

Abstract
The tumor microenvironment consists of tumor cells, extracellular matrix, blood vessels, and non-tumor cells such as fibroblasts and immune cells. Crosstalk among components of this cellular ecosystem can transform non-malignant cells and promote tumor invasion and metastasis. Evidence is accumulating that the transcription factor STAT2, a downstream effector of type I interferon (IFN-I) signaling, can either inhibit or promote tumorigenesis depending on the unique environment presented by each type of cancer. STAT2 has long been associated with the canonical JAK/STAT pathway involved in various biological processes including reshaping of the tumor microenvironment and in antitumor immunity. This dichotomous tendency of STAT2 to both inhibit and worsen tumor formation makes the protein a curious, and yet relatively ill-defined player in many cancer pathways involving IFN-I. In this review, we discuss the role of STAT2 in contributing to either a tumorigenic or anti-tumorigenic microenvironment as well as chemoresistance.
Keywords: Interferons, STAT2, Tumor, Microenvironment, Chemoresistance
1. Introduction
The signal transducer and activator of transcription 2 (STAT2) plays a significant role in the transcriptional response to type I interferons (IFN-I). Since its discovery more than 25 years ago, STAT2, together with STAT1, is recognized as a central transcription factor and downstream effector in IFN-I signaling pathway. IFN-I belong to a family of cytokines that are well-documented for inducing antiviral, antitumor and immunomodulatory activities. Years of extensive research corroborates the importance of IFN-I in engaging the immune system within the tumor microenvironment to restrict tumor growth [1]. And yet, tumors have developed mechanisms to evade or inactivate the immune system to escape recognition [2].
2. Canonical IFN-I signaling pathway
In response to viral infection, DNA damage, and Toll-like receptor activation, IFN-I (comprised of 13 IFN-α species, IFN-β and IFN-τ, IFN-κ, IFN-ω, IFN-σ and IFN-ζ) are synthesized and secreted by virtually all cell types [3]. All IFN-I bind to a shared heterodimeric IFN-I receptor (IFNAR) that is ubiquitously expressed in varying levels among cell types. IFNAR consists of two subunits, IFNAR1 and IFNAR2. Phosphorylation of the cytoplasmic tails of IFNAR1 and IFNAR2 by tyrosine kinases TYK2 and JAK1, respectively, provide docking sites for STAT1 and STAT2, allowing their tyrosine phosphorylation, dimerization, and association with interferon regulatory factor 9, IRF9. Together, these three proteins form the heterotrimeric IFN stimulated gene factor 3 (ISGF3) complex, which translocates to the nucleus and binds the IFN-stimulated response element (ISRE) found in the promoters of interferon stimulated genes (ISGs, Fig. 1). It is worth mentioning that in the initial response to IFN-I stimulation, while ISGF3 is the major transcriptional complex, homodimers of STAT1 and STAT3 can also form and drive gene transcription from the gamma associated sequence (GAS) gene promoters.
Fig. 1. Canonical and non-canonical type I Interferon (IFN-I) signaling.
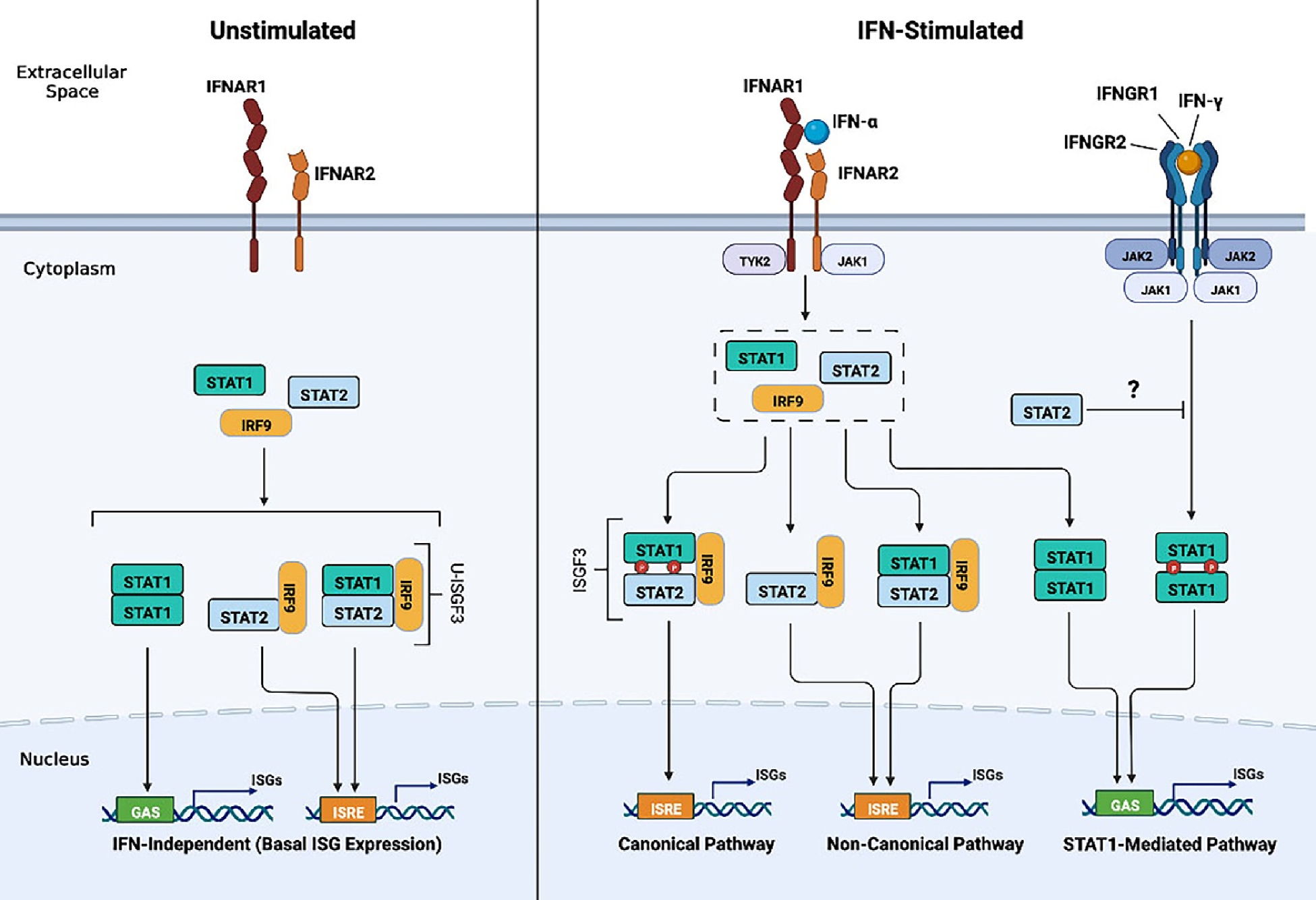
In the absence of IFN-I, non-canonical unphosphorylated STAT1, STAT2, and IRF9 assemble to form the unphosphorylated (U)-ISGF3 complex and STAT2/IRF9 to drive low level expression of ISGs (left). In the presence of IFN-I, IFN-I binds to its receptor and causes the phosphorylation of STAT1 and STAT2 by JAK1 and TYK2 tyrosine kinases (center). STAT1/STAT2 heterodimers associate with IRF9 to form the canonical ISGF3 complex. ISGF3 binds the ISRE in the promoters of ISGs to initiate the first transcriptional and robust response to IFN-I. IFN-I stimulation increases the expression of STAT1, STAT2 and IRF9 resulting in the formation of U-ISGF3 and STAT2/IRF9 to drive the non-canonical pathway and second and prolonged IFN-I response that is associated with chemoresistance and proliferative effects (center). Additionally, STAT2 can interfere with IFN- γ induced STAT1 mediated transcriptional responses by impeding its nuclear translocation (right).
3. Non-Canonical IFN-I signaling pathway
The canonical view is that IFN-activated ISGF3 activates the expression of genes with antitumor properties. However, there is new evidence pointing that IFN-I transcriptional responses can also be driven by STAT2 /IRF9 heterodimers, independently of STAT1 (as shown in Fig. 1) [4–6]. Furthermore, treatment of IFN-I induces high levels of STAT1, STAT2 and IRF9 resulting in the formation of unphosphorylated ISGF3 (U-ISGF3) which drives the late response of IFN-I signaling [7]. IFN-I-induced STAT2 mediated responses -as part of the ISGF3 complex-are classically regarded as antiproliferative, antiangiogenic, and proapoptotic. Yet emerging studies find STAT2 to be overexpressed in several human cancers of the skin, head and neck, kidney, lung, ovary, and endometrium and associated with either better or worse outcome [8–12]. Additionally, persistent STAT2 activity can cause hyperinflammation and be detrimental to the host [13,14]. In this review, we discuss both the potential tumorigenic and antitumorigenic facets of IFN-I in the tumor microenvironment as it relates to STAT2 and associated ISGs and implications in the development of chemoresistance.
4. Type I IFN in the clinical setting
In 1986, the FDA approved the use of IFN-I (specifically IFN-β2a) for the treatment of hairy cell leukemia. IFN-I was later recommended for other cancer types following several studies that showed the therapy reduced tumor growth in pre-clinical pan-cancer animal models [15]. Today, the wide use of IFN-I in the clinic is limited because of severe adverse reactions and only recommended for specific cancer types as adjuvant therapy [16,17]. Nevertheless, studies of primary and metastatic tumors in animal models of cancer (i.e., melanoma, colorectal, fibrosarcoma, liver) continue to show the importance of endogenous IFN-I in tumor immunosurveillance, anti-angiogenesis, and control of metastasis.
The timing, dosage and frequency of administration is key as IFN-I can suppress metastasis depending on the metastatic stage it is administered at [1]. Epithelial-mesenchymal transition is a key transition point from primary tumor to metastasis mediated by genes that regulate cellular adhesion and invasion [18]. A combination of in vitro and in vivo studies conducted with the highly metastatic human bladder carcinoma cell line 253 J B-VR showed that IFN-I increases the expression of epithelial cadherin and downregulates the expression of metalloproteinase MMP9 causing a phenotype that is less invasive. Daily lower dose of IFN-α treatment of established tumors implanted orthotopically was more effective than infrequent treatment with a higher dose in reducing the growth of the primary tumor and restricted metastasis [19].
The presence of an IFN-I transcriptional signature in the tumor microenvironment for certain cancer types (i.e., melanoma and breast cancer) is gaining traction as a favorable prognostic marker of therapeutic response whereby tumors show infiltration of immune cells. These so-called “hot tumors” are more amenable to responding better to immunotherapy [20]. Conversely, tumors that have impaired IFN-I signaling are described as “cold tumors” with a deficiency in lymphocyte infiltration and associated with poor prognosis and resistance to immune checkpoint inhibitors (CTLA-4, PD-L1) [21,22]. The idea of designing a next generation form of IFN-I that is less toxic with potent antitumor activity has not been completely abandoned. Most recently, a masked type of IFN-I (ProIFN) was engineered that consists of the N-terminal ligand binding domain of IFNAR2 fused to the N-terminal end of IFN-I through a protease cleavage linker [23]. The Fc domain of human IgG1 was linked to the construct to extend its half-life. Activation of ProIFN is performed by tumor-enriched proteases. In pre-clinical primary tumor models of melanoma, lung, colorectal and breast cancer, systemic administration of ProIFN increased CD8 + T cells in the tumor microenvironment and suppressed metastasis without adverse effects. This novel strategy of delivering IFN-I to the tumor site may prove of clinical value to overcome resistance to immune checkpoint blockade inhibitors and improve the therapeutic benefits of immunotherapies. However, this prospect warrants careful evaluation as persistent IFN-I signaling can be detrimental and induce resistance to immunotherapy and targeted therapies [24,25].
5. The role of STAT2/type I IFN in cancer
5.1. STAT2 in tumor cell motility and invasion
The invasive behavior of cancer cells is largely defined by the tumor microenvironment. Tumor cells show plasticity to invade and migrate by switching between invasion strategies [26]. Individual tumor cells can invade using the epithelial mesenchymal-transition (EMT) mode. Epithelial cells lose cell polarity and cell–cell junctions, acquire mesenchymal markers and become motile. Consequently, tumor cells attain an elongated morphology and invade through proteolytic degradation of the extracellular matrix (ECM) in conjunction with interactions between the ECM and integrins. The other type of invasion is tumor cells transitioning to the amoeboid mode, where tumor cells adopt a rounded shape that enables them to squeeze through pores in the ECM and migrate.
Higher levels of STAT1 and STAT2 were first described in HT1080 human fibrosarcoma cells when induced to undergo mesenchymal-amoeboid transition (MAT) under specific in vitro conditions [27]. Human melanoma cell lines were also studied due to their inherent invasion plasticity [28]. Interestingly, only IFN-β, and not IFN-ω nor IFN-γ, induced the amoeboid phenotype with an accumulation and phosphorylation of STAT1 and STAT2. IRF9, the main stabilizing protein in ISGF3, showed similar upregulation. Suppression of IRF9 led to the elongation of amoeboid cells, thus converting them back to the mesenchymal phenotype. IFN-β upregulated the expression of IL-6 IL-8, IFITM1, which are associated with inflammation and invasion. Thus, the IFN-I/ISGF3 pathway in melanoma played a key role in the induction of MAT, which shows that IFN-I can be pro-tumorigenic by promoting cancer invasion plasticity. Usage of this pathway could be of importance as amoeboid cells cannot degrade the ECM. However, unlike mesenchymal cells, amoeboid cells can easily escape through small gaps in the ECM, consume less energy, and remain viable during hypoxia and metabolic stress [29]. As a result of this finding, the IFN-I/ISGF3 pathway may be crucial in understanding why melanoma cells take on this amoeboid cell phenotype for invasive purposes and possible crosstalk between the mesenchymal and amoeboid phenotypes.
5.2. IFN-I/STAT2/STAT3 in cancer stem cells and inflammation
Cancer stem cells (CSCs) also known as tumor-initiating cells constitute a heterogeneous subpopulation of tumor cells characterized by the expression of stem-like markers and properties [30]. These cells are key in tumor maintenance; they have limitless self-renewal capacity; are chemoresistant and drive metastatic spread. Triple negative breast cancer is highly aggressive, notorious for responding poorly to chemotherapy and enriched with CSCs. This type of breast cancer was recently characterized by a repressed IFN-I/STAT2 gene signature [31]. This same study showed that human mammary epithelial cells (HMEC) that undergo EMT after transformation also showed repression of an IFN-I/STAT2 gene signature, acquisition of CSC properties and high motility [31]. IFN-β stimulation reversed these phenotypes in which cells recovered an epithelium-like morphology and lost expression of mesenchymal markers was repressed. What was surprising is that CSCs displayed high levels of unphosphorylated ISGF3 (U-ISGF3) which is associated with resistance to DNA damage [7].
IL-6 is a pro-inflammatory cytokine and an ISG with tumorigenic properties that contributes to tumor cell plasticity [32,33]. IL6-induced STAT3 activation and reliance on autocrine signaling of IL-6 is characteristic of many cancers. Unphosphorylated STAT2 association with IRF9 has been shown to boost the expression of IL-6 by pairing with NFκB family protein, p65. STAT2/IRF9 binds the ISRE and p65 binds the NFκB binding site on the IL-6 promoter [34]. STAT1 played no significant role as STAT1-null fibroblasts were dispensable in induction of IL-6 when stimulated with IFN-β. The same study also found elevated levels of STAT3 activation in several human lung and ovarian cancer cell lines expressing higher levels of U-STAT2 and IRF9 protein which correlated with autocrine IL-6 secretion. In this same study, an association between high STAT2 levels and poor survival in lung adenocarcinoma patients was noted. In a different study, STAT2 was identified as a tumor promoter in chemically induced animal models of colon and skin carcinogenesis [35]. In both models, STAT2 contributed to the production of soluble factors involved in tumor development and progression, including IL-6 and enhanced STAT3 activation. In this context, STAT2 displays tumorigenic activity which is opposite to the role it plays in driving the antitumor effects of IFN-I.
6. Prominent role of IFN-Activated STAT2 in tumor immunity
6.1. STAT2 in dendritic cell function and antigen Cross-Presentation
Conventional dendritic cells (cDCs) are central regulators of tumor immunity for their capacity to initiate immune responses by bridging innate and adaptive immunity [36]. cDCs can uptake and process exogenous antigen and present it with MHC class I molecules to CD8 + T cells, a process known as cross-presentation. Host IFN-I signaling in conventional cDCs is essential for cross-presentation of tumor antigens [37]. IFN-I-activated STAT2 is similarly important in this process and in tumor rejection [38–40]. An association between high STAT2 transcriptional signature in cDCs and overall increased survival was seen in metastatic melanoma patients. Furthermore, global Stat2 knockout (KO) mice display impaired cDC maturation [39]. These mice show accelerated tumor growth in a tumor transplantation model of murine B16 melanoma despite IFN-β administration [40]. The lack of antitumor response was explained by the inability of cDC to cross-present antigen to CD8 + T cells. Following TLR activation or IFN-α treatment, cDCs lacking STAT2 could not upregulate the expression of MHC I, CD40 and CD86 costimulatory molecules and ISGs (Cxcl10, Isg15, Irf7, and Mx1). The significance of STAT2 in cDCs was further confirmed with the use of conditional Stat2 KO mice with targeted deletion in cDCs [38]. It is worth noting that CXCL10 is an important chemokine in antitumor immunity, as it aids in the recruitment of T cells and natural killer cells to the tumor site with recent studies reporting that CXCL10 is required for the therapeutic efficacy of immune checkpoint blockade [41–44]. Furthermore, high expression of CXCL10 and STAT2 has been linked to increased survival among patients suffering from oral cancer due to greater activation of T cells [12].
6.2. IFN-I/STAT2 axis in immune checkpoint blockade
Immune checkpoint inhibitors (ICI), prevalent in treating various cancers that evade immune targeting via expression of immune checkpoint targets (PD-L1 and CTLA4) [45 46], were shown to have minimal effects in androgen receptor positive (AR+) prostate cancer (PCa) and estrogen receptor positive (ER+) breast cancer (BCa), which correlated with high expression of the transcription factor, FOXA1 [47]. All three IFN pathways (IFN-I, IFN-II, and IFN-III), which are crucial in the activation of an effective immune response, have shown synergy in ICI responsiveness, particularly IFN-I and IFN-II pathways, in cancers such as melanoma and neck squamous cell carcinoma [48,49]. An explanation for the role of high FOXA1 levels linked to poor therapeutic outcomes led to the identification of STAT2 as a binding partner of FOXA1. FOXA1 in complex with STAT2 prevented the binding of ISGF3 to DNA thus impairing IFN-I mediated antitumor responses. Of note, He et al. found that FOXA1 binding of STAT2 was independent of FOXA1 mutations prevalent in certain PCa and BCa, suggesting an alternate pro-tumorigenic pathway entirely. One could postulate that disruption of FOXA1/STAT2 interactions could restore IFN-I sensitivity and improve response to ICI.
6.3. IFN-I/STAT2 in the regulation of IFN- signaling and cancer
STAT2 can also play a role in regulating aberrant type II (IFN-γ) signaling which is important for immune cell trafficking and antitumor response despite being pro-inflammatory [4,50,51]. Loss of STAT2 in transgenic mice expressing IFN- α in the central nervous system led to increased lethality due to the development of medulloblastoma, thus suggesting that STAT2 is anti-tumorigenic [50]. In the absence of Stat2, not only decreased expression of ISGF3 driven ISGs was observed, but also a shift to expression of STAT1 dependent associated genes, a feature of IFN- γ signaling. Consequently, Stat2 KO mice showed an exacerbated recruitment of activated T cells to the brain and Ifng gene expression. Surprisingly, STAT2 deficiency led to the activation of the Sonic-Hedgehog (Shh) pathway, an alternate pathway known to be upregulated in medulloblastoma [52].
Other studies have shown that different populations of immune cells (T-cells, murine macrophages, immortalized B cell lines from a STAT2 deficient patient and human pluripotent stem cell-derived STAT2KO macrophages) [4,51] following treatment with IFN-α display transcriptional changes resembling an IFN- γ like response. For instance, MHC II expression was upregulated by IFN-α, which is canonically induced by IFN-γ. This analogous expression of MHC II proteins was brought about by increased STAT1 activation in conjunction with IRF1 to drive CIITA expression, the master regulator of MHC II expression. This aberrant and prolonged STAT1 activation was correlated with the absence of STAT2 to induce expression of SOCS-1, an IFN-I negative regulator. In fact, the notion that STAT2 is an inhibitor of STAT1 in multiple signaling pathways, independently of IFN-I, has been investigated (Fig. 1) [53]. Activated STAT1 in complex with unphosphorylated STAT2 prevented STAT1 from entering the nucleus. Taken together, STAT2′s wide ranging regulatory abilities could be responsible for the proper activation of the IFN- γ pathway in immune cells. This is important to note as low dose IFN- γ been associated with metastatic properties while high dose IFN- γ promotes tumor regression [54].
7. STAT2 protein stability in cancer
A deficiency in components of IFN-I signaling leads to defective antitumor and immune responses [55]. The progesterone receptor (PR) orchestrates a transcriptional program for the survival and proliferation of breast cancer cells and suppresses the expression of IFN-I target genes [56]. Recently, it was reported that IFN-I signaling can be inhibited in breast cancer cell lines (T47D and BT474) by PR activation due to degradation of STAT2 protein [57]. Ligand mediated activation of PR led to an increased interaction between STAT2 and PR, independently of STAT1. PR activation in the presence of IFN-α promoted STAT2 to be ubiquitinated and then degraded at a faster rate. Onapristone, a PR antagonist currently being tested in a Phase I clinical trial for late-stage breast cancer, reversed PR mediated STAT2 degradation. Only STAT2 knockdown impaired the occupancy of STAT1 and IRF9 on ISG promoters while STAT1 knockdown did not interfere with STAT2 and IRF9 binding to ISG promoters. From this study, it was clear that STAT2 and -not STAT1- was essential in mediating an IFN-I response. Furthermore, this study showed that post-translational modification of STAT2 (via ubiquitination) promoted by PR produces an immunosuppressive tumor microenvironment that impairs IFN-I signaling. Although not part of this study, it is worth assessing in a pre-clinical model of breast cancer whether inhibition of STAT2 degradation provided by pharmacological suppression of PR activation enhances the antitumor effects of IFN-I.
IFN-I is clinically used as adjuvant therapy for melanoma but not all patients benefit from this treatment for reasons that are not entirely clear. A recent paper provided a plausible explanation to this conundrum in human melanoma [8]. The tumor suppressor FBXW7 is a member of the F-box protein family within the ubiquitin–proteasome system that is involved in ubiquitination and degradation of target proteins [58]. In this study, STAT2 was identified as a binding partner and destruction target of FBXW7 by employing a melanoma culture system. Recognition of STAT2 by FBXW7 for degradation required the phosphorylation of STAT2 by GSK3α and GSK3β on several residues (S381, T385, S389 and S393) according to in vitro kinase assays but not confirmed by mass spectrometry analysis. Melanoma susceptibility pointed out to STAT2. Human melanoma tumors expressed elevated STAT2 mRNA and protein that inversely correlated with levels of FBXW7. Increased STAT2 expression was also associated with shortened survival [8]. Inversely, knockdown of STAT2 decreased cell proliferation while overexpression of mutant STAT2 not recognized by GSK3β enhanced tumor cell proliferation. One key piece of information that remains unknown and worth investigating is how IFN-I antitumor responses are shaped by loss of FBXW7 resulting in elevated STAT2 levels. Nonetheless, the FBXW7-STAT2 proteasome pathway provides first insight into STAT2 associated tumorigenic activity in melanoma.
8. STAT2 and chemoresistance
STAT2 and its associated ISGs are overexpressed in several chemoresistant cancer types. Gemcitabine resistance in pancreatic ductal adenocarcinoma (PDAC) cells and subsequent resistance to vesicular stomatitis virus (an oncolytic viral therapy) correlates with an accumulation of STAT1 and STAT2 as well as upregulation of a particular subset of ISGs: MX1, MX2, IFITM1, IFIT1, and IFI44 [59]. Interestingly, enhanced expression of these genes occurs without upregulation of IFN-α or IFN-β. Coincidentally, these five genes are also induced by U-ISGF3.
In addition to driving the late response to IFN-I signaling, U-ISGF3 is found to be induced by continuous exposure to low levels of IFN-β often observed in cancers [7]. The subset of ISGs stimulated by U-ISGF3 is nearly identical to the interferon-related DNA damage signature (IRDS) which is associated with resistance to chemotherapy and radiotherapy [60,61]. Furthermore, human colon carcinoma cells cultured as 3D tumor spheroids expressed this same IRDS-like subset of ISGs in response to cell crowding which in turn conferred resistance to cytotoxic drugs [62]. In this same study, the increased levels of STAT2 and IRF9 and ISG expression occurred independently of IFN-I. In this same cell overcrowding condition, knockdown of STAT1 did not abrogate the induction of STAT2 and ISGs (IFI27, IFITM1 and OAS1) suggesting that ISG expression was most likely driven by STAT2/IRF9.
STAT2 and IRF9 are also seen to drive resistance of melanoma to serine/threonine-protein kinase B-Raf inhibitors (BRAFi) which inhibit the protein product of the BRAF oncogene [63]. More specifically, STAT2 and IRF9 overexpression in subcutaneous xenograft melanoma model slowed BRAFi-induced tumor shrinking while their knockdown accelerated tumor regression. This same study revealed that STAT2/IRF9 reduces pyroptosis, a pro-inflammatory form of cell death regulated by the gasdermin family of pore-forming proteins (GSDME), by restraining the transcription of GSDME.
As upregulation of ISGs appears to be a common theme in chemoresistance, it is noteworthy to consider what happens when STAT2 activity is unrestrained in normal cells and whether this could lead to cellular transformation or enhanced tumorigenesis. To date, three children (two in the same family) have been identified with a germline homozygous missense mutation of Arginine 148 and diagnosed with type I interferonopathy [13,14], a rare monogenic autoinflammatory disease characterized by excessive IFN-I signaling [64]. This lethal mutation was found to cause hyperactive IFN-I-signaling resulting in an exaggerated immune response and severe systemic inflammation. Because inflammation is a major driver of cancer, these cases expose a phenotype associated with upregulated ISG expression that could be particularly relevant in cancer. Perhaps exploration of these non-cancer examples could reveal factors driving chemoresistance associated with hyperactive STAT2.
Differential phosphorylation of the JAK-STAT signaling pathway appears to play a role in breast cancer chemoresistance in a manner opposite to colon cancer chemoresistance. Chemoresistant breast cancer cells, MCF7Res (resistant to doxorubicin, a common chemotherapy drug in a variety of cancers) showed extensive kinase activity in the JAK-STAT pathway as well as increased STAT2 tyrosine phosphorylation [65]. Treatment of MCF7Res cells in vitro with the JAK inhibitor, Tofacitinib, restored sensitivity to doxorubicin. This study highlights an encouraging avenue of research in chemoresistance as kinase activity is highly druggable.
In a separate study of cisplatin-resistant human ovarian cancer organoids and OVACA433 tumor cells, fibrillin 1 (FBN1) and vascular endothelial growth factor receptor 2 (VEGFR2) were found to mediate the expression, tyrosine phosphorylation and nuclear translocation of STAT2 [66]. FBN1 via STAT2 directed the induction of genes involved in glycolysis and angiogenesis, both of which are known to support tumor growth and metastasis. In the absence of FBN1, VEGFR2 overexpression rescued transcription of this group of genes that was attenuated by STAT2 knockdown. Furthermore, loss of FBN1 reduced the growth of tumor xenografts and restored chemosensitivity. This finding suggests there is an uncharacterized mechanism by which STAT2 promotes glycolysis and vascular formation in chemoresistant cancer cells. Whether IFN-I takes part in this process is completely unknown.
9. Conclusion
IFN-I play a key role in the tumor microenvironment by restricting tumor growth. However, high endogenous induction of IFN-I and/or prolonged secondary response to IFN-I may be detrimental and risky if not properly regulated. What is clear is that IFN-I signaling in the immune compartment is essential for the efficacy of immunotherapy. IFN-I-activated STAT2 is important in shaping an adaptive T-cell killing antitumor response. Along those lines, the antitumor effects of IFN-I appear to vary depending on the cancer type and the invasive phenotype of the tumor cell itself. The protein stability of STAT2 can also dictate whether IFN-I-prolonged transcriptional responses will foster tumor growth. In its unphosphorylated form, when present at elevated levels, STAT2 can be pro-tumorigenic by inducing a transcriptional program linked to chemoresistance and hyperinflammation (summarized in Fig. 2). Further studies are warranted to identify the conditions in which IFN-I signaling should be restricted to improve therapeutic outcomes.
Fig. 2. IFN-I/STAT2 signaling exerts two opposing effects in the tumor microenvironment.
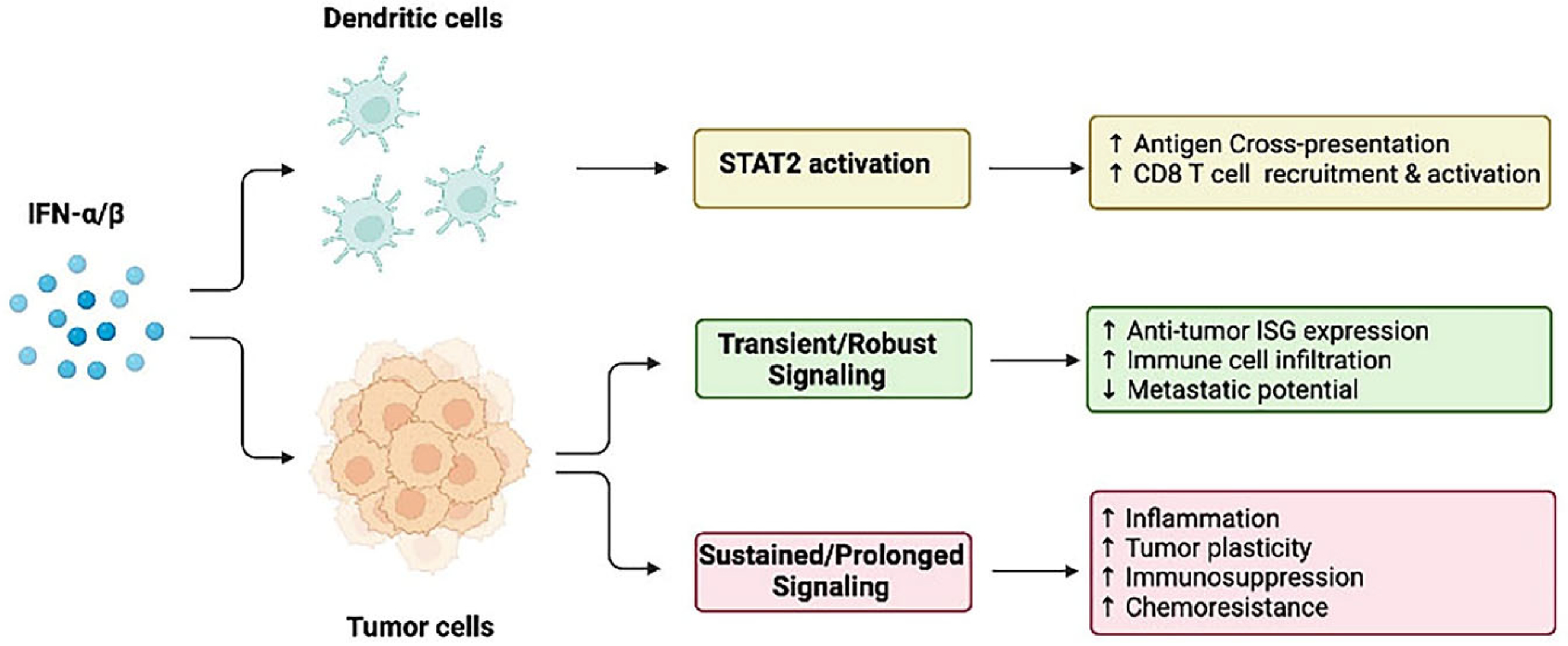
The initial IFN-I response mediated by STAT2 drives the expression of antitumor ISGs that are pivotal in DC maturation, generation of killer CD8 + T cells (top), and recruitment of immune cells to the tumor site to restrict tumor growth and metastasis (middle). In contrast, late IFN-I signaling sustained by unphosphorylated ISGF3 and STAT2/IRF9 promotes the expression of pro-inflammatory mediators and genes involved in chemoresistance and immunosuppression that confer tumor cell survival and disease progression.
Acknowledgement
Figures were created with Biorender.com.
Funding sources
This work was supported by the National Institutes of Health (R21 AR078350-01A1) and Department of Defense grant (W81XWH1810240) to A.M.G. This work was also partially supported by the National Cancer Institute (U54 CA221705) to K.D. and R.D. and T32 fellowship by NIGMS (GM142606-01) to J.C.
Abbreviations:
- IFN-I
type I interferons
- STAT
Signal Transducer and Activator of Transcription
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Jorge Canar: Writing – review & editing. Kennedy Darling: Writing – review & editing. Ryan Dadey: Writing – review & editing. Ana M. Gamero: Conceptualization, Writing – review & editing.
Data availability
No data was used for the research described in the article.
References
- [1].Ortiz A, Fuchs SY, Anti-metastatic functions of type 1 interferons: Foundation for the adjuvant therapy of cancer, Cytokine 89 (2017) 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Spranger S, Gajewski TF, Mechanisms of Tumor Cell-Intrinsic Immune Evasion, Ann. Rev. Cancer Biol. 2 (1) (2018) 213–228. [Google Scholar]
- [3].Mazewski C, Perez RE, Fish EN, Platanias LC, Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways, Front. Immunol. 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gothe F, Stremenova Spegarova J, Hatton CF, Griffin H, Sargent T, Cowley SA, James W, Roppelt A, Shcherbina A, Hauck F, Reyburn HT, Duncan CJA, Hambleton S, Aberrant inflammatory responses to type I interferon in STAT2 or IRF9 deficiency, Journal of Allergy and Clinical Immunology 150 (4) (2022) 955–964.e16. [DOI] [PubMed] [Google Scholar]
- [5].Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen Y-L, Chmielewski S, Kostyrko K, Wesoly J, Balint BL, Lee C-K, Bluyssen HAR, STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1, Biochem J 466 (3) (2015) 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, Gossenreiter T, Müller M, Novatchkova M, Decker T, A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription, Nat. Commun. 10 (1) (2019) 2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, Stark GR, IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage, The EMBO Journal 32 (20) (2013) 2751–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee C-J, An H-J, Kim S-M, Yoo S-M, Park J, Lee G-E, Kim W-Y, Kim DJ, Kang HC, Lee JY, Lee HS, Cho S-J, Cho Y-Y, FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation, Proc. Natl. Acad. Sci. U.S.A. 117 (1) (2020) 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang M, Chen H, Zhou L, Chen K, Su F, Expression profile and prognostic values of STAT family members in non-small cell lung cancer, Am J Transl Res 11 (8) (2019) 4866–4880. [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou L, Li Y, Li Z, Huang Q, Mining therapeutic and prognostic significance of STATs in renal cell carcinoma with bioinformatics analysis, Genomics 112 (6) (2020) 4100–4114. [DOI] [PubMed] [Google Scholar]
- [11].Zhou X-Y, Dai H-Y, Zhang H.u., Zhu J-L, Hu H, Signal transducer and activator of transcription family is a prognostic marker associated with immune infiltration in endometrial cancer, J Clin Lab Anal 36 (4) (2022) e24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huang Y-C, Huang J-L, Tseng L-C, Yu P-H, Chen S-Y, Lin C-S, High Expression of Interferon Pathway Genes CXCL10 and STAT2 Is Associated with Activated T-Cell Signature and Better Outcome of Oral Cancer Patients, Journal of Personalized Medicine 12 (2) (2022) 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, Rosain J, Buta S, Bousfiha A, Casanova J-L, Bustamante J, Bogunovic D, Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy, J. Exp. Med. 217 (5) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, Lovell SC, Shuttleworth VG, Brocklebank V, Corner B, Skelton AJ, Bondet V, Coxhead J, Duffy D, Fourrage C, Livingston JH, Pavaine J, Cheesman E, Bitetti S, Grainger A, Acres M, Innes BA, Mikulasova A, Sun R, Hussain R, Wright R, Wynn R, Zarhrate M, Zeef LAH, Wood K, Hughes SM, Harris CL, Engelhardt KR, Crow YJ, Randall RE, Kavanagh D, Hambleton S, Briggs TA, Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2, Sci Immunol 4 (42) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aricó E,Castiello L, Capone I, Gabriele L, Belardelli F, Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications, Cancers 11 (12) (2019) 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hemminki O, Perlis N, Bjorklund J, Finelli A, Zlotta AR, Hemminki A, Treatment of Advanced Renal Cell Carcinoma: Immunotherapies Have Demonstrated Overall Survival Benefits While Targeted Therapies Have Not, Eur Urol Open Sci 22 (2020) 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM, Interferon: cellular executioner or white knight? Curr Med Chem 14 (12) (2007) 1279–1289. [DOI] [PubMed] [Google Scholar]
- [18].Dongre A, Weinberg RA, New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer, Nat. Rev. Mol. Cell Biol. 20 (2) (2019) 69–84. [DOI] [PubMed] [Google Scholar]
- [19].Slaton JW, Karashima T, Perrotte P, Inoue K, Kim SJ, Izawa J, Kedar D, McConkey DJ, Millikan R, Sweeney P, Yoshikawa C, Shuin T, Dinney CP, Treatment with low-dose interferon-alpha restores the balance between matrix metalloproteinase-9 and E-cadherin expression in human transitional cell carcinoma of the bladder, Clin Cancer Res 7 (9) (2001) 2840–2853. [PubMed] [Google Scholar]
- [20].Brockwell NK, Parker BS, Tumor inherent interferons: Impact on immune reactivity and immunotherapy, Cytokine 118 (2019) 42–47. [DOI] [PubMed] [Google Scholar]
- [21].Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, Krishnan S, Allison JP, Sharma P, Hwu P, Komaki R, Overwijk WW, Gomez DR, Chang JY, Hahn SM, Cortez MA, Welsh JW, Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy, Cancer Res 77(4) (2017) 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, Puré E, Chatterji P, Rustgi AK, Diehl JA, Koumenis C, Rui H, Fuchs SY, Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment, Cancer Cell 31 (2) (2017) 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cao X, Liang Y, Hu Z, Li H, Yang J, Hsu EJ, Zhu J, Zhou J, Fu Y-X, Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance, Nat. Commun. 12 (1) (2021) 5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, Ferrere G, Becharef S, Vétizou M, Daillère R, Messaoudene M, Enot DP, Stoll G, Ugel S, Marigo I, Foong Ngiow S, Marabelle A, Prevost-Blondel A, Gaudreau P-O, Gopalakrishnan V, Eggermont AM, Opolon P, Klein C, Madonna G, Ascierto PA, Sucker A, Schadendorf D, Smyth MJ, Soria J-C, Kroemer G, Bronte V, Wargo J, Zitvogel L, Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade, Cell Res. 29 (10) (2019) 846–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].De Angelis C, Fu X, Cataldo ML, Nardone A, Pereira R, Veeraraghavan J, Nanda S, Qin L, Sethunath V, Wang T, Hilsenbeck SG, Benelli M, Migliaccio I, Guarducci C, Malorni L, Litchfield LM, Liu J, Donaldson J, Selenica P, Brown DN, Weigelt B, Reis-Filho JS, Park BH, Hurvitz SA, Slamon DJ, Rimawi MF, Jansen VM, Jeselsohn R, Osborne CK, Schiff R, Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer, Clin Cancer Res 27(17) (2021) 4870–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Panková K, Rösel D, Novotný M, Brábek J, The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells, Cell Mol Life Sci 67 (1) (2010) 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Čermák V,Gandalovičová A,Merta L, Harant K, Rösel D,Brábek J, High-throughput transcriptomic and proteomic profiling of mesenchymal-amoeboid transition in 3D collagen, Sci. Data 7 (1) (2020) 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gandalovičová A, Šůchová A-M,Čermák V,Merta L, Rösel D,Brábek J, Sustained Inflammatory Signalling through Stat1/Stat2/IRF9 Is Associated with Amoeboid Phenotype of Melanoma Cells, Cancers (Basel) 12 (9) (2020) 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hecht I, Bar-El Y, Balmer F, Natan S, Tsarfaty I, Schweitzer F, Ben-Jacob E, Tumor invasion optimization by mesenchymal-amoeboid heterogeneity, Sci Rep 5 (2015) 10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Atashzar MR, Baharlou R, Karami J, Abdollahi H, Rezaei R, Pourramezan F, Zoljalali Moghaddam SH, Cancer stem cells: A review from origin to therapeutic implications, J Cell Physiol 235 (2) (2020) 790–803. [DOI] [PubMed] [Google Scholar]
- [31].Doherty MR, Cheon HyeonJoo, Junk DJ, Vinayak S, Varadan V, Telli ML, Ford JM, Stark GR, Jackson MW, Interferon-beta represses cancer stem cell properties in triple-negative breast cancer, Proc. Natl. Acad. Sci. U.S.A. 114 (52) (2017) 13792–13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johnson DE, O’Keefe RA, Grandis JR, Targeting the IL-6/JAK/STAT3 signalling axis in cancer, Nat. Rev. Clin. Oncol. 15 (4) (2018) 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abaurrea A, Araujo AM, Caffarel MM, The Role of the IL-6 Cytokine Family in Epithelial-Mesenchymal Plasticity in Cancer Progression, Int. J. Mol. Sci. 22 (15) (2021) 8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nan J, Wang Y, Yang J, Stark GR, IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression, Proc Natl Acad Sci U S A 115 (15) (2018) 3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gamero AM, Young MR, Mentor-Marcel R, Bobe G, Scarzello AJ, Wise J, Colburn NH, STAT2 contributes to promotion of colorectal and skin carcinogenesis, Cancer Prev Res (Phila) 3(4) (2010) 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gardner A, Ruffell B, Dendritic Cells and Cancer Immunity, Trends Immunol. 37 (12) (2016) 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD, Type I interferon is selectively required by dendritic cells for immune rejection of tumors, J Exp Med 208 (10) (2011) 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Qiu CC, Kotredes KP, Cremers T, Patel S, Afanassiev A, Slifker M, Gallucci S, Gamero AM, Targeted Stat2 deletion in conventional dendritic cells impairs CTL responses but does not affect antibody production, Oncoimmunology 10 (1) (2020) 1860477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu J, Lee MH, Chakhtoura M, Green BL, Kotredes KP, Chain RW, Sriram U, Gamero AM, Gallucci S, STAT2 Is Required for TLR-Induced Murine Dendritic Cell Activation and Cross-Presentation, J Immunol 197 (1) (2016) 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yue C, Xu J, Tan Estioko MD, Kotredes KP, Lopez-Otalora Y, Hilliard BA, Baker DP, Gallucci S, Gamero AM, Host STAT2/type I interferon axis controls tumor growth, Int J Cancer 136 (1) (2015) 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ardighieri L, Missale F, Bugatti M, Gatta LB, Pezzali I, Monti M, Gottardi S, Zanotti L, Bignotti E, Ravaggi A, Tognon G, Odicino F, Calza S, MissoloKoussou Y, Ries CH, Helft J, Vermi W, Infiltration by CXCL10 Secreting Macrophages Is Associated With Antitumor Immunity and Response to Therapy in Ovarian Cancer Subtypes, Front. Immunol. 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, Sek K, Mardiana S, Gide TN, Quek C, Scolyer RA, Long GV, Wilmott JS, Loi S, Darcy PK, Beavis PA, Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade, Clin. Cancer Res. 26 (2) (2020) 487–504. [DOI] [PubMed] [Google Scholar]
- [43].Reschke R, Yu J, Flood BA, Higgs EF, Hatogai K, Gajewski TF, Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma, J. ImmunoTher. Cancer 9 (9) (2021) e003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu J, Li F, Ping Y, Wang L, Chen X, Wang D, Cao L, Zhao S, Li B, Kalinski P, Thorne SH, Zhang B, Zhang Y, Local production of the chemokines CCL5 and CXCL10 attracts CD8+ T lymphocytes into esophageal squamous cell carcinoma, Oncotarget 6 (28) (2015) 24978–24989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Darvin P, Toor SM, Sasidharan Nair V, Elkord E, Immune checkpoint inhibitors: recent progress and potential biomarkers, Exp. Mol. Med. 50 (12) (2018) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pardoll DM, The blockade of immune checkpoints in cancer immunotherapy, Nat Rev Cancer 12 (4) (2012) 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].He Y, Wang L, Wei T, Xiao Y-T, Sheng H, Su H, Hollern DP, Zhang X, Ma J, Wen S, Xie H, Yan Y, Pan Y, Hou X, Tang X, Suman VJ, Carter JM, Weinshilboum R, Wang L, Kalari KR, Weroha SJ, Bryce AH, Boughey JC, Dong H, Perou CM, Ye D, Goetz MP, Ren S, Huang H, FOXA1 overexpression suppresses interferon signaling and immune response in cancer, J. Clin. Investig. 131 (14) (2021) e147025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Davar D, Wang H, Chauvin JM, Pagliano O, Fourcade JJ, Ka M, Menna C, Rose A, Sander C, Borhani AA, Karunamurthy A, Tarhini AA, Tawbi HA, Zhao Q, Moreno BH, Ebbinghaus S, Ibrahim N, Kirkwood JM, Zarour HM, Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma, J Clin Oncol 36(35) (2018) Jco1800632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK, IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade, J Clin Invest 127 (8) (2017) 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang J, Pham-Mitchell N, Schindler C, Campbell IL, Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain, J Clin Invest 112 (4) (2003) 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao W, Cha EN, Lee C, Park CY, Schindler C, Stat2-dependent regulation of MHC class II expression, J Immunol 179 (1) (2007) 463–471. [DOI] [PubMed] [Google Scholar]
- [52].Malhotra A, Dey A, Prasad N, Kenney AM, Sonic Hedgehog Signaling Drives Mitochondrial Fragmentation by Suppressing Mitofusins in Cerebellar Granule Neuron Precursors and Medulloblastoma, Mol Cancer Res 14 (1) (2016) 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, Vinkemeier U, STAT2 Is a Pervasive Cytokine Regulator due to Its Inhibition of STAT1 in Multiple Signaling Pathways, PLoS Biol 14 (10) (2016) e2000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jorgovanovic D, Song M, Wang L, Zhang Y, Roles of IFN-γ in tumor progression and regression: a review, Biomarker Res. 8 (1) (2020) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, Swetter SM, Carlson RW, Fisher GA, Koong A, Holmes S, Lee PP, Impaired interferon signaling is a common immune defect in human cancer, Proceedings of the National Academy of Sciences 106(22) (2009) 9010–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Walter KR, Goodman ML, Singhal H, Hall JA, Li T, Holloran SM, Trinca GM, Gibson KA, Jin VX, Greene GL, Hagan CR, Interferon-Stimulated Genes Are Transcriptionally Repressed by PR in Breast Cancer, Mol Cancer Res 15 (10) (2017) 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Walter KR, Balko JM, Hagan CR, Progesterone receptor promotes degradation of STAT2 to inhibit the interferon response in breast cancer, OncoImmunology 9 (1) (2020) 1758547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang Z, Liu P, Inuzuka H, Wei W, Roles of F-box proteins in cancer, Nat. Rev. Cancer 14 (4) (2014) 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Goad DW, Bressy C, Holbrook MC, Grdzelishvili VZ, Acquired chemoresistance can lead to increased resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus, Mol. Ther. Oncolytics 24 (2022) 59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DSA, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, Roizman B, Bergh J, Pawitan Y, Vijver M.J.v.d., Minn AJ, An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer, Proceedings of the National Academy of Sciences 105(47) (2008) 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, Roizman B, Bergh J, Pawitan Y, van de Vijver MJ, Minn AJ, An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer, Proc Natl Acad Sci U S A 105 (47) (2008) 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kolosenko I, Fryknäs M, Forsberg ¨S, Johnsson P, Cheon H, Holvey-Bates EG, Edsbäcker E, Pellegrini P, Rassoolzadeh H, Brnjic S, Larsson R, Stark GR, Grand´er D, Linder S, Tamm KP, De Milito A, Cell crowding induces interferon regulatory factor 9, which confers resistance to chemotherapeutic drugs, Int. J. Cancer 136 (4) (2015) E51–E61. [DOI] [PubMed] [Google Scholar]
- [63].Wang D, Fu Z, Gao L, Zeng J, Xiang Y, Zhou L, Tong X, Wang X-Q, Lu J, Increased IRF9–STAT2 Signaling Leads to Adaptive Resistance toward Targeted Therapy in Melanoma by Restraining GSDME-Dependent Pyroptosis, J, Invest. Dermatol. (2022). [DOI] [PubMed] [Google Scholar]
- [64].Rodero MP, Crow YJ, Type I interferon–mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview, J. Exp. Med. 213 (12) (2016) 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nascimento AS, Peres LL, Fari AVS, Milani R, Silva RA, Jr C, da Costa Fernandes MP, Peppelenbosch CV, Ferreira-Halder WFZ, Phosphoproteome profiling reveals critical role of JAK-STAT signaling in maintaining chemoresistance in breast cancer, Oncotarget 8 (70) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang Z, Chen W, Zuo L, Xu M, Wu Y, Huang J, Zhang X, Li Y, Wang J, Chen J, Wang H, Sun H, The Fibrillin-1/VEGFR2/STAT2 signaling axis promotes chemoresistance via modulating glycolysis and angiogenesis in ovarian cancer organoids and cells, Cancer Commun. 42 (3) (2022) 245–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.