

Abstract
Emerging evidence in animal models of chronic kidney disease (CKD) implicates Aryl Hydrocarbon Receptor (AHR) signaling as a mediator of uremic toxicity. However, details about its tissue-specific and time-dependent activation in response to various renal pathologies remain poorly defined. Here, a comprehensive analysis of AHR induction was conducted in response to discrete models of kidney diseases using a transgenic mouse line expressing the AHR responsive-promoter tethered to a β-galactosidase reporter gene. Following validation using a canonical AHR ligand (a dioxin derivative), the transgenic mice were subjected to adenine-induced and ischemia/reperfusion-induced injury models representing CKD and acute kidney injury (AKI), respectively, in humans. Indoxyl sulfate was artificially increased in mice through the drinking water and by inhibiting its excretion into the urine. Adenine-fed mice showed a distinct and significant increase in β-galactosidase in the proximal and distal renal tubules, cardiac myocytes, hepatocytes and microvasculature in the cerebral cortex. The pattern of β-galactosidase increase coincided with the changes in serum indoxyl sulfate levels. Machine-learning based image quantification revealed positive correlations between indoxyl sulfate levels and β-galactosidase expression in various tissues. This pattern of β-galactosidase expression was recapitulated in the indoxyl sulfate-specific model. The ischemia/reperfusion injury model showed increase in β-galactosidase in renal tubules that persisted despite reduction in serum indoxyl sulfate and blood urea nitrogen levels. Thus, our results demonstrate a relationship between AHR activation in various tissues of mice with CKD or AKI and the levels of indoxyl sulfate. This study demonstrates the use of a reporter gene mouse to probe tissue-specific manifestations of uremia in translationally-relevant animal models and provide hypothesis-generating insights into the mechanism of uremic toxicity that warrant further investigation.
Keywords: Aryl hydrocarbon, uremia, chronic kidney disease, acute kidney injury
Graphical Abstract
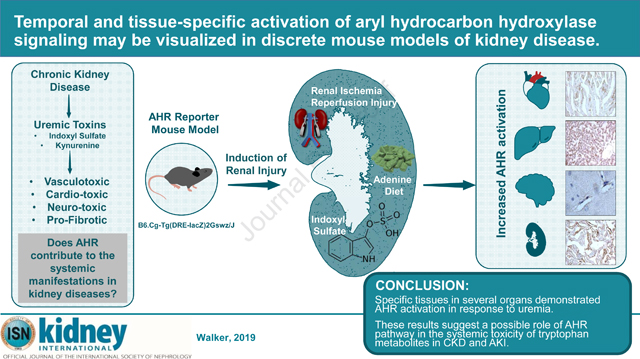
Introduction:
Chronic kidney disease (CKD) is a progressive disease resulting from irreversible alterations of kidney structure and function. Approximately 10% of the adult population in the United States and worldwide suffer from CKD, and the incidence of, and mortality due to, CKD is increasing1. CKD is characterized by the retention of a heterogeneous mix of metabolites, collectively referred to as uremic solutes, which are retained early in the blood of CKD patients and persist even after hemodialysis2,3. The systemic complications associated with CKD (grouped as uremic manifestations) are in part driven by these compounds called uremic toxins/solutes 4,5.
Of these uremic toxins, Indolic solutes (such as indoxyl sulfate (IS)) are particularly pathogenic and have protean manifestations in the vascular system such as inducing oxidative stress and suppressing fundamental endothelial functions6,7. Our work and that of others, in animal models and humans, implicate indolic uremic solutes contributing to the hyperthrombotic CKD milieu by activating Tissue Factor (TF) in the endothelial and vascular smooth muscle cells8–11. Indolic solutes have both pro-fibrotic and pro-inflammatory effects in cardiac tissue12,13 and also linked to toxicity to the microglia14. This evidence clearly demonstrates the multi system effects of indolic solutes.
The AHR pathway is emerging as a mediator of toxicity by indolic solutes8. AHR is a ligand-activated transcription factor, classically known to mediate effects of exposure to environmental toxins such as 2, 3, 7, 8-tetrachlorodibenzodioxin (TCDD)15. AHR is not only activated by exogenous chemicals, but also endogenous ligands, such as 6-formylindole[3, 2,-b]carbazole (FICZ), and kynurenine, and those generated by a combination of exogenous (microbiome) and endogenous (liver) metabolism such as IS 16,17. Research by our group and others showed that indolic solutes activate the AHR pathway in cultured cells as demonstrated by its nuclear translocation and induction of cell type-specific AHR target genes and followed by degradation of AHR protein8–10.
The AHR has been implicated independently in diseases related to the cardiovascular system18,19, and the CNS20. Given its activation in the uremic milieu, it is likely to contribute to some of the systemic manifestations of indolic uremic solutes. Despite the importance of the AHR pathway in uremic toxicity, there are no studies examining the kinetics and tissue-specific pattern of AHR activation in discrete models of renal diseases. We set out to examine these specific questions utilizing a transgenic mouse (B6.Cg-Tg(DRE-lacZ)2Gswz/J) with a β-galactosidase reporter gene downstream of two AHR dioxin response elements (DRE).
Results:
We confirmed the hemizygous status of the B6.Cg-Tg(DRE-lacZ)2Gswz/J transgenic mouse line using genotyping (Supplementary Table 1) and copy number of the Lacz transgene using quantitative real time polymerase chain reaction (qRT-PCR) (Supplementary Figure 1). The tissues from these mice subjected to adenine-induced CKD, I/R-induced AKI and IS-specific model models were examined at different time points (Figure 1).
Figure 1: Experimental design to examine AHR activation in discrete models of renal disease.

A combination of male and female B6.Cg-Tg(DRE-LacZ)2Gswz/J transgenic mice (DRE-LacZ) with a lacZ reporter gene downstream of two dioxin response elements (DRE) were subjected to four models. Transgenic mice treated with (TCDD) (n=4) served as a positive control. Transgenic mice (n=4) were subjected to a 0.25% adenine supplemented diet for 2 weeks in order to induce CKD10. Four transgenic mice on a normal mouse diet served as controls. An IS-specific model consisted of four transgenic mice that received IS + probenecid. Four mice exposed to probenecid alone served as controls. Renal I/R was used as a model of AKI. Nine transgenic male mice were used and sacrificed at specific time points after 23 minutes of bilateral renal ischemia and sacrificed at 24, 48, and 96 hours post-surgery. Three additional transgenic mice served as controls following a sham surgery.
Validation of the transgenic model
Previously this transgenic mouse was used to demonstrate the teratogenic effects of the AHR pathway by exposing pregnant mothers to TCDD21. While those studies provided a proof of activation of the transgene, they were performed in fetuses. Given the use of adult mice for all our disease models, we first validated this transgenic model in a group of 8–12 weeks old mice exposed to TCDD, a potent AHR agonist. Mice treated with vehicle served as controls. Activation of the AHR pathway in TCDD-treated mice was demonstrated by a significant elevation in β-galactosidase mRNA from the lysates of kidney, liver, and heart (Figure 2A). Compared to control animals, β-galactosidase mRNA was increased by 22.5% in the kidney (p=0.0481,) 26% in liver (p=0.0286), and 29.8% in the heart (p=0.0223) in TCDD-treated mice. These results demonstrated induction of the AHR pathway. In order to detect β-galactosidase protein in different tissues, we performed both β-galactosidase enzyme assays and immunohistochemistry using a pre-validated anti-β-galactosidase antibody22. The later method provided a distinct and specific signal and was used for the subsequent studies (Supplementary Figure 2).
Figure 2: Validation of transgenic mice.

(a). β-galactosidase mRNA was examined in whole tissue lysates from 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) treated and control transgenic mice. mRNA levels were normalized to GAPDH mRNA. Each data point represents the average of duplicate samples for each transgenic mouse used. (b). Frozen sections from TCDD and control treated transgenic animals were stained with an anti-β-galactosidase to examine tissue-specific expression. The β-galactosidase protein was localized to renal tubules (marked by a black asterisk) but not in glomeruli (black arrow), liver hepatocytes, and cardiac myocytes (black arrow). All data is represented as plus or minus the standard error of the mean. Images were taken at 200x magnification. Scale bar=50μM. V=vessel wall, L=lumen.
TCDD-treated mice showed β-galactosidase expression in several organs (Figure 2B) including renal tubules (Figure 2B, asterisk). However, a distinct absence of β-galactosidase was noted in the glomeruli (Figure 2B, arrowhead). Liver sections of TCDD-treated animals showed a uniform β-galactosidase expression in hepatocytes while patchy expression was noted in the cardiac myocytes (Figure 2B, arrowheads). AHR activation indicated by β-galactosidase expression in various tissues upon TCDD treatment validates this model.
AHR activation in an adenine-induced CKD model
We next examined AHR activation in an adenine-induced model, an established model of crystal-induced CKD characterized by extensive renal tubular damage, and interstitial fibrosis10. Among different animal models of CKD23, we used the adenine-induced CKD model due to the presence of several features of uremia in this model. Exposure to a 0.25% adenine diet for 2 weeks in mice resulted in a significant increase in blood-urea nitrogen (BUN) (Supplementary Figure 3A)10. AHR activation in adenine-induced CKD mice was confirmed by a significant upregulation of β-galactosidase mRNA in the kidney (37.5%), liver (22.7%), and heart (16.8%) lysates (Figure 3A).
Figure 3: AHR activation in an adenine-induced CKD model.

Transgenic mice were administered a diet supplemented with 0.25% adenine for 2 weeks. Transgenic mice on a normal diet served as controls. (a). β-galactosidase mRNA was examined in whole tissue lysates in transgenic mice and normalized to GAPDH mRNA. Each data point represents the average of duplicate samples for each transgenic mouse used. (b). Representative frozen sections stained with an anti-β-galactosidase antibody are shown. The β-galactosidase protein was localized to renal tubules (marked by a black asterisk), liver hepatocytes, and cardiac myocytes (black arrow). Additionally, β-galactosidase expression was seen in CNS microvessels (black arrowhead). (c). A color based image segmentation pipeline (see supplementary methods) was used to quantify the increase in β-galactosidase expression per tissue area in the kidney, liver, and heart. The number of microvessels in the CNS positive for β-galactosidase were counted and presented as the number of positive microvessels/mm2. All data is represented as plus or minus the standard error of the mean. Images of the kidney, liver, heart and aorta were taken at 200x magnification. Scale bar=50μM. Images in the CNS were taken at 400x magnification. Scale bar=25μM. V=vessel wall, L=lumen.
To estimate the extent of AHR activation within different tissues, a customized, color-based image segmentation pipeline was used to quantify the amount of β-galactosidase and was normalized to the tissue area (details in the supplementary method section and Supplementary Figure 4A)24. Our results showed a significant increase in β-galactosidase in the kidney (10.65-fold) of adenine-treated mice, predominantly expressed in the renal tubules and the peri-glomerular region (400x magnification, Supplementary Figure 5). A similar increase in β-galactosidase expression was observed in the liver (19.92-fold) and heart (7.32-fold) of transgenic mice exposed to adenine diet compared to transgenic mice on a normal diet (Figure 3C). For the aorta, the β-galactosidase signal intensity was measured specifically in the wall along a line extending from the intima to the adventitia (Supplementary Figure 4B). A 1.82-fold increase (p=0.0275) in β-galactosidase expression was observed in the aortic wall of adenine-induced CKD mice. Analysis of brain tissue revealed microvessels positive for β-galactosidase (Figure 3B). Further quantitation revealed an 8-fold increase in the average number of microvessels/mm2 positive for β-galactosidase in mice on the adenine diet (8.630±1.692) compared to the normal diet (1.425±1.145, p=0.048) (Figure 3C). A correlation analysis was performed to demonstrate the relationship between the levels of BUN or IS in CKD model with the extent of AHR activation in the tissues of adenine-exposed mice (Table 1). We observed a strong and significant correlation between the levels of BUN or IS with β-galactosidase protein expression in the organs. This data strengthens the relationship between AHR activation in various tissues with increased BUN (reflective of deterioration of renal function) or IS levels in CKD model. Taken together, these results demonstrate AHR activation within specific tissues of different organs in a CKD model.
Table 1:
Correlations between β-galactosidase expression and indoxyl sulfate and blood-urea nitrogen for organs of adenine exposed mice.
| Parameter | Spearman r coefficient | P value |
|---|---|---|
| BUN and IS | 0.9048 | 0.0046 |
| BUN and β-gal (Kidney) | 0.7143 | 0.0491 |
| BUN and β-gal (Liver) | 0.8264 | 0.0154 |
| BUN and β-gal (Heart) | 0.7381 | 0.0458 |
| BUN and β-gal (CNS) | 0.8189 | 0.0154 |
| IS and β-gal (Kidney) | 0.7619 | 0.0368 |
| IS and β-gal (Liver) | 0.7545 | 0.0368 |
| IS and β-gal (Heart) | 0.7381 | 0.0458 |
| IS and β-gal (CNS) | 0.7433 | 0.0480 |
β-gal=β-galactosidase. BUN= Blood-urea nitrogen. IS=Indoxyl sulfate.
Tissue specific activation of AHR in an Indoxyl Sulfate-specific model
To examine whether IS can recapitulate the pattern of AHR activation observed with adenine-induced CKD model, we used an IS-specific model as described previously25. This model is characterized by an increase in IS to a level similar to ESRD patients without alteration of renal function25.
AHR activation in various tissues in response to IS was confirmed by qRT-PCR, which showed significant increases in β-galactosidase mRNA in the liver, kidney, and heart (Figure 4A). Our quantitative analysis of different tissues revealed a significant increase in β-galactosidase protein expression by 8.76-fold in the kidneys, 14.21-fold in the liver and 6.37-fold in the heart (Figure 4C). Similarly to the adenine-induced CKD model, the IS-specific model also showed 26–30% (p=0.0286) increase in the β-galactosidase expression in the intima and media of aorta of IS-treated animals compared to controls (Figure 4C). There was a trend towards an increased number of β-galactosidase positive microvessels of CNS in mice treated with IS compared to controls (Figure 4B, arrow, 4C).
Figure 4: Tissue-specific expression of β-galactosidase expression in an IS-specific model.

(a). β-galactosidase mRNA was examined in whole tissue lysates in transgenic mice and normalized to GAPDH mRNA. (b). Representative frozen sections stained with an anti-β-galactosidase antibody are shown from kidney, liver, heart and CNS (c). A color based image segmentation pipeline was used to quantify the increase in β-galactosidase expression per tissue area in the kidney (black asterisk), liver, and heart (black arrowhead). The number of microvessels in the CNS positive for β-galactosidase were counted and presented as the number of positive microvessels/mm2 (black arrow). Each data point represents the average of 5 random fields of view analyzed for the percent positive tissue area for each transgenic mouse (n=4 control, n=4 IS). All data is represented as plus or minus the standard error of the mean. Images of the kidney, liver, heart and aorta are taken at 200x magnification. Scale bar=50μM. Images in the CNS were taken at 400x magnification. Scale bar=25μM. V=vessel wall, L=lumen.
We further probed the level of AHR activation in kidneys using cell-type specific markers for both proximal (aquaporin-1) and distal (uromodulin) tubules, and endothelial cells (CD31) (Supplementary Figure 6). Staining of consecutive kidney sections with β-galactosidase and the above markers showed the presence of β-galactosidase stain in the tubules positive for Aquaporin 1 or Uromodulin suggesting AHR activation in both proximal and distal renal tubules. Similar to the adenine-induced CKD model, the β-galactosidase signal was not observed in the glomeruli, but rather in the peri-glomerular region (Supplementary Figure 5). The endothelial cells of the arterioles within the kidneys stained with CD31 also were positive for β-galactosidase corroborating the findings in aortas of adenine- and IS-exposed mice (Supplementary Figure 6). A similar analysis in the adenine-induced CKD model could not be performed due to distorted tubules and extensive renal damage at the time of harvest. These results confirmed IS as an activator of the AHR pathway in various organs and could explain AHR activation observed in the adenine-induced CKD model.
Temporal activation of AHR in an I/R-induced AKI model
Since indolic solutes are also elevated in acute kidney injury (AKI) in patients26, we examined the AHR activation in an established mouse model of AKI, ischemic reperfusion injury (I/R). Since I/R models are known to have a distinct predilection for males27, this experiment was conducted in a group of male transgenic mice. I/R injury rapidly increased BUN at 24 and 48 hours post-surgery, and as expected, it decreased by 96 hours (Supplementary Figure 3C).
The kidneys of I/R experimental mice showed a significant increase in β-galactosidase mRNA by 34.3% (p=0.0301) and 29.0% (p=0.0328) at 24 hours and 48 hours post-I/R injury respectively (Figure 5A, left). Similarly, increased β-galactosidase mRNA was observed in the liver (Figure 5A, right). β-galactosidase mRNA was significantly increased 25.7% (p=0.0438) at 24 hours post-surgery and 27.0% (p=0.0435) at 48 hours post-surgery in kidney and liver, respectively. In contrast, no change in β-galactosidase mRNA was observed in the heart or CNS of I/R-mice.
Figure 5: AHR activation in renal ischemia reperfusion model of AKI.

AHR activation was examined in transgenic mice following 23 minutes of bilateral renal ischemia. Groups of mice were time sacrificed at 24 (A), 48 (B), and 96 (C) hours post-surgery. (a). β-galactosidase mRNA was examined in whole tissue lysates in transgenic mice and normalized to GAPDH mRNA. (b-e) Frozen sections of kidney and liver were stained with an anti β-galactosidase antibody. A color based image segmentation pipeline was used to measure the percentage of tissue area positive for β-galactosidase protein. Each data point represents the average of 5 random fields of view analyzed for the percent positive tissue area for each transgenic mouse (n=3 for each group; sham control, 24, 48, and 96 hours post-surgery, left graph is kidney and right graph is liver). All Data is represented as plus or minus the standard error of the mean. Images are taken at 200x magnification. Scale bar=50μM.
In the same vein, I/R injury resulted in a significant 9.91-fold (p=0.0079) and 14.27-fold (p=0.0195) increase in β-galactosidase protein expression in the tubules and hepatocytes, respectively, at 24 hours post-surgery (Figure 5B, 5E), which persisted up to 96 hours post I/R injury (Figure 5C–5E). The above results suggest that AKI-induced AHR activation is localized in kidney and liver and persisted despite a decrease in BUN and IS levels.
Relationship between kinetics of AHR activation and uremic solutes
Since CKD and AKI in humans are characterized by increased levels of uremic toxins5,28, we posited that the AHR activation in tissues is likely to coincide with the increase in the levels of uremic solutes. We examined specifically IS and kynurenine, in plasma, which are known to activate AHR signaling8,9. Plasma IS increased 4.79-fold in adenine-induced mice compared to mice on a normal diet (p=0.0286) (Figure 6A). A similar trend was observed with kynurenine (p=0.0286) (Figure 6A). In the IS-specific model, plasma IS levels increased 18.03-fold compared to control mice (Figure 6B, right). Plasma levels of kynurenine remained similar between the two groups (Figure 6B). The rise in IS coincided with the increased β-galactosidase protein expression in various organs in both these models (Figures 2 and 3). In the I/R model, the levels of IS significantly increased 1.76-fold 24 hours post-surgery (p=0.043) then rapidly declined (Figure 6C). There were no differences at any time point in levels of plasma kynurenine (Figure 6C). β-galactosidase expression persisted even with normalization of IS levels.
Figure 6: Concurrence between AHR ligands and AHR activation in CKD and AKI models.

Uremic solutes, in plasma, were determined used mass spectrometry. (a). Plasma samples from adenine-induced mice and controls analyzed for IS and Kynurenine are shown (b). Plasma samples from mice receiving IS and probenecid are shown (c). Plasma from mice at different time points after I/R injury were analyzed for IS and Kynurenine. All Data is represented as plus or minus the standard error of the mean.
Discussion:
In this study, we demonstrated tissue-specific activation of the AHR pathway in response to discrete rodent models of kidney diseases. The choice of models was driven by the hypothesis that focuses on AHR activation by indolic uremic solutes. Among various models of CKD23, we employed the adenine model since it is known to increase IS to a level corresponding to ESRD patients25. The time points selected for different models were driven by the prospects of obtaining a signal for β-galactosidase given the low transgene activity in this model21. For example, the adenine-induced model results in persistent accumulation of uremic toxins over a two week period after exposure to a 0.25% adenine diet29. The IS-specific model results in increased levels of IS analogous to different stages of CKD10. For both these models, AHR expression was examined at two time points. In general, AKI in humans and rodents results in compromised renal function and accumulation of indolic uremic solutes that improves with the recovery of renal function26. Therefore, we performed the kinetic analysis of AHR activation in organs at three different time points in the I/R injury model.
The data obtained by this investigation provides hypothesis-generating insights into uremic toxicity. Adenine and IS-exposure increased AHR activity in the proximal and distal renal tubules. AHR signaling regulates fundamental cellular processes such as apoptosis, cell proliferation and cell cycle by regulating BCL2, FasR and cell cycle kinases30,31. With the renal insult, tubular cell proliferation and survival are pivotal to maintain the functional renal tubular mass. Our results showing AHR activation in renal tubules by IS warrant further studies to examine the role of AHR in progressive tubular damage. Interestingly, adenine- and IS- exposed kidneys showed AHR activation in the peri-glomerular region (Supplementary Figure 5). While it is difficult to precisely discern the cell-type involved, the presence of an AHR signal in the peri-glomerular region is intriguing. Renal fibrosis involves different compartments such as the tubulointerstitial and peri-glomerular regions. AHR signaling has been implicated in the fibrosis of different organs. The AHR interacts with the p65-subunit of nuclear factor (NF)-κB transcription factors and also changes the chemokine profile through an AHR-IL-22 axis, altering fibrosis in the lung in cystic fibrosis 32–34. In fact, ARNT, a transcriptional co-activator of AHR, has been implicated in renal fibrosis and sought as a therapeutic target in CKD35. A possible connection between AHR and renal fibrosis is further supported by the reports that link the indolic solutes to renal fibrosis36, which may be mediated through AHR signaling. Taken together, further studies are needed to investigate the role of AHR signaling in the kidneys, especially since AHR perturbations have different effects in various organs.
AHR signaling is the main regulator of cytochrome P450 (CYP450). There are over 200 P450 proteins, of which AHR signaling regulates three members CYP1A1, CYP1A2, and CYP1B1, which are the AHR ligand metabolizing P450s37. AHR can potentially alter the pharmacokinetics of drugs metabolized specifically by the CYP1 family. Also, AHR activation can alter the pharmacokinetics of drugs through other mechanisms. For example, Santana et al demonstrated that IS, through AHR activation, regulates the transporter protein such as P-glycoprotein to influence the blood levels of immunosuppressive medications38. The effect of AHR activation on the pharmacokinetics of drugs remains an underexplored area and is relevant given polypharmacy in patients of CKD.
Cardiovascular disease constitute a major cause of mortality in the patients of CKD. The current results showing AHR activation in cardiac myocyte and vasculature have pathogenic implications. In a zebrafish model, AHR activation in cardiac myocyte exhibited a phenotype similar to congestive heart failure as well as reduced peripheral blood flow39. The relevance of these findings to CKD patients is important since CKD patients develop congestive heart failure with low ejection fraction and with preserved ejection fraction (HEpEF)40. CKD patients are at a higher risk of atherothrombotic vascular disease. AHR activation in aorta potentiall has functional consequences given its known role in atherosclerosis 41,42 and that accelerated atherosclerosis is a known manifestation of uremia 43,44. While previous studies had demonstrated the presence of AHR protein in the vessel wall or AHR activation in cultured endothelial and vascular smooth muscle cells, there was no direct in vivo evidence of cell-type specific activtion of AHR in vasculature in response to kidney disease models. In that regards, the current work supports the previous studies9,11,25, and provides an in vivo proof of AHR activation in the vessel wall in different models of CKD. It is noteworthy that AHR activation in vessel walls is associated with increased stiffness and an aging phenotype45. A similar phenomenon was observed by Nath et al in a mouse model of arteriovenous fistula in the uremic milieu46 raising a possiblity of influence of AHR pathway in dialysis access malfunction.
CKD patients of all stages are at an increased risk of cognitive decline47. In hemodialysis patients, the prevalence of cognitive impairment has been estimated at 30%–60%, at least twice that observed in age-matched controls48,49. While several factors are considered including white matter lesions, silent infarcts and microbleeds47, fundamental to all these pathological manifestations is vascular injury. Our observations of AHR activation in the endothelial cells of cerebral microvasculature in both the adenine-induced CKD and IS-specific model opens possible areas of investigation in the fundamentals of endothelial biology in CNS in the uremic milieu and AHR signaling.
AKI is considered to be a self-limiting event. However, emerging epidemiological studies showed profound long-term effects of a single episode of AKI50. For example, Ishani et al reported that the AKI was associated with a risk of ESRD that was 13 times higher than the patients without AKI, and the risk of ESRD was 40 times higher if the patients had AKI and pre- existing CKD. Our results in I/R model showed persistance activation of AHR signaling in the renal tubules even after reduction in the levels of BUN and IS (Figure 5 and 6, Supplementary Figure 3C). There can be alternative explanations for this phenomenon, such as involvement of other uremic solutes or increase in the inflammatory cytokines released during AKI known to activate AHR pathway. Irrespecitve of mechanism, it is conceivable that the persistant AHR activity may have untoward effect on survival of renal tubules.
A growing body of literature has shown that among different uremic solutes, tryptophan metabolites (such as IS) activate AHR signaling. Our results demonstrate AHR activation in several organs in the uremic milieu with a tissue-specific pattern, which may potentially explain some of the systemic manifestations of uremic toxicity. By illustrating the use of an in vivo tool, this study paves the way to targetable pathway.
Methods:
Animal Ethics:
Animals were maintained under the supervision of the Boston University Animal Core Facility following the Institutional Animal Care and Use Committee (IACUC) protocol number AN-15449.2016.03. Animals were caged in conventional housing with 3 males per cage and 5 females per cage.
Overview of Transgenic Mouse:
B6.Cg-Tg(DRE-lacZ)2Gswz/J mice (stock # 006229) were purchased from Jackson Laboratory, and housed and bred at the Boston University Animal Core Facility. This mouse line was originally donated by Dr. Thomas A. Gasiewicz, University of Rochester, School of Medicine and Dentistry. The mouse contains a lacZ reporter gene downstream of two dioxin response elements and chicken ovalbumin TATA box as previously described21. Male and female transgenic mice were randomized to experimental groups for models of chronic kidney disease. Male mice were used for the renal ischemia reperfusion experiments. experiments.
Genotyping of Transgenic Mice:
DNA was extracted from tail clippings for all mice used in these experiments following weaning at 21 days. Mice containing the lacZ transgene were identified through conventional polymerase chain reaction (PCR) using forward and reverse primers complementary to the transgene and an internal positive control (Table 1). Following identification of mice containing the transgene, extracted genomic DNA was used as a template for quantitative real-time polymerase chain reaction (qRT-PCR) in order to determine the copy number of the lacZ transgene. Genomic DNA was probed using a commercially available Taqman probe complementary to the lacZ gene from Thermofisher. All mice used in these experiments were homozygous for the transgene (Supplementary Figure 1).
Statistical Analysis:
Data was analyzed using a non-parametric Welch’s t-test or analysis of variance (ANOVA), as indicated. Significance was accepted as p<0.05. Data was presented as the mean plus or minus standard error of the mean. Correlations between BUN, IS, and quantification of β-galactosidase protein in all tissues were done using a non-parametric Spearman correlation.
Details for administration of TCDD to B6.Cg-Tg(DRE-lacZ)2Gswz/J, different models of kidney diseases (Adenine-induced CK, IS-specific, and I/R reperfusion modes), tissue collection, IHC, antibodies, image quantification, quantitative real-time PCR, BUN estimation and LC/MS method of uremic solutes are in the supplementary methods section.
Supplementary Material
All mice were genotyped using conventional PCR with the indicated primers. Genomic DNA from hemizygous mice was then used as a template for qRT-PCR using a commercially available Taqman probe complementary to the lacZ (green) gene in order to determine the copy number. Data was normalized to GAPDH (red). Representative Ct graph of three independent experiments is shown.
Dre-LacZ transgenic mice were administered TCDD and sacrificed after 4 days. Untreated transgenic mice served as controls. Fresh liver tissue was harvested and consecutive slides were stained with a β-galactosidase reporter gene staining kit (Sigma, cat# GALS-1LT) and pre-validated anti-β-galactosidase antibody (Supplementary Figure 2A). Consecutive sections of the liver from the same were stained with pre-validated anti-β-galactosidase antibody and isotype control to show specificity of the antibody used for the immunohistochemistry (IHC). Mice showed prominent stain of βgalactosidase protein using IHC (Supplementary Figure 2B). Representative images of liver tissue at 200X magnification are shown.
Renal function was assessed on terminal plasma from all mice by measuring blood-urea nitrogen (BUN). BUN was determined followed manufacturer’s protocol using a Quantichrom Urea Assay Kit (DIUR-100) from Bioassay Systems. In mice exposed to a 0.25% adenine supplemented diet, there was an approximate 4-fold increase in BUN compared to control (Supplementary Figure 3A). In the IS-specific model of CKD, BUN was not increased between mice treated with IS and control mice (Supplementary Figure 3B). In the renal ischemia reperfusion model of acute kidney injury (AKI), BUN significantly increased at 24hrs post-surgery and remained elevated at 48hrs while decreasing at 96hrs post-surgery (Supplementary Figure 3C).
A customized, color-based image segmentation pipeline was used to quantify the amount of βgalactosidase and expressed as percent positive tissue area for the kidney, liver, and heart in tissue from animal models of CKD and AKI. Briefly, RGB color images were converted to L*a*b* color space with the two dimensional a*b* space data used for further analysis with each image segmented into k=3 clusters and the percentage of tissue area positive for β-galactosidase analyzed (Supplementary Figure 4A). For the aorta, the amount of β-galactosidase protein was analyzed by measuring the pixel intensity of a grey scale image along a line from the intima to the adventitia. 5 lines were measured per section of the aorta (Supplementary Figure 4B).
Kidneys from both the adenine-induced model of CKD and the IS-solute specific model of uremia were stained with an antibody against β-galactosidase. High magnification imaging (400x) shows AHR activation, indicative of β-galactosidase expression, in the peri-glomerular fibrotic regions. For 200x magnification images, scale bar=50μM. For 400x magnification images, scale bar=25μM
To examine tissue specific localization of AHR in an IS-specific model of CKD, seriel sections from transgenic mice that received IS with probenacid for 4 days were stained with antibodies against β-galactosidase (AHR acitvation) and one of the following: uromodulin (distal tubules), aquaporin-1 (proximal tubule), and CD31 (endothelial cells). (a). β-galactosidase was found to be expressed in distal tubules marked by expression of uromodulin. (b). Additionally, proximal tubules marked by expression of aquaporin-1 were found to express β-galactosidase. (c). Kidney sections were stained with antibody against CD31, a marker of endothelial cells. We found expression of of β-galactosidase in the wall of vessels in the kidney marked by expresion of CD31.
Supplementary table S1: Primer Sequences for Identification of Transgenic Mice
Supplementary table S2: List of Primary Antibodies for Immunohistochemistry
Translational Statement:
Uremic solutes contribute to the pathogenesis of some of the systemic complications in the patients with chronic kidney disease (CKD). Among different solutes, tryptophan metabolites such as indolic solutes are the ligands of aryl hydrocarbon receptor (AHR), a ubiquitously expressed protein associated with several diseases. Here, using a reporter-gene mouse, we show tissue specific activation of AHR among different organs in models of CKD and AKI. Clinically, this work provides hypothesis-generating insights in the manifestations of uremia and strengthens the potential exploration of AHR inhibitors to reduce the organ-level toxicity of uremic solutes in the patients with CKD.
Acknowledgements:
We would like to thank the veterinary care staff at the Boston University Animal Core Facility for their help in maintaining the animal colonies. We would like to thank Boston University School of Medicine Cellular Imaging Core for their help with imaging the immunohistochemistry experimental slides. We also thank Teresa Russell (Boston University) for review of the manuscript.
Funding:
This work was funded in part by NCI R01CA175382, NIH R01 HL132325 and Evans Faculty Merit award (VCC); and T32 training grant in cardiovascular biology T32 HL007224-40 (JAW); T32 training grant in immunobiology of trauma T32 GM086308-06A1 (NA) and the Thrombosis to Hemostasis in Health and Disease Affinity Research Collaborative (Boston University, Evans Center For Interdisciplinary Biomedical Research).
Footnotes
Conflicts of Interest and Disclosures:
No conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Bowe B, Xie Y, Li T, et al. Changes in the us burden of chronic kidney disease from 2002 to 2016: An analysis of the global burden of disease study. JAMA Network Open. 2018;1:e184412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirich TL. Obstacles to reducing plasma levels of uremic solutes by hemodialysis. Semin Dial. 2017. [DOI] [PubMed] [Google Scholar]
- 3.Vanholder R, Glorieux G. Introduction: Uremic Toxicity - State of the Art 2014. Semin Nephrol. 2014;34:85–86. [DOI] [PubMed] [Google Scholar]
- 4.Vanholder R, De Smet R, Lameire N. Protein-bound uremic solutes: the forgotten toxins. Kidney Int Suppl. 2001;78:S266–270. [DOI] [PubMed] [Google Scholar]
- 5.Duranton F, Cohen G, De Smet R, et al. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 2012;23:1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou L, Jourde-Chiche N, Faure V, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5:1302–1308. [DOI] [PubMed] [Google Scholar]
- 7.Ying Y, Yang K, Liu Y, et al. A uremic solute, P-cresol, inhibits the proliferation of endothelial progenitor cells via the p38 pathway. Circ J. 2011;75:2252–2259. [DOI] [PubMed] [Google Scholar]
- 8.Kolachalama VB, Shashar M, Alousi F, et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J Am Soc Nephrol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shivanna S, Kolandaivelu K, Shashar M, et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J Am Soc Nephrol. 2016;27:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shashar M, Belghasem ME, Matsuura S, et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gondouin B, Cerini C, Dou L, et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013;84:733–744. [DOI] [PubMed] [Google Scholar]
- 12.Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J. 2010;31:1771–1779. [DOI] [PubMed] [Google Scholar]
- 13.Yisireyili M, Shimizu H, Saito S, Enomoto A, Nishijima F, Niwa T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013;92:1180–1185. [DOI] [PubMed] [Google Scholar]
- 14.Adesso S, Magnus T, Cuzzocrea S, et al. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front Pharmacol. 2017;8:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henry EC, Welle SL, Gasiewicz TA. TCDD and a putative endogenous AhR ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol Sci. 2010;114:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka H, Sirich TL, Meyer TW. Uremic Solutes Produced by Colon Microbes. Blood Purif. 2015;40:306–311. [DOI] [PubMed] [Google Scholar]
- 17.Velasquez MT, Centron P, Barrows I, Dwivedi R, Raj DS. Gut Microbiota and Cardiovascular Uremic Toxicities. Toxins (Basel). 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehrabi MR, Steiner GE, Dellinger C, et al. The arylhydrocarbon receptor (AhR), but not the AhR-nuclear translocator (ARNT), is increased in hearts of patients with cardiomyopathy. Virchows Arch. 2002;441:481–489. [DOI] [PubMed] [Google Scholar]
- 19.Vasquez A, Atallah-Yunes N, Smith FC, et al. A role for the aryl hydrocarbon receptor in cardiac physiology and function as demonstrated by AhR knockout mice. Cardiovasc Toxicol. 2003;3:153–163. [DOI] [PubMed] [Google Scholar]
- 20.Juricek L, Coumoul X. The Aryl Hydrocarbon Receptor and the Nervous System. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willey JJ, Stripp BR, Baggs RB, Gasiewicz TA. Aryl hydrocarbon receptor activation in genital tubercle, palate, and other embryonic tissues in 2,3,7, 8-tetrachlorodibenzo-p-dioxin-responsive lacZ mice. Toxicol Appl Pharmacol. 1998;151:33–44. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Xie T, Zhang D, Leung PS. GPR120 protects lipotoxicity-induced pancreatic beta-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin Sci (Lond). 2019;133:101–116. [DOI] [PubMed] [Google Scholar]
- 23.Lim BJ, Yang HC, Fogo AB. Animal models of regression/progression of kidney disease. Drug Discov Today Dis Models. 2014;11:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shashar M, Siwak J, Tapan U, et al. c-Cbl mediates the degradation of tumorigenic nuclear β-catenin contributing to the heterogeneity in Wnt activity in colorectal tumors. Oncotarget. 2016;7:71136–71150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shashar M, Belghasem ME, Matsuura S, et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci Transl Med. 2017;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Hao G, Pan Y, et al. Serum indoxyl sulfate is associated with mortality in hospital-acquired acute kidney injury: a prospective cohort study. BMC Nephrol. 2019;20:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei Q, Dong Z. Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol. 2012;303:F1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herget-Rosenthal S, Glorieux G, Jankowski J, Jankowski V. Uremic toxins in acute kidney injury. Semin Dial. 2009;22:445–448. [DOI] [PubMed] [Google Scholar]
- 29.Jia T, Olauson H, Lindberg K, et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013;14:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaher H, Fernandez-Salguero PM, Letterio J, et al. The involvement of aryl hydrocarbon receptor in the activation of transforming growth factor-beta and apoptosis. Mol Pharmacol. 1998;54:313–321. [DOI] [PubMed] [Google Scholar]
- 31.Mohammadi S, Seyedhosseini FS, Behnampour N, Yazdani Y. Indole-3-carbinol induces G1 cell cycle arrest and apoptosis through aryl hydrocarbon receptor in THP-1 monocytic cell line. J Recept Signal Transduct Res. 2017;37:506–514. [DOI] [PubMed] [Google Scholar]
- 32.Beamer CA, Shepherd DM. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin Immunopathol. 2013;35:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puccetti M, Paolicelli G, Oikonomou V, et al. Towards Targeting the Aryl Hydrocarbon Receptor in Cystic Fibrosis. Mediators Inflamm. 2018;2018:1601486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weidenbusch M, Rodler S, Song S, et al. Gene expression profiling of the Notch-AhR-IL22 axis at homeostasis and in response to tissue injury. Biosci Rep. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haase VH. ARNT as a Novel Antifibrotic Target in CKD. Am J Kidney Dis. 2019;73:281–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mutsaers HA, Stribos EG, Glorieux G, Vanholder R, Olinga P. Chronic Kidney Disease and Fibrosis: The Role of Uremic Retention Solutes. Front Med (Lausanne). 2015;2:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. 2005;338:311–317. [DOI] [PubMed] [Google Scholar]
- 38.Santana Machado T, Poitevin S, Paul P, et al. Indoxyl Sulfate Upregulates Liver P-Glycoprotein Expression and Activity through Aryl Hydrocarbon Receptor Signaling. J Am Soc Nephrol. 2018;29:906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanham KA, Plavicki J, Peterson RE, Heideman W. Cardiac myocyte-specific AHR activation phenocopies TCDD-induced toxicity in zebrafish. Toxicol Sci. 2014;141:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segall L, Nistor I, Covic A. Heart failure in patients with chronic kidney disease: a systematic integrative review. Biomed Res Int. 2014;2014:937398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr DH. Another important biological function for the aryl hydrocarbon receptor. Arterioscler Thromb Vasc Biol. 2011;31:1247–1248. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Nishimura N, Kuo V, et al. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25:1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massy ZA, Ivanovski O, Nguyen-Khoa T, et al. Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J Am Soc Nephrol. 2005;16:109–116. [DOI] [PubMed] [Google Scholar]
- 45.Eckers A, Jakob S, Heiss C, et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Sci Rep. 2016;6:19618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nath KA, O’Brien DR, Croatt AJ, et al. The murine dialysis fistula model exhibits a senescence phenotype: pathobiological mechanisms and therapeutic potential. Am J Physiol Renal Physiol. 2018;315:F1493-F1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bugnicourt JM, Godefroy O, Chillon JM, Choukroun G, Massy ZA. Cognitive disorders and dementia in CKD: the neglected kidney-brain axis. J Am Soc Nephrol. 2013;24:353–363. [DOI] [PubMed] [Google Scholar]
- 48.Fazekas G, Fazekas F, Schmidt R, Kapeller P, Offenbacher H, Krejs GJ. Brain MRI findings and cognitive impairment in patients undergoing chronic hemodialysis treatment. J Neurol Sci. 1995;134:83–88. [DOI] [PubMed] [Google Scholar]
- 49.Graham JE, Rockwood K, Beattie BL, et al. Prevalence and severity of cognitive impairment with and without dementia in an elderly population. Lancet. 1997;349:1793–1796. [DOI] [PubMed] [Google Scholar]
- 50.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys. 2006;450:67–77. [DOI] [PubMed] [Google Scholar]
- 52.Hesketh EE, Czopek A, Clay M, et al. Renal ischaemia reperfusion injury: a mouse model of injury and regeneration. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang KP, Lee JE, Lee AS, et al. Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep. 2014;9:2061–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lima-Posada I, Portas-Cortes C, Perez-Villalva R, et al. Gender Differences in the Acute Kidney Injury to Chronic Kidney Disease Transition. Sci Rep. 2017;7:12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang A, Rijal K, Ng SK, Ravid K, Chitalia V. A mass spectrometric method for quantification of tryptophan-derived uremic solutes in human serum. J Biol Methods. 2017;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All mice were genotyped using conventional PCR with the indicated primers. Genomic DNA from hemizygous mice was then used as a template for qRT-PCR using a commercially available Taqman probe complementary to the lacZ (green) gene in order to determine the copy number. Data was normalized to GAPDH (red). Representative Ct graph of three independent experiments is shown.
Dre-LacZ transgenic mice were administered TCDD and sacrificed after 4 days. Untreated transgenic mice served as controls. Fresh liver tissue was harvested and consecutive slides were stained with a β-galactosidase reporter gene staining kit (Sigma, cat# GALS-1LT) and pre-validated anti-β-galactosidase antibody (Supplementary Figure 2A). Consecutive sections of the liver from the same were stained with pre-validated anti-β-galactosidase antibody and isotype control to show specificity of the antibody used for the immunohistochemistry (IHC). Mice showed prominent stain of βgalactosidase protein using IHC (Supplementary Figure 2B). Representative images of liver tissue at 200X magnification are shown.
Renal function was assessed on terminal plasma from all mice by measuring blood-urea nitrogen (BUN). BUN was determined followed manufacturer’s protocol using a Quantichrom Urea Assay Kit (DIUR-100) from Bioassay Systems. In mice exposed to a 0.25% adenine supplemented diet, there was an approximate 4-fold increase in BUN compared to control (Supplementary Figure 3A). In the IS-specific model of CKD, BUN was not increased between mice treated with IS and control mice (Supplementary Figure 3B). In the renal ischemia reperfusion model of acute kidney injury (AKI), BUN significantly increased at 24hrs post-surgery and remained elevated at 48hrs while decreasing at 96hrs post-surgery (Supplementary Figure 3C).
A customized, color-based image segmentation pipeline was used to quantify the amount of βgalactosidase and expressed as percent positive tissue area for the kidney, liver, and heart in tissue from animal models of CKD and AKI. Briefly, RGB color images were converted to L*a*b* color space with the two dimensional a*b* space data used for further analysis with each image segmented into k=3 clusters and the percentage of tissue area positive for β-galactosidase analyzed (Supplementary Figure 4A). For the aorta, the amount of β-galactosidase protein was analyzed by measuring the pixel intensity of a grey scale image along a line from the intima to the adventitia. 5 lines were measured per section of the aorta (Supplementary Figure 4B).
Kidneys from both the adenine-induced model of CKD and the IS-solute specific model of uremia were stained with an antibody against β-galactosidase. High magnification imaging (400x) shows AHR activation, indicative of β-galactosidase expression, in the peri-glomerular fibrotic regions. For 200x magnification images, scale bar=50μM. For 400x magnification images, scale bar=25μM
To examine tissue specific localization of AHR in an IS-specific model of CKD, seriel sections from transgenic mice that received IS with probenacid for 4 days were stained with antibodies against β-galactosidase (AHR acitvation) and one of the following: uromodulin (distal tubules), aquaporin-1 (proximal tubule), and CD31 (endothelial cells). (a). β-galactosidase was found to be expressed in distal tubules marked by expression of uromodulin. (b). Additionally, proximal tubules marked by expression of aquaporin-1 were found to express β-galactosidase. (c). Kidney sections were stained with antibody against CD31, a marker of endothelial cells. We found expression of of β-galactosidase in the wall of vessels in the kidney marked by expresion of CD31.
Supplementary table S1: Primer Sequences for Identification of Transgenic Mice
Supplementary table S2: List of Primary Antibodies for Immunohistochemistry