

An emerging hallmark of cancer is cellular metabolic reprogramming to adapt to varying cellular environments. Throughout the process of metastasis cancer cells gain anchorage independence which confers survival characteristics when detached from the extracellular matrix (ECM). Previous work demonstrates that the bioactive metabolite of vitamin D, 1α,25-dihydroxyvitamin D (1,25[OH]2D), suppresses cancer progression, potentially by suppressing the ability of cells to metabolically adapt to varying cellular environments such as ECM detachment. The purpose of the present study was to determine the mechanistic bases of the effects of 1,25(OH)2D on cell survival in ECM-detached conditions. Pretreatment of MCF10A-ras breast cancer cells for 3 d with 1,25(OH)2D reduced the viability of cells in subsequent detached conditions by 11%. Enrichment of 13C5-glutamine was reduced in glutamate (21%), malate (30%), and aspartate (23%) in detached compared to attached MCF10A-ras cells. Pretreatment with 1,25(OH)2D further reduced glutamine flux into downstream metabolites glutamate (5%), malate (6%), and aspartate (10%) compared to detached vehicle treated cells. Compared to attached cells, detachment increased pyruvate carboxylase (PC) mRNA abundance and protein expression by 95% and 190%, respectively. Consistent with these results, 13C6-glucose derived M+3 labelling was shown to preferentially replenish malate and aspartate, but not citrate pools, demonstrating increased PC activity in detached cells. In contrast, 1,25(OH)2D pretreatment of detached cells reduced PC mRNA abundance and protein expression by 63% and 56%, respectively, and reduced PC activity as determined by decreased 13C6-glucose derived M+3 labeling in citrate (8%) and aspartate (50%) pools, relative to vehicle-treated detached cells. While depletion of PC with doxycycline-inducible shRNA reduced detached cell viability, PC knockdown in combination with 1,25(OH)2D treatment did not additionally affect the viability of detached cells. Further, PC overexpression improved detached cell viability, and inhibited the effect of 1,25(OH)2D on detached cell survival, suggesting that 1,25(OH)2D mediates its effects in detachment through regulation of PC expression. These results suggest that inhibition of PC by 1,25(OH)2D suppresses cancer cell anchorage independence.
1. Introduction
Metastasis accounts for the majority of all cancer-related deaths [1]. For breast cancer, the 5-year survival rate is 99% in patients with a localized diagnosis but is 28% when diagnosed with metastatic disease [2]. A growing body of evidence recognizes preventative actions of vitamin D against breast cancer development, with a potential to specifically inhibit promotion and metastasis [3–5]. Thus, it is critical to identify agents and their underlying mechanisms that suppress specific steps of metastasis to inhibit disease progression, and vitamin D may be a promising candidate.
Vitamin D is either acquired from the diet or synthesized from 7-dehydrocholesterol in the skin upon ultraviolet B exposure from the sun [6]. Two sequential hydroxylation steps are required for synthesis of the primary hormonal and active form in the blood, 1α,25-dihydroxyvitamin D (1,25[OH]2D). The action of 1,25(OH)2D is mediated through binding to the vitamin D receptor (VDR) which, once bound to 1,25[OH]2D, heterodimerizes with the retinoid X receptor. The heterodimer subsequently binds to vitamin D response elements (VDREs) in the promoter regions of vitamin D-regulated genes. Several potential mechanisms by which 1,25(OH)2D prevents breast cancer tumor formation and subsequent cancer progression have been identified, including increased cancer cell cycle arrest and apoptosis [7, 8]; however, the role of 1,25(OH)2D in regulating cellular bioenergetics as a mechanism for inhibiting cancer progression has not been elucidated.
Survival and growth of normal epithelial cells are dependent on attachment to the extracellular matrix (ECM). In a primary tumor, cellular changes permitting “anchorage independence,” which enables cancer cell survival when detached from the ECM, promotes tumor growth and metastasis [9]. In non-transformed cells, integrins mediate attachment to the matrix and, upon detachment, initiate signaling that induce anoikis, a form of programmed cell death [10]. For example, integrins reduce activation of focal adhesion kinase (FAK), leading to reduced downstream signaling through pro-survival signaling pathways such as mitogen activating protein kinase (MAPK) and phosphoinositide-3 kinase (PI3K) [11]. Oncogenic transformation, such as those mediated by mutation to RAS or amplification of ERBB2, promotes cancer cell anchorage independence which enables their survival when detached from the ECM by maintaining activation of survival signaling pathways such as PI3K [12, 13]. Thus, oncogenic transformation sustains detached cell viability through stimulation of signaling pathways, which confer inhibition of anoikis and alterations in cellular metabolism.
Evidence supports that metabolic reprogramming plays an important role in maintaining survival of ECM-detached cancer cells. For example, nonmalignant epithelial cells decrease energy metabolism pathways upon detachment [9]. However, detached MCF10A human breast epithelial cells with overexpression of the ERBB2 oncogene increase glucose and glutamine flux into the tricarboxylic acid (TCA) cycle compared to detached untransformed MCF10A cells [14]. Additionally, overexpression of FAK increases glycolysis in detached pancreatic ductal adenocarcinoma cells [15]. Further, detached MDA-MB-231 breast cancer cells increase the expression of antioxidant enzymes, which is required for fatty acid oxidation and the subsequent production of adenosine triphosphate (ATP) [16]. These results suggest that oncogenic transformation enables adaptations in cellular energy metabolism in ECM-detached conditions.
Reprogrammed energy metabolism is an emerging hallmark of cancer [17], and evidence supports that 1,25(OH)2D regulates energy metabolism in malignant breast epithelial cells [18–20]. One mechanism by which the hormone may regulate cellular energy metabolism is the enzyme pyruvate carboxylase (PC). PC mediates the conversion of pyruvate to the tricarboxylic acid (TCA) cycle intermediate oxaloacetate, which is a precursor for reactions involved in energy production, fatty acid synthesis, and oxidative stress protection. Previous research demonstrates that treatment with 1,25(OH)2D reduced lipid accumulation and fatty acid synthesis in MCF10CA1a breast cancer cells through direct downregulation of PC expression through a VDRE in the PC promoter [20, 21]. In addition, PC is also involved in oxidative stress protection in MCF10A-ras cells, and 1,25(OH)2D treatment acted as a prooxidant by reducing PC expression [21]. Thus, 1,25(OH)2D suppression of PC may alter cellular adaptability in steps in metastasis.
Thus, 1,25(OH)2D is proposed to inhibit metastatic progression and suppresses the expression of PC, which is a critical regulator of cellular metabolism. However, the metabolic effects of 1,25(OH)2D treatment on breast cancer cells on the adaptability to specific steps in metastasis, such as ECM detachment, has yet to be determined. Thus, in the present study, it was hypothesized that in breast cancer cells, 1,25(OH)2D reduces expression of PC which reduces cell survival in ECM detached conditions.
2. Materials and methods
2.1. Cell culture
Harvey-ras transformed MCF10A-ras cells were a gift from Michael Kinch, Purdue University and MCF10CA1a cells were a gift from Julia Kirshner, Purdue University. MCF10A-ras cells were grown in an incubator set to 37°C in 95% air: 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) (Sigma, St. Louis, MO) with 5 mol/L glucose, 2.5 mol/L glutamine, and lacking sodium pyruvate. Media was supplemented with 5% horse serum (Gibco, Waltham MA) and 1% penicillin/streptomycin (Gibco) for MCF10CA1a and MCF10A-ras cells. MCF10A-ras cell media was further supplemented with 10 mg/L insulin (Sigma), 50 μg/L cholera toxin (Sigma), 20 μg/L epidermal growth factor (Sigma), and 50 mg/L hydrocortisone (Sigma). Non-metastatic M-Wnt murine mammary cancer cells [22] were grown in DMEM with 5 mol/L glucose, 2 mol/L glutamine, and no sodium pyruvate supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin. Cells were pretreated for 72 h with 10 nM 1,25(OH)2D (Biomol, Plymouth Meeting, PA) in 100% ethanol vehicle (final concentration <0.1%), with media changed every 24 h.
2.2. Poly-HEMA coated plates
Detached conditions were obtained using Poly(2-hydroxyethyl methacrylate) (Poly-HEMA, Sigma) coated plates. Seventy μL/cm2 of 20 mg/mL Poly-HEMA in 95% ethanol was used to coat 96-well plates and 100 mol/L dishes. Following addition of Poly-HEMA, the plates or dishes were dried overnight under ultraviolet light. Before use, Poly-HEMA coated plates and dishes were rinsed twice with sterile 1x calcium/magnesium-free phosphate-buffered saline (PBS).
2.3. MTT assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) assay according to the manufacturer’s instructions. Briefly, following 72-h pretreatment, cells were plated into Poly-HEMA coated 96-well plates at 20k cells/well with corresponding treatments. After 40 h, MTT solution was added, allowed to incubate for 2 h and cells solubilized in dimethylsulfoxide. Absorbance was measured at 570 nm with a spectrophotometer. Cell number, as measured by the trypan blue exclusion assay, correlated (R=0.998; P<.0001) with the results of the MTT assay from detached cells pretreated with 1,25(OH)2D or doxycycline.
2.3. Flow cytometry
Following treatment and placement into detached conditions, a cellular suspension of 1,000,000 cells was harvested into calcium-rich Annexin V binding buffer containing 5 μL Annexin V (Invitrogen, Carlsbad, CA) and 10 mg/mL propidium iodide (Sigma). A BD Fortessa LSR SORP Cell Analyzer with a 488 nm laser was used to analyze apoptosis in cells. Results were analyzed with FCS Express software (Pasadena, CA). Results are shown as the percent of total cells in each apoptotic stage.
2.4. RNA isolation and analysis
Pretreated cells were plated into 100 mol/L Poly-HEMA coated dishes at 1500k cells/dish. After 40 h, cells were harvested and isolated using Tri-Reagent (Molecular Research Center, Cincinnati, OH) following the manufacturer’s instructions. Reverse transcription of total RNA was performed using MMLV reverse transcriptase (Promega, Madison, WI). Real-time quantitative PCR was performed in a LightCycler 480 instrument using LightCycler 480 SYBR Green I Master Mix (Roche, Indianapolis, IN). mRNA abundance of target genes was normalized to 18S abundance using the comparative Ct method (2−ΔΔCt), and results represent arbitrary units [23].
2.5. Glucose and glutamine flux analysis
Following 40 h in attachment or detachment, pretreated cell media was changed to media containing either 100% (2.5 mol/L) 13C5-glutamine or 100% (5 mol/L) 13C6-glucose. Cells were incubated at 37°C for 2 h, followed by harvest into 70% ethanol at 70°C and the addition of 1 μg of the internal standard norvaline to each sample. Intracellular metabolites were extracted from the cells into ethanol by a 5-min incubation at 95°C. A bicinchoninic acid (BCA, ThermoFisher, Waltham, MA) assay was used to measure protein content in the cell pellet, and the supernatant dried. Methoxylamine hydrochloride in pyridine was used to derivatize metabolites as previously described [24]. Following derivatization, metabolites were analyzed with gas chromatography-mass spectrometry (GC-MS) using a TG-5MS gas chromatography column and Thermo TSQ 8000 triple quadrupole mass spectrometer. Mass spectra data were acquired using Chromeleon 7 software (ThermoFisher). Intracellular pool sizes were calculated by dividing the area under the curve for the total metabolite by norvaline and protein in the cell pellet to control for recovery and cell quantity, respectively. Mass isotopomer percent enrichment of the metabolite pool sizes was used to calculate 13C flux, and results are calculated relative to levels determined for vehicle treated attached cells. To account for the natural abundance of 13C, MS data was corrected using IsoCor software as previously described [25]. Averages of 4 samples in each treatment group were used for results representing pool size and mass isotopomer percent enrichment.
2.6. Western blot analysis
Following two washes with calcium/magnesium-free PBS, cells were harvested on ice into 1x radioimmunoprecipitation assay (RIPA) lysis buffer (Cell Signaling, Danvers MA) supplemented with 1% phosphatase (Sigma) and 1% phenylmethyl-sulfonyl fluoride protease inhibitors (Cell Signaling). To harvest detached cells, cells were pelleted by 5-min centrifugation at 1000 RPM and rinsed twice with calcium/magnesium-free PBS. Pelleted detached cells were resuspended in 25 μL of RIPA lysis buffer. Attached and detached cells were sonicated for 15 min and centrifuged for 15 min at 12,000 revolutions per min. The supernatant was collected, and protein quantified using the BCA assay. A 4–15% gradient polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA) was used to resolve 20 μg of protein by electrophoresis. Primary antibodies for PC and actin were purchased from Sigma. Proteins were transferred to a nitrocellulose membrane (Bio-Rad Laboratories) and antigen-antibody complexes were detected by the Li-Cor Odyssey imaging system. Results show PC protein abundance normalized to actin levels. Table 1
Table 1.
Primers used for qRT-PCR
| 18S | Forward: 5′ - TTAGAGTGTTCAAAGCAGGCCCGA-3′ Reverse: 5′ - TCTTGGCAAATGCTTTCGCTC-3′ |
| ASCT2 | Forward: 5′ - TGAACATCCTGGGCTTGGTAG-3′ Reverse: 5′ - AGCAGGCAGCACAGAATGTAC-3′ |
| GLUD1 | Forward: 5′ - TTAGAGTGTTCAAAGCAGGCCCGA-3′ Reverse: 5′ - TCTTGGCAAATGCTTTCGCTC-3′ |
| GOT1 | Forward: 5′ -CAACTGGGATTGACCCAACT −3′ Reverse: 5′ - GGAACAGAAACCGGTGCTT −3′ |
| GOT2 | Forward: 5′ -ACCCATGTGGAAATGGGACC −3′ Reverse: 5′ - GACTTCGCTGTTCTCACCCA-3′ |
| GLUT1 | Forward: 5′ -TATCGTCAACACGGCCTTCACTGT −3′ Reverse: 5′ - CACAAAGCCAAAGATGGCCACGAT −3′ |
| HK2 | Forward: 5′ -CTGCAGCGCATCAAGGAGAACAAA −3′ Reverse: 5′ - ACGGTCTTATGTAGACGCTTGGCA −3′ |
| LDHA | Forward: 5′ - TGGTCCAGCGTAACGTGAACATCT −3′ Reverse: 5′ -TTGCAACCGCTTCCAATAACACGG −3′ |
| PC | Forward: 5′ - ATGTTGCCCACAACTTCAGCAAGC −3′ Reverse: 5′ - AGTTGAGGGAGTCAAACACACGGA-3′ |
2.7. PC knockdown and overexpression
PC knockdown in cells utilized TRIPZ lentiviral human shRNA which targeted PC and was doxycycline (Dox) inducible, purchased from GE Dharmacon (Lafayette, CO). PC overexpression utilized Dox inducible lentiviral plasmids containing the human PC gene (NM_000920.2) were purchased from VectorBuilder (Chicago, IL). HEK293T cells were transduced with lentiviral plasmids using polyethylamine, and lentiviral particles harvested. Transfection of MCF10A-ras cells took place for 48 h with 10 μg/mL polybrene, followed by puromycin (5 mg/mL) selection. PC overexpressing, stably transfected cells were selected with puromycin followed by hygromycin (450 ug/mL). PC knockdown or overexpression was verified by PCR analysis of PC mRNA abundance following treatment with Dox (0.5 μg/mL) for 3 d (Figs. 5A and 6A) and previously verified by western blot [21]. PC knockdown or overexpressing cells were pretreated with Dox (0.5 ug/mL) for 3 d with an additional cotreatment of vehicle or 1,25(OH)2D where indicated.
Fig. 5.
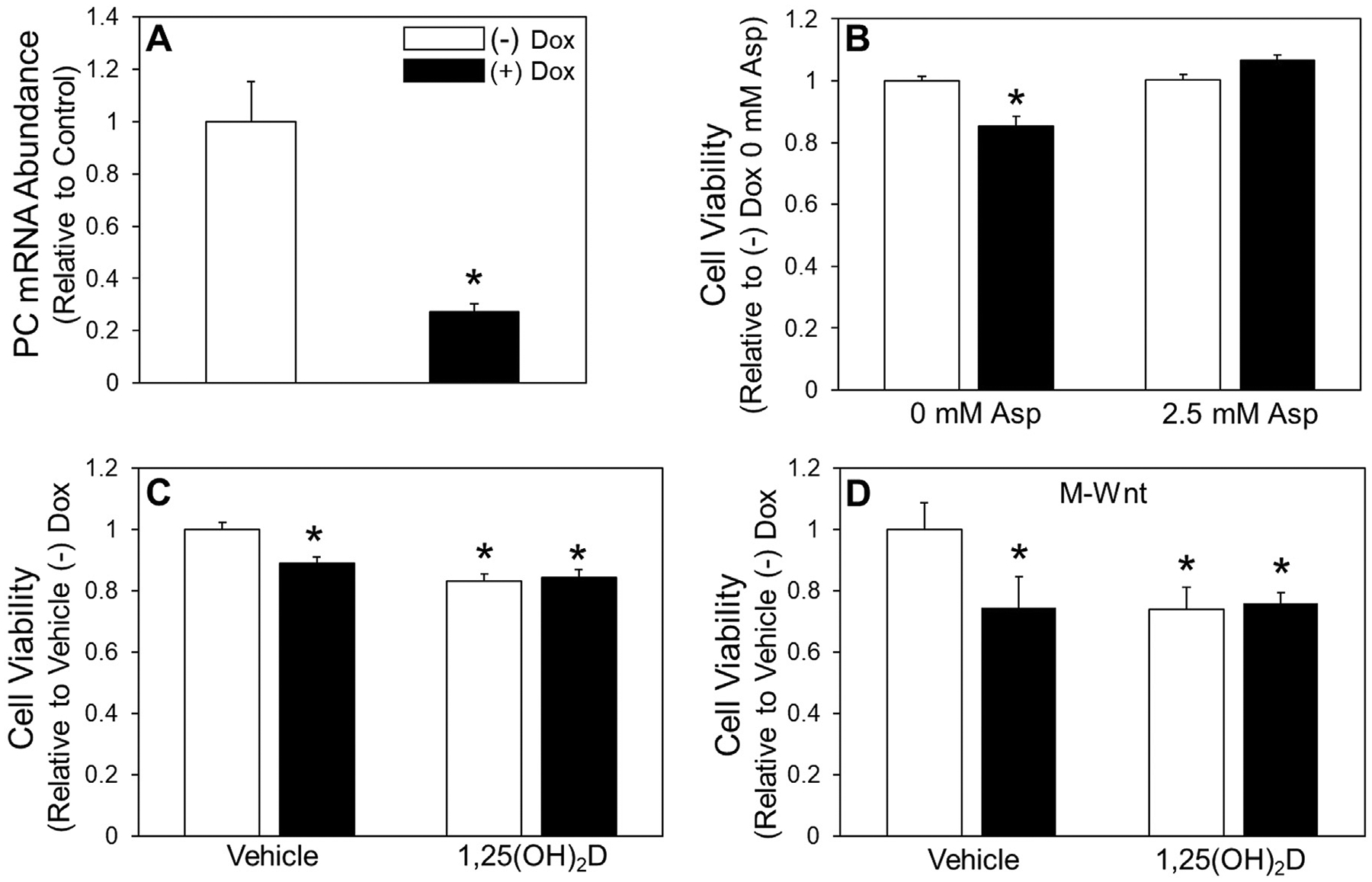
Detached cell viability with PC depletion and 1,25(OH)2D treatment. (A) (A) mRNA abundance of PC in attached shPC Dox-inducible MCF10A-ras cells treated with and without 0.5 μg/mL Dox was assessed. (B) Viability with 2.5 mol/L aspartate (Asp) supplementation of detached Dox-inducible PC knockdown MCF10A-ras cells treated with and without Dox. (C) Viability of 3-d pretreatment with 1,25(OH)2D of detached Dox-inducible PC knockdown MCF10A-ras and (D) M-Wnt cells. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle attached. All experiments were repeated in duplicate with n≥3.
Fig. 6.
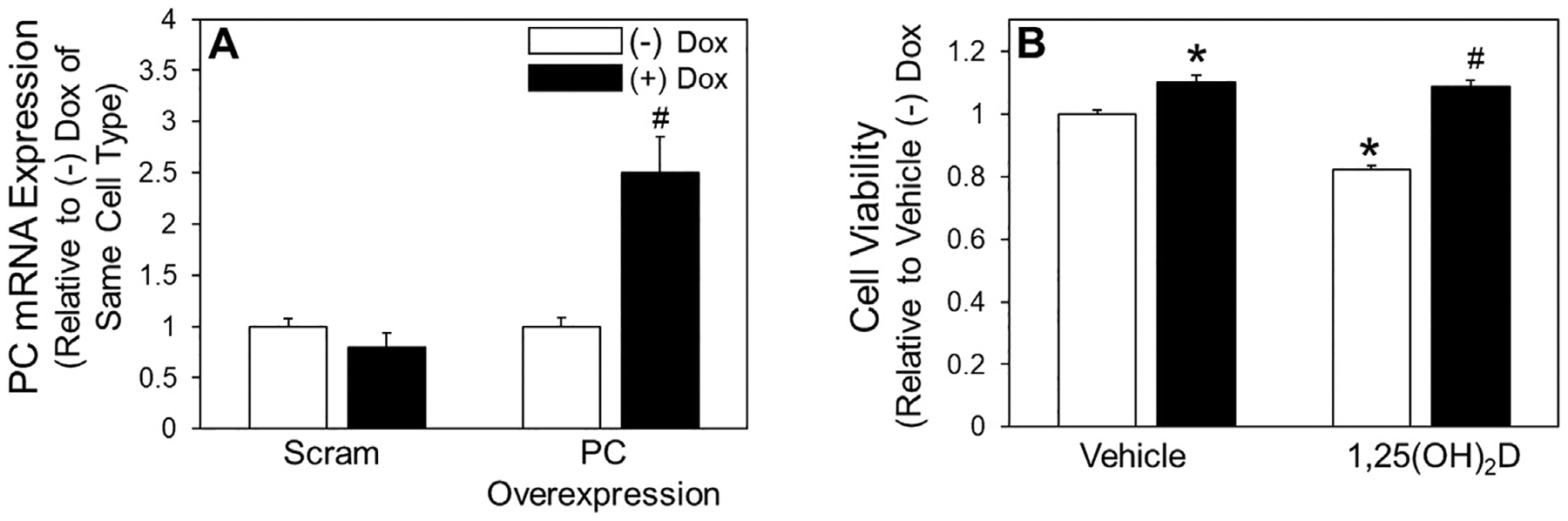
Effect of pyruvate carboxylase overexpression on viability of detached cells treated with 1,25(OH)2D. (A) mRNA abundance of PC in attached Dox-inducible PC overexpressing and Dox-inducible scram MCF10A-ras cells treated with 0.5 μg/mL Dox was assessed. (B) Following 3-d vehicle or 1,25(OH)2D treatment MTT was used to assess cell viability of detached Dox-inducible PC overexpressing MCF10A-ras cells. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle (−) Dox of the same cell type and symbol (#) indicates P<.05 relative to Dox treatment of same cell type or relative to 1,25(OH)2D (−) Dox treated cells. All experiments were repeated in duplicate with n≥3.
2.8. Statistical analysis
Results are displayed using mean ± S.E.M. Independent samples t’test was used to calculate comparisons between two groups, with P-values <.05 considered significant.
3. Results
3.1. 1,25(OH)2D treatment reduces survival in detached breast cancer cells
To evaluate the response of the MCF10A-ras cells to ECM detachment, cell viability was measured over a time course of 0, 24, 40, and 48 h (Fig. 1A). Cell viability decreased with time in detached conditions up to 40 h, with an increase at 48 h, suggesting growth of the surviving detached cells after 40 h. Given that 40 h was the time point with the lowest level of viability, this time point was selected for further study.
Fig. 1.
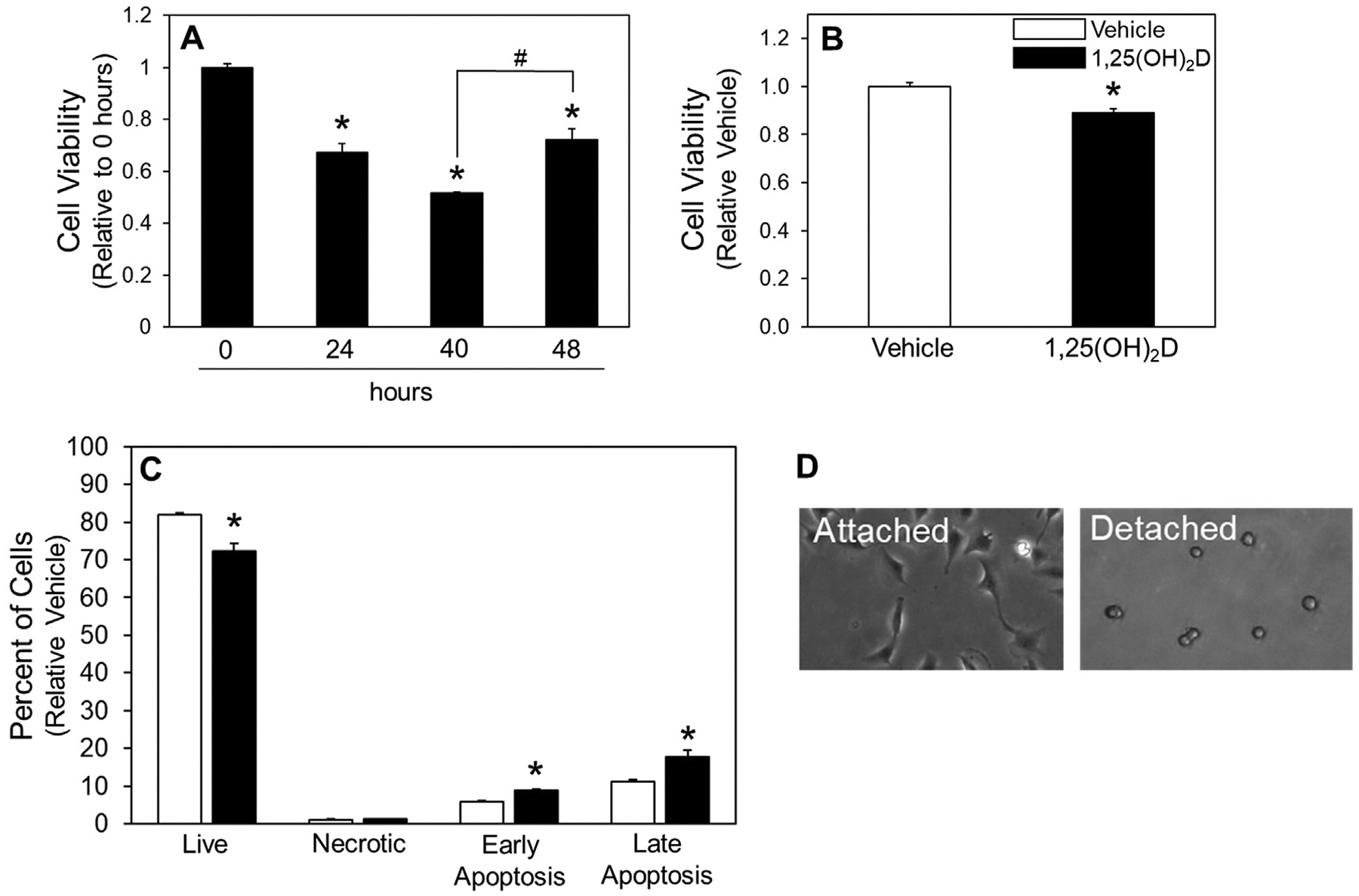
Effect of 1,25(OH)2D on detached cell viability. (A) Viability of MCF10A-ras cells was assessed by MTT after 0, 24, 40, and 48 h in detached conditions, and (B) following 3-d vehicle or 1,25(OH)2D pretreatment viability was assessed after 40 h in detached conditions by MTT assay, and (C) flow cytometry. (D) Cell morphology of attached and detached MCF10A-ras cells. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle attached and symbol (#) indicates P<.05 relative to vehicle detached of same cell group. All experiments were repeated in duplicate with n≥3.
The effect of 1,25(OH)2D on viability of detached cells was determined. Following pretreatment with 1,25(OH)2D, MCF10A-ras cell viability was decreased compared to vehicle-treated cells (Fig. 1B). In addition, 1,25(OH)2D pretreatment resulted in significantly fewer live cells, and significantly more cells undergoing apoptosis (Fig. 1C). Cell morphology changes between attached and detached cells are shown in Fig. 1D. Together these results demonstrate that 1,25(OH)2D reduces the viability of detached MCF10A-ras cells by inducing apoptosis.
3.2. Effects of detachment and 1,25(OH)2D treatment on glutamine metabolism
Given that previous studies demonstrated an effect of 1,25(OH)2D on regulating energy metabolism in breast cancer cell models [19–21], 1,25(OH)2D regulation of glutamine metabolism in detached cells was examined. While detachment reduced intracellular glutamate, there was no effect of detachment on the aspartate pool size (Fig. 2A). Interestingly, treatment with 1,25(OH)2D resulted in reduced intracellular pools of glutamate and aspartate in attached cells, but no further reduction was observed in detached cells (Fig. 2A). Tracer analysis using 13C5-glutamine show that detachment decreased glutamine flux into glutamate, malate, and aspartate, determined by decreased M+5 and M+4 labeling patterns, respectively, demonstrating reduced TCA cycle anaplerosis from glutamine (Fig. 2B-–H). Pretreatment with 1,25(OH)2D further reduced flux of the glutamine tracer to glutamate, malate, and aspartate in detached cells (Fig. 2B–H). In addition, the relative contribution of glutamine to oxidative (compared to reductive) metabolism was determined in all treatment groups by comparing the ratio of M+4/M+5 labelling patterns of citrate in cells supplemented with the 13C5-glutamine tracer (Fig. 2I). Detached vehicle-treated cells and attached cells treated with 1,25(OH)2D had reduced M+4/M+5 citrate, with a further decrease observed in detached 1,25(OH)2D treated cells, suggesting reduced oxidative metabolism through the TCA cycle with detachment and 1,25(OH)2D treatment. Thus, these results demonstrate that detachment and 1,25(OH)2D treatment reduced glutamine flux through the forward, oxidative, TCA cycle in detached cells.
Fig. 2.
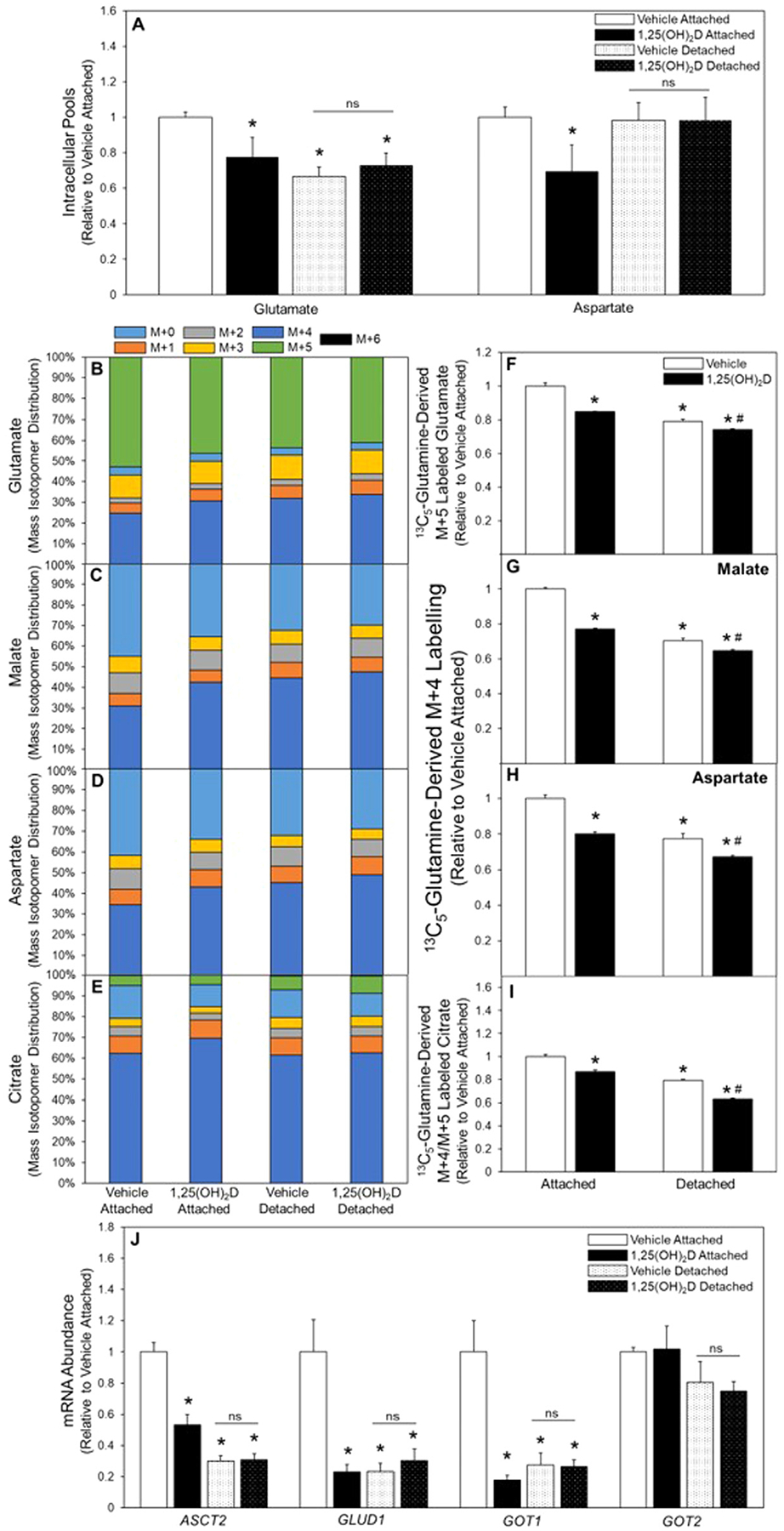
Glutamine metabolism in attached and detached cells treated with 1,25(OH)2D. (A) Following 3-d vehicle or 1,25(OH)2D pretreatment intracellular pool sizes of glutamate and aspartate were measured, and (B) 13C5-glutamine was used to determine M+5 labeling of glutamate, (C) M+4 malate, (D) aspartate, and (E) citrate in attached and detached MCF10A-ras cells. (F) 13C5-Glutamine incorporation into M+5 labelled glutamate, (G) M+4 labelled malate, (H) M+4 labelled aspartate, and (I) citrate oxidative/reductive contribution of glutamine determined by M+4/M+5 citrate in attached and detached vehicle and 1,25(OH)2D treated MCF10A-ras cells. (J) mRNA abundance of enzymes in glutamine metabolism was assessed in vehicle and 1,25(OH)2D treated attached and detached MCF10A-ras cells. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle attached and symbol (#) indicates P<.05 relative to vehicle detached. All mRNA abundance experiments were repeated in duplicate with n=3, flux and pool size experiments were completed once with n=4.
Given 1,25(OH)2D’s role in regulating gene expression and its effects on glutamine metabolism in both attached and detached cells, mRNA expression of glutamine metabolizing enzymes was measured. Detachment significantly reduced mRNA abundance of the neutral amino acid exchanger, ASCT2, which mediates extracellular glutamine transport into the cell (Fig. 2J). In addition, 1,25(OH)2D reduced ASCT2 mRNA abundance in attached cells but had no further reduction in detached cells (Fig. 2J), consistent with previous work from our lab demonstrating a direct regulation of 1,25(OH)2D on ASCT2 expression through a VDRE in the ASCT2 gene [19]. Moreover, lower mRNA abundance was measured in detached cells for glutamate metabolizing enzymes including glutamate dehydrogenase (GLUD1) and the cytosolic glutamate oxaloacetate transaminase (GOT) 1 (Fig. 2J). In contrast, mRNA abundance of the mitochondrial isoform GOT2 was unchanged in detached cells (Fig. 2J). Further, while 1,25(OH)2D-treatment reduced expression of GLUD1 and GOT1 in attached cells, in detached cells treated with 1,25(OH)2D no further reduction in mRNA abundance of GLUD1 or GOT1 and 2, enzymes was observed (Fig. 2G). While glutamine flux in detachment was decreased by 1,25(OH)2D treatment, mRNA abundance of key glutamine metabolizing enzymes was unchanged, suggesting that 1,25(OH)2D regulates glutamine metabolism in detached cancer cells through a mechanism distinct from direct transcriptional regulation of genes that express glutamine metabolizing proteins.
3.3. Effects of detachment and 1,25(OH)2D treatment on glucose metabolism
To explore glucose metabolism in ECM detached conditions, the effects of 1,25(OH)2D treatment and detachment on glycolysis was determined. Interestingly, 1,25(OH)2D treatment of attached cells reduced intracellular pool sizes of both pyruvate and lactate, and detachment alone reduced pool sizes by 51% and 50%, respectively (Fig. 3A). Universally labeled 13C6-glucose flux, which results in pyruvate and lactate labeled with three 13C’s (M+3), was decreased following detachment, as well as in in attached cells in response to 1,25(OH)2D treatment relative to attached vehicle-treated cells (Fig. 3B–E). Treatment with 1,25(OH)2D did not change pyruvate or lactate M+3 labelling of detached cells relative to vehicle treated detached cells (Fig. 3D and 3E). Decreased mRNA abundance of glycolytic enzymes hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA) was observed in detached cells relative to attached cells (Fig. 3F). However, compared to attached cells, mRNA abundance of glucose transporter 1 (GLUT1) was unchanged following detachment (Fig. 3F). In accordance with the 13C6-glucose flux results, 1,25(OH)2D did not affect mRNA abundance of glucose metabolizing enzymes in detached cells (Fig. 3F). These results suggest that detached cells reduce glucose metabolism through glycolysis compared to their attached counterparts, and that glycolysis is not further regulated by 1,25(OH)2D in detached cells.
Fig. 3.
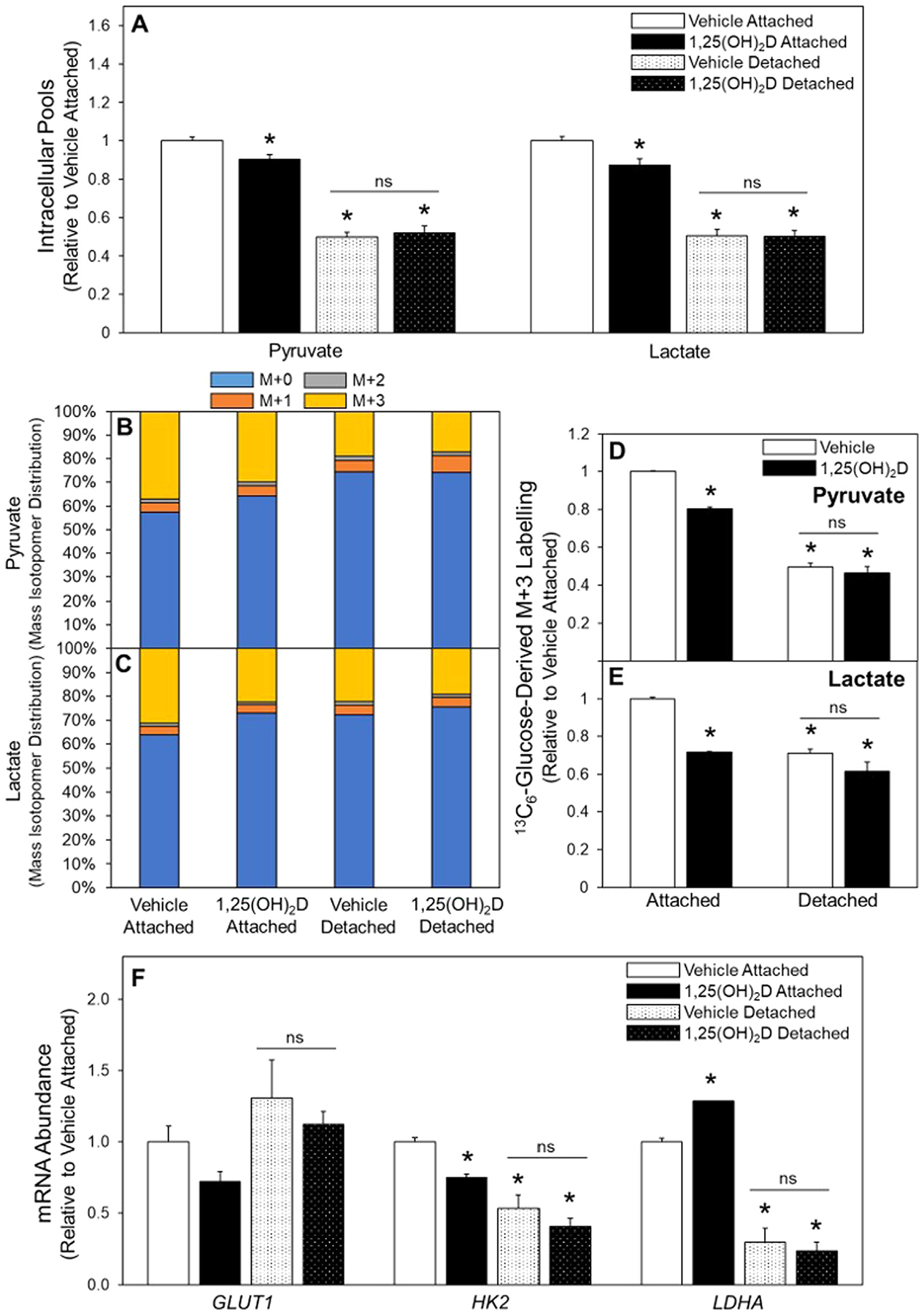
Glucose metabolism in attached and detached cells treated with 1,25(OH)2D. (A) Following 3-d vehicle or 1,25(OH)2D pretreatment intracellular pools of pyruvate and lactate were measured and (B) 13C6-Glucose was used to determine labeling of pyruvate and (C) lactate. (D) Following incubation with 13C6-glucose, M+3 labelling of pyruvate and (E) lactate were determined in all treatment groups. (F) mRNA abundance of enzymes in glucose metabolism was assessed in attached and detached MCF10A-ras cells. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle attached and symbol (#) indicates P<.05 relative to vehicle detached. All mRNA abundance experiments were repeated in duplicate with n=3, flux and pool size experiments were completed once with n=4.
The TCA cycle is a central component of cellular metabolism, thus the effects of 1,25(OH)2D treatment of detached cells on the TCA cycle was explored. Compared to attached cells, detachment decreased pool sizes for all TCA cycle intermediates measured, except citrate which was unchanged (Fig. 4A). Additionally, while 1,25(OH)2D treatment in attached cells reduced intracellular pools of citrate, fumarate, and malate it had no further effect on detached pool sizes (Fig. 4A). Glucose-derived carbon incorporation into the TCA cycle intermediates citrate, fumarate, and malate were measured using 13C6-glucose tracer analysis (Fig. 4B–D). Two primary routes of pyruvate into the TCA cycle are pyruvate dehydrogenase (PDH) or PC activity. Glucose-derived pyruvate metabolism through PDH activity results in enrichment of the downstream M+2 labelling of citrate. Relative to vehicle treated attached, vehicle treated detached cells had reduced M+2 labelled citrate suggesting reduced PDH activity (Fig. 4F). Additionally, 1,25(OH)2D treatment of attached cells reduced M+2 labelled citrate, however, there was no further effect of 1,25(OH)2D in detached cells (Fig. 4F). Taken together, these results demonstrate that detached cells have depleted pool sizes of TCA cycle intermediates, although citrate pools were maintained, and these effects were not further exacerbated by 1,25(OH)2D treatment.
Fig. 4.
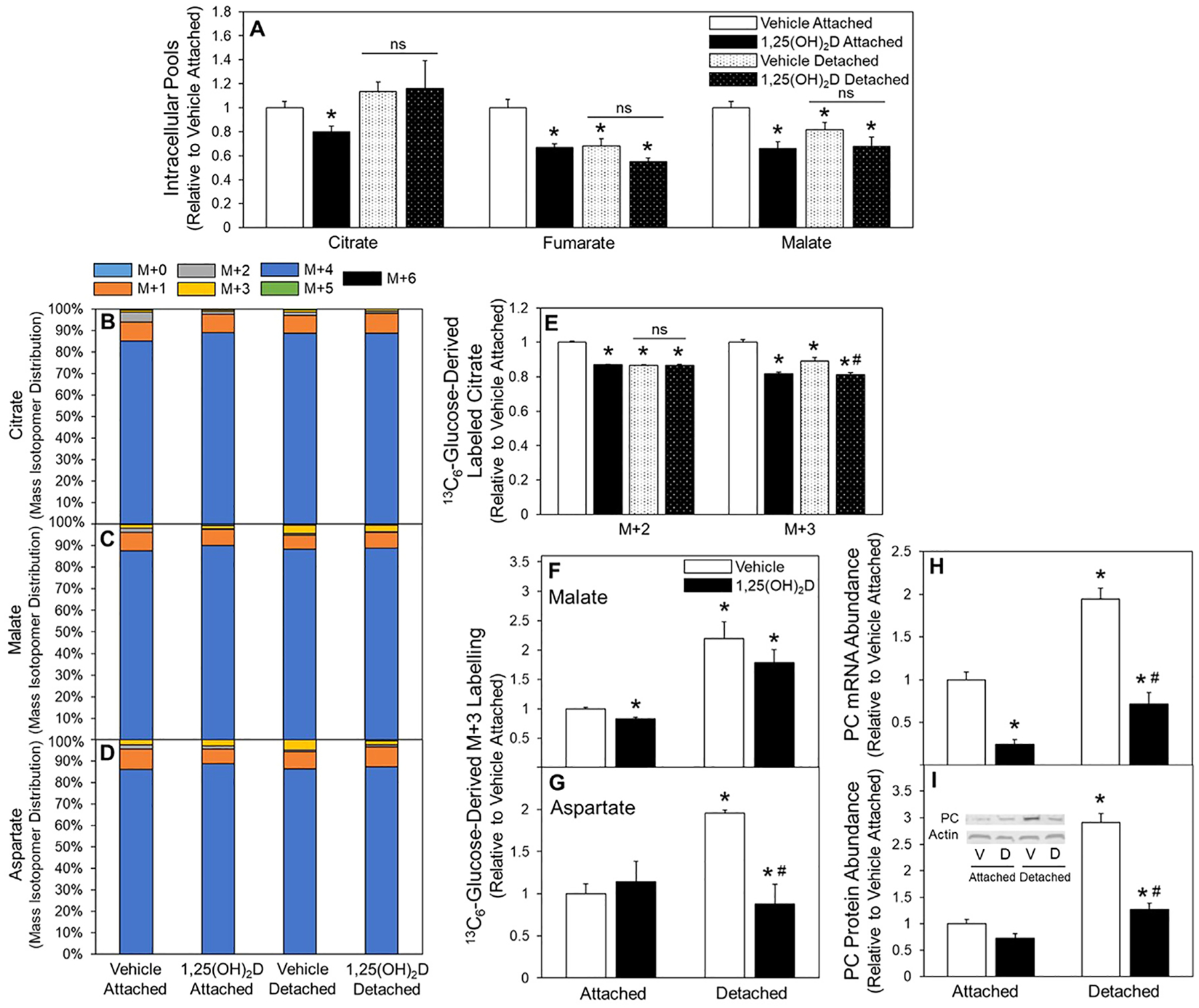
Effect of 1,25(OH)2D on the TCA cycle of attached and detached cells. (A) Following 3-d vehicle or 1,25(OH)2D pretreatment citrate, fumarate, and malate intracellular pool sizes were assessed and (B) 13C6-Glucose incorporation into citrate, (C) malate, (D) and aspartate were measured. (E) M+2 and M+3 labeled of citrate, (F) M+3 labelled malate, and (G) M+3 labeled aspartate were determined in attached and detached MCF10A-ras cells. (H) mRNA and (I) protein abundance of PC in vehicle and 1,25(OH)2D pretreated attached and detached MCF10A-ras cells was assessed. Results are expressed as ±SEM. Symbol (*) indicates P<.05 relative to vehicle attached and symbol (#) indicates P<.05 relative to vehicle detached. All mRNA and protein abundance experiments were repeated in duplicate with n=3, flux and pool size experiments were completed once with n=4.
Replenishment of the oxaloacetate pool to support the TCA cycle is in part mediated by PC activity, which synthesizes oxaloacetate from pyruvate and bicarbonate; thus, PC activity was also assessed. The flux of 13C6-glucose through the PC reaction results in M+3 labelling of citrate, malate, and aspartate. There was significantly decreased enrichment of the M+3 labeled citrate in detached compared to attached vehicle treated cells (Fig. 4E). However, detachment significantly increased M+3 labelling in malate and aspartate, demonstrating detachment increases PC activity and preferential utilization of PC-derived oxaloacetate into these metabolites rather than citrate (Fig. 4F and G). Treatment with 1,25(OH)2D reduced enrichment of M+3 citrate in attached cells and detached cells relative to their vehicle counterparts (Fig. 4E). Malate M+3 enrichment was reduced by 1,25(OH)2D treatment only in attached cells (Fig. 4F). In contrast, aspartate M+3, which was nearly doubled in detached cells, was reduced 50% by 1,25(OH)2D treatment (Fig. 4G). Compared to attached cells, detached cells significantly increased PC mRNA abundance (Fig. 4H) and protein expression (Fig. 4I), both of which were reduced with 1,25(OH)2D treatment in detached cells. In sum, 1,25(OH)2D reduced PC expression and activity in detached MCF10A-ras cells. These data demonstrate in detached cells a preferential utilization of PC-derived oxaloacetate into malate and aspartate, rather than citrate, and decreased PC expression and activity in detached cells by 1,25(OH)2D determined by reduced M+3 labelled aspartate.
3.4. Regulation of PC by 1,25(OH)2D reduces viability of detached breast cancer cells
Given that 1,25(OH)2D downregulates PC in detached cells, the impact of this regulation on survival in detached conditions was evaluated. Dox-inducible shRNA PC knockdown and PC overexpressing MCF10A-ras cell lines were utilized to measure cell viability in detached conditions with or without 1,25(OH)2D treatment (Figs. 5 and). PC depletion resulted in decreased viability in detached conditions (Fig. 5B). Supplementation with aspartate, the metabolite most enriched by PC activity in detached cells, resulted in rescued cell viability following PC depletion (Fig. 5B). Additionally, PC depletion reduced cell viability to a similar extent as 1,25(OH)2D treatment with no additional effect on detached cell viability with 1,25(OH)2D treatment and PC knockdown together in both MCF10A-ras cells and non-metastatic murine mammary cancer M-Wnt cells (Fig. 5C and 5D). Further, PC overexpression increased detached cell viability, and inhibited the 1,25(OH)2D mediated decrease in detached cell viability (Fig. 6B). These data demonstrate that 1,25(OH)2D’s negative effects on detached MCF10A-ras cell viability are dependent on PC downregulation.
4. Discussion
While breast cancer survival rates remain high when diagnosed early, it is estimated that between 25–50% of breast cancer patients will form metastatic tumors [2, 26]. Therefore, it is critical to identify safe and effective compounds that prevent metastasis, as it results in the vast majority of breast cancer-related deaths [2]. Throughout carcinogenesis, metastatic cancer cells acquire anchorage independence and reprogram energy metabolism, both of which support survival and growth at the primary and secondary site [9]. The results of the present study demonstrate that the downregulation of PC expression by the active metabolite of vitamin D, 1,25(OH)2D, reduces cell survival of detached breast cancer cells.
Recent studies have implicated PC in the promotion of breast to lung metastasis. For example, Christen et al. observed an increase in PC expression in metastatic breast tumors in the lung compared to the primary breast tumor [27]. In addition, PC knockdown in the primary tumor significantly decreased the number of breast to lung metastatic outgrowths in vivo [28]. Further support is provided by analysis of the Molecular Taxonomy of Breast Cancer International Consortium dataset where amplification of PC was detected in 16–30% of breast cancer patients, and PC amplification was associated with decreased patient survival consistent with PC’s role in promoting metastatic breast cancer [28]. PC activity replenishes the TCA cycle intermediate, oxaloacetate, which likely supports cancer progression through energy production, oxidative stress protection, and synthesis of building blocks such as amino and fatty acids required for proliferation [20, 21]. Studies also suggest that PC expression confers metastatic properties, as PC expression was positively associated with greater invasive and migratory capabilities, and PC inhibition reduced invasion and migration in various breast cancer models [29]. Interestingly, PC expression in cancer-associated fibroblasts was demonstrated to enhance TCA cycle metabolism and amino acid biosynthesis leading to increased collagen production and tumorigenesis [30]. Cumulatively, these results demonstrate that PC expression is associated with a metastatic phenotype.
Although others have reported 1,25(OH)2D’s long-term negative effects on ECM detached cancer cell survival, a mechanism was not elucidated. For example, Pervin et al observed that SKBR3 breast cancer cells grown in detachment for 4 and 7 d had reduced mammosphere formation with VDR overexpression, and that 1,25(OH)2D treatment was sufficient to reduce sphere formation [31]. In addition, 1,25(OH)2D treatment reduced mammosphere formation in metastatic SUM159 breast epithelial cells grown in detached conditions for 5 d [32]. Further, treatment with 1,25(OH)2D reduced mammosphere formation after secondary and tertiary passaging, suggesting that 1,25(OH)2D treatment may prevent tumor resurgence. While the long-term reduction of detached mammosphere formation by 1,25(OH)2D is promising, the mechanism by which 1,25(OH)2D regulates detached cancer growth remained unknown. The present study identifies 1,25(OH)2D mediated downregulation of PC as the mechanism by which 1,25(OH)2D decreases viability in detached transformed breast epithelial cells. While the 1,25(OH)2D-mediated reduction in viability of breast cancer cells is modest (11–15%), the effect of this on metastatic outcomes is yet to be determined and should be investigated in future studies. The current results demonstrate that detachment reduces most of the metabolic processes measured, except for metabolic flux through and protein expression of PC which was increased (Fig. 7A). In addition, 1,25(OH)2D treatment downregulates PC, resulting in reduced carbon flux through the PC reaction and viability of detached breast cancer cells (Fig. 7B). Growing evidence suggests PC activity provides cancer cells with the metabolic flexibility necessary to maintain cell survival in adverse environments with changing nutrient statuses throughout tumorigenesis and metastasis, and 1,25(OH)2D’s negative regulation of PC likely contributes to its anticancer effects [33, 34].
Fig. 7.
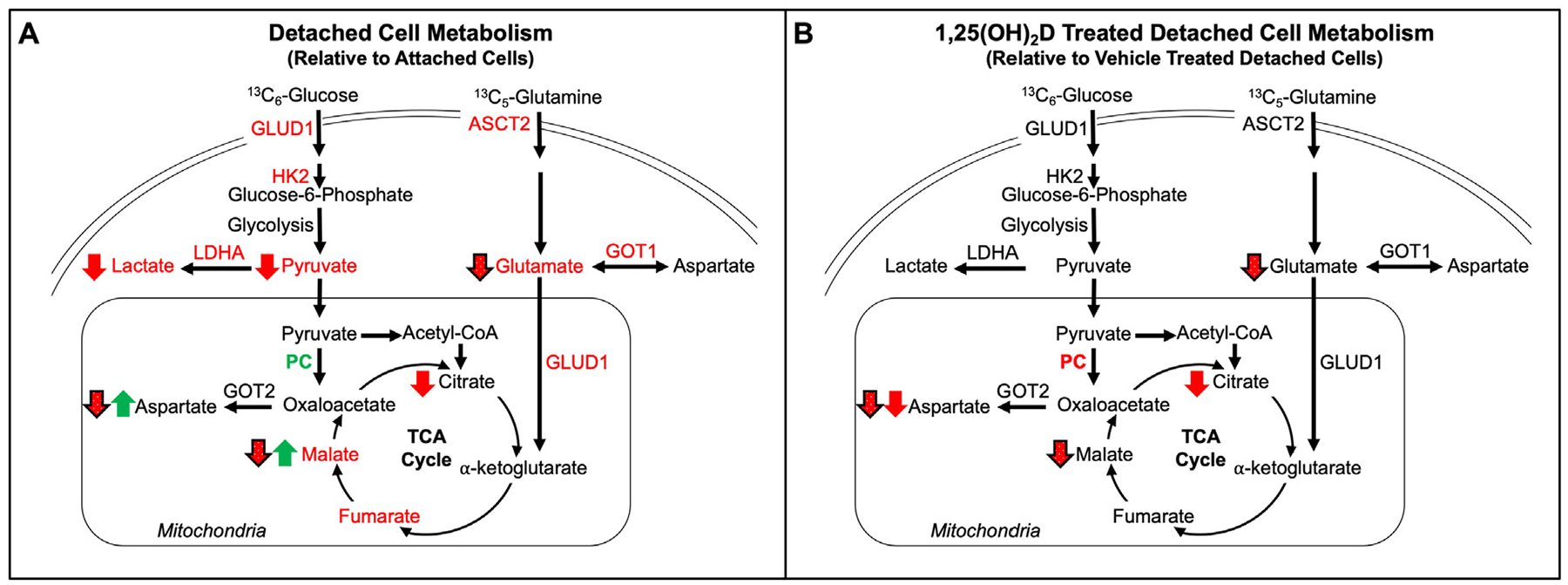
Summary of the metabolic effects of detachment and 1,25(OH)2D. (A) Effects of detached relative to attached and (B) 1,25(OH)2D treatment detached, relative to vehicle treated detached, MCF10A-ras cells on 13C6-glucose-derived or 13C5-glutamine-derived carbon flux. Pool sizes are indicated by green (increased) or red (decreased) font. Green upward arrow indicates a significant increase and red downward arrow indicates a significant decrease, and solid arrows indicate 13C6-glucose and black outlined dotted arrows indicate 13C5-glutamine flux. mRNA abundance in green font indicates a significant increase and red font indicates a significant decrease.
Although the current results show a reduction in glutamine metabolism by decreased 13C5-glutamine flux to glutamate, malate, and aspartate with 1,25(OH)2D, a 1,25(OH)2D target gene was not identified (Fig. 7B). Gene regulation is the predominant mechanism by which 1,25(OH)2D mediates its effects in target tissues. However, mRNA abundance of key glutamine-metabolizing enzymes was unchanged with 1,25(OH)2D treatment in detached cells, suggesting that 1,25(OH)2D regulates glutamine metabolism indirectly. A potential mechanism may involve reduced signaling through the adenosine monophosphate-activated protein kinase (AMPK) pathway which regulates energy metabolism as previously described [35]. The results from the current study complement earlier studies in attached MCF10A-ras cells where 1,25(OH)2D treatment reduced glutamine metabolism through direct downregulation of the glutamine transporter ASCT2; however, this effect was not demonstrated in detached cells [19].
In summary, these findings demonstrate that detachment of MCF10A-ras cells leads to a decreased flux of glucose and glutamine into the TCA cycle compared to their attached counterparts. However, PC expression and activity is increased in detached cells, and this effect is reversed with 1,25(OH)2D treatment resulting in decreased detached cell viability. These results indicate that PC plays a critical role in promoting the survival of cells in matrix-detached conditions and that PC is regulated in these conditions by 1,25(OH)2D.
Acknowledgments
The authors acknowledge the use of the facilities of the Bindley Bioscience Center, a core facility of the NIH-funded Indiana Clinical and Translational Sciences Institute.
Funding
This work was supported by the Purdue University Center for Cancer Research (P30CA023168); IU Simon Comprehensive Cancer Center (P30CA082709); Walther Cancer Foundation; Indiana Clinical Translational Science Institute NIH/NCRR (#TR000006) and the National Institute of Health (R01CA232589 and R35CA197627).
Footnotes
Declaration of Competing Interests
The authors declare that there are no conflicts of interest.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
- [1].Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med 2019;8:5574–6. doi: 10.1002/cam4.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin 2021;71. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- [3].Tice JA, Halalau A, Burke H. Vitamin D does not prevent cancer or cardiovascular disease: the vital trial. J Gen Intern Med 2020. doi: 10.1007/s11606-020-05648-x. [DOI] [PubMed] [Google Scholar]
- [4].Williams JD, Aggarwal A, Swami S, Krishnan AV, Ji L, Albertelli MA, et al. Tumor autonomous effects of vitamin D deficiency promote breast cancer metastasis. Endocrinology 2016;157:1341–7. doi: 10.1210/en.2015-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Horas K, Zheng Y, Fong-Yee C, Macfarlane E, Manibo J, Chen Y, et al. Loss of the vitamin D receptor in human breast cancer cells promotes epithelial to mesenchymal cell transition and skeletal colonization. J Bone Miner Res 2019;34:1721–32. doi: 10.1002/jbmr.3744. [DOI] [PubMed] [Google Scholar]
- [6].Tripkovic L, Lambert H, Hart K, Smith CP, Bucca G, Penson S, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr 2012;95:1357–64. doi: 10.3945/ajcn.111.031070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer 2014;14:342–57. doi: 10.1038/nrc3691. [DOI] [PubMed] [Google Scholar]
- [8].Simboli-Campbell M, Narvaez CJ, van Weelden K, Tenniswood M, Welsh J. Comparative effects of 1,25(OH)2D3 and EB1089 on cell cycle kinetics and apoptosis in MCF-7 breast cancer cells. Breast Cancer Res Treat 1997;42:31–41. doi: 10.1023/a:1005772432465. [DOI] [PubMed] [Google Scholar]
- [9].Grassian AR, Coloff JL, Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harb Symp Quant Biol 2011;76:313–24. doi: 10.1101/sqb.2011.76.010967. [DOI] [PubMed] [Google Scholar]
- [10].Schwartz MA. Integrins, oncogenes, and anchorage independence. J Cell Biol 1997;139:575–8. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 2013;1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- [12].Kang JS, Krauss RS. Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mol Cell Biol 1996;16:3370–80. doi: 10.1128/MCB.16.7.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Grassian AR, Schafer ZT, Brugge JS. ErbB2 stabilizes epidermal growth factor receptor (EGFR) expression via Erk and Sprouty2 in extracellular matrix-detached cells. J Biol Chem 2011;286:79–90. doi: 10.1074/jbc.M110.169821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, Brugge JS. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev 2011;25:1716–33. doi: 10.1101/gad.16771811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang J, Gao Q, Zhou Y, Dier U, Hempel N, Hochwald SN. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene 2016;35:1926–42. doi: 10.1038/onc.2015.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res 2013;73:3704–15. doi: 10.1158/0008-5472.CAN-12-2482. [DOI] [PubMed] [Google Scholar]
- [17].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [18].Zheng W, Tayyari F, Gowda GAN, Raftery D, McLamore ES, Shi J, et al. 1,25-dihydroxyvitamin D regulation of glucose metabolism in Harvey-ras transformed MCF10A human breast epithelial cells. J Steroid Biochem Mol Biol 2013;138:81–9. doi: 10.1016/j.jsbmb.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou X, Zheng W, Nagana Gowda GA, Raftery D, Donkin SS, Bequette B, et al. 1,25-Dihydroxyvitamin D inhibits glutamine metabolism in Harvey-ras transformed MCF10A human breast epithelial cell. J Steroid Biochem Mol Biol 2016;163:147–56. doi: 10.1016/j.jsbmb.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilmanski T, Buhman K, Donkin SS, Burgess JR, Teegarden D. 1α,25-dihydroxyvitamin D inhibits de novo fatty acid synthesis and lipid accumulation in metastatic breast cancer cells through down-regulation of pyruvate carboxylase. J Nutr Biochem 2017;40:194–200. doi: 10.1016/j.jnutbio.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wilmanski T, Zhou X, Zheng W, Shinde A, Donkin SS, Wendt M, et al. Inhibition of pyruvate carboxylase by 1α,25-dihydroxyvitamin D promotes oxidative stress in early breast cancer progression. Cancer Lett 2017;411:171–81. doi: 10.1016/j.canlet.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].O’Flanagan CH, Rossi EL, McDonell SB, Chen X, Tsai Y-H, Parker JS, et al. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 2017;3:26. doi: 10.1038/s41523-017-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- [24].Long CP, Antoniewicz MR. High-resolution 13C metabolic flux analysis. Nat Protoc 2019;14:2856–77. doi: 10.1038/s41596-019-0204-0. [DOI] [PubMed] [Google Scholar]
- [25].Millard P, Letisse F, Sokol S, Portais J-C. IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 2012;28:1294–6. doi: 10.1093/bioinformatics/bts127. [DOI] [PubMed] [Google Scholar]
- [26].Welsh J Induction of apoptosis in breast cancer cells in response to vitamin D and antiestrogens. Biochem Cell Biol 2022;72:537–45. doi: 10.1139/o94-072. [DOI] [PubMed] [Google Scholar]
- [27].Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep 2016;17:837–48. doi: 10.1016/j.celrep.2016.09.042. [DOI] [PubMed] [Google Scholar]
- [28].Shinde A, Wilmanski T, Chen H, Teegarden D, Wendt MK. Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res 2018;20:76. doi: 10.1186/s13058-018-1008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate carboxylase is up-regulated in breast cancer and essential to support growth and invasion of MDA-MB-231 cells. PLoS One 2015;10:e0129848. doi: 10.1371/journal.pone.0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schwörer S, Pavlova NN, v Cimino F, King B, Cai X, Sizemore GM, et al. Fibroblast pyruvate carboxylase is required for collagen production in the tumour microenvironment. Nat Metab 2021;3:1484–99. doi: 10.1038/s42255-021-00480-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pervin S, Hewison M, Braga M, Tran L, Chun R, Karam A, et al. Down-regulation of vitamin D receptor in mammospheres: implications for vitamin D resistance in breast cancer and potential for combination therapy. PLoS One 2013;8:e53287. doi: 10.1371/journal.pone.0053287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shan NL, Wahler J, Lee HJ, Bak MJ, das Gupta S, Maehr H, et al. Vitamin D compounds inhibit cancer stem-like cells and induce differentiation in triple negative breast cancer. J Steroid Biochem Mol Biol 2017;173:122–9. doi: 10.1016/j.jsbmb.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sheeley MP, Andolino C, Kiesel VA, Teegarden D. Vitamin D regulation of energy metabolism in cancer. British Journal of Pharmacology 2021. doi: 10.1111/bph.15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kiesel VA, Sheeley MP, Coleman MF, Cotul EK, Donkin SS, Hursting SD, et al. Pyruvate carboxylase and cancer progression. Cancer Metab 2021;9:20. doi: 10.1186/s40170-021-00256-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Santos JM, Khan ZS, Munir MT, Tarafdar K, Rahman SM, Hussain F. Vitamin D3 decreases glycolysis and invasiveness, and increases cellular stiffness in breast cancer cells. J Nutr Biochem 2018;53:111–20. doi: 10.1016/j.jnutbio.2017.10.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.