

Abstract
How genetic and environmental risk factors interact to trigger the development of post-traumatic stress disorder (PTSD) remains largely unknown. Seah et al. model stress hypersensitivity as a potential mechanism by examining transcriptomic responses to glucocorticoids in neurons derived from individuals with PTSD.
PTSD occurs in some individuals who undergo direct exposure to intense and terrifying experiences such as combat. Genetic studies of PTSD, especially large-scale genome-wide association studies (GWASs), are starting to reveal genetic variations that predispose an individual to develop PTSD1,2. However, the molecular and cellular mechanisms that explain individual susceptibility to PTSD remain largely unknown, which hinders risk prediction and more tailored treatment of PTSD. A major challenge is that, like other neuropsychiatric disorders, PTSD risk is polygenic, probably involving hundreds of genes each with a small effect. Moreover, many disease risk genes may elicit their biological effects only upon exposure to environmental risk factors, such as stress. It is imperative to develop a human cell-based model for dissecting the complex neurobiological mechanisms that underlie PTSD and other stress-related psychiatric conditions.
In this issue of Nature Neuroscience, Seah and colleagues3 address this challenge by using neurons derived from induced pluripotent stem cells (iPS cells) from patients with PTSD in a Petri dish as a cellular model to study gene–environment interactions in the context of PTSD (Fig. 1). With hydrocortisone (HCort; a glucocorticoid) as a stimulus to mimic some effects of the environmental stressor, the authors demonstrated that the transcriptomes of ‘stressed’ neurons, but not baseline (unstimulated) neurons, exhibited different patterns in cells derived from donors with PTSD as opposed to those without. Interestingly, this PTSD-specific hypersensitive transcriptomic response to glucocorticoid stimulation in iPS cell-derived neurons was not observed in peripheral blood mononuclear cells (PBMCs) from a largely overlapping group of combat veterans. Through an integrative analysis of the ‘stressed’ neural transcriptomes with previous data (post-mortem brain gene expression and GWAS results), the authors also identified co-expression gene networks and specific genes that might mediate the PTSD-specific hypersensitive neuronal response to glucocorticoid stimulation. These findings highlight the importance of cell type-specific and context-dependent gene regulation in linking static genomic risk factors to clinical phenotypes triggered by environmental factors in PTSD and other neuropsychiatric disorders.
Fig. 1 |. Modelling PTSD gene–environment interactions using PBMCs and iPS cell-derived glutamatergic NGN2-induced neurons.
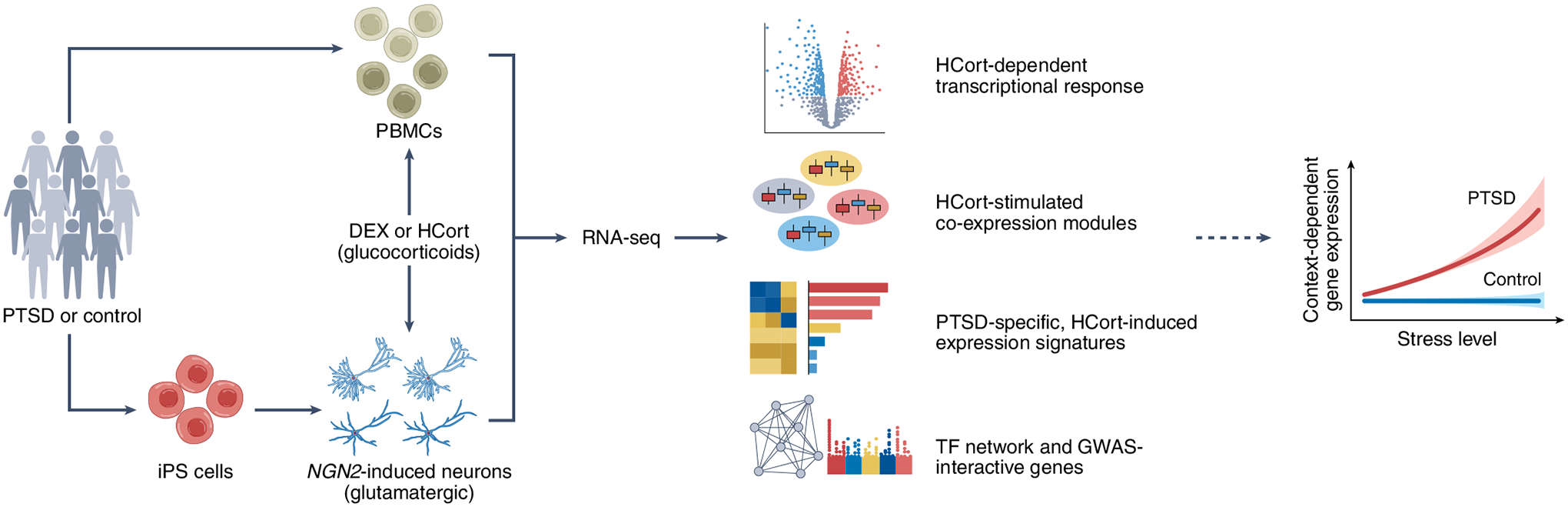
The authors started their analysis by isolating PBMCs and differentiating glutamatergic NGN2-induced neurons from iPS cells from healthy donors and donors diagnosed with PTSD. Both PBMCs and glutamatergic neurons were stimulated by glucocorticoids (DEX or HCort) to mimic stress and subjected to RNA sequencing (RNA-seq) analysis. Combined with post-mortem brain data from individuals with PTSD, the transcriptomic analysis revealed robust glucocorticoid hypersensitivity in glutamatergic neurons derived from donors with PTSD, suggesting strong context-dependent gene regulation.
Because most studies of the pathophysiology of PTSD have focused on PBMCs, the authors started by analyzing the differential transcriptional responses to dexamethasone (DEX; also a glucocorticoid)3 of individual-derived PBMCs from individuals with PTSD and control participants who had also been exposed to trauma. Increasing DEX concentrations resulted in growing numbers of differentially expressed genes (DEGs), which were concordant with their previous study on a much smaller sample size4. Interestingly, despite PBMCs being a cell type further removed from the brain, gene ontology (GO) analysis of the DEGs identified ‘synapse’ and more broad terms under the immune category as enriched GO terms. However, whereas the DEX-induced DEGs were enriched in genes associated with multiple neuropsychiatric disorders, no enrichment was observed for PTSD. Moreover, the DEX-induced genes did not show any differential expression between donors with and without PTSD. These results demonstrate the limitations of using PBMCs as a model to recapitulate PTSD-specific molecular signatures.
Although the disease-relevant cell type(s) for PTSD remain unidentified, it is known that trauma-induced perturbations in glucocorticoid signaling alter glutamatergic neural activity5. The authors therefore studied the transcriptional response to HCort in NGN2-induced glutamatergic neurons6 derived from iPS cells from control donors and those with PTSD (Fig. 1). NGN2-induced glutamatergic neurons have been successfully used in the functional interpretation of neuropsychiatric GWAS risk variants7,8. The uniqueness of the experimental design here was that the neurons used for HCort perturbation were from a sizable iPS cell cohort consisting of about 20 donors with PTSD and 20 without whose PBMCs were also subjected to the DEX stimulation, which enabled direct comparison of results from both cell types to assess cell type-specific transcriptomic responses to the ‘stressor’ — glucocorticoids.
The initial analysis of whether glucocorticoid stimulation of glutamatergic neurons without considering PTSD diagnosis as a factor was sufficient to recapitulate a PTSD-specific brain gene expression signature yielded mixed results. HCort exposure led to changes in the expression of thousands of genes in glutamatergic neurons without eliciting non-specific cell toxicity and death. Whereas the HCort-upregulated genes in glutamatergic neurons were enriched for the risk gene sets of multiple psychiatric disorders, they were not for PTSD, similar to PBMCs. Also, as in PBMCs, no DEGs were identified between NGN2-induced neurons from control donors and those with PTSD under baseline conditions.
However, when Seah and colleagues3 looked up differentially responsive genes (DRGs) in HCort-stimulated neurons between donors with or without PTSD, they uncovered a remarkable number of PTSD-specific DRGs. In total, about 740 genes showed significant diagnosis-by-HCort interactive effects, and overwhelmingly more genes showed PTSD-associated hyper-responsivity than hypo-responsivity (n = 312 and 84, respectively). Moreover, the authors evaluated the spatial association of genes with significant interactive effects and PTSD GWAS risk variants by mapping the significance (P value) of interactive genes against that of GWAS meta-analysis results and subsequently found shared peaks (that is, genes) on chromosomes 10, 17, and 19. The detection of a PTSD-diagnosis-specific transcriptomic signature only in HCort-stimulated glutamatergic neurons strongly suggests the need for research paradigms examining the functional impacts of genetic risk factors of PTSD and other neuropsychiatric disorders in the context of disorder-relevant environmental stimuli.
In an appropriate cellular model, stimulation can unmask the biological effects of otherwise static genetic risk factors, which would be undetectable under baseline conditions. Such context-dependent regulatory effects of disease risk variants have been better established for some immune disorders; for instance, cytokine stimulation of pancreatic β-cells unmasks abundant novel regulatory sequences, and remarkably, only stimulated enhancers are enriched for type 1 diabetes GWAS variants9. For neuropsychiatric disorders, the challenges lie in the lack of clarity on the disease-relevant cell types and environmental stimuli. The success of Seah and colleagues3 in uncovering PTSD-specific transcriptomic signatures in HCort-treated glutamatergic neurons obviously benefits from PTSD being a disorder for which an environmental risk factor (that is, a traumatic stressor) has been clearly defined and a proxy cellular stressor (that is, glucocorticoid stimulation) has been well-studied. Correspondingly, it may be more challenging to model environmental risk factors for other neuropsychiatric disorders in cell culture. Nonetheless, even without clearly defined environmental risk factors, stimuli relevant to the pathophysiology of neuropsychiatric disorders may still help to shed light on how polygenic risk factors interact with environmental factors. For instance, perturbation of lymphoblastic cell lines derived from peripheral blood cells of individuals with schizophrenia (SCZ) and controlled by dopamine, a neurotransmitter that has been implicated in SCZ, enabled the identification of SCZ-specific DRGs enriched for SCZ GWAS risk variants10. As another example, stimuli such as membrane-depolarizing levels of potassium chloride (KCl) can induce neuronal activity that mimics the in vivo effects of social experiences or stress and thereby elicit activity-dependent transcriptional and epigenetic changes in iPS cell-derived GABAergic neurons that are enriched for heritability of autism spectrum disorders (ASD)11. Future mapping of context-dependent regulatory sequences and variants that modulate gene expression and chromatin accessibility (that is, expression quantitative trait loci and chromatin accessibility quantitative trait loci) upon stimulation using an adequately powered iPS cell cohort holds the promise of uncovering specific genetic risk loci that may mediate gene–environment interactions in neuropsychiatric disorders.
Another strength of the work by Seah and colleagues3 was to leverage the ‘stressed’ transcriptomic data to predict transcription factors (TFs) that may drive glucocorticoid hyper-responsivity in PTSD. Identifying the TF driver or master regulators of disease risk genes is a new concept in psychiatry research12. The idea is that polygenic risk genes tend to function in networks that are co-regulated in disease-relevant biological processes. The authors identified 38 TFs that targeted the PTSD hyper-responsive genes and assembled co-expression gene networks regulated by specific TFs. Although the current study falls short of confirming FKBP5 as a stress-responsive molecular hub13 or discussing whether a strong PTSD GWAS risk gene, TCF4 (refs.1,2), was among those PTSD hyper-responsive TFs, it is noteworthy that one of the identified PTSD-specific driver TFs, YY1, has been recently shown to be a transcriptional regulator of chronic stress-induced cortical gene expression changes and maladaptive behavior in mice14. Such master regulators may represent promising therapeutic targets for PTSD.
Studying gene–environment interactions for neuropsychiatric disorders remains a substantial challenge. It would be an over-simplification to consider that the glucocorticoid-’stressed’ transcriptional responses in glutamatergic neurons reflect the cellular and molecular events that lead to the development of PTSD a priori. The complexity also stems from the uncertainty of the involved types of cells; for instance, PTSD GWAS1,2 strongly implicate striatal GABAergic inhibitory neurons as relevant cell types for PTSD. Moreover, a recent study showed that the stress-specific responses may occur at both somatic and mitochondrial genome levels15. Nevertheless, modeling the ‘stressed’ transcriptomes in a Petri dish3 takes us a step closer to comprehending why some individuals are more likely to develop PTSD than others. Further studies that integrate single-cell multi-omic, genetic, and clinical phenotypic data, in combination with cutting-edge 3D neural culture systems such as brain organoids and CRISPR–Cas9-induced genetic perturbation and/or disease-relevant cellular stimuli, will provide a deeper mechanistic understanding of PTSD and other neuropsychiatric disorders, which may lead to more tailored clinical interventions and therapeutic treatments.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Seriès P Nat. Neurosci 22, 334–336 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Gelernter J et al. Nat. Neurosci 22, 1394–1401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seah C et al. Nat. Neurosci 10.1038/s41593-022-01161-y (2022). [DOI] [Google Scholar]
- 4.Breen MS et al. Transl. Psychiatry 9, 201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Popoli M, Yan Z, McEwen BS & Sanacora G Nat. Rev. Neurosci 13, 22–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y et al. Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang S et al. Science 369, 561–565 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schrode N et al. Nat. Genet 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramos-Rodríguez M et al. Nat. Genet 51, 1588–1595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan J et al. Transl. Psychiatry 8, 158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boulting GL et al. Nat. Neurosci 24, 437–448 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doostparast Torshizi A et al. Sci. Adv 5, eaau4139 (2019). [Google Scholar]
- 13.Matosin N, Halldorsdottir T & Binder EB Biol. Psychiatry 83, 821–830 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Kwon DY et al. Nat. Commun 13, 55 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Replogle JM et al. Cell 185, 2559–2575.e28 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]