

Abstract
It is poorly understood how the tumor immune microenvironment influences disease recurrence in localized clear cell renal cell carcinoma (ccRCC). Here we perform whole-transcriptomic profiling of 236 tumors from patients assigned to the placebo-only arm of a randomized, adjuvant clinical trial for high-risk localized ccRCC. Unbiased pathway analysis identifies myeloid-derived interleukin-6 (IL-6) as a key mediator. Furthermore, a novel myeloid gene signature strongly correlates with disease recurrence and overall survival on uni- and multivariate analysis and is linked to TP53 inactivation across multiple datasets. Strikingly, effector T cell gene signatures, infiltration patterns, and exhaustion markers were not associated with disease recurrence. Targeting immunosuppressive myeloid inflammation with an adenosine A2A receptor antagonist in a novel, immunocompetent, Tp53-inactivated mouse model significantly reduces metastatic development. Our findings suggest myeloid inflammation promotes disease recurrence in ccRCC, is targetable, and provide a potential biomarker-based framework for the design of future immuno-oncology trials in ccRCC.
Introduction
The majority of patients (~75%) with clear cell renal cell carcinoma (ccRCC) initially present with localized disease for which surgical resection followed by active surveillance constituted the standard of care, prior to the recent FDA approval of adjuvant pembrolizumab.(1) Up to 40–50% of these patients, however, will subsequently develop disease recurrence.(2,3) Clinicopathologic risk stratification systems such as the Mayo Clinic stage, size, grade, and necrosis (SSIGN) and UCLA integrated staging system (UISS) have been developed to help identify patients at high risk of relapse(2,3). Additionally, three validated RNA gene signature-based tools, ClearCode34(4), the cell cycle proliferation (CCP) score(5) and the Recurrence Score(6) can also predict recurrence after nephrectomy. There is a growing understanding that ccRCC is composed of highly heterogenous subtypes which can be distinguished by genomic and transcriptomic features reflective of the stromal and immune microenvironment.(7) Indeed, alterations in frequent ccRCC driver genes such as BAP1, SETD2 and TP53 are correlated with worse recurrence-free and cancer-specific survival(8–11) and these mutations can be associated with distinct tumor microenvironments (TMEs).(12,13) Furthermore, biomarker analyses of pretreatment samples from recent clinical trials have revealed correlations between TME gene signatures related to angiogenesis or tumor-infiltrating immune cells, and outcomes to systemic therapies targeting tumor vasculature and immune checkpoint receptors, respectively.(7,14–16) We hypothesized that such microenvironmental features might also influence metastatic development of localized disease, and identification of these factors may support the design of future mechanism-based adjuvant trials.
Utilizing multimodal profiling of the placebo arm of a large adjuvant ccRCC trial(17), we find that metastatic recurrence in ccRCC is largely independent of T cell response, and rather is associated with a myeloid inflammatory state, which can be linked to TP53 inactivation across numerous data sets. We further develop a first of a kind immunocompetent, metastatic, Tp53-inactivated ccRCC mouse model and demonstrate that targeting myeloid populations via adenosine A2A receptor antagonism impedes metastatic development.
Results
Differential gene expression analysis identifies oncogenic and immune pathways associated with disease recurrence
A total of 236 out of 769 patients from the placebo arm of the PROTECT(17) trial (adjuvant pazopanib vs placebo for high risk localized ccRCC) had clinical, pathologic, immunohistochemical, and tumor whole-genome microarray data available for analysis. Demographic and clinicopathologic characteristics of the patient population are listed in Supplementary Table 1. We performed differentially expressed gene (DEG) analysis in patients who developed disease recurrence (n=88) vs those who did not (n=148). We identified 765 DEGs in tumors associated with disease recurrence (Fig. 1A, Supplementary Table 2). Gene set enrichment analysis (GSEA) demonstrated several oncogenic pathways known to be upregulated in ccRCC enriched in patients with disease recurrence including epithelial to mesenchymal transition (EMT), MYC targets, G2M checkpoint, and MTORC1 signaling (Fig. 1B). Multiple immune and inflammatory signaling pathways in tumors of patients with disease recurrence were also upregulated including interferon gamma, TNFα, inflammatory response, and IL6 (Fig. 1B). Unbiased Ingenuity pathway analysis (IPA) revealed IL6 as a key component among the molecular pathways enriched in said tumors (Fig. 1C).
Figure 1. Multiple oncogenic and inflammatory pathways upregulated in recurrent tumors converge on IL6.
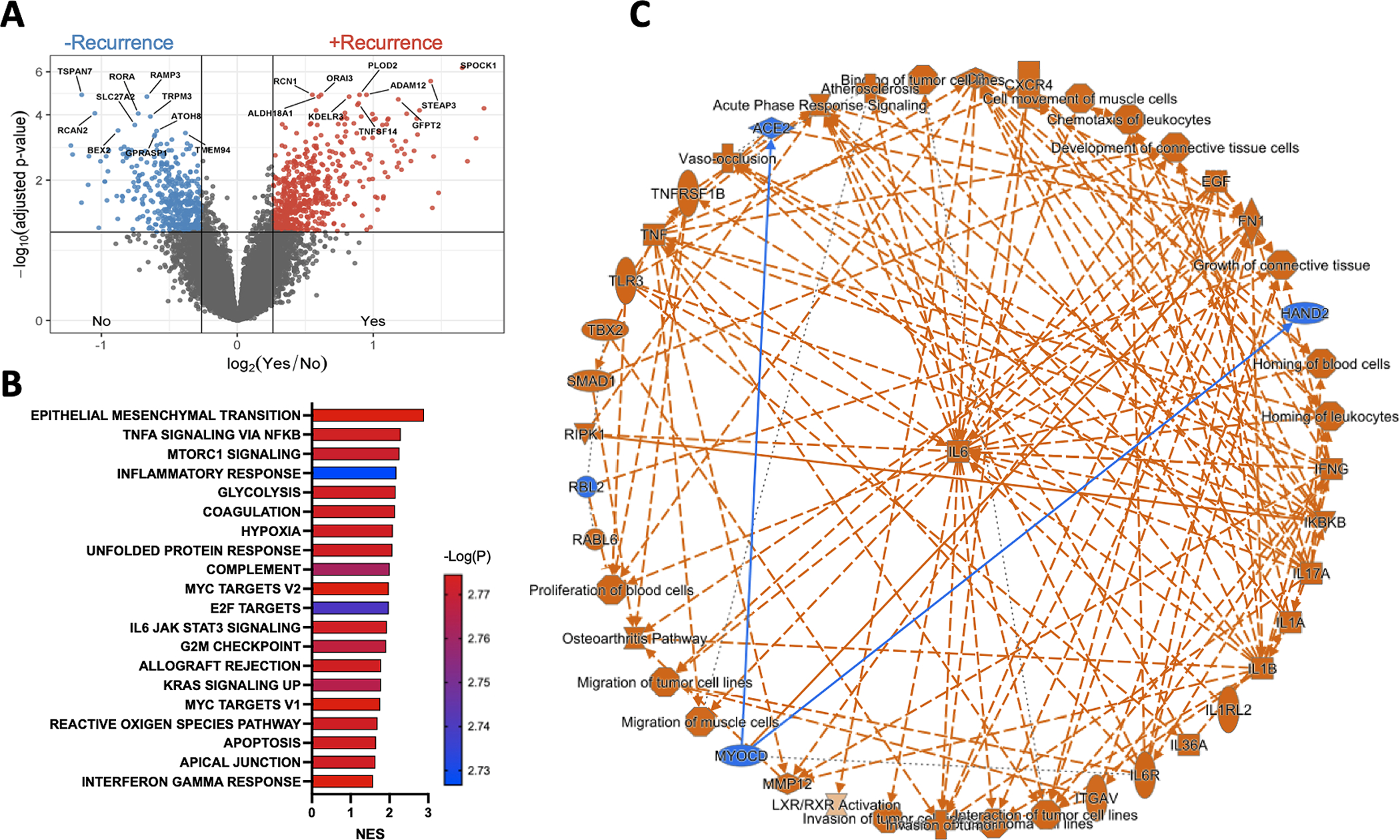
(A) Volcano plot demonstrating differentially expressed genes in recurrent vs non-recurrent tumors. (B) Bar graph displaying the top 20 GSEA analysis of hallmark gene sets, based on p-value, comparing recurrent vs non-recurrent tumors. Pathways are ranked by normalized enrichment scores (NES) and all shown pathways have adjusted p<0.05. (C) IPA graphical summary of core analysis highlighting IL6 as a common component among upregulated pathways in recurrent tumors.
Immune deconvolution links myeloid inflammation and IL6 pathway to metastasis
Recent single-cell RNAseq analysis of ccRCC tumors revealed enrichment of IL6 expression primarily in myeloid, vascular endothelial, and CA9+ tumor cell populations.(18) We therefore performed immune deconvolution using gene signatures reflective of myeloid inflammation that have recently been associated with responses to systemic therapy in the metastatic disease setting(15,16,19,20) to our cohort. Both the adenosine and myeloid signatures were significantly enriched in tumors of patients with subsequent disease recurrence (Fig. 2A). Adenosine signature groups defined by quartiles were significantly and adversely associated with median disease-free survival (DFS) (Log-rank test; p=1.79E-08; Fig. S1A) and median overall survival (Log-rank test; p=3.71E-06; Fig. S1B) with scores in the highest quartile demonstrating the worst DFS (HR 5.37; 95% CI, 2.73–10.6; p=1.00E-6; Q1 as reference) and OS (HR 5.39; 95% CI, 2.04–14.2; p=6.8E-04; Q1 as reference). Similarly, patients who harbored higher myeloid scores demonstrated decreased DFS (Log-rank test; p=2.20E-08, Fig. S1C) and worse OS (Log-rank test; p=7.69E-05, Fig. S1D) with the highest quartile scores associated with the worst DFS (HR 7.09; 95% CI, 3.28–15.3; p=6E-07) and OS (HR 6.39; 95% CI, 2.18–18.7; p=7.1E-04; Q1 as reference). Given the similarities between these two signatures (Fig. S1E), we sought to determine which of the included target genes (9 in total with 5 shared, 4 private) most strongly associated with survival outcomes in ccRCC. To this end, we performed a Cox regression analysis on expression levels of the nine genes comprising the adenosine and myeloid signatures in the TCGA pan-cancer dataset(21) with respect to OS. Out of the 32 malignancies examined in the TCGA dataset, seventeen had expression of at least one gene significantly associated with OS (Fig. 2B). Within the TCGA-KIRC cohort, we found upregulation of IL-1β, CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, and IL-6 were associated with significantly decreased OS (Fig. 2C). These genes were then combined to create a new composite myeloid inflammatory signature, termed MSK inflammatory (MSKI) (Fig. S1E, 2D). As expected, MSKI ssGSEA scores grouped by quartile were significantly associated with OS (Log-rank test; p=1.53E-07) in the TCGA-KIRC cohort (Fig. S1F). MSKI scores were also associated with worse DFS (Log-rank test; p=5.44E-03; Fig. 2E) with the worst outcomes seen in tumors with the highest quartile of enrichment scores (DFS: HR 2.38; 95% CI, 1.24–4.59; p = 9.45E-03, OS: HR 3.08; 95% CI, 1.96–4.85; p=1.1E-06; Q1 as reference). Within the PROTECT cohort, the MSKI gene signature was significantly enriched in recurrent vs non-recurrent tumors (Fig. 2F) with higher enrichment scores associated with worse DFS (Log-rank test; p=4.00E-11; Fig. 2G) and OS (Log-rank test; p=4.76E-07; Fig. S1G). We then applied the MSKI signature to an external validation cohort of 84 patients with localized, untreated ccRCC, termed the MOFFITT cohort (see Supplementary Table 3 for clinicopathologic demographics) and confirmed a significant association with DFS (Log-rank p=0.0199; Fig. 2H) as well as a non-significant OS trend (Log-rank p = 0.108; Fig. S1H).
Figure 2. Myeloid inflammatory gene signatures predict worse disease outcomes in RCC.
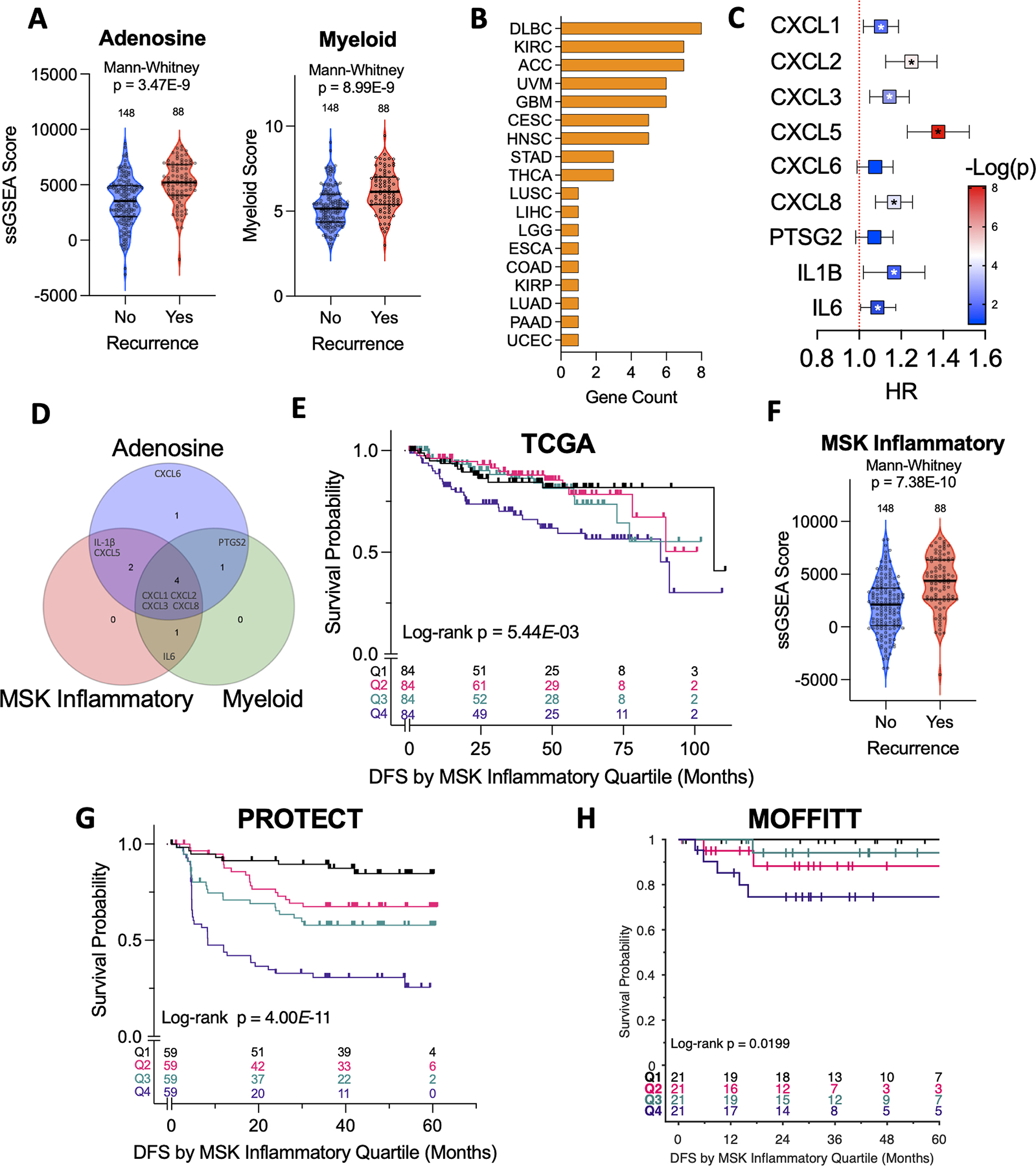
(A) Violin plots of enrichment scores for Adenosine and Myeloid gene signatures in recurrent vs non-recurrent tumors in the PROTECT cohort. (B) Bar graph of gene counts by cancer type that were significantly associated with overall survival on Cox regression in TCGA dataset. (C) Forest Plot of Cox regression analysis by listed genes for KIRC cohort (*p < 0.05). (D) Venn diagram highlighting number of genes shared by the listed signatures. (E) Kaplan-Meier analysis demonstrating impact of MSK inflammatory gene expression (based on quartiles of enrichment scores) on DFS in the TCGA-KIRC cohort. (F) Violin plots of enrichment scores for the MSK inflammatory signature in recurrent vs non-recurrent tumors in the PROTECT cohort. (G,H) Kaplan-Meier analysis demonstrating impact of MSK inflammatory gene expression (based on quartiles of enrichment scores) on DFS in the (G) PROTECT and (H) MOFFITT cohorts.
Lymphoid infiltration and exhaustion markers fail to predict disease recurrence
Intratumoral CD8+ cell infiltration has been associated with conflicting outcomes in patients with metastatic(22,23) and localized ccRCC(24). Gene signatures reflective of lymphoid infiltration(18) including Effector T cell (Teff)(15) and JAVELIN Renal 101 Immuno (Javelin)(16) have been associated with response rates to ICB + anti-VEGF therapies in metastatic ccRCC (mccRCC). However, deconvolved scores for Teff (Mann-Whitney test; p=0.2099) and Javelin (Mann-Whitney test; p=0.2014) signatures did not significantly differ between recurrent vs non-recurrent tumors in the PROTECT cohort (Fig. 3A). Quantitative IHC demonstrated these effector signatures were significantly correlated with intratumoral CD8+ cell counts (Fig. 3B), which also did not differ between tumors with/without subsequent disease recurrence in the PROTECT (Mann-Whitney test; p=0.1453; Fig. 3C) and MOFFITT (Mann-Whitney test; p=0.27; Fig. S2A) cohorts.
Figure 3. Lymphoid inflammation is not associated with disease recurrence.
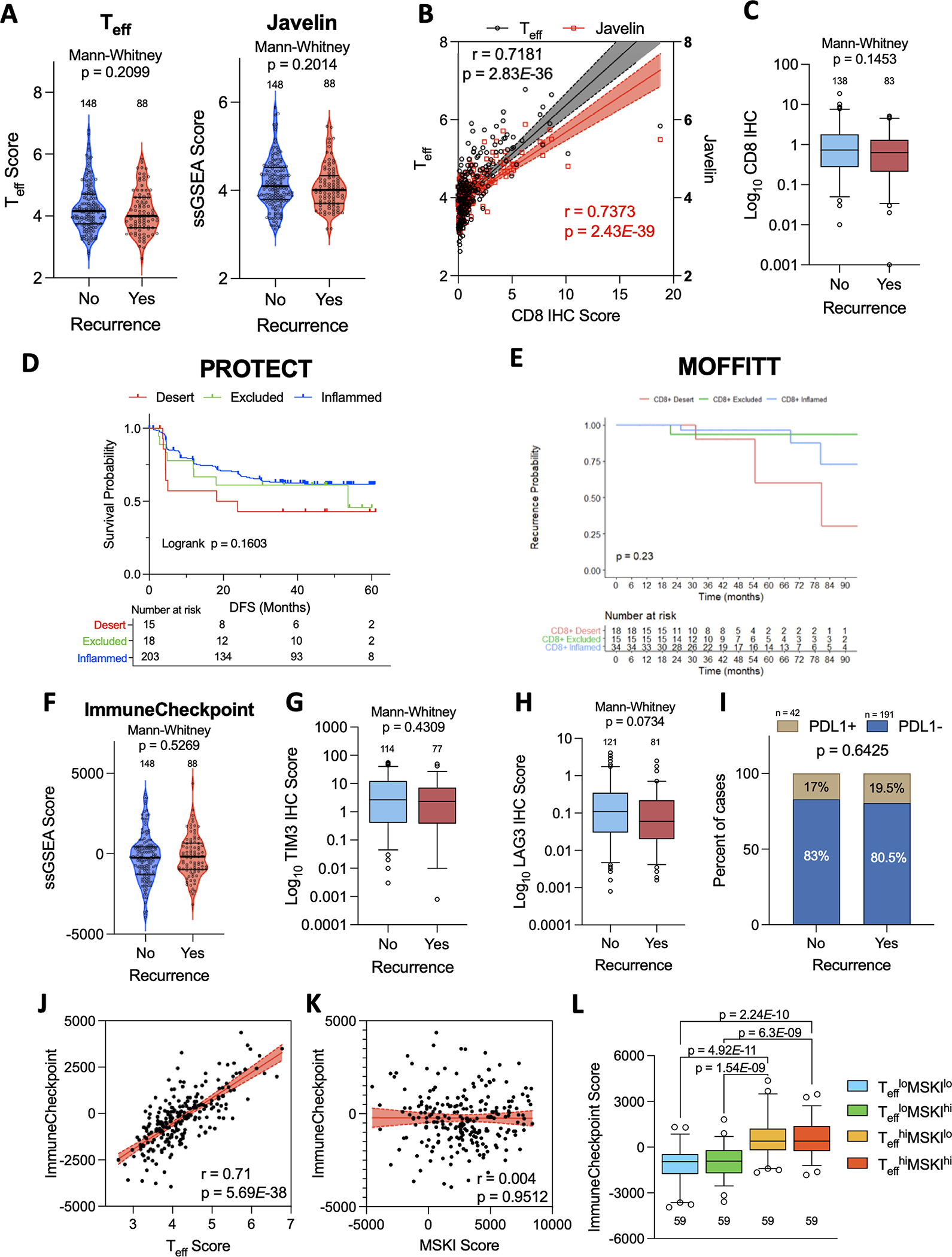
Violin plots of enrichment scores for (A) Effector T cell (Teff) and (B) Javelin gene scores in recurrent vs non-recurrent tumors. (B) Scatterplot showing relationship between CD8 IHC scores and Teff or Javelin signature scores. Spearman correlation coefficients with p-values obtained from two-sided tests shown. Shaded areas represent 95% confidence intervals. (C) Overall intratumoral CD8 IHC scores in recurrent vs non-recurrent tumors. (D,E) Kaplan-Meier analysis of CD8 infiltration patterns on DFS in the (D) PROTECT and (E) MOFFITT cohorts. (F,G) Violin plots of enrichment scores for (F) ImmuneCheckpoint GES in recurrent vs non-recurrent tumors. (G,H,I) IHC scores for (G) TIM3, (H) LAG3, and (I) PD-L1 in recurrent vs non-recurrent tumors. PD-L1 positivity defined as >1% tumor cells labeled. (J,K) Scatterplots showing correlation between the ImmuneCheckpoint score and (J) Teff score and (K) MSKI GES. (L) Box plots demonstrating difference in median ImmuneCheckpoint score and Teff high vs low (Teffhi vs Tefflo) and MSKI high vs low (MSKIhi vs MSKIlo) groups.
Further, we found no association between Teff levels (Teffhi vs Tefflo) and DFS (Log-rank test p=0.1449; Fig. S2B) or OS (Log-rank test p=0.6243; Fig. S2C). In the biomarker analysis of the IMmotion150 trial(15), efficacy of PD-L1 directed monotherapy in Teffhi tumors was limited in the presence of high myeloid signatures, suggestive of myeloid-mediated T cell suppression. In a Kaplan-Meier analysis on Teff and Myeloid subgroups (Teffhi vs Tefflo; Myeloidhi vs Myeloidlo) we found the Myeloid scores to be the primary driver of outcomes (DFS: Log-rank test p = 1.19E-07; Fig. S2D; OS: Log-rank test p = 1.58E-04; Fig. S2E), irrespective of high and low Teff scores.
We also analyzed IHC data for patients in the PROTECT (n = 236) and MOFFITT (n = 84) cohorts to assign distinct patterns of CD8+ cell infiltration including ‘inflamed’, ‘desert’, and ‘excluded’. Kaplan-Meier analysis revealed no association between these CD8+ infiltration patterns and DFS in the PROTECT (Log-rank test; p=0.1603; Fig. 3D) and MOFFITT (Log-rank test; p=0.76; Fig. 3E) cohorts. Co-expression of inhibitory immune checkpoint molecules on CD8+ cells including PD-1, TIM3, and LAG3 reflect an ‘exhausted’ phenotype that has been associated with worse disease outcomes.(25,26) Therefore, we compared ImmuneCheckpoint (Fig. 3F) and Immunosuppression(27) (Fig. S2F) scores but found no significant difference between recurrent and non-recurrent tumors in the PROTECT cohort. IHC markers of exhaustion including TIM3 (Mann-Whitney test; p=0.4309; Fig. 3G) and LAG3 (Mann-Whitney test; p=0.0734; Fig. 3H) expression also did not correlate with disease recurrence. There was no significant association between tumor PD-L1 status and disease recurrence (Fig. 3I). We noted a strong correlation between the ImmuneCheckpoint scores and Teff scores (Spearman, r=0.71; p=5.69E-38; Fig. 3J), but none between the ImmuneCheckpoint scores and MSKI scores (Spearman, r=0.004; p=0.9512; Fig. 3K). Accordingly, on subgroup analysis, the ImmuneCheckpoint scores (Fig. 3L) and LAG3 IHC scores (Fig. S2G) were only influenced by the degree of lymphocytic (Teffhi vs Tefflo), but not myeloid (MSKIhi vs MSKIlo) infiltration. Together, these data indicates that lymphocytic infiltrates were not associated with disease recurrence.
Angiogenic pathway is downregulated in recurrent tumors
The ubiquitous loss of function alterations of the Von-Hippel-Lindau (VHL) gene in ccRCC results in stabilization of hypoxia-inducible factors (HIF) and subsequent transcriptional upregulation of pro-angiogenic genes. The study of this canonical pathway in ccRCC led to the successful development of anti-angiogenic based therapies, and indeed, upregulation of angiogenesis genes associates with added antitumor effect from TKI therapy in the metastatic setting.(7,15,16,19)
In our cohort, we found that patients with lower angiogenesis scores had increased disease recurrence (Mann-Whitney test; p=4.28E-03; Fig. S3A) and, on Kaplan-Meier analysis, displayed inferior DFS (Log-rank test; p=4.85E-03; Q1: HR 3.02; 95% CI, 1.57–5.79; p=8.91E-04; Q4 as reference; Fig. S3B) and shorter OS (Log-rank test; p=9.58E-04; Q1: HR 4.10; 95% CI, 1.66–10.2; p=0.0022; Q4 as reference; Fig. S3B). Similarly, in the TCGA-KIRC cohort, we found lower angiogenesis score was associated with decreased OS (Log-rank test; p=1.65E-6) but not DFS (Log-rank test; p=0.607; Fig S3C). With notably smaller sample size, we did not detect significant associations between angiogenesis scores and DFS (Log-rank p=0.114) or OS (Log-rank p = 0.0601) in the MOFFITT cohort (Fig. S3D). However, there was a visible trend towards worse DFS and OS in tumors with the lowest quartile of angiogenesis score.
Integrated clinical and genomic models reveal independent predictive value of myeloid inflammation on clinical outcomes
Given our univariate findings for gene signatures reflecting myeloid inflammation (pro-metastasis) and tumor angiogenesis (anti-metastasis) gene signatures with respect to disease outcomes, we sought to integrate both TME features into and put them in context with established clinical and genomic strata. On univariate analysis, the MSKI score had no significant associations with tumor stage or grade (Kruskal-Wallis test, p=0.1649, Mann-Whitney test, p=0.099, respectively; Fig. 4A), however angiogenesis scores were significantly lower in high vs low grade tumors (Mann-Whitney test; p=4.41E-05; Fig. 4B), again without correlation with stage. We failed to detect associations between either MSKI or angiogenesis score and modified prognostic UISS scores (Fig. S4A–B). Next, we applied the validated genomic-based risk scores ClearCode34(4) and the CCP score(5) to the PROTECT cohort. Consistent with previous findings, the ClearCode34 poor risk group (ccB) had significantly shorter DFS than those in the good risk group (ccA) (Log-rank test; p=1.53E-06; Fig. S4C) and patients with high CCP scores from their tumors had shorter DFS than those with low CCP scores (Log-Rank test; p=2.15E-04; Fig. S4D). We noted significantly higher MSKI and lower angiogenesis scores in ccB vs ccA tumors (Mann-Whitney test, p=4.33E-10, p=8.01E-13, respectively; Fig. 4C). The MSKI scores were noted to have a positive correlation with CCP scores (Spearman, r=0.35; p=3.86E-08; Fig. 4D) and a negative correlation with angiogenesis scores (Spearman, r=−0.15; p=0.0179; Fig. 4D). Angiogenesis scores exhibited a negative correlation with CCP scores (Spearman, r=−0.39; p=8.0E-10; Fig. 4D). Taken together, tumors from patients with subsequent relapse of disease were more likely to be characterized as ccB group, had higher MSKI scores, but lower angiogenesis scores. We then integrated these four variables into a multivariate COX regression model and found that only the MSKI gene signature remained significantly associated with DFS (HR 1.28; 95% CI, 1.16–1.40; p=5.69E-07; Fig. 4E) and OS (HR 1.23; 95%CI, 1.09–1.39; p=7.81E-04; Fig. 4F). We also used 2-year timepoint receiving operating characteristic (ROC) curves and c-indices to assess the classification for both OS and DFS multivariable models. Models were run with and without MSKI as a covariate in order to determine the additive effects of MSKI, and we found that the addition of MSKI improved the c-index from 0.6 to 0.73 for DFS (Fig. 4G) and 0.66 to 0.72 for OS (Fig. 4H).
Figure 4. Univariate and multivariate analysis of relationships between TME gene signatures, previously validated risk models, and disease outcomes.
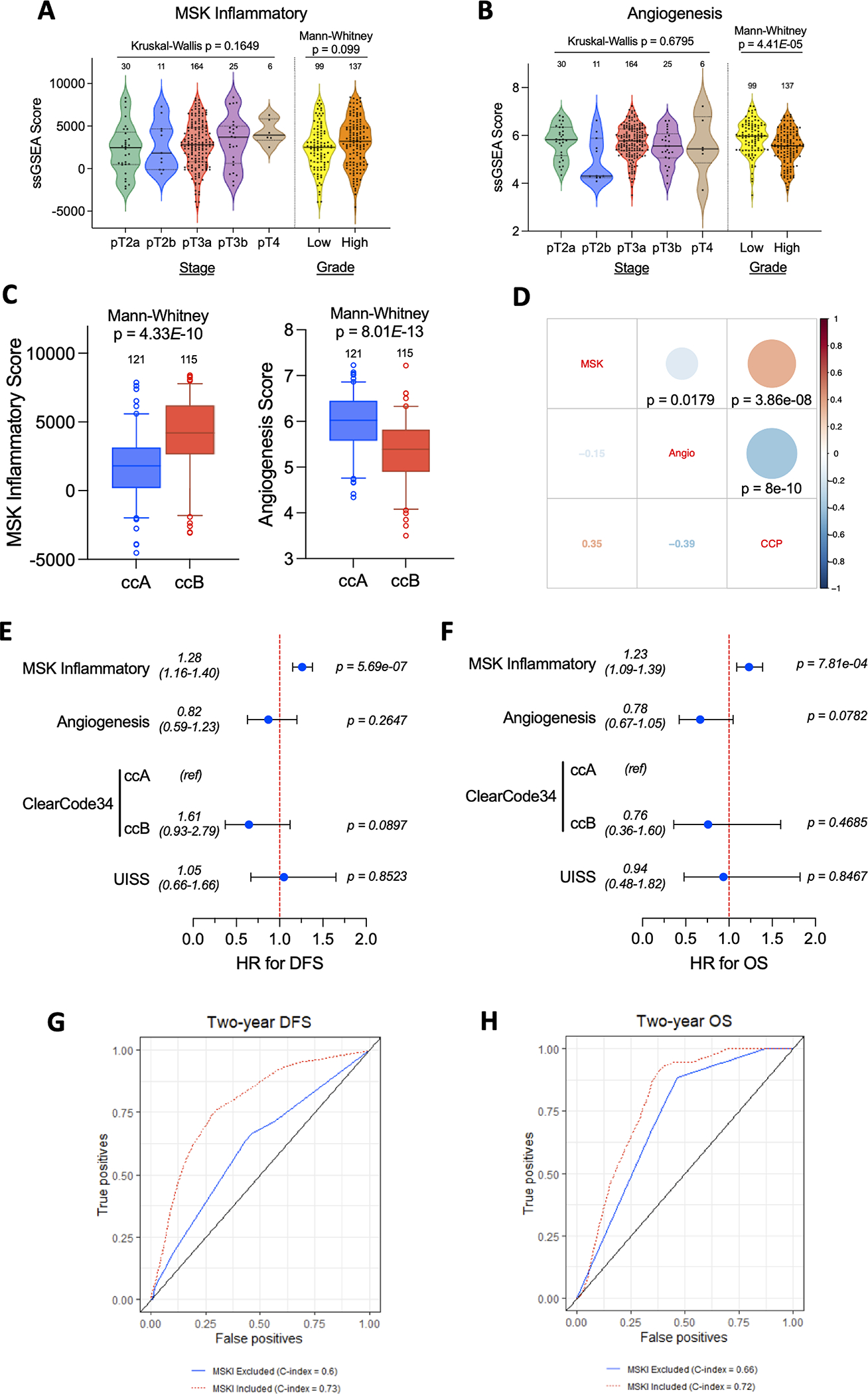
(A,B) Violin plots of (A) MSK inflammatory and (B) Angiogenesis score by tumor stage and grade. (C) Box plots of MSK Inflammatory and Angiogenesis score by ClearCode34 groups. (D) Correlation matrix displaying Spearman correlation coefficients (actual values in lower left-hand corner) with p-values obtained from two sided tests. (E,F) Forest plots displaying results of multivariate Cox regression for the listed variables with respect to (E) DFS and (F) OS. Two-year timepoint receiving operating characteristic (ROC) curves and c-indices to for both (G) DFS and (H) OS multivariable models. The DFS full model includes MSKI, ClearCode34, and UISS. The OS full model includes MSKI, CCP, and UISS.
Cell populations underlying and intratumoral heterogeneity of the MSK Inflammatory and angiogenesis gene signatures
To determine which intratumoral cell populations were contributing to the prognostic gene signatures in our cohort, we performed fluorescence-activated cell sorting (FACS) and RNA sequencing on 127 samples of 15 immune populations and non-CD45 cells isolated from ccRCC tumors, normal kidney and peripheral blood, from a total of 21 patients (Fig. S5 for flow gating strategy). In line with the cell populations underlying the adenosine and myeloid signatures reported in a recent single cell RNAseq analysis(18) we observed the highest MSKI median enrichment scores in myeloid cell populations including dendritic cells, neutrophils, macrophages, and monocytes (Fig. 5A).
Figure 5. Intratumoral heterogeneity and cell populations underlying the MSK Inflammatory signature.
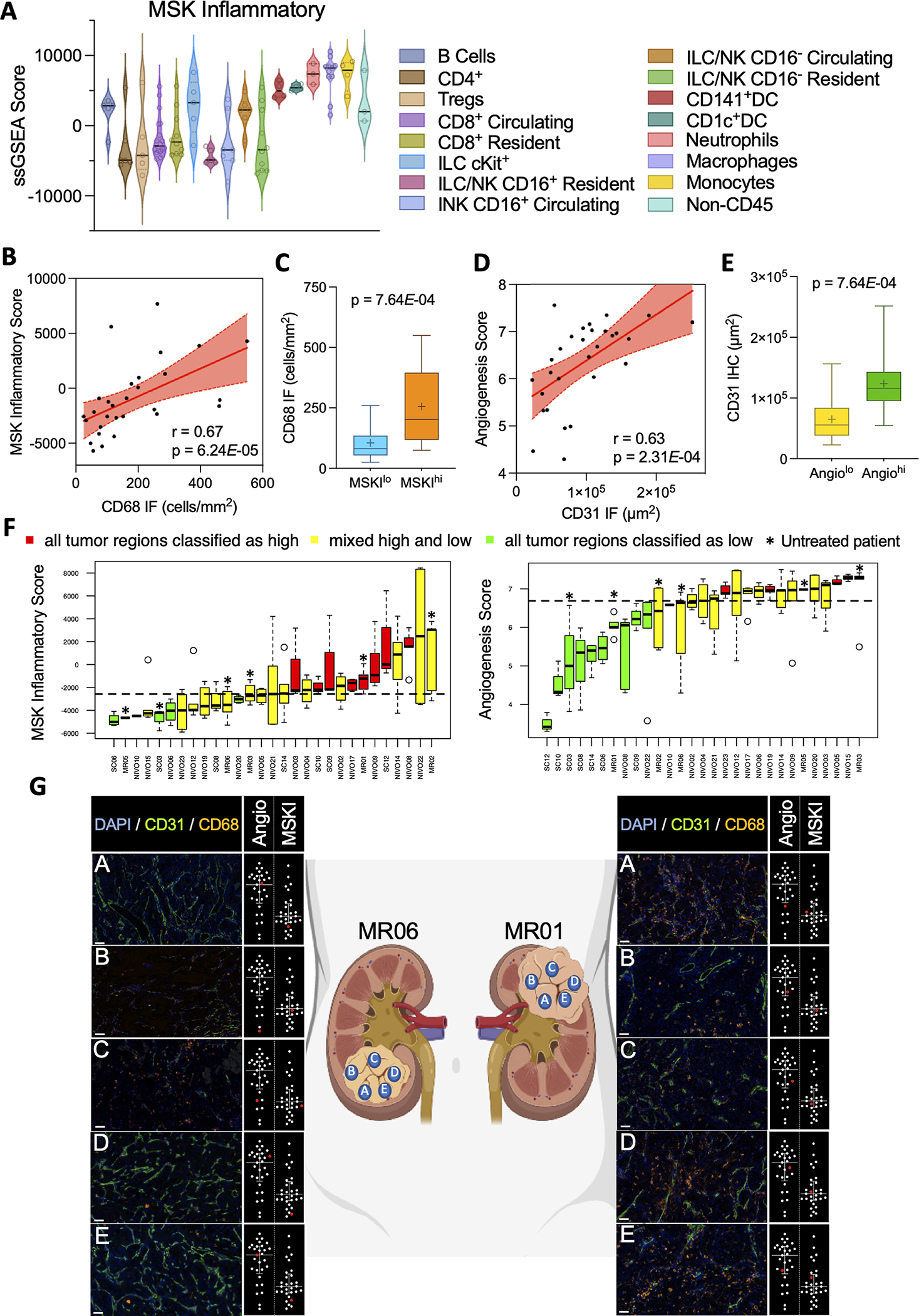
(A) Violin plots of MSK Inflammatory signature scores from fluorescence-activated cell sorted immune cell populations from ccRCC tumors. (B) Scatterplot showing correlations between MSK Inflammatory score and CD68 IF cell counts. Shaded areas represent 95% confidence intervals. (C) Box plots of CD68 IF cell counts in MSK Inflammatory high (MSKhi) vs MSK Inflammatory low (MSKIlo) tumors (based on median score) (D) Scatterplot showing correlations between Angiogenesis score and CD31 IF area derived from paired ccRCC tumor samples. Shaded areas represent 95% confidence intervals. (E) Box plots of CD31 IF areas in angiogenesis high (Angiohi) vs angiogenesis low (Angiolo) tumors (based on median score). (F) Box plots of MSK Inflammatory and angiogenesis score from multiregional tumor samples (n = 2–7 regions per tumor from total 29 patients). Each bar represents one individual patient. Dotted line represents the median enrichment score (G) Representative IF images corresponding to signature score dot plot (highlighted in red in inset) demonstrating intratumoral heterogeneity from two multiregional tumor samples. Scale bar = 50 μm
Multiregional tumor profiling studies have revealed a striking degree of intratumoral heterogeneity (ITH) in genomic alterations that have been associated with metastatic potential and clinical outcomes.(28–30) We therefore wondered whether our transcriptomic signatures were also vulnerable to ITH. To explore this, we analyzed a cohort of 29 ccRCC patients undergoing surgical resection at MSKCC where each patient had a median of 5 regions per tumor (range 2–7) collected for analysis. We first performed immunofluorescence (IF) staining in order to validate the MSKI and angiogenesis signatures at the protein level. We found strong positive correlations between the number of cells expressing the pan-macrophage marker CD68 and the MSKI signature (Spearman, r=0.67; p=6.24E-05; Fig. 5B) and significantly higher CD68 IF cell counts in MSKIhi versus MSKIlo tumors (Mann-Whitney test, p=7.64E-04; Fig. 5C). This suggests that although the MSKI signature is expressed by most myeloid cells (Fig. 5A), the signature may be predominantly capturing tumor-associated macrophages (TAMs) due to their high abundance in tumors. As expected, staining of the epithelial marker CD31 was also strongly correlated with angiogenesis scores (Spearman, r=0.63; p=2.31E-04; Fig. 5D), with higher CD31 IF scores in Angiohi versus Angiolo tumors (Mann-Whitney test, p=7.64E-04; Fig. 5E).
We then explored the ITH of MSKI and angiogenesis scores by performing RNAseq on this cohort. Overall, there was not a marked degree of ITH with respect to these microenvironmental features (Fig. 5F). We found that all regions analyzed in 48.3% and 58.6% of tumors were uniformly above or below the median enrichment score for the MSKI and angiogenesis signatures, respectively. Representative images of CD31 and CD68 IF and the corresponding angiogenesis and MSKI scores, respectively, for two multiregional tumor samples are highlighted in Figure 5G.
Development of a novel electroporation-derived ccRCC syngeneic model
We next sought to validate the importance of myeloid inflammation in RCC recurrence in a mouse model. Traditional germline-modified genetically engineered mouse models (GEMMs) most accurately recapitulate natural malignant disease progression, however it can take many months to both cross the various alleles sufficient for tumor formation and expand the experimental colony. Additionally penetrance in RCC models is not optimal, and tumor onset time can be highly variable between mice taking a minimum of 2–5 months following tumor suppressor gene deletion or oncogene activation(31–33). Lastly, of the existing ccRCC GEMMs, only one has been reported to develop metastases but this occurs at a very low frequency(31–34), thus limiting studies on mRCC.
To generate a more tractable and biologically relevant metastatic ccRCC model, the left kidney of wildtype (WT) C57Bl/6 mice were externalized, followed by subcapsular injection of a 50μl cocktail of plasmids encoding a transposase system to stably integrate EF-1ɑ promoter-driven c-myc overexpression, transient expression of Cas9 and sgRNA guides targeting Vhl, Rb1 and Tp53. We chose to model this genetic combination due the extensive molecular similarities of a GEMM encompassing inducible kidney-specific deletion of Vhl, p53, and Rb1 (KVpR) to human ccRCC.(32) Tp53 is also enriched in metatatic cohorts(7,16,19,35) and is associated with a myeloid high phenotype.(7) Additionally C-myc is amplified in 8–15% of human ccRCCs and associated with development of metastases.(32,36) Following plasmid injection, the kidney and injection site was immediately electroporated via electrode tweezers, and the wound surgically closed. At 48.7 weeks post-electroporation a large tumor in the left kidney was detected by MRI, dissected, dissociated, and cultured to derive a syngeneic cell line (Fig. 6A−B). No overt metastases were detected.
Figure 6. Novel electroporation-derived ccRCC syngeneic model is metastatic and transcriptomically resembles human stromal/proliferative ccRCC molecular subtype.
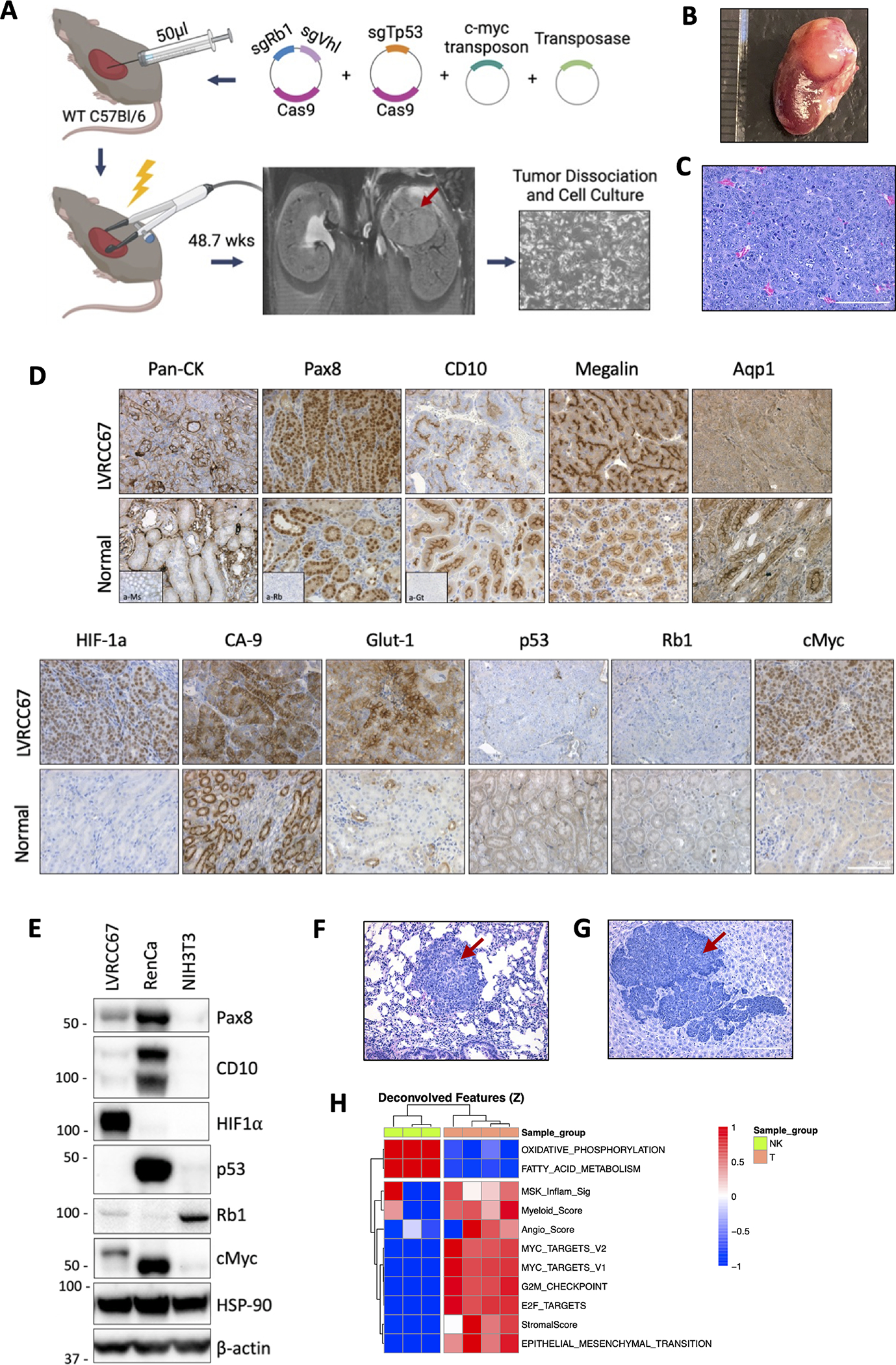
(A) Schematic of ccRCC syngeneic cell line development using electroporation of somatic tissues. Photo (B) and H&E (C) and IHC (D) of the parental kidney tumor and surrounding normal kidney. (E) Western blot of the EP-derived LVRCC67 cell line, RenCa and NIH3T3 cells are used as controls. H&E of lung (F) or liver (G) metastatic nodules following subcutaneous injection of LVRCC67 cells into WT C57Bl/6 mice. (H) Heatmap of ssGSEA scores in LVRCC67 tumors compared with normal kidney cortex samples from WT C57Bl/6 mice. Red arrows indicate tumor area. Scale bar 100μm. Schematic was made with BioRender.
Novel syngeneic tumors molecularly resemble ccRCC and spontaneously metastasize to distant organs
Gene editing of the parental tumor and derived cell line were confirmed by sanger sequencing (Fig. S6A). A 1 base-pair (bp) deletion was detected in all three targeted genes at the expected cut site, confirming that the tumor was electroporation-derived rather than spontaneous. Histologically, the parental tumor was high grade and highly dedifferentiated (Fig. 6C). Staining for neuroendocrine markers synaptophysin and chromogranin A were negative (Fig. S6B) despite areas of histological large cell neuroendocrine features (Fig. 6C). Since ccRCC originates from kidney proximal tubule cells(37) and VHL loss is used as a clinical diagnostic marker of ccRCC in poorly differentiated tumors, we confirmed by IHC that the tumor was epithelial (pan-CK+) and of kidney (Pax8+) origin (Fig. 6D). Two of three proximal tubule markers (CD10+, Megalin+, Aquaporin 1−) were present, and deletions at the protein level of Vhl (via downstream upregulation of HIF-1ɑ, carbonic anhydrase-9 and Glut-1), p53 and Rb1 plus myc overexpression were confirmed (Fig. 6D). We successfully derived a cell line from this tumor, LVRCC67, which grows rapidly in vitro and retains these molecular markers (Fig. 6A, Fig. 6E).
Whole exome sequencing of LVRCC67 confirmed CRISPR-mediated 1bp deletion in Vhl, Tp53 and Rb1, and failed to detect additional oncogenic events (Supplementary Table 4). However, there were large deletions in chromosome 10 as well as substantial copy number alterations in small regions of chromosome 13 (Fig. S6C and Supplemental Table 5). When injected subcutaneously into WT C57Bl/6 mice, LVRCC67 tumors retain their dedifferentiated histology (Fig. S6D). However, unlike the parent tumor, metastases to the lung (Fig. 6F) and/or liver (Fig. 6G) were readily observed in 80% (4/5) of mice and these metastatic nodules also exhibited dedifferentiated morphologies.
Syngeneic tumors resemble an aggressive stromal/proliferative human ccRCC molecular subtype
A recent study analyzing 823 advanced ccRCC tumors identified 7 molecular subtypes with distinct biological signatures(7) and differential prognosis(38). In order to compare our mouse model with these molecular clusters we performed bulk RNA sequencing of subcutaneous LVRCC67 tumors and healthy normal kidney cortex tissue, and looked for enrichment of hallmark gene set signatures from the molecular signatures database (MSigDb)(39) as well as TME gene signatures. We observed that our mouse tumors had strong upregulation of gene signatures related to cell cycle (myc targets, G2M checkpoint, E2F targets), EMT and stroma, as well as genes associated with anabolic metabolism (higher FAS/Pentose phosphate, reduced oxidative phosphorylation and reduced fatty acid metabolism; Fig. 6H, Fig. S6E). Tumors were angiogenic, consistent with Vhl-loss-mediated pseudohypoxia and known roles of p53 in regulating angiogenesis(40). We also observed enrichment of myeloid and MSKI signatures, together suggesting the model strongly resembles one of the 7 molecular subtypes (the stromal/proliferative molecular cluster 6, referred to as SPMC hereon) reported in the above mentioned study of human ccRCC(7). Our model also aligns well genomically with this molecular subtype, as TP53 mutations were highest in this cluster (29%)(7). Although CDKN2A/B alterations were enriched in SPMC tumors but not in our mouse tumors (Supplementary table 4−5), this is consistent with the finding that CDKN2A/B and TP53 mutations were largely non-overlapping.(7)
Patients with SPMC tumors have a particularly poor prognosis compared with other molecular subtypes(38). In line with this, pathways associated with this aggressive tumor type (EMT, cell cycle, myeloid) also seemed to be enriched in recurrent tumors within the PROTECT cohort (Fig. 1B). We therefore clustered PROTECT cohort tumors by recurrence status and MSKI gene expression quartiles and looked specifically at expression of gene signatures used to define the 7 molecular subtypes described in the IMMOTION151 analysis (Fig. S7). Indeed, the gene expression profiles defining SPMC tumors (described above for mouse tumors) were enriched in recurrent tumors and within the highest MSKI quartile. Our data suggests that the aggressiveness of this analogous molecular subtype may be driven by a myeloid inflammatory program.
Given the enrichment of TP53 alterations within SPMC tumors and the p53-driven foundation of our metastatic mouse model, we wondered whether there was also an association between TP53 mutations and MSKI score, which predicts recurrence. Although genomic data was unavailable for the PROTECT cohort, we confirmed in two previously described clinical cohorts(19,41) that TP53 mutations were indeed associated with significantly higher MSKI scores (JAVELIN101 p=0.006, COMPARZ p=0.033; Fig. S8A–B).
Adenosine receptor but not PD-1 inhibition attenuates ccRCC metastasis
We next leveraged our mRCC model to test whether targeting adenosine/myeloid-mediated inflammation and the MSKI-high environment could prevent metastases. To do this, mice were injected subcutaneously with LVRCC67 cells followed by daily constitutive treatment from day 2 with Ciforadenant (CPI-444)(14), a small molecule high affinity selective inhibitor of the immunosuppressive adenosine A2A receptor (A2AR), or twice weekly treatment of anti-PD-1. Primary tumor volumes were measured, and mice were sacked on day 35 to assess metastatic burden.
CPI-444 significantly reduced primary tumor growth (Fishers LSD test; p=1.4E-05) whereas anti-PD-1 treated mice had a heterogenous response that was not overall statistically significant (Fishers LSD test; p=0.0858; Fig. 7A). Upon dissection, only 27% (4/15) of CPI-444-treated mice, compared with 65% (9/14) of control and 71% (5/7) of anti-PD-1-treated mice had macroscopic metastases (fisher’s exact test; control compared with CPI-444 p=0.065; Fig. 7B). Additionally, CPI-444 but not anti-PD-1-treated mice had a lower abundance of metastatic nodules per mouse (Wilcoxon rank sum test; control versus CP444 p=0.024, or anti-PD-1 p=0.91; Fig. 7C).
Figure 7. Adenosine receptor, but not PD-1 inhibition attenuates spontaneous ccRCC metastasis.
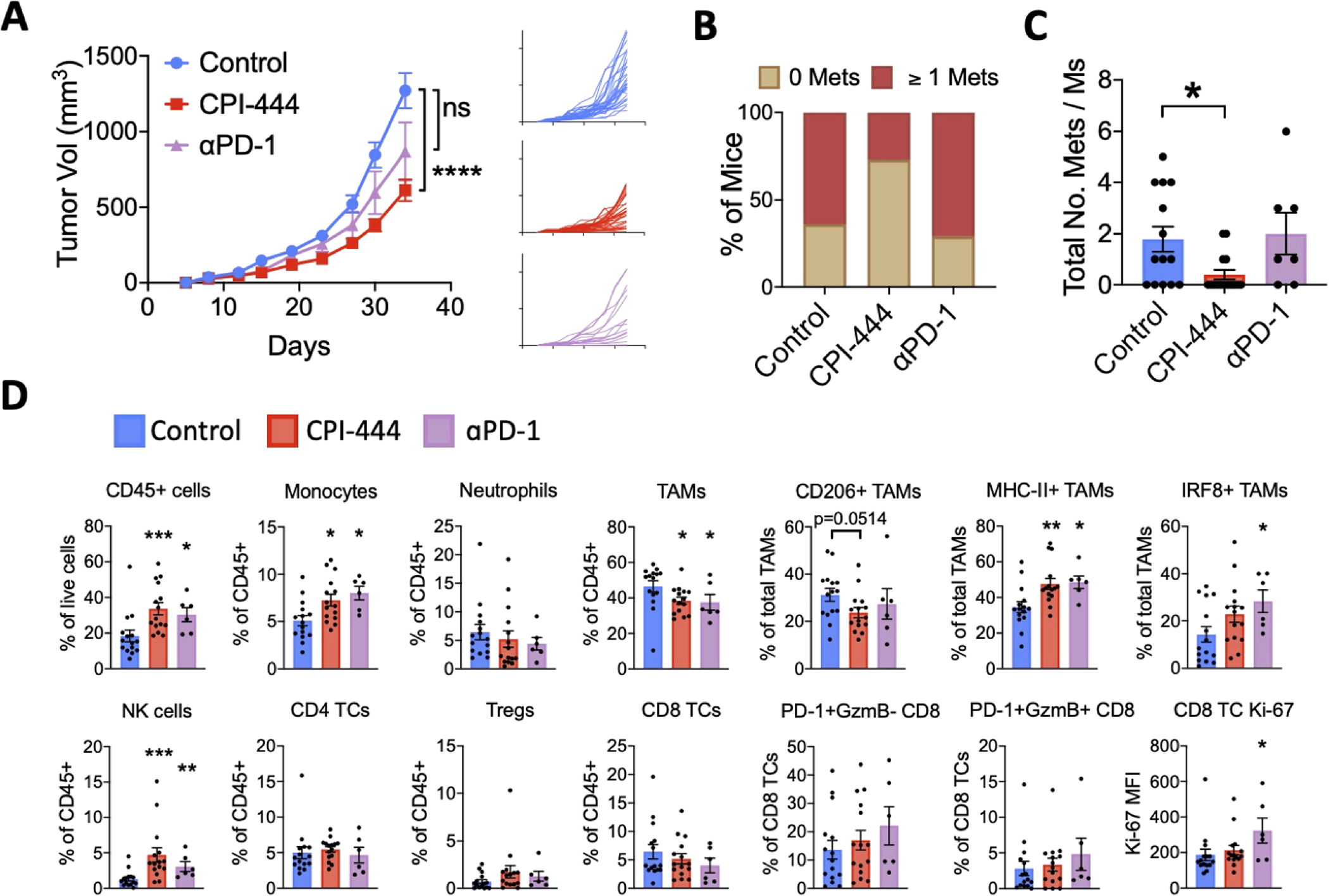
Subcutaneous LVRCC67 tumor-bearing mice were treated from day 2 post-injection with either 100mg/kg daily of the adenosine A2A receptor inhibitor CPI-444 self-administered via medicated chow (n=15), or 100μg of anti-PD-1 therapeutic antibody (n=7) delivered intraperitoneally twice weekly, or control chow plus 100μg of IgG isotype control antibody (n=15). (A) Average (left panel) and individual (right panel) primary tumor volumes are shown. On day 35 mice were sacked and the number of macroscopically visible metastatic nodules in liver and lung were counted (B-C). Primary tumors were dissociated and immune cells were analyzed by flow cytometry (D). Control versus treatment group tumor volumes were compared using a two-way ANOVA with Geisser-Greenhouse correction and Fisher’s LSD post hoc test. The number of metastases and immune populations were compared using Wilcoxon rank sums test. All analyses compared treatment groups to the control group. *p<0.05, **p<0.01, ***p=<0.001, ****p=<0.0001. Error bars show mean ± SEM. Tumor-associated macrophages, TAM; T cells, TCs; Median fluorescence intensity, MFI.
Due to insufficient metastatic tumor tissue, we used primary tumors to assess drug-related changes to the immune TME using flow cytometry (Fig. 7D, S9A–B). Control tumors were myeloid dominated, mostly by TAMs which constitute almost 50% of CD45+ cells. Other myeloid cells included low levels of monocytes and neutrophils. Importantly, tumor infiltrating CD8+, CD4+ and regulatory T cells (Tregs) were found in low abundance. Together this validates our RNAseq data that suggests LVRCC67 tumors are myeloid-rich and T cell-low, similar to SPMC human ccRCCs.
As expected, we found that overall CD45+ cell infiltration was significantly enhanced by both CPI-444 and anti-PD1 treatment (Fig. 7D). We observed a striking increase in natural killer (NK) cells following CPI-444, and to a lesser degree following anti-PD1 (Fig. 7D). Adenosine is known to affect the proliferation, maturation, and cytotoxicity of NK cells(42) and may therefore be an important mechanism of tumor control. Flow sorted populations show that NK/innate lymphocyte cell (ILC) populations can also express the adenosine signature, but at lower levels than myeloid cells (Fig. 5A).
Both drugs significantly increased the percentage of CD11b+Ly6C+ monocytes suggesting enhanced recruitment from blood, however total CD11b+F4/80+CD80+ TAMs were significantly reduced (Wilcoxon rank sums test; p=0.00150 and p=0.0433 for CPI-444 or anti-PD-1 versus control respectively; Fig. 7D). Further TAM phenotyping showed a reduction of M2-like CD206+ TAMs and enrichment of M1-like TAMs that express MHC-II and IRF8, a transcription factor necessary for antigen presentation(43), in line with adenosine-mediated immunosuppression of TAMs(42). The majority of CD8+ T-cells were neither activated (PD-1+granzyme B+) nor exhausted (PD-1+granzymeB−) suggestive of blood circulating or bystander naïve populations (Fig. 7D). In accordance with a T cell-cold tumor, we did not see substantial and consistent changes in T cell activation, exhaustion or proliferation following CPI-444 or anti-PD-1 treatment (Fig. 7D).
We noticed that within the control group some mice were more prone to metastases than others and wondered whether there were underlying immune TME differences. We observed the same upward shifts in NK cells and M1-like TAMs in control mice without metastases and no T cell-associated changes (Fig. S9C). Taken together, our preclinical data supports a T cell-independent, TAM- and NK-mediated mechanism of recurrence, consistent with our clinical data, and suggests that the drugs may be enhancing pre-existing anti-metastatic immunity.
Discussion
In this report we describe the results of a comprehensive transcriptomic analysis integrating clinicopathologic, flow cytometric, and immunofluorescence data from patients with high-risk localized ccRCC receiving placebo on a randomized phase 3 trial (PROTECT), the TCGA, and smaller, independent cohorts. Our unbiased IPA core analysis identified IL6 as a key component of the various upregulated molecular pathways in recurrent vs non-recurrent tumors. Elevated serum and intratumoral IL6 have been associated with higher rates of lymphatic and distant metastases(44,45) and worse disease outcomes in ccRCC.(46–50) In human RCC samples, myeloid-derived cytokines are positively correlated with intratumoral MDSCs, and blockade of IL1β and IL6 in mice decreased tumor MDSC infiltration, increased antitumor T cell responses, and decreased tumor growth.(51,52)
We found a striking association between gene signatures representative of myeloid inflammation and disease outcomes post nephrectomy that was validated in two independent cohorts and maintained significant prognostic value on multivariate analysis. Our findings are supported by a recent study identifying an association of C1Q+TREM2+APOE+ TAMs with recurrence in ccRCC in a small cohort.(53) Since our MSKI signature captures, but is not specific to this C1Q+ TAM population(18), future functional validation using higher resolution tools are needed. Although the basis of our signature is not novel, the genes comprising the adenosine and myeloid signatures were refined via TCGA analysis so that only the most relevant genes were combined to create the MSKI signature. This is the first reported application of these signatures in the localized disease recurrence setting.
Our analysis assessing tumor CD8 T cell density, lymphocyte infiltration patterns, exhaustion states and tumor PD-L1 expression, failed to detect an association with disease outcomes post nephrectomy. There have been conflicting data on the prognostic significance of intratumoral CD8 cell counts in ccRCC(24,25,54,55) and our data adds to the studies that did not find an association with tumor recurrence. A recent single cell RNA sequencing (scRNAseq) study reported increasing interactions between M2-like TAMs and exhausted CD8 T cells with advancing ccRCC disease stage(26) and the IMMOTION150 trial also suggested that myeloid cells can suppress ICB-induced anti-tumor T cell responses in ccRCC.(15) However, in the present study myeloid abundance neither impacted T cell exhaustion nor the prognostic ability of Teff signatures. In line with this, Senbabaoglu et al.(56) reported two T cell-enriched clusters in the TCGA cohort (termed TCa and TCb), of which the TCb cluster had significantly greater macrophage infiltration and worse cancer-specific survival whereas TCa clusters had similar survival to non- and poorly-infiltrated groups, suggesting that T cells may not be prognostic. No p53 mutations were reported in the 13 patients profiled by scRNAseq(26) perhaps highlighting that metastatic progression of the SPMC tumor subset is not governed by T cells and potentially point toward myeloid regulation of earlier events in the metastatic cascade such as stromal remodelling or EMT(57).
Patients with highly angiogenic tumors in the PROTECT cohort were less likely to develop metastatic disease and had more favorable OS compared to poorly angiogenic tumors. While we did not detect any significant associations between angiogenesis and disease recurrence in the TCGA-KIRC cohort, we noted poorly angiogenic tumors had significantly worse OS. Interestingly, analyses from recent clinical trials in mccRCC suggest that angiogenesis signatures may constitute a biomarker of response to anti-VEGF agents.(7,15,16,19) Taken together with our data showing enrichment of angio-low tumors in mccRCC patients, which are intrinsically less responsive to anti-VEGF agents, this may provide an explanation for the lack of efficacy of previous anti-VEGF TKI adjuvant trials.
In order to facilitate rapid basic and translational mRCC research we generated a relevant, syngeneic model of mccRCC on a pure C57Bl/6 background using electroporation to deliver CRISPR/Cas9 vectors to murine organs in vivo. These methods have been successful in initiating tumor development in a number of different cancer types including liver, pancreatic, prostate and ovarian.(58–62) This is the first report of this approach in kidney and we experienced very low tumor penetrance. Tumor induction in murine kidneys has been extremely challenging, likely owing to its intrinsic low cell turnover, limited regenerative capacity and tumor-suppressor rather than oncogene-driven nature(63,64). Although we were fortunate to generate a syngeneic cell line, further optimization of the electroporation approach is greatly needed to enable rapid and flexible genome editing as originally intended. Nevertheless, our model offers a molecularly-relevant alternative to the only available RCC syngeneic line, RenCa, which does not harbor Vhl loss, the most frequent initiating genomic event in ccRCC(64). LVRCC67 tumors transcriptomically resemble a TP53-mutation enriched molecular subtype of human ccRCC characterized by high cell proliferation, EMT, myeloid cells and poor prognosis. Encouragingly the genomically-matched KVpR autochthonous mouse model has a similar transcriptomic and immune profile(65) supporting that key aspects of the tumorigenesis process in our implantation model may be conserved.
Our data supports a potential role for TP53 loss in driving metastases in ccRCC. Although there was no significant difference in p53 genomic alterations in primary tumors between ccRCC patients with or without metastases in the recently described MSK-MET cohort(66), multiple IHC-based studies have shown TP53 alterations to be significantly correlated with poor disease-free survival in RCC (67–73). We found striking transcriptional similarities between our p53-driven mouse model and human TP53-enriched myeloid-dominant SPMS tumors, and similar enriched pathways within recurrent tumors of the PROTECT cohort. We observed enrichment of cell cycle and EMT pathways in recurrent human and mouse tumors, which may reflect the reported roles of p53 in regulating these processes(74,75).
We linked TP53 mutations with higher MSKI scores in the COMPARZ and JAVELIN101 cohorts. Several studies in autochthonous mouse models(75–77) suggest a direct role of tumor cell p53 loss in immunosuppressive myeloid recruitment, differentiation and function. This may occur through enhanced release of myeloid chemokines from p53-null tumor cells including CXCL17, IL-6, CXCL3 and CXCL5, three of which contribute to the MSKI gene signature(76,77). Additionally, IL-1β (also a component of the MSKI signature) release from TAMs can be induced by p53 loss-mediated Wnt autocrine signaling, which subsequently promotes ɣδ T cell-mediated expansion of systemic cKIT+ immature neutrophils that facilitate metastasis, with no reported changes in CD8 and CD4 T cells.(78) It is possible that this tumor cell p53-TAM-neutrophil axis also governs metastases in RCC since high neutrophil-to-lymphocyte (NLR) ratio ͣɣis a known prognostic factor.(79)
Using our SPMC-like mouse model we evaluated the efficacy of myeloid-inhibition on disease recurrence. Fong et al., have shown that patients with high adenosine signatures have greater responses to the adenosine A2AR inhibitor CPI-444(14). Indeed CPI-444 reduced primary and metastatic tumor burden in our MSKI-rich tumors. Importantly, durability of response needs to be evaluated in future experiments where the primary tumor is resected enabling longer follow-up times.
Interestingly we observed a lack of efficacy and associated changes in T cell phenotypes following anti-PD-1 treatment in mice. Molecular subgroup analyses indicate that ccRCC patients harboring the SPMC subtype of tumors are not responsive to first-line atezolizumab plus bevacizumab.(38) This raises concerns that despite overall efficacy with anti-PD-1 monotherapy seen in the KEYNOTE-564 trial(1), this subtype may also preclude response to ICB in the adjuvant setting. However, whether SPMC tumors also respond poorly to other more efficacious ICB VEGF combination approaches remains to be determined, although there does not seem to be an association between TP53 mutations and response to avelumab and axitinib(16).
Our data suggests that targeting the adenosine pathway may be beneficial in this myeloid high and angiogenesis low setting. In our study we used an adenosine A2AR antagonist but adenosine signaling via the immunosuppressive A2B receptor or other components involved in adenosine regulation may constitute a resistance mechanism. Our mouse model provides a valuable means to test additional adenosine-pathway inhibitors that are currently in early phases of clinical development(42), for RCC adjuvant therapy.
Our study has limitations. While the PROTECT cohort received no prior systemic therapies, overall survival analyses may be confounded by treatment given in the metastatic disease setting. We were not able to obtain sequencing data from patients enrolled on the treatment arm of the PROTECT trial. However, we do speculate that, based on our prior transcriptomic analysis from the COMPARZ trial(80), myeloid high tumors would likely see worse outcomes compared to myeloid low tumors in patients receiving pazopanib. In the MOFFITT cohort the MSKI score was significantly associated with recurrence but only trending for OS, potentially due to the relatively smaller patient population, necessitating validation in larger independent cohorts. Although the PROTECT cohort has a skewed distribution of T stages with no pT1 tumors and consisting of 73.9% pT3a tumors, the MSKI signature was prognostic in the TCGA cohort suggesting relevance beyond adjuvant trial populations.
In summary this study highlights the crucial independent role of myeloid inflammation in the development of metastatic ccRCC. In contrast to the pervading paradigm, this axis does not seem to be reliant on CD8 T cell effector function, nor regulated through well-established immune checkpoints. We posit that a substantial patient subgroup with TP53 altered treatment refractory ccRCC tumors may derive benefit from blockade of pleiotropic immunosuppressive adenosine signaling, and we provide a valuable preclinical model to advance our understanding and treatment of this aggressive subtype. More generally, this work emphasizes the importance of molecular subtyping for patient selection in future adjuvant trials.
Methods
PROTECT Cohort.
The phase 3 PROTECT (Pazopanib As Adjuvant Therapy in Localized/Locally Advanced RCC After Nephrectomy) study enrolled >1500 patients with resected pT2 (high grade), ≥pT3 or N1 (any grade) clear cell RCC with 1:1 randomization to 1 year of pazopanib vs. placebo. None of the patients had received any prior systemic therapy. As previously reported(17), the study failed to demonstrate disease free survival (DFS) benefit of adjuvant pazopanib. All patients provided archival tumor tissue at the time of enrollment. We analyzed tumor tissue, demographic and clinical data from patients enrolled on the placebo arm of the trial, which was conducted in accordance with the Declaration of Helsinski. All patients provided written informed consent for participation in the clinical trial.(17)
MOFFITT Cohort.
We obtained 84 tumor samples from 84 patients with RCC, from the years 2004–2018, through protocols approved by the institutional review board (H. Lee Moffitt Cancer Center and Research Institute’s Total Cancer Care protocol; Advarra IRB Pro00014441). Samples were obtained via surgical excision. None of the patients had received any prior systemic therapy. Written informed consent was obtained from all tissue donors. Patients were included if they had clinically localized ccRCC, provided written consent to the molecular characterization of their tissue and did not receive any systemic therapy prior to tissue collection.
Patient tumor samples then underwent microdissection and bulk RNA sequencing. The TruSeq RNA Exome kit (Illumina) for 50 million 100–base pair paired-end reads was utilized, and RNA sequence reads were aligned to the human reference genome in a splice-aware fashion using Spliced Transcripts Alignment to a Reference (STAR)(81), allowing for accurate alignments of sequences across introns. Aligned sequences were assigned to exons using the HTseq package(82) to generate initial counts by region. Normalization, expression modeling, and difference testing were performed using DESeq2(83).
MSKCC Cohort.
This cohort was composed of 21 patients undergoing partial or radical nephrectomy at with pathologic features consistent with those required for eligibility on the PROTECT trial.(17) Briefly, patients were > 18 years of age with nonmetastatic clear cell or predominantly clear cell ccRCC histology, underwent partial or radical nephrectomy and had intermediate or high-risk disease as defined by the SSIGN system. Formalin-fixed paraffine-mbedded (FFPE) tumor blocks were collected for histopathologic and immunofluorescence analyses.
TCGA-KIRC Cohort.
Data were abstracted from 534 patients with ccRCC profiled by TCGA with RNAseq and OS data available.(84) Of these, 63% (336) patients had RNAseq and DFS data available. Data were downloaded from form the NIH Genomic Data Commons (https://gdc.cancer.gov).
MSKCC Multiregional RNA Sequencing Cohort.
This cohort consisted of 2–7 tumor regions from 29 patients obtained from the operating room during nephrectomy. At the time of specimen extraction, samples 1–1.5 cm in largest dimension were obtained by the treating surgeon (A. A. H.) from spatially distinct tumor regions (at least 1 cm apart).
RNA sequencing alignment.
RNAseq raw read sequences were then aligned against human genome assembly hg19 (Feb.2009/GRCh37, https://genome.ucsc.edu/cgi-bin/hgGateway?db=hg19) or mm10 (Dec.2011/GRCm38, https://genome.ucsc.edu/cgibin/hgGateway?db=mm10) for human or mouse samples respectively by STAR 2-pass(81) alignment. RNAseq gene level count values were computed by using the R package GenomicAlignments(85) over aligned reads with UCSC KnownGene(86) in hg19 or mm10 as the base gene model. The Union counting mode was used and only mapped paired reads were considered. Fragments per kilobase million (FPKM) values were then computed from gene level counts by using fpkm function from the R package DESeq2.(83)
Microarray Analyses.
Two hundred thirty-six patients had microarray and clinicopathologic data available for analyses. RNA from the primary tumors was extracted by AltheaDx in 2013 using the Qiagen RNAeasy FFPE kit with a modified deparaffinization step. Gene-expression profiles were derived via Affymetrix GeneChip HTA 2.0 (Affymetrix). Microarray RNA data were normalized to log2 value. Probes without corresponding gene symbol found were excluded from further analysis. For genes matched with multiple probes, the probe with maximum median absolute deviation (MAD) was chosen for representing the expression of the gene. The log2 normalized expression values were used for immune deconvolution analyses.
RNA Expression Based Gene Signatures.
Gene signatures were defined as follows: Angiogenesis(15): VEGFA, KDR, ESM1, PECAM1, ANGPTL4, and CD34; Teff (15): CD8A, EOMES, PRF1, IFNG, and CD274; Myeloid(15): IL6, CXCL1, CXCL2, CXCL3, CXCL8, and PTGS2; Javelin(16): CD3G, CD3E, CD8B, THEMIS, TRAT1, GRAP2, CD247, CD2, CD96, PRF1, CD6, IL7R, ITK, GPR18, EOMES, SIT1, NLRC3, CD2, CD96, PRF1, CD244, KLRD1, SH2D1A, CCL5, XCL2, CST7, GFI1, KCNA3, and PSTPIP1; Adenosine(14): CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL8, IL-1B, PTGS2; MSK Inflammatory: CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, IL6, IL1B; ImmuneCheckpoint: CD274, CTLA-4, HAVCR2, LAG3, PDCD1, PDCD1LG2, TIGIT; Immunosuppression(27): CD274, IDO1, FASLG, CTLA-4, PDCD1, LAG3, HAVCR2, PDCD1LG2, IL10, TNF, TGFB1, IL12A, PTGS2. SsGSEA takes the sample gene expression values as the input and computes an overexpression measure for the given gene list of immune cell type relative to all other genes in the transcriptome. The specific method of deriving Angiogenesis, Teff, Myeloid and Javelin scores was according to their corresponding publications. Other TME feature gene enrichment scores were quantified using the ssGSEA implementation through the R package “gsva”.(87,88) ESTIMATE was used to calculate an immune score, which is the estimate of immune cell component in tumor tissue and is calculated through the “estimate” R package(89) based on given gene expression profile in FPKM or normalized log2 transformed values.
Analysis of Differentially Expressed Genes.
The R package “limma” (version 3.32.10) was used for DEG.(90) Limma returned empirical Bayes moderated-t P values and adjusted P values (Q-value) to correct for multiple comparisons testing using the Benjamini–Hochberg method to control the false discovery rate (FDR). Genes with mean expression (log2 value) greater than 1, a nominal P value less than 0.05 and fold change greater than 20% were subjected to IPA analysis.
Ingenuity Pathway Analysis.
IPA (Ingenuity) was used for running Canonical Pathway, Upstream Regulators, Diseases and Biofunctions analyses over the genes differentially expressed between patients with a disease recurrence and those without disease recurrence.
Gene Set Enrichment Analysis.
DEG analysis results were used in GSEA(91) analyses against the MSigDB Hallmark gene sets.(39)
FACS cell sorting.
Sorted cell populations were derived from 21 patients with renal cell carcinoma with a total of 127 unique samples representing 16 distinct flow sorted populations. Of these, 106 were sorted from tumor tissues and 21 monocyte samples were sorted from peripheral blood (n=20) or normal kidney (n=1). For bulk RNA sequencing, immune populations were sorted on Aria II (Becton Dickinson) into Trizol LS and flash frozen. RNA was extracted with chloroform. Isopropanol and linear acrylamide were added, and the RNA was precipitated with 75% ethanol. 0.649–1 ng total RNA with RNA integrity numbers 6.8–10 underwent amplification using SMART-Seq v4 Ultra Low Input RNA Kit (Clontech #63488). 15ng amplified cDNA was used to prepare libraries with KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using 8 cycles of PCR. Samples were barcoded and run on a HiSeq 2500 in 50bp/50bp paired end run using HiSeq3000/4000 SBS Kit (Illumina) for 30–40 million paired reads. The raw FASTQ files were processed as described above.
Immunohistochemistry for PROTECT Cohort.
Evaluation of multiple exploratory markers (targets) using IHC analysis was done using the following primary antibodies: CD8 (C8/144B; Dako), CD68 (KP-1; Ventana), PD-L1 (28–8; Dako), TIM-3 (D5D5RTM; Novartis) and LAG-3 (17B4; Novus Biologicals). The Ventana Benchmark XT autostainer was used with the following detection systems: Ultraview DAB (CD8, CD68), Optiview DAB with (TIM-3) or without (LAG-3) amplification. Envision DAB detection system was used for PD-L1 staining using the Dako Link 48 platform. PD-L1 IHC positivity was determined as a percentage of stained tumor cells (TPS). Batch analysis of CD8 IHC, CD68 IHC, LAG3 IHC and TIM3 IHC were used to determine the amount of IHC staining by measuring the area (Marker area) of the IHC-stained cells or structures in the center and periphery of a tumor. All IHC-stained slides were evaluated by a pathologist using the universal immune cell marker scoring algorithm. The output of this analysis is the relative surface area (%) of the tumor with a marker-positive immune cell density that belongs to one of the 8 density bins, and for each of the 8 density bins (8 data fields per IHC slide). The density bins are linked to reference images that are used by the pathologist and generated during extensive IHC assay validation. There are 4 density bins for intra-stromal marker-positive immune cells and 4 density bins for intra-epithelial marker-positive immune cells. In addition, the immune phenotype was scored as ‘desert’, ‘inflamed’, ‘excluded’ in the IHC slides. CD8 cell density measures for the intratumoral and stromal compartments were divided into high and low based on a median cutoff. ‘Excluded’ tumors were defined as high stromal and low intratumoral CD8 cell densities. ‘Desert’ tumors were defined as low stromal and intratumoral CD8 cell densities. The rest, including high stromal/high intratumoral and low stromal/high intratumoral were defined as ‘inflamed.’ In addition, and only for TIM3, the percentage of marker-positive tumor cells was estimated.
Immunofluorescence for MSKCC Cohort.
The Immunofluorescence detections of CD31 and CD68 were performed at Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery Ultra processor (Ventana Medical Systems.Roche-AZ). After 32 min of heat and CC1 (Cell Conditioning 1, Ventana cat #950-500) retrieval, the tissue sections were blocked first for 30 min in Background Blocking reagent (Innovex, catalog#: NB306). A mouse monoclonal anti-CD31 antibody (Ventana-Roche, cat#760-4378) was used in prediluted. The incubation with the primary antibody was done for 5 hours, followed by biotinylated anti-mouse secondary (Vector Labs, MOM Kit BMK-2202) in 5.75ug/mL Blocker D, Streptavidin- HRP and TSA Alexa488 (Life Tech, cat#B40932) prepared according to manufacturer instruction in 1:150 for 16 min. A mouse monoclonal IgG1 anti-CD68 antibody (DAKO, cat#M0814) was used in 0.02 ug/ml concentrations. The incubation with the primary antibody was done for 5 hours followed by biotinylated goat anti-mouse secondary (Vector Labs, MOM Kit BMK-2202) in 5.75ug/mL. Blocker D, Streptavidin- HRP and CF 543 (Biotium, cat#92172) were prepared according to manufacturer instruction in 1:500 for 16 min. All slides were counterstained in 5ug/mL DAPI [dihydrochloride(2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride] (Sigma D9542), for 5 minutes at room temperature, mounted with anti-fade mounting medium Mowiol [Mowiol 4-88 (CALBIOCHEM code: 475904)] and coverslipped.
Slides were scanned with Pannoramic P250 Flash scanner (3DHistech, Hungary) using 20x/0.8NA objective lens. Digitized images were viewed using CaseViewer software (3DHistech, Hungary), where 2–3 representative regions of interest (ROIs), each 2mm2 in area, were selected from each tumor and then exported as .tif files. These regions were then analyzed using ImageJ/Fiji software (v2.1.0; NIH, USA).(92)
Immunofluorescence for Moffitt Cohort
Prior to IF, tissue blocks were prepared from macrodissected RCC tumor specimens, and an experienced genitourinary pathologist (JD) reviewed each slide obtained from formalin-fixed paraffin-embedded tissue samples and annotated 3 spatially distinct regions of interest (ROIs) from the tumor core, and 3 ROIs from the stroma. The tumor-core ROIs studied were selected regions with high tumor cell content, and without evidence of grossly necrotic tissue.
Tissue samples were then stained using the PerkinElmer OPAL 7 Color Automation Immunohistochemistry Kit (PerkinElmer, Waltham, MA) on the BOND RX Autostainer (Leica Biosystems, Vista, CA). In brief, tissue slides were stained using antibodies against CD8 (CD8 (C8/144B) Mouse mAb, Agilent Dako, Santa Clara, Ca, USA; ID: GA623; RRID: AB_2075537).
All subsequent steps, including deparaffinization, antigen retrieval, and staining, were performed using the OPAL manufacturer’s protocol. Pan-cytokeratin (PCK) and 4′,6-diamidino-2-phenylindole (DAPI) counterstaining were applied to all slides, and imaging was performed using the Vectra3 Automated Quantitative Pathology Imaging System (PerkinElmer). Importantly, PCK was applied only after confirmation of tumor content by examination of H and E images for each ROI. Multilayer TIFF images were exported from InForm (PerkinElmer) and loaded into HALO (Indica Labs, Albuquerque, NM) for quantitative image analyses34. The size of the ROIs was standardized at 1356 × 1012 pixels, with a resolution of 0.5 μm/pixel, for a total surface area of 0.343 mm2. Using HALO, for each staining marker a positivity threshold within either the nucleus or cytoplasm was set, and the entire image set was analyzed. From this analysis, generated data included the total cell count, positive cell counts for CD8, fluorescence intensity of every individual cell, percent of cells that were positive for CD8, and Cartesian coordinates for their location in the ROI image. Median cutoffs for CD8 cell density (cells per mm2) in the tumor core and stroma zones were determined. Tumors with high stromal and low tumor CD8 cell density were defined as ‘excluded’, low stromal and low tumor CD8 cell density were defined as ‘desert’, and the remainder were considered ‘inflamed’ (i.e. high tumor and low stromal CD8 counts, high tumor and high stromal CD8 counts).
Animal Studies.
All mouse experiments were approved by the Memorial Sloan Kettering Cancer Center (MSKCC; New York, NY) Internal Animal Care and Use Committee. Mice were maintained under specific pathogen-free conditions, and food and water were provided ad libitum. Mice were purchased from Jackson Laboratory.
Electroporation-derived syngeneic model.
To generate EP tumors, sgRNAs targeting Vhl (CCCGGTGGTAAGATCGGGTA), Tp53 (ACCCTGTCACCGAGACCCC) and Rb1 (TGCGCGGGGTCGTCCTCCCG) were cloned into a pX330 CRISPR/Cas9 vector (Addgene #42230). A pX330 vector containing two sgRNA cassettes were cloned using previously described approach.(61) These recombinant pX330 plasmids (20μg each) were mixed with a c-myc-PT3EF1a transposon vector (5μg), and a Sleeping Beauty transposase (SB13; 1μg) in sterile saline for injection. All plasmids were purified using low endotoxin kits (Plasmid Plus kits; Qiagen). The SB13 and c-myc-PT3EF1a transposon vector were gifts from Dr Josef Leibold (MSKCC) via Dr. Xin Chen (University of California); Addgene plasmid # 92046). The pX330 vector was a gift from Feng Zhang of the Broad Institute (Addgene plasmid # 42230).
For surgery, mice were anaesthetized using isofluorane and given the analgesics buprenorphine, meloxicam and bupivacaine according to institutional guidelines. With the mouse in right lateral recumbency a ~1cm left paracostal (vertical) incision was made through the skin and peritoneum in the flank just caudal to the last rib. The kidney was gently exteriorized using blunt forceps. A 50μl plasmid solution was injected using a 30G needle inserted at a shallow angle just below the renal capsule so that a visible bleb is formed between the renal capsule and parenchyma. The injection site was electroporated using the NEPA21 electro-kinetic transformation system (Nepa Gene, Bulldog Bio) with CUY661-3X7 electrode tweezers using two consecutive positive poring pulses with 50V, 30ms duration, 450ms interval, 10% decay rate, followed by five consective positive transfer pulses with 30V, 50ms duration, 450ms intervals, 40% decay rate, followed by five consecutive negative transfer pulses with the same settings. The kidney was replaced into the abdominal cavity and the wounds were opposed. Mice were monitored for tumor development by manual palpation and confirmed by ultrasound and MRI imaging.
The LVRCC67 cell line was derived by dissociation of the primary tumor using 0.2mg/ml liberase TM (Roche) and 10μg /ml DNase I (DN25, Sigma) in PBS at 37°c with shaking for 30mins. Cells were washed and passed through 45μm cell strainer and cultured in previously described K1 media(65). Cells were passaged twice weekly at a 1:8 dilution. Cells were tested for mycoplasma using the MycoAlert kit (Lonza; latest date tested 4/4/22) and early passage cells (P2-P5) were used in all experiments. Genome editing in the EP-derived tumor and cell line was confirmed by cut-site PCR and sanger sequencing followed by sequence deconvolution using Synthego the ICE CRISPR Analysis Tool (Synthego; https://ice.synthego.com/#/).
For the syngeneic transplantation model, 5×105 LVRCC67 cells in 100μl of PBS were injected subcutaneously into both flanks of 6–8-week-old male C57Bl/6/J mice (Jackson Laboratories). Mice were randomized by cages to receive either 100mg/kg CPI-444 (4000mg/kg medicated chow; produced and irradiated by Research Diets, provided by Corvus Pharmaceuticals), control chow (Teklad 2018 standard diet), IgG isotype control (2A3, Bioxcel, 100μg), or anti-PD-1 (RMP1-14, Bioxcel 100μg) antibodies administered intraperitoneally in 100μl of PBS twice weekly. Tumor volumes were measured twice weekly using digital calipers and calculated using the formula π/6 × (length × width)3/2. Mice were euthanized with carbon dioxide prior to necropsy. Metastatic tumor burden was determined by gross examination of liver, lungs, kidney and spleen.
Immunohistochemistry for mouse tumors.
To characterize the LVRCC67 parental tumor, formalin-fixed paraffin embedded sections were dewaxed in xylene, rehydrated through graded ethanols into ddH20, and antigen retrieval was performed using an autoclave for 15mins in preheated citrate-based antigen unmasking solution (except for chromogranin A which required Tris/ETDA-based antigen retrieval solution; Vector Laboratories). Endogenous peroxidases were quenched using BLOXALL® Blocking Solution (Vector Laboratories) and non-specific binding was blocked using 2.5% normal serum matched to the species of the secondary antibody. For rabbit primary antibodies (Pax8 #10336-1-AP Proteintech 1:1000, HIF-1ɑ clone D1S7W Cell Signaling Technology 1:400, Aqp1 #Ab15080 Abcam 1:500, c-myc clone Y69 Abcam 1:100, CA-9 clone EPR23055-5 Abcam 1:500, Megalin #Ab76969 Abcam 1:1000, Glut1 #Ab14683 Abcam 1:500, Synaptophysin clone YE269 Abcam 1:400, Chromogranin-A clone EPR22537-248 1:1000) the ImmPRESS® Excel Amplified Polymer Staining Kit was used as recommended by the manufacturer. For mouse primary antibodies (Pan-CK clone AE1/AE3 1:1000, p53 clone 1C12 Cell Signaling Technology 1:100, Rb1 clone G3245 BD Pharmingen 1:100), the VECTASTAIN® Elite® ABC-HRP Universal Kit was used according to manufacturer instructions with biotin/avidin block, but also included an additional 1hour block with mouse-on-mouse block (Goat F(ab) Anti-Mouse IgG H&L; 1:50 dilution; Abcam) prior to addition of primary antibodies. For goat primary antibodies (CD10 #PA5-47075 Invitrogen 1:1000), the VECTASTAIN® Elite® ABC-HRP Kit was used but biotinylated rabbit-anti-goat secondary (1:200; Vector Laboratories) was used in place of the biotinylated universal antibody. Finally, slides were dehydrated through a reverse ethanol gradient into xylene, mounted and cover slipped in Cytoseal™ XYL (Thermo Scientific). Representative brightfield images were captured using a Zeiss microscope fitted with an AxioCam HRC Camera.
Western Blotting.
For validation of gene deletions and kidney cell origin of LVRCC67 cell line, whole cell extracts were prepared using CellLytic M (Sigma) and sonication for 10mins in a 4°c water bath. After centrifugation at 21,000rpm, supernatants were mixed with 6x loading dye containing 2-mercaptoethanol and run on pre-cast Novex 4–12% Bis-Tris gels (Invitrogen) for 90mins at 130V using the BioRad Transblot system. Proteins were transferred onto methanol-activated PVDF membranes for 1hour using 100V. Membranes were blocked with 5% milk and incubated at 4°c overnight in 5% BSA containing primary antibodies (Pax8 #10336-1-AP Proteintech 1:2500, HIF-1ɑ clone D1S7W Cell Signaling Technology 1:1000, c-myc clone Y69 Abcam 1:1000, Rb1 #9301S Cell signaling Technology 1:500, p53 clone 1C12 Cell Signaling Technology 1:1000, CD10 #PA5-47075 Invitrogen 1:2000, HSP-90 clone C45G5 Cell signaling Technology 1:3000, β-actin clone D6A8 1:5000) followed by secondary antibody incubation the following day for 1hour at room temperature.
Mouse RNA Sequencing
For mouse RNAseq, three normal kidney cortexes and four snap frozen tumors collected at experimental endpoint were submitted to the Integrated Genomics Core for isolation of RNA, library preparation using Illumina TruSeq stranded mRNA (polyA) and sequenced to a depth of 40–50million 100-bp paired-end reads per sample. RNAseq raw read sequences were then aligned as described above.
Flow Cytometry.
Resected tumors were collected in DMEM + 10% FBS on ice, washed in PBS and digested in PBS containing 0.2mg/ml Liberase TM (Roche) and 10μg /ml DNase I (DN25, Sigma) at 37°c with shaking for 30mins. The digestion reaction was quenched using an equal volume of 10mM EDTA PBS, followed by straining through 70μm and 45μm cell strainers and RBC lysis (Ack lysis buffer; Gibco). Single cell suspensions were stained with viability dye (Ghost Dye™ Violet 510; Tonbo Bioscience) and Fc block (TruStain FcX™; BioLegend) followed by surface antibodies. Surface antibodies were fixed and cells were permeabilized to allow subsequent staining of intracellular antibodies using Foxp3/transcription factor staining buffer kit (Tonbo Bioscience) according to manufacturer instructions. The following antibodies were used: CD80-FITC 16-10A1 1:100, CD206-PCPCy5.5 C068C2 1:200, Ly6C-APC-Cy7 AL-21 1:300, IRF8-PE V3GYWCH 1:200, Ly6G-PE-Cy7 1A8 1:800, F4/80-PE-CF594 T45-2342 1:200, MHC-II-BV605 M5/114.15.2 1:300, CD45-BV650 30-F11 1:500, CD11b-BV711 M1/70 1:400, PD-1-FITC RMP1-30 1:200, TCRɣδ-PerCPeFluor710 eBioGL3 1:200, Granzyme B-APC GB11 1:100, CD8ɑ-APC-Cy7 53–6.7 1:200, TCRβ-PE eBio4B10 1:100, NK1.1-PE-Cy7 PK136 1:300, CD4-PE-TexasRed RM5-5 1:200, Foxp3-BV421 FJK-16S 1:200, CD45-BV605 30-F11 1:200, Ki-67-BV650 B56 1:75. Stained single-cell suspensions were analyzed using the Fortessa flow cytometric analyzer (BD) and FlowJo software (Treestar). The gating strategies are shown in Fig. S5.
Whole exome sequencing.
DNA from LVRCC67 cell line pellet or normal kidney of a WT C57Bl/6 mouse was extracted and sequenced at 150x or 100x respectively. Libraries were prepared using Roche’s KAPA Hyper chemistry and Twist’s exome capture by the Integrated Genomics Operation at MSKCC. FASTQ files were aligned to mm10 reference genome with bwamem v.0.7.15. /). Further base-quality score recalibration and duplicate-read removal were performed using the Genome Analysis Toolkit (GATK) version 4.2.1.0 following raw reads alignments guidelines.(93) Somatic mutations were called with VarScan v.2.4.3, Strelka v.2.9.10, Platypus 0.8.1, Mutect2 (part of GATK 4.2.4.1) and SomaticSniper 1.0.5.0 (for SNVs only). For SNVs and INDELS, the following filtering parameters were applied: called by at least two out of 5 callers, covered by >50 reads in tumors and >7 reads in normals, with >10 reads supporting the mutation, mutant allele fraction ≥10% and <1% reads supporting the mutation in the normal (Tcov >10 and Tac >4 and Taf ≥0.04 and Ncov >7 and Naf ≤0.01). Variants with Tcov < 20 or Tac < 4 are marked as low_confidence. Indels reside in the blacklisted regions (https://www.encodeproject.org/annotations/ENCSR636HFF/) and low mappability regions (such as repeat maskers) are excluded. Copy-number variations were assessed using FACETS v.0.6.0(94). The fraction of the copy-number-altered genome was defined as the fraction of the genome with either nondiploid copy number or evidence of loss of heterozygosity.
Statistical Analysis.
Differences between groups were tested using Fisher exact test and χ2 test (SAS 9.4, SAS Institute Inc.) for categorical variables and the Wilcoxon rank sum test (SAS 9.4 & R package stats version 4.1.0) for continuous variables between sample groups. The Kruskal–Wallis test (R package “stats” & SAS 9.4) was used for comparisons between three or more groups of continuous variables. Patients who were lost to follow-up or alive at the time of the study end date were treated as censored events. Survival curves were calculated according to the Kaplan–Meier method, and differences between curves were assessed using the log-rank test. The hazard ratio (HR) estimates and 95% confidence intervals (CI) were determined by the Cox proportional hazards regression modeling (SAS 9.4) and MatSurv.(95) All statistical analyses were post hoc; statistical significance was set at p < 0.05. Illustrations made using BioRender.com.
Supplementary Material
Statement of Significance.
Improved understanding of factors that influence metastatic development in localized ccRCC are greatly needed to aid accurate prediction of disease recurrence, clinical decision making and future adjuvant clinical trial design. Our analysis implicates intratumoral myeloid inflammation as a key driver of metastasis in patients and a novel immunocompetent mouse model.
Acknowledgements
We would like to thank Corvus Pharmaceuticals for kindly providing the CPI-444 medicated and control chow, as well as the valuable services and advice received from the integrated genomics, flow cytometry, small animal imaging and molecular cytology core facilities at MSKCC.
This study was supported by NIH 5P30CA008748-56 MSK Cancer Center Support Grant (AAH), NIH 5R01CA258886 (AAH), Department of Defense KC180165 (AAH), Cycle for Survival (AAH), Weiss Family Funds (AAH), Oncology Research Information Exchange Network (ORIEN) Avatar Project in collaboration with the H. Lee Moffitt Cancer Center & Research Institute and supported by the Tissue Core, Biostatistics and Bioinformatics Shared Resource Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute; an NCI designated Comprehensive Cancer Center (P30-CA076292) (BJM), OMF CA233944 (SWL), German Research Foundation (DFG) under Germany’s excellence strategy (EXC 2180 – 390900677) (JL), Shulamit Katzman Endowed Postdoctoral Research Fellowship (JL).
Footnotes
Disclosures
T.A.C. is a co-founder of Gritstone Oncology and holds equity in An2H. T.A.C. acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai. T.A.C. has served as an advisor for Bristol-Myers, MedImmune, Squibb, Illumina, Eisai, AstraZeneca, and An2H. T.A.C. and V. L. hold ownership of intellectual property on using tumor mutation burden to predict immunotherapy response, with pending patent, which has been licensed to PGDx. R.J.M served in a consultancy or advisory role for Pfizer, Novartis, Merck, Genentech/Roche, Eisai and Exelixis, and received research funding from Bristol-Myers Squibb, Merck, Pfizer, Genentech/Roche, Eisai, Exelixis, and Novartis. M.H.V. reports honoraria from Novartis, consulting/advisory role for Alexion Pharmaceuticals, Bayer, Calithera Biosciences, Corvus Pharmaceuticals, Exelixis, Eisai, GlaxoSmithKline, Natera, Novartis and Pfizer, research funding from Pfizer, Bristol-Myers Squibb and Genentech/Roche, and travel, accommodations, and expenses from Eisai, Novartis and Takeda. J.M., M.M., A.R., and A.S. are employees of Novartis. A.A.H. has served in an advisory role for Merck. D.B.S. has consulted for/received honoraria from Pfizer, Loxo/Lilly Oncology, Vividion Therapeutics, FORE Therapeutics, Scorpion Therapeutics and BridgeBio. M.G. is a shareholder and an employee of Illumina Inc. The rest of the authors have no conflicts to disclose.
Data Availability
The data from the PROTECT cohort are available on request from the relevant Data Access Committee [e.g. https://ega-archive.org/dacs/EGAS00001006344]. Access to the TCGA cohort clinicopathologic, genomic, and transcriptomic data is detailed in our methods section. Data from the MSKCC and MOFFITT cohorts are available upon request from the corresponding author.
References
- 1.Choueiri TK, Tomczak P, Park SH, Venugopal B, Ferguson T, Chang Y-H, et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. N Engl J Med. 2021;385:683–94. [DOI] [PubMed] [Google Scholar]
- 2.Leibovich BC, Blute ML, Cheville JC, Lohse CM, Frank I, Kwon ED, et al. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer. 2003;97:1663–71. [DOI] [PubMed] [Google Scholar]
- 3.Zisman A, Pantuck AJ, Wieder J, Chao DH, Dorey F, Said JW, et al. Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J Clin Oncol. 2002;20:4559–66. [DOI] [PubMed] [Google Scholar]
- 4.Brooks SA, Brannon AR, Parker JS, Fisher JC, Sen O, Kattan MW, et al. ClearCode34: A prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol. 2014;66:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan TM, Mehra R, Tiemeny P, Wolf JS, Wu S, Sangale Z, et al. A Multigene Signature Based on Cell Cycle Proliferation Improves Prediction of Mortality Within 5 Yr of Radical Nephrectomy for Renal Cell Carcinoma. Eur Urol. 2018;73:763–9. [DOI] [PubMed] [Google Scholar]
- 6.Rini BI, Escudier B, Martini JF, Magheli A, Svedman C, Lopatin M, et al. Validation of the 16-gene recurrence score in patients with locoregional, high-risk renal cell carcinoma from a phase III trial of adjuvant sunitinib. Clin Cancer Res. 2018;24:4407–15. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell. 2020;38:803–817.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. 2013;19:3259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manley BJ, Zabor EC, Casuscelli J, Tennenbaum DM, Redzematovic A, Becerra MF, et al. Integration of Recurrent Somatic Mutations with Clinical Outcomes: A Pooled Analysis of 1049 Patients with Clear Cell Renal Cell Carcinoma. Eur Urol Focus. 2017;3:421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Y, Sahin M, Sinha S, Wang Y, Nargund AM, Lyu Y, et al. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat Cancer. 2022;3:188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapur P, Peña-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013;14:159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T, Lu R, Kapur P, Jaiswal BS, Hannan R, Zhang Z, et al. An empirical approach leveraging tumorgrafts to dissect the tumor microenvironment in renal cell carcinoma identifies missing link to prognostic inflammatory factors. Cancer Discov. 2018;8:1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X De, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat Commun. 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020;10:40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat Med. 2020;26:1733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motzer RJ, Haas NB, Donskov F, Gross-Goupil M, Varlamov S, Kopyltsov E, et al. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma. J Clin Oncol. 2017;35:3916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell. 2021;39:662–677.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: Data from a randomized phase III trial. Cancer Discov. 2019;9:510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martini JF, Plimack ER, Choueiri TK, McDermott DF, Puzanov I, Fishman MN, et al. Angiogenic and immune-related biomarkers and outcomes following axitinib/pembrolizumab treatment in patients with advanced renal cell carcinoma. Clin Cancer Res. 2021;26:5598–608. [DOI] [PubMed] [Google Scholar]
- 21.Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choueiri TK, Figueroa DJ, Fay AP, Signoretti S, Liu Y, Gagnon R, et al. Correlation of PD-L1 tumor expression and treatment outcomes in patients with renal cell carcinoma receiving sunitinib or pazopanib: Results from COMPARZ, a randomized controlled trial. Clin Cancer Res. 2015;21:1071–7. [DOI] [PubMed] [Google Scholar]
- 23.Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: Influence of tumor origin. Clin Cancer Res. 2013;19:4079–91. [DOI] [PubMed] [Google Scholar]
- 24.Weiss JM, Gregory Alvord W, Quiñones OA, Stauffer JK, Wiltrout RH. CD40 expression in renal cell carcinoma is associated with tumor apoptosis, CD8(+) T cell frequency and patient survival. Hum Immunol. 2014;75:614–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giraldo NA, Becht E, Vano Y, Petitprez F, Lacroix L, Validire P, et al. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin Cancer Res. 2017;23:4416–28. [DOI] [PubMed] [Google Scholar]
- 26.Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021;39:632–648.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI insight. 2016;1:e85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell. 2018;173:595–610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu YF, Cohn S, Christie A, McKenzie T, Wolff N, Do QN, et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov. 2017;7:900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med. 2017;23:869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey ST, Smith AM, Kardos J, Wobker SE, Wilson HL, Krishnan B, et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat Commun. 2017;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nargund AM, Pham CG, Dong Y, Wang PI, Osmangeyoglu HU, Xie Y, et al. The SWI/SNF Protein PBRM1 Restrains VHL-Loss-Driven Clear Cell Renal Cell Carcinoma. Cell Rep. 2017;18:2893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell. 2018;173:581–594.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Robertson G, et al. Comprehensivemolecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science (80- ). 2018;361:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motzer RJ, Powles T, Atkins MB, Escudier B, McDermott DF, Alekseev BY, et al. Final Overall Survival and Molecular Analysis in IMmotion151, a Phase 3 Trial Comparing Atezolizumab Plus Bevacizumab vs Sunitinib in Patients With Previously Untreated Metastatic Renal Cell Carcinoma. JAMA Oncol. 2021;10065:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assadian S, El-Assaad W, Wang XQD, Gannon PO, Barres V, Latour M, et al. P53 Inhibits̀ Angiogenesis By Inducing the Production of Arresten. Cancer Res. 2012;72:1270–9. [DOI] [PubMed] [Google Scholar]
- 41.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17:611–29. [DOI] [PubMed] [Google Scholar]
- 43.Nixon B, Kuo F, Liu M, Capistrano K, Do M, Franklin R, et al. IRF8 Governs Tumor-Associated Macrophage Control of T Cell Exhaustion. BioRxiv. 2020; [Google Scholar]
- 44.Takenawa J, Kaneko Y, Fukumoto M, Fukatsu A, Hirano T, Fukuyama H, et al. Enhanced expression of interleukin-6 in primary human renal cell carcinomas. J Natl Cancer Inst. United States; 1991;83:1668–72. [DOI] [PubMed] [Google Scholar]
- 45.Dosquet C, Schaetz A, Faucher C, Lepage E, Wautier JL, Richard F, et al. Tumour necrosis factor-α, interleukin-1β and interleukin-6 in patients with renal cell carcinoma. Eur J Cancer. 1994;30:162–7. [DOI] [PubMed] [Google Scholar]
- 46.Fu Q, Chang Y, An H, Fu H, Zhu Y, Xu L, et al. Prognostic value of interleukin-6 and interleukin-6 receptor in organ-confined clear-cell renal cell carcinoma: a 5-year conditional cancer-specific survival analysis. Br J Cancer. 2015;113:1581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gudbrandsdottir G, Aarstad HH, Bostad L, Hjelle KM, Aarstad HJ, Bruserud Ø, et al. Serum levels of the IL-6 family of cytokines predict prognosis in renal cell carcinoma (RCC). Cancer Immunol Immunother. 2021;70:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: A retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13:827–37. [DOI] [PubMed] [Google Scholar]
- 49.May JY, Negrier S, Combaret V, Attali S, Goillot E, Mercatello A, et al. Serum Level of Interleukin 6 as a Prognosis Factor in Metastatic Renal Cell Carcinoma. Cancer Res. 1992;52:3317–22. [PubMed] [Google Scholar]
- 50.Wang Y, Zhang Y. Prognostic role of interleukin-6 in renal cell carcinoma: a meta-analysis. Clin Transl Oncol. 2020;22:835–43. [DOI] [PubMed] [Google Scholar]
- 51.Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol. 2012;42:2060–72. [DOI] [PubMed] [Google Scholar]
- 52.Najjar YG, Rayman P, Jia X, Pavicic PG, Rini BI, Tannenbaum C, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1b, IL8, CXCL5, and Mip-1α. Clin Cancer Res. 2017;23:2346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell. 2021;184:2988–3005.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakano O, Naito Y, Nagura H, Ohtani H, Nakano O, Sato M, et al. Proliferative activity of intratumoral CD8+ T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–6. [PubMed] [Google Scholar]
- 55.Bromwich EJ, McArdle PA, Canna K, McMillan DC, McNicol A-M, Brown M, et al. The relationship between T-lymphocyte infiltration, stage, tumour grade and survival in patients undergoing curative surgery for renal cell cancer. Br J Cancer. 2003;89:1906–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Güç E, Pollard JW. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity. 2021;54:885–902. [DOI] [PubMed] [Google Scholar]
- 58.Maresch R, Mueller S, Veltkamp C, Öllinger R, Friedrich M, Heid I, et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park JS, Lim KM, Park SG, Jung SY, Choi HJ, Lee DH, et al. Pancreatic cancer induced by in vivo electroporation-enhanced sleeping beauty transposon gene delivery system in mouse. Pancreas. 2014;43:614–8. [DOI] [PubMed] [Google Scholar]
- 61.Leibold J, Ruscetti M, Cao Z, Ho YJ, Baslan T, Zou M, et al. Somatic tissue engineering in mouse models reveals an actionable role for WNT pathway alterations in prostate cancer metastasis. Cancer Discov. 2020;10:1038–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paffenholz SV, Salvagno C, Ho YJ, Limjoco M, Baslan T, Tian S, et al. Senescence induction dictates response to chemo- and immunotherapy in preclinical models of ovarian cancer. Proc Natl Acad Sci U S A. 2022;119:e2117754119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benigni A, Morigi M, Remuzzi G. Kidney regeneration. Lancet. 2010;375:1310–7. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell. 2018;173:611–623.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen B, Fong C, Luthra A, Smith SA, DiNatale RG, Nandakumar S, et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell. 2022;185:563–575.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zigeuner R, Ratschek M, Rehak P, Schips L, Langner C. Value of p53 as a prognostic marker in histologic subtypes of renal cell carcinoma: a systematic analysis of primary and metastatic tumor tissue. Urology. United States; 2004;63:651–5. [DOI] [PubMed] [Google Scholar]
- 68.Cho DS, Joo HJ, Oh DK, Kang JH, Kim YS, Lee KB, et al. Cyclooxygenase-2 and p53 expression as prognostic indicators in conventional renal cell carcinoma. Yonsei Med J. 2005;46:133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phuoc NB, Ehara H, Gotoh T, Nakano M, Yokoi S, Deguchi T, et al. Immunohistochemical Analysis with Multiple Antibodies in Search of Prognostic Markers for Clear Cell Renal Cell Carcinoma. Urology. 2007;69:843–8. [DOI] [PubMed] [Google Scholar]
- 70.Klatte T, Seligson DB, LaRochelle J, Shuch B, Said JW, Riggs SB, et al. Molecular signatures of localized clear cell renal cell carcinoma to predict disease-free survival after nephrectomy. Cancer Epidemiol Biomarkers Prev. 2009;18:894–900. [DOI] [PubMed] [Google Scholar]
- 71.Gayed BA, Youssef RF, Bagrodia A, Kapur P, Darwish OM, Krabbe LM, et al. Prognostic role of cell cycle and proliferative biomarkers in patients with clear cell renal cell carcinoma. J Urol. 2013;190:1662–7. [DOI] [PubMed] [Google Scholar]
- 72.Zheng K, Zhu W, Tan J, Wu W, Yang S, Zhang J. Retrospective analysis of a large patient sample to determine p53 and Ki67 expressions in renal cell carcinoma. J BUON. 2014;19:512–6. [PubMed] [Google Scholar]
- 73.Shvarts O, Seligson D, Lam J, Shi T, Horvath S, Figlin R, et al. P53 is an independent predictor of tumor recurrence and progression after nephrectomy for patients with localized Renal Cell Carcinoma: Implications for surveillance and adjuvant clinical trials. J Clin Oncol. 2004;22:4546–4546. [Google Scholar]
- 74.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell. 2008;134:657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang G, et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med. 2018;24:165–75. [DOI] [PubMed] [Google Scholar]
- 77.Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020;30:481–496.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572:538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valero C, Lee M, Hoen D, Weiss K, Kelly DW, Adusumilli PS, et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat Commun. 2021;12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: Data from a randomized phase III trial. Cancer Discov. 2019;9:510–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brugarolas J Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2014;32:1968–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Depristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Creed J, Gerke T, Berglund A. MatSurv: Survival analysis and visualization in MATLAB. J Open Source Softw. 2020;5:1830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data from the PROTECT cohort are available on request from the relevant Data Access Committee [e.g. https://ega-archive.org/dacs/EGAS00001006344]. Access to the TCGA cohort clinicopathologic, genomic, and transcriptomic data is detailed in our methods section. Data from the MSKCC and MOFFITT cohorts are available upon request from the corresponding author.