen, Y.‐S. , Zhang, T. , Gillings, M. R. , & Su, J.‐Q. (2017). Continental‐scale pollution of estuaries with antibiotic resistance genes. Nature Microbiolory, 2, 16270. 10.1038/nmicrobiol.2016.270 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Sampling information and sample characteristics.
Supplementary Table 2. Correlation between sample characteristics and particle concentrations of EVs.
Supplementary Table 3. The proportion of EVs‐associated DNA fragments from 200 bp to 1000 bp.
Supplementary Table 4. Metagenomic data of e ach sample after quality control.
Supplementary Table 5. Biological proportion and classification of samples at the phylum level.
Supplementary Table 6. Detailed information of species that were enriched in EVs.
Supplementary Table 7. Detailed information of the differential KEGG profiles secreted by major contributors Firmicutes and Proteobacteria between EVs and habitats.
Supplementary Table 8. Detailed information of enzyme encoding genes that were enriched in EVs.
Supplementary Table 9. Detailed information of the differential enzyme encoding genes secreted by major contributors Firmicutes and Proteobacteria between EVs and habitats.
Extended Data Fig. 1| Representative transmission electron microscopy of EVs from wastewater samples ( JM, YD, XL, QP, BZ ) and soil samples (JG, LYZ, HL, NH, ZS).
Extended Data Fig. 2| Quantitation and characterization of EVs‐associated DNA.
Extended Data Fig. 3| Quantitation of 16S rDNA copies of fecal EVs derived from differential centrifugation (DC) and density gradient centrifugation (DGC), respectively.
Extended Data Fig. 4| Summary of a bioinformatic workflow.
Extended Data Fig. 5| Composition of microbiota and EVs in different habitats.
Extended Data Fig. 6| Diversity and distribution of functional categories in the habitat samples and corresponding EVs samples.
Extended Data Fig. 7| Characterization of ARG and MGE profiles in the microbiota from each habitat.
Extended Data Fig. 8| Characterization of ARG profiles.
Data Availability Statement
Raw sequencing data has been uploaded to Science Data Bank https://datapid.cn/31253.11.sciencedb.01118.