

Abstract
Objective:
Gastroesophageal cancers (GEC) are resistant to therapy and lead to poor prognosis. The cancer stem cells (CSCs) and anti-apoptotic pathways often confer therapy resistance. We sought to elucidate the anti-tumor action of a BCL-2 inhibitor, AT101 in GEC in vitro, in vivo, and in a clinical trial.
Methods:
Extensive preclinical studies in vitro and in vivo were carried out to establish the mechanism action of AT101 on targeting CSCs and anti-apoptotic proteins. A pilot clinical trial in GEC patients was completed with AT-101 added to standard chemoradiation.
Results:
Overexpression of BCL-2 and MCL-1 was noted in gastric cancer tissues (GC). AT-101 induced apoptosis, reduced proliferation and tumor sphere formation in MCL-1/BCL-2 high GC cells. Interestingly, AT101 dramatically down-regulated genes (YAP-1/Sox9) that control CSCs in GEC cell lines regardless of BCL-2/MCL-1 expression. Addition of docetaxel to AT-101 amplified its anti-proliferation and induced apoptosis effects. In vivo studies confirmed the combination of AT101 and docetaxel demonstrated stronger antitumor activity accompanied with significant decrease of CSCs biomarkers (YAP1/SOX9). In a pilot clinical trial, 13 esophageal cancer (EC) patients received AT101 orally concurrently with chemoradiation. We observed dramatic clinical complete responses and encouraging overall survival in these patients. Clinical specimen analyses revealed that AT-101 dramatically reduced the expression of CSCs genes in treated EC specimens indicating antitumor activity of AT101 relies more on its anti-CSCs activity.
Conclusions:
Our preclinical and clinical data suggest that AT-101 overcomes resistance by targeting CSCs pathways suggesting a novel mechanism of action of AT101 in GEC patients.
Keywords: Gastroesophageal cancer, stemness pathways, BCL-2, MCL-1, AT101, apoptosis, docetaxel
Introduction
Gastroesophageal cancer (GEC) including esophageal cancer (EC) and gastric cancer (GC) continue to be a health care burden globally with nearly 1.5 million new cases each year.1 Patients are often diagnosed in advanced stage and survival is poor despite some advances made in the recent years.2, 3 Even when these cancers are localized, the prognosis continues to be poor with a few patients being cured.3 The understanding of molecular biology of GC and EC remains limited.4, 5 In the clinic, these tumors frequently have primary resistance or secondary resistance displayed after a short initial response. Immune modulation (example, checkpoint inhibition) is showing some positive results, however, considerably more progress needs to be made for these patients.2, 6
Mounting evidence documents that cancer stem cells (CSCs) are responsible for mediating resistance in these tumors2, 7–12 CSCs have the ability to self-renew, mediate resistance, and seed metastases.13, 14 CSCs can be controlled through the stemness pathways, such as Hippo/YAP, TGF-β, Notch, and Wnt, among others. Unlike in normal stem cells, these stemness pathways in CSCs are highly deregulated. Currently available treatment modalities target mostly mature and proliferating tumor cells without affecting the tumor-initiating CSCs. Many patients initially experience a positive treatment response, but then resistant cancer cells repopulate the tumor bed. Recently, by comprehensive analysis of singe cell sequencing data in pre- and post- treatment breast cancer tissues, Kim C et al found that pre-existing resistant clones (presumed CSCs) do expand after treating triple-negative breast cancer patients15. The evidence suggests that there may be enrichment drug-resistant CSCs also referred to adoptive resistance when sensitive clones die, preexisting resistant clones expand and accelerate disease progression8, 15. Targeting CSCs particularly in combination with conventional cytotoxics focusing on the bulk tumor could result in improvement in the long-term outcome.
Hippo pathway coactivator, YAP1 is reported to regulate CSCs properties16 and confers aggressive phenotype and therapy resistance.7, 9, 17 YAP1 overexpression and its activation (nuclear localization) correlate with poor outcome in several tumor types.18 Overexpression of YAP1 in cancer cell lines can promote EMT and enhances invasion in vitro.19 In transgenic mice, tissue specific expression of YAP1 in liver results in tissue overgrowth and tumor formation.20 Furthermore, the 11q13 locus containing YAP1 has been reported amplified in EC.21 Recently, we and others have demonstrated that both YAP1 and its target SOX9 are highly up-regulated in both EC and GC tissues compared with premalignant and normal tissues16, 22. YAP1 regulates SOX9 and endows non-tumorigenic cells and tumor cells with CSC properties16, and mediates tumorigenicity and chemo/radiation-resistance.7, 17, 23, 24 Other investigators have suggested that YAP1 is a mediator of resistance to multiple antitumor agents and often the terminal node of many oncogenic pathways.23 These data strongly suggest that YAP1 may be potentially an attractive target in both EC and GC.25
Additionally, anti-apoptotic proteins, such as MCL-1 and BCL-2, mediate resistance in these and other tumors.26–28 In some hematologic malignancies, anti-apoptotic proteins (such as BCL-2) have been successfully targeted but the work in solid tumors is limited. AT-101 is a levorotatory enantiomer of gossypol acetic acid, R(−)-gossypol acetic acid, and a natural BH3-mimetic molecule. AT-101 has demonstrated anti-tumor activity as a single agent or in combination with conventional anticancer therapies in B-cell lymphoma, prostate cancer, ovarian cancer, non-small cell lung cancer.29–31 AT-101 binds to the BH3 motif of all major anti-apoptotic proteins (Mcl-1, BCL-2 and BCL -xl) with high affinity, which is more effective than other BH3-mimetic molecules, such as ABT199 (target BCL-2 only) and ABT263 or ABT737 (target BCL-2, BCL -xl and BCL l-w).32, 33 Clinical development of AT-101 was underway but now halted.31, 34
In this study, we provide evidence from in vitro experiments, followed by in vivo validation and a pilot clinical trial to demonstrate that AT101 can target the CSC pathways (in addition to anti-apoptotic pathway) and demonstrate strong anti-tumor effects and improve patient survival when combined with chemoradiation. Interestingly, we found that AT101 had antitumor activity and favorable clinical response even when BCL-2 is down regulated in EC cells and EC patients which is well described.35 This indicates that the anti-tumor effects of AT101 in EC/GC rely on targeting CSCs.
Material and Methods
Patients and ethics statement
Ethical approval for this study was obtained from the Institutional Review Board of The University of Texas MD Anderson Cancer Center. All patients who volunteered to provide research specimens signed an approved written consent document.
Clinical trial:
An open-label, Phase 1/ 2 study of AT101 and chemoradiotherapy in patients with locally advanced esophageal or gastroesophageal junction cancer were approved by the IRB, was carried out at MDACC only and registered as NCT00561197 in ClinicalTrials.gov. The primary endpoint was to establish the maximum tolerated dose (MTD) of AT-101 combined with standard chemoradiation. Adult patients with locally advanced EC (histologic confirmation of adeno or squamous histology) who were not deemed suitable for surgery (due to tumor geography or co-morbidities) were eligible. Patients had to have a performance status of 0 or 1 and were 18 years of age or older. Near normal organ function and ability to swallow pills was required. Patients with metastatic cancer were excluded. Patients who were not eligible for radiation to mid-thoracic region were also excluded. Chemotherapy included docetaxel (20 mg/m2 as bolus once a week × 5) and fluorouracil (225–300 mg/m2 as low-dose continuous infusion daily from Monday through Friday × 5). Radiation dose was 50.4 Gy in 28 fractions given by 3-D CT planning radiation technique. Based on the prior clinical experience with AT-101, we chose to study only two dose levels (10 mg total dose daily was the starting dose and 20 mg was the next but final dose if 10 mg was well tolerated by the previous cohort of up to 6 patients). AT-101 was taken orally Monday through Friday of each week of chemoradiation. Dose limiting toxicity was defined in a standard manner and 3+3 study design was used. Weekly electrocardiogram and troponin levels were monitored. Upon completion of chemoradiation, patient had 6–8 weeks of recovery time and had an imaging study and endoscopic examination and biopsy. Response to biochemoradiation was assessed as clinical complete response (cCR; post-chemoradiaiton negative biopsies and physiologic uptake on PET) or non-cCR. Patients were then followed every 3 months for 1 year and every 6–12 months for the next 4 years. Patients who developed actionable local recurrences were discussed in the multidisciplinary conference for group decisions and implementation.
The more detail materials and methods can be found from online Supplemental Materials and Methods.
Results
Expression of anti-apoptotic proteins BCL-2/MCL-1 in human EC and GC cell lines and tissues
To determine if anti-apoptotic protein MCL-1 and BCL-2 were over-expressed in GEC cell lines, 8 EC cell lines (Flo-1, SKGT-4, BE3, OE33, JHESO, OACP,YES-6 and KATO-TN) as well as two Barrett’s cell lines (CP-A and CP-C) and 9 GC cell lines (N87, YCC1 and YCC2, AGS, KATO III, GT-5 MKN-45, SNU1, SNU16 were tested by western blotting. In EC cells, we found that MCL-1 was increased compared to premalignant Barrett’ cells in most of EC cell lines, while BCL-2 was decreased in majority EC cell lines (Figure 1A), while in GC cell lines, 2/3 of GC cell lines including AGS, KATO III and SNU1 GC cell lines were over-expressed BCL-2 and MCL-1 anti-apoptotic proteins (Figure 1B).
Figure 1. Expression of anti-apoptotic proteins BCL-2 and MCL-1 in human EC and GC cell lines and tissues.
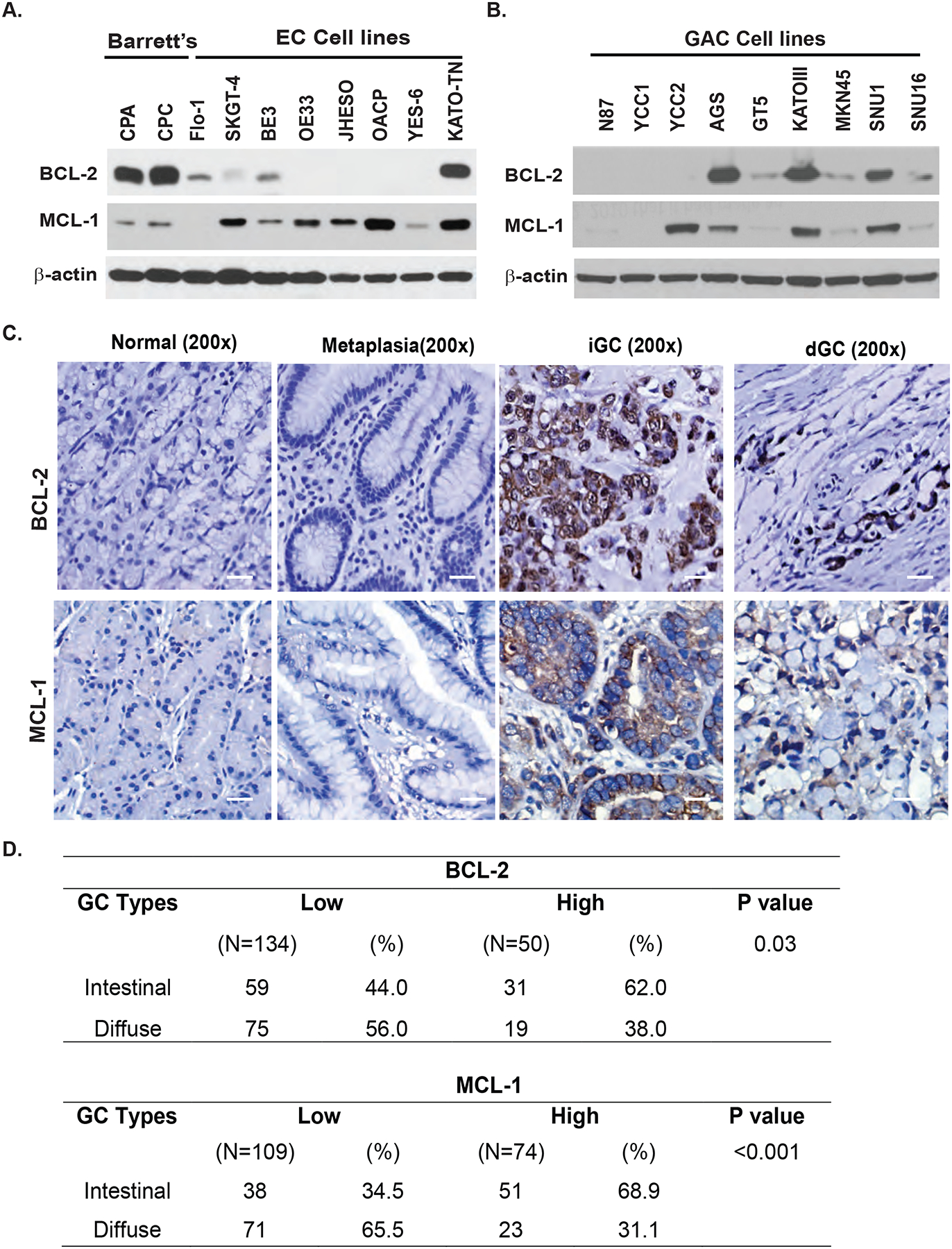
A. Expression of BCL-2 and MCL-1in two premalignant Barretts’ cell lines and eight EC cell lines detected by immunoblotting as described in Materias&Methods. B. Expression of BCL-2 and MCL-1 in 9 GC cell lines detected by immunoblotting as described in Materias&Methods. C. GC tissue microarray slides were immunohistochemically stained using BCL-2 and MCL-1 antibodies as described in Materials & Methods. Representative BCL-2 and MCL-1 staining are shown in normal, metaplasia intestinal GC (iGC) and diffused GC (dGC) tissues. Scale bar: 20μm. D. Statistical analysis of BCL-2 and MCL-1 expression and association with two GC types (iGC vs dGC) in GC TMA tissues. There is significant difference between intestinal and diffused type of GC for BCL-2 and MCL-1 expression.
To determine the expression status of BCL-2 and MCL-1 in GC and GE junction tumor tissues, a tissue micro array TMA comprised of 184 cases of gastroesophageal adenocarcinoma and some adjacent normal tissues were constructed and stained with both anti-MCL-1 and anti-BCL-2 monoclonal antibodies using standard immunohistochemistry36. Expression of both MCL-1 and BCL-2 were increased in tumor tissues compared with normal gastric epithelial and metaplasia tissues (Figure 1C). The positive rate of MCL-1 and BCL-2 were 55% and 27% respectively in all tested cases with dominant staining in intestinal type gastric adenocarcinoma for both MCL-1 and BCL-2 (Figure 1D). The positive staining for MCL-1 was observed in the cytoplasm of cancer cells but BCL-2 was found strongly in both cytoplasm and nuclear. BCL-2 was also positive in the cytoplasm and nuclear membrane of the infiltrated lymphocytes. With both anti-apoptotic proteins expressed in tumor tissues, we hypothesized that the therapy targeting anti-apoptotic proteins may benefit patients with GC.
AT101 potently inhibits tumor cell growth and induces apoptosis in GC cells
As indicated in Figure 1B, expression of MCL-1 and BCL-2 was relatively high in AGS, KATO III and SNU1 three GC cell lines, while it was relatively low in GT-5 for both MCL-1 and BCL-2 and most EC cell lines had decreased BCL-2 expression. To determine if AT101 has potential therapeutic value in GC/EC cell lines, the over-expressed MCL-1 and BCL-2 GC cell lines (AGS, KATO III, SNU1) and the low-expressed GC cell line GT-5 were treated with AT101 at different concentrations for 3 days and 6 days and then cell viability was assessed by the MTS assay. AT101 inhibited cells growth in a dose-dependent manner in all the four GC cell lines (Figure 2A). The inhibitory effects of AT101 on GC cells not fully dependent on the expression of level of both BCL-2 and MCL-1 (Figure 2A). The IC50 of AT101 on AGS and KATO-III cell lines with high BCL-2 are similar to that of GT5 and JHESO cell lines with low/none BCL-2 expression (Supplemental Figure 1). To determine whether the growth inhibition observed in GC cells is associated with specific changes in cell cycle distribution, we analyzed cell cycle using DNA flow cytometry. AGS and KATO III were treated with AT101 at 1μM, 5μM, 10μM or with DMSO as a negative control. We observed AT101 significantly increased sub-G1 phase and apoptosis, while decease in S-phase and G1 phase along with the increasing dosage of AT101 in both AGS and KATO-III cells (Figures 2B&2C).
Figure 2. AT101 inhibits GC cell growth and induce apoptosis.
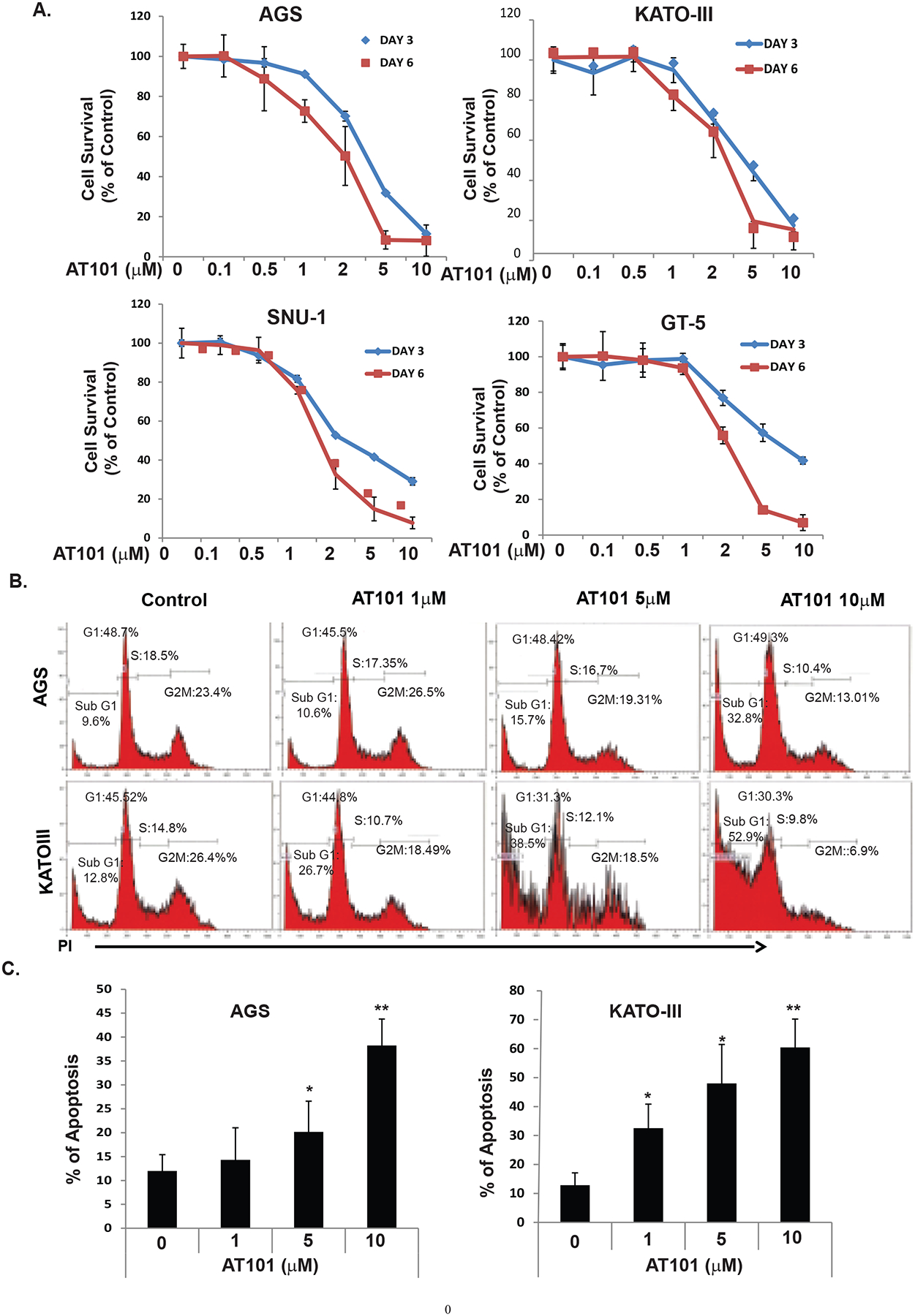
A. Four GC cell lines-AGS, KATOIII, SNU1 and GT5 cells were treated with AT101 at different dosage as indicated for three days and six days then cell growth was determined using MTS as indicated in Materials and Methods. B. The AGS and KATOIII cells were seeded onto 6-well plates and treated with 0.1% DMSO (as control) or with AT101 at 1–10μM for 48 hours and then fixed and stained for DNA with propidium iodide and then analyzed for DNA histograms and cell cycle phase distribution by flow-cytometry using a FACS Calibur instrument. C. AGS and KATOIII cells were treated with 0.1% DMSO (as control) or AT10 at different dosage indicated, and then determined the apoptosis index by flow cytometry, which indicated that the apoptosis index was significantly increased in a dose dependent manner.
Novel anti-tumor targets of AT101 identified using RPPA proteomic array and gene set enriched analysis (GSEA)
To decipher the novel mechanisms by which AT101 inhibited the growth of GC cell lines, we performed RPPA to evaluate 300 proteins expression on AGS cells treated with AT101 1μM. The Gene set enriched analysis (GSEA) demonstrated that many cancer stemness signaling was significantly down-regulated such as YAP1 in Hippo signaling and β-catenin in Wnt signaling in addition of down-regulation of anti-apoptotic proteins-BCL-2, BCL2L1, and BIRC2, the major target of AT101(Figure 3A). The detail gene list with the rank matric scores for each gene in the representative three different pathways were shown in Supplemental Figure 2. Additionally, we confirmed that expression of YAP1 and SOX9 that control CSCs signaling increased in most EC and GC cell lines compared to premalignant Barrett’ cells (Figure 3B). Western blotting further confirmed that AT101 strongly decreased the expression of CSCs genes (β-Catenin/YAP1/SOX9) and anti-apoptotic proteins- MCL-1/BCL-2 in a dosage-dependent manner in AGS and KATO III GC cells (Figure 3C). Similarly, AT101 also suppressed these stemness genes including β-Catenin, YAP1, SOX9, and BM1–1 in JHESO and SKGT-4 EC tumor cells (Figure 3D). Using real time quantitative RT-PCR, we validated that AT101 significantly reduced the mRNA levels of YAP1/SOX9 and MCL-1/ BCL-2 (Figure3E) in AGS cells. Furthermore, using immunofluorescence assay, we observed AT101 reduced the nuclear expression of YAP1 and SOX9 and decreased membrane expression of EpCAM and cytoplasmic expression of ALDH1 (Figure 3F) in JHESO cells (Figure 3F). Similarly, AT101 decreased the expression of YAP1 and SOX9 as well as c-MYC and MCL-1 in the AGS GC cells (Supplemental Figure 3A). Functionally, radiation resistant Flo-1 cells (Flo-1 XTR), enhanced tumor sphere forming capacity and enriched CSCs biomarkers, but both were significantly reduced by AT101(Supplemental Figure 3B). Luciferase assay revealed that AT101 dramatically reduced YAP1-mediated YAP1/Tead transcriptional activity and SOX9 promoter transcriptional activity in both AGS and KATO-III cells (Supplemental Figures 3C & 3D). This indicated that AT101 exerted its anti-tumor effects through down-regulation of the YAP1/SOX9 axis.
Figure 3. AT101 inhibits cell growth by suppressing MCL-1/BCL-2 and YAP/SOX9 axis.
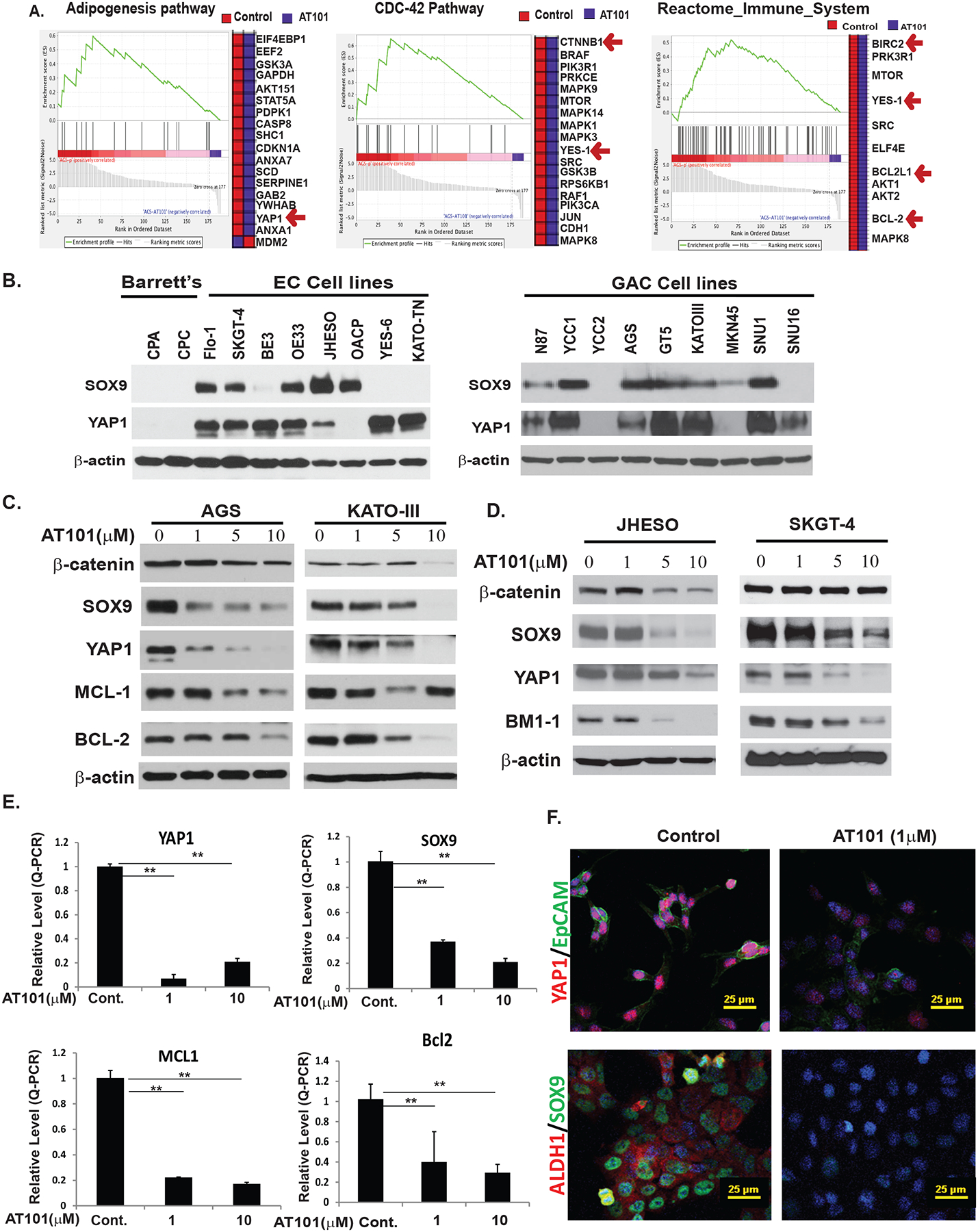
A. RPPA proteomic analysis on AT101 treated AGS cells and the fold changes of genes affected by AT101 in AGC GC cells. B. Expression of YAP1 and SOX9 in two premalignant Barretts’ cell lines and eight EC cell lines and nine GC cell lines were detected by immunoblotting as described in Materials & Methods. Overexpression of both CSCs related genes YAP1 and SOX9 in EC and GC cell lines compared to premalignant cells. C. Down-regulation of antiapoptotic proteins-MCL-1/BCL2 and stem cell signaling-YAP/SOX9 and β-catenin in AGS and KATOIII cells treated with different dosage of AT101 determined by immunoblotting (left, middle panel). D. Down-regulation of proteins controlling stem cell signaling-YAP, SOX9, β-catenin and BMI-1 in both JHESO and SKGT-4 EAC cells treated with different dosage of AT101 by west blotting as indicated. E. AT101 strongly inhibits transcription of YAP1, SOX9, MCL-1 and BCL2 detected by real-time-PCR. **P<0.05. F. Expression of stem cell markers YAP1, EpCAM, SOX9 and ALDH1 were dramatically decreased upon treatment of AT101 at 1μM using confocal immunofluorescent staining in JHESO cells. Scale bar:25 μm.
To further evaluate if AT101 downregulation of CSCs markers and indeed target CSCs functionally, we performed ALDH1 labeling using an ALDEFLUOR detection kit (STEMCELL Technologies) as ALDH1 is a reliable marker for CSCs population in gastrointestinal track37. As shown in Supplemental Figure 4, AT101 dramatically reduced ALDH1+ population in both AGS and JHESO cells from a relative lower dose (1μM) and in dose dependent manner (Supplemental Figure 4A). Further, AT101 suppressed tumor sphere formation in both AGS and JHESO cells (Supplemental Figure 4B). Most importantly, when we sorted ALDH1+ and ALDH1− cells from JHESO cell line with higher population of ALDH1+ in comparison with other cell lines (Supplemental Figure 5), we observed that AT101 has more inhibitory effects on ALDH1+ cells than that of ALDH1− cells indicating AT101 did preferentially target ALDH1+ CSCs population (Supplemental Figures 4C&4D).
AT101 sensitizes docetaxel in suppression of GC cell growth and induces cell apoptosis in vitro
Docetaxel is an important cytotoxic to treat EC/GC since 2006.38 To determine if AT101 enhances antitumor effects of docetaxel in GC cells, we treated GC cells with docetaxel plus AT101 in a relative low dose. As expected, AT101 significantly enhanced docetaxel on inhibition of both AGS and KATO-III cells (Figure 4A). Cell cycle analysis in treated AGS and KATO-cells using docetaxel alone or in combination of AT101 and docetaxel; we noted that docetaxel alone arrested GC cells in G2M phase, the combination with AT101 significantly increased population of the Sub-G1 phase in both AGS and KATO-cells (Figure 4B). This indicated that AT101 propelled cells arrested in G2M into apoptosis. Further analysis found that the combination treatment of AT101 with docetaxel strongly induced cell apoptosis in AGS and KATO III cells compared to control or treatment either alone (Figure 4C). Proteolytic cleavage of PARP is a reliable marker of apoptosis. The combination treatment potentiated AT101-mediated PARP and caspase 3 cleavages in both AGS and KATO III cells (Figure 4D). To further confirm the hypothesis that effects of AT101 on GEC cell proliferation and apoptosis were due to the inhibition of both CSCs and anti-apoptotic signaling, AGS and KATO III GC cells and JHESO EC cells were treated with AT101, docetaxel, or combination. As shown in Figure 4E, the combination treatment of AT101 and docetaxel dramatically down-regulated genes control CSCs such as YAP1, SOX9 and β-catenin as well as BCL-2/MCL-1 in GC cells; while the anti-proliferation effects of AT101 in JHESO EC cells are more dependent on inhibition of CSCs genes-YAP1/SOX9 due to low expression of BCL-2. Collectively, our findings implied that AT101 combination with docetaxel synergistically induces cell apoptosis and sensitizes docetaxel in GC cells through targeting both CSCs signaling and anti-apoptotic pathways in GC and targeting CSCs in EC cells.
Figure 4. AT101 sensitizes Docetaxel in inhibition of GC cell growth and promote apoptosis.
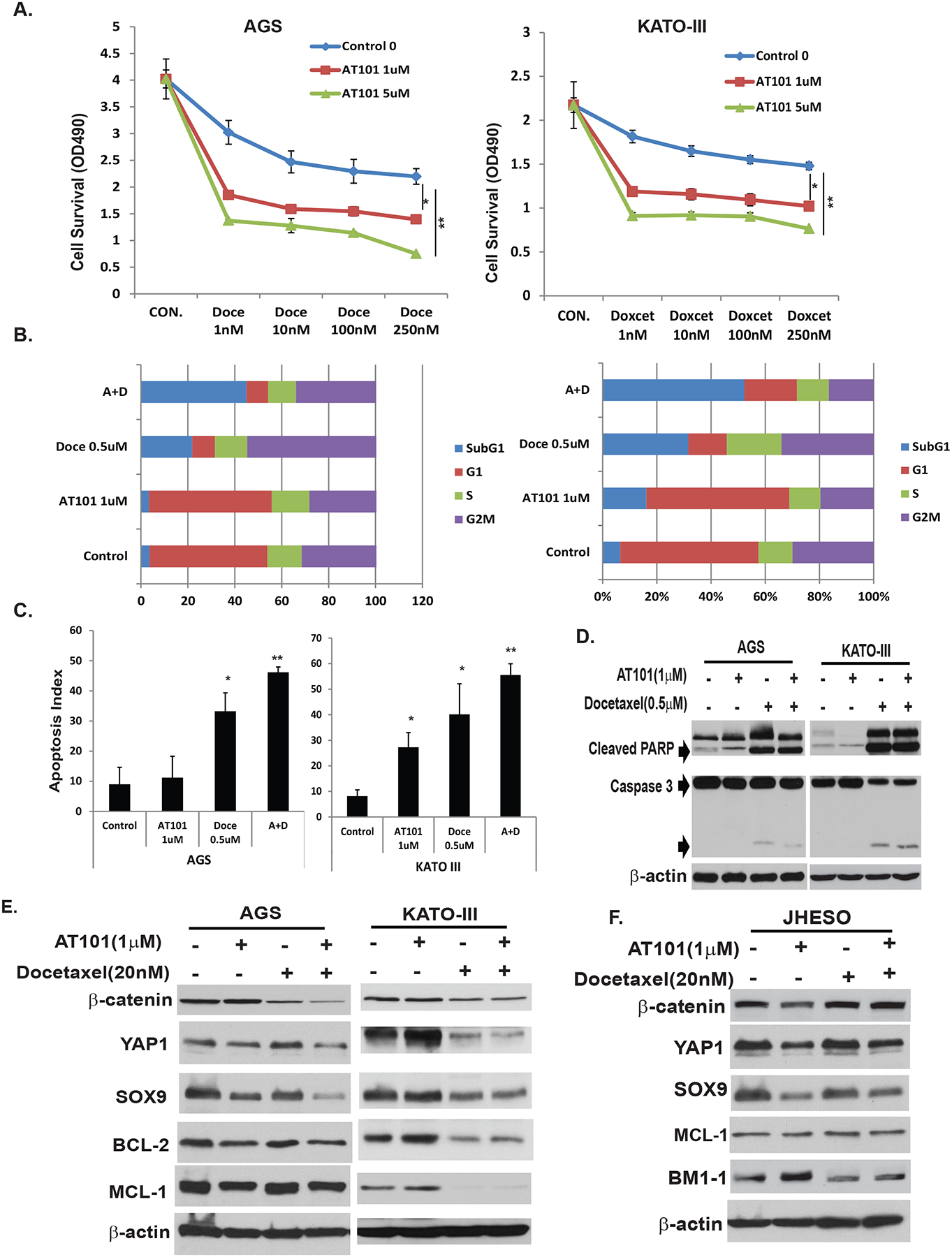
A. AGS and KATOIII GC cells were treated with docetaxel alone at dosage of 1nM to 250nM or in combination with AT101 at 1μM and 5μM respectively for 5 days, cell growth was determined using MTS as indicated in Materials and Methods. B. AGS and KATO-III cells were seeded onto 6-well plates and treated with 0.1% DMSO (as control) or with AT101 1μM or Docetaxcel 0.5μM or in combination for 48 hours and then fixed and stained for DNA with propidium iodide and then analyzed for DNA histograms and cell cycle phase distribution by flow-cytometry using a FACS Calibur. Results demonstrated that AT101 significantly sensitize docetaxel in inhibition of cell growth in a dose dependent manner and the combination of AT101 and docetaxel strongly increase cells in sub-G1. C. AGS and KATOIII cells were treated with 0.1% DMSO (as control) or AT101 or/and docetaxel at the dosage indicated, and then determined the apoptosis index by flow cytometry. Results indicated that the apoptosis index was significantly increased when cells treated with the AT101 in combination with docetaxel. D. Cell total lysates were isolated from AGS and KATO-III cells treated with 0.1% DMSO (as control) or AT101 or/and docetaxel at the dosage indicated for 48 hours, total and cleaved PARP and caspase 3 were determined by immunoblotting as described in material & Methods; E&F. Cell total lysates were isolated from AGS, KATO-III GC cells and JHESO EAC cells treated with 0.1% DMSO (as control) or AT101 or/and docetaxel at the dosage indicated for 48 hours, expression of stem cell signaling proteins-YAP1,SOX9 and β-catenin and antiapoptotic proteins MCL-1/BCL-2 were determined by immunoblotting as described in Materials & Methods.
Strong antitumor effect of AT101 in combination of docetaxel in vivo xenograft model
Nude mice bearing JHESO cell xenografts were divided randomly into 4 groups and then treated with control (PBS), AT101 alone, docetaxel alone, or in combination as described in Materials & Methods. At the end of three-week dosing schedule, tumor weight and volume and mice body weights were measured (Figures 5A–5D). Results from in vivo experiments demonstrated that mice treated with AT101 alone greatly reduced tumor volume and weight in vivo, while the mice treated with AT101 in combination with docetaxel, the significant reduction of tumor weights and tumor volumes were observed compared with either agent alone (Figures 5A & 5C), while mice body weights did not change significantly (Figure 5D). In addition, the level of YAP1 and SOX9 in mice tumors was dramatically diminished by the combination treatment of AT101 and docetaxel (Figures 5E & 5F). Thus, AT101 in combination of docetaxel had strong antitumor effects in vivo and these effects were, at least in part, due to the inhibition of CSC genes-YAP1/ SOX9. To further evaluate the combination effects of AT101 with chemo and radiation, we performed the triple combination of AT101, docetaxel and radiation compared to either alone in our JHESO xenograft model as indicated in Supplemental Figure 6. We applied radiation one time at 10Gy, 90 second at the second week of 3-week treatment (Supplemental Figure 6A). We found that triple combination had better antitumor activity than either alone (Supplemental Figure 6B&6C). Interestingly, we found the triple combination had the best inhibitory effects on stem cell markers-YAP1, SOX9 and EpCAM as well as proliferation marker KI67 (Supplemental Figure 6C).
Figure 5. Effects of AT101 in combination of Docetaxel in tumor growth in Vivo.
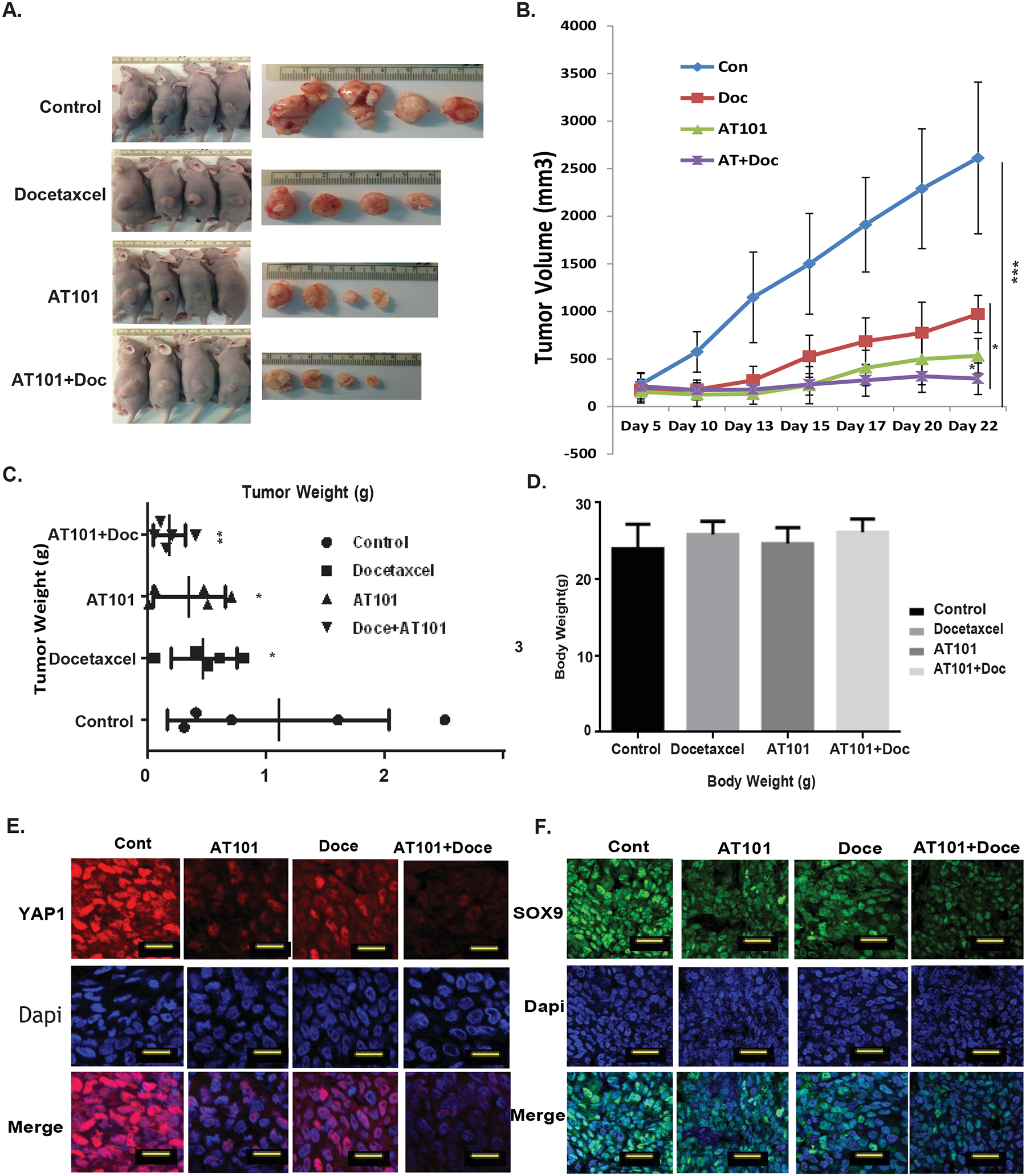
A. JHESO cells (1.5×106) were injected subcutaneously in nude mice, each mouse have two sites (left, right) injections; 5 mice/group and treated with either AT101 alone, docetaxel alone or in combination as described in Materials&Methords. B-D. After three week treatment, tumor Volume (B), tumor weight (C) and mouse body weight (D) in each group were measured and calculated as described in Materials & Methods. *P<0.05;**P<0.01. E. Immunofluorescent staining for YAP1 and SOX9 was performed in mouse tumor tissues derived from AGS xenograft nude mice. Scale bar:25 μm.
Clinical trial:
A total of 13 patients were enrolled. Patient characteristics are listed in Supplemental Table 1. The median age was 61 years and 10 had adenocarcinoma. The median performance status was 1 (range, 1). Most had poorly differentiated histology (69%). The first 7 patients (one patient could not continue therapy for none-drug related reason) received the starting dose of 10 mg daily dose of AT-101 (6 completed) and because no DLT was observed, the next 6 patients received AT-101 at 20 mg daily dose.
The most common adverse symptoms were gastrointestinal tract (GIT) related, including vomiting, anorexia, odynophagia. A total of 9 Serious Adverse Events (SAEs) were encountered irrespective of relationship to AT-101 (atrial fibrillation, failure to thrive, high fevers, bacteremia, confusion, chest pain, and GIT perforation). The bowel perforation led to death of one patient, but it was not considered study drug related. Troponin I levels were elevated in 4 patients (related to AT-101); study drug was held temporarily in 3 patients, but none had Electro Cardiogram (ECG) abnormalities or cardiac symptoms. One patient, however, was challenged with AT-101 and troponin levels rose again resulting in prompt discontinuation of AT-101. No AEs required dose reduction. No Dose Limiting Toxicity (DLT) was experienced/established. However, further dose escalation was not allowed based on liver toxicity from higher dose in other trials. Therefore, the recommended phase 2 dose of AT-101 was 20 mg daily when combined with chemoradiation.
11 of 13 patients (two did not complete therapy) achieved cCR. The median duration of cCR was 12 months (range, 3 months to 59 months). Salvage surgery could be performed in only 4 patients.
At this analysis, 5 of 13 patients had expired. The median overall survival (OS) was not reached at a median follow up time of 2 years. Figure 6A shows serial positron emission tomography of two patients who achieved cCR and had prolonged survival. The median progression free survival (PFS) time was 52 weeks with recurrences occurring in 10 of 13 patients (Figure 6B). Two patients who had a surgical salvage (residual tissue was available for pharmacodynamics) other two post-surgery patients didn’t have sufficient residual cancer for further studies. None of the clinical variables correlated with OS or PFS.
Figure 6. Clinical trial outcome and analysis of patients’ tissues of clinical trial on AT101.
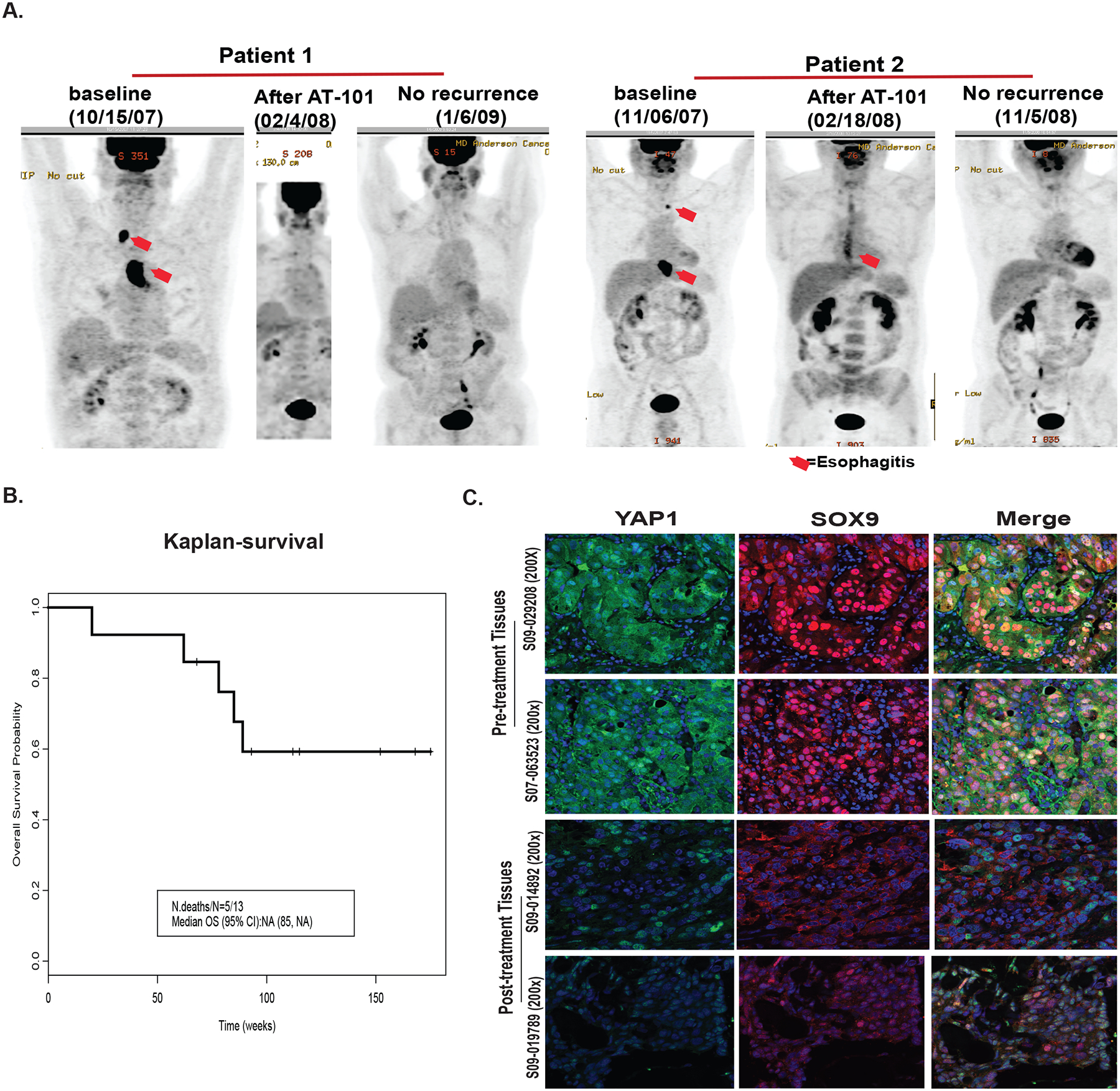
A. CT-IMAGING of patients before and after AT101 and chemoradiation treatment in two representative patients with cCR; B. Survival of 13 patients treated with AT101 plus the standard chemoradiation therapy. C. immunohistochemistry staining of YAP1 and SOX9 on pre-or post tumor tissues from two individual patients with partial response.
Biomarker data from clinical trial: We were able to assess YAP-1 and SOX-9 as representative CSC biomarkers in 8 patients (Figure 6C). All untreated tissues demonstrated overexpression of YAP-1 and SOX-9 and the merged images confirmed colocation of both CSC biomarkers. In two patients who underwent salvage surgery and had residual post-treatment tissues, we observed overexpression of YAP-1 and SOX-9 in untreated specimens but in the post-treatment tissues, there was downregulation of YAP-1 and SOX-9 confirming our preclinical data in human specimens suggesting that AT-101 can directly downregulate CSC signaling-YAP1/SOX9 axis.
Discussion
Localized gastroesophageal cancers are notoriously resistant to combined modality therapy. We reported that 218 gastroesophageal cancer patients who had no evidence of clinical cancer after chemoradiation (negative biopsies and negative imaging studies), 69% has residual resistant cancer in the surgical specimens.39 There is also a correlation between the volume/stage of the residual cancer and patient outcome.40, 41 Thus the level of resistance of the primary cancer dictates the degree of metastatic potential. When localized cancer cannot be removed surgically, the 5-year survival is only 28% to 33% and the median survival is short.42,43. The progress against esophageal cancer has been unimpressive even with the advent of multidisciplinary approaches and checkpoint inhibition.14, 44 All patients are currently treated by empirical approaches (patients with similar stage of cancer get the same treatment and without regard to genetic makeup of the tumor).
In this study, using in vitro experiments, followed by in vivo validation and a pilot clinical trial, we provide evidence that AT101 has strong antitumor activity and improve patient OS when combined with chemoradiation. We further define its mechanism of action is through targeting the CSC pathways (in addition to anti-apoptotic pathway). Interestingly, we found that AT101 exerts its antitumor activity and favorable patient response even when BCL-2 is down regulated in EC cells and EC patients indicating that AT101 targets cells with CSCs properties and had better inhibitory effects in tumor growth when combing with chemo agent docetaxel. Triple combination of AT101 with chemo and radiation even had the best anti-tumor activity than either treatment alone which may account for the good outcome for AT101 clinical trial with chemo and radiation.
The mechanisms of chemoradiation resistance are likely to be complex and multiple pathways participate.45–49 The TCGA analysis has uncovered that facts that squamous cell esophageal cancer (is more similar to head and neck cancer) is dramatically different in its genetic profile compared to adenocarcinoma.5 We and others have previously reported the role of CSC biomarkers’ association with radiation resistance23, 26, 50. In fact, we have previously demonstrated that YAP1 and its target SOX9 are highly increased in EC tumor tissues compared to normal tissues16, 36. Similarly, EC cell lines have more abundant expression of YAP1 and SOX9 compared to normal premalignant BE cells (Figures 3B). We have recently documented that YAP1 mediated CDK6 expression and activation confers radiation resistance in esophageal cancer.24 Here, through various approaches, we demonstrated that AT101 strongly suppress YAP1 and SOX9 expression and transcription in both EC and GC cells.
We note that BCL-2 is lost when BE progresses to adenocarcinoma35. BCL-2 levels are reduced or lost in seven out of eight EC cells compared to BE cells. Therefore, a pan-BLC-2 inhibitor should not be very effective, however, AT-101, turned out to be a highly active agent in the preclinical and clinical settings regardless of BCL-2 level. This suggested that an anti-apoptotic pathway molecule must have been overcoming therapy resistance through another mechanism. We observed that AT-101 was acting through the CSC pathway to reduce resistance and our pharmacodynamics studies also support this notion. Where as in GC, where BCL-2 protein is preserved, AT-101 acted on both anti-apoptotic and anti-CSC pathways.
Using cell surface biomarkers to define CSCs has limitations when compared to in vivo lineage tracing, even though a recent study showed consistency between cell surface biomarkers and in vivo lineage tracing in colon cancer51. To reduce such limitations, we, used multiple biomarkers, including genes controls CSCs such as YAP1, SOX9 and β-catenin and BMI-1 and a metabolic biomarker (i.e., ALDH1), a common CSC biomarker in the literature37, 52, 53 and together with in vitro functional assays (i.e., tumor sphere formation assay) to determine the effects of AT101 on CSCs signaling and functions. We acknowledge that cancer stemness function might be regulated by tumor microenvironment (TME) and intrinsic tumor cell plasticity. The work of Vermeulen et al54 and several others55–57 clearly indicates the importance of TME on CSCs. Role of intrinsic plasticity of tumor cells on clonogenicity and metastases has been emphasized55, 56. Here we demonstrated AT101 appears to target CSCs by abrogating YAP1/SOX9/β-catenin signaling in addition to suppress antiapoptotic signaling; however, we did not study mechanisms by which AT101 might have influenced CSC functions through TME interactions.
Our clinical data are very heartening, as this group of patients has very poor prognosis. However, in the phase I study, majority of the patient did well, and their survival was much longer than expected. The phase 2 study could not be completed because the sponsor stopped development of AT-101 due to failure of a pivotal trial in acute lymphocytic leukemia. However, AT-101 deserves to be studied further.
In conclusion, we report the novel finding of a pan-BCL-2 inhibitor being very active even when BCL-2 is downregulated and the novel mechanism of action is against the CSC pathway.
Supplementary Material
What is already known on this subject?
Gastroesophageal cancer including esophageal cancer and gastric cancer continue to be a health care burden globally and patients are often diagnosed in advanced stage and survival is poor. The cancer stem cells (CSCs) and anti-apoptotic pathways often confer therapy resistance. AT-101, a natural BH3-mimetic molecule and a pan-BCL-2 Inhibitor has demonstrated anti-tumor activity in some hematologic malignancies.
What are the new findings?
In this study, we provide evidence from in vitro experiments, followed by in vivo validation and a pilot clinical trial to demonstrate that AT101 can target the CSC pathways (in addition to anti-apoptotic pathway) and demonstrate strong anti-tumor effects and improve patient survival when combined with chemoradiation. Interestingly, we found that AT101 had antitumor activity and favorable clinical response even when BCL-2 is down regulated in EC cells and EC patients. This indicates that the anti-tumor effects of AT101 in Gastroesophageal cancer rely more on targeting CSCs.
How might it impact on clinical practice in the foreseeable future?
Our results establish the molecular foundation for a pan BCL-2 inhibitor, AT101 had antitumor activity and favorable clinical response in gastroesophageal cancer even when BCL-2 is downregulated and the novel mechanism of action is against the CSC pathway. This study provides a strong rationale for developing a large clinical trial for AT101 in combination with current chemotherapy such as docetaxel agent in gastroesophageal cancer.
Acknowledgement
We appreciate Sarah Bronson, scientific editor from Department of Scientific publications of MDACC for her excellent edition on English of this manuscript and Dr. Veera Baladandayuthapani for his statistical support. Funding supports for this study was in part by the donations received from the Caporella, Dallas, Sultan, Park, Smith, Frazier, Oaks, Sultan, Vansteklenberg, Planjery, and Cantu Families, the Stupid Strong Foundation, the V foundation, the Schecter Private Foundation, Rivercreek Foundation, Kevin Fund, Myer Fund, Dio Fund, Milrod Fund, and the Multidisciplinary Research Grants provided by the University of Texas M. D. Anderson Cancer Center, Houston, USA; Public Health Service Grant DF56338 which supports the Texas Medical Center Digestive Diseases Center (Song S); UTMDACC IRG(3-0026317,Song S). CA160433 and CA170906 from the Department of Defense (Song S). CA129906, CA127672, CA138671, and CA172741 from the National Cancer Institute and CA150334 and CA160445 from the Department of Defense (JAA).
Abbreviations:
- GEC
Gastroesophageal cancers
- GC
Gastric cancer
- EC
Esophageal cancer
- PC
Peritoneal carcinomatosis/Malignant ascites
- CSCs
Cancer stem cells
- cCR
clinical complete response
- MTD
maximum tolerated dose
- SAEs
Serious Adverse Events
- ECG
Electro Cardiogram
- DLT
Dose Limiting Toxicity
- OS
overall survival
- PFS
progression free survival
- TME
Tumor microenvironment
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Ajani JA, Lee J, Sano T, et al. Gastric adenocarcinoma. Nat Rev Dis Primers 2017;3:17036. [DOI] [PubMed] [Google Scholar]
- 3.Ajani JA, D’Amico TA, Almhanna K, et al. Gastric Cancer, Version 3.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2016;14:1286–1312. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research N, Analysis Working Group: Asan U, Agency BCC, et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017;541:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017. [DOI] [PubMed] [Google Scholar]
- 7.Song S, Honjo S, Jin J, et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res 2015;21:2580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ajani JA, Song S, Hochster HS, et al. Cancer stem cells: the promise and the potential. Semin Oncol 2015;42 Suppl 1:S3–17. [DOI] [PubMed] [Google Scholar]
- 9.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 2015;15:73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medema JP. Targeting the Colorectal Cancer Stem Cell. N Engl J Med 2017;377:888–890. [DOI] [PubMed] [Google Scholar]
- 11.Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 2017;16:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adorno-Cruz V, Kibria G, Liu X, et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res 2015;75:924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ajani JA, D’Amico TA, Almhanna K, et al. Esophageal and esophagogastric junction cancers, version 1.2015. J Natl Compr Canc Netw 2015;13:194–227. [DOI] [PubMed] [Google Scholar]
- 15.Kim C, Gao R, Sei E, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018;173:879–893 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song S, Ajani JA, Honjo S, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res 2014;74:4170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin L, Sabnis AJ, Chan E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet 2015;47:250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KW, Lee SS, Hwang JE, et al. Development and Validation of a Six-Gene Recurrence Risk Score Assay for Gastric Cancer. Clin Cancer Res 2016;22:6228–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overholtzer M, Zhang J, Smolen GA, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A 2006;103:12405–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camargo FD, Gokhale S, Johnnidis JB, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007;17:2054–60. [DOI] [PubMed] [Google Scholar]
- 21.Lockwood WW, Thu KL, Lin L, et al. Integrative genomics identified RFC3 as an amplified candidate oncogene in esophageal adenocarcinoma. Clin Cancer Res 2012;18:1936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang W, Tong JH, Chan AW, et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin Cancer Res 2011;17:2130–9. [DOI] [PubMed] [Google Scholar]
- 23.Keren-Paz A, Emmanuel R, Samuels Y. YAP and the drug resistance highway. Nat Genet 2015;47:193–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Xu Y, Liu B, et al. YAP1-Mediated CDK6 Activation Confers Radiation Resistance in Esophageal Cancer - Rationale for the Combination of YAP1 and CDK4/6 Inhibitors in Esophageal Cancer. Clin Cancer Res 2019;25:2264–2277. [DOI] [PubMed] [Google Scholar]
- 25.Song S, Ajani JA, Honjo S, et al. Hippo coactivator YAP1 upregulates SOX9 and endows stem-like properties to esophageal cancer cells. Cancer Res 2014; 74(15):4170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ajani JA, Wang X, Song S, et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol Oncol: 2014;8(1):142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delbridge AR, Grabow S, Strasser A, et al. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 2016;16:99–109. [DOI] [PubMed] [Google Scholar]
- 28.Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 2016;16:539–48. [DOI] [PubMed] [Google Scholar]
- 29.Paoluzzi L, Gonen M, Gardner JR, et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 2008;111:5350–8. [DOI] [PubMed] [Google Scholar]
- 30.Balakrishnan K, Burger JA, Wierda WG, et al. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood 2009;113:149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ready N, Karaseva NA, Orlov SV, et al. Double-blind, placebo-controlled, randomized phase 2 study of the proapoptotic agent AT-101 plus docetaxel, in second-line non-small cell lung cancer. J Thorac Oncol 2011;6:781–5. [DOI] [PubMed] [Google Scholar]
- 32.Boiani M, Daniel C, Liu X, et al. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. J Biol Chem 2013;288:6980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai A, Zeitlin BD, Visioli F, et al. Metronomic dosing of BH3 mimetic small molecule yields robust antiangiogenic and antitumor effects. Cancer Res 2012;72:716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linton A, Pond G, Clarke S, et al. Glasgow prognostic score as a prognostic factor in metastatic castration-resistant prostate cancer treated with docetaxel-based chemotherapy. Clin Genitourin Cancer 2013;11:423–30. [DOI] [PubMed] [Google Scholar]
- 35.Raouf AA, Evoy DA, Carton E, et al. Loss of Bcl-2 expression in Barrett’s dysplasia and adenocarcinoma is associated with tumor progression and worse survival but not with response to neoadjuvant chemoradiation. Dis Esophagus 2003;16:17–23. [DOI] [PubMed] [Google Scholar]
- 36.Song S, Maru DM, Ajani JA, et al. Loss of TGF-beta adaptor beta2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res 2013;73:2159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ajani JA, Wang X, Song S, et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol Oncol 2014;8:142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Cutsem E, Moiseyenko VM, Tjulandin S, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 2006;24:4991–7. [DOI] [PubMed] [Google Scholar]
- 39.Cheedella NK, Suzuki A, Xiao L, et al. Association between clinical complete response and pathological complete response after preoperative chemoradiation in patients with gastroesophageal cancer: analysis in a large cohort. Ann Oncol; 2013;24(5):1262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chirieac LR, Swisher SG, Ajani JA, et al. Posttherapy pathologic stage predicts survival in patients with esophageal carcinoma receiving preoperative chemoradiation. Cancer 2005;103:1347–55. [DOI] [PubMed] [Google Scholar]
- 41.Taketa T, Sudo K, Correa AM, et al. Post-chemoradiation surgical pathology stage can customize the surveillance strategy in patients with esophageal adenocarcinoma. J Natl Compr Canc Netw 2014;12:1139–44. [DOI] [PubMed] [Google Scholar]
- 42.Sudo K, Xiao L, Wadhwa R, et al. Importance of surveillance and success of salvage strategies after definitive chemoradiation in patients with esophageal cancer. J Clin Oncol 2014;32:3400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harada K, Wu CC, Wang X, et al. Total Lesion Glycolysis Assessment Identifies a Patient Fraction With a High Cure Rate Among Esophageal Adenocarcinoma Patients Treated With Definitive Chemoradiation. Ann Surg; 2020;272(2):311–318 [DOI] [PubMed] [Google Scholar]
- 44.Doi T, Piha-Paul SA, Jalal SI, et al. Safety and Antitumor Activity of the Anti-Programmed Death-1 Antibody Pembrolizumab in Patients With Advanced Esophageal Carcinoma. J Clin Oncol 2018;36:61–67. [DOI] [PubMed] [Google Scholar]
- 45.Mistry IN, Thomas M, Calder EDD, et al. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int J Radiat Oncol Biol Phys 2017;98:1183–1196. [DOI] [PubMed] [Google Scholar]
- 46.Haffty BG, Glazer PM. Molecular markers in clinical radiation oncology. Oncogene 2003;22:5915–25. [DOI] [PubMed] [Google Scholar]
- 47.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15:81–94. [DOI] [PubMed] [Google Scholar]
- 48.Barker HE, Paget JT, Khan AA, et al. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 2015;15:409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23:1124–1134. [DOI] [PubMed] [Google Scholar]
- 50.Wadhwa R, Wang X, Baladandayuthapani V, et al. Nuclear expression of Gli-1 is predictive of pathologic complete response to chemoradiation in trimodality treated oesophageal cancer patients. Br J Cancer 2017;117:648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goto N, Fukuda A, Yamaga Y, et al. Lineage tracing and targeting of IL17RB(+) tuft cell-like human colorectal cancer stem cells. Proc Natl Acad Sci U S A 2019;116:12996–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007;1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Douville J, Beaulieu R, Balicki D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev 2009;18:17–25. [DOI] [PubMed] [Google Scholar]
- 54.Lenos KJ, Miedema DM, Lodestijn SC, et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat Cell Biol 2018;20:1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dirkse A, Golebiewska A, Buder T, et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat Commun 2019;10:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fumagalli A, Oost KC, Kester L, et al. Plasticity of Lgr5-Negative Cancer Cells Drives Metastasis in Colorectal Cancer. Cell Stem Cell 2020;26:569–578 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Heijden M, Miedema DM, Waclaw B, et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc Natl Acad Sci U S A 2019;116:6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.