

Abstract
The Kelch-like ECH-associated protein 1 (KEAP1) - nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway senses reactive oxygen species and regulates cellular oxidative stress. Inhibiting KEAP1 to activate the NRF2 antioxidant response has been proposed as a promising strategy to treat chronic diseases caused by oxidative stress. Here, we developed a proteolysis targeting chimera (PROTAC) that depletes KEAP1 from cells through the ubiquitin-proteasome pathway. A previously developed KEAP1 inhibitor and thalidomide were incorporated in the heterobifunctional design of the PROTAC as ligands for KEAP1 and CRBN recruitment, respectively. Optimization of the chemical composition and linker length resulted in PROTAC 14 which exhibited potent KEAP1 degradation with low nanomolar DC50 in HEK293T (11 nM) and BEAS-2B (<1 nM) cell lines. Furthermore, PROTAC 14 increased the expression of NRF2 regulated antioxidant proteins and prevented cell death induced by reactive oxygen species. Together, these results established a blueprint for further development of KEAP1-targeted heterobifunctional degraders and will facilitate the study of the biological consequences of KEAP1 removal from cells. This approach represents an alternative therapeutic strategy to existing treatments for diseases caused by oxidative stress.
Keywords: PROTAC, KEAP1-NRF2 pathway, Antioxidant, ROS, Oxidative stress
Graphical abstract

Highlights
-
•
A potent and selective PROTAC degrader of KEAP1 was developed.
-
•
PROTAC 14 induced upregulation of the NRF2 mediated antioxidant response.
-
•
PROTAC 14 showed potent activity in the lung epithelial BEAS-2B cell line.
-
•
PROTAC 14 rescued BEAS-2B cells from ROS induced death.
1. Introduction
Cellular oxidative stress occurs when cells are exposed to excessive reactive oxygen species (ROS) and as a consequence, the cellular redox balance cannot be maintained [1]. Chronic exposure to oxidative stress leads to diverse pathologic conditions such as respiratory, autoimmune, and neurodegenerative disorders [2]. Cells are equipped with a sophisticated antioxidant system to combat and eliminate harmful ROS and maintain redox homeostasis [3]. The Kelch-like ECH-associated protein 1 (KEAP1) - nuclear factor erythroid 2-related factor 2 (NRF2) pathway is the principal cellular mechanism that regulates redox balance [4].
KEAP1 is a cysteine-rich protein that acts as an oxidant sensor and a negative regulator of NRF2 [4]. KEAP1 consists of three functional domains, namely the broad complex, tramtrack and a bric-à-brac (BTB) domain, the intervening region (IVR), and the Kelch domain [5]. The BTB domain is responsible for KEAP1 dimerization and interacts with Cullin3/Rbx1 to form an E3 ligase [[6], [7], [8]]. The IVR domain bridges the BTB and the Kelch domain in the KEAP1 complex, enabling the dimerized Kelch domains to recognize and capture cytosolic NRF2 [9]. The Kelch domain adopts a classic β-propeller structure and in the KEAP1 homodimeric complex, interacts with the high-affinity ETGE or low-affinity DLG motifs in the Neh2 domain of the NRF2 protein [10].
Under basal conditions, NRF2 is sequestered by a homodimer of KEAP1/Cullin3/Rbx1 E3 ligase, resulting in NRF2 polyubiquitination and proteasomal degradation [11]. Under oxidative stress, multiple cysteines in KEAP1 are modified by ROS, leading to a conformational change in KEAP1 which prevents NRF2 binding and ubiquitination. NRF2 then accumulates in the cytosol and translocates into the nucleus. In the nucleus, NRF2 binds to antioxidant response elements (ARE) with other co-factors to initiate antioxidant gene expression [12]. Therefore, inhibiting KEAP1 to activate NRF2 signaling has been proposed as a potential therapeutic avenue to treat chronic disorders caused by oxidative stress [13].
To this extent, several electrophilic small molecules that covalently modify Cys151 in the KEAP1 BTB domain have been clinically developed but all displayed adverse off-target effects, probably due to the promiscuous potential of these compounds to react with cysteines on other proteins [14]. To overcome the off-target promiscuity, direct and non-covalent inhibitors of KEAP1-NRF2 interaction have been developed that activate NRF2 signaling both in vitro and in vivo, but to date, none of them have proceeded to clinical trials [14].
Instead of inhibiting the active site of the target protein, removal of the whole protein from cells is an alternative strategy for therapeutic development [15]. In the past two decades, targeted protein degradation using proteolysis-targeting chimeras (PROTAC) has received great interest from both academic researchers and pharmaceutical industries [16]. PROTACs eliminate their target proteins by harnessing the cellular ubiquitin-proteasome system (UPS). A heterobifunctional PROTAC consists of a ligand that binds to the protein of interest (POI) conjugated to an E3 ligase ligand. The E3 ligase ligand recruits an E3 ligase to mediate the ubiquitination of the POI. The E3 ligase complex then dissociates and the ubiquitinylated POI is subsequently degraded by the proteasome [17].
In this study, we designed and characterized a PROTAC to target KEAP1 for proteasomal degradation. The PROTAC exploits a known non-covalent KEAP1 inhibitor (1) which we linked to thalidomide, the ligand for cereblon (CRBN)/Cullin4/Rbx1 E3 ligase. Initially, a series of PROTACs were conceived and synthesized with varying linker lengths to assess their ability to degrade KEAP1. The PROTAC with the optimal linker was then characterized for its ability to degrade KEAP1 in various cell lines and activate an NRF2-mediated antioxidant response in cellular assays.
2. Methods and materials
2.1. Chemistry
Chemistry experimental is included in the supplementary information section.
2.2. Biology
2.2.1. Cell culture conditions
HEK293T, HCA7, A549, and BEAS-2B cell lines were obtained from the ATCC. The HepG2-ARE reporter cell line was purchased from BPSBioscience (BPSBioscience, USA). HEK293T, HCA7, A549 cells were cultured in DMEM medium (Gibco, Invitrogen Corp., USA) with 10% fetal bovine serum (FBS) (Gibco, Invitrogen Corp., USA) and penicillin/streptomycin (Gibco, Invitrogen Corp., USA). BEAS-2B cells were cultured in LHC-9 medium (Gibco, Invitrogen Corp., USA) with penicillin/streptomycin. HepG2-ARE reporter cells were cultured in DMEM medium supplied with 1% non-essential amino acids (Gibco, Invitrogen Corp., USA), 1 mM Na pyruvate (Gibco, Invitrogen Corp., USA), penicillin/streptomycin plus 600 μg/mL of Geneticin (Roche, Switzerland). All cell lines were maintained in a humidified incubator at 37 °C with 10% CO2.
2.2.2. Expression and purification of recombinant human KEAP1-Kelch domain
BL21(DE3) cells were transformed with a pOPINK plasmid encoding the His-GST-KEAP1 Kelch domain (312–624) and grown in 1 L YT cultures to OD600 0.8 in a shaking incubator at 37 °C. The temperature was reduced to 20 °C for 45 min and KEAP1 expression was induced by the addition of 0.5 mM IPTG (Gold Biotechnology, USA). Cultures were harvested by centrifugation after overnight incubation at 20 °C. For purification, cells were resuspended in high-salt binding buffer (50 mM Tris pH 8.0, 500 mM NaCl, 10% sucrose, 10% glycerol) supplemented with leupeptin, lysozyme, DNaseI, MgCl2, and PMSF and lysed by sonication. Lysates were cleared by centrifugation at 40,000 g for 30 min at 4 °C. Protein purification was initiated by passing supernatant over Ni-NTA His-bind resin (Merck Millipore, USA) in gravity flow columns equilibrated with high-salt binding buffer. Ni-NTA His-bind resin was washed with 3 × 10 mL of high-salt binding buffer, and 1 × 6 mL of high-salt binding buffer supplemented with 10 mM imidazole. The protein was then eluted with high-salt binding buffer supplemented with 300 mM imidazole and further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 pg column (Cytiva, USA) in 10 mM HEPES pH 7.9, 100 mM NaCl. Protein samples were concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C.
2.2.3. Homogeneous time resolved fluorescence (HTRF) assay
The HTRF assay was performed in a Greiner white 1536-well plate (Interpath, Australia) with buffer containing 25 mM HEPES, pH 7.4, 100 mM NaCl, 0.005% BSA, 0.1% Tween-20, and 1 mM TECP. In the assay, 0.4 nM GST-KEAP1 Kelch domain, 1.25 nM biotin labeled NRF2 peptide (82QLQLDEETGEFL93) and a serial dilution of test compounds was mixed and incubated at room temperature for 1 h. Thereafter, the HTRF mix (37.5 ng/mL anti-GST terbium (Tb) and 180 ng/mL streptavidin-d2) was added and incubated for 2 h. Signal was measured on a PheraSTAR (BMG LABTECH, Germany) using an HTRF module (excitation: 337 nm; emission 1: 665 nm; emission 2: 620 nm).
2.2.4. Immunoblotting
Sample preparation and immunoblotting were performed as previously reported [18]. Briefly, cells were harvested and lysed followed by electrophoresis using SDS PAGE (Invitrogen and 4–12%). Samples were then transferred to a nitrocellulose membrane (Cytiva, USA) and blocked with 5% skim milk for 1 h at room temperature. Proteins were detected by the following antibodies: anti-KEAP1 (sc-365626), anti-GCLM (sc-55586), and horseradish peroxidase (HRP) conjugated anti-β-actin (sc-47778) purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Anti-CRBN (D8H3S) was purchased from Cell Signaling Technology (Cell Signaling Technology, USA), anti-NQO1 (ab34173) from Abcam (Abcam, USA), and anti–HO–1 (MA1112) was purchased from Invitrogen (Invitrogen, USA). HRP conjugated anti-mouse (NA931) and anti-rabbit (NA934) secondary antibodies were purchased from Amersham (Amersham, USA).
2.2.5. ARE-luciferase activity assay
HepG2-ARE cells were seeded into a 96-well plate at a density of 1 × 104 cells/well and incubated overnight. Cells were then treated with the indicated concentration of compounds for 48 h and then lysed. Luciferase activity was measured using the Luciferase Reporter Assay Kit (Promega, USA) and a luminometer (Promega, USA) as previously described [19].
2.2.6. Quantitative RT-PCR
RNA was extracted from BEAS-2B cells using Qiagen RNeasy Plus Kits (Qiagen, Germany) following the manufacturer's instructions. The RNA was reverse transcribed into cDNA using a SuperScript IV kit (Thermo Fisher, USA). Quantitative RT-PCR was performed using SyberGreen (Bioline, Australia) on the QuantStudio 12 K Flex Real-Time PCR System (Thermo Fisher, USA). Relative RNA levels were normalized to the 18S internal control using the delta-delta-cT statistical method. Primers for RT-PCR are listed in Table S1.
2.2.7. MS proteomics
BEAS-2B cells were treated with DMSO, 0.13 μM of PROTAC 14, or inhibitor 2 for 48 h. Samples were then harvested and lysed with 5% SDS (in 50 mM TEAB) for label-free proteomics. BCA assay was used to confirm protein concentration and 20 μg of protein per sample was processed via micro S-traps (Protifi) as described by the manufacturer (with the exception that chloroacetamide was used for alkylation) and digested with trypsin/Lys-C. Samples were cleaned using SDB pre-packed stage tips (GLSicences) and approximately 100 ng of peptide was injected onto a custom Dionex-PAL nanoflow pump connected to a timsTOF pro (Bruker). Peptides were separated using a 48 min gradient (solvent A, 0.1% formic acid; solvent B, 99.9% acetonitrile/0.1% formic acid) on a C18 analytical column (IonOpticks, Aurora Elite 15 cm × 75 μm ID, 1.7 μm AUR3-15075C18). Data-independent PASEF acquisition was performed (100–1700 m/z scan range, 0.6–1.6 Vs/cm2, and 100 ms ramp and accumulation time) and library-free searching was performed with DIA-NN (v1.8) using the reviewed Homo sapiens uniprot database (UP000005640). MS1 and mass accuracy was set to 15 ppm and the precursor FDR was set at 1%. Protein quantification was performed using the DIA-NN in-built MaxLFQ algorithm (using a TopN = 3). Downstream data processing and analysis was performed using the DEP package in R. Protein abundance in 14 and 2 treatment samples was compared to the DMSO control group. Proteins with more than 1.5-fold change and adjusted P value less than 0.01 were considered significant.
2.2.8. Flow cytometric detection of cell viability
BEAS-2B cells were seeded in a 48-well plate at a density of 1.2 × 104 cells/well and incubated overnight. Cells were then pre-treated with the indicated concentration of compounds for 24 h, followed by 50 μM tert-butyl hydroperoxide (tBHP) treatment, a chemical that can generate ROS rapidly in cells, for another 24 h. Cells were then harvested and stained with 1 μg/mL propidium iodide (PI). Data were acquired using a BD FACSymphony flow cytometer and analyzed using BD FlowJo v10 software. Cells were first gated on FSC-A and SSC-A to exclude debris and then gated on FSC-H and FSC-A to exclude doublets. Viability was assessed as %PI negative cells.
2.2.9. PAMPA membrane permeability assay
This assay was performed by BioDuro-Sundia using the following standard protocol. Working solutions of each compound were prepared from 10 mM stock solution in DMSO diluted to a final concentration of 10 μM in PBS buffer (pH 7.4, 1% DMSO). 1% (w/v) lecithin/dodecane was added to the donor side of the Multi Screen Filter Plate, then 10 μM control or test compound working solution is added. The receiver side of the Multi Screen Filter Plates was filled with PBS buffer containing 1% DMSO. The plates were kept at room temperature for 24 h. Samples were collected from the donor and receiver sides. The donor sides samples were diluted 20-fold with PBS (1% DMSO). All receiver and diluted donor side samples were mixed with ACN/MeOH (1:1, v/v) containing 25 ng/mL terfenadine and 50 ng/mL tolbutamide as internal standards. Samples were vortexed and then centrifuged at 4 °C. An aliquot of the supernatant was transferred to a 0.65 mL tube for LC-MS/MS analysis. The MS detection was performed using a SCIEX API 4000 instrument. Each compound was analyzed by reversed phase HPLC using a Kinetex 2.6μ C18 100 Å column (3.0 mm × 30 mm, Phenomenex). Mobile phase – Solvent A: water with 0.1% formic acid, solvent B: ACN with 0.1% formic acid. Analysis of the compound was determined on the basis of the peak area ratio (compound area to IS area) for the two sides; LogPe = Log (C*-Ln(1-[drug]acceptor/[drug]equilibrium)) where C = VD*VA/((VD + VA)*Area*time) and % Recovery = (Total compound mass in donor and receiver compartments at the end of the incubation/Initial compound mass in the donor compartment) x 100.
3. Results
3.1. Design and binding affinity of KEAP1 PROTACs
Davies et al. disclosed the KEAP1 inhibitor (1, Fig. 1A) bearing a 3-phenylpropanoic acid scaffold. It displayed tight KEAP1 binding and potent induction of the NRF2 antioxidant response both in vitro and in vivo [20]. We selected this compound as the parental KEAP1 recruitment warhead for our PROTAC design. Instead of using the enantiopure molecule 1, we employed the racemic version 2 (R,S-stereochemistry at the 3-position of the propanoic acid) (Fig. 1B), in the design of the KEAP1 targeted PROTAC, due to synthetic tractability. To ensure the feasibility of our approach, we synthesized and evaluated compounds 1 and 2 in a KEAP1-Kelch domain – NRF2 peptide HTRF biochemical assay. As expected, the racemic compound 2 was 2-fold less potent than the enantiopure compound 1 (1, IC50 0.037 μM; 2, IC50 0.077 μM), supporting our decision to utilize the racemic version 2 as the KEAP1 ligand in the design of the PROTAC. The co-crystal structure of compound 1 and the KEAP1-Kelch domain revealed the methoxy group on the 7-position of benzotriazole ring was solvent exposed, suggesting this would be a suitable position for conjugation of the E3 ligase ligand via a linker (Fig. 1C) [20]. The CRBN E3 ligase has been widely exploited in the development of PROTACs targeting other proteins of interest and its ligands are well characterized [16]. We therefore incorporated thalidomide as the CRBN recruitment ligand in our KEAP1-targeted PROTAC (Fig. 1D).
Fig. 1.

Design of a CRBN-based KEAP1 degrader. (A) The structure and activity of the KEAP1 inhibitor 1 reported by Davies et al. [20]. (B) Activity of the synthesized racemic KEAP1 inhibitor 2 incorporated in the KEAP1 degrader. (C) KEAP1 inhibitor 1 co-crystalized with the KEAP1-Kelch domain [20]. The red circle highlights the linker attachment site used in the design of a PROTAC. (D) Design and structure of CRBN based KEAP1 degraders used in this study. IC50 (SD) values shown represent 3 independent experiments using recombinant KEAP1-Kelch domain in a HTRF biochemical assay. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To synthesize the series of PROTACs bearing different linker lengths and types, intermediate 25 and thalidomide functionalized with a fluoro or carboxylic acid in the 4-position 40–41 were employed as key intermediates (Scheme 1). Intermediate 25 was synthesized using a procedure adapted from literature [20,21] starting from phenol 18. Phenol 18 was alkylated and brominated to give 19 followed by methylation of the amino group and reduction of the nitro group to give the amine 20. Diazotization of 20 and a Heck reaction gave the cinnamoyl benzotriazole intermediate 22. Conjugate addition of the boronic acid to 22 gave the aryl derivative 23, followed by a Mitsunobu reaction, and deprotection of the benzyl ether gave intermediate 25. Intermediate 25 was then O-alkylated with various alkyl halide linkers bearing an azide or Cbz-protected amines to afford 26, 28, 30, 32, 34, 36, and 38. The azide or Cbz-amine groups were then subjected to hydrogenation conditions to give the corresponding amines 27, 31, 33, 35, 37, and 39. Finally, the thalidomide component was installed via an amide coupling or a SnAr reaction with thalidomide derivatives 40–41, and then the tert-butyl group was deprotected to afford the PROTACs 3–14.
Scheme 1.

General synthetic pathway to generate PROTACs. Reaction conditions: (a) K2CO3, BnBr, DMF, 0–20 °C; (b) AcOH, NaOAc, Br2, 20 °C; (c) NaH, MeI, DMF, 0–20 °C; (d) Fe, AcOH, 65 °C; (e) NaNO2, 10% aq. H2SO4; (f) t-butyl acrylate, DIPEA, P(o-tolyl)3, Pd(OAc)2, DMF, 95 °C; (g) [RhCl(cod)]2, (3-(hydroxymethyl)-4- methylphenyl)boronic acid, Et3N, dioxane, H2O, 95 °C; (h) PPh3, DIAD, THF, 0–20 °C; (i) Pd/C, H2, MeOH, 20 °C; (j) K2CO3, alkyl halide, DMF, 80 °C; (k) Pd/C, H2, MeOH; (l) 40, HATU, DIPEA, DCM, 20 °C; (m) TFA, DCM, 20 °C; (n) 41, DIPEA, DMF 80 °C.
The binding affinity of synthesized PROTACs to the KEAP1-Kelch domain was evaluated using the HTRF biochemical assay (Table 1). In general, the attachment of a linker conjugated to a CRBN ligand did not significantly impair the binding affinity of the resulting PROTACs to KEAP1 (0.059–0.15 μM). The exceptions were derivatives 4, 5, and 12, which showed a greater than 3-fold reduction in KEAP1 binding affinity compared to inhibitor 2. The inhibitory activity against KEAP1 supported our PROTAC design strategy and warranted further characterization of these analogs in cell-based assays to assess KEAP1 degradation.
Table 1.
Activity of KEAP1 PROTACs against the KEAP1-Kelch domain.
| Cmpd | Linker | X | IC50 (SD) μMa |
|---|---|---|---|
| 2 | – | – | 0.077 (0.003) |
| 3 | 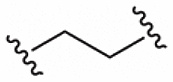 |
NH | 0.150 (<0.001) |
| 4 |  |
NH | 0.267 (0.012) |
| 5 | 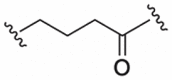 |
O | 0.223 (0.006) |
| 6 | 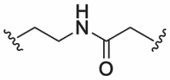 |
O | 0.147 (0.006) |
| 7 | 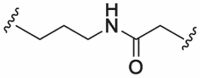 |
O | 0.143 (0.012) |
| 8 | 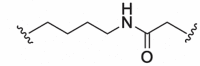 |
O | 0.066 (0.006) |
| 9 | 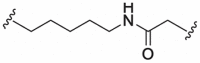 |
O | 0.086 (0.002) |
| 10 | 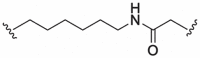 |
O | 0.127 (0.006) |
| 11 | NH | 0.120 (<0.001) | |
| 12 | O | 0.563 (0.029) | |
| 13 | O | 0.059 (0.001) |

IC50 (SD) values represent 3 independent experiments against recombinant KEAP1-Kelch domain in a HTRF biochemical assay.
3.2. KEAP1 degradation by PROTACs in HEK293T cells
The ability of the synthesized PROTACs to degrade KEAP1 was assessed in HEK293T cells. Cells were treated with 1 μM PROTAC for 5 or 24 h, and KEAP1 levels were then analyzed by immunoblotting (Fig. 2A and S1). PROTACs possessing a longer linker length (>7 atoms, 8 to 13) resulted in a robust reduction in KEAP1 protein levels at both time points compared to PROTACs with shorter linker lengths (3 to 7). To study their efficacy in more detail, PROTACs 8 to 13 were evaluated at three lower concentrations (Fig. 2B and S2). PROTACs 8 (7-atom linker) and 10 (9-atom linker) were the most efficient at degrading KEAP1. Considering atom efficiency, we selected PROTAC 8 for further optimization.
Fig. 2.

Evaluation of PROTACs with different linkers on the degradation of KEAP1 in HEK293T cells. HEK293T cells were treated with DMSO vehicle control or 1 μM of compounds (A) at the indicated concentrations (B) for 5 and 24 h. Western blots are representative of 3 independent experiments. Repeats are shown in Figs. S1 and S2.
Linker composition has been reported to affect the chemical stability of thalidomide-based PROTACs in cells [22]. While PROTAC 8 had the strongest effect on KEAP1 degradation at 24 h, it became less effective at longer time points of 48 h and 72 h, suggesting the stability of 8 was compromised in cells over time (Fig. 3 and S3). This data is consistent with previous reports that thalidomide-based heterobifunctional degraders conjugated via oxy-acetamide linkers are susceptible to hydrolysis in cells within 24 h [22]. To overcome instability, we synthesized PROTAC 14, bearing a 7-atom linker and an amino-carbon linkage replacing the oxy-acetamide linkage to thalidomide (Fig. 3A and S3). PROTAC 14 had an IC50 of 46 nM in the KEAP1 HTRF biochemical assay, consistent with the activity of PROTAC 8 (IC50 66 nM). A direct comparison of PROTACs 8 and 14 showed PROTAC 14 degraded KEAP1 more efficiently at 48 and 72 h compared to 8 (Fig. 3B and S3). At 24 h, PROTAC 14 also degraded KEAP1 more efficiently at lower concentrations than 8. These data suggest the cellular stability of PROTAC 14 is enhanced, resulting in improved cellular degradation of KEAP1. Consequently, PROTAC 14 was selected for further characterization.
Fig. 3.

A comparison of the KEAP1 degradation efficiency of compounds 8 and 14. (A) Structures of compounds 8 and 14. (B) HEK293T cells were treated with DMSO vehicle control or 8 and 14 at the indicated concentrations for 5, 24, 48, and 72 h. Immunoblots are representative of 3 independent experiments. Repeats are shown in Fig. S3.
3.3. KEAP1 degradation mechanism of PROTAC 14
The molecular mechanism underlying PROTAC 14-mediated degradation of KEAP1 was verified in a competition experiment. HEK293T cells were pre-treated with the KEAP1 inhibitor 2 or thalidomide (TOM) to compete with PROTAC 14 for KEAP1 or CRBN binding, respectively. As shown in Fig. 4 and S4, KEAP1 degradation induced by 14 was significantly suppressed by the addition of excess 2 or thalidomide. These results demonstrate that simultaneous binding of PROTAC 14 to KEAP1 and CRBN is required for KEAP1 degradation. In addition, pretreatment with the proteasome inhibitor MG132 prevented KEAP1 degradation, confirming degradation mediated by 14 was dependent on the cellular proteasomal machinery.
Fig. 4.

Characterization of the targeted KEAP1 degradation capacity of compound 14. (A) HEK293T cells were pre-treated with DMSO, 10 μM of thalidomide (TOM), 10 μM of proteasome inhibitor MG132, or 1 μM of inhibitor 2 for 1 h. Cells were then treated and incubated with 14 for 5 h. (B) HEK293T cells were treated with DMSO vehicle control or compound 14 at the indicated concentrations for 24 h. Immunoblots are representative of 3 independent repeats. Repeats are shown in Figs. S4 and S5.
It is known that PROTACs can form unproductive binary complexes at excessive concentrations via saturated binding to the target protein or the E3 ligase, a phenomenon termed the hook effect [23]. To this extent, we evaluated PROTAC 14 in a dose titration experiment (0.01–8 μM) in HEK293T cells. A clear hook effect was observed with 14 at concentrations higher than 1 μM and where the maximum degradation (Dmax = 94%) was observed (Fig. 4B and S5). The observed DC50 for KEAP1 in HEK293T cells was 11 nM (Fig. S5).
3.4. Activation of antioxidant response in cell lines induced by 14
We next examined the ability of PROTAC 14 to activate the antioxidant response in different cell lines. In this study, we included compound 15 bearing an N-methylated glutarimide moiety, as a negative control for CRBN recruitment, and tert-butyl ester 16 as a negative control for KEAP1 binding, in addition to the covalent KEAP1 inhibitor sulforaphane (Sul). The KEAP1 inhibitor 2 was included as a benchmark positive control along with its negative control counterpart tert-butyl ester 17 (Fig. S6). HEK293T and HCA7 cell lines treated with PROTAC 14 for 48 h displayed robust degradation of KEAP1 which was not observed with other tested compounds. However, PROTAC 14 did not degrade KEAP1 in the lung cancer cell line A549, which harbors a G333C mutation in the KEAP1-Kelch domain [24] preventing the PROTAC 14 from binding (Fig. 5A and S7). Given that KEAP1 is the recruitment component of the Cullin3/Rbx1 E3 ligase complex, we assessed whether CRBN was able to be degraded by KEAP1 in these cell lines. Immunoblotting showed no change in CRBN levels upon treatment with PROTAC 14, suggesting CRBN E3 ligase is outcompeting KEAP1 in this cellular system (Fig. 5A). In addition, changes in PROTAC linker length and composition (compounds 3–11) did not affect CRBN levels in HEK293T cells (Fig. S8).
Fig. 5.

Impact of PROTAC 14 on activation of the KEAP1/NRF2 signaling pathway. (A) HEK293T, HCA7, BEAS-2B, and A549 cells were treated with compounds at the indicated concentrations for 48 h. Immunoblots are representative of 3 independent experiments. Repeats are shown in Fig. S7. (B) HepG2 ARE-luciferase reporter cells were treated with compounds at the indicated concentrations for 48 h and luminescence was measured. Fold change in induction is relative to the DMSO control group. (C) BEAS-2B cells were treated with 0.13 μM of compounds for 48 h. The expression levels of the antioxidant signature genes were analyzed by RT-qPCR. For B and C, data shown are from three independent experiments, each of which was performed with technical triplicates. Data shown represent mean and S.D. Unpaired t-test was performed. (*) p < 0.05; (****) p 0.0001.
To measure the effect on antioxidant gene expression we monitored the protein levels of NRF2 downstream targets, NQO1 and HO-1 in the HEK293T and HCA7 cell lines following PROTAC 14 treatment. NQO1 and HO-1 were both upregulated by 14 in a dose-dependent manner, but to a lesser degree than observed with KEAP1 inhibitor 2 (Fig. 5A and S7). Compound 15 and sulforaphane upregulated NQO1, but not HO-1, expression. As expected, control compounds 16 and 17 showed no impact on KEAP1, NOQ1, or HO-1 expression.
To confirm the observed antioxidant response, PROTAC 14 was benchmarked against inhibitor 2 in a HepG2 cell line expressing an NRF2-responsive luciferase reporter, with an antioxidant response element (ARE) upstream of the firefly luciferase gene (Fig. 5B). PROTAC 14 treatment resulted in a dose-dependent increase in luciferase signal with a maximum 3.5-fold induction at 1 μM compared to the DMSO control group. In comparison, the KEAP1 inhibitor 2 showed a 5- to 6-fold increase in luciferase expression and was approximately 2-fold more potent than PROTAC 14.
We next investigated the impact of KEAP1 degradation in BEAS-2B cells, a human non-tumorigenic lung epithelial cell line [25]. Notably, KEAP1 was more effectively degraded by PROTAC 14 in BEAS-2B cells with DC50 < 1 nM (Fig. 5A, S7, and S5), compared to KEAP1 degradation in HEK293T and HCA7 cell lines. Furthermore, PROTAC 14 was equipotent to KEAP1 inhibitor 2, as evidenced by the increased expression of both NQO1 and GCLM at all tested concentrations (GCLM was measured in this cell line as HO-1 was not detectable by immunoblotting). The increased potency of PROTAC 14 in the BEAS-2B cell line could be due to the reduced basal levels of KEAP1 expression compared to HEK293T and HCA7 cell lines (Fig. S9).
To confirm the effect of PROTAC 14 on the expression of antioxidant genes, quantitative RT-PCR was employed to determine the mRNA levels of NQO1, TALDO1, and GCLM in the BEAS-2B cell line. 48 h treatment with PROTAC 14 or KEAP1 inhibitor 2 at 0.13 μM, resulted in similar levels of mRNA induction, with a 4-fold induction of NQO1 and a 2-fold induction of TALDO1 and GCLM (Fig. 5C). These findings highlight the antioxidant potential of PROTACs targeting KEAP1 for degradation in a lung epithelial model.
3.5. Proteomics study of PROTAC 14
Label-free quantitative proteomics was performed to identify the impact of KEAP1 degradation relative to KEAP1 inhibition in the BEAS-2B cell line. The proteomic results showed that PROTAC 14 significantly reduced the abundance of KEAP1, while other Kelch domain proteins such as KBTBD4, KLHDC2, and KLHL11 were not impacted, suggesting PROTAC 14 (and the inhibitor 2 from which it was derived) was highly selective for the KEAP1-Kelch domain (Fig. 6). PROTAC 14 significantly upregulated proteins commonly involved in the antioxidant response including GCLC, GCLM, SRXN1, ALDH3A1, PIR, TXNRD1, and ARK1C family proteins (Fig. 6A). Treatment with the KEAP1 inhibitor 2 did not affect KEAP1 expression and presented a similar pattern of antioxidant protein regulation, but some proteins although differentially expressed, did not achieve significance (Fig. 6B). Notably, NRF2 was not detected in the proteomic experiment, presumably due to its relative instability and short half-life [26].
Fig. 6.

KEAP1 degradation or inhibition induces anti-oxidant protein expression. Volcano plots showing differentially expressed proteins following treatment with (A) PROTAC 14, and (B) inhibitor 2. BEAS-2B cells were treated with DMSO, PROTAC 14, or inhibitor 2 for 48 h, lysed and processed for proteomic analysis. Antioxidant proteins that were significantly changed following PROTAC 14 or inhibitor 2 treatment relative to the DMSO control, are highlighted as black dots (>1.5-fold abundance change and adjusted P value less than 0.01). Proteins that showed a significant change but were not directly related to antioxidant response are labeled with yellow dots. For B, antioxidant proteins that showed more than 1.5-fold change, but were not significant, are labeled as enlarged grey dots. KEAP1, ZFP91, and ABHD4 are labeled with green, orange, and purple colors, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
PROTAC 14 was also found to regulate proteins involved in pathways not related to the antioxidant response (Fig. 6). For example, SQSTM1, another binding partner of KEAP1 [27], was upregulated on treatment with 14. FTL and IREB2 proteins were other examples and participate in iron metabolism but have no known direct connection with the KEAP1-NRF2 signaling pathway [28]. ABHD4, a mitochondrial protein involved in lipid metabolism, was also significantly upregulated by PROTAC 14, whereas ABHD4 was not upregulated in BEAS-2B cells on treatment with inhibitor 2 (Fig. 6). ABHD4 is upregulated in NRF2 hyperactivated tumors [29], but the exact relationship with KEAP1 and the antioxidant pathway remains to be elucidated. Off-target degradation of ZFP91 induced by PROTAC 14 was also observed (Fig. 6A). ZFP91 is an established substrate of thalidomide-based immunomodulatory drugs (IMiDs) [30] and this observation is consistent with other previously reported CRBN-based PROTACs [31]. Levels of TK2, CALM1, TPD52L1, and PARP16 were also impacted by PROTAC 14 (Fig. 6A), but not the KEAP1 inhibitor 2 (Fig. 6B), possibly due to off-target degradation or an undefined connection with KEAP1-NRF2 signaling.
3.6. Effects of anti-tBHP induced cell death by PROTAC 14 in BEAS-2B cells
We next determined whether KEAP-1 degradation was sufficient to prevent ROS-induced cell death. tert-butyl hydroperoxide (tBHP) was chosen as the oxidative stress agent to impair BEAS-2B cell viability. BEAS-2B cells were pre-treated with PROTAC 14 or inhibitor 2 for 24 h and then exposed to 50 μM of tBHP for 24 h and cell viability was measured by flow cytometry (Fig. 7). PROTAC 14 and inhibitor 2 were equal potent between 500 and 16 nM but PROTAC 14 was less effective at concentrations lower than 16 nM, while the KEAP1 inhibitor 2 still maintained greater than 80% cell viability at similar concentrations. This data is consistent with the potent activation of the NRF antioxidant pathway observed for 2 and 14 (Fig. 5).
Fig. 7.

KEAP1 degradation or inhibition rescues BEAS-2B cells from ROS induced cell death. BEAS-2B cells were pre-treated with the indicated compounds at various concentrations for 24 h and then exposed to 50 μM tBHP for an additional 24 h. Cells were harvested and the % of viable cells was analyzed by PI staining and flow cytometry. Data shown are from three independent assays, with each assay shown as a different symbol. Each assay was performed with single data points. Error bars represent average and S.D. Unpaired t-test was performed. (*) p < 0.05, (***) p < 0.001, (****) p < 0.0001.
3.7. PAMPA evaluation of PROTACs 8 and 14
PROTACs typically have lower membrane permeability than the progenitor compounds on which they are designed [[32], [33], [34]]. Permeability is also a common issue encountered with the design of KEAP1 inhibitors [35,36] due to the requirement for a polar and ionizable carboxylic acid group that mimics the ETGE (and DLG) moiety of the NRF2 substrate [14]. To determine whether cell permeability is a contributing factor to the decreased cell activity of the KEAP1 PROTACs 8 and 14 compared to the KEAP1 inhibitor 2, we performed a parallel artificial membrane permeability assay (PAMPA). The data shows that the KEAP1 inhibitor 2 and PROTACs 8 and 14 all have limited passive permeability in the PAMPA assay (Pe < 0.005 10−6 cm/s) (Table S2). This suggests that passive cell permeability is not a contributing factor to the cell activity of the PROTACs 8 and 14 in comparison to the KEAP1 inhibitor 2. The low permeability shown by all three compounds is somewhat surprising given the potent activity shown in cell assays (Sections 3.2 to 3.7). Active cell membrane transport mechanisms are not measured by PAMPA and therefore could be contributing to cell activity differences between the KEAP1 inhibitor 2 and PROTACs 8 and 14.
4. Discussion
Inhibition of KEAP1 and subsequent activation of the NRF2 antioxidant response is an established and promising avenue to treat chronic diseases induced by oxidative stress. Currently, strategies are focused on the development of small molecules that either covalently interact with key cysteines on KEAP1 or directly disrupt the interaction between the KEAP1 Kelch domain and NRF2 [14]. In addition to traditional inhibitors, the PROTAC technology developed over the past two decades provides another strategy that not only inhibits but also deletes the target protein from cells [15]. At the time that this research was completed, Du et al. reported on a CRBN-based KEAP1 heterobifunctional degrader but did not investigate the consequence of KEAP1 depletion on the NRF2 antioxidant pathway in cell assays [37]. In our research, we synthesized and characterized a series of CRBN-based PROTACs targeting KEAP1 for proteasomal degradation and performed various assays to evaluate the effect of KEAP1 degradation on the NRF2 signaling axis. Through medicinal chemistry efforts and optimization of PROTAC linker type and length, a potent heterobifunctional degrader of KEAP1 (14) was identified. PROTAC 14 showed potent degradation of KEAP1 in HEK293T and BEAS-2B cell lines (DC50 11 nM, Dmax 94% @ 1 μM and DC50 < 1 nM, Dmax 90% @ 62.5 nM, respectively) and was benchmarked against the parental inhibitor 2 in different cell lines comparing the activation of the NRF2 signaling pathway. In HEK293T and HCA7 cell lines, 14 effectively decreased KEAP1 protein levels, but NRF2 mediated gene expression was inferior compared to the KEAP1 inhibitor 2. However, in the lung epithelial BEAS-2B cells, PROTAC 14 induced NRF2 antioxidant gene expression to a similar level as the KEAP1 inhibitor 2. The proteomics data confirmed that PROTAC 14 effectively reduced KEAP1 protein levels in cells and significantly increased the expression of proteins related to the NRF2 antioxidant response. Lastly, PROTAC 14 was shown to potently rescue BEAS-2B cells from ROS-induced cell death following treatment with tBHP comparable to 2.
The PROTACs investigated were less potent binders to KEAP1 than the parent KEAP1 inhibitor 2, as assessed via HTRF assay. In theory, PROTACs act by degrading the targeted protein via a catalytic mechanism, and therefore 14 would be expected to elicit equivalent or enhanced potency in activating the NRF2 antioxidant response compared to 2. However, PROTAC 14 was consistently less effective than the KEAP1 inhibitor 2 in HEK239T and HCA7 cells. We show that the passive cellular permeability of the KEAP1 inhibitor 2 and the PROATCs 8 and 14 as measured by PAMPA (Table S2) was not contributing to the cell activity differences observed. Considering, the low passive cell membrane permeability, compound 2 and PROTACS 8 and 14 have remarkedly potent cell activity, and therefore it cannot be discounted that an active cell membrane transport mechanism (not measured by PAMPA) is contributing to cell activity and the differences observed with 14 (and 8) in comparison to 2. It is also possible that PROTAC 14 is degraded by cellular enzymes, however, compared to PROTAC 8 which has a cell stability liability, PROTAC 14 remains functional up to 72 h (Fig. 3). Furthermore, the formation of a ternary complex between PROTAC, E3 ligase, and the target protein is also a key factor in the observed degradative efficiency of a PROTAC. The ability of PROTAC 14 to induce an active ternary complex remains to be elucidated in further studies via SPR or X-ray crystallography and will be key to the future optimization of a KEAP1 targeted PROTAC.
We observed that PROTAC 14 was more potent in BEAS-2B cells compared to other cell lines. We rationalized that this could be due to the low basal expression level of KEAP1 in BEAS-2B cells (Fig. S9), which may not require a high intracellular concentration of 14 to induce NRF2 activation and potentially counterbalances the low cellular permeability and intracellular concentration of 14. This observation highlights how the endogenous expression level of the target protein can impact the efficacy of a PROTAC.
KEAP1 forms a Cullin-Ring E3 ligase [11], and thus theoretically, may be able to degrade CRBN when both are recruited by the PROTAC. However, we did not observe CRBN degradation with our KEAP1-CRBN PROTACs (Fig. 5 and S8), suggesting that the CRBN E3 ligase complex was dominant over the KEAP1/Cul3/Rbx1 complex. This data is consistent with the results from Du et al. [37] showing KEAP1 is degraded by both KEAP1-CRBN and KEAP1-VHL PROTACs. Interestingly, the opposite was found for a CRBN-VHL PROTAC which degraded CRBN and not VHL [[38], [39], [40], [41]], while the MDM2-CRBN PROTAC degraded MDM2 [42]. It is unknown why CRBN and VHL E3 ligases dominate over KEAP1, but KEAP1 was shown to have a modest degradative capacity as the E3 component in a PROTAC to degrade bromodomain and focal adhesion kinase proteins [21,37]. It is postulated that KEAP1 dimerization is required to achieve optimal degradative capacity, as seen with ubiquitination of the native KEAP1 substrate NRF2, with the PROTAC or ternary PROTAC complex needing to engage with both juxtaposed KEAP1-kelch molecules in the homodimeric degradative E3 complex [8,9]. Above all, further optimization of the linker to form an active ternary complex may be required to fully harness the E3 degradative ability of KEAP1 in a PROTAC strategy.
As proof-of-principle, we have demonstrated that PROTAC 14 can efficiently degrade KEAP1 in cells, however, further optimization is required to enhance cell permeability, target selectivity, and pharmacological properties such as metabolism that are an important considerations for efficacy in animal models. The optimization may include rigidifying the linker by introducing heterocyclic scaffolds such as piperazine and piperidines, a strategy that has been used previously to improve cell membrane permeability [43,44]. Carboxylic acid isosteres or a prodrug approach previously applied to KEAP1 inhibitors [35,36] could also be introduced to mask the polar carboxylic acid group to improve cell membrane permeability. Although the stability of the CRBN ligand was improved in our study, the thalidomide ligand could be replaced by other phenyl glutarimide analogs which have been reported to have greater metabolic stability [45]. To overcome undesirable off-target consequences of the CRBN ligand, recruitment of the VHL E3 ligase could be considered in the design of a KEAP1 heterobifunctional degrader [46]. Du et al. have shown that VHL-based PROTAC at high concentrations can degrade KEAP1 [37], proving a VHL-based concept but also suggesting further optimization is required. The incorporation of a VHL ligand may also favor the formation of an active ternary complex between the E3 ligase and KEAP1 to improve degradative efficiency. The switch to a VHL ligand will also enable optimization to be guided by biophysical and structural data [43], whereas this is challenging with CRBN because of the technical difficulties we experienced with the expression of recombinant CRBN constructs.
In conclusion, the KEAP1 targeted PROTAC designed here is a useful tool for studying the biological consequences of KEAP1 degradation. This work established the possibility of degrading KEAP1 as an alternative to existing KEAP1 therapeutic strategies that covalently or directly inhibit KEAP1. Further development of the KEAP1 bifunctional degrader is required to fully understand the implications of KEAP1 degradation in vitro and in animal models, working toward a new treatment for oxidative stress-induced disorders.
Conflict of interest
All authors have approved the final version of the manuscript and declare no conflict of interest related to this work.
Declaration of competing interest
All authors have approved the final version of the manuscript and declare no conflict of interest related to this work.
Acknowledgments
This work was supported by the Australian Cancer Research Foundation and the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. The laboratory of R.F. is supported by The Galbraith Family Charitable Trust and the K & M Foundation for Women. R.F. and B.C.L received grant funding from the Australian Government via the MRFF Frontier program (Grant ID: RFRHPI000269). B.E.S. is a Corin Centenary Fellow. G.B. is supported by an Australian Government Research Training Program scholarship and H.C. is supported by a Research Scholarship from the University of Melbourne.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102552.
Abbreviations
- ABHD4
Alpha/beta-hydrolase domain containing 4
- ALDH3A1
Aldehyde dehydrogenase 3A1
- ARE
Antioxidant response element
- ARK1C
Aldo/keto reductase family1 member C1
- BSA
Bovine serum albumin
- BTB
Broad complex, tramtrack and bric-à-brac
- CALM1
Calmodulin 1
- CRBN
Cereblon
- E3
Ubiquitin ligase
- FACS
Fluorescence-activated cell sorting
- FTL
Ferritin light chain
- GCLM
Glutamate-cysteine ligase modifier subunit
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HO-1
Heme oxygenase-1
- HRP
Horse radish peroxidase
- IMiDs
Immunomodulatory drugs
- IREB2
Iron responsive element binding protein 2
- IVR
Intervening region
- KBTBD4
Kelch repeat and BTB domain containing 4
- KEAP1
Kelch-like ECH-associated protein 1
- KLHDC2
Kelch domain containing 2
- KLHL11
Kelch like family member 11
- NQO1
NAD(P)H:quinone oxidoreductase 1
- NRF2
Nuclear factor erythroid 2–related factor 2
- PAMPA
Parallel artificial membrane permeability assay
- PARP16
Poly (ADP-ribose) polymerase family member 16
- PI
Propidium iodide
- PIR
Pirin
- POI
Protein of interest
- PROTAC
Proteolysis-targeting chimeras
- RBX1
RING-box protein 1
- RING
Really interesting new gene
- ROS
Reactive oxygen species
- SQSTM1
Sequestosome 1
- SRXN1
Sulfiredoxin 1
- TALDO1
Transaldolase 1
- tBHP
tert-butyl hydroperoxide
- TK2
Thymidine kinase 2
- TOM
Thalidomide
- TPD52L1
Tumor protein D52 like 1
- TXNRD1
Thioredoxin reductase 1
- UPS
Ubiquitin–proteasome system
- VHL
Hippel–Lindau tumour suppressor
- ZFP91
Zinc finger protein 91
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pizzino G., Irrera N., Cucinotta M., Pallio G., Mannino F., Arcoraci V., Squadrito F., Altavilla D., Bitto A. Oxidative stress: harms and benefits for human health. Oxid. Med. Cell. Longev. 2017;2017 doi: 10.1155/2017/8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He L., He T., Farrar S., Ji L., Liu T., Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017;44(2):532–553. doi: 10.1159/000485089. [DOI] [PubMed] [Google Scholar]
- 4.Dayalan Naidu S., Dinkova-Kostova A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020;10(6) doi: 10.1098/rsob.200105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakabayashi N., Dinkova-Kostova A.T., Holtzclaw W.D., Kang M.I., Kobayashi A., Yamamoto M., Kensler T.W., Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U. S. A. 2004;101(7):2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albagli O., Dhordain P., Deweindt C., Lecocq G., Leprince D. The BTB/POZ domain: a new protein-protein interaction motif common to DNA- and actin-binding proteins. Cell Growth Differ. 1995;6(9):1193–1198. [PubMed] [Google Scholar]
- 7.Cleasby A., Yon J., Day P.J., Richardson C., Tickle I.J., Williams P.A., Callahan J.F., Carr R., Concha N., Kerns J.K., Qi H., Sweitzer T., Ward P., Davies T.G. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One. 2014;9(6) doi: 10.1371/journal.pone.0098896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canning P., Cooper C.D.O., Krojer T., Murray J.W., Pike A.C.W., Chaikuad A., Keates T., Thangaratnarajah C., Hojzan V., Marsden B.D., Gileadi O., Knapp S., von Delft F., Bullock A.N. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 2013;288(11):7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canning P., Sorrell F.J., Bullock A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015;88(Pt B):101–107. doi: 10.1016/j.freeradbiomed.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong K.I., Katoh Y., Kusunoki H., Itoh K., Tanaka T., Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell Biol. 2006;26(8):2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi A., Kang M.I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baird L., Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell Biol. 2020;40(13) doi: 10.1128/MCB.00099-20. e00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuadrado A., Rojo A.I., Wells G., Hayes J.D., Cousin S.P., Rumsey W.L., Attucks O.C., Franklin S., Levonen A.L., Kensler T.W., Dinkova-Kostova A.T. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019;18(4):295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H., Wang Y., You Q., Jiang Z. Recent progress in the development of small molecule Nrf2 activators: a patent review (2017-present) Expert Opin. Ther. Pat. 2020;30(3):209–225. doi: 10.1080/13543776.2020.1715365. [DOI] [PubMed] [Google Scholar]
- 15.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017;16(2):101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou Y., Ma D., Wang Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019;37(1):21–30. doi: 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottis P., Crews C.M. Proteolysis-targeting chimeras: induced protein degradation as a therapeutic strategy. ACS Chem. Biol. 2017;12(4):892–898. doi: 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- 18.Chen H., Wu Y., Li K., Currie I., Keating N., Dehkhoda F., Grohmann C., Babon J.J., Nicholson S.E., Sleebs B.E. Optimization of phosphotyrosine peptides that target the SH2 domain of SOCS1 and block substrate ubiquitination. ACS Chem. Biol. 2022;17(2):449–462. doi: 10.1021/acschembio.1c00884. [DOI] [PubMed] [Google Scholar]
- 19.Chen H., Ware T.M.B., Iaria J., Zhu H.J. Live cell imaging of the TGF- beta/smad3 signaling pathway in vitro and in vivo using an adenovirus reporter system. JoVE. 2018;137 doi: 10.3791/57926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies T.G., Wixted W.E., Coyle J.E., Griffiths-Jones C., Hearn K., McMenamin R., Norton D., Rich S.J., Richardson C., Saxty G., Willems H.M., Woolford A.J., Cottom J.E., Kou J.P., Yonchuk J.G., Feldser H.G., Sanchez Y., Foley J.P., Bolognese B.J., Logan G., Podolin P.L., Yan H., Callahan J.F., Heightman T.D., Kerns J.K. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016;59(8):3991–4006. doi: 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 21.Wei J., Meng F., Park K.S., Yim H., Velez J., Kumar P., Wang L., Xie L., Chen H., Shen Y., Teichman E., Li D., Wang G.G., Chen X., Kaniskan H.U., Jin J. Harnessing the E3 ligase KEAP1 for targeted protein degradation. J. Am. Chem. Soc. 2021;143(37):15073–15083. doi: 10.1021/jacs.1c04841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bricelj A., Dora Ng Y.L., Ferber D., Kuchta R., Muller S., Monschke M., Wagner K.G., Kronke J., Sosic I., Gutschow M., Steinebach C. Influence of linker attachment points on the stability and neosubstrate degradation of cereblon ligands. ACS Med. Chem. Lett. 2021;12(11):1733–1738. doi: 10.1021/acsmedchemlett.1c00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreau K., Coen M., Zhang A.X., Pachl F., Castaldi M.P., Dahl G., Boyd H., Scott C., Newham P. Proteolysis-targeting chimeras in drug development: a safety perspective. Br. J. Pharmacol. 2020;177(8):1709–1718. doi: 10.1111/bph.15014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh A., Misra V., Thimmulappa R.K., Lee H., Ames S., Hoque M.O., Herman J.G., Baylin S.B., Sidransky D., Gabrielson E., Brock M.V., Biswal S. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10) doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddel R.R., Ke Y., Gerwin B.I., McMenamin M.G., Lechner J.F., Su R.T., Brash D.E., Park J.B., Rhim J.S., Harris C.C. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48(7):1904–1909. [PubMed] [Google Scholar]
- 26.He F., Ru X., Wen T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020;21(13) doi: 10.3390/ijms21134777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 28.Chen X., Yu C., Kang R., Tang D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.590226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacher S.E., Slattery M. Gene regulatory effects of disease-associated variation in the NRF2 network. Curr Opin Toxicol. 2016;1:71–79. doi: 10.1016/j.cotox.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An J., Ponthier C.M., Sack R., Seebacher J., Stadler M.B., Donovan K.A., Fischer E.S. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun. 2017;8 doi: 10.1038/ncomms15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S., Wang J., Hamman B.D., Ishchenko A., Crews C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018;25(1):78–87 e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cecchini C., Pannilunghi S., Tardy S., Scapozza L. From conception to development: investigating PROTACs features for improved cell permeability and successful protein degradation. Front. Chem. 2021;9 doi: 10.3389/fchem.2021.672267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foley C.A., Potjewyd F., Lamb K.N., James L.I., Frye S.V. Assessing the cell permeability of bivalent chemical degraders using the chloroalkane penetration assay. ACS Chem. Biol. 2020;15(1):290–295. doi: 10.1021/acschembio.9b00972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott D.E., Rooney T.P.C., Bayle E.D., Mirza T., Willems H.M.G., Clarke J.H., Andrews S.P., Skidmore J. Systematic investigation of the permeability of androgen receptor PROTACs. ACS Med. Chem. Lett. 2020;11(8):1539–1547. doi: 10.1021/acsmedchemlett.0c00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu M., Zhang X., Zhao J., You Q., Jiang Z. A hydrogen peroxide responsive prodrug of Keap1-Nrf2 inhibitor for improving oral absorption and selective activation in inflammatory conditions. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pallesen J.S., Narayanan D., Tran K.T., Solbak S.M.Ø., Marseglia G., Sørensen L.M.E., Høj L.J., Munafò F., Carmona R.M.C., Garcia A.D., Desu H.L., Brambilla R., Johansen T.N., Popowicz G.M., Sattler M., Gajhede M., Bach A. Deconstructing noncovalent kelch-like ECH-associated protein 1 (Keap1) inhibitors into fragments to reconstruct new potent compounds. J. Med. Chem. 2021;64(8):4623–4661. doi: 10.1021/acs.jmedchem.0c02094. [DOI] [PubMed] [Google Scholar]
- 37.Du G., Jiang J., Henning N.J., Safaee N., Koide E., Nowak R.P., Donovan K.A., Yoon H., You I., Yue H., Eleuteri N.A., He Z., Li Z., Huang H.T., Che J., Nabet B., Zhang T., Fischer E.S., Gray N.S. Exploring the target scope of KEAP1 E3 ligase-based PROTACs. Cell Chem Biol. 2022;29(10):1470–1481. doi: 10.1016/j.chembiol.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Girardini M., Maniaci C., Hughes S.J., Testa A., Ciulli A. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019;27(12):2466–2479. doi: 10.1016/j.bmc.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim K., Lee D.H., Park S., Jo S.H., Ku B., Park S.G., Park B.C., Jeon Y.U., Ahn S., Kang C.H., Hwang D., Chae S., Ha J.D., Kim S., Hwang J.Y., Kim J.H. Disordered region of cereblon is required for efficient degradation by proteolysis-targeting chimera. Sci. Rep. 2019;9(1) doi: 10.1038/s41598-019-56177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powell C.E., Du G., Bushman J.W., He Z., Zhang T., Fischer E.S., Gray N.S. Selective degradation-inducing probes for studying cereblon (CRBN) biology. RSC Med Chem. 2021;12(8):1381–1390. doi: 10.1039/d0md00382d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinebach C., Kehm H., Lindner S., Vu L.P., Köpff S., López Mármol Á., Weiler C., Wagner K.G., Reichenzeller M., Krönke J., Gütschow M. PROTAC-mediated crosstalk between E3 ligases. Chem. Commun. 2019;55(12):1821–1824. doi: 10.1039/c8cc09541h. [DOI] [PubMed] [Google Scholar]
- 42.Li Y., Yang J., Aguilar A., McEachern D., Przybranowski S., Liu L., Yang C.-Y., Wang M., Han X., Wang S. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 2019;62(2):448–466. doi: 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farnaby W., Koegl M., Roy M.J., Whitworth C., Diers E., Trainor N., Zollman D., Steurer S., Karolyi-Oezguer J., Riedmueller C., Gmaschitz T., Wachter J., Dank C., Galant M., Sharps B., Rumpel K., Traxler E., Gerstberger T., Schnitzer R., Petermann O., Greb P., Weinstabl H., Bader G., Zoephel A., Weiss-Puxbaum A., Ehrenhofer-Wolfer K., Wohrle S., Boehmelt G., Rinnenthal J., Arnhof H., Wiechens N., Wu M.Y., Owen-Hughes T., Ettmayer P., Pearson M., McConnell D.B., Ciulli A. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019;15(7):672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han X., Wang C., Qin C., Xiang W., Fernandez-Salas E., Yang C.Y., Wang M., Zhao L., Xu T., Chinnaswamy K., Delproposto J., Stuckey J., Wang S. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019;62(2):941–964. doi: 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- 45.Min J., Mayasundari A., Keramatnia F., Jonchere B., Yang S.W., Jarusiewicz J., Actis M., Das S., Young B., Slavish J., Yang L., Li Y., Fu X., Garrett S.H., Yun M.K., Li Z., Nithianantham S., Chai S., Chen T., Shelat A., Lee R.E., Nishiguchi G., White S.W., Roussel M.F., Potts P.R., Fischer M., Rankovic Z. Phenyl-glutarimides: alternative cereblon binders for the design of PROTACs. Angew Chem. Int. Ed. Engl. 2021;60(51):26663–26670. doi: 10.1002/anie.202108848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diehl C.J., Ciulli A. Discovery of small molecule ligands for the von Hippel-Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem. Soc. Rev. 2022;51:8216–8257. doi: 10.1039/d2cs00387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.