

Abstract
Cytochrome P450cam (CYP101A1) catalyzes the hydroxylation of d-camphor by molecular oxygen. The enzyme-catalyzed hydroxylation exhibits a high degree of regioselectivity and stereoselectivity, with a single major product, d-5-exo-hydroxycamphor, suggesting that the substrate is oriented so as to facilitate this specificity. In previous work, we used an elastic network model (ANM) and perturbation-response scanning (PRS) to show that normal deformation modes of the enzyme structure are highly responsive not only to the presence of substrate but to substrate orientation as well. The current work examines the effects of mutations near the active site on substrate localization and orientation. The investigated mutations were designed to promote a change in substrate orientation and/or location that might give rise to different hydroxylation products, while maintaining the same carbon and oxygen atom balances as in the wild type enzyme. Computational experiments and parallel in vitro site-directed mutations of CYP101A1 were made to examine reaction products and enzyme activity. 1H-15N TROSY-HSQC correlation maps were used to compare the computational results with detectable perturbations in the enzyme structure and dynamics. We found that all of the mutant enzymes retained the same regio- and stereospecificity of hydroxylation as the WT enzyme, with varying degrees of efficiency, which suggests that large portions of the enzyme have been subjected to evolutionary pressure to arrive at the appropriate sequence-structure combination for efficient 5-exo hydroxylation of camphor.
Keywords: enzyme substrate recognition, molecular dynamics, nuclear magnetic resonance
Introduction
The P450 superfamily of enzymes catalyze a variety of oxidation reactions of diverse substrates in a vast array of organisms (3). These reactions, especially those involved in biosynthetic or catabolic processes, often show high regio- and stereoselectivity. Despite this diversity, the superfamily exhibits a highly conserved tertiary structure, with a conserved iron heme ligated on the proximal face by a cysteinyl thiolate ligand. We are interested in the sequence-function relationships that control substrate selectivity and binding orientation, with an eye towards the potential for computer-assisted enzyme engineering. Cytochrome P450cam (CYP101A1) is the archetype of the P450 superfamily and has figured prominently in investigations of P450 activity and its relationship with the enzyme structure. CYP101A1 catalyzes the 5-exo-hydroxylation of d-camphor, the first step of camphor catabolism by the soil bacterium Pseudomonas putida. The reaction is shown in Figure 1.
Figure 1:
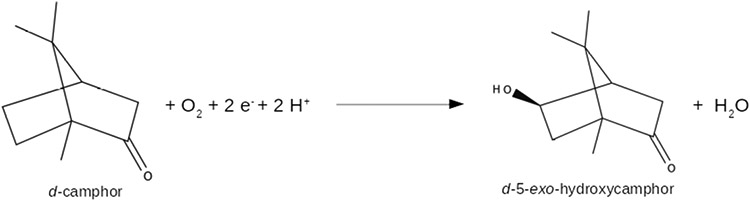
Regio and stereospecific hydroxylation of camphor catalyzed by CYP101A1.
The specificity of the reaction implies that camphor is bound to CYP101A1 in a preferred orientation, or at least only one orientation that is productive. Our groups have previously shown that in order to attain the correct substrate orientation, CYP101A1 must be bound to its physiological redox partner and effector, the iron–sulfur protein putidaredoxin (Pdx) (4, 5). Using nuclear magnetic resonance (NMR) it was found that camphor orientation in the absence of Pdx differs from that seen in the crystal structure (6). Applying residual dipolar couplings (RDCs) as experimental restraints in “soft annealing” molecular dynamics simulations, we generated an ensemble of solution structures of camphor-bound CYP101A1 (1). The best-fit structure from that ensemble was deposited in the PDB database as entry 2L8M (Figure 2). A similarly-derived structure of the substrate-free enzyme (PDB entry 2LQD) provided insight into those regions of the structure that are most perturbed by substrate binding (7). We then investigated the mechanical response map of the CYP101A1 polypeptide and identified secondary structural features that are responsive to substrate binding (5, 8). In the current work, we continue the investigation and expand our analysis of substrate orientation in CYP101A1, including the effects of mutations that were designed based on our previous results. We describe the effects of several mutations near the CYP101A1 active site as determined by solvated molecular dynamics (MD) simulations. Our analysis was focused on exploring how these mutations affect protein dynamics, and if such scenarios might result in a different preferred substrate orientation, thereby altering regioselectivity of the hydroxylation reaction. To support the computational work, the corresponding mutations were generated in vitro as well as in silico, and the effects determined by standard enzyme turnover assays, as well as NMR mapping of structural perturbations.
Figure 2.
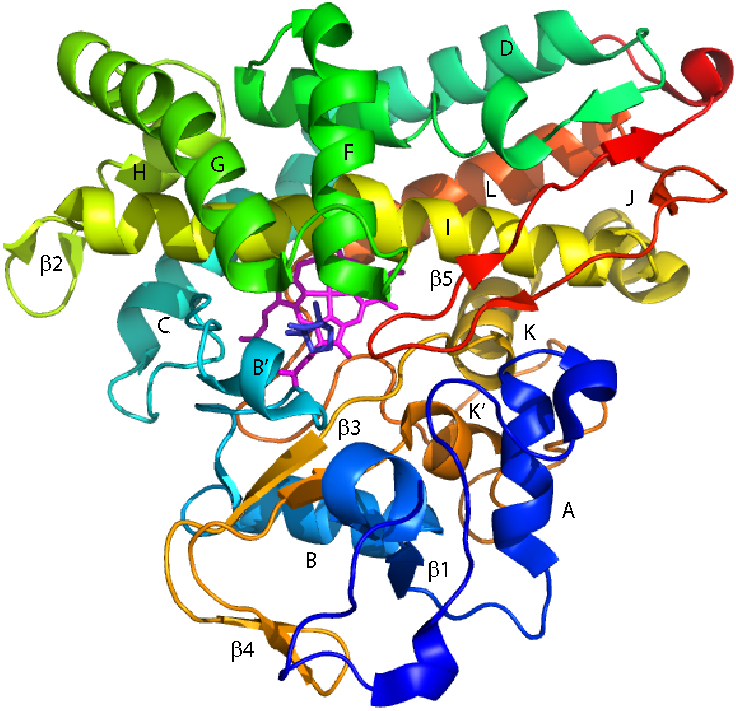
Solution structure of CYP101A1 (PDB entry 2L8M (1)), with secondary structural features referred to in the text labeled according to the scheme of Raag and Poulos (2). Heme is shown as magenta sticks, and camphor as blue. Residue numbers are as follows: A helix (V38–E47), β1 (L53–A65), B helix (R67–D77), B′ helix (P89–E94), C helix (Q108–V119), D helix (D125–R143), E helix (F150–G168), F helix (P170–T185), G helix (T192–K214), H helix (A219–A224), β2 (Q227–P232), I helix (T234–A265), J helix (S267–R277), K helix (R280–F292), β3 (G298–L301 and Q317–L319), β4 (Y305–L312), K′ helix (P321–L327), L helix (Q360–I378), β5 (S382–V405).
Materials and Methods
Molecular Dynamics Simulations.
All simulations were performed using the Amber 20 pmemd.cuda implementation (9) mounted on the Extreme Science and Engineering Discovery Environment (XSEDE) node Comet at the San Diego Supercomputer Center. The starting structure for all simulations was the NMR-derived solution structure of the reduced camphor- and carbonmonoxy-bound form of CYP101A1, PDB entry 2L8M, in which the carbon atom of the carbon monoxide (CMO) is bound via a dative covalent bond to the heme iron (1). All systems were prepared for MD using tleap (10). For each system, the enzyme was placed in a rectangular TIP3P water box with a 10 Å cushion, under periodic boundary conditions, and sufficient K+ and Cl− ions were added to neutralize charge (11) and approximate the concentration of KCl used for NMR measurements (0.1M KCl). The Amber force field ff14SB (12) was used to parameterize the simulations, with the exception of the CMO (carbonmonoxy)-heme complex. The heme was represented in a reduced hexacoordinate low spin state. The AMBER database and Antechamber were used to parametrize the heme, with an explicit bond between Fe and Cys357. Heme and Cys357 parameters were provided by the Estrin group (13, 14). The distal position was occupied by CMO and the covalent bond between the carbon atom of CMO and iron was harmonically restrained with a 1000 kcal/mol/Å2 force constant to maintain a correct distance and angle parallel to the heme normal during the simulation. Simulations were performed with a 10 Å cutoff applied to electrostatic interactions under the Particle Mesh Ewald (PME) summation method. Covalent bonds involving hydrogen were maintained at their equilibrium distance using the SHAKE algorithm (15). The system energy was first minimized by steepest descent, followed by conjugate gradient minimization, for 10000 cycles. The system was then heated to 298 K using a Langevin thermostat (16) with collision frequency of 2 ps−1, followed by NPT equilibration using a Berendsen barostat (17) with semi-isotropic pressure scaling for 100 ps, and an NVT equilibration of 20 ns. After equilibration, a 600 ns production run was performed using an NVT ensemble, at 298 K, with coordinates and velocities saved every 10 ps. Five independent simulations were performed for each system, starting with different random initial velocities.
MD trajectory analyses were performed on a local Linux computer implementing the software CPPTRAJ (18). Global stability of the simulated systems was evaluated by RMSD calculations, fitted to an average structure, and the first 50 ns of each simulation were considered as equilibration time and excluded from subsequent calculations. Therefore, the five independent simulations of each system provide an ensemble trajectory of 2.75 μs, which was resubjected to RMSD fitting to generate an average structure.
The selection of structures for further characterization was made by implementation of a k-means algorithm clustering analysis to group structures based upon the CAM(C5)-CMO(C) distance (19). This algorithm groups structures based on a given number k of clusters establishing k centroids that minimizes the distance between the cluster elements with respect to the cluster centroid in an iterative fashion. The clustering was made over the five independent trajectories of each complex together, sampling every 10 frames. Six k clusters were chosen: The choice was made by testing several k values for each system and comparing quality parameters. The initial centroids were set randomly, and the algorithm was run until clusters no longer changed after a maximum of 500 iterations were reached.
Generation of in silico mutations.
The locations of mutations described here are shown in Figure 3. Mutant 3M was constructed by making the amino acid change V247A-V295I-V396I in the NMR-derived 2L8M structure. After production MD of the 3M complex was completed, frame 5001 from the first run was saved and used as template for making the mutations A247G-G248A to construct the 4M mutant (V247G-G248A-V295I-V396I). The swap F87Y-Y96F implemented to give rise to the 2S mutant was also made in the 2L8M structure. For construction of the 4M2S mutant, a representative structure of 4M was chosen by clustering analysis, sampling the five equilibrated trajectories, ensembled in a 2.75 μs simulation, each 10 frames and using the RMSD as clustering parameter. Finally, the swap F87Y-Y96F was made over this representative structure (frame 34165).
Figure 3.
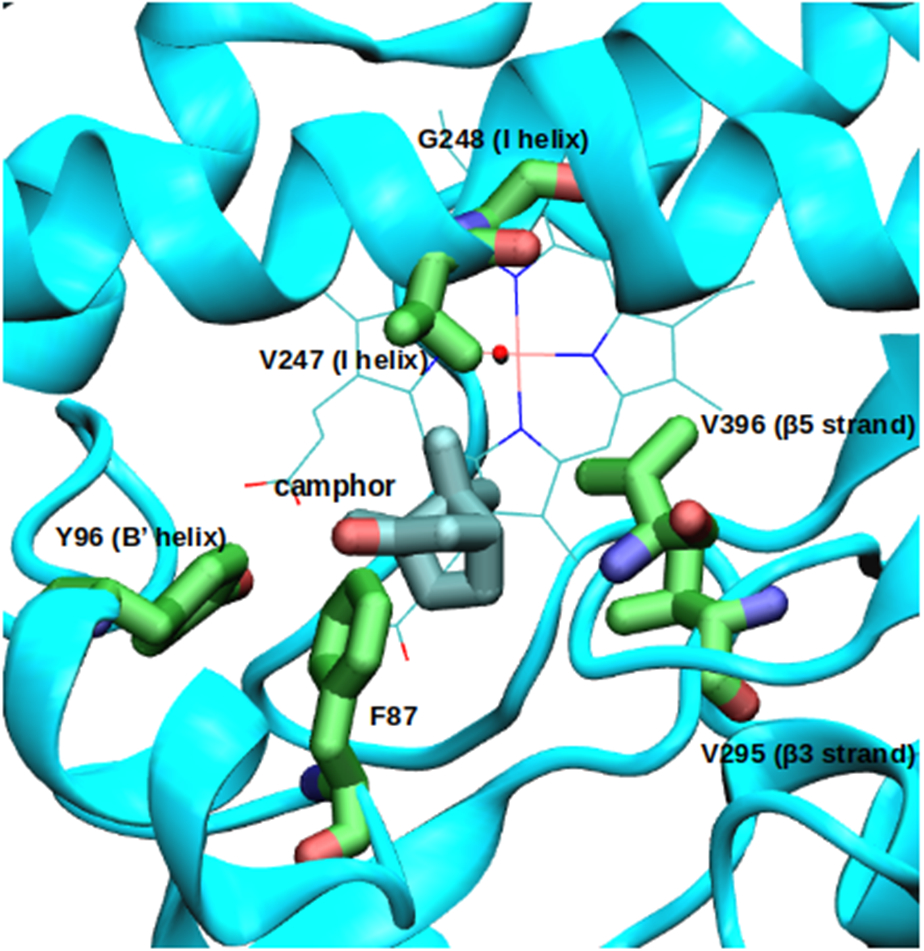
Mutation sites in the CYP101A1 structure (PDB entry 2L8M).
Simulations with harmonic restraints for fixing protein coordinates.
Three different sets of initial coordinates were chosen from the 2.75 μs equilibrated trajectory of each enzyme, and a 100 ns simulation in a NVT ensemble was produced as described above. The protein, the heme and the carbon monoxide were kept fixed by the application of a 1 kcal/mol/Å2 harmonic potential.
In vitro experiments
Site-directed mutations in CYP101A1.
The pET-28a(+)/CYP101A1 construct pLR1 kindly provided by Dr. Lingyun Rui was used for all site-directed mutagenesis and expression experiments. Multisite mutations 3M, 2S and 4M2S were performed using the Gibson Assembly® Ultra kit (Codex DNA, Inc.), following the suggested protocols. PCR amplification used Phusion™ High-Fidelity DNA polymerase (Thermo Scientific) and oligonucleotides obtained from Eurofins MWG Operon (Huntsville, AL) (see Supplementary Material for oligos used). The 4M mutant was made using the Q5® site-directed mutagenesis kit (New England Biolabs) in the background of the 3M mutant. All mutations were confirmed by Sanger sequencing of mini-prepped DNA from XL1 Blue or DH5α transformants.
Unlabeled expression and purification of CYP101A1.
Wild type and mutant pLR1 plasmids were transformed into E. coli strain BL21(DE3) and inoculated into starter cultures supplied with kanamycin at appropriate concentrations. After overnight growth, starter cultures were transferred to 1L of LB supplied with kanamycin, allowed to grow in shaker flasks (37°C at 220 rpm) until an OD600 of 0.8 was reached, at which point porphyrin precursor δ-aminolevulinic acid (70mg) was added. Protein expression induced with IPTG (1mM), and camphor (1mM) was also added. Expression was carried out at 28°C at 150 rpm for 18 hours. Cells were pelleted by centrifugation at 2,700 x g at 4°C. Pellets were resuspended in 50 mM Tris·HCl pH 7.4, 50 mM KCl, 1mM camphor. Cells were lysed by French press (Thermo) at 16,000 psi and lysate cleared by centrifugation at 20,000 x g at 4°C. The supernatant was filtered through a 0.45 μm PVDF filter and applied to DEAE column pre-equilibrated with 50 mM Tris·HCl, 50 mM KCl, 1mM camphor pH 7.4. Fractions were eluted with a linear gradient of 50 mM KCl to 300 mM KCl, fractions with A417/A280 > 0.2 were combined and concentrated with Amicon Ultra-15 (Millipore).
Enzyme assays.
Samples were prepared containing 0.5μM CYP101A1, 5μM putidaredoxin (Pdx), 0.5μM putidaredoxin reductase (PdR), and 1mM camphor in 50mM KPi, 100mM KCl, pH 7.4. To initiate reaction, NADH was added to a final concentration of 160μM. NADH consumption was monitored spectroscopically as absorbance at 340 nm decreased. Reactions were assumed to be complete when no further change in the 340 nm absorption was observed. The aqueous assays were then extracted with dichloromethane and dried over anhydrous MgSO4. Gas chromatography (GC) was performed on a 30m HP-5ms capillary column (Agilent) with a temperature program run initially from 50°C for 1 minute, ramp rate at 13°C per minute to 240°C for 5 minutes. Retention time for camphor 7.55 min, 5-exo-hydroxy camphor 10.23 min. GC peak analysis was performed using an Agilent 5975C VL quadrupole mass spectrometer. Turnover efficiencies were calculated based on the relative peak areas of camphor and hydroxycamphor, with WT turnover taken as the maximum. Assays were performed in triplicate.
15N-labeled expression of CYP101A1 mutants.
E. coli strain BL21(DE3) was transformed as described above and grown in 5mL of LB supplemented with kanamycin at 37°C at 220 rpm to an OD600 of 0.6, after which the growth was scaled up to 120mL of LB to OD600 of 0.6. At this point, cells were pelleted and transferred to 1L of M9 minimal media with 15NH4Cl as the sole nitrogen source. The culture was grown to OD600 of 0.8 and δ-aminolevulinic acid (70mg) was added. Protein expression was induced with IPTG (1mM), and camphor (1mM) was also added. Expression was carried out at 28°C at 150 rpm for 18 hours. Cells were collected and purified similarly to unlabeled purification.
NMR Spectroscopy.
Prior to NMR experiments, samples were exchanged into 50mM KPi pH 7.4, 100mM KCl, 1mM camphor, 90% H2O/10% D2O. The protein was transferred to a septum-sealed reaction vial and flushed with carbon monoxide. Approximately 4μL of freshly prepared 250mM Na2S2O4 in 1M KPi pH 8 was added in 1μL aliquots to reduce the protein in a sealed vial purged with carbon monoxide. Reduction was monitored by eye, with the fully reduced protein a bright cherry red. The reduced and CO-bound protein was transferred anaerobically to a susceptibility-matched NMR tube (Shigemi). NMR data were acquired on a Bruker Avance NEO spectrometer operating at 800.13 MHz (1H) and 81.08 MHz (15N) at 25°C. Data acquisition, processing and analysis were performed using the Topspin software package (Bruker Biospin, Inc.). 1H, 15N TROSY-HSQC spectra were acquired as 2048 (1H) x 128 (15N) complex points datasets with 16 scans.
Results
While the reaction mechanism for the formation of d-5-exo-hydroxycamphor is complex (20), the step expected to determine the regio- and stereochemistry of the hydroxylation is the abstraction of the 5-exo hydrogen from camphor by the nominal Fe(V)=O species (Compound I). Although it is known that further re-arrangement from the initial enzyme-substrate complex is required prior to the abstraction, we have found that the most occupied camphor orientation in equilibrated MD simulations places the carbon C5 in a position such that an unhindered displacement places the substrate in a conformation suitable for abstraction.
We investigated the effects of active site mutations on substrate location and orientation in CYP101A1 combining in silico and in vitro essays. All of the mutations described herein were designed to change the positions of active site carbons (or oxygens) without changing the atom balance (that is, the same number of atoms of each type are present in each mutant as in the WT enzyme) (Figure 3).
Swap mutation F87Y-Y96F (2S).
Given the role played in maintaining substrate position by the hydrogen bond between the Tyr 96 hydroxyl group (B’ helix) and the camphor carbonyl oxygen evident from crystallographic and NMR-derived structures as well as from our simulations, we wished to see if re-orientation of the substrate could be forced by changing hydrogen bonding options. Our intent was to break the hydrogen bond that is maintained between the carbonyl oxygen of the camphor and the Tyr 96 OH group, in order to force a new hydrogen bond by making the swap F87Y-Y96F. This swap was implemented in the mutant 2S. We found that this mutant is active in vitro producing the same product as the WT enzyme, 5-exo-hydroxycamphor, with somewhat less efficiency (60% turnover efficiency relative to WT, as judged by substrate/product ratios after complete consumption of the reducing agent, NADH. See Table 1 and Supplementary Material for details). Unlike the nearly complete shift to high spin (λmax = 390nm) of WT, only around half of the camphor-bound 2S (51%) exhibited high spin at equilibrium.
Table 1.
In vitro efficiencies and spin state equilibria of mutants in the turnover of substrate (d-camphor) relative to WT based on comparative substrate/product peak areas in GC analysis of extracts. See Supplementary Material for details.
| Mutation | Mutations | Product observed |
Turnover efficiency (with respect to WT) |
Substrate- bound spin state (390/418 nm), WT at 1.0 |
|---|---|---|---|---|
| 3M | V247A-V29SI-V396I | 5-exo-hydroxy | 39% | 0.45 |
| 4M | V247G-G248A-V295I-V396I | 5-exo-hydroxy | 13 % | 0.2 |
| 2S | F87Y-Y96F | 5-exo-hydroxy | 60 % | 0.51 |
| 4M2S | F87Y-Y96F-V247G-G248A-V295I-V396I | 5-exo-hydroxy (traces) | <1 % | 0.1 |
V247A-V295I-V396I (3M) mutation.
Val 295 (on the β3 strand) is a primary contact for the camphor geminal methyl groups (C8 and C9). It was hoped that V295I mutation would rotate the geminal methyl groups clockwise closer to the I helix position of Val 247 from their preferred orientation in the WT active site. The V396I mutation (on the turn between two strands of the β5 sheet) was expected to help drive this rotation as well. As this rotation would be expected to move the C8 and C9 methyl groups, the V247A mutation was generated in order to accommodate new positions of the C8 and C9 methyl groups. This mutant, which we call 3M, proved to be significantly unstable in vitro; a loss of heme upon purification was noted, and the enzyme was modestly active in turnover assays, producing 5-exo-hydroxycamphor as the only product (see Table 1). While the WT enzyme typically exhibits an almost complete ferric heme Soret band shift from a low-spin (λmax=418 nm) to a high-spin (λmax=390 nm) in the optical spectrum upon binding of d-camphor, the 3M mutant´s shift to high spin in the presence of camphor is ~45%.
V247G-G248A-V295I-V396I (4M) mutation.
The 4M mutant was designed in response to the experimentally observed instability of the 3M mutant. Comparative sequence analysis of related cytochromes P450 identified glycine as the most common residue at position 247, which in 3M is alanine (and valine in WT). However, with glycine at position 247, the most frequent residue found at position 248 is alanine. Given that residue 248 in WT CYP101A1 is glycine, we incorporated a swap mutation ¢››in the 3M mutant (V247A-V295I-V396I→A247G-G248A) to obtain the 4M mutant V247G-G248A-V295I-V396I. This mutant was found to be functional in vitro and more stable than 3M. Although considerably less efficient than WT, with only 20% fraction of high-spin, once again 5-exo-hydroxycamphor was the only reaction product observed.
2S mutation in the background of 4M (4M2S).
The 2S swap described above was made in the presence of the earlier mutations, yielding 4M2S with the substitutions F87Y-Y96F-V247G-G248A-V295I-V396I. The in vitro assay showed only trace amounts of 5-exo-hydroxy product detectable by GC/MS, although NADH consumption was observed, indicating that the use of reducing equivalents is significantly uncoupled from product formation. Likewise, less than 10% high-spin form was noted in the camphor-bound form.
Molecular dynamics simulations and analyses.
All simulations were performed as described in the Methods section, and analyses were performed on trajectories acquired after equilibration. We first characterized the distance between camphor (CAM) C5 atom and carbon monoxide (CMO) C atom, which provides a proxy for the oxygen in the Fe=O complex. Figure 4 shows the distribution of the distance between CAM(C5) and CMO(C) for each of the complexes in the equilibrated simulations. To visualize the substrate, we performed a k-means clustering analysis [20] to choose representative structures and characterize some of the peaks seen in the distance distribution. These structures are presented with the distributions in Fig. 3. It can be seen that the distance between CAM(C5) and CMO(C) tracked during the simulations covers a wide range. In the WT simulations the range goes from 3.0 Å to 8.3 Å with three distances (3.9 Å, 5.6 Å and 6.9 Å) significantly populated. The shorter distances, near the maximum around 3.98 Å, are assumed to be the most likely for the reaction to take place after Pdx-enforced displacement.
Figure 4.
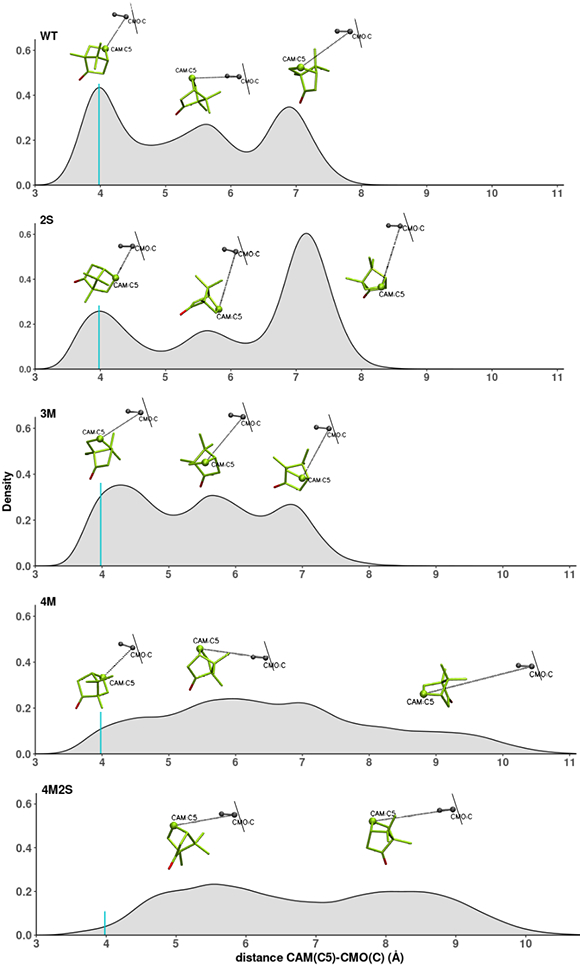
Distribution of distance between camphor CAM(C5) and C atom of carbon monoxide (CMO(C)). The turquoise vertical line shows the maximum in the WT distribution, around 3.98 Å. From top to bottom: WT, 2S, 3M, 4M and 4M2S. A representative structure is shown above the peaks of each distribution.
For both 2S and 3M, the same range of distances are covered, and three maxima can be distinguished. The distributions for these two mutants share some similarity with the WT distribution. In the case of 2S, there is a maximum that coincides with the WT maximum at 3.9 Å, but is less populated than in the WT. For 3M, this maximum is shifted with respect to WT to around 4.25 Å.
In the case of mutants 4M and 4M2S, the range of C5-CMO(C) distances covered is larger than the WT and the distribution is more diffuse. In the 4M distribution, there is some population at the WT maximum of 3.98 Å, but it is not a maximum and the population is relatively low, and is even lower for 4M2S. The 4M and 4M2S distributions are much less localized than in the other mutants, suggesting that, for these mutants, there is no clearly preferred substrate orientation. For the reader's convenience, links to videos comparing WT and mutant C5-CMO(C) distance progressions are provided in the Supplementary Material.
Displacement or reorientation?
Following the WT simulations and taking into account the structures represented in Fig. 4, we note that the distribution of C5-CMO(C) distances are the result of camphor reorientation rather than a net displacement of its center of mass (CM). In the WT, camphor spends much of the simulation tethered via the hydrogen bond between the carbonyl oxygen of the camphor and the hydroxyl group of Y96, and the movement described by the substrate suggests the precession of a rigid body. This raises the question of whether the observed range of C5-CMO(C) distances for the mutants shown in Figure 4 is due to a displacement of the substrate CM or to a change in the camphor orientations, or a combination of both. To address this issue, we defined an internal angle θ to describe the rigid body movement, and tracked this angle throughout the simulation (Figure 5). The angle θ is defined by an internal vector that connects the camphor CM and carbon C5, and the vector between camphor CM and CMO(C). We correlated changes in θ with the distance between camphor C5 and CMO(C) on one hand, and distance between camphor CM and CMO(C) on the other. The results for the WT and the four mutants are shown in Figure 6.
Figure 5.

Angle θ is defined between an internal vector connecting the camphor center of mass (CM) and C5, and a vector that connects camphor CM and CMO(C).
Figure 6.

2D density plots, regions in yellow represent the most populated states after equilibration. Angle θ on the vertical axis against distance between camphor center of mass (CM) and CMO(C) on the horizontal axis. From left to right: top: WT, 2S and 3M, bottom: 4M and 4M2S.
The density plots in Fig. 6 show the angle θ against the distance between camphor CM and CMO(C) and give us an insight on camphor rotation and localization. For the first three enzymes represented, WT, 2S and 3M, we can see that camphor CM spends much of the simulation time anchored in a preferred location, around 6 Å from CMO(C), meaning that the distribution presented in Figure 3 for these enzymes is mainly due to camphor reorientation without CM displacements. Furthermore, the angle θ= ~30° is favored in the WT, while in the mutant 2S, the most populated value of θ is ~120°. For mutant 3M, maxima are observed around 30° and 90° as well as a weaker maximum at ~110°. For mutant 4M, the population density is more diffuse, showing that camphor CM is able to change its location, but the most populated states are still ~6 Å from CMO(C). The most frequent values of θ are around 30° and around 90°, which is populated in the 3M but not in WT. Lastly, the distribution observed for mutant 4M2S is not as diffuse as in 4M, but the most populated states are around a longer CAM(CM) - CMO(C) distance (7 Å) with θ ~ 120°. It is thus established that in 4M and 4M2S the changes on camphor orientation are accompanied by CM position changes as well.
The role of hydrogen bonding in maintaining camphor orientation.
As shown in the previous section, the variation of C5-CMO(C) distances observed for the WT are due to changes in substrate orientation rather than CM movements. Camphor spends much of the simulation time tethered via the hydrogen bond between its carbonyl oxygen and the hydroxyl group of Tyr 96 (58% of the simulation time). This interaction has been proposed to contribute strongly to the regioselectivity of the hydroxylation (21).
For the 2S mutant, the expected reorientation due to a hydrogen bond between CAM(O) and Tyr 87-(OH) was not observed. Instead, a hydrogen bond forms between the hydroxyl group of Thr101 on the B’-C loop and the CAM carbonyl oxygen. This interaction is found in 49.6% of the equilibrium conformations, and helps maintain the substrate orientation seen in Figure 7 for the 2S mutant.
Figure 7.

Hydrogen bond pattern in WT and 2S. Left: WT enzymatic complex. The hydrogen bond between the carbonyl oxygen of the camphor and the hydroxyl group of Tyr 96 is thought to be critical for camphor orientation. Right: 2S enzymatic complex. A hydrogen bond between the carbonyl oxygen of the camphor and Thr 101 side chain hydroxyl group.
In the case of 3M, the MD trajectories indicate that the combination of mutations promote a change in substrate movement somewhat closer to the I helix, as expected, and the anchorage provided by the hydrogen bond with Tyr96 is maintained 41% of the simulated time. In vitro experiments show that this mutant is still functional and gives rise to 5-exo-hydroxycamphor as the only detectable product. This is consistent with the observation that camphor is still oriented as in the WT complex for a significant fraction of the time (Figs. 4 and 6).
For the 4M mutant, camphor is found closer to I helix than in 3M, as a consequence not only of the V295I change, but also, to the V247G-G248A change, that seems to affect protein dynamics. The 4M enzyme is functional in vitro, and though much less efficient than the WT enzyme (13% of product relative to WT for the same NADH consumption), 5-exo-hydroxycamphor is the only reaction product observed. Consistently, camphor is still anchored by the Tyr96 hydrogen bond but is found in a smaller fraction of time (17%) placing the C5 close to the CMO(C).
The 4M2S mutation proved to be the most disruptive, both in simulations and in vitro: As shown in Fig. 6, the camphor CM is found on average at longer distances from the CMO(C) than in the other enzymes. The camphor-Thr101 hydrogen bond populated in 2S was also observed for 4M2S in 37% of equilibrated frames.
Coupled movements of the enzyme and substrate.
The next question that emerges is as to whether the substrate location changes observed for mutants 4M and 4M2S are due to an unanticipated enlargement of the active site or primarily due to a change in the active site dynamics. To investigate these possibilities, we ran a series of short MD simulations applying harmonic positional restraints to the polypeptide, allowing only camphor to move. These simulations were started from three different initial structures selected from the unrestrained dynamics and allowed to proceed for 100 ns. The harmonic restraint was then removed, and an additional 100 ns simulation performed. In Figure 8 the angle θ is represented against the distance between camphor CM and CMO(C) corresponding to these simulations. As can be seen by inspection of the trajectory, the distributions change considerably when the protein is allowed to move. If protein motion is restricted, the camphor CM remains close to the initial position. On the other hand, when the restriction is removed, camphor moves more freely, covering a wide range of conformations. From this result, we infer that camphor CM displacements are coupled to protein motions. While the mutations generated do not change the number of carbon or oxygen atoms in the enzyme active site, it was pointed out by a reviewer of the original manuscript that changes in packing and secondary structure placement may change the volume accessible to the rigid camphor molecule, and selected distance comparisons between active site residues in the WT and mutants suggest that such distortions are occurring (SI Fig. S2). However, estimates based on Connelly surface areas indicate that active site volumes are not changing significantly as a function of mutation (SI Table S1).
Figure 8.

Distance between camphor center of mass CM and CMO(C) (horizontal axis) and angle θ (vertical). From top to bottom: WT, 2S, 3M, 4M and 4M2S. The results for the restricted protein trajectories are shown in black, while the colored points represent the results after removal of positional restraints on the protein. The red point within the black points shows the coordinates of the initial structure for each MD run.
Perturbation response scanning (PRS) of mutants and WT enzymes.
In a previous publication [7], we described the use of perturbation response scanning (PRS) to characterize the role of the enzyme structure in determining the positioning and orientation of the bound substrate in CYP101A1. Briefly, the application of a unit force vector with random direction at the Cα carbon of each residue is correlated with observed perturbations, resulting in a map of those residues most responsive to perturbations elsewhere in the protein, as well as those residues, "influencers", at which the applied force is most detectable in responsive regions. A more complete description of the methodology used for PRS is available in Ref. [7]. Two results from that work are relevant to this study. First, in the substrate-bound enzyme, responsive regions were detected across the entire enzyme structure, including the junctions between the B' and C helices, the C-D and G-H loops, the K'-β meander link, as well as the β3-β4 and β5-L helix connections (see Fig. 2), with vectors connecting these sites either crossing or passing near to bound substrate. In the absence of substrate, fewer responsive regions were detected, and the responses themselves were weaker and more diffuse. The conclusion we reached is that large portions of the enzyme structure are involved in "detecting" the presence of bound substrate, not only residues in the active site, and that bound substrate forms a critical linkage in the transmission of mechanical perturbations. Secondly, the application of an artificial restraint reorienting substrate away from the preferred orientation (that is, the most common orientation observed in unrestrained simulations), resulted in dramatic changes in the PRS patterns observed, with residues or regions previously unresponsive showing significant responsiveness, while regions responsive in the absence of the restraint becoming less responsive (or unresponsive). From these results, we concluded that significant portions of the enzyme structure are sensitive not only to substrate binding, but also to the orientation of substrate in the active site.
To connect the previous findings to the present work, we performed similar PRS studies. PRS heat maps generated for the mutant enzymes described in the current work are provided in the Supplementary Material. Comparison of those maps with that of the WT enzyme indicates that, while the overall pattern of responsive residues for all of the mutant enzymes are similar to what is observed in the WT, with the exception of the 4M mutant, the responsive regions are generally less well-defined and less sensitive than in WT, and, in turn, "influencer" residues are not as well-differentiated in the mutants as in the WT. The 4M mutant, while recapitulating the overall response pattern of WT, appears to be somewhat more responsive than WT in the PRS analysis, indicating that the Ala-Gly swap in the I helix has a significant effect on the mechanical coupling pathways.
Structural perturbations - NMR characterization of mutants.
The MD results presented above provide insight into initial substrate orientation and placement and also show that the mutations cause changes in enzyme dynamics and that the substrate dynamics is coupled to protein motions. We now associate enzyme activity variations with these dynamical changes. The dynamical changes described are obviously the result of the mutations and we wished to see if such changes could be detected experimentally. To this end, we used the assigned 1H,15N TROSY-HSQC correlation spectrum of the WT CYP101A1(4, 22) to detect structural perturbations (as evidenced by chemical shift changes) or local dynamics (detectable from changes in linewidth or in resonance multiplicity). Figure 9 shows the distribution of perturbations caused by each set of mutations. These perturbations range from small chemical shift changes to severe line broadening, in some cases to the point of the loss of an observable signal for a residue. Such line broadening is indicative of conformational changes occurring on the ms time scale. Not surprisingly, the least degree of perturbation is observed in the 2S mutant (which is also the most enzymatically active) and the greatest degree of perturbation is observed in the 4M2S mutant. However, even in the case of the 2S mutant, perturbations are widely distributed, indicating that effects due to mutation are transmitted over large regions of the structure.
Figure 9.

Distribution of NMR-detected perturbations in the four mutants described (upper left, 2S; upper right, 3M; lower left, 4M; lower right, 4M2S). Color coding: yellow, slight resonance shifts (< 0.1 ppm in either the 1H or 15N dimension); orange, significant shifts (> 0.1 ppm in either dimension); red, strongly broadened or not observable. The positions of the mutations are indicated in blue for each structure. All structures are identical, and in approximately the same orientation as in Figure 2.
Perturbations to amide NH correlations, as monitored here, reflect local changes in the electronic environments of the N-H bond, most of which are involved in hydrogen bonding interactions, either in regular secondary structures or at junctions between secondary structures and turns. Chemical shift changes therefore reflect a change in the distribution of states being sampled (that is, time-averaged changes in local conformations are being detected). If unbroadened, these signals represent the weighted average of all contributions to the local environment that exchange fast on the chemical shift timescale. As conformational exchange slows down, lines broaden, and the loss of a correlation indicates relatively slow conformational exchange between states on a time scale similar to the chemical shift differences between the sampled conformations.
Discussion
The persistence of 5-exo-hydroxycamphor as the only detected product in turnover assays with the mutant enzymes (albeit all with lower efficiencies than WT) despite the relatively dramatic changes the mutants introduce in the active site was initially surprising. An interesting combined experimental and computational study by Das et al. using l-camphor in place of the native substrate d-camphor noted that, although there is increased uncoupling with the non-native substrate, the hydroxylation product is still the 5-exo-hydroxy product (23). Although we cannot rule out the possibility that the 5-endo hydrogen is abstracted rather than the 5-exo in the mutant assays, product retention times in GC/MS experiments are constant regardless of the mutation, and such a change would be expected to influence the sublimation point of the products to some extent, a parameter that is closely related to GC retention times. Furthermore, daughter ion fragmentation patterns in the mass spectra are identical within experimental error across the analyses, and again, a different stereochemistry of hydroxylation at C5 might be expected to modify preferred fragmentations, at least slightly. While Atkins and Sligar detected small amounts of hydroxylation occurring at other positions with the Y96F mutant P450cam (21), we did not detect any of these alternate products in our assays. We note that our approach of maintaining the atom balance of WT in all of our mutations may be the difference between the two cases.
Spin state equilibrium as a measure of "goodness of fit".
We have recently proposed that the spin state equilibrium detected spectroscopically by shifts of the heme Soret band upon substrate binding to P450 is the result of substrate-mediated changes in allowed heme vibrational modes (24, 25). The mutations examined in this work all inhibit the high-spin shift upon substrate binding to a greater or lesser extent, suggesting that the arrangement of residues in the WT enzyme is evolutionarily optimized for the degree of spin state shift induced by substrate, as well as stabilizing a preferred substrate orientation. Indeed, the observed extent of substrate-induced spin state shift for the mutants relative to WT agrees qualitatively with the fraction of time that C5 spends in close proximity to CMO(C) (see Table 1 and Fig. 4). Still, given the lesser degree of mechanical coupling detected by PRS in most of the mutants relative to the WT, it may also be that the tight coupling of active site dynamics to other regions of the protein is required to drive the heme conformation favoring the high spin state.
Potential roles for chemoselectivity and/or steric hindrance in regiochemical selection.
A comparison of distances between substrate carbon atoms in the populated orientations in WT and the 2S mutant is shown in Figure 10, with Figure 11 showing the correlation between the fraction of time that C5 is in close approach (< 4 Å) to CMO(C) and measured turnover efficiency. As can be seen, the two are strongly correlated.
Figure 10.
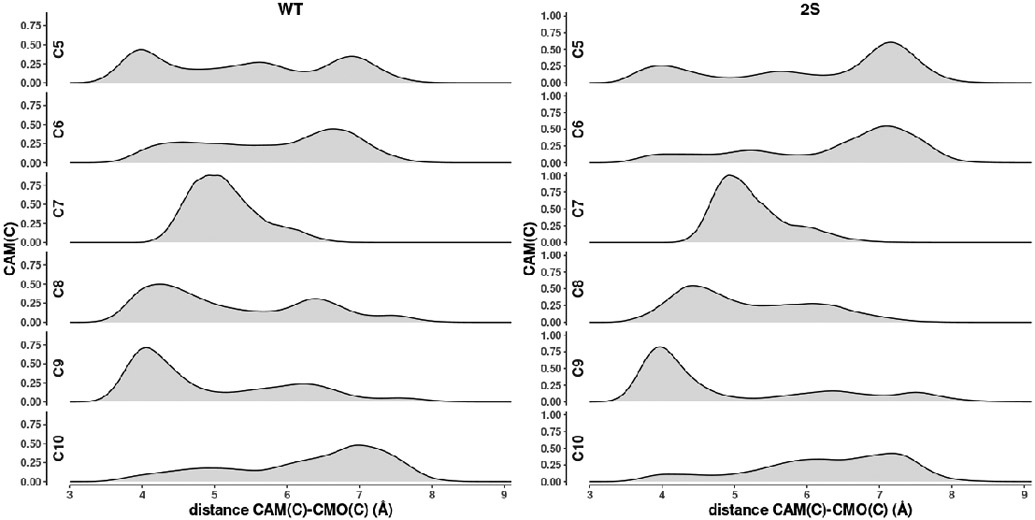
Distance distributions of carbon atoms of substrate camphor relative to carbonmonoxy carbon (CMO(C)) in WT and 2S mutants. C8, C9 and C10 are methyls (CH3), C5 and C6 are methylenes (CH2).
Figure 11.

Turnover efficiency (%) relative to WT plotted as a function of the percentage of time substrate spends with C5-CMO(C) distance less than 4 Å.
It can be seen in WT that the closest approach to the heme CMO(C) is by C5, the site of hydroxylation, with a methyl group C9 spending a substantial fraction of the simulation time only slightly further away (~4.2 Å at the distribution maximum). In the case of 2S, C9 is now closer to the CMO(C) (~3.9 Å) than C5 (~4.1 Å) and spends much more of the simulation time in that position, yet the observed hydroxylation reaction for 2S remains at C5, albeit with lower efficiency. Two reasons can be put forward for this observation. First, it is thermodynamically more costly to generate a primary radical (at a methyl group, C9) than at a methylene (C5) by hydrogen abstraction, the first step in the oxidation (26). A second consideration is that, as we have noted previously, the rate-determining step in the CY101A1 reaction cycle is the conformational change driven by the binding of Pdx: If the orientation of substrate placing C9 close to the heme interferes sterically with the Pdx-driven conformational change, that orientation would represent a dead end, and only the less-populated but productive orientation placing C5 near to the CMO(C) will allow the conformational change to occur.
Finally, Gelb and co-workers noted that there is only a small intermolecular isotope effect due to replacement of either the 5-endo or 5-exo substrate proton with deuterium, while there was a significant intramolecular effect, suggesting the possibility of heavy atom motion in the transition state (27). The synthesis of the 5-deuterated camphor used in that study involved the formation of a fused three-membered ring involving carbons 3, 4 and 5. As such, it is possible that transient interactions (involving heavy atom movement) between those atoms upon hydrogen abstraction from the 5-position could result in additional stabilization for abstraction from the 5-position not available to other positions in the camphor molecule.
"Dynamic cavities" and the role of mechanical linkage in substrate placement and orientation.
One interesting observation is that the swap A247G-G248A leading from 3M to 4M appears to have a considerable impact on enzyme dynamics, as 4M is the only mutant to have stronger mechanical coupling than the WT enzyme in some regions as visualized in PRS heat maps. In particular, the residues in the G-H loop on the proximal face of the I helix opposite the swap site are more sensitive than in WT, as are residues in the B'-C and C-D loops, also located in this quadrant of the enzyme structure. We note that the camphor CM moves closer to the I helix than in 3M, which can be rationalized by the glycine now in a substrate contact position occupied by valine in WT, which may increase the degree of mechanical linkage between camphor and the N-terminal half of the I helix by allowing closer approach. However, the lack of an orientational preference and the more diffuse CM positioning ( Fig.7) in 4M would suggest that this increased coupling may be counterproductive to proper substrate orientation by allowing the substrate cavity to "slide" along the I helix without favoring any particular orientation. Interestingly, the same strong coupling is not observed in the 4M2S mutant, suggesting that the presence of the native hydrogen bonding scheme in 4M provides some directionality to the mechanical coupling between enzyme and substrate.
Conclusions and future directions.
This combined in vitro/silico examination of the role of active site residues in determining the regiochemistry of hydroxylation of d-camphor by CYP101A1 originated in an attempt to establish an enzyme engineering protocol for manipulating regioselectivity in the cytochrome P450 superfamily. We hoped that by maintaining the atom balance in the active site while manipulating the positions of substrate contacts, we would force a reorientation of substrate that would favor hydroxylation at a new location. Our expectation was that the active site cavity would not change in size, but a new orientation might be favored over that which places C5 close to the CMO(C). While we believe that the active site cavity size does in fact remain fairly constant based on our current results (SI, Table S1), it is clear that other factors are in play that determine the preferred orientation. We find significant the fact that PRS patterns, which were shown to be highly sensitive to substrate orientation in our previous work, remain largely similar between WT and mutants. To us, this observation emphasizes the importance of such mechanical coupling pathways in determining substrate orientation and regioselectivity. As a result, we expect that any successful re-engineering of a cytochrome P450 will need to take long-range mechanical coupling into account. We note that Arnold and co-workers have found that re-engineering of cytochromes P450 via directed evolution involves mutations remote from the enzyme active site (28, 29).
Recent experimental and computational results by Follmer and Poulos suggest that a secondary substrate binding site on CYP101A1 may be occupied at physiologically relevant camphor concentrations, this site being located on the proximal side of the I helix, near the C-D loop (30, 31). Given that we have previously noted significant effects of mutations in this region on both enzyme efficiency and spin state equilibrium (32), it will be of interest to see how conformational dynamics, and in particular, PRS patterns might be affected by inclusion of camphor in this site in future simulations.
Supplementary Material
Acknowledgements
This work was supported in part by NIH grant R01-GM130997 (TCP, co-PI) and grant PICT-2016-4209 of Agencia Nacional de Promoción Científica y Tecnológica. NRW acknowledges partial support from NIH training grant T32GM007596 to Brandeis University. TL was supported by a Brandeis SURF summer fellowship. Access to the XSEDE network was provided through the US National Science Foundation (MCB-200206, TCP and EKA, co-PIs)).
Abbreviations used:
- GC/MS
tandem gas chromatography/mass spectrometry
- IPTG
isopropyl-β-thiogalactoside
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- PCR
polymerase chain reaction
- PRS
perturbation response scanning
- TROSY-HSQC
transverse relaxation enhanced heteronuclear single-quantum coherence spectroscopy
Footnotes
Accession information.
Cytochrome P450cam (CYP101A1) is NCBI accession P00183.2, also used by UniProtKB and SwissProt.
Supporting Information
Supporting information includes distance between CAM(C5) and CMO(C) (Figure S1) and supplementary videos in .mp4 format, active site volumes (Figure S2), PRS heat maps (Figures S3-S6) and influence profiles (Figures S7-S10), a list of oligonucleotides used for Gibson assembly and Quikchange mutagenesis, UV-visible spectra of oxidized WT and mutant P450cam used for spin state shift estimates (Figures S11-S14) and turnover efficiencies of WT and mutant P450cam. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.Asciutto EK, Dang M, Pochapsky SS, Madura JD, and Pochapsky TC (2011) Experimentally restrained molecular dynamics simulations for characterizing the open states of cytochrome P450(cam), Biochemistry 50, 1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raag R, and Poulos TL (1989) Crystal structure of the carbon monoxide-substrate-cytochrome P450CAM ternary complex, Biochemistry 28, 7586–7592. [DOI] [PubMed] [Google Scholar]
- 3.Pochapsky TC (2021) A dynamic understanding of cytochrome P450 structure and function through solution NMR, Current Opinion in Biotechnology 69, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pochapsky SS, Pochapsky TC, and Wei JW (2003) A model for effector activity in a highly specific biological electron transfer complex: The cytochrome P450cam–putidaredoxin couple, Biochemistry 42, 5649–5656. [DOI] [PubMed] [Google Scholar]
- 5.Asciutto EK, Madura JD, Pochapsky SS, OuYang B, and Pochapsky TC (2009) Structural and dynamic implications of an effector-induced backbone amide cis–trans isomerization in cytochrome P450cam, Journal of Molecular Biology 388, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei JY, Pochapsky TC, and Pochapsky SS (2005) Detection of a high-barrier conformational change in the active site of cytochrome P450(cam) upon binding of putidaredoxin, Journal Of The American Chemical Society 127, 6974–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asciutto EK, Young MJ, Madura JD, Pochapsky SS, and Pochapsky TC (2012) Solution structural ensembles of substrate-free cytochrome P450cam, Biochemistry 51, 3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asciutto EK, and Pochapsky TC (2018) Some surprising implications of NMR-directed simulations of substrate recognition and binding by cytochrome P450cam (CYP101A1), Journal of Molecular Biology 430, 1295–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, and Walker RC (2012) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, Journal of Chemical Theory and Computation 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Case D, Aktulga HM, Belfon K, Ben-Shalom I, Brozell S, Cerutti D, Cheatham T, Cruzeiro V, Darden T, Duke R, Giambasu G, Gilson M, Gohlke H, G^tz A, Harris R, Izadi S, Izmailov S, Jin C, Kasavajhala K, and Kollman P (2021) Amber 2021. [Google Scholar]
- 11.Jorgensen WL, and Madura JD (1983) Quantum and statistical studies of liquids .25. Solvation and conformation of methanol in water, Journal of the American Chemical Society 105, 1407–1413. [Google Scholar]
- 12.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB, Journal of Chemical Theory and Computation 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bikiel DE, Boechi L, Capece L, Crespo A, De Biase PM, Di Lella S, Lebrero MCG, Marti MA, Nadra AD, Perissinotti LL, Scherlis DA, and Estrin DA (2006) Modeling heme proteins using atomistic simulations, Physical Chemistry Chemical Physics 8, 5611–5628. [DOI] [PubMed] [Google Scholar]
- 14.Marti MA, Crespo A, Capece L, Boechi L, Bikiel DE, Scherlis DA, and Estrin DA (2006) Dioxygen affinity in heme proteins investigated by computer simulation, Journal of Inorganic Biochemistry 100, 761–770. [DOI] [PubMed] [Google Scholar]
- 15.Ryckaert J-P, Ciccotti G, and Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, Journal of Computational Physics 23, 327–341. [Google Scholar]
- 16.Grest GS, and Kremer K (1986) Molecular dynamics simulation for polymers in the presence of a heat bath, Physical Review A 33, 3628–3631. [DOI] [PubMed] [Google Scholar]
- 17.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984) Molecular dynamics with coupling to an external bath, The Journal of Chemical Physics 81, 3684–3690. [Google Scholar]
- 18.Roe DR, and Cheatham TE (2013) PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, Journal of Chemical Theory and Computation 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 19.Hartigan JA (1975) Clustering algorithms, John Wiley & Sons, Inc., Hoboken, NJ. [Google Scholar]
- 20.Wong LL, Westlake CG, and Nickerson DP (1997) Protein engineering of cytochrome P450(cam), Metal Sites in Proteins and Models 88, 175–207. [Google Scholar]
- 21.Atkins WM, and Sligar SG (1988) The Roles of active-site hydrogen bonding in cytochrome P450cam as revealed by site-directed mutagenesis, Journal of Biological Chemistry 263, 18842–18849. [PubMed] [Google Scholar]
- 22.OuYang B, Pochapsky SS, Dang M, and Pochapsky TC (2008) A functional proline switch in cytochrome P450(cam), Structure 16, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das B, Helms V, Lounnas V, and Wade RC (2000) Multicopy molecular dynamics simulations suggest how to reconcile crystallographic and product Formation data for camphor enantiomers bound to cytochrome P-450cam, Journal of Inorganic Biochemistry 81, 121–131. [DOI] [PubMed] [Google Scholar]
- 24.Pochapsky TC, Wong N, Zhuang Y, Futcher J, Pandelia M-E, Teitz DR, and Colthart AM (2017) NADH reduction of nitroaromatics as a probe for residual ferric form high-spin in a cytochrome P450, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1866, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karunakaran V, Denisov I, Sligar SG, and Champion PM (2011) Investigation of the low frequency dynamics of heme proteins: Native and mutant cytochrome P450(cam) and redox partner complexes, J. Phys. Chem. B 115, 5665–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nunes PM, Estácio SG, Lopes GT, Costa Cabral BJ, Borges dos Santos RM, and Martinho Simões JA (2008) C–H Bond Dissociation Enthalpies in Norbornane. An Experimental and Computational Study, Organic Letters 10, 1613–1616. [DOI] [PubMed] [Google Scholar]
- 27.Gelb MH, Heimbrook DC, Malkonen P, and Sligar SG (1982) Stereochemistry and deuterium-isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system, Biochemistry 21, 370–377. [DOI] [PubMed] [Google Scholar]
- 28.Chen K, and Arnold FH (2020) Engineering new catalytic activities in enzymes, Nature Catalysis 3, 203–213. [Google Scholar]
- 29.Arnold FH (2018) Directed evolution: Bringing new chemistry to life, Angewandte Chemie International Edition 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skinner SP, Follmer AH, Ubbink M, Poulos TL, Houwing-Duistermaat JJ, and Paci E (2021) Partial opening of cytochrome P450cam (CYP101A1) is driven by allostery and putidaredoxin binding, Biochemistry 60, 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Follmer AH, Tripathi S, and Poulos TL (2019) Ligand and redox partner binding generates a new conformational state in cytochrome P450cam (CYP101A1), Journal of the American Chemical Society 141, 2678–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colthart AM, Tietz DR, Ni Y, Friedman JL, Dang M, and Pochapsky TC (2016) Detection of substrate-dependent conformational changes in the P450 fold by nuclear magnetic resonance, Scientific Reports 6, 22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.