

Abstract
Background and Objectives
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of unknown etiology and poorly understood pathophysiology. There is no specific biomarker either for diagnosis or prognosis. The aim of our study was to investigate differentially expressed proteins in the CSF and serum from patients with ALS to determine their role in the disease process and evaluate their utility as diagnostic or prognostic biomarkers.
Methods
We performed mass spectrometry in the CSF from 3 patients with ALS and 3 healthy controls (HCs). The results were compared with motor cortex dysregulated transcripts obtained from 11patients with sporadic ALS and 8 HCs. Candidate proteins were tested using ELISA in the serum of 123 patients with ALS, 30 patients with Alzheimer disease (AD), 28 patients with frontotemporal dementia (FTD), and 102 HCs. Patients with ALS, AD, and FTD were prospectively recruited from January 2003 to December 2020. A group of age-matched HCs was randomly selected from the Sant Pau Initiative on Neurodegeneration cohort of the Sant Pau Memory Unit.
Results
Nucleotide-binding oligomerization domain–containing protein 2 (NOD2) and osteopontin (Spp1) were differentially expressed in the CSF and the motor cortex transcriptome of patients with ALS compared with that in HCs (p < 0.05). NOD2 and Spp1 levels were significantly higher in sera from patients with ALS than in HCs (p < 0.001). Receiver operating characteristic analysis showed an area under the curve of 0.63 for NOD2 and 0.81 for Spp1. NOD2 levels were significantly lower in patients with AD and FTD than in patients with ALS (p < 0.0001), but we found no significant differences in Spp1 levels between patients with ALS, AD (p = 0.51), and FTD (p = 0.42). We found a negative correlation between Spp1 levels and ALS functional rating scale (r = −0.24, p = 0.009).
Discussion
Our discovery-based approach identified NOD2 as a novel biomarker in ALS and adds evidence to the contribution of Spp1 in the disease process. Both proteins are involved in innate immunity and autophagy and are increased in the serum from patients with ALS. Our data support a relevant role of neuroinflammation in the pathophysiology of the disease and may identify targets for disease-modifying treatments in ALS. Further longitudinal studies should investigate the diagnostic and prognostic value of NOD2 and Spp1 in clinical practice.
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disease characterized by selective loss of upper and lower motor neurons.1 Clinical presentation is highly variable, depending on the site of onset and the degree of upper and lower motor neuron involvement, with some patients presenting only upper or only lower motor neuron signs at onset. This variability and the absence of a diagnostic biomarker can make diagnosis difficult. Survival is also highly heterogeneous, and no reliable prognostic biomarker for ALS has been reported to date.2,3
Although the pathophysiology of ALS remains unknown, it has been suggested that an altered protein homeostasis may play a role in the pathogenesis of the disease. The neuropathologic hallmark of ALS is the aberrant cytoplasmic aggregation of mislocalized proteins, mainly transactive response DNA binding protein 43 (TDP-43) and fused in sarcoma (FUS), in degenerating neurons.4 The presence of stress granules and neuroinflammation are also consistent features in the CNS of patients with ALS.5,6
One explanation for the formation of cytoplasmic protein aggregates is a defective autophagy process.7 Aberrant accumulation of misfolded proteins could work as danger-associated molecular patterns (DAMPs) and may trigger a combined innate/adaptive immune response with direct activation of inflammation pathways.8 Neurodegeneration involves neuroinflammatory components that may accelerate ALS progression and accentuate disease severity.9 Inflammasomes are multiprotein complexes that are responsible for detecting and eliminating pathogen-associated molecular patterns (PAMPs) and DAMPs, and they play critical roles in neurodegenerative diseases. Inflammasomes can also be activated by abnormal protein aggregation,10 and the resulting overexpression of proinflammatory cytokines can aggravate the chronic inflammatory state.11-13
In this study, we performed a proteomic analysis of the CSF from patients with ALS and controls to identify potentially dysregulated proteins in ALS. The investigation of protein biomarkers involved in neuroinflammatory and neurodegenerative pathways could help to understand the pathophysiologic mechanisms involved in the initiation and progression of the disease. The identification of diagnostic or prognostic biomarkers could also be useful in clinical practice and may provide important insights into the clinical and pathologic heterogeneity by tracking various aspects of pathophysiology.
Methods
In this study, we differentiated 2 phases. In the discovery phase, we performed a proteomic analysis of the CSF from patients with ALS and controls to identify dysregulated proteins in patients with ALS in comparison with those in controls. In the second phase, the candidate biomarkers selected were tested in a validation cohort including patients with ALS, controls, and individuals with other neurodegenerative disorders.
Discovery Phase
Study Population and Sample Preparation for Proteomic Analysis
The CSF from 3 patients with ALS (mean age 67.7 ± 2.3 years; 2 females) and 3 healthy controls (HCs), age matched and sex matched, were used to analyze differentially expressed proteins in the CSF in both groups. Patients with ALS were selected from our Motor Neuron Diseases (MND) Clinic and fulfilled El Escorial revised criteria for definite ALS, and controls were randomly selected from the Sant Pau Initiative on Neurodegeneration (SPIN) cohort (SPIN cohort: santpaumemoryunit.com/our-research/SPIN-cohort/).14 Samples of CSF were obtained through lumbar puncture using previously published methods.15 CSF samples were collected following international consensus recommendations. CSF samples were stored immediately at −80°C until analysis.
Proteins present in 1 mL of CSF were precipitated using 2-D Clean up kit (GE Healthcare) following the manufacturer's instructions. Protein pellets were resuspended in radioimmunoprecipitation assay buffer supplemented with protease inhibitors. The protein concentration was measured using the reducing agent and detergent compatible Protein Assay (BioRad). Fifty micrograms of protein was loaded into a 12% acrylamide gel.
In-Gel Digestion and Liquid Chromatography–Mass Spectrometry Analysis
In-gel digestion protocol and liquid chromatography–mass spectrometry (LC-MS) analysis protocol were both performed as previously reported.16
Protein Identification and Quantitative Differential Analysis
Protein identification and quantitative differential analysis protocols were adapted from previous reports.17,18 Progenesis QI for proteomics software v3.0 (Nonlinear dynamics, UK) was used for MS data analysis using default settings. The results from each of the 3 gel fractions were analyzed independently. LC-MS runs were automatically aligned to an automatically selected reference sample—with manual supervision—and automatically normalized to all features. Features were considered for identification and quantification only if they were within the 400–1,500 m/z range, had a 15- to 80-minute retention time, and showed positive charges between 2 and 5. Peak lists were generated by Progenesis and analyzed using the Mascot search engine (v2.2, Matrix Science, UK). Protein identification was performed using the SwissProt-human database (2017_01: 20,171 entries). Precursor mass tolerance was set to 10 ppm and fragment mass tolerance to 0.8 Da. Oxidized methionine was considered as variable amino acid modification and carbamidomethylation of cysteines as fixed modification. Trypsin was selected as the enzyme allowing up to 2 missed cleavages. The significant threshold for peptide identification was set to p < 0.05 for the probability-based Mascot score and a minimum ions score of 20. Finally, the 3 fractions were combined into a single Progenesis dataset. Label-free protein abundance quantification was based on the sum of the peak areas within the isotope boundaries of peptide ion peaks. Only unique nonconflicting peptides were used for quantification. To allow comparison of sample runs, the abundance of each protein was normalized to all proteins. Only proteins that were quantified and identified with at least 2 unique peptides, a p value <0.05, and a fold change >|2| were considered meaningful for subsequent analyses.17,18
RNA sequencing
The differentially expressed proteins detected by mass spectrometry were compared with the results obtained using total RNA sequencing data derived from the motor cortex of 11 patients with sporadic ALS and 8 HCs. Proteins overlapping with significantly differentially expressed genes were prioritized for validation.19 Human motor cortex (Brodmann area 4) samples were provided by the Neurologic Tissue Bank of the Biobanc-Hospital Clinic-IDIBAPS. Samples from patients with ALS fulfilled El Escorial revised criteria for definite ALS during life and presented TDP43 inclusions in the motor cortex at postmortem examination. None of the patients showed symptoms of cognitive or behavior impairment nor had a family history of ALS or dementia, and C9orf72 hexanucleotide repeat expansion was not present in any of the cases. Demographic and clinical data (sex, age at onset, disease duration, and site of onset) have been previously described.19
Validation Phase
Study Population and Classification
Patients with ALS were prospectively recruited from the MND Clinic at Hospital de la Santa Creu i Sant Pau between January 2003 and December 2020. We included patients of both sexes, older than 18 years, and categorized as probable laboratory-supported or definite ALS according to El Escorial revised criteria.2
Patients with Alzheimer disease (AD) with pathophysiologic evidence in the CSF and patients with frontotemporal dementia (FTD) were selected from the SPIN cohort.14 All patients underwent comprehensive cognitive and behavioral assessments. According to this assessment, patients with ALS were classified to one of 2 groups: ALS with no cognitive or behavioral impairment (ALS no-CBi) and ALS-FTD fulfilling criteria for FTD. Patients with ALS-FTD were included in the ALS group for the main group comparisons. In accordance with current diagnostic criteria, patients with FTD were classified into 3 clinical groups: those presenting a behavioral variant of frontotemporal dementia (bvFTD), those with a semantic variant of primary progressive aphasia,20 and those with a nonfluent agrammatic primary progressive aphasia.11,21
We performed an active screening for signs suggestive of motor neuron involvement in patients with FTD. If present, they were referred to the MND clinic for further clinical and neurophysiologic evaluation. If motor neuron involvement was confirmed during follow-up, they were switched to the ALS group and analyzed within this group.11
A group of age-matched cognitively normal controls was randomly selected from the SPIN cohort.14 We registered the revised ALS functional rating scale (ALSFRS-R) during serum sampling and calculated the ALS progression rate as originally described.11,22
All patients provided written informed consent before participation. The study was approved by the local ethics committee and was conducted in compliance with the Declaration of Helsinki.
Enzyme-Linked Immunosorbent Assays
Nucleotide-binding oligomerization domain–containing protein 2 (NOD2) levels in the CSF and serum of patients and controls were detected using human NOD2 ELISA (My Biosource, San Diego, CA), following the manufacturer's instructions.
Scavenger receptor cysteine-rich type 1 protein M130 (CD163), C-X-C motif chemokine 16 (CXCL16), and Human Osteopontin (Spp1) ELISAs were purchased from Biotechne R&D Systems (Minneapolis, MN). All samples were analyzed in duplicate.
Statistical Analysis
We selected the proteins that had an analysis of variance test with a p < 0.05 and a >2-fold change. Multiple comparisons were analyzed using a 1-way analysis of variance test, followed by the Kruskal-Wallis post hoc test. A Shapiro-Wilk test showed that the distribution of NOD2 and Spp1 values departed significantly from normality (W = 0.8, p < 0.0001 and W = 0.94, p < 0.0001, respectively). Based on this outcome, a nonparametric test (the Mann-Whitney U test) was used to compare the protein concentration between patient groups. We evaluated the discrimination power between patients with ALS and HCs by means of the corresponding receiver operating characteristic (ROC) curve for both proteins. The Spearman rank correlation was computed to assess the relationship between NOD2 and Spp1 and total ALSFRS-R, time from blood sampling and disease onset, ALS progression rate, and survival. Data analysis and graphical representation was performed using the STATA 13 software for Windows (StataCorp LP, College Station, TX).
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Hospital de la Santa Creu I Sant Pau Ethics Committee. The CSF and serum samples were obtained and registered according to standard protocols and previous written informed consent by the patients. Brain tissue samples were provided by the Neurologic Tissue Bank of the Biobanc-Hospital Clinic-IDIBAPS. All participants or their relatives had given their written informed consent to use the sample for this study.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Sample Composition of the Validation Cohort
We included 123 patients with ALS, 28 patients with FTD, 30 patients with AD dementia, and 102 HCs. Twenty-three of 123 patients with ALS had associated FTD. Eleven patients had genetically confirmed ALS (6 C9orf72, 2 SOD1, 1 FUS, 1TBK1, and 1 TARDBP). Among patients with FTD, we included 16 cases of bvFTD, and 14 were primary progressive aphasia. Two patients with FTD later developed motor neuron involvement during follow-up and were moved to the ALS group for analysis.
Proteomic Analysis
CSF analysis using mass spectrometry detected 801 proteins, 450 of which were identified with more than 1 peptide homology and 25 were found to be differentially expressed proteins (p < 0.05). If we restrict the number of recognized peptides to more than 2, 16 proteins were found upregulated in patients with ALS compared with HCs.
Four of the 16 proteins, the nucleotide-binding oligomerization domain-containing protein 2 (NOD2), the scavenger receptor cysteine-rich type 1 protein M130 (CD163), the C-X-C motif chemokine 16 (CXCL16), and the osteopontin (Spp1) showed significant differential gene expression changes in RNA sequencing data from the motor cortex of patients with sporadic ALS when compared with HCs (p < 0.05 and log2–fold change >0.45)19 (Figure 1).
Figure 1. Box Plot Showing the Normalized RNA Expression Values of the 4 Genes Encoding the Prioritized Proteins in the Motor Cortex of Patients With Sporadic ALS and Healthy Controls Obtained Through RNA Sequencing.

*p < 0.05; **p < 0.01 ***p < 0.001. sALS = sporadic amyotrophic lateral sclerosis.
We performed commercially available ELISAs to detect the expression of NOD2, CD163, CXCL16, and Spp1 in the CSF and serum from patients with ALS and HCs. None of these candidate proteins were detected by ELISA in the CSF in either group. For this reason, we performed a preliminary study analyzing protein levels in the serum from 20 randomly selected patients and 20 age-matched and sex-matched HCs. No significant differences were observed in CD163 or in CXCL16 expression in either group. However, we observed significant differences between the 2 groups in both NOD2 and Ssp1 expression. Thus, we increased the number of samples analyzed to 123 serum samples from patients with ALS and 102 control serum samples. We also analyzed the expression of NOD2 and Spp1 in the serum of 28 patients with FTD and 30 patients with AD.
NOD2 Analysis
NOD2 levels in serum differed significantly between patients with ALS (273.6 pg/mL, SD 257.8) and healthy individuals (164.6 pg/mL, SD 181.9) (p = 0.0007). ROC analysis showed an area under the curve (AUC) of 0.63 (Figure 2A). Statistically significant differences were observed between patients with ALS and those with FTD (57.8 pg/mL, SD 78.2) (p < 0.0001) or AD (75.0 pg/mL, SD 126.2) (p < 0.0001) (Figure 3A). We found no correlation between NOD2 levels and ALSFRS-R (p = 0.66), time from blood sampling to disease onset (p = 0.82), survival (p = 0.57), and ALS progression (p = 0.71).
Figure 2. Analysis of Diagnostic Utility of Serum Oligomerization Domain–Containing Protein 2 (NOD2) and Osteopontin (Spp1) Through Receiver Operating Characteristic (ROC) Curves.

Sensitivity and specificity of serum (A) oligomerization domain–containing protein 2 (NOD2) and (B) osteopontin (Spp1) levels in patients with ALS and healthy controls. ALS = amyotrophic lateral sclerosis; HC = healthy control.
Figure 3. Levels of Nucleotide-Binding Oligomerization Domain–Containing Protein 2 (NOD2) and Osteopontin (Spp1) Across Diagnostic Groups (Controls, n = 102; ALS, n = 123; AD, n = 30; and FTD, n = 28).

Group differences between serum biomarker raw levels of (A) NOD2 and (B) Spp1. Error bars represent the 10th to 90th percentile range of data, and circles represent outliers. *<0.001. AD = Alzheimer disease; ALS = amyotrophic lateral sclerosis; FTD = frontotemporal dementia; HC = healthy control.
Spp1 Analysis
Spp1 expression showed statistically significant differences between patients with ALS (38.7 ng/mL, SD 21.0) and healthy individuals (mean 17.8 ng/mL, SD 14.0) (p < 0.0001). ROC analysis showed an AUC of 0.81 (Figure 2B). No significant differences were observed in Spp1 levels between patients with ALS and patients with AD (34.0, SD 12.7, p = 0.51) and those with FTD (39.9, SD 14.7, p = 0.42) (Figure 3A). We found a negative correlation between Spp1 levels and ALSFRS-R (r = −0.24, p = 0.009) (Figure 4). No correlation was found between Spp1 levels and time from blood sampling to disease onset (p = 0.30), survival (p = 0.25) and ALS progression (p = 0.90).
Figure 4. Correlation Between the Raw Osteopontin (Spp1) Levels and the ALSFRS-R Score at the Moment of Sample Extraction in the ALS Group.
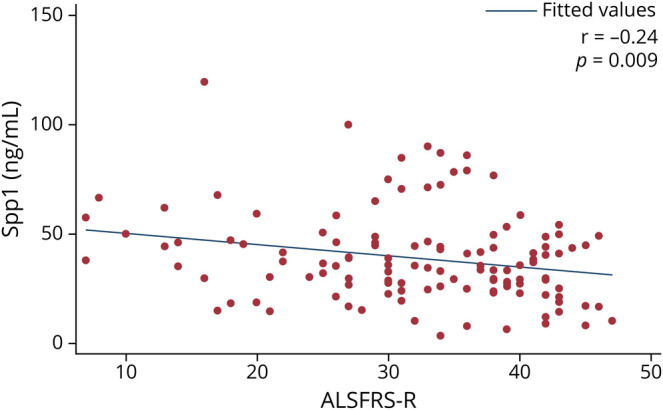
ALS = amyotrophic lateral sclerosis; ALSFRS-R = Revised ALS functional rating scale.
Discussion
Our CSF discovery–based screening revealed 16 proteins were differentially expressed in patients with ALS compared with that in HCs. Four of these 16 proteins were also significantly upregulated in the transcriptome of the motor cortex from patients with ALS. We therefore prioritized these 4 proteins for further analyses. Our results were validated by ELISA in sera samples and showed differentially increased levels in 2 of these proteins, NOD2 and Spp1. No significant differences were found in CXCL16 and CD163 expression in the serum from patients with ALS compared with those from HCs. NOD2 serum levels were significantly higher in patients with ALS than in those with other neurodegenerative diseases. NOD2 gene expression was also upregulated in the motor cortex from patients with ALS when compared with that in controls. NOD2 belongs to the nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs) family. It can interact with multiple proteins and modulates immune responses in a stimuli-dependent manner. The NLR family, together with the toll-like receptor family and retinoic acid-inducible gene I (RIG-I)–like helicase receptor family (including RIG-I), belongs to pathogen recognition receptors that are activated either by evolutionary conserved PAMPs or by DAMPs. RIG-I is an RNA helicase that recognizes viral double-stranded RNA and initiates an innate immune response through mitochondrial antiviral signaling (MAVS). NOD2 senses single-stranded RNA from viruses and interacts with MAVS, activating interferon (IFN) regulatory factor–3 and expression of type I IFN. Both NOD2 and RIG-I exhibit broad-spectrum anti-PAMPs and DAMPS. NOD2 activation mediates numerous interactions and responses that include regulation of inflammasome, production of proinflammatory cytokines, production of type I IFNs, and triggering autophagy.23
Cytoplasmic protein aggregates of TDP-43 and stress granules, formed by ribonucleoproteins are pathologic markers of ALS.5,24 RIG-I has been described as a direct target of TDP-43 because its mRNA contains (uracil guanine)3-5 repeats, which are known binding motifs of TDP-43.25 Similarly, in silico analysis of NOD2 mRNA showed that it is rich in conserved uracil guanine/guanine uracil rich repeats interspaced by other nucleotides, a major factor influencing the interaction of NOD2 mRNA and TDP-43. These RNA molecules from cytoplasmic aggregates could activate NOD2, triggering the neuroinflammatory response observed in patients with ALS. Neurodegeneration involves a neuroinflammatory component that accelerates ALS progression and accentuates disease severity,9 and NOD2 could have a role in the inflammatory process observed in patients with ALS. These results suggest NOD2 has a specific role of in ALS pathophysiology because it is not elevated in other neurodegenerative diseases.
We also found high levels of Spp1 in the serum of patients with ALS. Spp1 is a multifunctional, proinflammatory cytokine that upregulates the expression of IFN-γ and interleukin 12 and is essential in type I immunity.26 Expression of Spp1 can be upregulated by proinflammatory cytokines, under hypoxic conditions or in response to stress or cellular injury.27-29
Spp1 could induce autocrine and paracrine signaling by binding to the cell surface receptors integrin αvβ2 and/or CD44 and transduce cell matrix signaling into cell regulating proliferation, survival, migration, and angiogenesis.27 CD44 acts as a ligand receptor by interacting with Spp1. It has been implicated in the pathogenesis of inflammation in the CNS, contributing to the recruitment of inflammatory cells. It is expressed in most astrocytes and in a portion of microglia.30,31 During the pathogenesis of ALS, astrocytes and microglia are activated,32 contributing to the progression of disease.30
Although the role of Spp1 in ALS is largely unknown, CSF levels of this protein have been proposed as a potential biomarker in ALS.33,34 A recent study showed that Spp1 is overexpressed in perivascular fibroblasts from the spinal cord of patients with ALS.35 The authors also found that high plasma levels of Spp1 correlated with shorter survival.35 By contrast, we found that Spp1 serum levels in our ALS group correlated with more severe disease (lower ALSFRS-R scores) during sample collection. However, this correlation was weak. In addition, we did not find a correlation either with the disease progression rate or survival. Longitudinal studies with follow-up samples would be necessary to evaluate Spp1 level use in clinical practice. Although Spp1 levels were found to be increased in patients with ALS in comparison with HCs, we did not find differences in Spp1 levels in patients with ALS when compared with levels in patients with other neurodegenerative diseases such as AD and FTD. It therefore seems that increased levels of Spp1 are not a distinct finding of ALS alone, but rather a neurodegeneration biomarker. Further prospective studies are needed to elucidate the role of upregulation of Spp1 and its utility in the diagnosis and prognosis of the disease.
Although the differences in serum levels of NOD2 between our patients and controls were significant, ROC analysis suggests the value of this protein as a diagnostic marker is limited. NOD2 levels were also significantly higher in patients with ALS than in the groups with other neurodegenerative diseases, but we cannot conclude from this that NOD2 provides a definitive practical utility to differentiate ALS from other diseases. However, these results suggest a different pathophysiologic pathway is involved in neurodegeneration. Similarly, we found Spp1 levels in patients with ALS were higher than those in HCs. In this case, ROC analysis showed an AUC of 0.82, indicating it can discriminate between the 2 groups with good sensitivity and specificity and suggesting that Spp1 may be a good biomarker in the diagnostic process of ALS.
The main strengths of this study are the prospective design and the detailed phenotype description that includes a standardized cognitive and behavioral assessment in all patients. The homogenous assessment applied within a single center and the inclusion of other neurodegenerative diseases allowed us to investigate the role of biomarkers in detail in different settings. The main limitation of the study is that although we found significant differences between patients with ALS and controls in the serum levels of NOD2 and Spp1, we did not find serum levels of Spp1 differed significantly between patients with ALS and those with other neurodegenerative conditions, and we observed a moderate overlap between groups. Although this overlap might limit the value of Spp1 and NOD2 as diagnostic markers, our results may help understand the role of neuroinflammation in the pathophysiology of neurodegenerative diseases. In addition, although our results showed differentially expressed NOD2 and Spp1 in CSF tested by mass spectrometry and upregulation of NOD2 and SPP1 genes in brain tissue of patients with ALS by RNA sequencing, unfortunately, we were unable to detect our candidate biomarkers using ELISA in the CSF. This would have been desirable for a more detailed understanding of their possible role in the underlying physiopathogenic mechanism and disease process. However, we were able to detect these proteins in sera, which is an easier option to use in clinical practice.
Our discovery-based approach allowed us to identify a novel biomarker that points to a relevant role of neuroinflammatory activity in the pathophysiology of ALS and adds evidence to the involvement of Spp1 in the disease process. Nevertheless, the precise role of inflammation in neurodegenerative disorders can be difficult to clarify. Our data do not prove a causative role of inflammation in neurodegeneration. In fact, it could be a response to an active degenerative process. However, our results provide evidence of dysregulated pathways that can be a potential target for therapeutic interventions.
Our data suggest that an impaired innate immunity, impaired autophagy, and protein homeostasis can facilitate protein aggregation, thereby influencing disease progression. These findings shed light on the cell signaling mechanisms involved in ALS. These mechanisms may serve as potential targets for therapeutic strategies and may promote the development of disease-modifying treatments for ALS, aimed at modulating neuroinflammation.
Further longitudinal studies, including other neurodegenerative disorders, such as Parkinson disease, are needed to elucidate the clinical utility of NOD2 and Spp1 levels as diagnostic and prognostic biomarkers in clinical practice and to understand their role in the disease process and progression of ALS.
Acknowledgment
The authors thank the patients and their relatives for their support for this study. The authors dedicate this work to our master and colleague Isabel Illa.
Glossary
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
ALS functional rating scale
- AUC
area under the curve
- bvFTD
behavioral variant of frontotemporal dementia
- DAMPs
danger-associated molecular patterns
- ELISA
Enzyme-Linked Immunosorbent Assays
- FTD
frontotemporal dementia
- FUS
fused in sarcoma
- HCs
healthy controls
- IFN
interferon
- MAVS
mitochondrial antiviral signaling
- MNDs
motor neuron diseases
- NLRs
nucleotide-binding domain leucine-rich repeat-containing receptors
- NOD2
nucleotide-binding oligomerization domain–containing protein 2
- PAMPs
pathogen-associated molecular patterns
- RIG-I
retinoic acid–inducible gene I
- ROC
receiver operating characteristic
- SPIN
Sant Pau Initiative on Neurodegeneration
- TAR
transactivating response
- TDP-43
TAR DNA binding protein–43
Appendix. Authors
Study Funding
This work was supported by grants from the Fondo de Investigación Sanitaria (FIS), Instituto de Salud Carlos III (PI15/01618 and PI 19/01543 to R. Rojas-Garcia and PI21/00791 to I. Illan-Gala), and cofunded by European Union (ERDF/ESF, “A way to make Europe”/“Investing in your future”); CERCA programme/Generalitat de Catalunya; FUNDELA (Por un mundo sin ELA, 2019 to O. Dols-Icardo); A. Carbayo is supported by the Rio Hortega contract (cm21/00057) from Instituto de Salud Carlos III. O. Dols-Icardo is a recipient of a grant by The Association for Frontotemporal Degeneration (Clinical Research Postdoctoral Fellowship, AFTD 2019–2021); E. Cortés-Vicente and I. Illan-Gala are supported by the Juan Rodés Contract (JR19/00037 and JR20/0018 respectively) from Instituto de Salud Carlos III. I. Illan-Gala is also an Atlantic Fellow for Equity in Brain Health at the Global Brain Health Institute (GBHI) and is supported with funding from the Alzheimer's Association (GBHI ALZ UK-21-720973 and AACSF-21-850193). N. de Luna, E. Cortés-Vicente, J. Turon-Sans, L. Querol, and R. Rojas-Garcia are members of the ERN Euro-NMD.
Disclosure
N. de Luna, A. Carbayo, O. Dols-Icardo, J. Turon-Sans, D. Reyes-Leiva, D. Reyes-Leiva, I. Illán-Gala, I. Jericó, I. Pagola-Lorz, C. Lleixà, L. Querol, and S. Rubio-Guerra report no disclosures. D. Alcolea participated in advisory boards for Fujirebio-Europe and Roche Diagnostics and received speaker honoraria from Fujirebio-Europe, Roche Diagnostics, Nutricia, Krka Farmacéutica S.L., Zambon S.A.U. and Esteve Pharmaceuticals. S.A. D. Alcolea declares a filed patent application (WO2019175379 A1 Markers of synaptopathy in neurodegenerative disease). J. Fortea, A. Lleó, E. Cortés-Vicente, and R. Rojas-García report no disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162-172. doi: 10.1056/nejmra1603471. [DOI] [PubMed] [Google Scholar]
- 2.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord. 2000;1(5):293-299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 3.Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12(3):310-322. doi: 10.1016/s1474-4422(13)70036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74(1):20-38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aksoy YA, Deng W, Stoddart J, et al. Stressed out: the role of FUS and TDP-43 in amyotrophic lateral sclerosis. Int J Biochem Cell Biol. 2020;126:105821. doi: 10.1016/j.biocel.2020.105821. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Front Immunol. 2017;8:1005. doi: 10.3389/fimmu.2017.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amin A, Perera ND, Beart PM, Turner BJ, Shabanpoor F. Amyotrophic lateral sclerosis and autophagy: dysfunction and therapeutic targeting. Cells. 2020;9(11):2413. doi: 10.3390/cells9112413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaelson N, Facciponte D, Bradley W, Stommel E. Cytokine expression levels in ALS: a potential link between inflammation and BMAA-triggered protein misfolding. Cytokine Growth Factor Rev. 2017;37:81-88. doi: 10.1016/j.cytogfr.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Beers DR, Zhao W, Neal DW, et al. Elevated acute phase proteins reflect peripheral inflammation and disease severity in patients with amyotrophic lateral sclerosis. Sci Rep. 2020;10(1):15295. doi: 10.1038/s41598-020-72247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guan Y, Han F. Key mechanisms and potential targets of the NLRP3 inflammasome in neurodegenerative diseases. Front Integr Neurosci. 2020;14:37. doi: 10.3389/fnint.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Illan-Gala I, Alcolea D, Montal V, et al. CSF sAPPβ, YKL-40, and NfL along the ALS-FTD spectrum. Neurology. 2018;91(17):e1619-e1628. doi: 10.1212/wnl.0000000000006383. [DOI] [PubMed] [Google Scholar]
- 12.Steinacker P, Verde F, Fang L, et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J Neurol Neurosurg Psychiatry. 2018;89(3):239-247. doi: 10.1136/jnnp-2017-317138. [DOI] [PubMed] [Google Scholar]
- 13.Thompson AG, Gray E, Thezenas ML, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol. 2018;83(2):258-268. doi: 10.1002/ana.25143. [DOI] [PubMed] [Google Scholar]
- 14.Alcolea D, Clarimón J, Carmona-Iragui M, et al. The Sant Pau Initiative on Neurodegeneration (SPIN) cohort: a dataset for biomarker discovery and validation in neurodegenerative disorders. Alzheimers Dement (N Y). 2019;5:597-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alcolea D, Martinez-Lage P, Sanchez-Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 2015;85(7):626-633. doi: 10.1212/wnl.0000000000001859. [DOI] [PubMed] [Google Scholar]
- 16.Barbero F, Michelini S, Moriones OH, et al. Role of common cell culture media supplements on citrate-stabilized gold nanoparticle protein corona formation, aggregation state, and the consequent impact on cellular uptake. Bioconjug Chem. 2022;33(8):1505-1514. doi: 10.1021/acs.bioconjchem.2c00232. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Berrocoso T, Llombart V, Colas-Campas L, et al. Single cell immuno-laser microdissection coupled to label-free proteomics to reveal the proteotypes of human brain cells after ischemia. Mol Cell Proteomics. 2018;17(1):175-189. doi: 10.1074/mcp.ra117.000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simats Oriol A. Potential Terapèutic de Nous Biomarcadors d'isquèmia Cerebral. Universitat Autònoma de Barcelona Departament de Bioquímica i Biologia Molecular; 2018. hdl.handle.net/10803/662614. [Google Scholar]
- 19.Dols-Icardo O, Montal V, Sirisi S, et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e829. doi: 10.1212/nxi.0000000000000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006-1014. doi: 10.1212/wnl.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(pt 9):2456-2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labra J, Menon P, Byth K, Morrison S, Vucic S. Rate of disease progression: a prognostic biomarker in ALS. J Neurol Neurosurg Psychiatry. 2016;87(6):628-632. doi: 10.1136/jnnp-2015-310998. [DOI] [PubMed] [Google Scholar]
- 23.Dominguez-Martinez DA, Nunez-Avellaneda D, Castanon-Sanchez CA, Salazar MI. NOD2: activation during bacterial and viral infections, polymorphisms and potential as therapeutic target. Rev Invest Clin. 2018;70(1):18-28. doi: 10.24875/ric.17002327. [DOI] [PubMed] [Google Scholar]
- 24.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995-1007. doi: 10.1016/s1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 25.MacNair L, Xiao S, Miletic D, et al. MTHFSD and DDX58 are novel RNA-binding proteins abnormally regulated in amyotrophic lateral sclerosis. Brain. 2016;139(1):86-100. doi: 10.1093/brain/awv308. [DOI] [PubMed] [Google Scholar]
- 26.Zhao K, Zhang M, Zhang L, et al. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci Rep. 2016;6(1):23771. doi: 10.1038/srep23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahmud FJ, Du Y, Greif E, et al. Osteopontin/secreted phosphoprotein-1 behaves as a molecular brake regulating the neuroinflammatory response to chronic viral infection. J Neuroinflammation. 2020;17(1):273. doi: 10.1186/s12974-020-01949-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sodhi CP, Phadke SA, Batlle D, Sahai A. Hypoxia and high glucose cause exaggerated mesangial cell growth and collagen synthesis: role of osteopontin. Am J Physiol Ren Physiol. 2001;280(4):F667-F674. doi: 10.1152/ajprenal.2001.280.4.f667. [DOI] [PubMed] [Google Scholar]
- 29.Uchibori T, Matsuda K, Shimodaira T, Sugano M, Uehara T, Honda T. IL-6 trans-signaling is another pathway to upregulate Osteopontin. Cytokine. 2017;90:88-95. doi: 10.1016/j.cyto.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto T, Imagama S, Hirano K, et al. CD44 expression in astrocytes and microglia is associated with ALS progression in a mouse model. Neurosci Lett. 2012;520(1):115-120. doi: 10.1016/j.neulet.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 31.Nikodemova M, Small AL, Smith SM, Mitchell GS, Watters JJ. Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol Dis. 2014;69:43-53. doi: 10.1016/j.nbd.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beers DR, Zhao W, Liao B, et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun. 2011;25(5):1025-1035. doi: 10.1016/j.bbi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varghese AM, Sharma A, Mishra P, et al. Chitotriosidase–a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin Proteomics. 2013;10(1):19. doi: 10.1186/1559-0275-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Neuhoff N, Oumeraci T, Wolf T, et al. Monitoring CSF proteome alterations in amyotrophic lateral sclerosis: obstacles and perspectives in translating a novel marker panel to the clinic. PLoS One. 2012;7(9):e44401. doi: 10.1371/journal.pone.0044401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manberg A, Skene N, Sanders F, et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat Med. 2021;27(4):640-646. doi: 10.1038/s41591-021-01295-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.