

Abstract
SUMOylation, the covalent conjugation of small ubiquitin-like modifier (SUMO) proteins to protein substrates, has been reported to suppress type I interferon (IFN1) responses. TAK-981, a selective small molecule inhibitor of SUMOylation, pharmacologically reactivates IFN1 signaling and immune responses against cancers. In vivo treatment of wild type mice with TAK-981 upregulated IFN1 gene expression in blood cells and splenocytes. Ex vivo treatment of mouse and human dendritic cells promoted their IFN1-dependent activation, and vaccination studies in mice demonstrated stimulation of antigen cross-presentation and T cell priming in vivo. TAK-981 also directly stimulated T cell activation, driving enhanced T cell sensitivity and response to antigen ex vivo. Consistent with these observations, TAK-981 inhibited growth of syngeneic A20 and MC38 tumors in mice, dependent upon IFN1 signaling and CD8+ T cells, and associated with increased intratumoral T and NK cell number and activation. Combination of TAK-981 with anti-PD1 or anti-CTLA4 antibodies improved the survival of mice bearing syngeneic CT26 and MC38 tumors. In conclusion, TAK-981 is a first in class SUMOylation inhibitor that promotes anti-tumor immune responses by activation of IFN1 signaling. TAK-981 is currently being studied in phase I clinical trials (NCT03648372, NCT04074330, NCT04776018 and NCT04381650) for the treatment of patients with solid tumors and lymphomas.
One sentence summary:
TAK-981, a first-in-class small molecule SUMOylation inhibitor, activates IFN1 signaling to promote anti-tumor immune responses.
Introduction
The recent development of therapeutics directed at mobilizing the immune system to fight cancer, in particular immune checkpoint inhibitors (ICIs), has revolutionized oncology clinical practice, delivering long-term benefit to a substantial number of patients by blocking inhibitory T cell checkpoint signals (1). Nevertheless, most cancer patients with advanced disease remain refractory or develop resistance to ICIs. Modulation of the innate immune response represents a complementary strategy for promoting antitumor immunity and potentially enabling response to ICIs (2). The Type I interferons (IFN1s, such as IFNα and IFNβ) are potent immunomodulatory molecules induced early in the innate immune response which act upon multiple immune cell types, including NK and T cells (3–6), to shape both innate and adaptive immunity. They also play a central role in propagating adaptive immune responses by promoting maturation of dendritic cells (DCs) and cross presentation of antigens to T cells (7–9).
Innate immune responses and the IFN1 pathway are modulated by a variety of mechanisms including protein SUMOylation (10), a reversible post-translational modification that regulates protein function by attachment of a small ubiquitin-like modifier (SUMO) protein to protein substrates (11). Notably, genetic inactivation of the SUMO pathway in immune cells resulted in enhanced basal expression of IFN1 and inflammatory cytokines, with marked sensitization to upregulation of IFN1 by pathogenic stimuli (12). Relief of SUMO-dependent transcriptional repression of the IFNβ gene was identified as one mechanism contributing to activation of the IFN1 pathway (12), consistent with the well-documented, typically inhibitory, association of SUMO with chromatin proteins (13–15). SUMO-dependent modulation of IFN1 gene expression is further supported by studies of genetic inactivation of SUMOylation in human monocytic THP1 cells (16).
The therapeutic potential of IFN1 for promoting anti-tumor immune responses has yet to be fully realized due to the dose limiting toxicity associated with systemic IFN1 administration (17, 18), as well as the loss of the IFN1 receptor (19). Similar concerns may limit approaches directed at stimulating IFN1 production through systemic administration of pattern recognition receptor (PRR) agonists (20, 21), such as cyclic dinucleotides (STING agonists) or TLR agonists. We sought to explore the potential for pharmacological inhibition of SUMOylation to stimulate IFN1-driven mechanisms of innate and adaptive anti-tumor immunity, hypothesizing that this approach could provide a mechanistically differentiated, and potentially more homeostatic and better tolerated, approach towards upregulating the IFN1 pathway. We have developed a small molecule SUMOylation inhibitor, TAK-981 (22), which forms an irreversible adduct with each of the three functional mammalian SUMO paralogues (SUMO 1, SUMO 2, and SUMO 3) when bound to the E1 SUMO activating enzyme (SAE). This prevents transfer of SUMO to the sole E2 conjugating enzyme, UBC9, and subsequent ligation to protein substrates. Central to the utility of TAK-981 was the ability to achieve a duration of in vivo SUMOylation inhibition sufficient to activate IFN1 signaling in immune cells without driving sustained IFN1 expression, to avoid the potential for suppression of immune responses associated with prolonged IFN1 signaling (23), and allow flexibility in design of combination schedules with complementary immune therapies. We describe here the characterization of the selective SAE inhibitor TAK-981 and assessment of its activity in promoting IFN1-dependent immune responses ex vivo in mouse and human immune cells and in syngeneic mouse tumor models.
Results
TAK-981 is a potent and selective inhibitor of SUMOylation in vitro and in vivo
TAK-981 (Fig. 1A) was identified by an iterative medicinal chemistry program applied to optimization of the pharmaceutical properties of the chemical series exemplified by the SAE inhibitor ML-792 (24), including improvements in physical properties and in particular in vivo activity. TAK-981 inhibited SAE in a manner analogous to ML-792, forming an irreversible adduct with SUMO protein in an enzyme-catalyzed, ATP dependent process, as shown by mass spectrometry analysis of SAE inhibition reaction mixtures, showing species whose sizes are consistent with covalent SUMO1-TAK-981 and SUMO2-TAK-981 adducts (fig. S1, red traces). Adduct formation was not detected in the absence of ATP (fig. S1, blue traces). TAK-981 inhibited SAE (IC50 0.6 nM), and showed strong selectivity for SAE relative to the closely related E1 enzymes NEDD8-activating enzyme (NAE), ubiquitin activating enzyme (UAE), or autophagy related 7 (ATG7) (Fig. 1B), and exhibited limited inhibition of kinases (table S1).
Fig. 1. TAK-981 is a potent and selective inhibitor of SUMOylation.
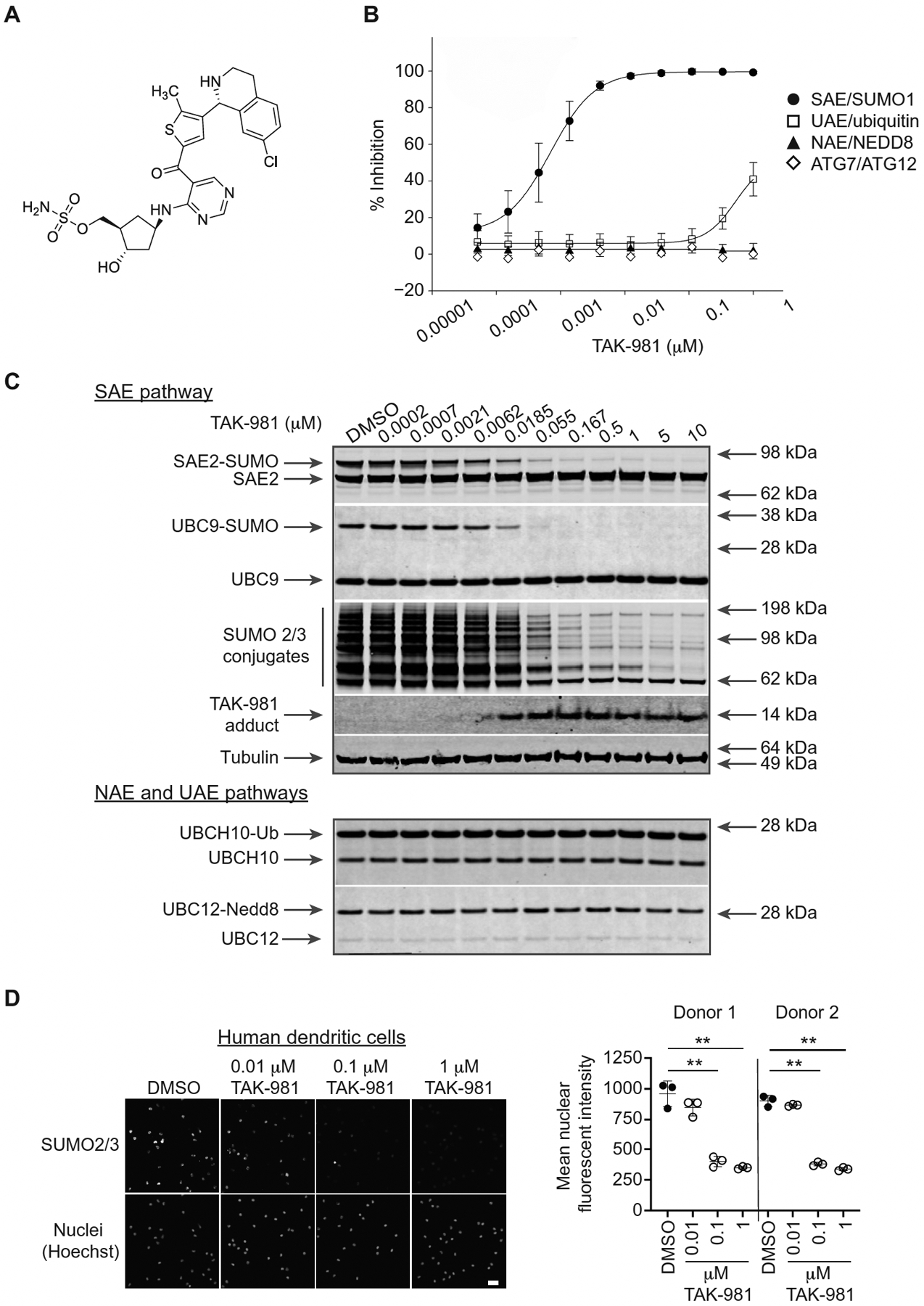
(A) Chemical structure of TAK-981, ((1R,2S,4R)-4-((5-(4-((1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl)-5-methylthiophene-2-carbonyl)pyrimidin-4-yl)amino)-2-hydroxycyclopentyl)methyl sulfamate. (B) E1–E2 homogeneous time-resolved fluorescence (HTRF) transthiolation assay to assess the activity of TAK-981 against recombinantly purified SAE, UAE, NAE and ATG7. Each of the E1 assays was tested at an ATP concentration near the Km for each enzyme. The points represent averages of nine replicate individual biochemical assays, except for ATG7 which represents a single assay. Data are shown as mean +/− SD. (C) Representative Western blot analysis (of n = 2 independent experiments) of HCT116 cells treated for 4h with increasing concentrations of TAK-981. The status of SUMO 2/3 protein conjugation to the E1 (SAE) and E2 (UBC9) enzymes, as well as the presence of SUMO 2/3 conjugates and formation of the TAK-981-SUMO adduct, was assessed. (D) Dose-dependent suppression of SUMO 2/3 conjugates in human DCs. DCs were isolated from the PBMCs of two donors, treated with TAK-981 or dimethylsulfoxide (DMSO) for 3h in triplicate, fixed and stained with anti-SUMO 2/3 antibody. Representative images are shown in which nuclei are visualized by Hoechst staining, and quantification of total nuclear SUMO 2/3 fluorescent signal intensity is shown. Scale bar, 25 μm. Individual values representing the mean signal from >200 nuclei, quantified for each triplicate, are shown, as are mean and SD. **P < 0.01, compared with DMSO control, by Welch’s t test.
The antibody MIL113 was raised against the TAK-981-SUMO adduct to provide a target engagement biomarker. Dose-dependent formation of the TAK-981-SUMO adduct (EC50 9.5 nM) was readily detected in HCT116 cells by MIL113 (Fig. 1C; table S2). Concomitant dose-dependent pathway inhibition, manifested as loss of global SUMOylation (EC50 26.1 nM), was demonstrated using SUMO 2/3 antibodies (Fig. 1C; fig. S2A). As a result of TAK-981-SUMO adduct formation, the formation of the high energy thioester conjugates between SUMO and SAE or UBC9, required for SUMO transfer through the enzymatic cascade, were inhibited (EC50 5 nM and 8.5 nM, respectively). Thioester conjugation of either ubiquitin or NEDD8 with their E2 enzymes (UBCH10 and UBC12, respectively) was unaffected at the highest drug concentration tested (10 μM). Comparable data were obtained in the mouse A20 lymphoma B cell line (fig. S2B). Finally, an immunofluorescent assay demonstrated that TAK-981 promoted robust and durable loss of nuclear SUMO 2/3 conjugates in HCT116 cells (table S3), mouse bone marrow-derived dendritic cells (fig. S2C), and human peripheral blood mononuclear cell (PBMC)-derived dendritic cells (Fig. 1D). Consistent with previous reports of SUMOylation inhibition (24, 25), sustained exposure of tumor cell lines to TAK-981 was associated with inhibition of cell cycle progression, mitotic block and endoreduplication (fig. S2, D and E), as demonstrated by propidium iodide staining showing decreased 2n DNA content and increased 4n DNA content, consistent with mitotic block, and appearance of 8n DNA content, consistent with endoreduplication, in HCT116 cells treated for 24–48h with either TAK-981 or ML-792 (fig. S2D). No alteration in cell cycle profiles was observed following more limited (4h) exposure to TAK-981 (fig. S2E).
In vivo analyses in Severe Combined Immunodeficiency (SCID) mice bearing subcutaneous OCI-Ly-10 human lymphoma xenografts demonstrated rapid and long-lasting formation of the TAK-981-SUMO adduct in tumor cells, associated with inhibition of SUMO 2 and SUMO3 conjugates for approximately 15–20h, followed by gradual recovery of SUMOylation, after a single 10 mg/kg dose of TAK-981 (fig. S3).
TAK-981 induces IFN1 signaling and protects the IFN1 pathway from intratumoral inactivation in syngeneic mouse tumors
Because SUMOylation has been reported to suppress IFN1 responses (12, 16), we sought to determine the effect of TAK-981 on the IFN1 pathway. Treatment of OCI-Ly-10 cells with TAK-981 resulted in phosphorylation of the key transcriptional factors downstream of the IFN1 receptor, STAT1 and STAT2 (fig. S4A). Similarly, mouse primary splenocytes exposed to TAK-981 responded with a stimulation of STAT1 phosphorylation which was not observed in splenocytes derived from mice lacking the IFNAR1 chain of the IFN1 receptor (Fig. 2A), demonstrating that TAK-981 stimulates the IFN1 signaling pathway in an IFNAR1-dependent manner.
Fig. 2. Activation of IFN1 signaling by TAK-981.
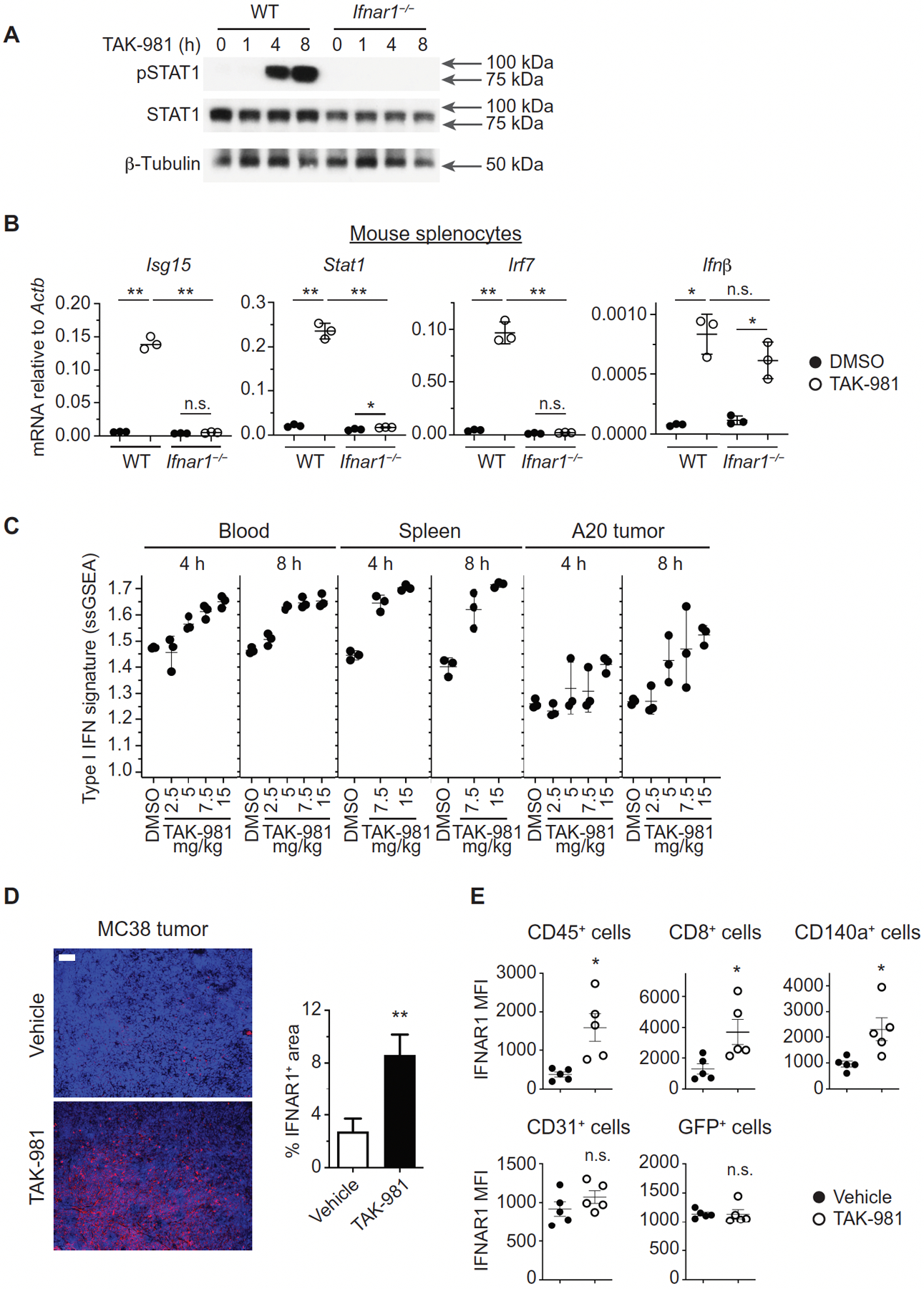
(A) Western blot analysis of STAT1 phosphorylation in splenocytes derived from WT and Ifnar1−/− C57BL/6 mice treated ex vivo with 100 nM TAK-981 for the indicated times. (B) Expression of interferon stimulated genes (ISGs) in splenocytes from WT and Ifnar1−/− mice exposed to TAK-981. Q-PCR analysis was carried out on splenocytes derived from WT and Ifnar1−/− C57BL/6 mice treated ex vivo with DMSO or 100 nM TAK-981 for 12h (n = 3 for each treatment). Horizontal lines represent mean and SD. *P < 0.05, **P < 0.01, n.s. not significant, compared with DMSO control by Welch’s t test. (C) TAK-981 dependent upregulation of IFN1 mRNA signature in peripheral blood, spleen and tumor of BALB/c mice bearing A20 tumors (n = 3 for each tissue and treatment). The plot depicts IFN1 ssGSEA scores in peripheral blood at 4h (P = 0.0011) and 8h (P = 0.0001), in spleen at 4h (P = 0.0002) and 8h (P = 0.0020), and in tumor at 4h (P = 0.0095) and 8h (P = 0.0033) after treatment with DMSO or the indicated doses of TAK-981. P values were calculated by Welch’s ANOVA test. (D) Regulation of IFNAR1 in the tumor microenvironment in response to TAK-981. Immunofluorescence analysis and quantification of IFNAR1 in MC38 tumors on day 4, following administration of vehicle or 15 mg/kg TAK-981 on days 1 and 3. Scale bar, 100 μm. Quantification is represented as the mean +/− SEM of 3–4 random fields in sections from each of 3 vehicle treated mice (n = 12 fields) and TAK-981 treated mice (n = 11 fields). **P < 0.01 compared with vehicle control by Welch’s t test. (E) Flow cytometric analysis of IFNAR1 cell surface expression on immune cells (CD145+), CD8+ T cells, fibroblasts (CD140a+), endothelial cells (CD31+), and tumor cells (GFP+) from MC38-OVA/GFP tumors harvested from C57BL/6 mice on day 4, following treatment with vehicle or 15 mg/kg TAK-981 on days 1 and 3 (n = 5 for each analysis). *P < 0.05, n.s. not significant, compared with vehicle control, calculated by Welch’s t test.
Consistent with STAT activation, a robust induction of mRNA expression of IFNβ and of IFN-stimulated genes (ISGs) was observed in both mouse splenocytes and human T cells treated with TAK-981 (Fig. 2B; fig. S4, B and C). This is in contrast to the modest transcriptional changes previously observed in solid tumor cell lines in response to inhibition of SUMOylation by ML-792 (24). Similar limited transcriptional changes are induced by TAK-981 in these solid tumor cell lines (fig. S4D), lacking a robust induction of IFN1 genes (fig. S4E). In mouse splenocytes, the induction of ISGs but not of IFNβ by TAK-981 was dependent on the integrity of IFNAR1 (Fig. 2B), indicating that induction of IFN1 and ensuing activation of IFNAR1 likely mediate stimulation of the IFN1 pathway by TAK-981.
In situ injection of TAK-981 microdoses into A20 mouse B cell lymphoma subcutaneous tumors using the Comparative In Vivo Oncology (CIVO) intratumoral microdosing platform (26) promoted expression of IFNβ (fig. S4F). Intravenous administration of TAK-981 to BALB/c or C57BL/6 mice bearing A20 or B16F10 syngeneic tumors, respectively, resulted in a strong induction of IFN1 pathway genes in peripheral blood leukocytes, spleen and A20 tumor tissues (Fig. 2C, fig. S4, G and H). Additional analysis using DAVID GO terms (fig. S4I) and clustering of biological processes using Cytoscape EnrichmentMap (fig. S4J) highlighted immune system process, cellular response to interferon beta, immune response, and response to lipopolysaccharide, as well as hemostasis and cholesterol homeostasis. The NFκB pathway was not among the top regulated pathways, but more detailed analysis demonstrated modulation of expression of some NFkB-regulated genes in blood and spleen (fig. S4K).
Single cell RNA sequencing (scRNA-Seq) analysis of A20 tumors indicated that the IFN1 signature was derived primarily from monocytes and T cells, not the B cell population which includes A20 tumor cells (fig. S4L), consistent with the marginal induction of an IFN1 signature in poorly immune cell-infiltrated B16F10, vs the substantial induction in robustly immune cell-infiltrated A20, tumors (Fig. 2C, fig. S4, G and H). In vitro treatment of the A20, B16F10, CT26 and MC38 mouse tumor cell lines with TAK-981 did not result in induction of the IFN1 pathway (fig. S4M). The overall degree of transcriptional modulation by TAK-981 was modest in the mouse tumor cell lines (fig. S4N), with only limited overlap observed in the transcriptional responses to TAK-981, as also observed for solid tumor cell lines (fig. S4, D and O).
Previous reports suggest that intratumoral stress stimuli and tumor-derived factors (including inflammatory cytokines and extracellular vesicles) can downregulate IFNAR1 and inhibit cellular responses to IFN1 (27–30). Immunofluorescent analysis revealed that treatment with TAK-981 resulted in robust increases in IFNAR1 protein in MC38 colorectal adenocarcinoma tumors (Fig. 2D). Subsequent flow cytometry analysis of MC38-OVA/GFP tumors showed that neither malignant nor endothelial cells exhibited increased IFNAR1 in response to TAK-981 administration (Fig. 2E, fig. S4P). However, TAK-981 increased IFNAR1 on intratumoral immune cells and tumor-associated fibroblasts. Downregulation of IFNAR1 on both immune cells (30) and cancer-associated fibroblasts (31) within the tumor microenvironment was previously demonstrated to enable rapid tumor growth. Collectively, these data indicate that TAK-981 acts to reactivate the IFN1 pathway in cells of the tumor microenvironment by a combination of stimulating IFN1 expression and upregulating IFNAR1. However we cannot rule out the possibility that increased immune cell IFNAR1 reflects, at least in part, recruitment of infiltrating T cells with higher intrinsic cell surface expression of IFNAR1, rather than upregulation of IFNAR1 on tumor resident T cells.
TAK-981 activates dendritic cells and T lymphocytes in vitro and in vivo
Analysis of blood samples from A20 lymphoma-bearing BALB/c mice treated with TAK-981 revealed induction of immunomodulatory cytokines and chemokines including IP-10 and IFNγ (fig. S5). Treatment with TAK-981 also resulted in transient depletion of circulating lymphocytes (fig. S6A), as previously described in response to IFN1 (32), with more rapid recovery of T cell than B cell numbers following cessation of dosing (fig. S6, B and C). Flow cytometric analysis of lymph nodes demonstrated a non-significant trend towards increased T cell, but not B cell, numbers in lymph nodes following treatment with TAK-981 (fig. S6, D and E), consistent with previous observations of IFN1-induced T cell redistribution (33).
Because DCs have been shown to serve as important targets for, and mediators of, the antitumorigenic effects of IFN1 (8, 18, 19), we assessed the effects of TAK-981 on these cells. Treatment with TK-981 activated DCs derived from both human PBMCs (Fig. 3A) and mouse bone marrow (BMDCs; fig. S7A), as evident by increased secretion of multiple cytokines including IFNα and IFNβ. Moreover, TAK-981 upregulated the costimulatory markers CD40, CD80 and CD86 on human DCs (Fig. 3B, fig. S7B), as also seen using a different SAE inhibitor, ML-792 (fig. S7, C and D). TAK-981 alone or in combination with lipopolysaccharide (LPS) also upregulated the costimulatory markers CD40 and CD86 on BMDCs; this effect could be attenuated by pre-treatment with an IFNAR1 neutralizing antibody (fig. S7, E and F), indicating dependence on IFN1 signaling for this activity ex vivo.
Fig. 3. TAK-981 activates dendritic cells ex vivo.
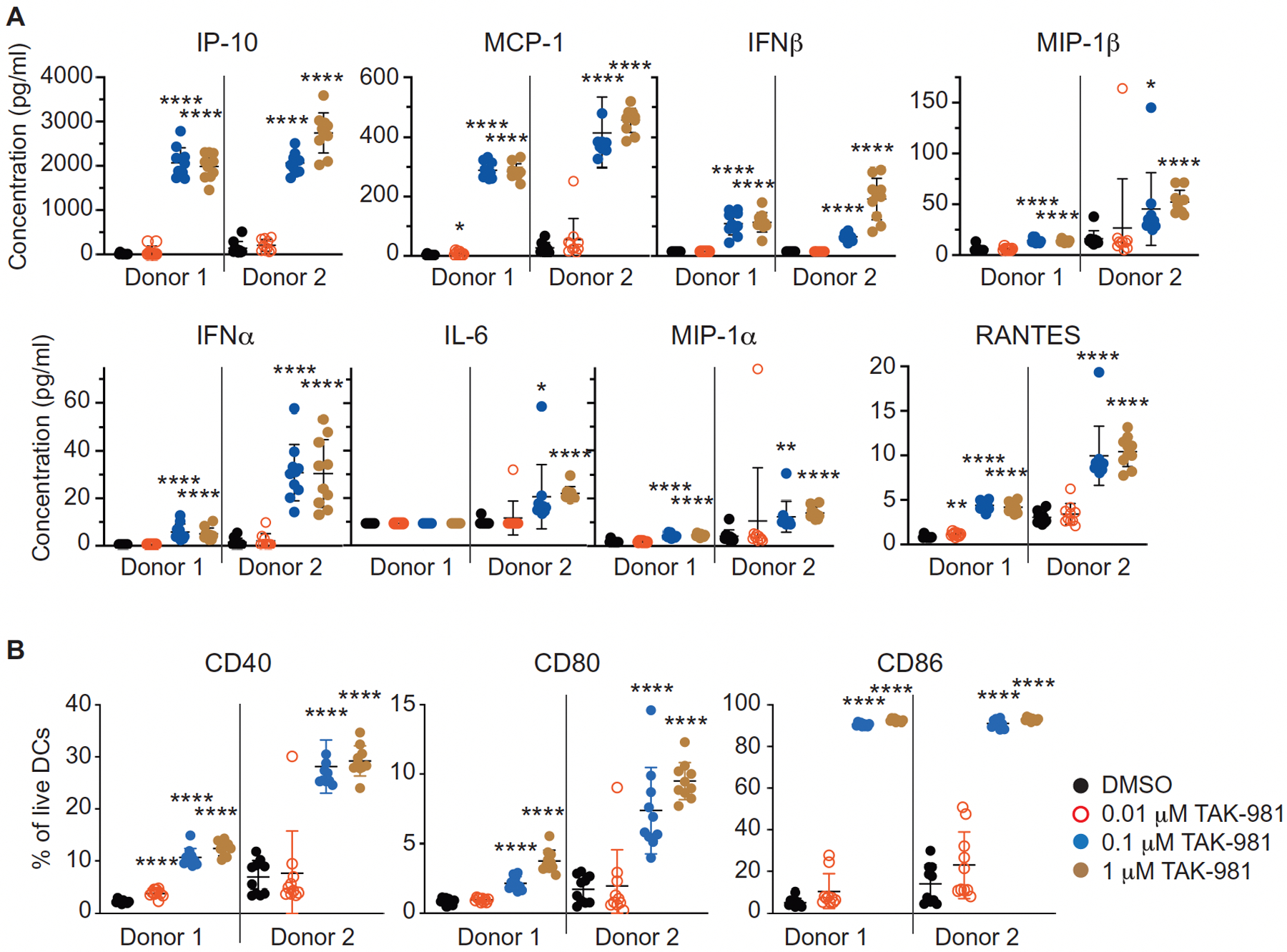
(A) Cytokines were measured in cell culture medium from human DCs, isolated from the peripheral blood of two healthy donors, following 16h of treatment with DMSO or the indicated concentrations of TAK-981 (n = 10 for each treatment). Horizontal lines represent mean and SD. *P < 0.05, **P < 0.01, ****P < 0.0001, compared with DMSO control, by Welch’s t test. (B) The percent of CD40, CD80 and CD86 positive DCs was assessed by flow cytometry of human DCs isolated from the peripheral blood of two healthy donors following 16h of ex vivo treatment with DMSO or the indicated concentrations of TAK-981 (n = 10 for each treatment). Horizontal lines represent mean and SD. ****P < 0.0001, compared with DMSO control, by Welch’s t test.
To examine DC activation by TAK-981 in vivo, BALB/c mice were injected subcutaneously with TAK-981, and lymph nodes adjacent to (brachial) and distal from (inguinal) the injection site were isolated 18h later. Increases in the intensity of the activation markers CD40 and CD86 were detected selectively in the proximal brachial lymph node following administration of TAK-981 (fig. S7, G and H). Using the same model, we also assessed the effects of TAK-981 on T-cell priming by activated DCs, using subcutaneous co-injection of ovalbumin protein with TAK-981. Co-administration of TAK-981 and ovalbumin resulted in the appearance of CD8+ T cells staining with the H-2Kb SIINFEKL tetramer, in the brachial lymph nodes adjacent to the injection site, and in the spleen (Fig. 4A, fig S7I). In addition, staining of DCs presenting the SIINFEKL peptide on MHC-1 could be detected in the brachial lymph node. These findings demonstrate enhanced antigen cross presentation and T-cell priming by DCs in response to TAK-981 in vivo.
Fig. 4. TAK-981 activates T cells ex vivo and promotes T cell priming in vivo.
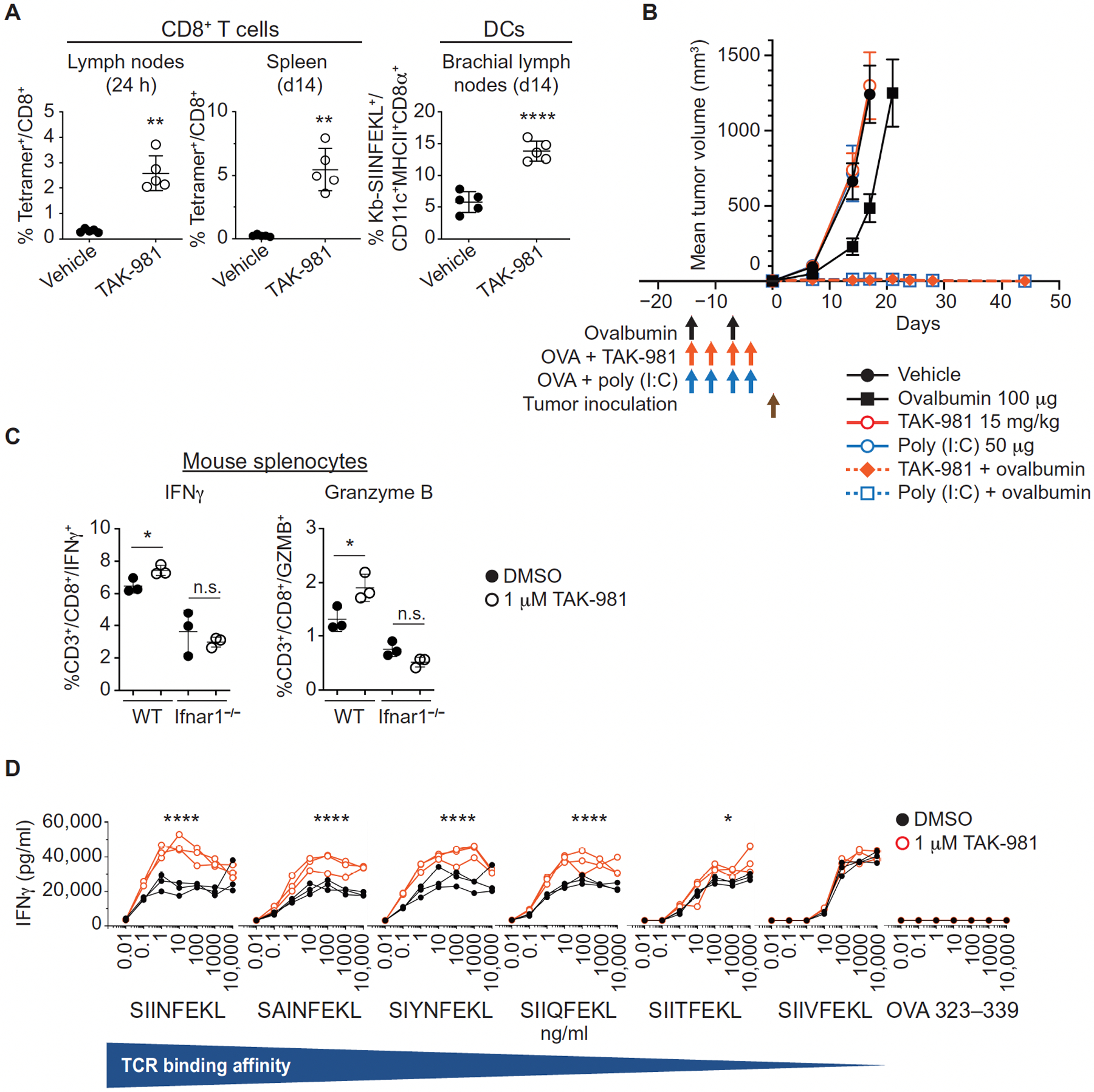
(A) Lymph nodes and spleens of C57BL/6 mice subcutaneously co-injected with ovalbumin protein +/− TAK-981 or vehicle (n = 5 for each treatment) were harvested 1 day after immunization, or administered a boost immunization on day 7 and harvested on day 14. Samples were analyzed by flow cytometry for Kb-SIINFEKL tetramer positive CD8+ T cells, and CD8α+ DCs loaded with the peptide SIINFEKL on the MHC 1 molecule H-2Kb. Horizontal lines represent mean and SD. **P < 0.01, ****P < 0.0001, compared with vehicle control, by Welch’s t test. (B) C57BL6 mice were subcutaneously injected with 100 μg ovalbumin alone on day -14 and -7, or, on a day -14, -11, -7, and -4 schedule: vehicle alone, or 100 μg ovalbumin combined with either 15 mg/kg TAK-981 or 50 μg poly (I:C), as also shown by the vertical arrows in the figure. B16F10-OVA tumors were implanted on day 0 and mice were monitored for tumor growth. Data is shown as mean tumor volume +/− SEM, n=16 mice/treatment arm except n=15 for ovalbumin combined with TAK-981. Mean tumor volumes for single agent treatment arms are shown up to the last day that the entire cohort was available for tumor measurement, before mice were removed due to humane endpoints for tumor size. (C) Ex vivo activation of T cells by TAK-981. Splenocytes derived from C57BL/6 WT and Ifnar1−/− mice were treated with DMSO or 100 nM TAK-981 for 15h (n = 3 for each treatment) and then with PMA/Ionomycin/GolgiStop for an additional 6h. IFNγ and Granzyme B were detected in CD45+CD3+CD8+ CTLs by FACS. Horizontal lines represent mean and SD. *P < 0.05, n.s. not significant, compared with DMSO control, by Welch’s t test. (D) CD8+ T cells derived from OT-I mice were treated ex vivo for 4h with DMSO or 1μM TAK-981 (n = 3 for each treatment) and subsequently co-cultured with splenocytes derived from C57BL/6J mice in the presence of OVA peptides, with varying TCR affinities, for 3 days, after which IFNγ in the medium was quantitated by ELISA. *P < 0.05, ****P < 0.0001, compared with DMSO control, by two-way ANOVA test with Geisser-Greenhouse correction (TAK-981 treatment × peptide concentration).
To assess whether TAK-981 could promote protective anti-tumor responses, C57BL6 mice were injected subcutaneously with ovalbumin alone, or in combination with TAK-981 or the TLR3 agonist poly (I:C), a potent adjuvant and inducer of IFN1, prior to challenge with B16F10-OVA tumors. Whereas vaccination with ovalbumin alone resulted in a slight tumor growth delay, addition of either TAK-981 or poly (I:C) to ovalbumin resulted in complete tumor rejection in all implanted mice (Fig. 4B). Subsequent re-challenge of these tumor-free mice demonstrated durable protection against growth of B16F10-OVA, but not B16F10, tumors in all mice previously treated with ovalbumin in combination with TAK-981, or ovalbumin in combination with poly (I:C) (fig. S7J). Delayed growth of B16F10 tumors was observed in mice treated with both TAK-981 and poly (I:C), relative to naïve mice, suggestive of antigen spread in the vaccinated mice.
Because IFN1s have been shown to provide a “third signal” in stimulating the clonal expansion of CD8+ T cells (6, 34, 35) and to increase the viability of activated anti-viral CD8+ T lymphocytes (36–39) and tumor-specific cytotoxic T lymphocytes (CTL) (30, 40), we assessed the effects of TAK-981 on CTL activation. Pretreatment of mouse T cells with TAK-981 (subsequently activated with PMA/ionomycin) led to a notable increase in the percentage of IFNγ or Granzyme B-expressing CD8+ T lymphocytes (Fig. 4C, fig. S7K). This increase was not observed in cells derived from Ifnar1−/− mice, again demonstrating a critical role of the IFN1-IFNAR1 pathway in TAK-981 activity. Ex vivo studies demonstrated that TAK-981 also increased the sensitivity, and response, of OT-I T cells to antigen (Fig. 4D), in an IFN1-dependent manner (fig. S7L). Treatment with TAK-981 did not notably enhance signaling downstream of the T cell receptor or CD28, signal 1 and signal 2, respectively, for T cell activation (fig. S7M).
Suppression of tumor growth by TAK-981 depends on the IFN1 pathway and adaptive immunity
In vivo administration of TAK-981 dose-dependently suppressed the growth of mouse syngeneic A20 lymphoma tumors, with two complete regressions (CRs) observed at the highest dose (7.5 mg/kg twice weekly) in the study shown (Fig. 5A). TAK-981 was well-tolerated in mouse models up to the maximum tolerated dose of 40 mg/kg, with mean body weight gain typically observed over the course of dosing (fig. S8A). Blockade of the IFN1 signaling pathway by administration of an IFNAR1-neutralizing antibody completely prevented TAK-981 antitumor activity in wild type mice bearing A20 tumors (Fig. 5B, fig. S8B) and suppressed the increase in plasma cytokines associated with TAK-981 administration (fig. S8C). Likewise, although lacking any notable antiproliferative activity against MC38 mouse colon carcinoma cells in vitro (fig. S8D), TAK-981 elicited single agent antitumor activity against MC38 tumors grown in immune competent mice, but not in Ifnar1−/− knockout mice (Fig. 5C, fig. S8E) or in ovalbumin-expressing MC38 tumors grown in Rag1−/− knockout mice (fig. S8F). These results are consistent with a central role for the IFN1 pathway in TAK-981 anti-tumor activity.
Fig. 5. TAK-981 promotes tumor growth inhibition dependent on IFNAR signaling and CD8+ T cells.
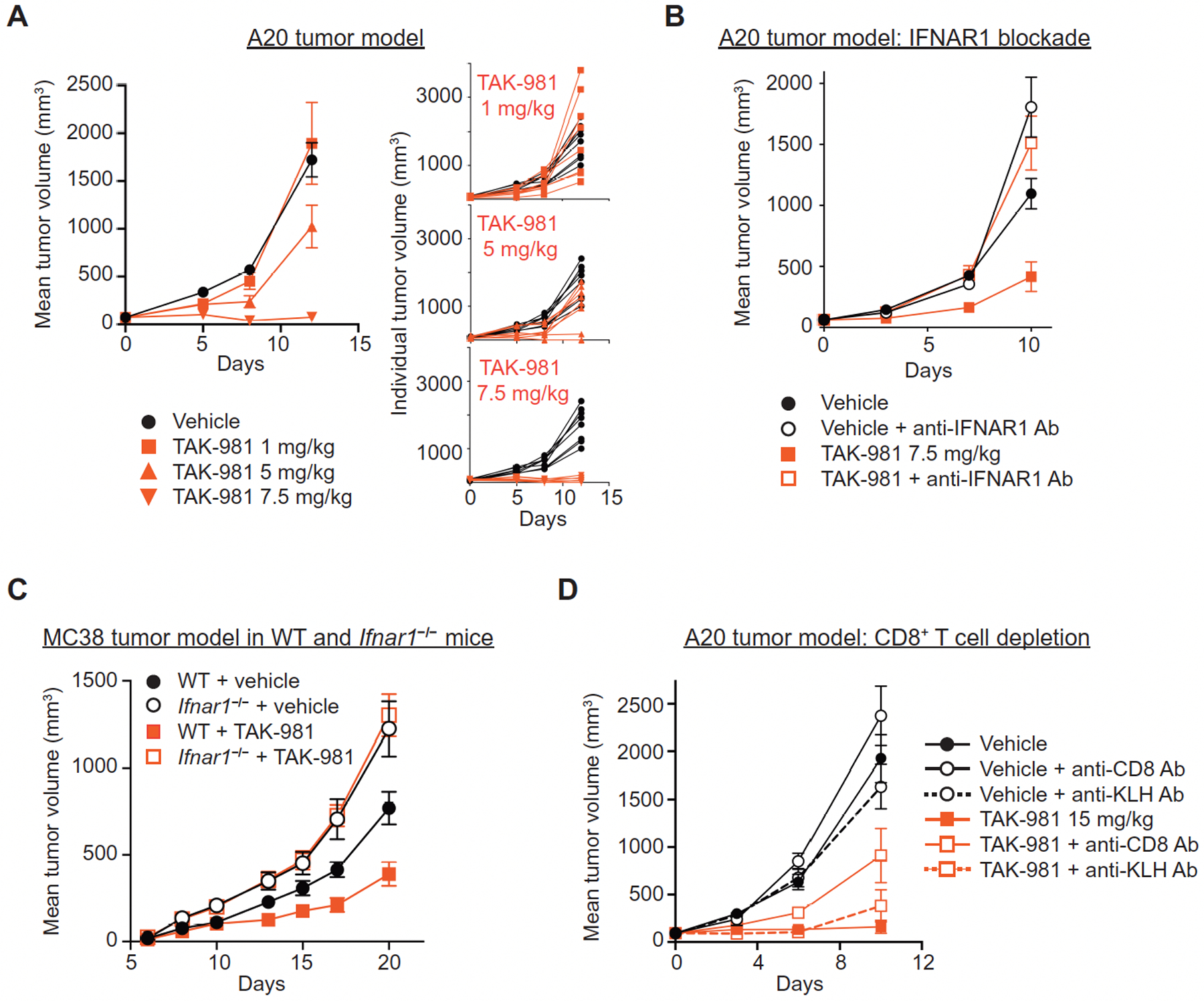
(A) Dose responsive tumor growth inhibition in the A20 tumor model. BALB/c mice bearing subcutaneous A20 tumors were treated intravenously (i.v.) with vehicle or the indicated concentrations of TAK-981 twice weekly for 2 weeks (days 0, 3, 7, 10). Data is shown as both mean tumor volume +/− SEM and individual tumor growth curves, n = 8 mice/treatment arm. P = 0.06 for tumor growth inhibition following treatment with 5 mg/kg TAK-981, compared to vehicle; P <0.001 for tumor growth inhibition following treatment with 7.5 mg/kg TAK-981, compared to vehicle. P values were determined by Welch’s t test. (B) BALB/c mice bearing subcutaneous flank A20 tumors were treated i.v. with vehicle or 7.5 mg/kg TAK-981 on days 0, 3 and 7, +/− anti-IFNAR1 antibody administered intraperitoneally (i.p.) 24h prior to dosing of vehicle or TAK-981. Data is shown as mean tumor volume +/− SEM, n=8 mice/treatment arm. P = 0.011 for tumor growth inhibition following treatment with TAK-981, compared to vehicle; P = 0.685 for tumor growth inhibition following treatment with vehicle + anti-IFNAR1 antibody, compared to TAK-981 + anti-IFNAR1 antibody. P values were determined by Welch’s t test. (C) MC38 tumor cells subcutaneously implanted into C57BL/6 wild type (WT) and C57BL/6 Ifnar1−/− mice were treated once weekly i.v. with vehicle or 15 mg/kg TAK-981 on days 8 and 15 after tumor implantation. Data is shown as mean tumor volume +/− SEM, n=6 mice/treatment arm for WT mice, n=7 mice/treatment arm for Ifnar1−/− mice. P = 0.025 for tumor growth inhibition following treatment with TAK-981 in WT mice, compared to vehicle; P = 0.299 for tumor growth inhibition following treatment with TAK-981 in Ifnar1−/− mice, compared to vehicle. P values were determined by Welch’s t test. (D) BALB/c mice bearing subcutaneous A20 tumors were treated i.v. with vehicle or 15 mg/kg TAK-981 on days 0, 3 and 7, +/− either anti-CD8 antibody or isotype control anti-KLH antibody administered i.p on days 0 and 7. Data is shown as mean tumor volume +/− SEM, n = 8 mice/treatment arm. P < 0.001 for tumor growth inhibition following treatment with TAK-981 compared to vehicle; P = 0.005 for tumor growth inhibition following co-administration of anti-CD8 antibody with TAK-981 compared to TAK-981 treatment alone; P = 0.237 following co-administration of control anti-KLH antibody with TAK-981 compared to TAK-981 treatment alone; P = 0.151 and 0.77, respectively, for tumor growth inhibition following co-administration of vehicle with either anti-CD8 or anti-KLH antibodies, compared to vehicle alone. P values were determined by Welch’s t test.
Depletion of CD8+ T cells resulted in a marked loss of TAK-981 efficacy in the A20 tumor model (Fig. 5D). A similar outcome was observed for A20 tumors grown in immune deficient Recombination Activating Gene 2 deficient (Rag2−/−) or Non-Obese Diabetic-SCID (NOD-SCID) mice (fig. S8G), indicating a role for the adaptive immune response in TAK-981 anti-tumor activity. Phenotypic analyses of tumor infiltrating lymphocytes (TILs) were carried out to explore the impact of TAK-981 treatment on the tumor environment. Anti-tumor responses to TAK-981 were typically variable in A20-tumor-bearing mice, therefore TAK-981-treated mice were segregated into groups of large and small tumors prior to harvest, as a means of enriching for responders and non-responders (fig. S9A). Flow analysis revealed that A20 tumors exhibiting substantial tumor growth inhibition in response to TAK-981 showed increases in T and NK cell numbers, and clear evidence of CD8+ and CD4+ T cell, as well as NK cell, activation, relative to vehicle and to large TAK-981-treated tumors (Fig. 6A, fig. S9, B to G). An increase in the number of activated intratumoral T cells by TAK-981 was recapitulated in MC38 tumors growing in wild type, but not in Ifnar1 deficient, mice (Fig. 6B,C, Fig. S9, H and I), supportive of a requirement for activation of the IFN1-IFNAR1 pathway for TAK-981-mediated anti-tumor activity.
Fig. 6. TAK-981 promotes T and NK cell tumor infiltration and activation.
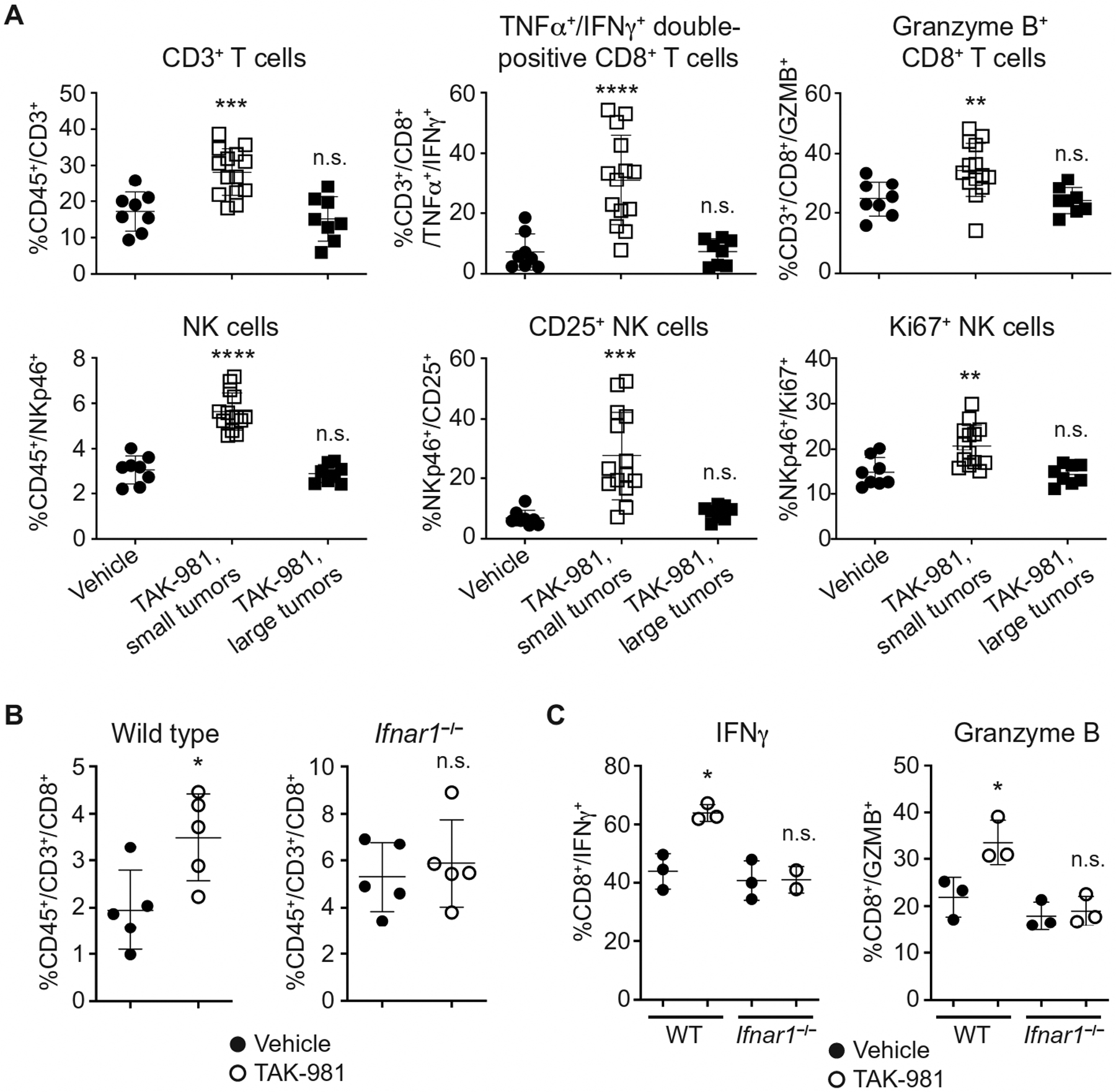
(A) Flow cytometric analysis of subcutaneous A20 tumors from BALB/c mice treated i.v. with vehicle (n = 8) or 7.5 mg/kg TAK-981 on days 0, 3 and 7. Tumors were harvested on day 9 and TAK-981-treated tumors were divided into small (TAK-981 responsive, n = 14) and large (non-responsive, n = 8) tumors, as described in fig. S9A, for flow analysis. **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. not significant, compared with vehicle control, by Welch’s t test. (B) IFN1-dependent CD8+ T cell infiltration into TAK-981-treated MC38 tumors. Flow cytometric analysis of MC38 tumor cells subcutaneously implanted into C57BL/6 WT and Ifnar1−/− mice. Mice were treated i.v. once weekly with vehicle or 15 mg/kg TAK-981 on days 8 and 15 after tumor implantation, n = 5 mice/treatment arm. Tumors were harvested on day 21, and CD8+ cell percentage in tumor tissues was measured by flow analysis. *P < 0.05, n.s. not significant, compared with vehicle control, by Welch’s t test. (C) IFN1-dependent CD8+ T cell activation in TAK-981-treated MC38 tumors. Flow cytometric analysis of MC38 tumor cells subcutaneously implanted into C57BL/6 WT and Ifnar1−/− mice. Mice were treated i.v. with vehicle or 15 mg/kg TAK-981 on days 10 and 13 after tumor implantation. Tumors were harvested on day 14, and IFNγ and Granzyme B were measured in tumor-derived CD45+CD3+CD8+ cells by flow analysis, n = 3 mice/treatment arm except for IFNγ in TAK-981 treated Ifnar1−/− mice, where n = 2. *P < 0.05, n.s. not significant, compared with vehicle control, by Welch’s t test.
In parallel studies, intratumoral injection of TAK-981 into A20 tumors using the CIVO platform resulted in a time-dependent accumulation of granzyme B+ CD8+ T cells around the injection site (fig. S9J). A localized cell death response was observed proximal to TAK-981 injection sites by 72h, suggestive of cytotoxic T cell-mediated tumor cell death.
TAK-981 induces stable antitumor immune responses and can overcome resistance to checkpoint inhibition
Intratumoral delivery of TAK-981 in vivo using the CIVO platform elicited tumor growth inhibition not only in injected tumors but also in distant uninjected tumors in mice implanted with A20 tumor cells on both flanks. Mice showing the most substantial inhibition of tumor growth in the injected tumors exhibited a comparable suppression of growth of uninjected contralateral tumors. In contrast, growth of uninjected contralateral tumors was not affected in mice with unresponsive injected tumors (fig. S10).
A20 tumor-bearing mice which showed CRs to systemically administered TAK-981 were further evaluated. Mice with CRs were challenged with A20 tumor cells implanted in the opposite flank from the original tumor implantation, and age-matched untreated control mice received identical inoculations. Although robust tumor growth was observed in control mice, no growth was observed in the previously treated mice (Fig. 7A). The specificity of this response was assessed by subsequently challenging the tumor-free survivors with CT26 tumors, whose rapid growth was comparable to that observed in untreated control mice (Fig. 7A), indicative of a tumor-specific immune response in TAK-981 treated mice. In a replicate study, A20 tumor-bearing mice which had achieved CRs and demonstrated resistance to re-challenge with A20 tumors subsequently demonstrated robust A20 tumor growth in response to a secondary re-challenge following depletion of CD8 T cells (Fig. 7B). These data suggest that animals cured by TAK-981 developed durable and tumor-specific T cell-dependent antitumor immunity.
Fig. 7. TAK-981 promotes a tumor-specific protective response and potentiates response to immune checkpoint blockade.
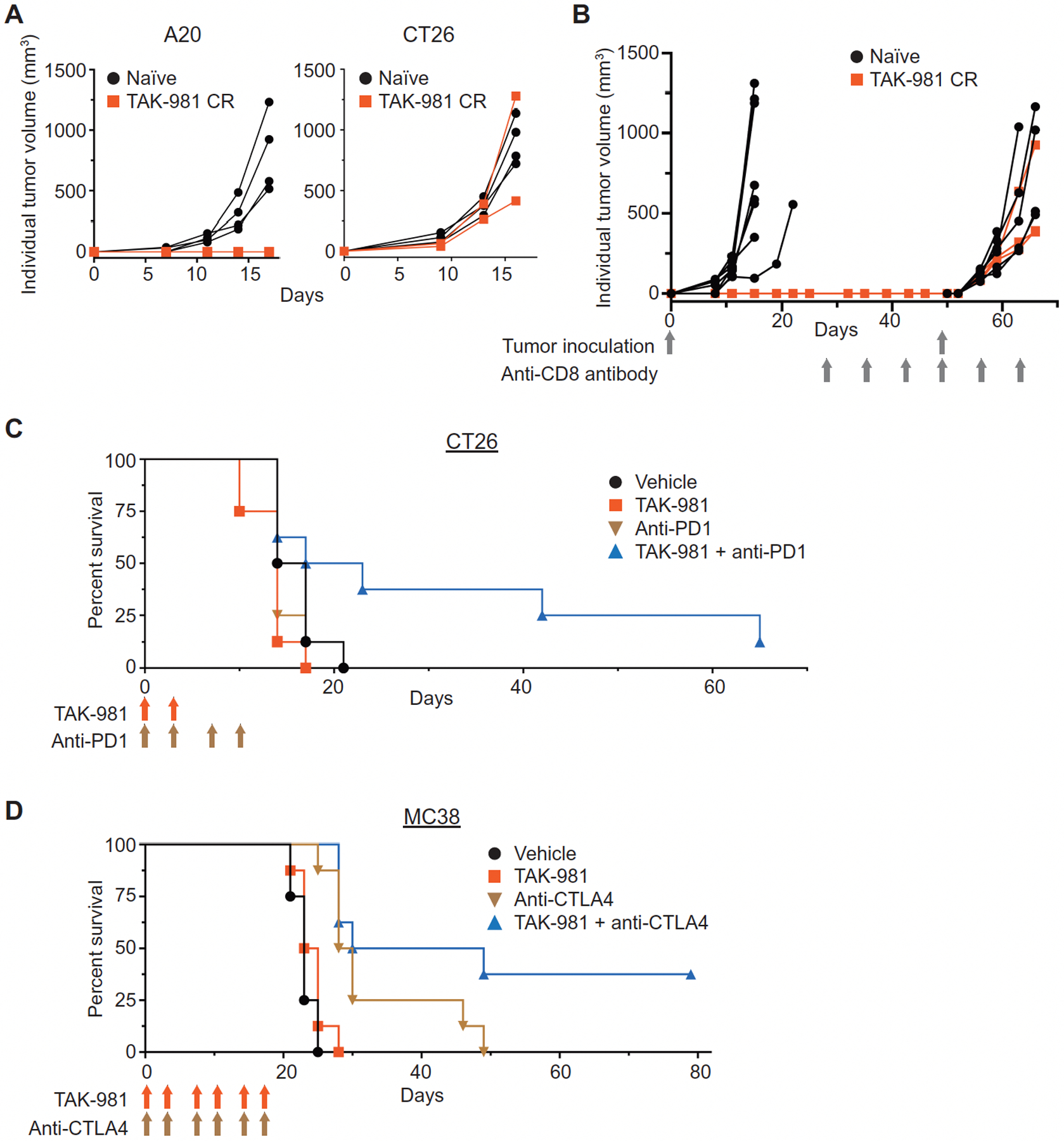
(A) Three A20 tumor-bearing mice that had achieved CRs in response to TAK-981 treatment, along with 4 age-matched naïve BALB/c mice, were inoculated subcutaneously with A20 cells on day 0. One of the re-challenged mice met a humane end-point (ascites) due to unknown causes and was removed from the study 11 days following re-challenge. The remaining two mice did not show any A20 tumor growth, and were subsequently subcutaneously inoculated with CT26 tumors, along with four age matched naïve BALB/c mice. (B) Three A20 tumor-bearing mice that had achieved CRs in response to TAK-981 treatment, from a different study than described in Fig. 7A, and 8 naïve age-matched BALB/c mice, were inoculated subcutaneously with A20 cells and monitored for tumor growth. No tumor growth was observed in mice that had previously achieved CRs, and these 3 mice, along with 6 naïve age matched BALB/c mice were treated i.p. with anti-CD8 antibody weekly, for 6 weeks, beginning on day 28, and inoculated subcutaneously with A20 cells on day 49, and monitored for tumor growth. (C) Survival curves for the combination of TAK-981 with anti-PD1 antibody. BALB/c mice inoculated subcutaneously with CT26 tumor cells were treated i.v. with vehicle on days 0, 3, 7 and 10, or with 7.5 mg/kg TAK-981 on day 0 and 3 (blue arrows), or with 10 mg/kg anti-mouse PD1 antibody on days 0, 3, 7 and 10 (green arrows), or with a combination of TAK-981 (day 0 and 3) and anti-PD1 antibody (days 0, 3, 7 and 10), n = 8 mice/treatment arm. P < 0.001 for survival of mice treated with both TAK-981 and anti-PD1 antibody, compared to treatment with either TAK-981 or anti-PD1 antibody as a single agent. P values were calculated by Weibull regression analysis. (D) Survival curves for the combination of TAK-981 with anti-CTLA4 antibody. C57BL/6 mice inoculated subcutaneously with MC38 tumor cells were treated with vehicle, TAK-981 (blue arrows), anti-mouse CTLA4 antibody (green arrows) or TAK-981 combined with anti-mouse CTLA4 on days 0, 3, 7, 10, 14 and 17, n = 8 mice/treatment arm. P < 0.001 for survival of mice treated with both TAK-981 and anti-CTLA4 antibody, compared to treatment with TAK-981 alone; P = 0.0019 for survival of mice treated with both TAK-981 and anti-CTLA4 antibody, compared to treatment with anti-CTLA4 antibody alone. P values were calculated by Weibull regression analysis.
These results encouraged us to explore combination of TAK-981 with immune checkpoint blockade. In the CT26 tumor model, which did not show single-agent responsiveness to either TAK-981 or an anti-mouse PD1 antibody, combination treatment resulted in a notable survival benefit (Fig. 7C). Similar observations were noted in the MC38 tumor model in combination with anti-mouse CTLA4 (Fig. 7D). Analysis of TILs in the CT26 model was carried out as described for TAK-981-treated A20 tumors. To enrich for tumors responding to the combination of TAK-981 and anti-PD1, combination-treated tumors were segregated into large and small tumor groups at the time of tumor harvest (fig. S11A). Flow analysis revealed that tumors with marked growth inhibition in response to treatment with the combination of TAK-981 and anti-PD1 showed enhanced activation of CD8+ T cells and NK cells, compared to either drug combination-treated tumors not showing growth inhibition, or tumors treated with either drug as single agent, or vehicle-treated tumors (fig. S11,B to E).
Discussion
We have described the pharmacological activity of TAK-981, a small molecule inhibitor of SUMOylation which forms an irreversible adduct with SUMO when bound to the E1 enzyme SAE, preventing transfer of SUMO to the E2 conjugating enzyme, UBC9, and to substrate proteins. This inhibitory mechanism is similar to that previously described by us for inhibitors of the E1 enzymes UAE (41) and NAE (42). Despite this similarity, TAK-981 is highly selective for inhibition of SAE relative to other E1 enzymes, and kinases. The salient response to TAK-981 treatment of syngeneic mouse tumor models in vivo, or isolated immune cells ex vivo, was induction of IFN1 expression and stimulation of IFN1 signaling, recapitulating previously published observations made using targeted knockout of UBC9 in immune cells (12, 16). The convergence of phenotypic responses arrived at by both pharmacological and genetic means makes a compelling argument in support of a key role for SUMOylation as a repressor of IFN1 responses. In this report we leveraged these observations to demonstrate that pharmacological inhibition of SUMOylation promoted activation of innate and adaptive immune responses in mice which were shown to drive single agent antitumor activity, and beneficially combine with ICIs. Induction of IFN1 signaling by TAK-981 was absent or minimal in the tumor cell lines examined in this paper, with the exception of OCI-Ly-10 (fig. S4A), consistent with previous observations of prevalent IFN1 pathway mutations in cancer cells (43).
We have recently reported on the in vitro activity of the tool compound SAE inhibitor, ML-792 (24). TAK-981 was derived from a medicinal chemistry program designed to generate a clinical candidate by optimizing the pharmaceutical properties of the chemical series which produced ML-792. In particular, TAK-981 was designed to exhibit enhanced retention of the TAK-981-SUMO adduct in the SAE active site relative to the ML-792-SUMO adduct, thus extending the limited duration of in vivo pathway inhibition achievable with ML-792 (22). TAK-981 demonstrated potent and durable inhibition of SUMO2 and SUMO3 conjugate formation in vivo for approximately 15–20h following a single 10 mg/kg dose. Interestingly, the TAK-981-SUMO adduct could be detected for a longer period, several days after a single dose. We hypothesize that the recovery of SUMOylation, beginning approximately 16–24h after TAK-981 treatment, reflects de novo synthesis of SAE which can freely initiate the SUMOylation cascade at a time when TAK-981 has been cleared from circulation (TAK-981 plasma half-life in the mouse is approximately 2.5h).
In our earlier work with ML-792, we studied the effect of prolonged drug treatment on solid tumor cancer cell line growth in culture, demonstrating that typically ≥48h of sustained drug exposure was required to achieve inhibition of tumor cell proliferation (24). In contrast, in the in vivo studies reported here, tumor cells experienced only transient drug exposure once or twice weekly, for periods of only a few hours prior to drug clearance. Under these conditions, we anticipate that anti-proliferative responses to periodic and temporally limited loss of SUMOylation are likely contributing minimally to antitumor activity. Consistent with these expectations, cell cycle analyses of the cell lines used for tumor models in these studies have shown no evidence of cell cycle block following brief (4h) periods of drug exposure, and in vivo studies using these models have revealed that tumor growth inhibition by TAK-981 was largely dependent on induction of an immune response initiated by activation of IFN1 signaling.
We have shown that TAK-981 augments responses of tumors to immune checkpoint blockade with both anti-PD1 and anti-CTLA4 antibodies, respectively. Based on the data reported here, this activity could be driven by both the ability of TAK-981 to promote a de novo adaptive immune response via IFN1-dependent DC activation and T cell priming, or by direct stimulation of CD8+ T cell activity. The IFN1-dependent mechanism of action of TAK-981 supports the exploration of additional mechanism-based combinations with immunomodulatory agents. For example, combination of TAK-981 with PRR agonists such as CDNs and TLR agonists is worthy of exploration, because SUMOylation inhibition has been shown to enhance responses to PRR pathway agonists (Decque et al (12) and this report).
Limitations of this study include elucidation of the mechanism by which TAK-981 activates IFN1 responses. Studies based on genetic inactivation of the SUMO pathway have identified relief of SUMO-dependent transcriptional repression of the IFNβ gene as one important mechanism (12). In addition, we have also observed increased cell surface expression of the IFN1 receptor IFNAR1 in the tumor microenvironment in response to treatment with TAK-981. The mechanistic underpinnings, and cell type specificities, of these responses to pharmacological inhibition of the SUMO pathway remain to be determined. In addition, although we have established a requirement for CD8+ T cells for TAK-981-dependent tumor growth inhibition, the potential contribution of other immune cell types, such as natural killer cells and macrophages, or effects on Treg number or function (44), represent interesting areas for future study. Furthermore, a variety of tumor intrinsic roles for SUMOylation in promoting tumorigenesis have been previously documented (45). The contribution of non-immune mediated antitumor mechanisms to the activity of TAK-981 cannot be excluded, and also provide an area for future investigation.
As documented by the data presented here, the small molecule SUMOylation inhibitor TAK-981 represents a means of stimulating antitumor immune responses through the reactivation of IFN1 signaling. Unlike the administration of TLR agonists or CDNs, which induce IFN1 expression downstream of PRR pathway activation, inhibition of SUMOylation has been reported to relieve transcriptional repression of IFNβ expression (12) resulting in enhanced basal expression of IFN1s and sensitization to induction. In addition, TAK-981 has been shown to upregulate IFNAR1, partial loss of which, in the immune cells within the tumor microenvironment, has been implicated in attenuating anti-tumor immune responses and promoting tumor growth (30). The activity of this first in class SAE inhibitor is currently being assessed in phase I clinical trials in adult patients with metastatic solid tumors, multiple myeloma and lymphomas (NCT03648372, NCT04381650, NCT04776018 and NCT04074330).
Materials and Methods
Study Design
The objective of this study was to assess the outcome of pharmacological inhibition of SUMOylation on induction of IFN1 signaling and promotion of IFN1-dependent antitumor immune responses, using the SAE inhibitor TAK-981. The potency and selectivity of TAK-981 was assessed biochemically in an E1–E2 transthiolation assay, and in cellular assays using Western blot hybridization and SUMO 2 and SUMO 3 immunofluorescence. Selectivity against kinases was determined in a full kinase panel. Induction of IFN1 signaling in immune cells by TAK-981 was investigated by ex vivo treatment of splenocytes isolated from C57BL/6 wild type and Ifnar1−/− mice, as well as in seven cultured tumor cell lines. Assessment of TAK-981-dependent immune cell activation was carried out in dendritic cells isolated from mouse bone marrow and human PBMCs, and in CD8 T cells derived from C57BL/6 wild type and Ifnar1−/− mice, and from OT-1 T cell receptor transgenic mice. Global transcriptional response to treatment with TAK-981 was assessed in plasma, spleen and A20 mouse B- cell lymphoma tumors or B16F10 mouse melanoma tumors in BALB/c or C57BL/6 mice, respectively.
To investigate TAK-981 dependent DC and T cell activation in vivo, non-tumor bearing C57BL/6 mice were vaccinated by subcutaneous injection with ovalbumin (OVA) in the presence or absence of TAK-981, and assessed for DC activation markers, SIINFEKL-presenting DCs, and Kb-SIINFEKL tetramer positive CD8+ T cells. To assess whether TAK-981 can promote a protective anti-tumor immune response, C57BL6 mice were vaccinated with OVA combined with either vehicle, TAK-981 or poly (I:C), over a period of 2 weeks prior to challenge with B16F10-OVA tumors. The in vivo anti-tumor activity of TAK-981 was assessed alone, in response to IFNAR1 blockade and CD8+ T cell depletion, and in combination with the immune checkpoint inhibitors anti-mouse PD1 and anti-mouse CTLA4. Mice were randomized to treatment groups after tumor establishment and sample sizes were determined based on variation in tumor growth for the models used (A20 mouse B cell lymphoma and CT26 mouse colon adenocarcinoma in BALB/c hosts, and MC38 mouse colon adenocarcinoma in C57BL/6 hosts). The number of mice in each treatment group is specified in the legends to the figures reporting each tumor growth study. For analysis of tumor immune cell modulation following treatment, A20 or CT26 tumors treated with single agent TAK-981 or TAK-981 combined with anti-PD1 were segregated into large and small tumors to enrich for responsive and non-responsive tumor populations prior to flow cytometry analysis. The number of experimental replicates is indicated in each figure legend. Ex vivo experiments did not have rigorous sample size planning. Many sample sizes were small as we anticipated that large and biologically meaningful effects would be identified even with small sample sizes. No data, including outlier values, were excluded from analyses, except for the correlation analyses shown in fig. S4D, and the volcano plots shown in fig. S4K, as documented in the legends of both figures.
Statistical analysis
For experiments where unpaired samples were compared between two conditions, two-sided Welch’s t-test was used to calculate the statistical significance between the conditions so that no variance equality nor directionality assumption was imposed. Although t-tests were originally based on normal distribution of samples, they are robust to nonnormality and can maintain nominal type-I error rates unless very serious outliers exist (46). Similarly, when more than two conditions were compared against each other, we used Welch’s ANOVA. In the experiments that investigated the sensitivities of OT-I T cells to OVA peptides, two-way ANOVA was used to test the interaction effect between peptide concentrations and TAK-981 treatment, or whether the sensitivity across peptide concentrations is different with and without TAK-981 treatment. Geisser-Greenhouse correction was used to adjust for lack of sphericity in the ANOVA analysis. For those in-vivo experiments where treatment groups have different follow-up durations, Welch’s t-test was not used because it would be unfair to compare tumor volumes at different follow-up times. Instead, the time-to-progression (TTP) was used. TTP is defined as the time it takes for the tumor volume to reach 1000 mm^3 as determined by exponential interpolation of available measurements. If the tumor volume did not reach 1000 mm^3 by the end of the experiment, the TTP was censored at the last tumor measuring day. Given the small sample size of the in-vivo experiments, we used Weibull regression to perform survival analysis on TTP. Hazard ratios and their corresponding P-values were calculated.
Supplementary Material
Acknowledgements:
We thank Hirotaki Mizutani, Tiger Hu, Matthew Duffey, Jianping Guo, Charles McIntyre, Rachel Gershman and Scott Freeze for synthesis and scale-up of TAK-981. We thank Chris Roy and Min Young Lee for help with RNA-Seq data analysis and Alan Copenhaver for help with flow data analyses. We are appreciative of the contributions by Marc Grenley, Joyoti Dey, Emily Beirne, Connor Burns, Sally Ditzler, and Angela Merrel to the CIVO data.
Funding:
We are thankful for support by the NIH/NCI R01 grants CA247803 to S.Y.F., CA240814 to S.Y.F., and CA092900 to S.Y.F.
Competing Interests:
E.S.L., S.G., K.S., M.K., K.X., X.H., J.M.G., H.I., J.M.G., Y.F., E.K., J.R., V.S., C.L., J.M., X.Y., D.E., S.L., K.G., G.S., S.M.P., M.A.M. and D.H. were employees of and stockholders in Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, while engaged in the research project. B.A.H. and R.A.K. are employees of Presage Biosciences, which received research funding from Takeda Pharmaceuticals.
The following patents are held by Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited: U.S. Pat. No. 9,683,003 B2 Heteroaryl compounds useful as inhibitors of SUMO activating enzyme, and 10,335,410 B2 Heteroaryl compounds useful as inhibitors of SUMO activating enzyme; Dylan England and Steve Langston are listed as co-inventors on these patents. Millennium Pharmaceuticals, Inc. has filed U.S. Pat. App. No. 17/258,845 Administration of SUMO-activating enzyme inhibitor, and PCT App. Pub. No. WO 2020/176,772 Administration of SUMO-activating enzyme inhibitor and checkpoint inhibitor; Sai M. Pulukuri Is listed as an inventor on these applications. Millennium Pharmaceuticals, Inc. has also filed PCT App. Pub. No. WO 2020/176,643 Compounds useful as adjuvants; Mithun Khattar is listed as an inventor on this application.
Footnotes
Data and Materials Availability:
All data associated with the study are present in the paper or supplementary materials. TAK-981 is available to academic researchers upon completion of a materials transfer agreement with Takeda Pharmaceutical Company Limited. RNA-Seq data are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE173118.
References
- 1.Hargadon KM, Johnson CE, Williams CJ, Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol 62, 29–39 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Galon J, Bruni D, Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Lee C-K, Rao DT, Gertner R, Gimeno R, Frey AB, Levy DE, Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol 165, 3571–3577 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A, Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrack P, Kappler J, Mitchell T, Type I interferons keep activated T cells alive. J. Exp. Med 189, 521–530 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF, Cutting Edge: Type I IFNs provide a third signal to CD8 T Cells to stimulate clonal expansion and differentiation. J. Immunol 174, 4465–4469 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD, Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208, 1989–2003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF, Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med 208, 2005–2016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre AL, Gajewsky TF,STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hannoun Z, Maarifi G, Chelbi-Alix MK, The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 29, 3–16 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Geiss-Friedlander R, Melchior F, Concepts in sumoylation: a decade on. Nat. Rev. Mol. cell Biol 8, 947 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Decque A, Joffre O, Magalhaes JG, Cossec JC, Blecher-Gonen R, Lapaquette P, Silvin A, Manel N, Joubert PE, Seeler JS, Albert ML, Amit I, Amigorena S, Dejean A, Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat. Immunol 17, 140–149 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ouyang J, Gill G, SUMO engages multiple corepressors to regulate chromatin structure and transcription. Epigenetics 4, 440–444 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Cubeñas-Potts C, Matunis MJ, SUMO: a multifaceted modifier of chromatin structure and function. Dev. Cell 24, 1–12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cossec JC, Theurillat I, Chica C, Búa Aguín S, Gaume X, Andrieux A, Iturbide A, Jouvion G, Li H, Bossis G, Seeler JS, Torres-Padilla ME, Dejean A, SUMO safeguards somatic and pluripotent cell identities by enforcing distinct chromatin states. Cell Stem Cell 23, 742–757.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Crowl JT, Stetson DB, SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proc. Natl. Acad. Sci 115, 6798–6803 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sleijfer S, Bannink M, Van Gool AR, Kruit WHJ, Stoter G, Side effects of interferon-α therapy. Pharm. World Sci 27, 423 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Parker BS, Rautela J, Hertzog PJ, Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 16, 131–144 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Fuchs SY, Hope and fear for interferon: The receptor-centric outlook on the future of interferon therapy. J. Interf. Cytokine Res 33, 211–225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith PL, Lombardi G, Foster GR, Type I interferons and the innate immune response - More than just antiviral cytokines. Mol. Immunol 42, 869–877 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Corrales L, Matson V, Flood B, Spranger S, Gajewski TF, Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 27, 96–108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langston SP, Grossman S, England D, Afroze R, Bence N, Bowman D, Bump N, Chau R, Chuang B-C, Claiborne C, Cohen L, Connolly K, Duffey M, Durvasula N, Freeze S, Gallery M, Galvin K, Gaulin J, Gershman R, Greenspan P, Grieves J, Guo J, Gulavita N, Hailu S, He X, Hoar K, Hu Y, Hu Z, Ito M, Kim M-S, Lane SW, Lok D, Lublinsky A, Mallender W, McIntyre C, Minissale J, Mizutani H, Mizutani M, Molchinova N, Ono K, Patil A, Qian M, Riceberg J, Shindi V, Sintchak MD, Song K, Soucy T, Wang Y, Xu H, Yang X, Zawadzka A, Zhang J, Pulukuri SM, Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J. Med. Chem 64, 2501–2520 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, Wherry EJ, Zhang NR, Ishwaran H, Hellmann MD, Wolchok JD, Kambayashi T, Minn AJ, Opposing functions of nterferon coordinate adaptive and innate immune responses to Ccncer immune checkpoint blockade. Cell 178, 933–948.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He X, Riceberg J, Soucy T, Koenig E, Minissale J, Gallery M, Bernard H, Yang X, Liao H, Rabino C, Shah P, Xega K, Yan Z-H, Sintchak M, Bradley J, Xu H, Duffey M, England D, Mizutani H, Hu Z, Guo J, Chau R, Dick LR, Brownell JE, Newcomb J, Langston S, Lightcap ES, Bence N, Pulukuri SM, Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol 13, 1164 (2017). [DOI] [PubMed] [Google Scholar]
- 25.He X, Riceberg J, Pulukuri SM, Grossman S, Shinde V, Shah P, Brownell JE, Dick L, Newcomb J, Bence N, Characterization of the loss of SUMO pathway function on cancer cells and tumor proliferation. PLoS One 10, 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klinghoffer RA, Bahrami SB, Hatton BA, Frazier JP, Moreno-Gonzalez A, Strand AD, Kerwin WS, Casalini JR, Thirstrup DJ, You S, Morris M, Watts KL, Veiseh M, Grenley MO, Tretyak I, Dey J, Carleton M, Beirne E, Pedro KD, Ditzler SH, Girard EJ, Deckwerth TL, Bertout JA, Meleo KA, Filvaroff EH, Chopra R, Press OW, Olson JM, A technology platform to assess multiple cancer agents simultaneously within a patient’s tumor. Sci. Transl. Med 7, 284ra58–284ra58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharya S, HuangFu W-C, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, Fuchs SY, Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses,. Oncogene 32, 4214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.HuangFu W-C, Qian J, Liu C, Liu J, Lokshin AE, Baker DP, Rui H, Fuchs SY, Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 31, 161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortiz A, Gui J, Zahedi F, Yu P, Cho C, Bhattacharya S, Carbone CJ, Yu Q, V Katlinski K, V Katlinskaya Y, Handa S, Haas V, Volk SW, Brice AK, Wals K, Matheson NJ, Antrobus R, Ludwig S, Whitside TL, Sander C, Tarhini AA, Kirkwood JM, Lehner PJ, Guo W, Rui H, Minn AJ, Koumenis C, Diehl JA, Fuchs SY, An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell 35, 33–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, Puré E, Chatterji P, Rustgi AK, Diehl JA, Koumenis C, Rui H, Fuchs SY, Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31, 194–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho C, Mukherjee R, Peck AR, Sun Y, McBrearty N, Katlinski KV, Gui J, Govindaraju PK, Puré E, Rui H, Fuchs SY, Cancer-associated fibroblasts downregulate type I interferon receptor to stimulate intratumoral stromagenesis. Oncogene 39, 6129–6137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U, Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108, 3253–3261 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Gresser I, Guy-Grand D, Maury C, Maunoury MT, Interferon induces peripheral lymphadenopathy in mice. J. Immunol 127, 1569–1575 (1981). [PubMed] [Google Scholar]
- 34.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S, CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol 176, 4525–4529 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Hervas-Stubbs S, Riezu-Boj JI, Gonzalez I, Mancheño U, Dubrot J, Azpilicueta A, Gabari I, Palazon A, Aranguren A, Ruiz J, Prieto J, Larrea E, Melero I, Effects of IFN-α as a signal-3 cytokine on human naïve and antigen-experienced CD8+ T cells. Eur. J. Immunol 40, 3389–3402 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, VonLaer D, Kalinke U, Vivier E, Jonjic S, Oxenius A, Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K, Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med 202, 637–650 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, Colonna M, Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11, 631–642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, Deenen R, Köhrer K, Rahbar R, Diefenbach A, Gibbert K, Löhning M, Höcker L, Waibler Z, Häussinger D, Mak TW, Ohashi PS, Lang KS, Lang PA, Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Hiroishi K, Tüting T, Lotze MT, IFN-α-expressing tumor cells enhance generation and promote survival of tumor-specific CTLs. J. Immunol 164, 567–572 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, Huck J, Shi J, Gavin J, Brownell J, Yang Y, Stringer B, Griffin R, Bruzzese F, Soucy T, Duffy J, Rabino C, Riceberg J, Hoar K, Lublinsky A, Menon S, Sintchak M, Bump N, Pulukuri SM, Langston S, Tirrell S, Kuranda M, Veiby P, Newcomb J, Li P, Wu JT, Powe J, Dick LR, Greenspan P, Galvin K, Manfredi M, Claiborne C, Amidon BS, Bence NF, A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med 24, 186 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Naryanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP, An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC, Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med 6, 821–825 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Ding X, Wang A, Ma X, Demarque M, Jin W, Xin H, Dejean A, Dong C, Protein SUMOylation Is required for regulatory T cell expansion and function. Cell Rep. 16, 1055–1066 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Seeler JS, Dejean A, SUMO and the robustness of cancer. Nat. Rev. Cancer 17, 184–197 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Sawilowsky SS, Hillman SB, Power of the independent samples t test under a prevalent psychometric measure distribution. J. Consult. Clin. Psychol (1992), doi: 10.1037/0022-006x.60.2.240. [DOI] [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hänzelmann S, Castelo R, Guinney J, GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14 (2013), doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Merico D, Isserlin R, Stueker O, Emili A, Bader GD, Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS One 5 (2010), doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zehn D, Lee SY, Bevan MJ, Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hather G, Liu R, Bandi S, Mettetal J, Manfredi M, Shyu W-C, Donelan J, Chakravarty A, Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 13s4, CIN.S13974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with the study are present in the paper or supplementary materials. TAK-981 is available to academic researchers upon completion of a materials transfer agreement with Takeda Pharmaceutical Company Limited. RNA-Seq data are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE173118.