

Abstract
Lack of response and acquired resistance continue to be limitations of targeted and immune-based therapies. Pyroptosis is an inflammatory form of cell death characterized by the release of inflammatory damage-associated molecular patterns (DAMPs) and cytokines via gasdermin (GSDM) protein pores in the plasma membrane. Induction of pyroptosis has implications for treatment strategies in both therapy-responsive, as well as resistance forms of melanoma. We show that the caspase-3 activator, raptinal, induces pyroptosis in both human and mouse melanoma cell line models and delays tumor growth in vivo. Release of DAMPs and inflammatory cytokines was dependent on caspase activity and GSDME expression. Furthermore, raptinal stimulated pyroptosis in melanoma models that have acquired resistance to BRAF and MEK inhibitor therapy. These findings add support to efforts to induce pyroptosis in both the treatment-naïve and resistant settings.
Introduction
Distinct mechanisms of programmed cell death exist (1). Apoptosis involves the condensation of chromatin and degradation of DNA followed by cell engulfment by macrophages. Extrinsic (FAS/TRAIL/TNF receptor) and intrinsic (mitochondrial) apoptotic pathways are typically considered immune inert. Both extrinsic and intrinsic apoptotic pathways trigger caspase cleavage events that result in the activation of executioner caspases-3, -6 and -7 (2–5). In contrast, inflammatory forms of cell death such as necroptosis and pyroptosis lead to the release of inflammatory cytokines and damaged-associated molecular patterns (DAMPs) (4,6,7). Pyroptosis is mediated by gasdermin (GSDM) family proteins, which are cleaved to form N-terminal fragments capable of binding membrane phospholipids and oligomerizing into pore structures (6,8,9). Although the ability of inflammatory cell death mechanisms to alter the tumor immune microenvironment is poorly understood, it is likely to be an important area of research given the advances in immune checkpoint inhibitor (ICI) therapy and the reliance of ICI efficacy on tumor-infiltrating immune cells (10). Findings have uncovered mechanistic links between apoptosis and pyroptosis. Specifically, caspase-3 cleaves GSDME after Asp-270 to create an N-terminal fragment with plasma membrane-targeting and pore-forming abilities (11–13).
Targeted therapy kinase inhibitors are often effective in cancers that show dependency on a driver oncogene; however, durable effects are limited by the acquisition of resistance. In cutaneous melanomas that harbor BRAF V600 mutations, BRAF inhibitor (BRAFi) plus MEK inhibitor (MEKi) therapy elicits responses in nearly all patients; however, these effects last for a median time of 13–15 months (14,15). BRAFi and MEKi are known to elicit effects on the tumor immune microenvironment. Studies from the Scolyer group showed an increase in CD4+ and CD8+ cells in tumor biopsies after 7 days of BRAFi treatment (16). Furthermore, the Wargo group showed increased expression of melanoma antigens such MART1, GP100, TYRP1 and TYRP2 on treatment with BRAFi (17). The Kawakami and Wargo groups showed that BRAFi plus MEKi treatment led to decreased expression of immune suppressive factors such as VEGF (18,19). Acquired resistant melanomas lack T cell infiltrates and are typically non-responsive to other treatment options.
Since pyroptosis is associated with the release of inflammatory DAMPs and cytokines that have the potential to alter the tumor immune microenvironment (20), we sought to activate this cell death mechanism in both treatment-naïve and therapy-resistant tumor cells. We tested raptinal, a caspase-3 activator, in melanoma cell models (21). We show that raptinal induces pyroptosis in human and mouse BRAF mutant melanoma cells and in a dose and time-dependent manner. Furthermore, raptinal delayed the growth of BRAF mutant melanomas in vivo. The effects of raptinal were caspase- and GSDME-dependent. In addition, raptinal treatment induced pyroptosis in BRAFi plus MEKi-resistant cell lines and delayed growth of resistant cells in vivo. Together, these findings provide further support for testing agents that induce inflammatory forms of cell death to inhibit tumor growth.
Materials and Methods
Cell lines
Mouse D4M3.A cells (derived from Tyr::CreER; BrafV600E, Pten lox/lox mice; donated in 2016 by Dr. Constance E. Brinckerhoff, Dartmouth University, Hanover, NH) were cultured in DMEM/F12 (GIBCO, Life Technologies, Grand Island, NY, USA) supplemented with 5% fetal bovine serum (FBS), and 1% L-glutamine (Corning Inc.). Mouse YUMM1.7 cells (derived from BrafV600E/WT, Pten Lox/Lox, Cdkn2−/− mice; donated in 2014 by Dr. Marcus Bosenberg, Yale University, New Haven, CT) were cultured in DMEMF-12 50/50 (GIBCO, Life Technologies) supplemented with 10% FBS, and 1% non-essential amino acids (Corning Inc.). Human A375 cells (purchased from ATCC in 2005) were cultured in DMEM (GIBCO, Life Technologies) supplemented with 10% FBS. Human WM35, WM793 and 1205Lu cells (donated by Dr. Meenhard Herlyn, The Wistar Institute, 2005) were cultured in MCDB 153 medium containing 20% Leibovitz-L15 medium, 2% FBS, 0.2% sodium bicarbonate (Corning Inc.), 5 μg/mL insulin. 1% penicillin/streptomycin (Corning Inc.) was added unless otherwise noted. All cells were grown at 37°C in a humidified incubator supplemented with 5% CO2. All melanoma cell lines described above were short tandem repeat (STR) analyzed, confirmed for BRAF/BrafV600E mutation by Sanger sequencing and routinely tested for mycoplasma contamination (most recent testing was January 2021 for mouse cells and November 2021 for human cells) with a MycoScope™ PCR Mycoplasma Detection Kit (Genlantis).
CRT cells were isolated from mouse tumors and cultured in the same medium as parental cells with the addition of PLX4720 (1 μmol/L) and PD0325901 (35 nmol/L) and utilized within 5 passages for experiments. A375 and YUMM1.7-derived CRT cells were published previously (20,22). The GSDME CRISPR/Cas9 knockout cells were previously described (20).
Western blot analysis and cell supernatant collection
Protein lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking in 5% bovine serum albumin (BSA), PVDF membranes were incubated with the indicated primary antibodies overnight at 4°C, washed in PBS/Tween and incubated with peroxidase-coupled secondary antibodies. Immunoreactivity was detected using horseradish peroxidase (HRP)-conjugated secondary antibodies (CalBioTech, Spring Valley, CA) and chemiluminescence substrate (Thermo Fisher Scientific, Rockford, IL) and a ChemicDoc MP imaging system (Bio-Rad).
For experiments involving supernatant, cells were cultured in FBS-free medium to avoid distortion of SDS-PAGE. Cell supernatants were harvested and centrifuged for 10 minutes at 500g at 4°C to remove cell debris. Resulting supernatants were concentrated 10x using Amicon Ultra 10K (Sigma-Aldrich) by centrifugation for 30 minutes at 4,500g at 4°C. Concentrates were mixed with sample buffer and analyzed via Western blotting as described above. Protein gel staining was performed using Ponceau red staining
Primary antibodies used were caspase-3 (Cell Signaling Technology Cat# 9664, RRID:AB_2070042), cleaved caspse-7 (Cell Signaling Technology Cat# 9491, RRID:AB_2068144), human cleaved caspase-8 (Cell Signaling Technology Cat# 9496, RRID:AB_561381), mouse cleaved caspase-8 (Cell Signaling Technology Cat# 8592, RRID:AB_10891784), human cleaved caspase-9 (Cell Signaling Technology Cat# 9505, RRID:AB_2290727), mouse cleaved caspase-9 (Cell Signaling Technology Cat# 9509, RRID:AB_2073476), PARP (Cell Signaling Technology Cat# 9542, RRID:AB_2160739), α-tubulin (Cell Signaling Technology Cat# 2144, RRID:AB_2210548), HMGB1 (Cell Signaling Technology Cat# 6893, RRID:AB_10827882), IL-1α (Santa Cruz Biotechnology Cat# sc-9983, RRID:AB_627789), vinculin (Santa Cruz Biotechnology Cat# sc-73614, RRID:AB_1131294), GSDME (Abcam Cat# ab215191, RRID:AB_2737000), IL-1β (GeneTex Cat# GTX74034, RRID:AB_378141), and Actin (Sigma-Aldrich Cat# A2066, RRID:AB_476693). Secondary antibodies used were as follows: goat anti-rabbit IgG (Thermo Fisher Scientific Cat# 31460, RRID:AB_228341), goat anti-rabbit IgG (Millipore Cat# 401315, RRID:AB_2617117), and goat anti-mouse IgG (Sigma-Aldrich Cat# 401215, RRID:AB_10682749).
LDH release assays
Membrane disruption was measured by LDH release assay using the CytoTox 96 cytotoxicity assay (Promega). FBS-free culture medium was collected and centrifuged at 4°C, 10 minutes at 500g to remove cell debris. LDH release was measured according to the manufacturer’s instructions. A maximum LDH release control was included in each experiment corresponding to cells treated with 1% Triton-X-100 for 30 minutes before harvest. The percent of LDH release was calculated by the ratio: 100x (Treated condition / Max LDH release). Each experiment was performed in triplicate and averages with SEM at each time point were plotted.
IncuCyte assays
Cells (8 × 103) were seeded in 96-well plates for 24 hours prior to treatment with the indicated drug. After that, cells were monitored in an IncuCyte® S3 system (Essen BioScience, Ann Arbor, MI, USA), acquiring images (objective 10x) every 30 minutes in 4 separate regions per well. Images were analyzed to identify cell membrane swelling induced by treatment.
Kinetic quantification of caspase-3/7 activity or SYTOX green using live-cell time-lapse imaging
Caspase-3/7 activity was assessed using the IncuCyte® caspase-3/7 green apoptosis assay reagent (Sartorius), and SYTOX green activity using the SYTOX™ Green Nucleic Acid Stain (Thermo Fisher Scientific). For SYTOX green experiments, cells were pre-incubated 30 minutes with 2.5 μM SYTOX green prior to corresponding treatment. Caspase-3/7 reagent was added at a final concentration of 5 mM with corresponding drug. Data were analyzed using IncuCyte analysis software to detect and quantify green cells/image (Caspase-3/7 or SYTOX green positive cells). The live-cell phase contrast images were used to calculate confluence using the IncuCyte® software. Accumulation of caspase-3/7 or SYTOX green over time was normalized to confluence of cells. Each condition was performed in triplicate and averages with SEM at each time point were plotted.
In vitro drug treatments
Raptinal (a rapid-caspase activator), Z-VAD-FMK (a pan-caspase inhibitor) and Z-DEVD-FMK (a caspase-3 inhibitor) were purchased from Selleck Chemicals LLC (Houston, TX) and solubilized in DMSO. PLX4720 (a BRAF inhibitor) and PD0325901 (a MEK inhibitor) were purchased from Selleck and solubilized in DMSO. Cells were treated with 5 μM of raptinal for A375 cells or 2.5 μM for all other cells studied. Cells were pre-treated with Z-VAD-FMK or Z-DEVD-FMK for 3 hours (100 μg/mL) before treatment with raptinal with indicated doses for indicated times. Cells were treated with 1 μM and 35 nM of BRAFi (PLX4720) and MEKi (PD0325901), respectively.
In vivo mouse studies
Male C57BL/6J mice (RRID:IMSR_JAX:000664) were purchased from The Jackson Laboratory and maintained in-house. Mice were between 6 and 7 weeks old at the time of tumor implantation. The Institutional Animal Care and Use Committee at Thomas Jefferson University reviewed and approved all studies (protocol# 01052). For tumor implantation, YUMM1.7 cells (3 × 106 cells/mL) or CRT47R (2.5 × 106 cells/mL) cells were resuspended in Hanks balanced salt solution (HBSS) and 100 μL of cell suspension was injected intradermally in the shaved right flank of the mouse. Tumor growth was monitored by measuring the length and width of the tumors using digital calipers and tumor volume was determined using the following formula: volume = (length × width2) × 0.52. When tumors reached 100 mm3 in volume, mice were randomized into groups receiving intraperitoneal injections of vehicle control or 20 mg/kg raptinal (adjusted to the heaviest mouse) as outlined previously (21) +/− BRAFi plus MEKi diet (200 ppm PLX4720 plus 7 ppm PD0325901).
Statistical analysis
For in vitro experiments, statistics were performed using Excel (Microsoft, Redmond, WA, USA), GraphPad Prism (RRID:SCR_002798), version 8.1, or R (version 4.1.2 [https://www.r-project.org/]). If data were normally distributed (as assessed by the Shapiro test), a two-tailed t test was used to analyze the data. If data were non-normally distributed, a Mann-Whitney test was performed instead. For in vivo experiments, statistics were performed using GraphPad Prism, version 9, or R (version 4.1.0 [https://www.r-project.org/]). If data were normally distributed (as assessed by the Kolmogorov-Smirnov test), a two-tailed t test was used to analyze the data. If data were non-normally distributed, a Mann-Whitney test was performed instead. To assess tumor doubling times, the tumor measurements were log transformed and a linear regression was used to model the rate of tumor growth as a function of time from the initial injection to the endpoint of 500 mm3. Finally, a log-rank (Mantel-Cox) test was used to compare Kaplan-Meier survival curves. A p value < 0.05 was considered significant and p values were represented by: * for p < 0.05, ** for p < 0.01, *** for p < 0.001 and **** for p < 0.0001. Results are expressed as mean or mean ± SEM unless otherwise indicated.
Data availability
The data generated for this study are available within the article and its supplementary data files.
Results
Raptinal induces pyroptosis in BRAF mutant human and mouse melanoma cells
We studied the actions of raptinal, a rapid caspase activator, in human (A375, WM35, WM793, and 1205Lu) and mouse (D4M3.A and YUMM1.7) BRAF mutant melanoma cells (Supp. Figure 1). Effects on pyroptotic markers such as the cleavage of GSDM proteins as well as the release of cytokines and DAMPs were characterized. By Western blot analysis, raptinal treatment induced a dose-dependent cleavage of GSDME, giving rise to the pore-forming ~35 kDa N-terminal fragment in all six melanoma cell lines that were tested (Figure 1A, Supp. Figure 2A). Furthermore, raptinal induced the release of the DAMPs, high–mobility group box 1 (HMGB1) and lactate dehydrogenase (LDH), as well as the cytokines, IL-1α or IL-1β, into the cell supernatant in a dose-dependent manner (Figures 1A and 1B, Supp. Figures 2A and 2B). The release of LDH, HMGB1 and IL-1α or IL-1β was induced when raptinal was used at 2.5 μM for most cell lines, although 5 μM was required for effects in A375 cells; hence, we used these concentrations for the respective cell lines in subsequent experiments.
Figure 1: Raptinal induces pyroptosis in human and mouse melanoma cells.
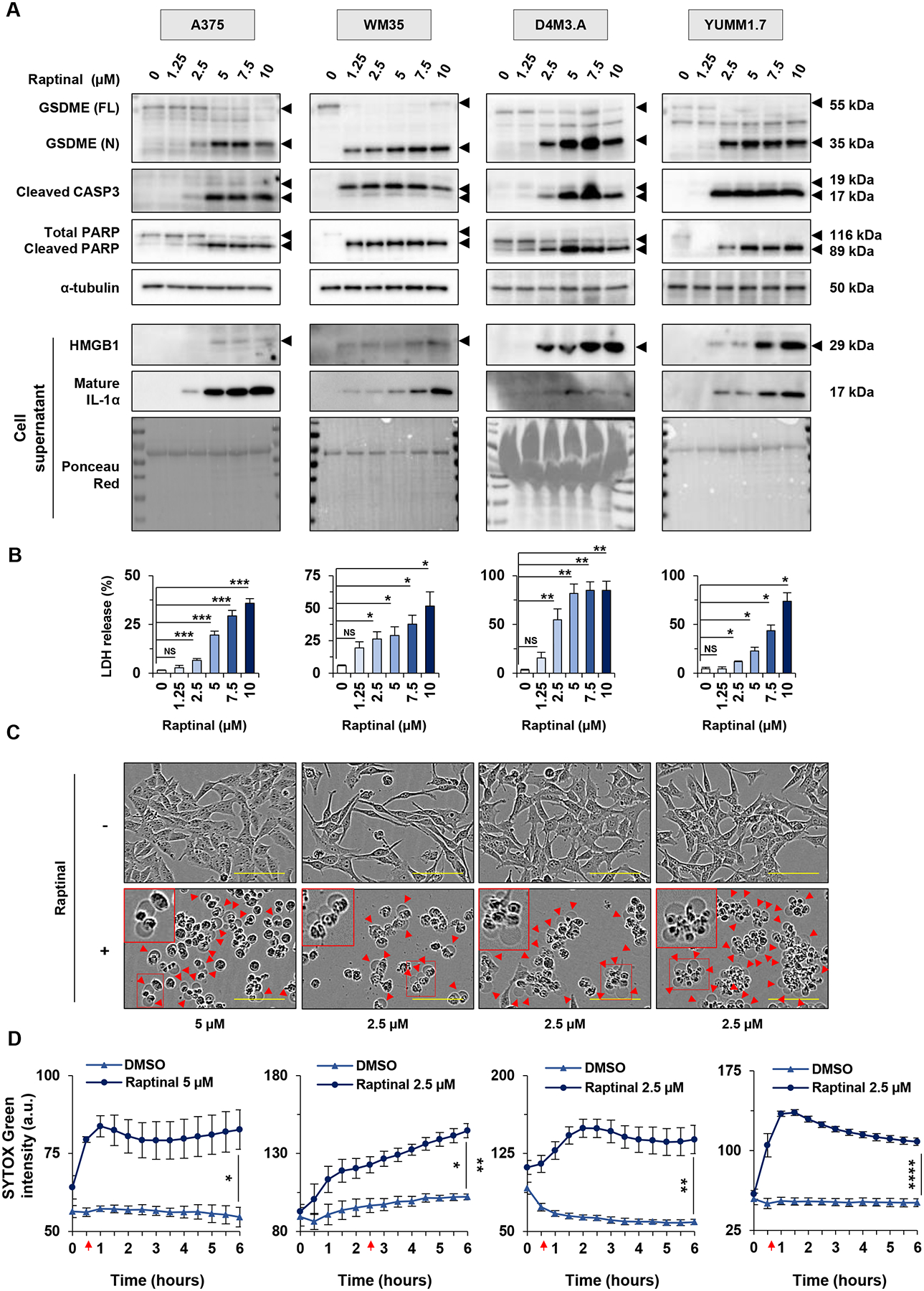
(A-B) Human melanoma cells (A375 and WM35) and mouse melanoma cells (D4M3.A and YUMM1.7), were treated in vitro for 3 hours with doses of raptinal ranging from 1.25 to 10 μM. DMSO was used as a vehicle control. (A) Levels of GSDME, cleaved caspase-3 and PARP in lysates, or HMGB1 and IL-1α in supernatants after raptinal treatment. α-tubulin or ponceau red staining are controls for protein lysates or supernatant total protein extracts, respectively. Full-length GSDME (FL) runs at 55 kDa and cleaved N-terminal GSDME (N) runs at 35 kDa. Total PARP runs at 116 kDa and cleaved PARP runs at 89 kDa. Western Blots shown are representative of at least three independent experiments. (B) Percentage of LDH release averaged from at least three independent experiments. Error bars are SEM. Significance was assessed by Student t test for WM35 and YUMM1.7 cells and by Mann-Whitney test for A375 and D4M3.A cells, NS: non-significant, *p<0.05, **p<0.01, ***p<0.001. (C-D) Cells were treated with 2.5 μM raptinal, except for A375 that were treated with 5 μM. (C) Cell morphology visualized with the IncuCyte S3 system at x10 objective. Plasma membrane bubble-like protrusions, a characteristic feature of pyroptosis, are indicated by red arrow and a magnification is show in the red frame. Scale bar: 100 μm. Cell morphology images shown are representative of at least three independent experiments. (D) SYTOX green fluorescence was measured over time with IncuCyte S3 system and normalized to cell confluence at each timepoint for cells treated with DMSO or raptinal. At least two replicate wells were performed in each experimental condition and all experiments were performed at least three times independently. Data are representative of at least three independent experiments. Error bars are SEM. Significance was assessed by Student t test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Red arrows indicate the time when the treated cells were significantly more fluorescent than untreated cells.
Raptinal treatment was associated with cleavage of caspase-3 and the apoptotic marker PARP (Figure 1A, Supp. Figure 2A). Cells treated with raptinal also exhibited plasma membrane bubble-like protrusions, a characteristic feature of pyroptosis (Figure 1C, Supp. Figure 2C). Next, we analyzed cell permeability using a fluorescent probe, SYTOX green, which enters the cells and stains nucleic acid when the plasma membrane is disrupted. We observed a time-dependent increase of fluorescence in cells treated with raptinal compared to untreated cells, although only weak effects were observed in WM793 and 1205Lu cells (Figure 1D, Supp. Figure 2D). Overall, these data show that raptinal induces features of pyroptosis in human and mouse BRAF mutant melanoma cells.
Raptinal delays melanoma growth in an in vivo mouse model
To test the impact of raptinal on tumor growth in vivo, YUMM1.7 cells were implanted intradermally into C57BL/6J mice. Once tumors reached a volume of approximately 100 mm3, mice were injected for four consecutive days with either vehicle control or raptinal via intraperitoneal injection. When compared to the control group, raptinal delayed tumor growth (Figure 2A) and significantly increased tumor doubling time (Figure 2B). Consistent with decreased tumor growth, raptinal treatment significantly improved mouse survival (Figure 2C). Raptinal treatment did cause a decrease in mouse weight with one animal losing more than 20% of its original weight; however, treated animals returned to their baseline weights by day 10 (Figure 2D). These data show that raptinal treatment delays tumor progression in an in vivo melanoma model.
Figure 2: Raptinal delays tumor growth in vivo.
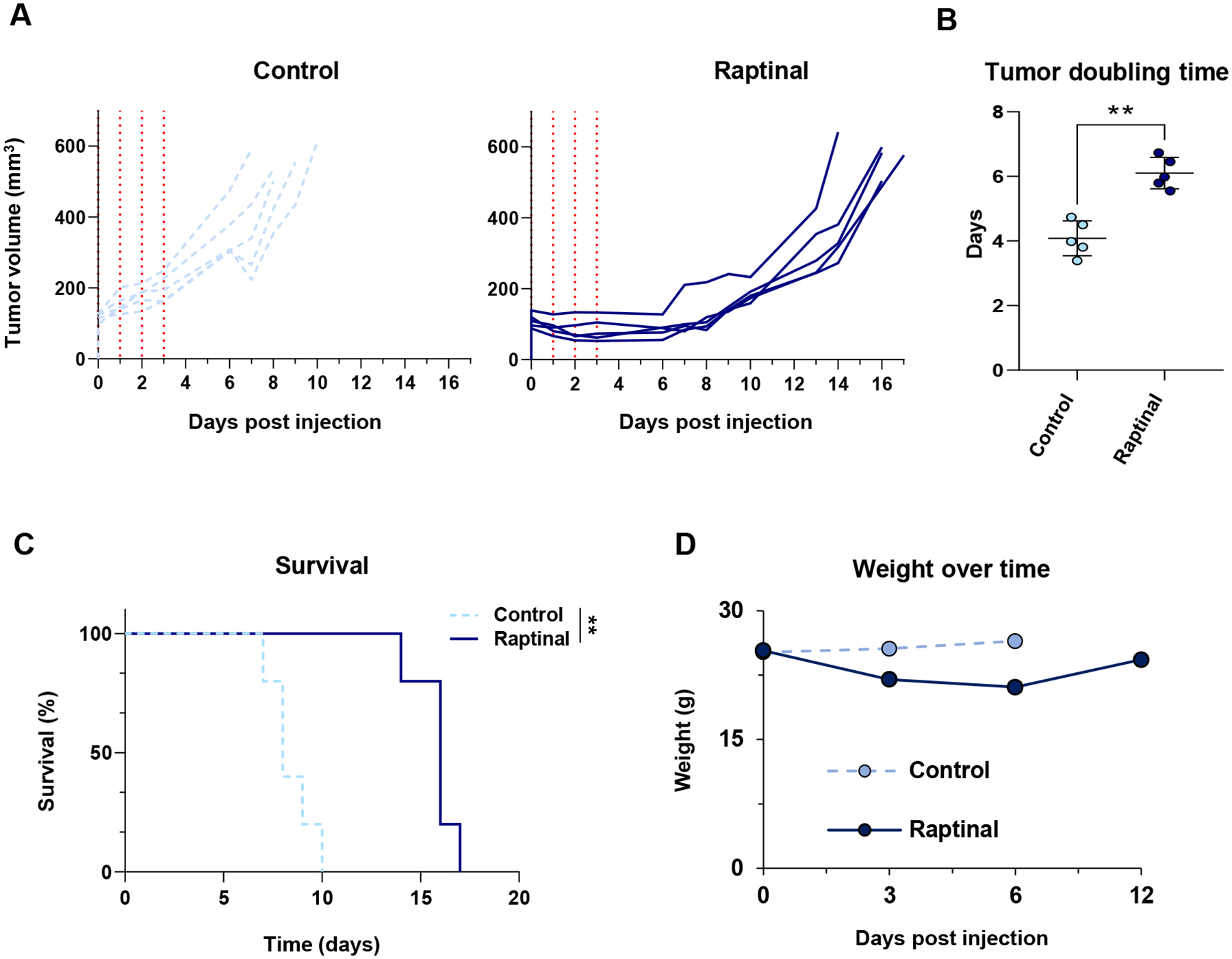
YUMM1.7 cells were implanted intradermally into C57BL/6J animals and allowed to grow to approximately 100 mm3. (A) Tracings of each tumor treated for four days with vehicle control (n=5) (light blue) or raptinal (n=5) (dark blue) are shown to the endpoint of 500 mm3. The vertical red dotted lines correspond to the days in which mice received injections. (B) Tumor growth was modeled as a function of time to assess average tumor doubling time from day 0 to the 500 mm3 endpoint. Each individual data point represents one animal used with the average and standard deviation displayed. Significance was assessed using the Mann-Whitney test, **p<0.01. (C) Kaplan-Meier survival curves comparing vehicle control to the raptinal group. A log rank (Mantel-Cox) test was used to determine significance, **p<0.01. (D) Mouse weights over time from the vehicle control and raptinal treatment arms.
Raptinal-induced pyroptosis is caspase-dependent in melanoma cells
Since cleavage of GSDME has been shown to be mediated by caspase-3 (11), we tested caspase dependence of raptinal-induced effects. In time course experiments and Western blot analyses, raptinal treatment induced the cleavage of multiple caspases including caspase-3, -7 and -8 in human and mouse melanoma cells (Figure 3A, Supp. Figure 3A). Similar effects were observed in caspase-3/7 fluorescence-based substrate assays (Supp. Figure 4A). We additionally observed the cleavage of GSDME followed by an accumulation of cytokines and LDH in cell supernatant (Figures 3A and 3B, Supp. Figures 3A and 3B). To determine the extent to which pyroptotic features induced by raptinal were caspase-dependent, cells were pre-treated with one of two independent caspase inhibitors, Z-VAD (a pan caspase inhibitor) and Z-DEVD (a caspase-3 inhibitor) (Supp. Figure 4B). Pre-treatment with Z-VAD effectively reversed raptinal-induced GSDME cleavage and the accumulation of cytokines and LDH in cell supernatant (Figures 3C and 3D, Supp. Figures 3C and 3D). By contrast, pre-treatment with Z-DEVD only partially reversed raptinal-induced pyroptotic features. These results show that raptinal induces pyroptosis by a mechanism likely involving multiple caspases.
Figure 3: Raptinal-induced pyroptosis is caspase-dependent in melanoma cells.
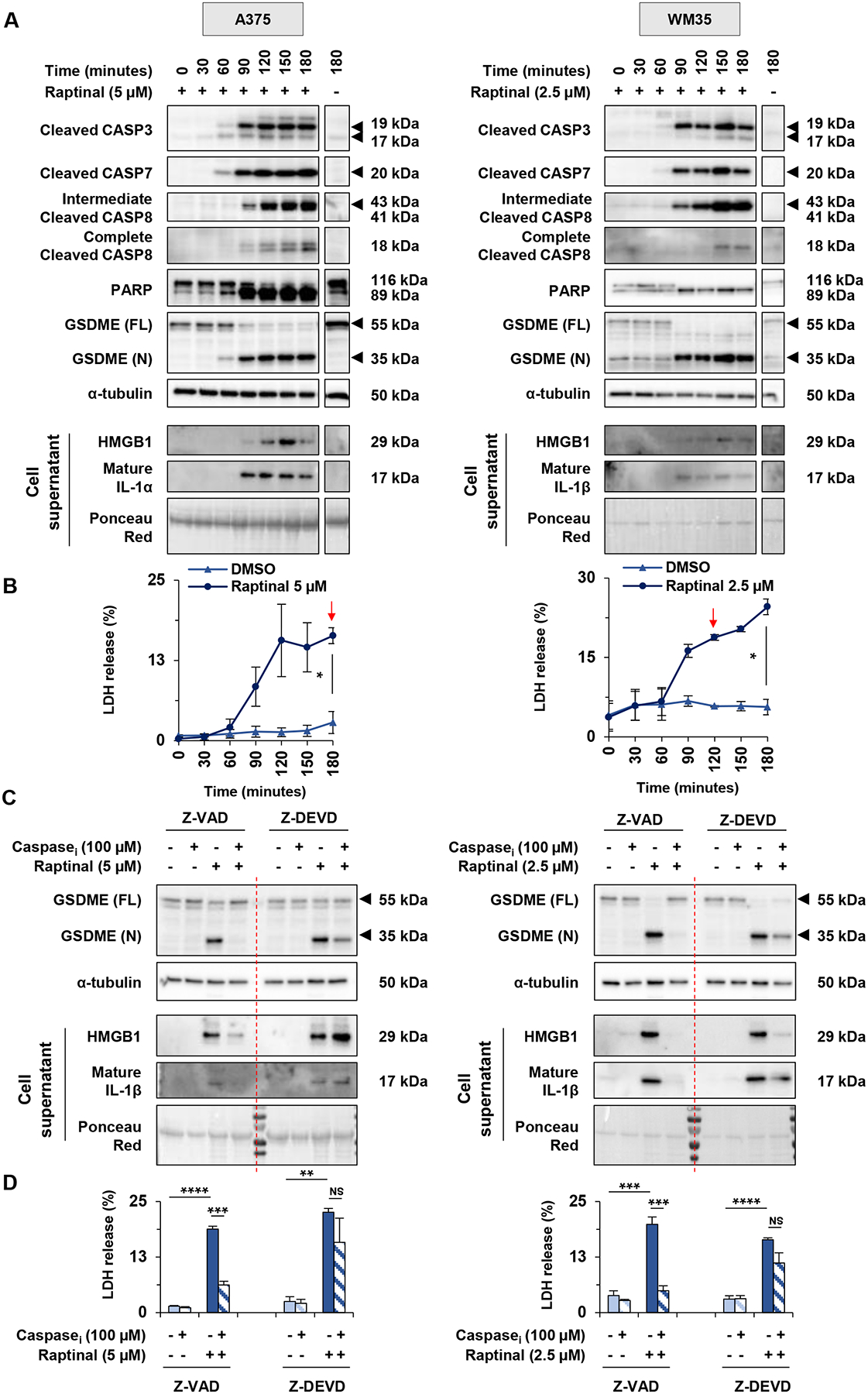
(A-B) A375 and WM35 were respectively treated with 5 μM and 2.5 μM of raptinal. Cell lysates and supernatants were harvested every 30 minutes for 3 hours. (A) Levels of cleaved caspase-3, cleaved caspase-7, cleaved caspase-8, PARP and GSDME in lysates, or HMGB1, IL-1α or IL-1β in supernatants. α-tubulin or ponceau red were used for lysate or supernatant loading controls, respectively. Cells treated with DMSO for 180 minutes were used as negative control (last lane). Western Blots shown are representative of three independent experiments. (B) Percentage of LDH release averaged from two independent experiments. Error bars are SEM. Significance was assessed by Student t test, *p<0.05. Red arrows indicate the time when the treated cells released significantly more LDH than untreated cells. (C-D) A375 and WM35 cells were pre-treated for 3 hours with Z-VAD-FMK, Z-DEVD-FMK or DMSO at 100 μM. After this pre-treatment, A375 were treated with 5 μM and WM35 with 2.5 μM of raptinal or DMSO for 3 hours. (C) Levels of GSDME in lysates, or HMGB1 and IL-1β in supernatants. Western blots shown are representative of three independent experiments. (D) Percentage of LDH release averaged from three independent experiments. Error bars are SEM. Significance was assessed by Student t test, NS: non-significant, **p<0.01, ***p<0.001, ****p<0.0001.
Raptinal-induced pyroptosis is GSDME-dependent in melanoma cells
Since multiple members exist within the GSDM family, we tested whether raptinal-induced pyroptosis is GSDME-dependent. We analyzed the ability of raptinal to induce pyroptosis in GSDME wild-type vs knockout cells. Analysis of cell supernatants showed reduced accumulation of HMGB1, IL-1α and IL-1β in GSDME knockout cells (Figure 4A, Supp. Figure 5A). Similarly, GSDME knockout cells exhibited significantly lower levels of LDH in the conditioned medium following raptinal treatment compared to control wild-type cells (Figure 4B). Cell membrane swelling was decreased in GSDME knockout cells treated with raptinal compared to wild-type cells (Figure 4C). Nonetheless, upstream cleavage of caspases and PARP remained detectable (Supp. Figure 5B). These data suggest that pyroptotic features induced by raptinal are, in part, GSDME-dependent in melanoma.
Figure 4: Raptinal-induced pyroptosis is GSDME-dependent in mouse melanoma cells.
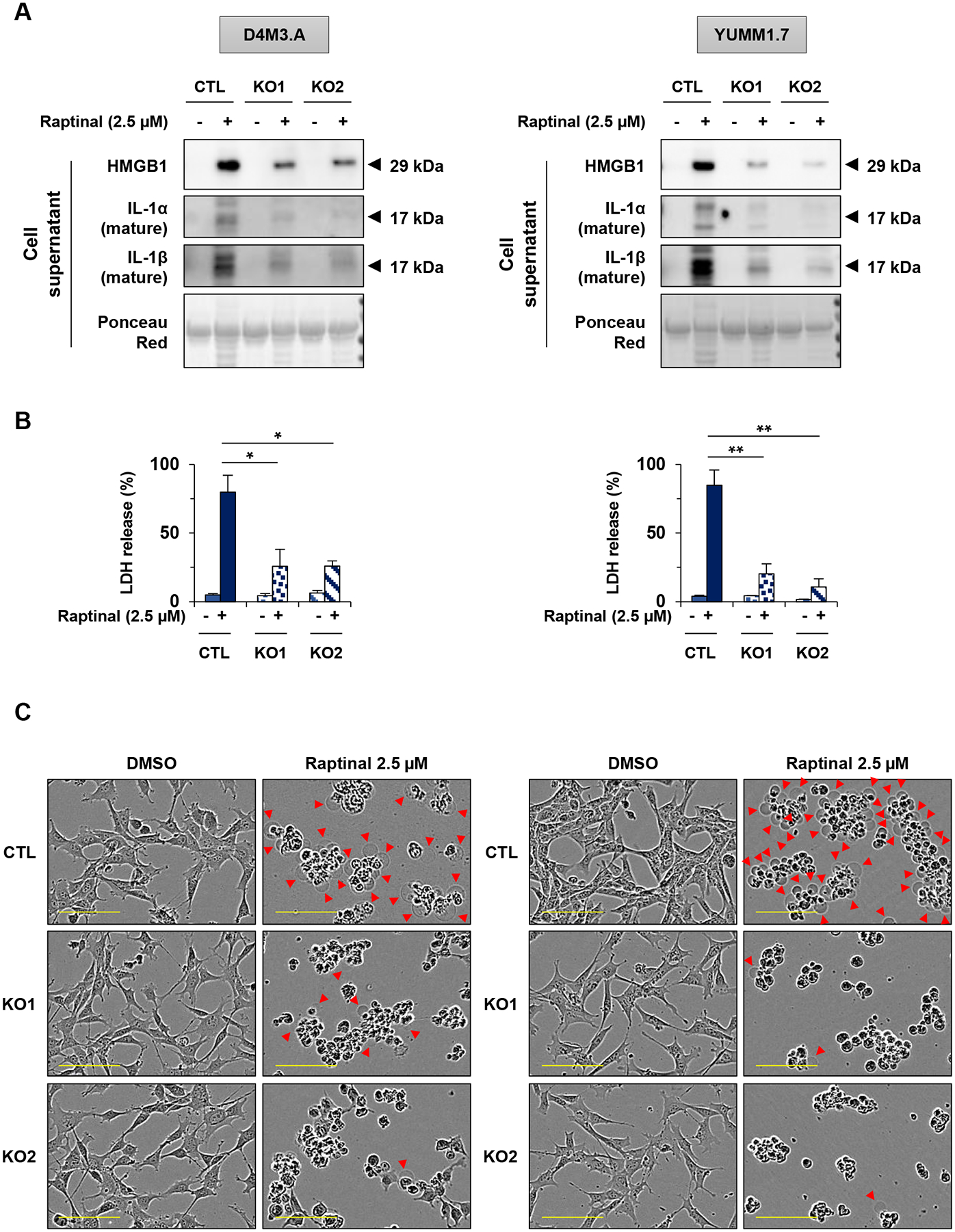
(A-C) D4M3.A and YUMM1.7 mouse melanoma cells were treated with 2.5 μM of raptinal for 3 hours (A) Levels of secreted HMGB1, IL-1α and IL-1β in supernatants from wild-type (noted CTL) and two clones of D4M3.A or YUMM1.7 GSDME knockout cells (noted KO1 and KO2). Ponceau red as protein loading. Western Blots shown are representative of three independent experiments. (B) Percentage of LDH release averaged from three independent experiments. Error bars are SEM. Significance was assessed by Student’s t test, *p<0.05, **p<0.01. (C) Cell morphology visualized with the IncuCyte S3 system at x10 objective. Plasma membrane bubble-like protrusions, a characteristic feature of pyroptosis, are indicated by red arrow. Scale bar: 100 μm. Cell morphology images shown are representative of at least three independent experiments.
Raptinal induces pyroptosis in BRAFi plus MEKi-resistant melanoma cells
A major limitation for BRAFi and MEKi targeted therapies in melanoma is acquired resistance in which cells show minimal caspase-3 cleavage following treatment. Resistance may be mediated through ERK1/2 pathway re-activation as well as epigenetic mechanisms (23–25). We tested whether raptinal could bypass resistance by inducing the cleavage of caspase-3 in two human A375-derived BRAFi plus MEKi resistant cell lines and in two mouse YUMM1.7-derived BRAFi plus MEKi resistant cells lines (denoted as CRT lines). As expected in parental A375 and YUMM1.7 cell lines, both BRAFi plus MEKi and raptinal were able to induce the cleavage of GSDME, caspase-3 and PARP (Figure 5A). By contrast, raptinal but not BRAFi plus MEKi was able to induce these events in both human and mouse resistant cells lines. Caspases-9, -7 and -8 were cleaved following BRAFi plus MEKi-treatment in parental cells but not in CRT cells, whereas raptinal treatment induced the cleavage of all these caspases in both parental and CRT cells (Supp. Figure 6A). Analysis of supernatants demonstrated that both BRAFi plus MEKi treatment and raptinal treatment led to the release of LDH and the accumulation of HMGB1, IL-1α or IL-1β in parental cells; however, only raptinal elicited these effects in CRTs (Figures 5A and 5B). Consistent with these results, we observed cell membrane swelling only in CRT cells treated with raptinal but not with BRAFi plus MEKi (Figure 5C). In addition, raptinal treatment but not BRAFi plus MEKi treatment increased SYTOX green fluorescence in YUMM1.7 CRTs cells (Supp. Figure 6B). Together, these results show that raptinal induces pyroptosis in targeted therapy-resistant melanoma cells in vitro.
Figure 5: Raptinal induces pyroptosis in BRAFi and MEKi resistant melanoma cells.
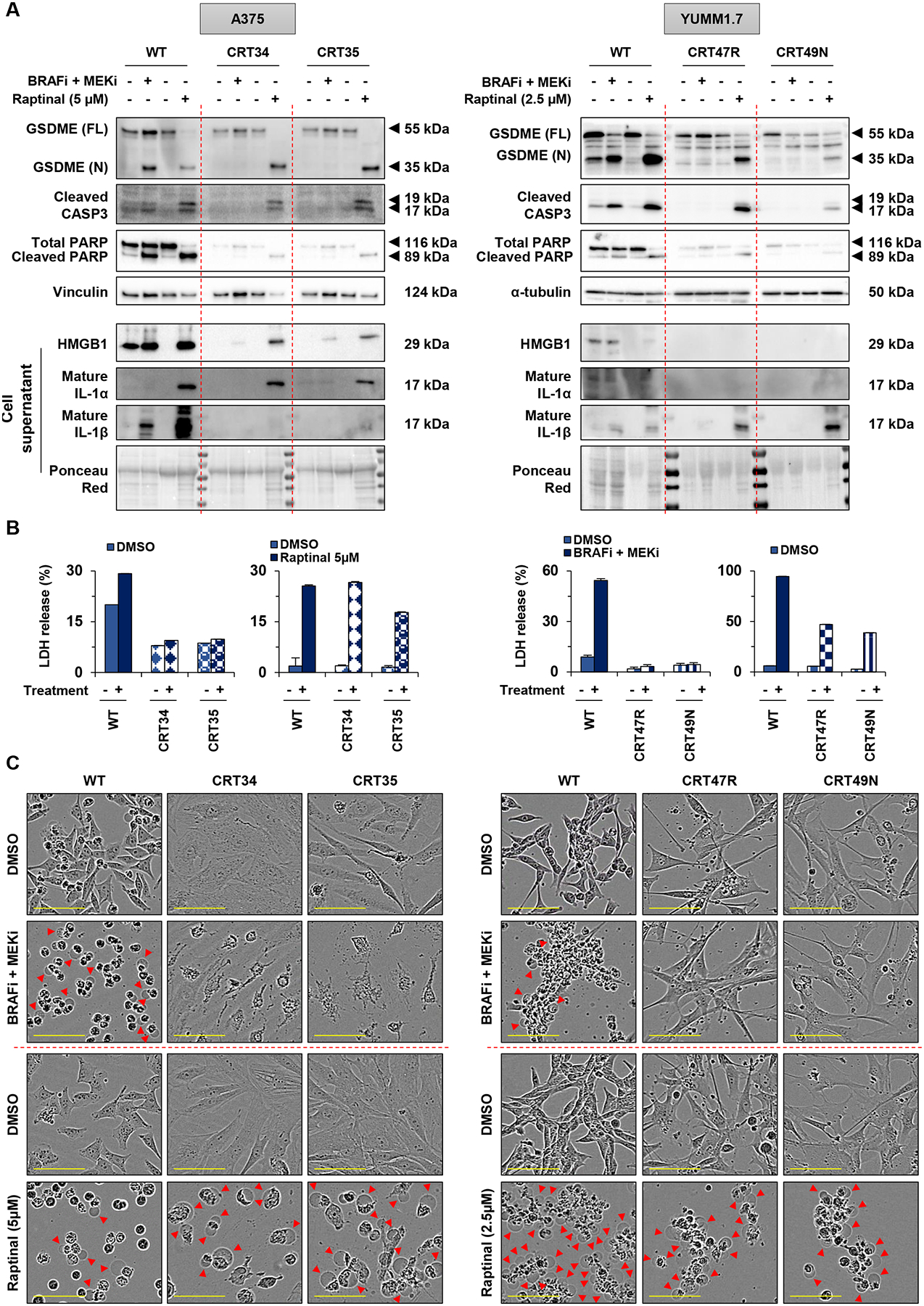
(A-C) A375 WT and CRT cells (CRT34 and CRT35) were treated with PLX4720 (1 μmol/L) and PD0325901 (35 nmol/L) for 48 hours and with raptinal 5 μM for 3 hours. YUMM1.7 WT and CRT cells (CRT47R and CRT49N) were treated with PLX4720 (1 μmol/L) and PD0325901 (35 nmol/L) for 24 hours and with raptinal 2.5 μM for 3 hours. (A) Levels of GSDME, cleaved caspase-3 and PARP in lysates, or HMGB1, IL-1α and IL-1β in supernatants. Vinculin, α-tubulin or ponceau red were utilized for loading controls. Western Blots shown are representative of three independent experiments. (B) Percentage of LDH release representative of two independent experiments. Three replicate wells were performed for each experimental condition. Error bars are SEM. (C) Cell morphology visualized with the IncuCyte S3 system at x10 objective. Plasma membrane bubble-like protrusions, a characteristic feature of pyroptosis, are indicated by red arrow. Scale bar: 100 μm. Cell morphology shown are representative of at least two independent experiments.
Raptinal delays growth of BRAFi plus MEKi-resistant melanoma cells in vivo
To determine if raptinal effects the growth of the CRTs in vivo, we implanted CRT47R cells subcutaneously into C57BL/6J mice. Once tumors reached a volume of approximately 100 mm3, mice were administered BRAFi plus MEKi diet until tumor volume reached an endpoint of 500mm3. Animals were also injected with either vehicle control or raptinal via intraperitoneal injection for four consecutive days at the start of BRAFi plus MEKi treatment. Tumor growth was significantly delayed in mice treated with raptinal compared with vehicle treated mice (Figure 6A) with an increase in both tumor doubling time (Figure 6B) and survival (Figure 6C). These data show that raptinal induction of pyroptosis reduces growth of tumors that become resistant to BRAF and MEK inhibitors in preclinical models.
Figure 6: Raptinal delays growth of BRAFi and MEKi resistant tumor cells in vivo.
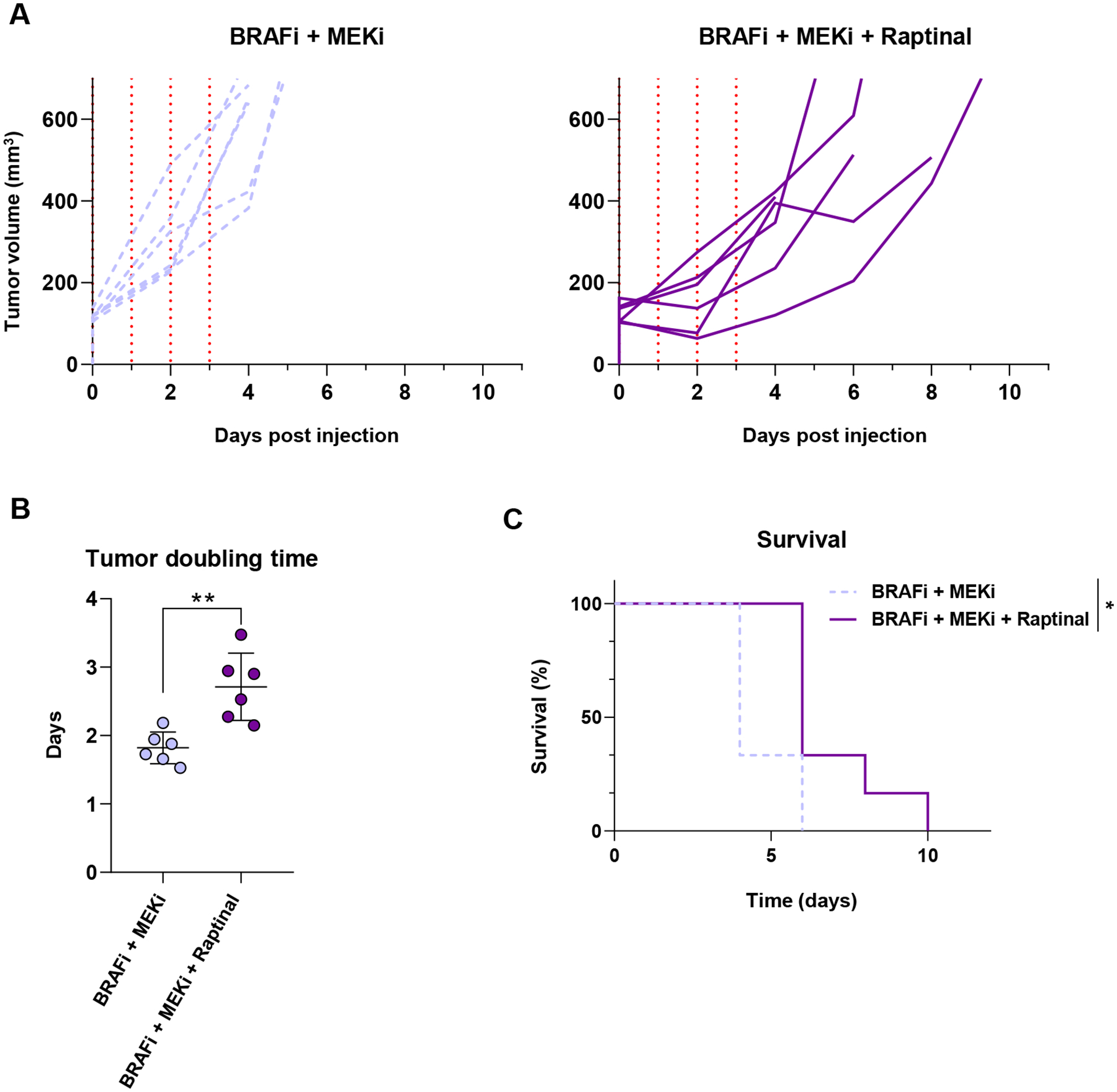
CRT47R cells were implanted intradermally into C57BL/6J animals and allowed to grow to approximately 100 mm3. (A) Tumor growth after treatment for four days with either vehicle (n=5) (light purple) or raptinal (n=5) (dark purple) are shown to the endpoint of 500 mm3. The vertical red dotted lines correspond to the days in which mice received injections. (B) Tumor growth was modeled as a function of time to assess average tumor doubling time from day 0 to the 500 mm3 endpoint. Each individual data point represents one animal used with the average and standard deviation displayed. Significance was assessed using the Mann-Whitney test, **p<0.01. (C) Kaplan-Meier survival curves comparing vehicle control to the raptinal group. A log rank (Mantel-Cox) test was used to determine significance, **p<0.01.
Discussion
Raptinal is a cell permeable compound capable of rapidly inducing caspase-3 cleavage. Based on the knowledge that caspase-3 is able to cause GSDME cleavage (11), we explored effects of raptinal on pyroptosis. We utilized BRAF mutant melanoma models; however, the induction of inflammatory forms of cell death may have important tumor intrinsic effects that change based on the driver mutation of the tumor or the tumor model used. Although raptinal was originally identified as an apoptosis-inducing agent (21), we show that raptinal treatment induces pyroptosis in cutaneous melanoma cells. These effects are mediated by GSDME and are dependent on caspase activity. Importantly, raptinal-mediated effects are evident in BRAFi and MEKi-resistant cell lines, and raptinal delays tumor progression in both BRAFi and MEKi-sensitive and -resistant in vivo melanoma models.
We show that raptinal treatment induces pyroptosis in cutaneous melanoma cells, as illustrated by GSDME cleavage and the release of HMGB1, IL-1α and IL-1β. The effects were dramatically reduced, but not eliminated completely, in GSDME knockout cells. It remains possible that raptinal-induced pyroptosis may be partially dependent on additional GSDM family members. It is known that the release of IL-1β can be mediated by GSDME in either sublytic and lytic phases when GSDMD expression is low/absent (26). Raptinal acts either via regulation of caspase-9 and downstream caspases and/or through cytochrome C release in a BAX/BAK/BOK-independent manner (27). Consistent with the broad effects of raptinal, we observed cleavage of caspases involved in both intrinsic and extrinsic apoptotic pathways. Furthermore, the cleavage of GSDME was prevented with pan-caspase inhibitors. Investigating the effects of raptinal in models with low GSDME and/or high expression of other GSDM family members will be key in determining the potential actions of raptinal in additional tumor types.
In vivo, raptinal promoted significant tumor growth delay in YUMM1.7 and CRT47R tumors. A limitation of our work is that the raptinal-treated tumors eventually grew out, which indicates the persistence of tolerant tumor cells. Thus, treatment strategies require further optimization. Raptinal also showed toxicity when injected for four consecutive days via intraperitoneal injection as assessed by the body weight of the animals. Future studies are needed to optimize dose and timing of raptinal as well as route of injection to increase efficacy and reduce systemic toxicity.
The precise effects of tumor cell pyroptosis on the immune microenvironment are unknown; however, BRAFi plus MEKi induced pyroptosis has been shown to increase CD8+ T cell activation in a melanoma model (20). YUMM1.7 tumors are poorly immunogenic and raptinal may not be sufficient to recruit immune cells to the tumor. A goal of our future work will be to combine raptinal with drugs like imiquimod that promote immune cell infiltration into the tumor (28–30) to induce a more profound anti-tumor response. Other experiments will also assess the efficacy of raptinal in high mutational burden models to determine if raptinal has better efficacy in tumors that have higher basal levels of immune infiltration (31,32).
In summary, we show that raptinal induces pyroptosis in both treatment-naïve and resistant melanoma cell lines. In the absence of GSDME, the effects of raptinal were associated with diminished release of inflammatory DAMPs and cytokines. Intriguingly, the screen used to identify raptinal as an apoptosis inducing agent was performed in a human leukemia cell line, and acute myeloid leukemia has the lowest expression of GSDME among the 32 cancer types profiled in TCGA. We speculate that in highly expressing tumors, GSDME may act as a switch between raptinal-induced apoptosis and pyroptosis (33). Thus, while raptinal may have the ability to promote cell death in other cancer models, the mode of cell death induced by raptinal may be cancer specific and depend on the expression level and activation status of pyroptosis mediators. Therefore, in addition to promoting cell death, raptinal may activate an immune response via the release of inflammatory cytokines and DAMPs in cancers with high expression of GSDME, such as melanoma. Overall, raptinal is a promising drug that warrants future study into its ability to induce inflammatory cell death and synergize with immunotherapy.
Supplementary Material
Implications:
Raptinal can rapidly induce pyroptosis in naïve and BRAFi plus MEKi-resistant melanoma, which may be beneficial for patients who have developed acquired resistance to targeted therapies.
Acknowledgments
This work is supported by grants from National Institutes of Health: CA182635 to A.E. Aplin; CA256945 to A.E. Aplin and E.S. Alnemri, and AR074564 to E.S. Alnemri. N. Wilski was supported by NCI T32 training grant, CA236736. The Sidney Kimmel Cancer Center Flow Cytometry, Translational Pathology and Meta-omics core facilities are supported by National Cancer Institute (NCI) Support Grant (P30 CA056036). We thank Dr. Scott Varney for valuable feedback on this manuscript.
Footnotes
Conflict of interest: A.E. Aplin reports ownership interest in patent number 9880150. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Kim EH, Wong S-W, Martinez J. Programmed Necrosis and Disease:We interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 2019;26:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Current Opinion in Cell Biology. 2003;15:725–31. [DOI] [PubMed] [Google Scholar]
- 3.Cullen SP, Martin SJ. Caspase activation pathways: some recent progress. Cell Death Differ. Nature Publishing Group; 2009;16:935–8. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. Nature Publishing Group; 2008;27:6194–206. [DOI] [PubMed] [Google Scholar]
- 6.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. Nature Publishing Group; 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- 10.Rosenbaum SR, Wilski NA, Aplin AE. Fueling the fire: Inflammatory forms of cell death and implications for cancer immunotherapy. Cancer Discov. 2021;11:266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. Nature Publishing Group; 2017;547:99–103. [DOI] [PubMed] [Google Scholar]
- 14.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012;367:1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N Engl J Med. Massachusetts Medical Society; 2012;367:107–14. [DOI] [PubMed] [Google Scholar]
- 16.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF Inhibitors Induce Marked T-cell Infiltration into Human Metastatic Melanoma. Clinical Cancer Research. 2012;18:1386–94. [DOI] [PubMed] [Google Scholar]
- 17.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA, et al. BRAF Inhibition Increases Tumor Infiltration by T cells and Enhances the Anti-tumor Activity of Adoptive Immunotherapy in Mice. Clin Cancer Res. 2013;19:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10:254–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palchaudhuri R, Lambrecht MJ, Botham RC, Partlow KC, Van Ham TJ, Putt KS, et al. A Small Molecule that Induces Intrinsic Pathway Apoptosis with Unparalleled Speed. Cell Rep. 2015;13:2027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez IM, Purwin TJ, Chervoneva I, Erkes DA, Nguyen MQ, Davies MA, et al. In vivo ERK½ reporter predictively models response and resistance to combined BRAF and MEK inhibitors in melanoma. Mol Cancer Ther. 2019;18:1637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Allen EM, Wagle N, Sucker A, Treacy D, Johannessen C, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. 2014;5:5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou B, Abbott DW. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 2021;35:108998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heimer S, Knoll G, Schulze-Osthoff K, Ehrenschwender M. Raptinal bypasses BAX, BAK, and BOK for mitochondrial outer membrane permeabilization and intrinsic apoptosis. Cell Death Dis. 2019;10:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinmann A, Funk JO, Schuler G, von den Driesch P. Topical imiquimod treatment of a cutaneous melanomametastasis. Journal of the American Academy of Dermatology. Elsevier; 2000;43:555–6. [PubMed] [Google Scholar]
- 29.Suzuki H, Wang B, Shivji GM, Toto P, Amerio P, Tomai MA, et al. Imiquimod, a topical immune response modifier, induces migration of Langerhans cells. J Invest Dermatol. 2000;114:135–41. [DOI] [PubMed] [Google Scholar]
- 30.Fehres CM, Bruijns SCM, van Beelen AJ, Kalay H, Ambrosini M, Hooijberg E, et al. Topical rather than intradermal application of the TLR7 ligand imiquimod leads to human dermal dendritic cell maturation and CD8+ T-cell cross-priming. European Journal of Immunology. 2014;44:2415–24. [DOI] [PubMed] [Google Scholar]
- 31.Lo JA, Kawakubo M, Juneja VR, Su MY, Erlich TH, LaFleur MW, et al. Epitope spreading toward wild type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci Transl Med. 2021;13:eabd8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenbaum SR, Knecht M, Mollaee M, Zhong Z, Erkes DA, McCue PA, et al. FOXD3 Regulates VISTA Expression in Melanoma. Cell Reports. Elsevier; 2020;30:510–524.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. Nature Publishing Group; 2020;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated for this study are available within the article and its supplementary data files.