

Abstract
Background
Routine use of whole‐cell pertussis (wP) vaccines was suspended in some countries in the 1970s and 1980s because of concerns about adverse effects. Following this action, there was a resurgence of whooping cough. Acellular pertussis (aP) vaccines, containing purified or recombinant Bordetella pertussis (B. pertussis) antigens, were developed in the hope that they would be as effective, but less reactogenic than the whole‐cell vaccines. This is an update of a Cochrane review first published in 1999, and previously updated in 2012. In this update, we included no new studies.
Objectives
To assess the efficacy and safety of acellular pertussis vaccines in children and to compare them with the whole‐cell vaccines.
Search methods
We searched CENTRAL (2013, Issue 12), MEDLINE (1950 to January week 2, 2014), EMBASE (1974 to January 2014), Biosis Previews (2009 to January 2014) and CINAHL (2009 to January 2014).
Selection criteria
We selected double‐blind randomised efficacy and safety trials of aP vaccines in children up to six years old, with active follow‐up of participants and laboratory verification of pertussis cases.
Data collection and analysis
Two review authors independently extracted data and assessed the risk of bias in the studies. Differences in trial design precluded a meta‐analysis of the efficacy data. We pooled the safety data from individual trials using a random‐effects meta‐analysis model.
Main results
We included six efficacy trials with a total of 46,283 participants and 52 safety trials with a total of 136,541 participants. Most of the safety trials did not report the methods for random sequence generation, allocation concealment and blinding, which made it difficult to assess the risk of bias in the studies. The efficacy of multi‐component (≥ three) vaccines varied from 84% to 85% in preventing typical whooping cough (characterised by 21 or more consecutive days of paroxysmal cough with confirmation of B. pertussis infection by culture, appropriate serology or contact with a household member who has culture‐confirmed pertussis), and from 71% to 78% in preventing mild pertussis disease (characterised by seven or more consecutive days of cough with confirmation of B. pertussis infection by culture or appropriate serology). In contrast, the efficacy of one‐ and two‐component vaccines varied from 59% to 78% against typical whooping cough and from 41% to 58% against mild pertussis disease. Multi‐component acellular vaccines are more effective than low‐efficacy whole‐cell vaccines, but may be less effective than the highest‐efficacy whole‐cell vaccines. Most systemic and local adverse events were significantly less common with aP vaccines than with wP vaccines for the primary series as well as for the booster dose.
Authors' conclusions
Multi‐component (≥ three) aP vaccines are effective in preventing whooping cough in children. Multi‐component aP vaccines have higher efficacy than low‐efficacy wP vaccines, but they may be less efficacious than the highest‐efficacy wP vaccines. Acellular vaccines have fewer adverse effects than whole‐cell vaccines for the primary series as well as for booster doses.
Plain language summary
Acellular vaccines for preventing whooping cough (pertussis) in children
Review question We aimed to answer the question of whether acellular pertussis vaccines are as effective as the whole‐cell vaccines at protecting children against whooping cough (pertussis), but with fewer side effects.
Background Whooping cough can be a serious respiratory infection in children and is caused by the bacterium Bordetella pertussis (B. pertussis). Vaccines made from killed whole B. pertussis, known as whole‐cell pertussis vaccines, can cause severe neurologic disorders and minor side effects, such as anorexia, drowsiness, fever, irritability, prolonged crying, vomiting and pain/redness/swelling/hardening at the injection site. This led to a fall in immunisation rates, which resulted in an increase in the number of cases of whooping cough. Acellular pertussis vaccines (containing more purified antigens of B. pertussis) were developed in the hope that they would be as effective but safer than the whole‐cell pertussis vaccines.
Search date We searched for trials published up to January 2014.
Study characteristics We included trials comparing the efficacy and safety of whole‐cell and acellular pertussis vaccines in children up to six years old.
Key results This updated review included six trials with 46,283 participants evaluating the efficacy and 52 trials with 136,541 participants assessing the safety of pertussis vaccines. Duration varied from 12 months to 27 months and from 3 days to 12 months for efficacy trials and safety trials, respectively. The efficacy of acellular vaccines with three or more components varied from 84% to 85% in preventing typical whooping cough (characterised by 21 or more consecutive days of severe coughing attacks with laboratory evidence of B. pertussis infection or contact with a household member who has culture‐confirmed pertussis) and from 71% to 78% in preventing mild pertussis disease (characterised by seven or more consecutive days of cough with laboratory evidence of B. pertussis infection). In contrast, the efficacy vaccines with one and two components varied from 59% to 78% in protecting against typical whooping cough and from 41% to 58% against mild pertussis disease. Most systemic and local side effects were significantly less common with acellular vaccines than with whole‐cell vaccines for the first doses and booster dose. We found that acellular pertussis vaccines with three or more components are more effective than low‐efficacy whole‐cell vaccines, but may be less effective than the highest‐efficacy whole‐cell vaccines. Acellular vaccines have fewer side effects than whole‐cell vaccines.
Implications for practice The implications of the findings of this review for clinical practice may be different in high‐income and low‐income countries. In high‐income countries, death from whooping cough is rare and parental acceptance is a major determinant of immunisation uptake. In these circumstances, the improved side effect profile of acellular vaccines argues in favour of their use, even though they might sacrifice some degree of effectiveness compared to the best whole‐cell vaccines. In low‐income countries, where the risk of pertussis is higher and cases are more likely to be fatal, greater weight needs to be given to vaccine efficacy. If an acellular vaccine has been shown to be less effective than a high‐efficacy whole‐cell vaccine it is intended to replace, the safety advantage of the acellular vaccine may be offset by increased mortality and morbidity due to a significantly higher rate of pertussis. However, most of the whole‐cell vaccines used in low‐income countries have not been adequately studied for efficacy and, therefore, it is not known where on the wide spectrum of whole‐cell vaccine efficacy an individual product lies.
Quality of evidence All included trials were randomised and double‐blind, that is, the participants had an equal chance of receiving either acellular or whole‐cell vaccines and both researchers and participants were unaware of the treatment assignment. However, most of trials did not report details of these methodological techniques. This may cast some uncertainty on the quality of evidence in this review.
Background
Description of the condition
Whooping cough, or pertussis, is a highly contagious disease caused predominantly by the fastidious Gram‐negative coccobacillus, Bordetella pertussis (B. pertussis). The disease can occur at any age but is more severe in infants, with most deaths occurring in this age group (Singh 2006). The disease in this age group is also more easily diagnosed because they present with whooping cough, which is characterised by paroxysmal coughing followed by an audible inspiratory whoop and occasionally vomiting. Infants often have a cough and apnoeic episodes, which can be severe and may require admission to hospital. Although habitually a persistent, relatively benign respiratory illness, pertussis can result in serious consequences, such as pneumonia, seizures, encephalopathy and death, especially among infants (Galanis 2006). Immunised children, adolescents and adults may not exhibit whooping cough. They may be asymptomatic or present with a cough lasting several weeks.
The reported incidence of pertussis should be interpreted cautiously, because the case definitions and surveillance system performance vary markedly between countries. In low‐income countries, case definition is mostly based on clinical confirmation due to limited access to laboratory facilities (Singh 2006). According to the World Health Organization (WHO) there are about 16 million pertussis cases annually worldwide among children, teenagers and adults, 95% of which are in low‐income countries, and about 195,000 children die of the disease (WHO 2010). Before the introduction of the pertussis vaccine in the 1940s, there were approximately 200,000 cases reported annually in the United States. Immunisations reduced disease rates and there were only 1010 reported cases in 1976 (Bamberger 2008). The incidence decreased from 157 per 100,000 population in the early 1940s to less than 1 per 100,000 in 1973 (Cherry 2012). It has been generally believed that pertussis particularly affected children under six years of age, but recent trends show that in countries that have achieved good control of pertussis, there is a change in the epidemiology of pertussis in the older age group. Several factors have been proposed as possible causes for the increasing incidence of pertussis disease, including waning immunity with subsequent atypical disease manifestations (Singh 2006). There is a frequent misconception that protection provided by childhood immunisation is lifelong. However, the protection provided by vaccination tends to reduce over time. Evidence shows that the proportion of susceptible children with infections in countries with good vaccination coverage (70%) can be estimated at 10% by one year, 60% by five years and 100% by 15 years (Singh 2006).
Since the 1980s, there has been a substantial increase in the number of cases reported, especially in high‐income countries. This has occurred in children and adolescents aged 6 to 10 years, after childhood immunisations were completed (Bamberger 2008; Singh 2006). In the USA, for example, there were 25,827 cases reported in 2004 and there has been a 19‐fold increase of pertussis cases in adolescents (Bamberger 2008; CDC 2011). Affected adolescents and adults act as reservoirs of the disease to the vulnerable population of infants, for whom the disease can be life‐threatening (Harnden 2009; WHO 2010).
Description of the intervention
After the isolation of B. pertussis in 1906, the possibility of vaccine development was considered. In 1933, Madsen reported some degree of protection in individuals who received a vaccine composed of suspended organisms in saline (Cherry 1996). Vaccines made from killed whole B. pertussis bacteria (whole‐cell pertussis vaccines ‐ wP) have been available since the 1940s. Today, wP vaccines are manufactured in many countries. Although their basic preparation procedures are similar, the vaccines frequently elicit markedly different immune responses to various B. pertussis antigens (Cherry 1996). The wP vaccines are based on regular cultures of selected B. pertussis strains that are killed, usually by being heating and treated with formalin. The methods used for the production vary between laboratories, therefore wP vaccines are relatively heterogeneous. Most wP vaccines are combined with diphtheria toxoid and tetanus toxoid. This combination has shown an efficacy of 80% and has been effective in reducing the incidence rates markedly in countries with good immunisation coverage (WHO 2010). The immune response to wP vaccines is directed against an array of antigens of whole bacterial cells. Significant differences in the immune responses to various antigens have been observed among the different wP vaccines. Unwanted components such as endotoxin cannot be eliminated during whole‐cell vaccine production, therefore an acceptable level of potency is inevitably associated with a greater incidence of adverse effects.
Concerns about possible relations of wP vaccines with neurological disorders led to the development of acellular pertussis (aP) vaccines in the 1970s and they were widely tested and used in Japan during the 1980s (Sato 1984). aP vaccines consist of recombinant or isolated, purified antigens of B. pertussis. They include antigens extracted by various methods, as well as those produced by genetic recombinant technology. Five antigens have been identified as appropriate vaccine components: pertussis toxin (PT), filamentous haemagglutinin (FHA), pertactin and fimbriae type 2 and 3 (FIM 2 and 3) (Jefferson 2003; Singh 2006). One or more of these components may be included in various combinations to produce the vaccine. Vaccines differ from each other, with regard to the bacterial clone used for primary antigen production, methods of cleansing and detoxification, included adjuvants and the use of preservatives (WHO 2010). The exact contribution of the different aP antigens to protection is not well established.
Vaccination strategies vary by health policies in each country. Over the last several years, many potential immunisation strategies have been proposed to improve pertussis control. Immunogenicity data indicate that a primary series should consist of three doses and that booster doses are necessary at ages two and four to six years (Cherry 1996). Most immunisation schedules consist of five to six intramuscular injections given from the age two months to 16 years (CDC 2009; Rodríguez‐Cobo 2008), but booster doses every 10 years throughout life have been suggested because protection provided by childhood immunisation is only partial and not lifelong (Forsyth 2004; Rodríguez‐Cobo 2008).
In the 1990s, safety concerns prompted a switch from wP to aP vaccines in most high‐income countries. However, wP vaccines remain the choice for the national childhood immunisation programmes in many low‐income countries, as they are considerably less expensive and highly effective against pertussis (Singh 2006; WHO 2010). In children older than six years of age, only aP vaccines should be used for vaccination (WHO 2010).
How the intervention might work
The mechanism of vaccine‐induced protection against B. pertussis is still not well understood. Induction of antibodies to the components of B. pertussis appears to be associated with protection by vaccines (Cherry 1998; Kerr 2000; Storsaeter 1998; Taranger 2000), and it is believed that anti‐B. pertussis IgG antibodies may play a key role in preventing bacterial adherence (Mills 1999). However, no consensus has been reached regarding the protective antigens. A household study nested in the Swedish vaccine efficacy trial (Storsaeter 1998) (SmithKline Beecham DTaP 2 vaccine, Connaught DTaP 5 vaccine, Connaught DTwP vaccine, SBT DT vaccine) found a correlation between clinical protection and levels of anti‐pertactin, anti‐FIM2/3 and anti‐PT antibodies in serum, listed in order of a decreasing degree of correlation. There was no correlation between anti‐FHA antibodies and clinical protection. Similar results were found in the German vaccine efficacy trial (Cherry 1998) (Lederle/Takeda DTaP vaccine, Lederle wP vaccine).
Cell‐mediated immunity has also been proposed as a possible protective mechanism of vaccines (Mills 1999; Plotkin 2008; Tran Minh 1998). Both human and animal studies demonstrate that aP and wP vaccines induce distinct T cell populations (Feunou 2010; Millis 1998; Mills 1999). Th1 responses are important for bacterial clearance following primary infection and in immunity induced with a wP vaccine, whereas Th2 cells play a more critical role in the protective mechanism of the aP vaccine (Mills 1999). It is believed that B. pertussis‐specific T cells (probably IL‐4 and IL‐5 secreting Th0/Th2 cells) are required for the induction of humoral response, whereas Th1 cells function in limiting the course of infection through enhanced bacterial uptake and killing by phagocytic cells (Mills 1999).
Why it is important to do this review
This systematic review was initially conducted by Tinnion 1999 to assess the efficacy and safety of aP vaccines in children and to compare them with wP vaccines. The review has been regularly updated (Zhang 2009; Zhang 2012). This review can provide high‐level evidence regarding the benefits and risks of vaccines against pertussis for health policy makers, as well as for paediatricians and parents.
Objectives
To assess the efficacy and safety of acellular pertussis vaccines in children and to compare them with the whole‐cell vaccines.
The following four questions were answered by the review.
Do acellular vaccines protect against pertussis?
If so, do different vaccines confer different levels of protection?
Do acellular vaccines protect against pertussis to the same degree as the whole‐cell vaccines, which they are intended to replace?
Do acellular vaccines cause fewer side effects than wP vaccines?
Methods
Criteria for considering studies for this review
Types of studies
Double‐blind randomised controlled trials (RCTs) of the efficacy of aP vaccines, with active follow‐up of participants and laboratory verification of pertussis cases.
Double‐blind RCTs of the safety of aP vaccines.
Active follow‐up was required to minimise the potential for bias in the recording of pertussis cases. Passive follow‐up (such as relying on parents to report cases spontaneously, or monitoring laboratory records of pertussis isolates) has been shown to lead to bias in case ascertainment. This occurs because disease in vaccinated individuals tends to be less severe than in unvaccinated ones and therefore less likely to come to the attention of a physician (Taranger 1997). Laboratory verification was required because case definitions of pertussis based on clinical criteria alone have been shown to lack specificity (Blackwelder 1991). There was no requirement for laboratory verification to be performed according to any particular method, because the most appropriate method will vary according to the composition of the vaccine under study.
We did not consider trials which only examined antibody response after immunisation in this review, as no particular antibody level has been found to correlate with the clinical efficacy of pertussis vaccines (Granoff 1997).
Types of participants
Children up to six years of age at time of study entry.
Types of interventions
We considered two types of interventions in the experimental (acellular vaccine) group.
aP vaccine: a vaccine containing purified, detoxified pertussis antigens. This includes antigens extracted from B. pertussis organisms by various purification methods, as well as those produced by genetic recombinant technology.
Diphtheria‐tetanus‐aP (DTaP) vaccine. An aP vaccine which also contains diphtheria and tetanus toxoids.
We included four types of control intervention.
wP vaccine: a vaccine containing killed, whole B. pertussis organisms. (An appropriate control where the acellular vaccine used in the experimental group contains only pertussis antigens).
Diphtheria‐tetanus‐wP vaccine (DTwP): a whole‐cell pertussis vaccine, which also contains diphtheria and tetanus toxoids. (An appropriate control if these antigens are present in the acellular vaccine used in the experimental group).
Placebo: a preparation containing no antigens or organisms. (An appropriate control where the acellular vaccine used in the experimental group contains only pertussis antigens).
DT: diphtheria‐tetanus (DT) toxoid vaccine. (An appropriate control if these antigens are present in the acellular vaccine used in the experimental group).
Types of outcome measures
Primary outcomes
The primary outcome measure was vaccine efficacy. Trials comparing acellular vaccines with a randomised placebo/DT group permitted the determination of absolute vaccine efficacy. This is the conventional parameter used to express effectiveness in vaccine trials and represents the percentage of potential disease cases prevented by the vaccine. Absolute vaccine efficacy is calculated as (1 ‐ RR) x 100%, where risk ratio (RR) equals the risk of disease in the vaccine group divided by the risk of disease in the placebo/DT group. Trials with no placebo/DT control do not permit the estimation of absolute efficacy but do allow an assessment of the comparative efficacy of the various vaccines within each trial (expressed as the RR of disease in the acellular compared to the whole‐cell group).
Estimates of pertussis vaccine efficacy may vary greatly according to the case definition used (Blackwelder 1991), and most studies report efficacy results for a range of case definitions. In this review, we examined two case definitions. The first, 'whooping cough', corresponds to the well‐recognised clinical syndrome of pertussis, characterised by protracted, paroxysmal cough. The second, 'pertussis disease', includes milder cases that do not fit the classical picture but may be important in the spread of infection. The criteria for these two case definitions were as follows.
Whooping cough: 21 or more consecutive days of paroxysmal cough with confirmation of B. pertussis infection by culture, appropriate serology or contact with a household member who has culture‐confirmed pertussis. This case definition is recommended by the WHO for use in pertussis vaccine trials (WHO 1991).
Pertussis disease: seven or more consecutive days of cough with confirmation of B. pertussis infection by culture or appropriate serology.
When studies did not report vaccine efficacy using these exact criteria, we reviewed efficacy for the case definitions that corresponded most closely to those above.
Where possible, we reviewed efficacy endpoints for the population who received all scheduled doses of the randomised vaccine ('per protocol' population) and for the population who received at least one dose of the randomised vaccine.
Secondary outcomes
The safety outcome measures were as follows.
Failure to complete all scheduled doses of the primary immunisation series because of adverse events.
Mortality due to any cause.
Mortality due to infection.
Encephalopathy.
Convulsions.
Hypotonic‐hyporesponsive episodes.
Selected minor adverse events generally considered to be associated with wP vaccines: anorexia, drowsiness, fever, irritability/fretfulness, prolonged crying, vomiting, injection site pain/tenderness, injection site redness and injection site swelling/induration.
Search methods for identification of studies
Electronic searches
The initial search was carried out in March 1997 and updated in March 1998 (see Appendix 1 for details of the search terms). The search was updated again in April 2009. See Appendix 2 for details of search terms.
For this update we searched the Cochrane Central Register of Controlled Trials (CENTRAL) (2013, Issue 12), which contains the Cochrane Acute Respiratory Infections Group's Specialised Register, MEDLINE (April 2009 to January week 2, 2014) and EMBASE (April 2009 to January 2014). We also searched Biosis Previews (2009 to January 2014) and CINAHL (2009 to January 2014) as we aimed to include a broader range of databases in order to identify potential studies.
We used the search strategy in Appendix 3 to search MEDLINE and CENTRAL. We combined the MEDLINE search with the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐ maximising version (2008 revision); Ovid format (Lefebvre 2011). We modified the search strategy to search EMBASE (Appendix 4), Biosis Previews (Appendix 5), and CINAHL (Appendix 6).
Searching other resources
We did not manually search the reference lists of retrieved papers for either initial or updated searches, and we made no systematic attempt to obtain unpublished articles. We did not limit the searches to English language reports because such limitation has been shown to be a potential source of bias (Egger 1997).
Data collection and analysis
Selection of studies
One of the original review authors identified and assessed trials in the first publication of this review (Tinnion 1999), to determine whether they satisfied the predefined inclusion criteria. The review author was not blinded to the trial authors or sources of the trial reports during study selection and data extraction.
Two review authors (LZ, SP) independently assessed the titles and abstracts of all studies identified by the searches to select new trials when updating this review. We obtained the full articles when they appeared to meet the inclusion criteria or there were insufficient data in the title and abstract to make a clear decision for their inclusion. We resolved any disagreement between the review authors about study inclusion by discussion.
Data extraction and management
We gave each study a unique identifier for use in RevMan 2012. When a single study was reported in several publications, we used the lead author of the publication containing the main efficacy or adverse event data in the identifier (for example, Decker 1995).
We extracted data into a database (MS Access 7.0) using pre‐prepared electronic forms, which had been refined after testing on a sample of trials. We then sorted the data for entry into RevMan 2012. Two review authors (LZ, SP) independently extracted data from the new trials included for update using a standardised data extraction form. We resolved any disagreement by discussion.
A number of studies reported the percentage of vaccine recipients with each adverse event, but not the actual number. To permit entry into RevMan 2012, we calculated the number of participants experiencing each event from the reported percentage and the number vaccinated. This practice was required only for the common, minor adverse events. It may have introduced a small rounding error in some instances but would not have materially affected the odds ratio (OR) and overall conclusions for these common events.
We excluded the data from the review if adverse event data for a particular dose were available for less than 80% of those who had received the dose. This was done because excessive loss to follow‐up could lead to a spurious reduction in the reported frequency of adverse events.
Study reports often graded adverse events according to severity. For the purposes of this review, we defined two (overlapping) severity categories. The primary category was the number of patients with any occurrence of the event under consideration. A secondary category was the number of patients with 'moderate to severe' grades of the event.
We applied the following rules during data extraction.
We defined primary series immunisation as the first series of up to three doses administered to children who had not previously been immunised against pertussis. We defined booster immunisation as doses administered at or after the age of 12 months to children who had completed a primary series.
When results for both pain and tenderness were reported, we used the result for pain. When results for both swelling and induration were reported, we used that for swelling.
We defined fever as a temperature of 38 ºC or greater. When a study did not report a result using this cut‐point, we entered the result for the cut‐point closest to 38 ºC but below 39 ºC. We defined moderate to severe fever as a temperature at or above 39 ºC. If a study did not report a result using this cut‐point, we recorded the result for the next highest cut‐point.
We defined moderate to severe redness and swelling/induration as reactions with a diameter of 2 cm or greater. When a study did not report a result using this cut‐point, we recorded the result for the next highest cut‐point.
We recorded data for deaths if the study report explicitly stated the number of deaths (or that there were no deaths), or if the absence of deaths could be confirmed by one or more of the following: (1) the number of withdrawals was stated and all were accounted for by causes other than death; (2) the report stated that there were no withdrawals, or that all participants completed the study; and (3) the report stated that there were no serious reactions and also defined death as a serious reaction. While the last requirement may seem superfluous, this was not the case, as one study stated that "there were no serious reactions to the vaccines" and then went on to report that there had been two convulsions and one death (Trollfors 1995).
We recorded data for encephalopathy, convulsions or hypotonic‐hyporesponsive episodes if: (1) the report explicitly stated the number of these reactions (or that there were none); or (2) the report stated that there were no serious reactions and defined the event under consideration (encephalopathy, convulsions or hypotonic‐hyporesponsive episodes) as a serious reaction.
Assessment of risk of bias in included studies
Two review authors (LZ, SP) independently assessed the risk of bias of each eligible RCT by using The Cochrane Collaboration's tool for assessing risk of bias (Higgins 2011). The tool contains seven domains: random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting and other sources of bias. We rated each domain as having 'low risk of bias', 'high risk of bias' or 'unclear risk of bias'.
Measures of treatment effect
We used risk ratio (RR) and 95% confidence interval (CI), rather than odds ratio (OR) and 95% CI, to estimate the risk of adverse events because the interpretation of the OR is more difficult, and it may produce inflated estimates of risk when the outcomes are frequent, as are minor adverse events.
Dealing with missing data
Selective reporting of outcomes in some included studies may result in missing data. We based the analysis on available data but the potential impact of missing data on the findings of the review is addressed in the Discussion section.
Assessment of heterogeneity
We assessed heterogeneity across studies using the Chi2 test. We took a conservative approach, as the test for heterogeneity has low statistical power (Petitti 1994), whereby heterogeneity was assumed if the P value for the test was less than 0.10. We addressed the possible causes of heterogeneity across studies in the Discussion section.
Assessment of reporting biases
Reporting biases, especially publication bias, may be expected to occur in the majority of systematic reviews. Unfortunately there is no reliable method to detect publication bias. We examined the possibility of selective reporting of outcomes and its potential impact on the findings of the review is addressed in the Discussion section.
Data synthesis
Due to the small number of efficacy studies and differences between them in dose schedule, vaccine characteristics, case definitions and background pertussis incidence, we considered a meta‐analysis of the efficacy data inappropriate. In any event, the adjusted absolute and relative vaccine efficacies in these studies were reported as percentages and RR, respectively. Data in this form cannot be entered into RevMan 2012.
We synthesised safety data using meta‐analysis routines available in RevMan 2012. In the original review, the fixed‐effect model was used for meta‐analysis when the endpoints were homogeneous and the random‐effects model was applied when there was significant heterogeneity across studies. However, this strategy is currently not recommended by The Cochrane Collaboration (Higgins 2011). In this updated review, we used only the Mantel‐Haenszel (random‐effects model) method for meta‐analysis because it is more appropriate than a fixed‐effect model and gives more conservative estimates with wider CIs when there is significant heterogeneity across studies. Otherwise, the two models generate similar results.
Subgroup analysis and investigation of heterogeneity
We analysed adverse events for the 'any' severity category. A few studies only contributed data in the 'moderate to severe' category for some reactions. We did not include these data in the analysis because we considered it inappropriate to combine them with the 'any' severity data.
We performed an analysis within the following Review Manager‐defined hierarchy of comparisons, outcomes and subcategories.
Comparisons: the two comparisons were (1) safety ‐ acellular versus whole‐cell vaccines; and (2) safety ‐ acellular vaccines versus placebo/DT.
Outcomes: each safety endpoint (as described in the Outcomes section of this review) constituted a separate outcome. We considered it inappropriate to combine data across event types (for example, 'children with any adverse event') because different studies examined different event subsets. Furthermore, data for different events were obtained from the same cohort of participants within the one study. Such data cannot be legitimately combined within Review Manager 5 (RevMan 2012), because the observations are not independent. Finally, the use of combined endpoints could lead to an increase in one type of adverse event (for example, convulsions) being offset and therefore hidden by a decrease in another type (for example, fever).
Subcategories: some events occurred only once in any individual, either by virtue of their nature (failure to complete the primary series and death), or because they consistently led to withdrawal from further vaccine doses (encephalopathy, convulsions and hypotonic‐hyporesponsive episodes). We analysed these events in terms of the number of participants experiencing the event out of the total receiving at least one dose of the vaccine. We analysed deaths and encephalopathy only for the primary series because follow‐up after single booster doses was generally too short to provide useful data for these potentially delayed events. We analysed convulsions and hypotonic‐hyporesponsive episodes separately for the primary series and boosters to determine if risk varied across these subcategories.
Minor adverse events often occurred more than once in any individual (after successive doses of vaccine). Data for these events were almost always reported as the number of children experiencing the event after each dose of the vaccine. Events occurring in the same individual are not independent, therefore they could not then be combined across doses. We analysed such data, therefore, using separate subcategories for each dose of the primary series and two subcategories of booster dose (acellular boosters in children who had previously been vaccinated with whole‐cell vaccines, and acellular boosters in children who had previously been vaccinated with acellular vaccines). We selected the booster subcategories on the basis of a preliminary reading of the literature, which suggested that when acellular vaccines are given after acellular priming, adverse events may be more common than when they are given after whole‐cell priming.
We could not incorporate data in the meta‐analysis for minor adverse events from studies that did not report results separately for each dose (for example, Greco 1996). We calculated a summary estimate of effect and 95% CI for each subcategory. We did not calculate an overall summary estimate for each type of adverse event for reasons given above.
Sensitivity analysis
We planned three sensitivity analyses, and conducted two. The first compared random‐ and fixed‐effect analyses to determine whether the conclusions were sensitive to model selection.
The second pre‐planned sensitivity analysis dropped any study which contributed more than 50% of the total weight to an endpoint. This was done in order to assess whether the summary estimate of effect was sensitive to the inclusion of individual, heavily weighted studies. The third pre‐planned sensitivity analysis would have dropped any study with inadequate allocation concealment, but we did not identify such studies from those included in the meta‐analyses.
Results
Description of studies
Results of the search
During the initial search, the MEDLINE database yielded 13,509 citations; of these, 156 were reports of possibly eligible trials. The CENTRAL database yielded 129 citations and we identified 61 as potentially eligible trials. The updated search (March 1998) yielded 172 citations; of these, 57 were reports of possibly eligible trials. From an examination of the retrieved publications, we identified 52 eligible studies, of which we included 45 in the primary review and excluded the remaining seven studies for various reasons (see Characteristics of included studies and Characteristics of excluded studies tables).
The new comprehensive search in May 2008 yielded a total of 1197 citations (451 for MEDLINE, 403 for EMBASE and 343 for CENTRAL), plus an additional 31 search results when the searches were updated in April 2009. We identified 10 additional studies, of which we included seven and excluded three studies in the updated review (see Characteristics of included studies and Characteristics of excluded studies tables).
For the 2012 update, we identified 75 search results when the search was run in May 2011 and a further 29 search results when the search was updated in January 2012; although we did not identify any new eligible studies. For this new 2014 update, we identified 54 citations when the search was run in January 2014; again we did not identify any new eligible trials.
Included studies
Studies included in the review of efficacy
We included six eligible RCTs of acellular vaccine efficacy in the review (AHGSPV 1988; Greco 1996; Gustafsson 1996; PVSG 1998; Simondon 1997; Trollfors 1995), all of which were identified by the initial search. The trial of Afari 1996 compared two formulations of an acellular vaccine with a whole‐cell vaccine but was ineligible because pertussis cases were not confirmed by any laboratory procedure. In any event, vaccine efficacy in this trial could not be calculated because no pertussis cases were reported in either the vaccine or control groups. Blennow 1988 reported the comparative efficacy of two acellular vaccines after randomised administration but did not employ active follow‐up. In addition, it inappropriately combined case data from three different vaccination schedules across two separate studies (Blennow 1988; Hedenskog 1987).
AHGSPV 1997 did not meet the active follow‐up criterion specified in this review but was a very large and well publicised study which merits some consideration. It was conducted in Sweden and involved the randomised immunisation of 82,892 infants using a British whole‐cell vaccine (Evans Medical, ex Wellcome), or one of three acellular vaccines: a two‐component (SKB), a three‐component (Chiron‐Biocine) or a four‐component (Connaught). Vaccines were given at three, five and 12 months to 72,698 infants and at two, four and six months to the remaining 10,194. Efficacy data were reported only for the three, five and 12‐month schedule. Follow‐up for the whole‐cell, three‐ and four‐component vaccines lasted for a mean of 22 months after the third dose. Part way through the trial, the two‐component acellular vaccine was shown (in Gustafsson 1996) to have an unacceptably low efficacy, so the blind was broken for infants who had received this vaccine; they were offered boosting with a three‐component vaccine, and efficacy data were only available for this group up to the time of the third dose.
Cases of whooping cough in AHGSPV 1997 were detected by surveillance of Swedish laboratories for reports of positive B. pertussis culture. Computer matching was performed on each report to determine if it originated from a study participant, and a nurse then contacted the family for clinical follow‐up. Based on the incidence of whooping cough with the two‐ and five‐component vaccines in Gustafsson 1996, this passive follow‐up method appeared to miss about 90% of cases amongst the study participants. Despite this, the RR for culture‐confirmed whooping cough (21 or more days of paroxysmal cough) in the five‐component versus the two‐component group was the same in both trials (0.25). This suggests that although the passive follow‐up in AHGSPV 1997 had a low sensitivity, it was not associated with differential case ascertainment, at least for this case definition.
Studies included in the review of safety
Fifty‐two included studies contributed safety data for one or more endpoints; of these we identified 45 in the initial search and seven in the updated search. The salient features of these trials, including details of the specific endpoints to which each trial contributed data, are summarised in the Characteristics of included studies table.
Excluded studies
We excluded 10 randomised trials from the review. The reasons for exclusion and study characteristics are summarised in the Characteristics of excluded studies table.
Risk of bias in included studies
The overall risk of bias is presented graphically in Figure 1 and summarised in Figure 2.
1.
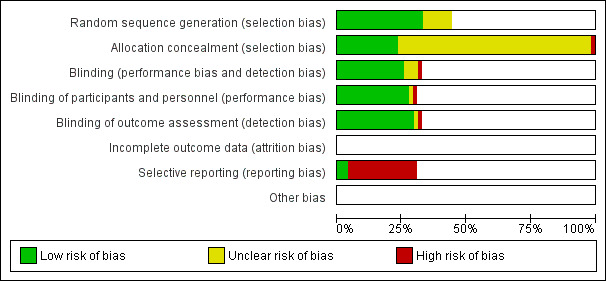
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
2.
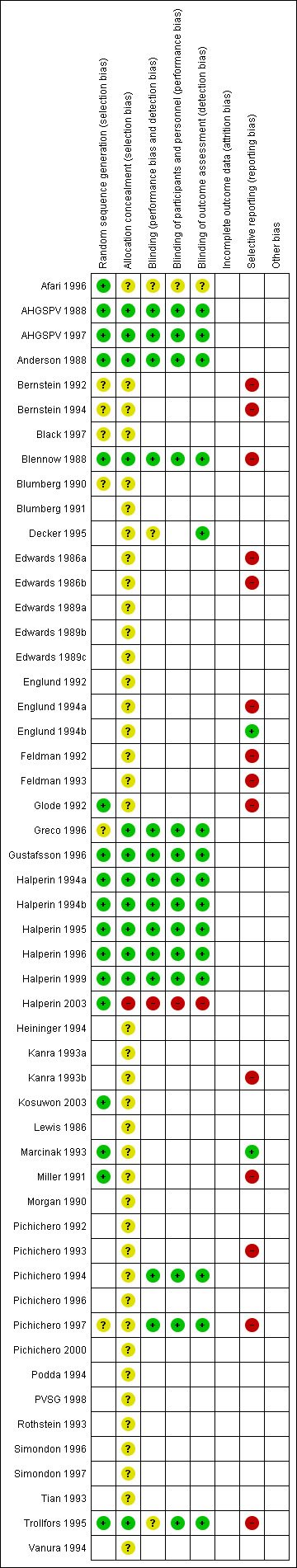
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Most of the safety trials did not describe the methods for random sequence generation and allocation concealment, which made it difficult to assess the risk of selection bias. However, selection bias may nevertheless have affected the assessment of adverse events at other than the first vaccine dose. This is because children were more likely to be withdrawn due to adverse events after whole‐cell than after acellular vaccines. The incidence of adverse events in the whole‐cell group after subsequent vaccine doses would be lower than if these children had remained in the study and this bias would tend to reduce the apparent difference in reactogenicity between the whole‐cell and acellular vaccines. Selection bias may also have occurred in Pichichero 1996, a booster dose study performed as a follow‐on to Pichichero 1993 and Pichichero 1994, in which parents of only 83% of the participants who had enrolled in the primary series studies elected to have their child continue into the booster study.
Blinding
To be eligible for inclusion, a claim of double‐blinding had to be made in the study report. However, most of the safety trials did not provide details about the methods of blinding.
Bias in case ascertainment may have occurred in Gustafsson 1996 and possibly Greco 1996, due to partial unblinding of the whole‐cell vaccine. It is uncertain in which direction such a bias may have operated. On the one hand, whole‐cell vaccines are generally perceived to be effective, so one might expect any bias to favour of the whole‐cell preparations. On the other hand, Europe has a history of withdrawal of wP vaccines due to inadequate efficacy, so observers in these European studies might have been biased against the whole‐cell vaccines. In any event, the codified, active case ascertainment procedures used in the included studies would have minimised the potential for observer bias from any source. Moreover, observer bias alone would seem an unlikely explanation for the very low efficacy of the whole‐cell vaccines in these two studies. Finally, unblinding of the whole‐cell vaccine groups would not have affected the acellular versus placebo/DT comparisons.
Pertussis in immunised children tends to have a milder clinical course than in those who are not immunised. Such milder cases may be selectively excluded, leading to over‐estimation of absolute vaccine efficacy (Fine 1997). Laboratory confirmation of cases did not eliminate this bias, because participants had to exhibit clinical symptoms (generally at least seven days of cough) before laboratory samples were collected. This problem should not occur in direct comparisons of acellular and whole‐cell vaccines.
The partial unblinding in Gustafsson 1996, Halperin 2003 and possibly Greco 1996 might have biased the assessment of adverse events in favour of the acellular preparations. However, the only summary odds ratios (ORs) that were materially altered in the sensitivity analysis, which excluded these studies, were those for drowsiness and vomiting ‐ endpoints which should be interpreted with caution due to other problems.
In all studies, detection bias could have arisen in the assessment of minor adverse event incidence because reactions to whole‐cell vaccines tended to be more severe than those to acellular vaccines (more severe reactions would be more likely to be noticed by the parents). However, the requirement for parents to look for specific events and complete diaries or forms at regular intervals would have reduced the potential for such differential reporting.
Incomplete outcome data
Follow‐up in all but one study was balanced across the vaccine groups and covered over 90% of available participants at each vaccine dose; the exception was Simondon 1996. The rates of withdrawal and loss to follow‐up were high in this study, which was included for safety only. Eighty‐one per cent of the acellular group and 87% of the whole‐cell group contributed data after the first dose. Data for minor adverse events were available for less than 80% of vaccinated individuals after the second and third doses, and so we excluded these doses from the review, in accordance with the pre‐defined data extraction rules. In addition, we conducted a sensitivity analysis in which this study was excluded entirely.
Active case ascertainment was a requirement for inclusion in the efficacy review. All but one of the studies included in the review of safety actively questioned parents at regular intervals regarding prospectively defined adverse events, using standardised forms or diaries; the exception was AHGSPV 1997. This large study, which enrolled over 80,000 infants, did not examine minor adverse events. Data for serious adverse events were collected from participating physicians and child health nurses, by weekly surveillance of admissions to hospitals within the study area (which covered much of Sweden) and by questioning parents at the time of each vaccine dose, and when the child was aged 18 months. It is possible that this type of follow‐up may have led to under‐reporting of serious adverse events. However, while reducing the power of the study to detect a significant difference, there is no reason to suspect that any such under‐reporting would have been differential, or introduced a systematic bias. Nevertheless, we conducted sensitivity analyses, eliminating this study from the endpoints to which it contributed, to determine if the summary ORs were materially affected.
Selective reporting
Some studies reported results for only a subset of the adverse events for which data had been collected. Vomiting was the endpoint most affected, with up to 11 studies collecting data but failing to report it (Bernstein 1992; Bernstein 1994; Blennow 1988; Edwards 1986a; Edwards 1986b; Feldman 1992; Feldman 1993; Halperin 1996; Marcinak 1993; Pichichero 1993; Pichichero 1997). Six studies collected data on drowsiness and did not report it (Edwards 1986a; Edwards 1986b; Kanra 1993a; Kanra 1993b; Pichichero 1993; Pichichero 1997), while data for anorexia were collected but not reported in three studies (Blennow 1988; Pichichero 1993; Trollfors 1995). Qualitative statements within some reports indicated that data for these events had been omitted because they had occurred with similar frequency in each vaccine group. In contrast, those studies which did include extractable data generally reported a significant difference. Accordingly, there is a possibility that inclusion of the missing data could significantly alter the pooled ORs for these endpoints.
Other potential sources of bias
1. Bias introduced by the methods of the review
a) Study selection and data extraction
The review author was not blinded to study authorship or journal of publication, allowing the potential for bias during study selection and data extraction. The use of predefined criteria for study inclusion and rules for data extraction helped to minimise this potential. While desirable, the need for review author blinding during study selection and data extraction is unproven. Berlin and co‐workers have shown that rigorous blinding had neither a clinically nor statistically significant effect on the summary OR in a random sample of five meta‐analyses (Berlin 1997).
b) Publication bias
It is well recognised that studies reporting statistically significant results are more likely to be published than those that do not (Mahid 2008). The review is potentially subject to bias arising from this source because the databases we searched for eligible trials only included published studies. While it would be desirable to identify unpublished studies and include these in the review, this was precluded by resource limitations. Although we may have missed small unpublished studies of vaccine safety in this manner, it seems unlikely that an efficacy or safety trial large enough to materially affect the conclusions of the review would have been suppressed from publication.
Publication bias also occurred within studies and took two forms. The first involved the selective reporting of events as previously mentioned. The second type of within‐study publication bias was the tendency to collect adverse event data at several time points and then report the time point which showed the maximum difference between vaccines. This is not a problem as long as one remains aware that the summary results reflect the maximum observable difference between vaccines and not the difference at a specific time point.
2. Confounding in the included studies
a) Confounding due to the use of erythromycin for post‐exposure prophylaxis of pertussis contacts
Erythromycin prophylaxis would not confound the assessment of vaccine safety. By reducing the number of pertussis cases observed in a study it could widen the CI for vaccine efficacy but as long as its use was not associated with one or other vaccine type, then it should not act as a confounder. Equivalent use would be expected in double‐blind studies but there is some doubt that all the efficacy studies in this review were truly blinded in respect to the whole‐cell vaccines. Gustafsson 1996 and possibly Greco 1996 used identifiable whole‐cell vaccines and in all studies the rate of adverse reactions following whole‐cell vaccines was appreciably higher than after the acellular vaccines or placebo/DT. These factors may have led to partial unblinding of the whole‐cell vaccines during the efficacy follow‐up period, which in turn may have allowed differential use of erythromycin prophylaxis.
Greco 1996 made no statement regarding erythromycin prophylaxis during the study. In Gustafsson 1996, prophylaxis was assumed to occur in accordance with Swedish guidelines, which recommend it only in infants under six months of age. Actual prophylaxis use was not documented in the trial, but if the assumption is valid then it should not have affected the main efficacy follow‐up period (which started after the third vaccine dose, at the age of six months). In another Swedish study, AHGSPV 1988, the first vaccination was scheduled at a minimum age of five months in order to avoid potential problems due to use of prophylaxis during the efficacy follow‐up period (which began at a minimum age of seven months). In any case, this study did not use a whole‐cell vaccine, and blinding of the two acellular vaccines and placebo was confirmed by a questionnaire. In a third Swedish study, Trollfors 1995, prophylaxis use was low (six per cent) and equivalent in the two study groups. Simondon 1997 and PVSG 1998 included no information on this topic. The study vaccines were adequately blinded at administration, but partial unblinding may have occurred during the efficacy assessment due to a higher incidence of adverse events in the whole‐cell arms. In theory this might have provided the opportunity for differential use of prophylaxis but the probability of such bias seems low.
Overall, erythromycin prophylaxis appears unlikely to have significantly confounded the estimation of vaccine efficacy.
b) Confounding due to the use of antipyretic/analgesic medication
The use of antipyretic/analgesic medication would be a potential confounding factor in the assessment of reactogenicity. Truly prophylactic use (i.e. use before a fever or pain appeared) should be non‐differential in a blinded study, but by lowering the incidence of fever and pain in each treatment group, it would reduce the ability to detect a difference between vaccines. Antipyretic/analgesic medication might also be used in a 'reactive' fashion after a low fever or mild pain had occurred, in order to prevent a higher fever or more severe pain. One would expect this type of use to be differential (more common with the more reactogenic vaccines). The result would be to reduce the apparent difference between vaccines in the incidence of severe fever or pain. It would also reduce the apparent difference in the incidence of systemic events that may be associated with a high fever, such as convulsions, irritability, anorexia, vomiting, drowsiness and withdrawal from the study.
No study report specifically stated that prophylactic use of antipyretic/analgesic medications was permitted. A few stated that it was not permitted, or that it was discouraged, while most made no statement. Similarly, only a few studies documented 'reactive' use of antipyretic/analgesic medication. It is worth noting that where such use was recorded, parents were asked to give the drug only when the child's temperature had reached a point higher than that used for defining fever in the analysis of reactogenicity, or if the child was in obvious pain or distress. This would have served to minimise the effect on the assessment of fever and other minor adverse events.
Allowance for undocumented antipyretic/analgesic use would strengthen the conclusion that acellular vaccines are less reactogenic that the whole‐cell vaccines, but weaken the finding of no significant difference between the acellular vaccines and placebo.
Effects of interventions
Primary outcome
Absolute vaccine efficacy ‐ whooping cough
Four of the eligible efficacy trials included a randomised placebo/diphtheria‐tetanus (DT) group and were able to determine the absolute efficacy of acellular vaccines against a case definition identical to, or closely approximating 'whooping cough', as specified in this review. These results are summarised below. 'All': efficacy (with 95% confidence interval (CI), where reported) in participants receiving all scheduled vaccine doses; "≥ 1 immunisation (immunis) dose": efficacy (with 95% CI, where reported) in participants receiving at least one dose of the randomised vaccine. See Characteristics of included studies table for information on the composition of each vaccine.
Study: Trollfors 1995 Dose schedule: 3, 5 and 12 months. Vaccine: Amvax [1] ‐ All: 71% (63 to 78); ≥ 1 immunisation dose: not reported.
Study: AHGSPV 1988 Dose schedule: 5 to 11, and 7 to 13 months. Vaccine: JNIH7 [1] ‐ All: 78% (57 to 88); ≥ 1 immunisation dose: not reported. Vaccine: JNIH6 [2] ‐ All: 78% (58 to 89), ≥ 1 immunisation dose: not reported.
Study: Greco 1996 Dose schedule: two, four and six months. Vaccine: SKB [3] ‐ All: 84% (76 to 89); ≥ 1 immunisation dose: 82% (73 to 87). Vaccine: Chiron‐Biocine [3] ‐ All: 84% (76 to 90); ≥ 1 immunisation dose: 84% (76 to 89). Vaccine: Connaught [W] ‐ All: 36% (14 to 52); ≥ 1 immunisation dose: 34% (13 to 50).
Study: Gustafsson 1996 Dose schedule: two, four and six months. Vaccine: SKB [2] ‐ All: 59% (51 to 66); ≥ immunisation dose: 59% (51 to 66). Vaccine: Connaught [5] ‐ All: 85% (81 to 89); ≥ immunisation dose: 84% (80 to 88). Vaccine: Connaught [W] ‐ All: 48% (37 to 58); ≥ immunisation dose: 48% (37 to 57).
In the trial of PVSG 1998, comparison with a non‐randomised controlled trial (RCT) DT control group gave an estimated absolute efficacy against whooping cough of 84% (95% CI 77% to 89%) for the whole‐cell vaccine and 79% (95% CI 72% to 85%) for the acellular vaccine. In Simondon 1997, a substudy examined the incidence of whooping cough in vaccine recipients who came into household contact with confirmed pertussis cases. In this substudy, comparison with an unvaccinated control group gave an estimated absolute efficacy of 96% for the whole‐cell vaccine and 85% for the acellular vaccine.
Relative vaccine efficacy ‐ whooping cough
Four eligible studies determined the relative risk of whooping cough after administration of acellular vaccines compared to that after whole‐cell vaccines. Results are summarised below. Risk ratios (RRs) are expressed in relation to the incidence in the whole‐cell vaccine group. A RR < 1.0 favours the acellular vaccine.
Study: Greco 1996 Dose schedule: two, four and six months. Whole‐cell comparator: Connaught [W]. Vaccine: SKB [3] ‐ All: 0.25 (0.17 to 0.36); ≥ 1 immunisation dose: 0.28 (0.20 to 0.39). Vaccine: Chiron‐Biocine [3P] ‐ All: 0.25 (0.17 to 0.36); ≥ 1 immunisation dose: 0.25 (0.17 to 0.36).
Study: Gustafsson 1996 Dose schedule: two, four and six months. Whole‐cell comparator: Connaught [W]. Vaccine: SKB [2] ‐ All: 0.83 (0.65 to 1.05); ≥ 1 immunisation dose: 0.83 (0.66 to 1.05). Vaccine: Connaught [5] ‐ All: 0.29 (0.21 to 0.40); ≥ 1 immunisation dose: 0.30 (0.22 to 0.42).
Study: PVSG 1998 Dose schedule: 2, 4 to 10, 6 to 12, and 15 to 18 months. Whole‐cell comparator: Lederle [W]. Vaccine: Lederle/Takeda [4] ‐ All: 2.1 (upper 1‐sided 95% CI = 3.3); ≥ 1 immunisation dose: not reported.
Study: Simondon 1997 Whole‐cell comparator: Pasteur‐Merieux [W]. Dose schedule: two, four and six months. Pasteur‐Merieux [2] ‐ All: 2.42 (1.35 to 4.34); ≥ 1 immunisation dose: 2.06 (1.25 to 3.39).
As previously noted, AHGSPV 1997 did not meet the active follow‐up criterion specified in this review, but the passive follow‐up method used in that trial (although low in sensitivity) did not appear to be associated with differential case ascertainment for the 'whooping cough' case definition. Results from this study are summarised below:
Study: AHGSPV 1997 Dose schedule: 3, 5 and 12 months. Whole‐cell comparator: Evans Medical [W]. Vaccine: Chiron‐Biocine [3] ‐ All: 2.55 (1.50 to 4.33); ≥ 1 immunisation dose: 1.84 (1.36 to 2.51). Vaccine: Connaught [5] ‐ All: 1.40 (0.78 to 2.52); ≥ 1 immunisation dose: 1.25 (0.90 to 1.75).
Thus, the efficacy of the five‐component vaccine against this case definition was not significantly different to that of the whole‐cell vaccine, whereas that of the three‐component vaccine was. While this suggests that the five‐component vaccine may have an advantage over the three‐component, it should be noted that the two acellular vaccines were not actually significantly different from each other (the 95% CIs overlapped). Furthermore, any conclusions from this study must be tempered by its reliance on passive follow‐up.
Absolute vaccine efficacy ‐ pertussis disease
Four trials determined absolute vaccine efficacy against pertussis disease, as defined in this review. These results are summarised below:
Study: Trollfors 1995 Dose schedule: 3, 5 and 12 months. Vaccine: Amvax [1] ‐ All: 54% (43 to 63); ≥ 1 immunisation dose: not reported.
Study: AHGSPV 1988 Dose schedule: 5 to 11, and 7 to 13 months. Vaccine: JNIH7 [1] ‐ All: 41% (12 to 60); ≥ 1 immunisation dose: not reported. Vaccine: JNIH6 [2] ‐ All: 58% (35 to 73); ≥ 1 immunisation dose: not reported.
Study: Greco 1996 Dose schedule: two, four and six months. Vaccine: SKB [3] ‐ All: 71% (61 to 78); ≥ 1 immunisation dose: not reported. Vaccine: Chiron‐Biocine [3] ‐ All: 71% (61 to 79); ≥ 1 immunisation dose: not reported. Vaccine: Connaught [W] ‐ All: 23% (1 to 40); ≥ 1 immunisation dose: not reported.
Study: Gustafsson 1996 Dose schedule: two, four and six months. Vaccine: SKB [2] ‐ All: 44% (35 to 52); ≥ 1 immunisation dose: 44% (35 to 52). Vaccine: Connaught [5] ‐ All: 78% (73 to 83); ≥ 1 immunisation dose: 78% (73 to 83). Vaccine: Connaught [W] ‐ All: 42% (30 to 51); ≥ 1 immunisation dose: 41% (30 to 50).
In PVSG 1998, comparison with a non‐RCT DT control group gave an estimated absolute efficacy against pertussis disease of 83% (95% CI 76% to 88%) for the whole‐cell vaccine and 72% (95% CI 62% to 79%) for the acellular vaccine.
Relative vaccine efficacy ‐ pertussis disease
One trial determined relative vaccine efficacy against pertussis disease, as defined in this review. The results of this trial are summarised below.
Study: Simondon 1997 Whole‐cell comparator: Pasteur‐Merieux (W). Dose schedule: two, four and six months. Pasteur‐Merieux [2] ‐ All: 1.54 (1.23 to 1.94); ≥ 1 immunisation dose: 1.43 (1.16 to 1.74).
Secondary outcomes
Comparison of acellular and whole‐cell vaccines
Seven additional trials identified by the updated search (2009) provided available data only for minor adverse events. Updated meta‐analysis including the data from these seven trials did not significantly alter the results of any endpoint (Appendix 7).
1. Failure to complete all scheduled doses of the primary immunisation series because of adverse events
The risk of failure to complete the primary series because of adverse events was significantly lower in acellular vaccine recipients compared to those immunised with whole‐cell vaccines (11 trials with a total of 108,909 participants, RR 0.23, 95% CI 0.12 to 0.43, P value < 0.00001) (Analysis 1.1).
1.1. Analysis.
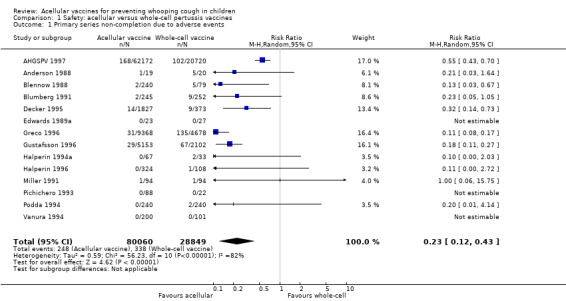
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 1 Primary series non‐completion due to adverse events.
2. Mortality due to any cause
The risk of death due to any cause did not differ significantly between acellular and whole‐cell recipients (nine trials with a total of 122,451 participants, RR 0.87, 95% CI 0.62 to 1.22, P value = 0.41) (Analysis 1.2).
1.2. Analysis.
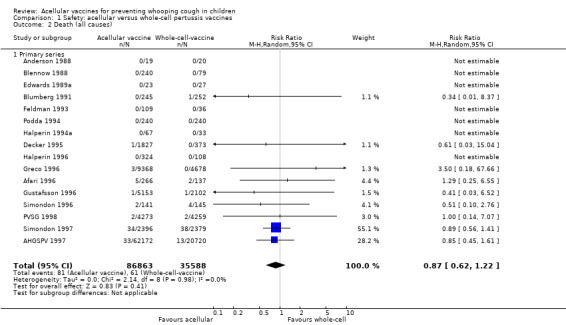
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 2 Death (all causes).
3. Mortality due to infection
The risk of death due to infection did not differ significantly between acellular and whole‐cell recipients (two trials with a total of 34,498 participants, RR 0.97, 95% CI 0.23 to 4.16, P value = 0.97) (Analysis 1.3).
1.3. Analysis.
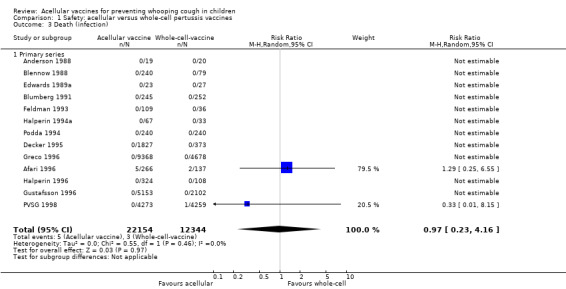
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 3 Death (infection).
4. Encephalopathy
No cases of encephalopathy after primary series immunisation were observed in 81,601 acellular and 32,161 whole‐cell vaccine recipients (Analysis 1.4).
1.4. Analysis.
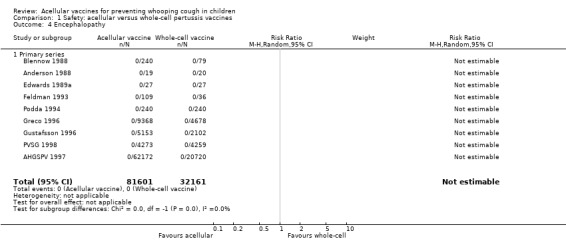
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 4 Encephalopathy.
5. Convulsions
The risk of convulsion after primary series immunisation was significantly lower in acellular vaccine recipients compared to those immunised with whole‐cell vaccines (nine trials with a total of 124,387 participants, RR 0.47, 95% CI 0.31 to 0.73, P value = 0.0007) (Analysis 1.5)
1.5. Analysis.
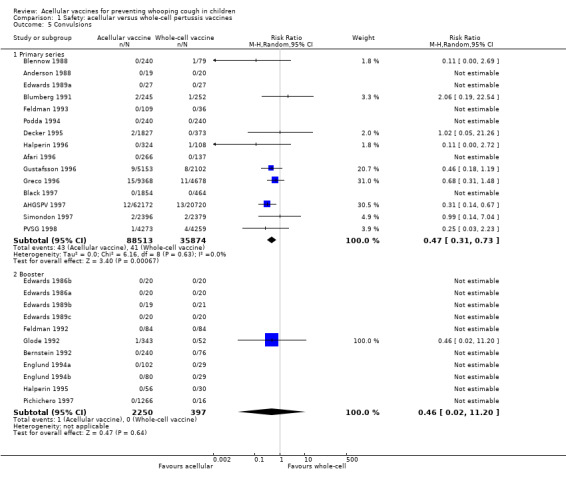
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 5 Convulsions.
6. Hypotonic‐hyporesponsive episodes
The risk of hypotonic‐hyporesponsive episodes after primary series immunisation was significantly lower in acellular vaccine recipients compared to those immunised with whole‐cell vaccines (six trials with a total of 121,573 participants, RR 0.26, 95% CI 0.08 to 0.81, P value = 0.02) (Analysis 1.6). No hypotonic‐hyporesponsive episodes were recorded after any booster dose in the 2171 acellular and 316 whole‐cell vaccine recipients for whom data were available (Appendix 8).
1.6. Analysis.
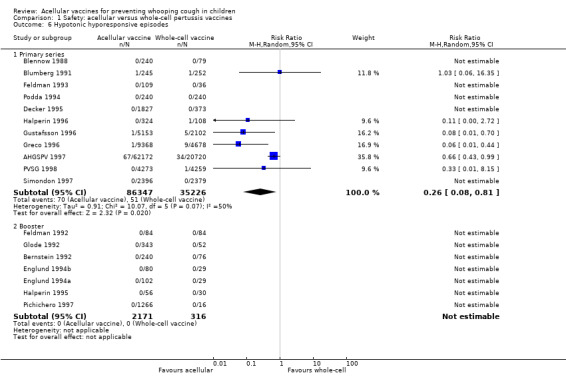
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 6 Hypotonic hyporesponsive episodes.
7. Minor adverse events
Minor adverse events (anorexia, drowsiness, fever, irritability/fretfulness, prolonged crying, vomiting, injection site pain/tenderness/redness/swelling) after most vaccine doses were significantly less common in acellular vaccine recipients than in those immunised with whole‐cell vaccines (Analysis 1.7 to Analysis 1.15).
1.7. Analysis.
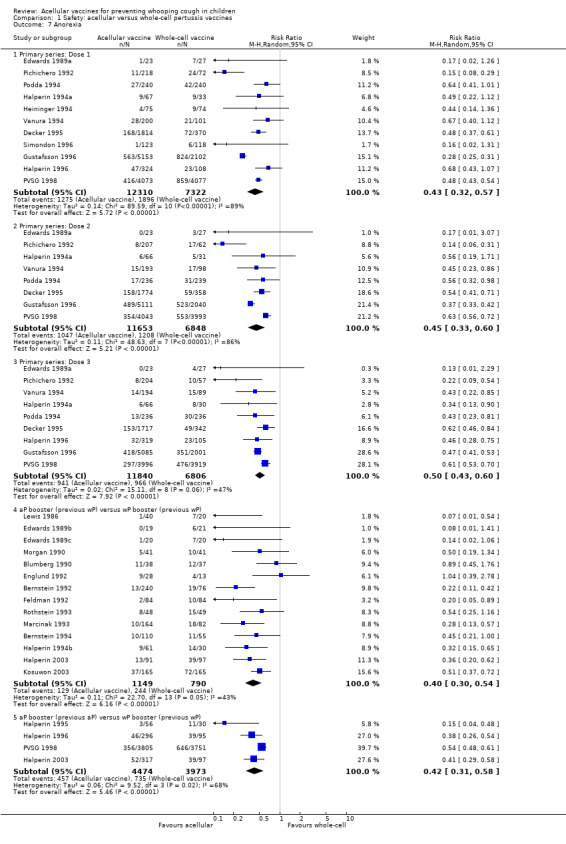
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 7 Anorexia.
1.15. Analysis.
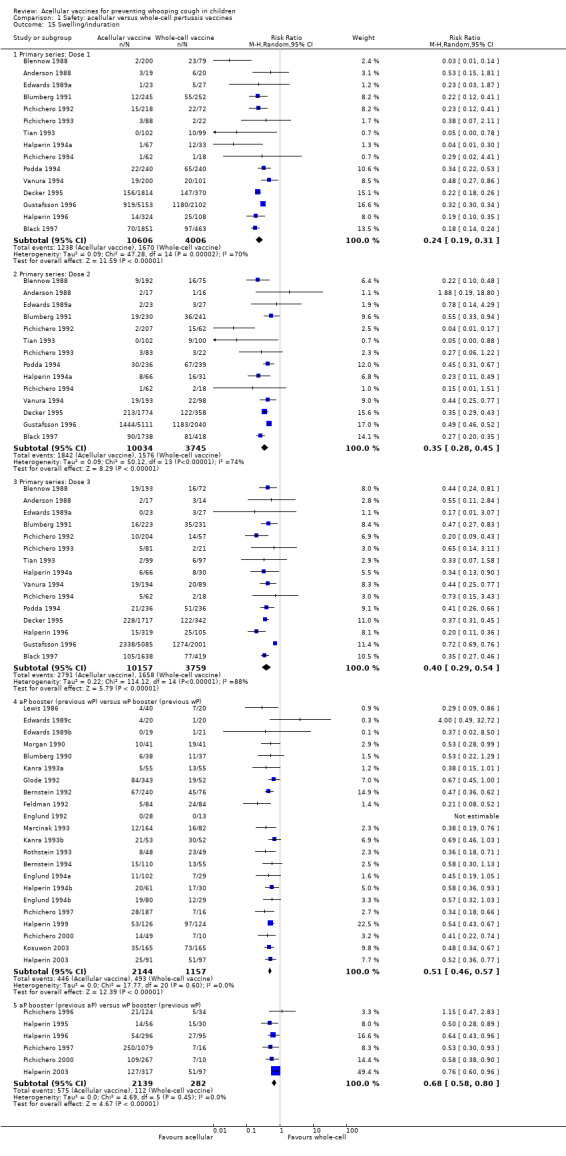
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 15 Swelling/induration.
We conducted two pre‐planned sensitivity analyses. The first compared random‐effects and fixed‐effect analyses to determine whether the conclusions were sensitive to model selection. This led to changes from a non‐significant difference (a random‐effects model) to a significant difference in favour of acellular vaccines for two endpoints: vomiting (the third primary dose and booster after acellular priming), and drowsiness (the first primary dose). The second repeated the random‐effects analysis after removing, from each endpoint, any study allocated more than 50% of the total weight in the original analysis. This led to changes from a significant difference in favour of acellular vaccines to a non‐significant difference for vomiting after the first and second primary series doses.
We performed an unplanned sensitivity analysis excluding AHGSPV 1997. This was the largest study in the review and differed from the others in that follow‐up was not fully active. Removal of this study did not change the significance or otherwise of any endpoints.
We performed a second unplanned sensitivity analysis excluding three studies with possible inadequate blinding (Greco 1996; Gustafsson 1996; Halperin 2003). This resulted in the summary OR for drowsiness changing from non‐significant to significant (in favour of acellular vaccines), while that for vomiting after the first and second primary series doses changed from significant to non‐significant.
No analysis of any endpoint resulted in a RR that significantly favoured whole‐cell vaccines.
We also examined the incidence of minor adverse events in cohorts of children after successive doses of acellular and whole‐cell vaccines. The incidence of anorexia and vomiting fell with successive doses of the primary series for both acellular and whole‐cell vaccines. The incidence of irritability and injection site pain/tenderness remained relatively constant, whereas the incidence of fever, local redness and swelling/induration increased markedly over the primary series of acellular vaccines (Appendix 9).
The incidence of minor adverse reactions in children boosted with acellular vaccines after whole‐cell priming was lower than if whole‐cell vaccines were used for every dose. However, an unexpected rise in the incidence of fever, irritability, local pain, redness and swelling was seen among children primed and boosted with acellular vaccines compared with those boosted with acellular vaccines after whole‐cell priming (Appendix 10).
Comparison of acellular vaccines and placebo/DT
The number of studies and participants in this comparison was considerably less than in the comparison of acellular with whole‐cell vaccines, with a maximum of four studies contributing data to any endpoint. No booster data were available (Appendix 11; Appendix 12).
1. Failure to complete all scheduled doses of the primary immunisation series because of adverse events
There was no statistically significant difference between acellular and placebo/DT recipients in terms of the risk of failure to complete the primary series because of adverse events (four trials with a total of 25,901 participants, RR 0.70, 95% CI 0.38 to 1.29, P value = 0.25) (Analysis 2.1).
2.1. Analysis.
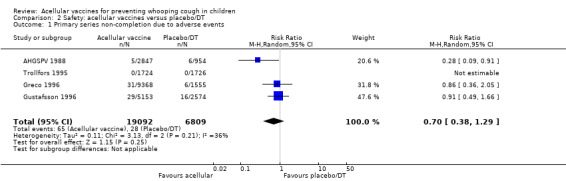
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 1 Primary series non‐completion due to adverse events.
2. Mortality due to any cause
The risk of death due to any cause did not differ significantly between acellular and placebo/DT recipients (four trials with a total of 25,901 participants, RR 1.08, 95% CI 0.26 to 4.42, P value = 0.91) (Analysis 2.2).
2.2. Analysis.
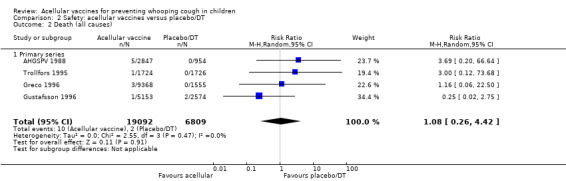
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 2 Death (all causes).
3. Mortality due to infection
The risk of death due to infection did not differ significantly between acellular and placebo/DT recipients (four trials with a total of 25,901 participants, RR 1.21, 95% CI 0.19 to 7.80, P value = 0.84) (Analysis 2.3).
2.3. Analysis.
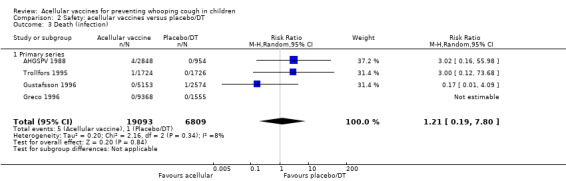
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 3 Death (infection).
4. Encephalopathy
No cases of encephalopathy after primary series immunisation were observed in 14,521 acellular and 4129 placebo/DT recipients (Analysis 2.4).
2.4. Analysis.
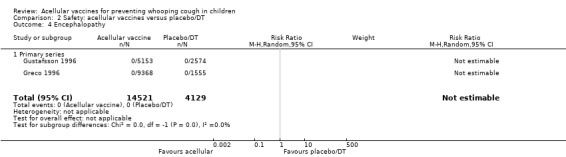
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 4 Encephalopathy.
5. Convulsions
There was no statistically significant difference between acellular and placebo/DT recipients in terms of the risk of convulsion after primary series immunisation (four trials with a total of 25,901 participants, RR 0.44, 95% CI 0.12 to 1.69, P value = 0.23) (Analysis 2.5)
2.5. Analysis.
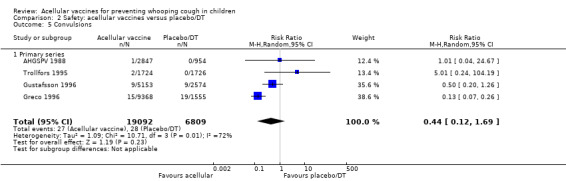
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 5 Convulsions.
6. Hypotonic‐hyporesponsive episodes
The risk of hypotonic‐hyporesponsive episodes after primary series immunisation did not differ significantly between acellular and placebo/DT recipients (four trials with a total of 25,901 participants, RR 0.29, 95% CI 0.02 to 5.13, P value = 0.40) (Analysis 2.6).
2.6. Analysis.
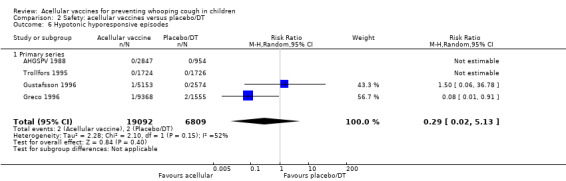
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 6 Hypotonic hyporesponsive episodes.
7. Minor adverse events
There was no statistically significant difference between acellular and placebo/DT in terms of the risk of minor adverse events (anorexia, drowsiness, fever, irritability/fretfulness, prolonged crying, vomiting, injection site pain/tenderness, injection site redness and injection site swelling/induration) after most vaccine doses (Analysis 2.7 to Analysis 2.15).
2.7. Analysis.
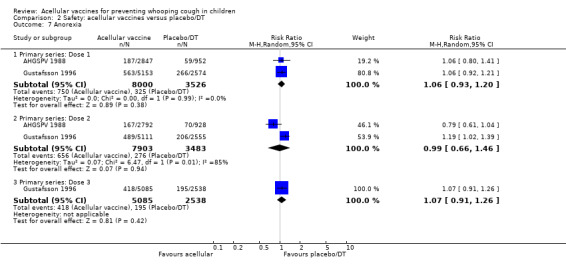
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 7 Anorexia.
2.15. Analysis.
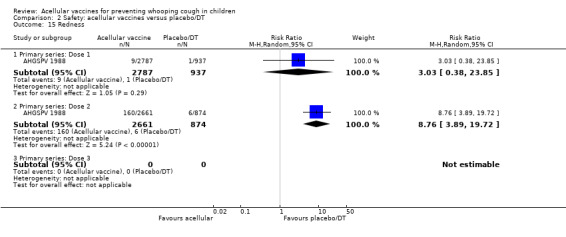
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 15 Redness.
Most endpoints in this comparison were homogeneous. For these, the fixed‐effect analysis differed from the random‐effects analysis in only three cases: the RRs for convulsions in the primary series and swelling/induration after the first two primary doses were significantly in favour of placebo/DT in the fixed‐effect analysis but non‐significant in the random‐effects analysis.
We conducted a second planned sensitivity analysis in which the random‐effects analysis was repeated after removing from each endpoint any study which had been assigned more than 50% of the total weight. A number of endpoints contained data from only a single study and were thus not evaluable after that study had been excluded. In addition, the RR for swelling/induration after the second dose became significant and after the third dose became non‐significant.
No analysis of any endpoint resulted in a RR which significantly favoured acellular vaccines over placebo/DT.
Discussion
Summary of main results
Efficacy
Due to a small number of efficacy trials and significant heterogeneity across studies regarding immunisation schedules, case definitions, follow‐up duration and background pertussis rates, it was not applicable to conduct a meta‐analysis in this review to estimate the pooled efficacy of acellular pertussis vaccines against whooping cough. However, some considerations could be made on the basis of the data from six included trials.
Firstly, comparisons across studies suggest that multi‐component (≥ three) acellular vaccines have a higher efficacy than one‐ and two‐component vaccines against both typical whooping cough (characterised by 21 or more consecutive days of paroxysmal cough with confirmation of B. pertussis infection by culture, appropriate serology or contact with a household member who has culture‐confirmed pertussis) and mild pertussis disease (characterised by seven or more consecutive days of cough with confirmation of B. pertussis infection by culture or appropriate serology) (WHO 1991). The efficacy of multi‐component vaccines varied from 84% to 85% in preventing typical whooping cough and from 71% to 78% in preventing mild pertussis disease (Greco 1996; Gustafsson 1996). In contrast, the efficacy of one‐ and two‐component vaccines varied from 59% to 78% against typical whooping cough and from 41% to 58% against mild pertussis disease (AHGSPV 1988; Trollfors 1995). The superiority of five‐component vaccines over two‐component vaccines in preventing typical whooping cough has been confirmed by direct comparison of such vaccines in the trials of Gustafsson 1996 and AHGSPV 1997. However, the data are insufficient to determine whether there is a clinically significant difference between three‐ and five‐component vaccines. Comparisons across trials do not confirm such a difference. A small and statistically non‐significant difference was observed in the trial of AHGSPV 1997, but incomplete case ascertainment would have reduced the statistical power of the study.
Secondly, not all whole‐cell vaccines are efficacious, as has traditionally been thought, as evidenced by the low efficacy of the Connaught whole‐cell vaccine used in the trials of Greco 1996 (efficacy: 36%; 95% confidence interval (CI) 14% to 52%) and Gustafsson 1996 (efficacy: 48%; 95% CI 37% 58%). This makes it more difficult to interpret the results of direct efficacy comparisons between acellular vaccines and whole‐cell vaccines across studies. The multi‐component (≥ 3) acellular vaccines are more effective than low‐efficacy whole‐cell vaccines, but may be less effective than the highest‐efficacy whole‐cell vaccines in preventing whooping cough (AHGSPV 1997; Greco 1996; Gustafsson 1996; PVSG 1998; Simondon 1997).
Thirdly, randomised controlled trials (RCTs) generally measure vaccine efficacy but not the effectiveness of a large‐scale vaccination programme. Compliance with immunisation is likely to be higher in these studies than would be expected in actual practice and 'real world' effectiveness would probably be correspondingly lower. Having said this, it is encouraging that where data were available for the population who did not complete all scheduled doses, efficacy was only marginally lower than in those who had received all vaccine doses. Moreover, the effectiveness of national vaccination programmes with acellular pertussis (aP) vaccines in preventing whooping cough in children has been shown in Japan, the United States and Canada, where such vaccines have been routinely used among infants and young children (Bettinger 2007; Bisgard 2005; Kuno‐Sakai 2004; Watanabe 2005). In contrast, recent research from Queensland, Australia (Sheridan 2012) and Oregon, United States (Liko 2013) suggests that children primed with acellular rather than whole‐cell pertussis vaccine are at a higher risk of subsequent pertussis. The authors raised the hypothesis that the recent pertussis epidemic in many high‐income countries might be related to the shift from whole‐cell pertussis (wP) to aP vaccines. However, further nationwide studies are needed to test this hypothesis.
Safety
The comparison of acellular vaccines with whole‐cell vaccines displays that the former have a better safety profile. The superiority of acellular vaccines over whole‐cell vaccines is evident for all selected minor reactions during the primary and booster doses. The incidence of primary series non‐completion due to adverse events is significantly lower for acellular vaccines than for whole‐cell vaccines. Acellular vaccines are also less likely to cause febrile convulsions and hypotonic‐hyporesponsive episodes during the primary series. A benefit has not been seen in respect to febrile convulsions after booster doses, hypotonic‐hyporesponsive episodes after booster doses or encephalopathy during the primary series, but the former was very uncommon, while the latter two were non‐existent with either type of vaccine. The risk of death due to any cause, and death due to infection, were similar between acellular and whole‐cell vaccine recipients.
The comparison of acellular vaccines with placebo/DT also reveals a good safety profile of such vaccines. There is no significant difference between acellular vaccines and placebo/DT in the incidence of severe or minor adverse events, with the exception of injection site swelling, which was significantly more common among recipients of acellular vaccines than placebo/DT after the third dose during the primary series. However, given the uncommon occurrence of severe adverse reactions, the statistical power of this meta‐analysis may be not enough to detect small but clinically relevant differences between acellular vaccines and placebo/DT. In this sense, continuing surveillance for rare severe adverse reactions of aP vaccines may be warranted.
Two points about minor adverse events of acellular vaccines deserve special comment. Firstly, a significant increase in incidence (number of events per 1000 recipients) from the first dose to the third dose during the primary series has been observed for some minor adverse reactions, such as fever (from 60 to 162), local redness (from 96 to 162) and swelling (from 117 to 275). In spite of this increase, the incidence of these minor adverse events among recipients of acellular vaccines is still lower than that among recipients of whole‐cell vaccines. Secondly, the incidence of adverse reactions in children boosted with acellular vaccines after whole‐cell priming is lower than if whole‐cell vaccines are used for every dose. Comparison across studies displays a rise in the incidence of some minor adverse reactions (fever, irritability, local pain, redness and swelling) among children primed and boosted with acellular vaccines compared with those boosted with acellular vaccines after whole‐cell priming. The rate of reported injection site reactions continues to increase after the fifth consecutive dose of aP vaccine at four to six years of age. Although redness and swelling increase to rates similar to those associated with five consecutive doses of whole‐cell vaccine, injection site tenderness after five does of acellular vaccine is significantly lower than after five doses of whole‐cell vaccine, and the vaccine is preferred by parents of these preschool‐aged children. These increases in injection site reactions may have limited clinical implications as the current evidence suggests an acceptable safety profile of acellular vaccines for booster doses among preschool children and adolescents who had been primed with acellular vaccines (Jacquet 2006; Pichichero 2005; Pichichero 2006).
Overall completeness and applicability of evidence
This review provides evidence to answer the questions posed in the Objectives section.
1. Do acellular vaccines protect against pertussis?
All aP vaccines reported in the efficacy trials protect against pertussis. Acellular vaccines with three or more components (multi‐component vaccines) are effective against both classical whooping cough and mild pertussis disease.
2. Do different types of acellular vaccine confer different levels of protection?
Currently available evidence suggests that multi‐component vaccines confer better protection against both classical whooping cough and mild pertussis infection than vaccines containing only one or two components. However, the clinical implication of any possible superiority of multi‐component vaccines over mono‐ and bivalent vaccines in the efficacy demonstrated by RCTs should be considered with caution. The effectiveness of vaccination programmes on a national scale for controlling infectious disease depends not only on the efficacy of the vaccine, but also other factors such as the vaccination schedule and adherence, and transportation and storage of the vaccine. Moreover, indirect effects in producing herd immunity in the population may also contribute to the effectiveness of large‐scale vaccination in controlling infectious diseases (Carlsson 2009; Stephens 2008). Therefore, successful control of pertussis infections by two‐component vaccines in Japan and in other countries (Carlsson 2009; Hviid 2004; Kuno‐Sakai 2004) does not necessarily exclude the potential additional benefits of large‐scale vaccination with multi‐component vaccines.
3. Do acellular vaccines protect against pertussis to the same degree as the whole‐cell vaccines which they are intended to replace?
The answer here remains unclear because the effectiveness of whole‐cell vaccines varies and there are limited efficacy and effectiveness data with different whole‐cell vaccines. Studies to date indicate that multi‐component acellular vaccines are more effective than low‐efficacy whole‐cell vaccines but may be less effective than the highest‐efficacy whole‐cell vaccines.
4. Do acellular vaccines cause fewer side effects than whole‐cell vaccines?
Acellular vaccines cause fewer side effects than whole‐cell vaccines during the primary immunisation series, when used as a booster in toddlers and four‐to‐six‐year‐olds who have previously received whole‐cell vaccines, and when used in toddlers after acellular priming.
Quality of the evidence
As previously mentioned, meta‐analysis was not applicable in this review to estimate the pooled efficacy. However, three large, double‐blind RCTs from Italy, Sweden and Germany (Greco 1996; Gustafsson 1996; PVSG 1998) provide high‐level evidence about effectiveness of multi‐component aP vaccines against whooping cough.
Regarding the safety profile, meta‐analysis of 52 studies provides robust evidence about the superiority of aP vaccines over whole‐cell vaccines. However, the heterogeneity of results across studies deserves special consideration.
Heterogeneity in the meta‐analysis of safety endpoints
Many of the safety endpoints in the comparison of acellular with whole‐cell vaccines displayed significant heterogeneity. This heterogeneity did not invalidate the finding of significant differences for various endpoints because the point estimates for the individual studies were almost always in favour of the acellular vaccines. In other words, the individual study results were quantitatively but not qualitatively different.
Possible explanations for the heterogeneity, which was sometimes substantial, include the following.
Differences between studies in the definition or recording of adverse events. For example, the threshold for fever differed slightly amongst studies, albeit within the confines of the definition specified in the review. Prolonged crying was defined as longer then one hour in some studies and longer than three hours in others. In some studies the mildest recorded category of redness or swelling was 'any', whereas in others it was a diameter of one centimetre. Some studies reported the incidence of events within a certain time period of each dose, whereas others reported the incidence at a specific time point.
The use of different immunisation schedules. Most studies used a two‐, four‐ and six‐month schedule for the primary series but others used a three‐, five‐ and 12‐month schedule. AHGSPV 1988 used a 5 to 11‐month, and 7 to 14‐month schedule. The difference in reactogenicity between acellular and whole‐cell vaccines has been shown to be less with the earlier dose schedule, primarily because the reactogenicity of whole‐cell vaccines increases with the age of administration (for example, in Miller 1997).
Differences in the reactogenicity of separate acellular vaccines. This is a possibility, although a number of head‐to‐head studies of acellular vaccines have not shown any consistent differences (for example, AHGSPV 1988; AHGSPV 1997; Decker 1995; Englund 1994a; Greco 1996; Pichichero 1997).
Differences in the reactogenicity of the whole‐cell vaccines used as controls.
Differences in the duration over which adverse events were recorded.
Differences in the susceptibility of different patient populations (for example, different racial groups) to adverse events after pertussis immunisation.
Given the large number of endpoints examined in the review and the conservative critical value for determining heterogeneity, approximately 10% of the endpoints would be expected to fail the test of homogeneity by chance alone.
A special comment is warranted about drowsiness after the first and second doses of the primary series. The statistical test displays an extreme heterogeneity for this endpoint and it strongly suggests inconsistent findings across studies. By visual inspection of the forest plot, we identified that Gustafsson 1996 was responsible for this extreme heterogeneity. In contrast with other studies, Gustafsson 1996 demonstrated the results in favour of whole‐cell vaccines. This is only one safety endpoint where the superiority of whole‐cell vaccines over acellular vaccines was reported. Partial unblinding of the whole‐cell vaccine arm occurred in this trial but the expected effect of unblinding would be to bias the assessment of adverse events in favour of the acellular preparations. We have no plausible explanations for this finding.
Potential biases in the review process
The potential sources and impact of bias and confounding have been addressed in the Risk of bias in included studies section. In addition to the potential bias and confounding, the following limitations of the review should also be noted.
The review examines the safety of acellular vaccines as a group and does not attempt to determine whether different vaccines have different safety profiles.
The absence of statistically significant differences between acellular vaccines and placebo/DT in respect to severe adverse events may be due to a lack of statistical power in the meta‐analysis, rather than a true lack of difference.
The review is based entirely on public domain information and did not use any confidential vaccine manufacturer data that might contribute to the decisions of regulatory agencies.
Agreements and disagreements with other studies or reviews
We searched CENTRAL, MEDLINE and EMBASE for systematic reviews of pertussis vaccines and identified 11 papers. Two could be compared to our review (Casey 2005; Jefferson 2003). Like us, Jefferson 2003 searched the literature without any publication year restriction, whereas Casey 2005 limited their search to efficacy studies from 1990 onwards. Unlike us, both reviews included observational studies. Jefferson 2003 and our review both included 52 studies; most but not all studies were common to Jefferson 2003 and our review.
Efficacy
All efficacy studies of acceptable quality were published between 1988 and 1997.
Casey 2005 retrieved eight efficacy trials; six of them were also included in our efficacy review. Of the remaining two, one was a case‐controlled study (Liese 1997) and the other a cohort study (Schmitt 1996).
Jefferson 2003 included Afari 1996 and Decker 1995, which we excluded due to the absence of laboratory‐confirmed pertussis cases. (Decker 1995 also lacked efficacy data). However, we included these studies in the safety review.
In contrast to Jefferson 2003, we did not study the absolute efficacy of whole‐cell vaccines and we did not perform meta‐analyses of efficacy studies due to significant heterogeneity.
Several studies of acellular vaccines with three to five components showed that the efficacy of the acellular vaccines was superior or equal to the whole‐cell comparator. Jefferson 2003 claims that AHGSPV 1997 "is the source of the widely‐held view that acellular vaccines are less effective than high‐efficacy whole‐cell vaccines". However, both Jefferson 2003 and our review included several studies with acellular vaccines inferior to whole‐cell vaccines, including a study of a four‐component acellular vaccine (PVSG 1998).
Safety
Jefferson 2003 included a systematic review of studies assessing the safety of DTP vaccines. We excluded some of these studies because of lack of sufficient data. However, the results of the reviews were largely similar.
Booster doses
Casey 2005 found that booster doses of all DTaP vaccines in 1% to 2% of individuals caused large, local injection reactions, in most cases probably IgE‐mediated. These reactions cause no permanent harm, and the authors of Casey 2005 argue that everybody should have access to a booster dose every 10 years throughout life.
Despite different designs, there are no significant disagreements between our review and the above mentioned two systematic reviews regarding efficacy and safety of aP vaccines.
Observational studies have also demonstrated the effectiveness of aP vaccines. Gustafsson 2006 (Swedish birth cohorts) showed that immunisation with aP vaccines at three, five and 12 months of age resulted in a reduction in the incidence of laboratory‐confirmed pertussis from 113 to 150 per 100,000 during 1992/1995, to 11 to 16 per 100,000 during 2001/2004. Bisgard 2005 (case‐control study of 1072 children in the United States) found that any combination of ≥ three DTwP/DTaP vaccine doses was highly protective against pertussis. As compared with 0 doses, the unadjusted vaccine effectiveness was 83.6%, 95.6% and 97.7% for one or two, three and ≥ four vaccine doses, respectively. In contrast with the findings of our review, this study showed that four‐component DTaP vaccine was less effective than two‐component DTaP vaccine, however, the potential biases related to study design might cast serious doubt on the validity of comparison between different vaccines.
Authors' conclusions
Implications for practice.
Multi‐component acellular vaccines are effective against both typical whooping cough and mild pertussis disease, with a good safety profile. However, the implications of this finding for clinical practice may be different in high‐income and low‐income countries. In high‐income countries, death from whooping cough is rare and parental acceptance is a major determinant of immunisation uptake. In these circumstances, the improved side effect profile of acellular vaccines argues in favour of their use, even though they might sacrifice some degree of effectiveness compared to the best whole‐cell vaccines. The available efficacy data favours the multi‐component acellular vaccines over the one‐ and two‐component vaccines in this regard.
In low‐income countries, where the risk of pertussis is higher and cases are more likely to be fatal, greater weight needs to be given to vaccine efficacy. If an acellular vaccine has been shown to be less effective than a high‐efficacy whole‐cell vaccine it is intended to replace, the reactogenicity advantage of the acellular vaccine may be offset by increased mortality and morbidity due to a higher breakthrough rate of pertussis. However, most of the whole‐cell vaccines used in low‐income countries have not been studied for efficacy or effectiveness and, therefore, it is not known where on the wide spectrum of whole‐cell vaccine efficacy an individual product lies. The more consistent manufacturing of the acellular vaccines may lead to more consistent efficacy of these products. However, the higher cost of acellular vaccines at this time would seem likely to affect their usefulness in low‐income countries.
Implications for research.
There are several gaps in our current knowledge of acellular pertussis vaccines. One of the more notable is that the optimal composition of these vaccines has yet to be defined. All vaccines in this review included some form of inactivated pertussis toxin, while those with more than one component all included filamentous haemagglutinin. Although the available multi‐component vaccines are more effective than those with one or two components, it has not yet been determined what the optimal composition is for the acellular vaccines.
More data are needed on the optimal timing of booster doses, school‐entry acellular boosting in children who have received acellular vaccines for all previous doses, the effectiveness of acellular vaccines in adults and what is the proper interval for subsequent doses of acellular pertussis vaccine in adulthood.
Ethical barriers to the inclusion of a placebo group, combined with the evidence that whole‐cell vaccines are not uniformly effective, will create problems for future efficacy studies. Such studies will need to include a self selected, non‐immunised and potentially biased control group, in order to provide an estimate of absolute vaccine efficacy. Further analyses of the data from existing placebo‐controlled studies, with the aim of determining characteristics of participants and their environment which affect vaccine efficacy, will permit future studies to improve these estimates of absolute efficacy by adjusting for such factors.
Finally, the lack of a laboratory correlate of efficacy means that the testing of new acellular pertussis vaccines currently requires prolonged and expensive clinical trials. Research into determining such a laboratory correlate should be a priority.
Feedback
Response to Zhang et al: Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 2011, Issue 1, 25 January 2011
Summary
A meta‐analysis initially done by Jefferson et al (1) of acellular (acP) vaccine efficacy and safety in children was revisited by Zhang et al in the Cochrane Library (2). Zhang correctly did not perform a formal meta‐analysis, stating that it was not appropriate due to the small number of efficacy trials and the significant heterogeneity across these trials with respect to immunization schedules, case definitions, follow‐up duration, products used, and background pertussis rates. The inappropriateness of applying a meta‐analysis methodology to these studies was previously highlighted by Desauziers et al (3) and Simondon (4) following Jefferson et al’s previous publication (1).
The review by Zhang et al concludes that “of the currently available acellular vaccines, multi‐component vaccines confer better protection, against both classical whooping cough and mild pertussis infection, than vaccines containing only one or two components.” Although it is intuitively attractive to adopt a “more components is better” position on acP vaccines, this strong conclusion on differential efficacy of acellular vaccines is not objectively supported by the reviewed trials nor by the data these trials generated. Nor is this conclusion supported by previously published commentaries or by the positions expressed by professional pediatric and national immunization policy bodies.
Zhang et al included in their review six controlled trials that were designed well enough to ensure high internal validity and one trial without active surveillance of cases. All of these trials were performed between 1988 and 1997. Only three of these trials were efficacy trials that directly compared different acellular vaccines in a double‐blind, randomized design, with an active follow‐up, and laboratory confirmation of cases. The majority of currently licensed pertussis vaccines have not been compared in head‐to‐head efficacy trials. Most critically there are no clinical trials that directly compare the efficacy of licensed 2‐acP vaccines with that of licensed 3‐acP or 5‐acP vaccines. In fact, the only study in which a 2‐acP vaccine was less efficacious than multi‐component acellular vaccines involved an experimental 2‐acP vaccine that was never licensed/registered, precisely because of its limited efficacy. Since other 2‐acP vaccines did demonstrate sufficient efficacy to be licensed, the more appropriate interpretation of the experimental 2‐acP vaccine data is that it is not the number of acP components but rather some other feature or combination of features (e.g. the source of the antigens, their relative concentrations, the processes for their purification and detoxification, or the nature and concentration of the adjuvants) of each individual vaccine that determine efficacy. The biasing potential of including data from the unlicensed, experimental 2‐acP vaccine is not clearly discussed in Zhang et al’s review.
The Zhang et al publication highlights the continuing debate over the relative merits of data from randomized controlled trials (RCTs) versus data from observational studies in evidence‐based reviews. Our belief is that, in the real world of vaccination policy decision making, these two different data sets are entirely complementary. Well‐designed RCTs offer higher internal validity, while equally well‐designed observational studies provide higher external validity and a better reflection of the real world. For example, a well‐designed case‐control study performed in Germany by Liese et al (5) reported that a licensed 2‐acP vaccine was 93% effective in prevention of 21 or more days of paroxysmal cough that was laboratory‐confirmed as pertussis. This study was excluded from the review because it was not a RCT. The exclusion of this study and the associated potential for bias should also have been discussed by Zhang et al.
Other published effectiveness data clearly support the effectiveness of both 1‐ and 2‐acP vaccines as well as other multi‐component acP vaccines. These data, with their high external validity, have been reported not only from Japan, the United States and Canada (6‐10) but also from France, Austria, Finland, Denmark, Sweden and Germany (11‐16), and underscore the effectiveness of national vaccination programs in preventing whooping cough in children, regardless of the number of acP components in the licensed vaccines used.
Observational studies might overestimate vaccine effectiveness, if persons included as cases in the studies had more severe pertussis than persons not included, or if the vaccination status would influence the likelihood of the diagnosis of pertussis in a coughing person. Nonetheless, none of these studies identified a lower effectiveness of 2‐acP licensed vaccines in pre‐school children compared to “multi‐component vaccines or whole cell vaccines” (9, 15). These results contradict the conclusions of Zhang et al and as a result they should have been discussed.
The studies included in the review did not use the same follow‐up duration but these differences are not addressed even though they contribute to the heterogeneity of the data. The impact of duration of follow‐up on efficacy estimates has been well illustrated. In one study (17), a highly effective whole cell vaccine and a 2‐acP vaccine demonstrated equivalent efficacies until 18 months of age with a relative risk: 1.16 (95%CI 0.77‐1.72). Only after 18 months of age was the whole cell vaccine shown to retain higher efficacy (relative risk: 1.76 [95%CI 1.33‐2.33]) than the acellular vaccine in the absence of a booster dose in the two groups.
The inappropriateness of relating acP vaccine efficacy to the number of pertussis components has been addressed by both American and European regulatory authorities (18, 19) and by international expert panels charged with comparing results between studies (20‐22). These groups have concluded that the available data support the practical policy guidance that all licensed acellular vaccines are highly effective.
In summary, the overall body of available data supports a conclusion that all licensed acP vaccines, regardless of the number of pertussis components, have proven highly effective. Despite the intrinsic attractiveness of such a conclusion, these data do not support the Zhang et al conclusion that 1‐ and 2‐acP vaccines are less effective than those with more acellular components. The dangers of this conclusion coming from such respected authors include confusion in the vaccination community and the inappropriate preference for one group of vaccines over another. This in turn might lead to negative impacts on vaccine supply and immunization rates.
1Sanofi Pasteur, Lyon France
References:
1. Jefferson T, Rudin M, DiPietrantonj C. Systematic review of the effects of pertussis vaccines in children. Vaccine. 2003 May 16;21(17‐18):2003‐14.
2. Zhang L, Prietsch SOM, Axelsson I, Halperin SA. Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 2011, Issue 1. Art. No.: CD001478. DOI:10.1002/14651858.CD001478.pub4.
3. Desauziers E, Hessel L, Decker MD, Caro JJ, Liese JG. Systematic review of the effects of pertussis vaccines in children. Vaccine. 2004 Jul 29;22(21‐22):2681‐4; author reply 2685.
4. Simondon F. Systematic review of the effects of pertussis vaccines in children. Vaccine. 2004 Aug 13;22(23‐24):2965.
5. Liese JG, Stojanov S, Belohradsky BH. [Pertussis vaccination with acellular vaccines. Tolerance‐‐effectiveness‐‐current vaccination recommendations]. Fortschr Med. 1997 Aug 30;115(24):22‐7.
6. Kuno‐Sakai H, Kimura M. Safety and efficacy of acellular pertussis vaccine in Japan, evaluated by 23 years of its use for routine immunization. Pediatr Int. 2004 Dec;46(6):650‐5.
7. Watanabe M, Nagai M. Acellular pertussis vaccines in Japan: past, present and future. Expert Rev Vaccines. 2005 Apr;4(2):173‐84.
8. Okada K, Ohashi Y, Matsuo F, Uno S, Soh M, Nishima S. Effectiveness of an acellular pertussis vaccine in Japanese children during a non‐epidemic period: a matched case‐control study. Epidemiol Infect. 2009 Jan;137(1):124‐30.
9. Bisgard KM, Rhodes P, Connelly BL, Bi D, Hahn C, Patrick S, Glodé MP, Ehresmann KR; Centers for Disease Control and Prevention, Atlanta. Pertussis vaccine effectiveness among children 6 to 59 months of age in the United States, 1998‐2001. Pediatrics. 2005 Aug;116(2):e285‐94.
10. Bettinger JA, Halperin SA, De Serres G, Scheifele DW, Tam T. The effect of changing from whole‐cell to acellular pertussis vaccine on the epidemiology of hospitalized children with pertussis in Canada. Pediatr Infect Dis J. 2007 Jan;26(1):31‐5.
11. Bonmarin I, Levy‐Bruhl D, Baron S, Guiso N, Njamkepo E, Caro V; Renacoq. Pertussis surveillance in French hospitals: results from a 10 year period. Euro Surveill. 2007 Jan 20;12(1).
12. Rendi‐Wagner P, Kundi M, Mikolasek A, Vécsei A, Frühwirth M, Kollaritsch H. Hospital‐based active surveillance of childhood pertussis in Austria from 1996 to 2003: estimates of incidence and vaccine effectiveness of whole‐cell and acellular vaccine. Vaccine. 2006 Aug 14;24(33‐34):5960‐5.
13. Elomaa A, He Q, Minh NN, Mertsola J. Pertussis before and after the introduction of acellular pertussis vaccines in Finland. Vaccine. 2009 Sep 4;27(40):5443‐9.
14. Hviid A, Stellfeld M, Andersen PH, Wohlfahrt J, Melbye M. Impact of routine vaccination with a pertussis toxoid vaccine in Denmark. Vaccine. 2004 Sep 9;22(27‐28):3530‐4.
15. Carlsson RM, Trollfors B. Control of pertussis‐‐lessons learnt from a 10‐year
surveillance programme in Sweden. Vaccine. 2009 Sep 25;27(42):5709‐18.
16. Juretzko P, von Kries R, Hermann M, Wirsing von König CH, Weil J, Giani G. Effectiveness of acellular pertussis vaccine assessed by hospital‐based active surveillance in Germany. Clin Infect Dis. 2002 Jul 15;35(2):162‐7.
17. Simondon F, Preziosi MP, Yam A, Kane CT, Chabirand L, Iteman I, Sanden G, Mboup S, Hoffenbach A, Knudsen K, Guiso N, Wassilak S, Cadoz M. A randomized double‐blind trial comparing a two‐component acellular to a whole‐cell pertussis vaccine in Senegal. Vaccine. 1997 Oct;15(15):1606‐12.
18. Purvis WV. Letter to all manufacturer of acellular pertussis vaccines. Center for Biologics Evaluation and Research, Food and Drug Administration, Washington; 25 June 1998.
19. European regulatory authorities’ view on proving safety and immunogenicity of combine vaccines—general aspects—recent experiences. In: Viral hepatitis, vol. 10.2 June 2002.
20. Informal consultation on control of pertussis with whole cell and acellular vaccines. CVI/WHO, Geneva; 18‐19 May 1998.
21. Halperin SA. Acellular pertussis vaccines: the next step. Dev Biol Stand 1997;89:363‐5.
22. Orenstein WA. Rapporteur’s summary of the symposium. Dev Biol Stand 1997;89:397‐403.
Reply
We thank Philippe P. J. André and David R. Johnson from Sanofi Pasteur (Lyon, France) for their interests and comments on our review. Their main concern is one of the review’s conclusions “Of the currently available acellular vaccines, multi‐component vaccines confer better protection against both classical whooping cough and mild pertussis infection than vaccines containing only one or two components”. We recognise that this conclusion is based on indirect evidence since the majority of currently licensed pertussis vaccines have not been compared in head to head efficacy trials as André and Johnson pointed out.
André and Johnson’s comments are likely to provoke an old and ongoing debate on the comparative efficacy of acellular pertussis vaccines with different antigen components which is ironic since Sanofi Pasteur markets both two and five component acellular pertussis vaccines. This question has been well addressed by Patrick Olin in 1997 (1). In this commentary, Olin stated that “Contrary to the position taken in most commentaries, analysis of the results of the four placebo‐controlled trials of two one‐component, two two‐component, two three‐component and one five‐component vaccine unequivocally demonstrate the multi‐component vaccines to have better protective efficacy against both mild and typical pertussis than one‐ and two‐component vaccines”. The results from another recent systematic review with 49 randomised controlled trials (RCTs) and 3 cohort studies are also consistent with the findings of our review (2). The current evidence on comparative efficacy of acellular pertussis vaccines with different antigen components has been cited by the most recent WHO position paper on pertussis vaccines (3).
We are conscious that the clinical implication of any possible superiority of multi‐component vaccines over mono‐ and bivalent vaccines in the efficacy demonstrated by RCTs needs to consider the transferability of this conclusion to whole countries and vaccine delivery systems. The effectiveness of vaccination programmes on a national scale for controlling infectious disease depends not only on the efficacy of the vaccine but also other factors such as the vaccination schedule and adherence, and transportation and storage of the vaccine. Moreover, indirect effects in producing herd immunity in the population may also contribute to the effectiveness of large‐scale vaccination in controlling infectious diseases (4,5). Therefore, successful control of pertussis infections by two‐component vaccines in Japan and in other countries (5‐7) does not necessarily exclude the potential additional benefits of large‐scale vaccination with multi‐component vaccines.
This is a systematic review of RCTs, so we did not included observational studies in the review. In the next update, we will devote a special paragraph in the discussion to compare the results from RCTs and observational studies, and to address the potential contribution of follow‐up duration for the heterogeneity across the studies, as suggested by André and Johnson. We will also modify the conclusion to be “Currently available evidence suggests that multi‐component vaccines confer better protection against both classical whooping cough and mild pertussis infection than vaccines containing only one or two components”.
References:
1. Olin P. Commentary: The best acellular pertussis vaccines are multicomponent. Pediatr Infect Dis J 1997;16(5):517‐519.
2. Jefferson T, Rudin M, DiPietrantonj C. Systematic review of the effects of pertussis vaccines in children. Vaccine 2003; 21(17‐18):2003‐2014.
3. WHO. Pertussis vaccines: WHO position paper. Weekly Epidemiol Record 2010;85:385‐400.
4. Stephens DS. Vaccines for the unvaccinated: Protecting the herd. JID 2008;197:643‐645.
5. Carlsson RM, Trollfors B. Control of pertussis‐‐lessons learnt from a 10‐year surveillance programme in Sweden. Vaccine. 2009;27(42):5709‐5718.
6. Kuno‐Sakai H, Kimura M. Safety and efficacy of acellular pertussis vaccine in Japan, evaluated by 23 years of its use for routine immunization. Pediatr Int. 2004;46(6):650‐655.
7. Hviid A, Stellfeld M, Andersen PH, Wohlfahrt J, Melbye M. Impact of routine vaccination with a pertussis toxoid vaccine in Denmark. Vaccine. 2004;22(27‐28):3530‐3534.
Contributors
Philippe P. J. André, MD, PhD1 David R. Johnson, MD, MPH1
What's new
| Date | Event | Description |
|---|---|---|
| 20 September 2014 | Amended | Abstract and Plain language summary corrected in reporting the efficacy of one‐ and two‐component vaccines against typical whooping cough and against mild pertussis disease. |
History
Review first published: Issue 2, 1999
| Date | Event | Description |
|---|---|---|
| 20 January 2014 | New search has been performed | Searches updated. We did not identify any new trials for inclusion or exclusion. |
| 20 January 2014 | New citation required but conclusions have not changed | Our conclusions remain unchanged. |
| 9 January 2012 | New search has been performed | Searches conducted. No new included or excluded trials found |
| 17 May 2011 | New citation required but conclusions have not changed | Our conclusions remain unchanged in this update |
| 18 April 2011 | Feedback has been incorporated | Feedback comment added to review |
| 21 April 2009 | New search has been performed | Searches conducted. Three trials were excluded and seven additional trials were included |
| 21 April 2009 | New citation required but conclusions have not changed | New team of review authors updated this previously withdrawn review |
| 13 December 2007 | Amended | Converted to new review format |
| 23 March 2006 | Amended | Review withdrawn from The Cochrane Library, Issue 3, 2006 |
| 22 January 1998 | New search has been performed | Searches conducted |
Notes
This review was previously withdrawn from The Cochrane Library in Issue 3, 2006. The electronic searches had been conducted in 1998 and the lead author was unable to update the review. In May 2008 this review was taken over and updated by a new team of review authors.
Acknowledgements
Thanks to Sarah Thorning, Trials Search Co‐ordinator of the Cochrane Acute Respiratory Infections (ARI) Group, for performing the updated search and to Elizabeth Dooley, Managing Editor of the Cochrane ARI Group, for continuous support. Owen Tinnion and Mark Hanlon were responsible for the first published version of this review.
Appendices
Appendix 1. Initial search 1997 and 1998
The initial search was carried out in March 1997 and updated in March 1998, covering the period up to and including January 1998. The databases used were MEDLINE (Medline SilverPlatter CD‐ROM) and the Cochrane Central Register of Controlled Trials (CENTRAL). We searched CENTRAL using the term 'pertus* OR whoop*'. We searched MEDLINE using the following strategy:
1. explode PERTUSSIS‐VACCINE / all subheadings 2. explode BORDETELLA‐PERTUSSIS / all subheadings 3. explode WHOOPING‐COUGH / all subheadings 4. PERTUS* 5. WHOOP* 6. #1 or #2 or #3 or #4 or #5
The files downloaded from MEDLINE SilverPlatter were screened for randomised controlled trials using the RCT FILTER.
Appendix 2. April 2009 updated search strategies
In the 2009 updated review we searched the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2009, Issue 2), which contains the Cochrane Acute Respiratory Infections Group's Specialised Register, MEDLINE (1950 to April week 2 2009) and EMBASE.com (1974 to April 2009).
We used the following search strategy to search MEDLINE and CENTRAL. The MEDLINE search was combined with the Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐ maximising version (2008 revision); Ovid format (Lefebvre 2011). We modified the search strategy to search Embase.com (see below).
MEDLINE (Ovid)
1 exp Pertussis Vaccine/ 2 pertussis vaccin*.tw. 3 Whooping Cough/ 4 whoop*.tw. 5 Bordetella pertussis/ 6 pertuss*.tw. 7 1 or 2 or 3 or 4 or 5 or 6 8 exp Vaccines, Acellular/ 9 (acellular adj5 vaccin*).tw. 10 8 or 9 11 7 and 10
EMBASE.com
1. 'pertussis vaccine'/de 2. 'diphtheria pertussis tetanus vaccine'/de 3. 'diphtheria pertussis poliomyelitis tetanus vaccine'/de 4. 'diphtheria pertussis tetanus haemophilus influenzae type b hepatitis b vaccine'/de 5. 'diphtheria pertussis tetanus haemophilus influenzae type b vaccine'/de 6. 'bordetella pertussis'/de 7. 'pertussis'/de 8. 'dpt vaccine':ti,ab 9. pertuss*:ti,ab 10. whoop*:ti,ab 11. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 12. 'acellular vaccine'/de 13. (acellular:ti,ab AND vaccin*:ti,ab) 14. #12 OR #13 15. #11 AND #14 16. 'randomized controlled trial'/de 17. 'controlled clinical trial'/de 18. 'single blind procedure'/de 19. 'double blind procedure'/de 20. 'phase 3 clinical trial'/de 21. random*:ti,ab 22. placebo*:ti,ab 23. 'clinical trial':it 24. 'randomized controlled trial':it 25. (singl*:ti,ab OR doubl*:ti,ab OR trebl*:ti,ab OR tripl*:ti,ab) AND (mask*:ti,ab OR blind*:ti,ab) 26. 'controlled clinical trial':ti,ab 27. 'controlled clinical trials':ti,ab 28. #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 29. ('nonhuman'/exp OR 'animal'/exp) NOT 'human'/exp 30. #28 NOT #29 31. #15 AND #30
Appendix 3. MEDLINE search strategy
1 exp Pertussis Vaccine/ 2 pertussis vaccin*.tw. 3 Whooping Cough/ 4 whoop*.tw. 5 Bordetella pertussis/ 6 pertuss*.tw. 7 1 or 2 or 3 or 4 or 5 or 6 8 exp Vaccines, Acellular/ 9 (acellular adj5 vaccin*).tw. 10 8 or 9 11 7 and 10
Appendix 4. EMBASE search strategy
#14. #10 AND #13 269 17 May 2011 #13. #11 OR #12 861,917 17 May 2011 #12. random*:ab,ti OR placebo*:ab,ti OR factorial*:ab,ti OR crossover*:ab,ti OR 'cross over':ab,ti OR 'cross‐over':ab,ti OR volunteer*:ab,ti OR assign*:ab,ti OR allocat*:ab,ti OR ((singl* OR doubl*) NEAR/1 blind*):ab,ti AND [embase]/lim 822,212 17 May 2011 #11. 'randomized controlled trial'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp AND [embase]/lim 241,991 17 May 2011 #10. #6 AND #9 1,393 17 May 2011 #9. #7 OR #8 1,451 17 May 2011 #8. (acellular NEAR/5 vaccin*):ab,ti AND [embase]/lim 1,321 17 May 2011 #7. 'acellular vaccine'/de AND [embase]/lim 271 17 May 2011 #6. #1 OR #2 OR #3 OR #4 OR #5 25,318 17 May 2011 #5. pertuss*:ab,ti OR whoop*:ab,ti AND [embase]/lim 19,626 17 May 2011 #4. 'bordetella pertussis'/de AND [embase]/lim 4,046 17 May 2011 #3. 'pertussis'/de AND [embase]/lim 5,903 17 May 2011 #2. 'diphtheria pertussis tetanus vaccine'/de OR 'diphtheria pertussis tetanus hepatitis b vaccine'/de OR 'diphtheria pertussis tetanus haemophilus influenzae type b vaccine'/de OR 'diphtheria pertussis tetanus haemophilus influenzae type b hepatitis b vaccine'/de OR 'diphtheria pertussis poliomyelitis tetanus vaccine'/de OR 'diphtheria pertussis poliomyelitis tetanus hepatitis b vaccine'/de OR 'diphtheria pertussis poliomyelitis tetanus haemophilus influenzae type b hepatitis b vaccine'/de AND [embase]/lim 5,402 17 May 2011 #1. 'pertussis vaccine'/de AND [embase]/lim 4,338 17 May 2011
Appendix 5. Biosis Previews (Thomson ISI)
Topic=(pertuss* or whoop* or bordetella*) AND Topic=(acellular vaccin*)
Refined by: Topic=(random* or placebo* or clinical trial* or singl* blind* or doubl* blind*)
Appendix 6. CINAHL (Ebsco)
S18 S7 and S16 S17 S7 and S16 S16 S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 S15 (MH "Quantitative Studies") S14 (MH "Placebos") S13 TI placebo* or AB placebo* S12 TI random* or AB random* S11 TI ( singl* blind* or doubl* blind* or tripl* blind* or trebl* blind* or singl* mask* or doubl* mask* or tripl* mask* or trebl* mask* ) or AB (singl* blind* or doubl* blind* or tripl* blind* or trebl* blind* or singl* mask* or doubl* mask* or tripl* mask* or trebl* mask* ) S10 TI clinic* trial* or AB clinic* trial* S9 PT clinical trial S8 (MH "Clinical Trials+") S7 S5 and S6 Search modes S6 TI acellular N5 vaccin* or AB acellular N5 vaccin* S5 S1 or S2 or S3 or S4 S4 TI ( whoop* or pertuss* ) or AB ( whoop* or pertuss* ) S3 (MH "Bordetella Pertussis") S2 (MH "Whooping Cough") S1 (MH "Pertussis Vaccine+")
Appendix 7. Comparison of results between the first published (primary) review and the updated reviews
Minor adverse events: acellular vaccines versus whole‐cell vaccines
| Outcomes |
Number of trials Primary Update |
Sample size (n) Primary Update |
Effect size ‐ RR (95% CI) Primary Update |
|
Anorexia Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
10 11 7 8 8 9 11 14 3 4 |
13,942 19,632 18,232 18,501 18,385 18,646 1380 1939 8033 8447 |
0.47 (0.35 to 0.64) 0.43 (0.32 to 0.57) 0.50 (0.37 to 0.67) 0.45 (0.33 to 0.60) 0.52 (0.45 to 0.61) 0.50 (0.43 to 0.60) 0.35 (0.24 to 0.51) 0.40 (0.30 to 0.54) 0.41 (0.26 to 0.64) 0.42 (0.31 to 0.58) |
|
Drowsiness Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
11 12 7 8 8 9 10 14 2 3 |
20,200 20,490 19,039 19,308 19,169 19,430 1695 2254 7953 8367 |
0.74 (0.50 to 1.09) 0.72 (0.50 to 1.03) 0.67 (0.43 to 1.03) 0.61 (0.41 to 0.93) 0.56 (0.40 to 0.79) 0.56 (0.40 to 0.77) 0.43 (0.34 to 0.53) 0.48 (0.41 to 0.56) 0.50 (0.45 to 0.55) 0.49 (0.44 to 0.54) |
|
Fever Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
18 19 15 17 15 17 19 25 5 8 |
22,977 23,267 21,652 22,001 21,390 21,731 2513 3381 9012 9879 |
0.17 (0.13 to 0.21) 0.17 (0.13 to 0.20) 0.31(0.25 to 0.37) 0.31 (0.26 to 0.37) 0.34 (0.29 to 0.38) 0.34 (0.30 to 0.38) 0.33 (0.24 to 0.45) 0.33 (0.26 to 0.43) 0.38 (0.21 to 0.68) 0.35 (0.22 to 0.55) |
|
Irritability/fretfulness Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
13 15 10 12 11 13 13 16 4 6 |
20,337 20,707 19,080 19,429 19,170 19,511 1978 2596 9165 9856 |
0.50 (0.43 to 0.58) 0.48 (0.42 to 0.56) 0.51 (0.43 to 0.60) 0.48 (0.41 to 0.56) 0.55 (0.49 to 0.62) 0.53 (0.47 to 0.59) 0.32 (0.23 to 0.45) 0.36 (0.28 to 0.47) 0.48 (0.44 to 0.51) 0.48 (0.44 to 0.51) |
|
Prolonged crying Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
7 8 5 6 6 7 4 7 2 2 |
16,849 17,184 16,078 16,347 16,248 16,545 955 996 7943 7943 |
0.14 (0.12 to 0.18) 0.15 (0.11 to 0.19) 0.29 (0.24 to 0.35) 0.29 (0.24 to 0.35) 0.32 (0.24 to 0.44) 0.33 (0.24 to 0.46) 0.23 (0.10 to 0.55) 0.21 (0.10 to 0.48) 0.48 (0.22 to 1.02) 0.27 (0.02 to 3.12) |
|
Vomiting Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
7 8 6 7 5 6 5 6 1 1 |
11,160 11,450 10,716 10,985 10,552 10,813 703 744 86 86 |
0.78 (0.67 to 0.90) 0.77 (0.66 to 0.88) 0.71 (0.60 to 0.85) 0.62 (0.45 to 0.86) 0.80 (0.66 to 0.97) 0.69 (0.46 to 1.04) 0.35 (0.15 to 0.82) 0.50 (0.22 to 1.11) 1.07 (0.10 to 11.34) 1.07 (0.10 to 11.34) |
|
Pain/tenderness Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
10 13 8 11 9 12 16 21 3 5 |
11,683 14,180 10,710 13,186 10,938 13,333 2433 3051 1572 2263 |
0.21 (0.16 to 0.28) 0.20 (0.16 to 0.25) 0.19 (0.16 to 0.22) 0.18 (0.15 to 0.22) 0.21 (0.19 to 0.25) 0.20 (0.17 to 0.24) 0.40 (0.31 to 0.51) 0.43 (0.36 to 0.53) 0.36 (0.26 to 0.49) 0.43 (0.32 to 0.58) |
|
Redness Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
11 13 10 12 11 13 16 22 3 5 |
6783 7253 6078 6427 6291 6632 2187 3055 1572 2263 |
0.30 (0.22 to 0.40) 0.30 (0.23 to 0.39) 0.42 (0.32 to 0.55) 0.39 (0.29 to 0.51) 0.48 (0.41 to 0.56) 0.47 (0.41 to 0.54) 0.52 (0.43 to 0.63) 0.51 (0.44 to 0.59) 0.67 (0.51 to 0.89) 0.65 (0.52 to 0.80) |
|
Swelling/induration Primary dose 1 Primary dose 2 Primary dose 3 aP booster (pre wP) aP booster (pre aP) |
13 15 12 14 13 15 17 22 4 6 |
14,242 14,612 13,430 13,779 13,575 13,916 2433 3301 1730 2421 |
0.24 (0.19 to 0.31) 0.24 (0.19 to 0.31) 0.38 (0.30 to 0.47) 0.35 (0.28 to 0.45) 0.41 (0.30 to 0.57) 0.40 (0.29 to 0.54) 0.51 (0.44 to 0.59) 0.51 (0.46 to 0.57) 0.61 (0.47 to 0.80) 0.68 (0.58 to 0.80) |
CI: confidence interval RR: risk ratio
Appendix 8. Incidence of severe adverse events: acellular vaccines versus whole‐cell vaccines
| Outcomes | Number of trials |
Acellular vaccines n/N* Incidence # |
Whole‐cell vaccines n/N Incidence |
RR 95% CI |
| Primary series non‐completion due to adverse events | 11 | 248/80,060 3.09 | 338/28,849 11.72 | 0.23 (0.12 to 0.43) |
|
Death (infection) Primary series |
13 |
5/22,154 0.23 |
3/12,344 0.24 |
0.97 (0.23 to 4.16) |
|
Death (all causes) Primary series |
16 |
81/86,863 0.93 |
61/35,588 1.71 |
0.87 (0.62 to 1.22) |
|
Encephalopathy Primary series |
9 |
0/81,601 0.0 |
0/32,161 0.0 |
‐ |
|
Convulsions Primary series Booster |
15 11 |
43/88,513 0.49 1/2250 0.44 |
41/35,874 1.14 0/397 0.0 |
0.47 (0.31 to 0.73) 0.46 (0.02 to 11.2) |
|
Hypotonic hyporesponsive episodes Primary series Booster |
11 7 |
70/86,347 0.81 0/2171 0.0 |
51/35,226 1.45 0/316 0.0 |
0.26 (0.08 to 0.81) ‐ |
*n/N: number of events/number of recipients # Incidence: number of events per 1000 recipients CI: confidence interval RR: risk ratio
Appendix 9. Incidence of minor adverse events over the primary series: acellular vaccines versus whole‐cell vaccines
| Outcomes |
Dose 1 Acellular Whole‐cell n/N* n/N Incidence# Incidence Number of trials |
Dose 2 Acellular Whole‐cell n/N n/N Incidence Incidence Number of trials |
Dose 3 Acellular Whole‐cell n/N n/N Incidence Incidence Number of trials |
| Anorexia | 1275/12,310 1896/7322 103.57 258.95 11 |
1047/11,653 1208/6848 89.85 176.40 8 |
941/11,840 966/6806 79.48 141.93 9 |
| Drowsiness | 3427/12,813 2435/7677 267.46 317.18 11 |
2087/12,134 1500/7174 171.99 209.09 9 |
1548/12,301 1231/7129 125.84 172.67 10 |
| Fever | 898/14,910 3900/8357 60.23 466.67 19 |
1809/14,167 3427/7834 127.69 437.45 17 |
2270/14,024 3439/7707 161.87 446.22 17 |
| Irritability/fretfulness | 3645/12,875 4488/7832 283.11 573.03 15 |
3763/12,144 3695/7285 309.86 507.21 12 |
3482/12,268 3228/7243 283.83 445.67 13 |
| Prolonged crying | 116/10,364 395/6820 11.19 57.92 8 |
161/9867 248/6480 16.32 38.27 6 |
68/10,096 94/6449 6.74 14.58 7 |
| Vomiting | 530/8126 269/3324 65.22 80.93 8 |
356/7885 189/3100 45.15 60.97 7 |
322/7788 148/3025 41.35 48.93 7 |
| Pain/tenderness | 733/10,279 1840/3901 71.31 471.67 13 |
710/9642 1606/3544 73.64 453.16 11 |
712/9769 1358/3564 72.88 381.03 12 |
| Redness | 512/5348 620/1805 95.74 343.49 13 |
599/4821 551/1606 124.25 343.09 12 |
807/4972 583/1660 162.31 351.20 13 |
| Swelling/induration | 1238/10,606 1670/4006 116.73 416.87 15 |
1842/10,034 1576/3745 183.58 420.83 14 |
2791/10,157 1658/3759 274.79 441.07 15 |
*n/N: number of events/number of recipients # Incidence: number of events per 1000 recipients
Appendix 10. Incidence of minor adverse events in boosters: acellular vaccines versus whole‐cell vaccines
| Outcomes |
aP booster (previous wP) versus wP booster (previous wP) Acellular Whole‐cell n/N* n/N Incidence# Incidence Number of trials |
aP booster (previous aP) versus wP booster (previous wP) Acellular Whole‐cell n/N n/N Incidence Incidence Number of trials |
| Anorexia | 129/1149 244/790 112.27 308.86 14 |
457/4474 735/3973 102.15 184.99 4 |
| Drowsiness | 203/1453 292/801 139.71 364.54 13 |
532/4419 913/3948 120.39 231.26 3 |
| Fever | 191/2184 305/1197 87.45 254.80 24 |
1320/5838 2058/4041 226.10 509.28 8 |
| Irritability/fretfulness | 314/1728 445/868 181.71 512.67 17 |
1256/5837 1379/4019 215.18 343.12 6 |
| Prolonged crying | 7/666 20/330 10.51 60.61 6 |
9/4105 15/3838 2.19 3.91 2 |
| Vomiting | 18/551 13/193 32.67 67.36 6 |
2/56 1/30 35.71 33.33 1 |
| Pain/tenderness | 620/2018 661/1033 307.23 639.88 21 |
691/2015 203/248 342.93 818.55 5 |
| Redness | 503/1980 523/1075 254.04 486.51 21 |
692/2015 135/248 343.42 544.35 5 |
| Swelling/induration | 446/2144 493/1157 208.02 426.10 22 |
575/2139 112/282 268.82 397.16 6 |
*n/N: number of events/number of recipients # Incidence: number of events per 1000 recipients
Appendix 11. Incidence of severe adverse events: acellular vaccines versus placebo/DT
| Outcomes | Number of trials |
Acellular vaccine n/N* Incidence# |
Placebo/DT n/N Incidence |
RR 95% CI |
| Primary series non‐completion due to adverse events | 4 | 65/19,092 3.40 | 28/6809 4.11 | 0.70 (0.38 to 1.29) |
|
Death (infection) Primary series |
4 |
5/19,093 0.26 |
1/6809 0.15 |
1.21 (0.19 to 7.80) |
|
Death (all causes) Primary series |
4 |
10/19,092 0.52 |
2/6809 0.29 |
1.08 (0.26 to 4.42) |
|
Encephalopathy Primary series |
2 |
0/14,521 0.0 |
0/4129 0.0 |
‐ |
|
Convulsions Primary series |
4 |
27/19,092 1.41 |
28/6809 4.11 |
0.44 (0.12 to 1.69) |
|
Hypotonic hyporesponsive episodes Primary series |
4 |
2/19,092 0.10 |
2/6809 0.29 |
0.29 (0.02 to 5.13) |
*n/N: number of events/number of recipients # Incidence: number of events per 1000 recipients CI: confidence interval RR: risk ratio
Appendix 12. Incidence of minor adverse events over the primary series: acellular vaccines versus placebo/DT
| Outcomes |
Dose 1 Acellular Placebo/DT n/N* n/N Incidence# Incidence Number of trials |
Dose 2 Acellular Placebo/DT n/N n/N Incidence Incidence Number of trials |
Dose 3 Acellular Placebo/DT n/N n/N Incidence Incidence Number of trials |
| Anorexia | 750/8000 325/3526 93.75 92.17 2 |
656/7903 276/3483 83.00 79.24 2 |
418/5085 195/2538 82.20 76.83 1 |
| Drowsiness | 1696/7557 792/3397 224.43 233.15 2 |
1145/7334 549/3286 156.12 167.07 2 |
718/5085 350/2538 141.20 137.90 1 |
| Fever | 557/7762 230/3493 71.76 65.85 3 |
1053/7476 502/3377 140.85 148.65 3 |
1135/5070 552/2584 223.87 213.63 2 |
| Irritability/fretfulness | 2373/8000 1084/3526 296.63 307.43 2 |
2716/7903 1267/3483 343.67 368.53 2 |
1848/5085 958/2538 363.42 377.46 1 |
| Prolonged crying | 132/7999 50/3526 16.50 14.18 2 |
181/7903 78/3483 22.90 22.39 2 |
55/5085 26/2538 10.82 10.24 1 |
| Vomiting | 513/8000 209/3526 64.13 59.27 2 |
370/7903 185/3483 46.82 53.12 2 |
241/5085 131/2538 47.39 51.62 1 |
| Pain/tenderness | 458/7940 226/3511 57.68 64.37 2 |
630/7772 289/3430 81.06 84.26 2 |
511/5085 253/2538 100.49 99.68 1 |
| Redness | 9/2787 1/937 3.23 1.07 1 |
160/2661 6/874 60.13 6.86 1 |
‐ ‐ |
| Swelling/induration | 933/8042 391/3610 116.02 108.31 3 |
1549/7874 663/3527 196.72 187.98 3 |
2340/5184 1030/2632 451.39 391.34 2 |
*n/N: number of events/number of recipients # Incidence: number of events per 1000 recipients
Data and analyses
Comparison 1. Safety: acellular versus whole‐cell pertussis vaccines.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary series non‐completion due to adverse events | 14 | 108909 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.12, 0.43] |
| 2 Death (all causes) | 16 | 122451 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.62, 1.22] |
| 2.1 Primary series | 16 | 122451 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.62, 1.22] |
| 3 Death (infection) | 13 | 34498 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.23, 4.16] |
| 3.1 Primary series | 13 | 34498 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.23, 4.16] |
| 4 Encephalopathy | 9 | 113762 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 Primary series | 9 | 113762 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Convulsions | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Primary series | 15 | 124387 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.31, 0.73] |
| 5.2 Booster | 11 | 2647 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.02, 11.20] |
| 6 Hypotonic hyporesponsive episodes | 18 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Primary series | 11 | 121573 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.08, 0.81] |
| 6.2 Booster | 7 | 2487 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Anorexia | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Primary series: Dose 1 | 11 | 19632 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.32, 0.57] |
| 7.2 Primary series: Dose 2 | 8 | 18501 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.33, 0.60] |
| 7.3 Primary series: Dose 3 | 9 | 18646 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.43, 0.60] |
| 7.4 aP booster (previous wP) versus wP booster (previous wP) | 14 | 1939 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.30, 0.54] |
| 7.5 aP booster (previous aP) versus wP booster (previous wP) | 4 | 8447 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.31, 0.58] |
| 8 Drowsiness | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Primary series: Dose 1 | 12 | 20490 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.45, 0.68] |
| 8.2 Primary series: Dose 2 | 9 | 19308 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.35, 0.60] |
| 8.3 Primary series: Dose 3 | 10 | 19430 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.40, 0.77] |
| 8.4 aP booster (previous wP) versus wP booster (previous wP) | 13 | 2254 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.41, 0.56] |
| 8.5 aP booster (previous aP) versus wP booster (previous wP) | 3 | 8367 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.44, 0.54] |
| 9 Fever | 46 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Primary series: Dose 1 | 19 | 23267 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.13, 0.20] |
| 9.2 Primary series: Dose 2 | 17 | 22001 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.26, 0.37] |
| 9.3 Primary series: Dose 3 | 17 | 21731 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.30, 0.38] |
| 9.4 aP booster (previous wP) versus wP booster (previous wP) | 24 | 3381 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.26, 0.43] |
| 9.5 aP booster (previous aP) versus wP booster (previous wP) | 8 | 9879 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.22, 0.55] |
| 10 Irritability/fretfulness | 33 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Primary series: Dose 1 | 15 | 20707 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.42, 0.56] |
| 10.2 Primary series: Dose 2 | 12 | 19429 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.41, 0.56] |
| 10.3 Primary series: Dose 3 | 13 | 19511 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.47, 0.59] |
| 10.4 aP booster (previous wP) versus wP booster (previous wP) | 17 | 2596 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.28, 0.47] |
| 10.5 aP booster (previous aP) versus wP booster (previous wP) | 6 | 9856 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.44, 0.51] |
| 11 Prolonged crying | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Primary series: Dose 1 | 8 | 17184 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.11, 0.19] |
| 11.2 Primary series: Dose 2 | 6 | 16347 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.24, 0.35] |
| 11.3 Primary series: Dose 3 | 7 | 16545 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.24, 0.46] |
| 11.4 aP booster (previous wP) versus wP booster (previous wP) | 6 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.10, 0.48] |
| 11.5 aP booster (previous aP) versus wP booster (previous wP) | 2 | 7943 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.02, 3.12] |
| 12 Vomiting | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 Primary series: Dose 1 | 8 | 11450 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.66, 0.88] |
| 12.2 Primary series: Dose 2 | 7 | 10985 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.45, 0.86] |
| 12.3 Primary series: Dose 3 | 7 | 10813 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.46, 1.04] |
| 12.4 aP booster (previous wP) versus wP booster (previous wP) | 6 | 744 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.22, 1.11] |
| 12.5 aP booster (previous aP) versus wP booster (previous wP) | 1 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.10, 11.34] |
| 13 Pain/tenderness | 35 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Primary series: Dose 1 | 13 | 14180 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.16, 0.25] |
| 13.2 Primary series: Dose 2 | 11 | 13186 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.15, 0.22] |
| 13.3 Primary series: Dose 3 | 12 | 13333 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.17, 0.24] |
| 13.4 aP booster (previous wP) versus wP booster (previous wP) | 21 | 3051 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.36, 0.53] |
| 13.5 aP booster (previous aP) versus wP booster (previous wP) | 5 | 2263 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.32, 0.58] |
| 14 Redness | 35 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Primary series: Dose 1 | 13 | 7153 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.23, 0.39] |
| 14.2 Primary series: Dose 2 | 12 | 6427 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.29, 0.51] |
| 14.3 Primary series: Dose 3 | 13 | 6632 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.41, 0.54] |
| 14.4 aP booster (previous wP) versus wP booster (previous wP) | 21 | 3055 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.44, 0.59] |
| 14.5 aP booster (previous aP) versus wP booster (previous wP) | 5 | 2263 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.52, 0.80] |
| 15 Swelling/induration | 39 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Primary series: Dose 1 | 15 | 14612 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.19, 0.31] |
| 15.2 Primary series: Dose 2 | 14 | 13779 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.28, 0.45] |
| 15.3 Primary series: Dose 3 | 15 | 13916 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.29, 0.54] |
| 15.4 aP booster (previous wP) versus wP booster (previous wP) | 22 | 3301 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.46, 0.57] |
| 15.5 aP booster (previous aP) versus wP booster (previous wP) | 6 | 2421 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.58, 0.80] |
1.8. Analysis.
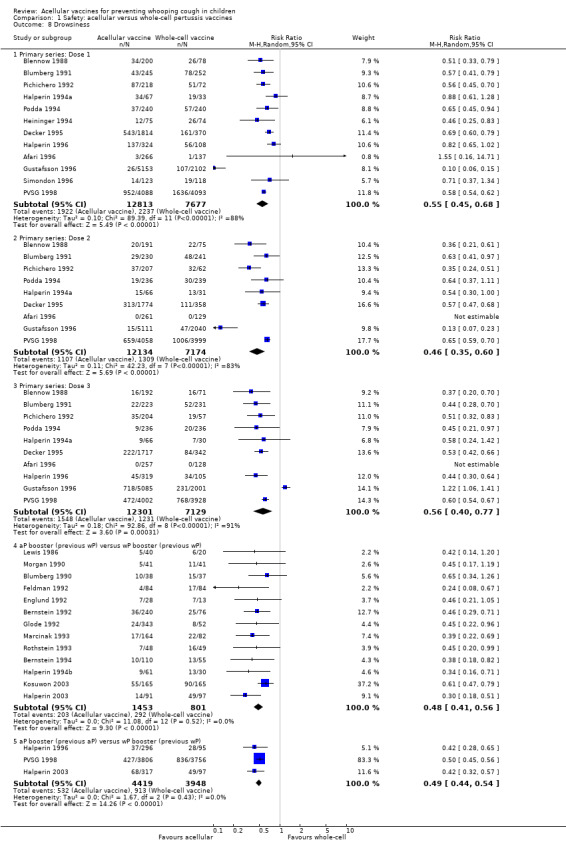
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 8 Drowsiness.
1.9. Analysis.
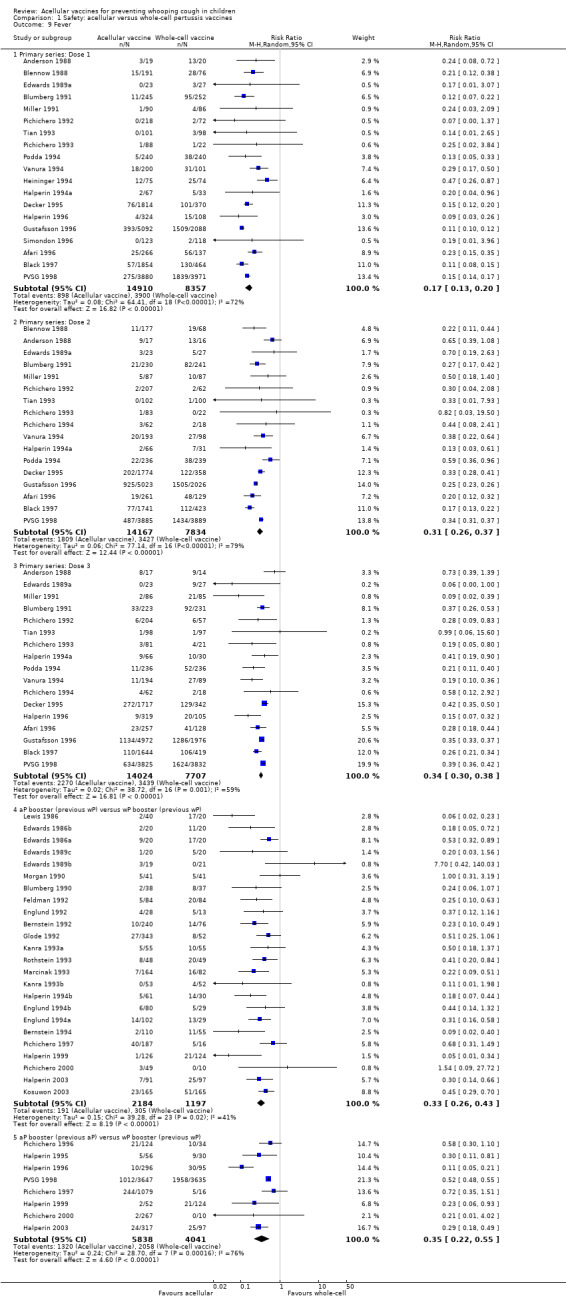
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 9 Fever.
1.10. Analysis.
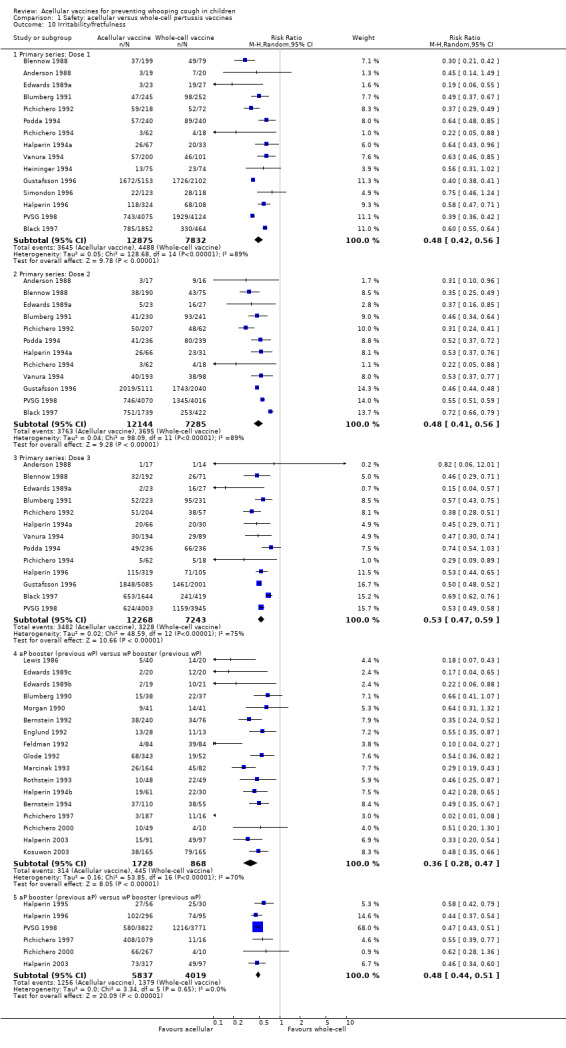
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 10 Irritability/fretfulness.
1.11. Analysis.
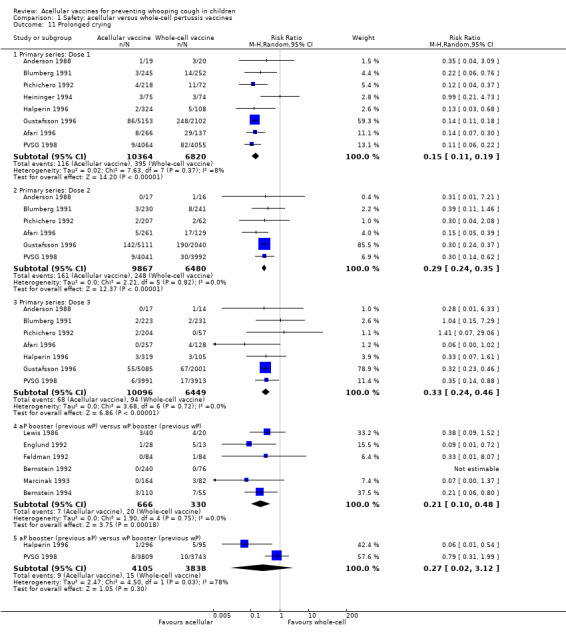
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 11 Prolonged crying.
1.12. Analysis.
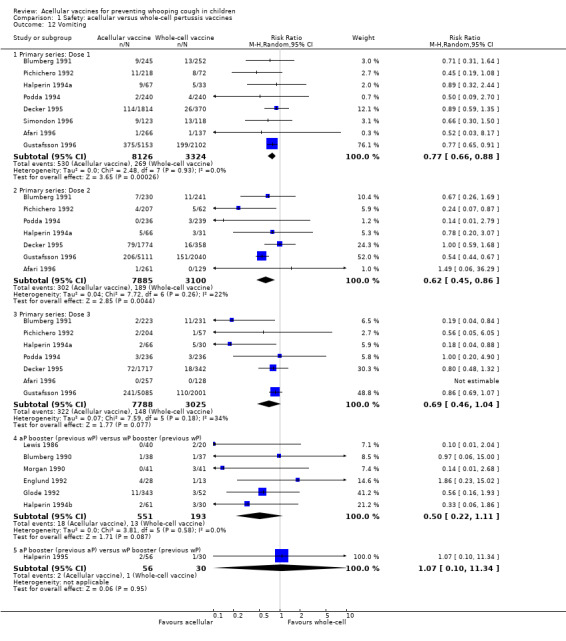
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 12 Vomiting.
1.13. Analysis.
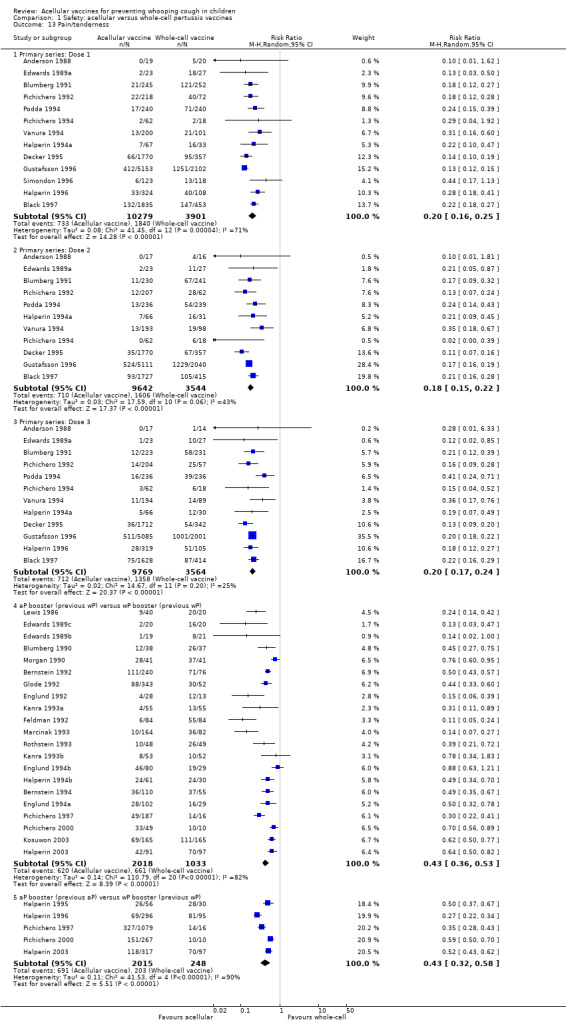
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 13 Pain/tenderness.
1.14. Analysis.
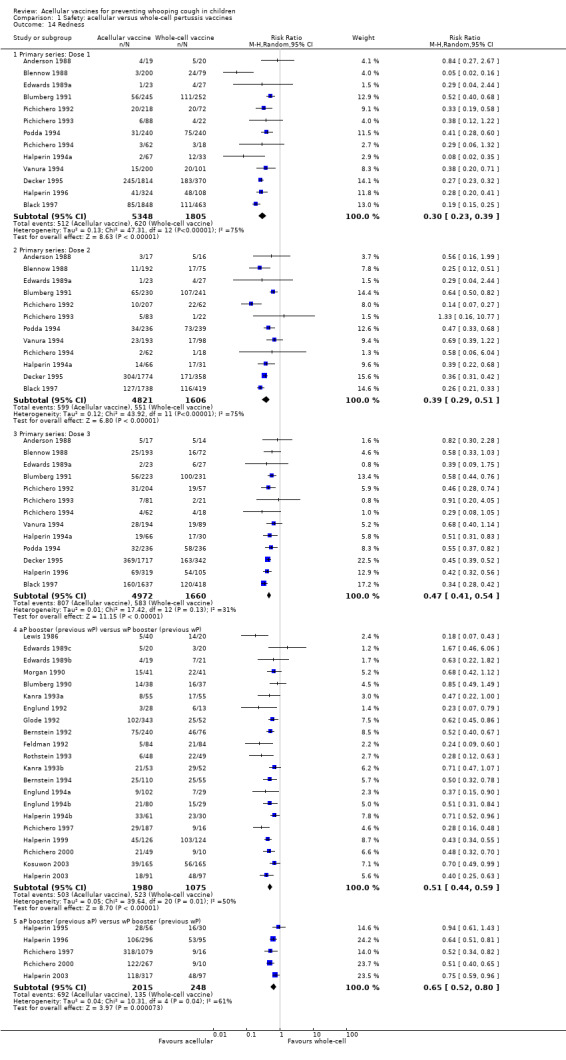
Comparison 1 Safety: acellular versus whole‐cell pertussis vaccines, Outcome 14 Redness.
Comparison 2. Safety: acellular vaccines versus placebo/DT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Primary series non‐completion due to adverse events | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.38, 1.29] |
| 2 Death (all causes) | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.26, 4.42] |
| 2.1 Primary series | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.26, 4.42] |
| 3 Death (infection) | 4 | 25902 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.19, 7.80] |
| 3.1 Primary series | 4 | 25902 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.19, 7.80] |
| 4 Encephalopathy | 2 | 18650 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 Primary series | 2 | 18650 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Convulsions | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.12, 1.69] |
| 5.1 Primary series | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.12, 1.69] |
| 6 Hypotonic hyporesponsive episodes | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.02, 5.13] |
| 6.1 Primary series | 4 | 25901 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.02, 5.13] |
| 7 Anorexia | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Primary series: Dose 1 | 2 | 11526 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.93, 1.20] |
| 7.2 Primary series: Dose 2 | 2 | 11386 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.66, 1.46] |
| 7.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.91, 1.26] |
| 8 Drowsiness | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Primary series: Dose 1 | 2 | 10954 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.96, 1.11] |
| 8.2 Primary series: Dose 2 | 2 | 10620 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.91, 1.09] |
| 8.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.91, 1.15] |
| 9 Fever | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Primary series: Dose 1 | 3 | 11255 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.73, 1.90] |
| 9.2 Primary series: Dose 2 | 3 | 10853 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.91, 1.11] |
| 9.3 Primary series: Dose 3 | 2 | 7654 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.94, 1.13] |
| 10 Irritability/fretfulness | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Primary series: Dose 1 | 2 | 11526 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.93, 1.05] |
| 10.2 Primary series: Dose 2 | 2 | 11386 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.93, 1.03] |
| 10.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.91, 1.02] |
| 11 Prolonged crying | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Primary series: Dose 1 | 2 | 11525 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.71, 2.34] |
| 11.2 Primary series: Dose 2 | 2 | 11386 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.83, 1.40] |
| 11.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.66, 1.68] |
| 12 Vomiting | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 Primary series: Dose 1 | 2 | 11526 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.94, 1.30] |
| 12.2 Primary series: Dose 2 | 2 | 11386 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| 12.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.75, 1.13] |
| 13 Pain/tenderness | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Primary series: Dose 1 | 2 | 11451 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.78, 1.32] |
| 13.2 Primary series: Dose 2 | 2 | 11202 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.81, 1.51] |
| 13.3 Primary series: Dose 3 | 1 | 7623 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.87, 1.16] |
| 14 Swelling/induration | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Primary series: Dose 1 | 3 | 11652 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.62, 2.68] |
| 14.2 Primary series: Dose 2 | 3 | 11401 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.54, 8.01] |
| 14.3 Primary series: Dose 3 | 2 | 7816 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [1.07, 1.20] |
| 15 Redness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Primary series: Dose 1 | 1 | 3724 | Risk Ratio (M‐H, Random, 95% CI) | 3.03 [0.38, 23.85] |
| 15.2 Primary series: Dose 2 | 1 | 3535 | Risk Ratio (M‐H, Random, 95% CI) | 8.76 [3.89, 19.72] |
| 15.3 Primary series: Dose 3 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
2.8. Analysis.
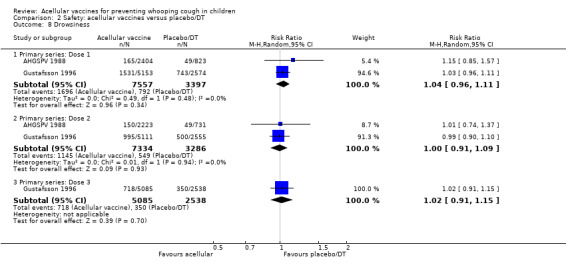
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 8 Drowsiness.
2.9. Analysis.
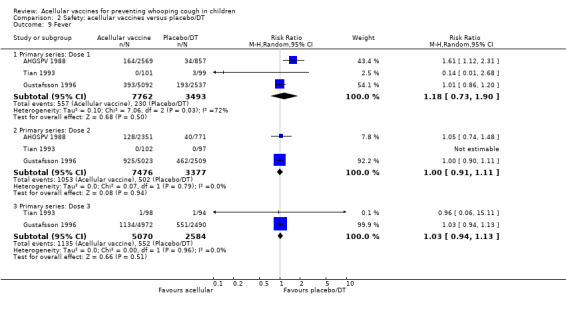
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 9 Fever.
2.10. Analysis.
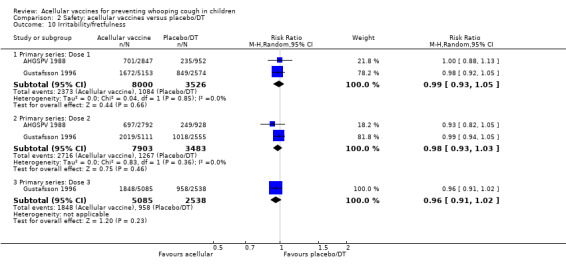
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 10 Irritability/fretfulness.
2.11. Analysis.
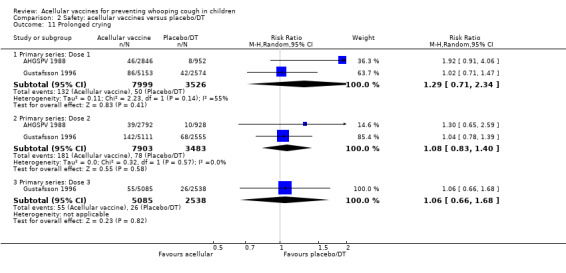
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 11 Prolonged crying.
2.12. Analysis.
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 12 Vomiting.
2.13. Analysis.
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 13 Pain/tenderness.
2.14. Analysis.
Comparison 2 Safety: acellular vaccines versus placebo/DT, Outcome 14 Swelling/induration.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Afari 1996.
| Methods | Site: Ghana Design: parallel‐group RCT | |
| Participants | Included: age 6 weeks Excluded: neurological disorder; serious disease; birth weight < 2 kg | |
| Interventions | Primary series (aP versus wP)
Number randomised: 266 aP, 137 wP Dose schedule: 3 doses (6, 10, 14 weeks) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Local adverse events excluded (results only for all local adverse event types combined). No statement on antipyretic/analgesic use. In this review, results are combined for the 2 aP vaccine formulations | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Using a computer program (EPI Info) |
| Allocation concealment (selection bias) | Unclear risk | On‐site computer assignment but file locking not reported. Vaccines visually distinguishable |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Parents and nurses collecting efficacy and adverse event data were blinded but blinding details not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Parents and nurses collecting efficacy and adverse event data were blinded but blinding details not stated |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Parents and nurses collecting efficacy and adverse event data were blinded but blinding details not stated |
AHGSPV 1988.
| Methods | Site: Sweden
Design: DB parallel‐group RCT Active case ascertainment (monthly telephone). Case incidence adjusted for follow‐up duration by actuarial method. Parents recorded adverse events in diary |
|
| Participants | Included: age 5 to 11 months Excluded: suspected progressive neurological disease; failure to thrive; renal failure; cardiac failure; prior pertussis or pertussis immunisation |
|
| Interventions | Primary series (aP versus placebo)
Number randomised: 2837 aP, 954 placebo Dose schedule: 2 doses (entry + 8 to 12 weeks later) Concurrent vaccines: DT and IPV (not within 1 week before or 2 weeks after aP or wP) |
|
| Outcomes |
|
|
| Notes | Macrolide prophylaxis not used (not recommended in Sweden for age > 6 months and efficacy follow‐up in this study started at minimum age of 7 months). Reactive antipyretic/analgesic use allowed. Blinding of study nurses confirmed by questionnaire | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centrally generated random sequence |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
AHGSPV 1997.
| Methods | Site: Sweden
Design: DB parallel‐group RCT Passive case ascertainment (based on laboratory reports of pertussis and questionnaire of parents at 18‐month visit). Case incidence adjusted for follow‐up duration by Cox proportional hazard regression. Follow‐up of serious adverse events by active weekly surveillance in study area hospitals, reports from participating physicians and child health nurses, plus questioning of parents at each trial dose and when the child was 18 months old |
|
| Participants | Included: age 2 months Excluded: progressive neurological disease; uncontrolled epilepsy; infantile spasm; failure to thrive; renal failure; cardiac failure; immunosuppression; prior pertussis | |
| Interventions | Primary series (aP versus wP)
Number randomised: 62172 aP, 2072 wP Dose schedules: 3 doses (3, 5, 10 months ‐ 88% of vaccinees; 2, 4, 6 months ‐ 12% of vaccinees) Concurrent vaccines: HiB and IPV |
|
| Outcomes |
|
|
| Notes | There was good evidence that the passive case ascertainment led to significant under‐reporting of cases. It is possible (not discussed in study report) that under‐reporting of serious adverse events may have occurred but there are no grounds to suspect that under‐reporting would affect the acellular and whole‐cell groups differently Deaths recorded for this study are due to any cause and include at least 1 due to injury (vaccine group not specified) No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centrally generated random sequence |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Anderson 1988.
| Methods | Site: USA Design: DB parallel‐group RCT |
|
| Participants | Included: age 2 months; healthy Excluded: not stated | |
| Interventions | Primary series (aP versus wP) 1. DTaP: Wyeth/Takeda[4] 2. DTwP: Wyeth[W] Number randomised: 19 aP, 20 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated | |
| Outcomes |
|
|
| Notes | Reactive analgesic/antipyretic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation |
| Allocation concealment (selection bias) | Low risk | Vaccines in coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Bernstein 1992.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 4 to 6 years; healthy; completed DTwP primary series and 15‐ to 24‐month booster Excluded: personal or family history of developmental delay or neurological disorder; previous aP or DTaP; previous contraindicating reaction to DTwP | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number randomised: 240 aP, 76 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Prophylactic antipyretic/analgesic use not allowed. Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation at study site, but details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment details not reported |
| Selective reporting (reporting bias) | High risk | Data for vomiting collected but not reported |
Bernstein 1994.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Age: 15 to 20 months; healthy; completed DTwP primary series Excluded: personal or family history of developmental delay or neurological disorder; previous DTaP; previous contraindicating reaction to DTwP | |
| Interventions | BOOSTER (wP.aP versus wP.wP)
Number randomised: 110 aP, 22 wP Dose schedule: 1 dose (15 to 20 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for vomiting collected but not reported (stated to be "infrequent" and "not significantly different") |
Black 1997.
| Methods | Site: USA Design: parallel‐group RCT | |
| Participants | Included: age 2 months Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number randomised: 2000 aP, 498 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccines: HiB (separate injection site) and OPV |
|
| Outcomes |
|
|
| Notes | Booster dose of Chiron/Biocine[3] or Lederle/Takeda[4] at age 15 to 18 months. Data for this dose are not included as there was no DTwP or placebo control SIDS was recorded in 4/2000 (0.2%) DTaP and 1/498 (0.2%) DTwP recipients in the first year of life. Death due to other causes was not studied Late onset fever (> 3 days after dose) occurred in both groups, with peak percentage slightly higher in DTaP group (approximately 5% versus 4.5% in DTwP group) but the overall percentage with fever over the 14‐day follow‐up was lower in the DTaP group No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Blennow 1988.
| Methods | Site: Sweden Design: parallel‐group RCT | |
| Participants | Included: age 6 months Excluded: contraindication to DTP vaccine as per ref Anon 1977 | |
| Interventions | Primary series (aP versus wP ‐ see notes)
Number randomised: 121 aP, 119 P, 79 wP Dose schedule: 3 doses (6, 7, 8 months) Concurrent vaccine: DT and polio vaccines not given within 2 weeks of dose |
|
| Outcomes |
|
|
| Notes | The whole‐cell series was given as 3 doses of whole‐cell vaccine. The acellular series was given as 3 doses of acellular vaccine, or 2 doses of acellular vaccine plus 1 dose of placebo (replacing the 1st, 2nd or 3rd dose of acellular vaccine). In this review, data for series non‐completion, deaths and serious adverse events relate to all acellular‐containing vaccine regimens. The acellular primary series non‐completion (due to adverse events) data include 1 infant who withdrew after receiving placebo at the 1st dose. Data for minor adverse events at each dose are recorded only for those patients who actually received the acellular vaccine at that dose Data for anorexia and vomiting collected but not reported No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence key concealed from study personnel and parents |
| Allocation concealment (selection bias) | Low risk | Vaccines indistinguishable |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
| Selective reporting (reporting bias) | High risk | Data for anorexia and vomiting collected but not reported |
Blumberg 1990.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 17 to 24 months; healthy; completed DTwP primary series Excluded: not stated | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 38 aP, 37 wP Dose schedule: 1 dose (17 to 24 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Blumberg 1991.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 2 months; healthy Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number randomised: 245 aP, 252 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | 1 death in DTwP arm due to accident (strangulation by pacifier cord) 4th dose of DTaP given at 18 months to all children. 4th dose not included in review because no DTwP control group for that dose. Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Decker 1995.
| Methods | Site: USA
Phased parallel‐group RCT (2 to 3 vaccines per phase) Parents recorded adverse events on forms |
|
| Participants | Included: age 6 to 12 weeks; healthy Excluded: born < 36 weeks gestation; immune system disease; major congenital malformation; serious chronic disease; developmental delay; neurological disease; convulsions; other contraindications to DTP vaccine as per ref CID 1991a | |
| Interventions | Primary series (aP versus wP)
Number randomised: 1827 aP, 373 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: HiB |
|
| Outcomes |
|
|
| Notes | In this review, safety results are combined for all acellular vaccines. Irritability and pain were reported only for the moderate/severe category At an additional retrospective medical record review 1 year after dose 3 (not all subjects studied), convulsions were documented in 1.1% DTaP recipients compared to 0.7% DTwP Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Vaccine vials labelled with letter codes instead of type/manufacturer details but unable to ascertain from study report whether vaccinators remained unaware of what each code represented |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Vaccinators took no further part in the study and did not participate in follow‐up data collection, so outcome assessment was double‐blind |
Edwards 1986a.
| Methods | Site: USA Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 18 to 24 months; completed DTwP primary series Excluded: not stated | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 20 aP, 20 wP Dose schedule: 1 dose (18 to 24 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for drowsiness and vomiting collected but not reported (stated to be "rare"). Data for irritability, pain, redness and induration only reported for moderate/severe category |
Edwards 1986b.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 4 to 6 years; completed DTwP primary series and 15‐ to 24‐month booster Excluded: not stated | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number randomised: 20 aP, 20 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for drowsiness and vomiting collected but not reported (stated to be "rare’'). Data for irritability, pain, redness and induration only reported for moderate/severe category |
Edwards 1989a.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: infants Excluded: previous DTP vaccination | |
| Interventions | Primary series (aP versus wP)
Number randomised: 23 aP, 27 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Follow‐up for deaths probably longer than until 2 weeks after dose 3 but not clearly stated to be so No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Edwards 1989b.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 18 to 24 months; completed DTwP primary series Excluded: not stated | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 19 aP, 21 wP Dose schedule: 1 dose (18 to 24 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Edwards 1989c.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 4 to 6 years; completed DTwP primary series and 18‐ to 24‐month booster Excluded: not stated | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number randomised: 20 aP, 20 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Englund 1992.
| Methods | Site: Houston, USA
Design: parallel‐group RCT Parents were interviewed by telephone. Parents recorded adverse events in diary for 14 days after immunisations |
|
| Participants | Included: ages 16 to 20 months, who had received primary immunisation with DTwP Excluded: not stated | |
| Interventions | Booster (aP versus wP)
Number randomised: 28 aP, 13 wP Dose schedule: 1 dose (16 to 20 months) Concurrent vaccines: none |
|
| Outcomes |
|
|
| Notes | Prolonged crying was studied as increased crying. Use of analgesic/antipyretic was allowed during the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Englund 1994a.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 16 to 21 months; healthy; completed DTwP primary series Excluded: known immune deficiency; receipt of blood products in past month; contraindication to DTwP as specified in ref CID 1991a; physician did not recommend DTwP | |
| Interventions | Booster (wP.aP versus wP.wP)
Number studied: 102 aP, 29 wP Dose schedule: 1 dose (16 to 21 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | The number of 16‐ to 24‐month old children randomised to aP or wP was not stated. A total of 258 children aged 16 to 24 months or 4 to 6 years were randomised in Englund 1994a and Englund 1994b combined: 192 to aP and 66 to wP. 240 of these contributed safety data Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for irritability collected but not reported |
Englund 1994b.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 4 to 6 years; healthy; completed DTwP primary series and 15‐ to 24‐month booster Excluded: known immune deficiency; receipt of blood products in past month; contraindication to DTwP as specified in ref CID 1991a; physician did not recommend DTwP | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number studied: 80 aP, 29 wP Dose schedule: 1 dose (16 to 21 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Number of 4 to 6‐year old children randomised to aP or wP not stated. A total of 258 children aged 16 to 24 months or 4 to 6 years were randomised in Englund 1994a and Englund 1994b combined: 192 to aP and 66 to wP. 240 of these contributed safety data. Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | Low risk | Data for irritability collected but not reported |
Feldman 1992.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 15 to 24 months; healthy; completed DTwP primary series Excluded: immune dysfunction; major congenital malformation; serious chronic disorder; developmental delay; receipt of immunoglobulin within past 3 months | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 84 aP, 78 wP Dose schedule: 1 dose (15 to 24 months) Concurrent vaccine: OPV; HiB 4 weeks after study vaccine |
|
| Outcomes |
|
|
| Notes | Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for vomiting collected but not reported |
Feldman 1993.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 2 months; healthy Excluded: born < 36 weeks gestation; immune disorder; major congenital malformation; serious chronic disease; ongoing immunoglobulin therapy; pertussis infection; personal or family history of neurological disorder or developmental delay | |
| Interventions | Primary series (aP versus wP)
Number randomised: 109 aP, 36 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: OPV |
|
| Outcomes |
|
|
| Notes | 1 infant did not complete the primary series due to an adverse event (high pitched cry) but the report does not state whether this infant received aP or wP. Deaths were not specifically reported but all infants either completed the study up to the final dose or withdrew due to reasons other than death Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for vomiting collected but not reported |
Glode 1992.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 17 to 24 months; healthy; full‐term; completed DTwP primary series Excluded: acute or chronic illness; history of diphtheria, tetanus or pertussis; known contraindication to DTP | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 345 aP, 52 wP Dose schedule: 1 dose (17 to 24 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Results for DTaP lots combined Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | On‐site randomisation |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for prolonged crying collected but not reported |
Greco 1996.
| Methods | Site: Italy Design: DB parallel‐group RCT Passive and active case ascertainment (parent report and monthly telephone). Case incidence adjusted for follow‐up duration by use of incidence density. Parents recorded adverse events in diary |
|
| Participants | Included: age 6 to 12 weeks; weight > 3rd percentile Excluded: history of seizures or central nervous system disease; known/suspected immunological deficit; major congenital abnormality; failure to thrive; renal failure; prior pertussis or pertussis vaccination | |
| Interventions | Primary series (aP versus wP versus DT)
Number randomised: 9368 aP, 4678 wP, 1555 DT Dose schedule: 3 doses (6 to 12, 13 to 20, and 21 to 28 weeks) Concurrent vaccine: OPV and hepatitis B vaccine with doses 1 and 2. Booster dose of DT to all subjects at 12 months of age |
|
| Outcomes |
|
|
| Notes | No statement on macrolide prophylaxis. Possible partial unblinding of DTwP (not discussed in article but study used same DTwP as Gustafsson96) No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Gustafsson 1996.
| Methods | Site: Sweden
Design: DB parallel‐group RCT Passive and active case ascertainment (parent report and telephone every 6 to 8 weeks). Case incidence adjusted for follow‐up duration by use of Cox proportional hazards regression. Parents recorded adverse events in diary |
|
| Participants | Included: age 2 to 3 months Excluded: progressive neurological disease; failure to thrive; renal failure; cardiac failure; uncontrolled epilepsy; infantile spasms; immunosuppression; prior pertussis or pertussis vaccination | |
| Interventions | Primary series (aP versus wP versus DT)
Number randomised: 5153 aP, 2102 wP, 2574 DT Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: inactivated polio vaccine and HiB at least 2 weeks after study vaccine |
|
| Outcomes |
|
|
| Notes | Macrolide prophylaxis not used (not recommended in Sweden for age > 6 months and main efficacy follow‐up in this study started at age 6 months) DTwP unavailable for first 2 months of study. Only DTaP and DT randomised during that period. Questionnaire of study nurses showed partial post‐allocation unblinding of DTwP (harder to re‐suspend and more likely to be followed by adverse events). Pre‐allocation concealment was determined to be adequate (see text for reasons) Redness reported only for moderate/severe category Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centrally generated randomisation in blocks of 12 or 16 |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical vials. At each study site, vaccines administered in numbered order |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 1994a.
| Methods | Site: Canada
Design: DB parallel‐group RCT Adverse events recorded by structured telephone interview of parents |
|
| Participants | Included: age 2 months; healthy Excluded: children with contraindications to pertussis vaccine as specified in ref NACI 1989 | |
| Interventions | Primary series (aP versus wP)
Number randomised: 67 aP, 33 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: HiB |
|
| Outcomes |
|
|
| Notes | The 2 acellular vaccines had differing amounts of PT and FH. In this review, results for the 2 acellular vaccine formulations are combined Antipyretic/analgesic prophylaxis was discouraged but not prohibited |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 1994b.
| Methods | Site: Canada
Design: DB parallel‐group RCT Adverse events recorded by structured telephone interview of parents |
|
| Participants | Included: age 17 to 19 months; healthy Excluded: children with contraindications to pertussis vaccine as specified in ref NACI 1989 | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 61 aP, 30 wP Dose schedule: 1 dose (17 to 19 months) Concurrent vaccine: Hib |
|
| Outcomes |
|
|
| Notes | The 2 acellular vaccines had differing amounts of PT, FH, Prn and Fim2,3. In this review, results for the 2 acellular vaccine formulations are combined Antipyretic/analgesic prophylaxis was discouraged but not prohibited | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 1995.
| Methods | Site: Canada
Design: DB parallel‐group RCT. Subjects had been randomised and received primary series in Halperin 1994a Adverse events recorded by structured telephone interview of parents |
|
| Participants | Included: age 17 to 19 months; healthy; primary series DTaP or DTwP given in Halperin 1994a Excluded: children with contraindications to pertussis vaccine as specified in ref NACI 1989 | |
| Interventions | Booster (aP.aP versus wP.wP)
Number studied: 56 aP, 30 wP Dose schedule: 1 dose (17 to 19 months) Concurrent vaccine: Hib |
|
| Outcomes |
|
|
| Notes | The 2 acellular vaccines had differing amounts of PT and FH. In this review, results for the 2 acellular vaccine formulations are combined. DTaP or DTwP primary series was given in Halperin 1994a. Randomisation took place in that study, parents and investigators remained blinded and children received the same vaccine in Halperin 1995 as they had received in Halperin 1994a Antipyretic/analgesic prophylaxis was discouraged but not prohibited | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 1996.
| Methods | Site: Canada
Design: DB parallel‐group RCT Adverse events recorded by structured telephone interview of parents |
|
| Participants | Included: age 2 to 3 months; healthy Excluded: children with contraindications to pertussis vaccine | |
| Interventions | Primary series + booster (aP.aP versus wP.wP)
Number randomised: 324 aP, 108 wP 4 doses (2, 4, 6, 17 to 19 months) Concurrent vaccine: Hib |
|
| Outcomes |
|
|
| Notes | Antipyretic/analgesic prophylaxis was discouraged but not prohibited | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 1999.
| Methods | Site: Canada
Design: DB parallel‐group RCT Adverse events recorded by structured telephone interview of parents |
|
| Participants | Included: age 4 to 6 years; healthy; primary series DTaP or DTwP given in Halperin 1994 or Halperin 1996 Excluded: children with contraindications to pertussis vaccine as specified in ref NACI 1989 | |
| Interventions | Booster (aP versus wP)
Number studied: 178 aP, 178 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: IPV |
|
| Outcomes |
|
|
| Notes | Antipyretic/analgesic prophylaxis was discouraged but not prohibited | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | Low risk | Vaccines in identical, coded vials |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Halperin 2003.
| Methods | Site: Canada
Design: parallel‐group RCT Computer‐generated randomisation list kept in a locked file. Study personnel collecting telephone reactogenicity data were blinded as to which vaccine the subject had received |
|
| Participants | Included: age 4 to 6 years; healthy Excluded: children with contraindications to pertussis vaccine | |
| Interventions | Booster (aP versus wP)
Number randomised: 408 aP, 97 wP 1 dose (4 to 6 years) Concurrent vaccines: Hib; IPV |
|
| Outcomes |
|
|
| Notes | Only participants who had received 4 previous doses of DTwP were blinded to which vaccine they received for the fifth dose, because of different vaccine container formats. Antipyretic/analgesic prophylaxis was discouraged but not prohibited | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation list kept in a locked file |
| Allocation concealment (selection bias) | High risk | Different vaccine container formats |
| Blinding (performance bias and detection bias) All outcomes | High risk | Study personnel collecting telephone reactogenicity data were blinded as to which vaccine the subject had received. Only participants who had received 4 previous doses of DTwP were blinded to which vaccine they received for the fifth dose |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Only participants who had received 4 previous doses of DTwP were blinded to which vaccine they received for the fifth dose |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Only participants who had received 4 previous doses of DTwP were blinded to which vaccine they received for the fifth dose |
Heininger 1994.
| Methods | Site: Germany
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 2 to 4 months; healthy Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number randomised: 75 aP, 74 wP Dose schedule: 3 doses (2 to 4 months, then 2 doses at 6‐week intervals) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Excluded for adverse events after 2nd and 3rd doses (number vaccinated/studied at these doses was uncertain due to inconsistencies in tabulated data). Redness and induration were reported only for the moderate/severe category No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Kanra 1993a.
| Methods | Site: Turkey
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 15 to 20 months; completed DTwP primary series Excluded: history of pertussis or progressive neurological disease; chronic drug therapy | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 55 aP, 55 wP Dose schedule: 1 dose (15 to 20 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Data for drowsiness, irritability (restlessness), "unusual" crying and "gastrointestinal symptoms" collected but not reported. Report states "no serious events" but serious events not defined No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Kanra 1993b.
| Methods | Site: Turkey
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 4 to 6 years; completed DTwP primary series and 15‐ to 24‐month booster Excluded: history of pertussis or progressive neurological disease; chronic drug therapy | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number studied: 53 aP, 52 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Total 108 randomised, data for 105 No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for drowsiness, irritability (restlessness), "unusual" crying and "gastrointestinal symptoms" collected but not reported |
Kosuwon 2003.
| Methods | Site: Thailand
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 4 to 6 years old; healthy, who had received 4 doses of DTwP at 2, 4, 6 and 18 months Excluded: history of diphtheria or tetanus at any time, confirmed pertussis in the previous 5 years, if received vaccines not foreseen in the protocol within 30 days prior to study start or after receiving a study vaccine, history of allergic disease or reactions by any component of the vaccine or previously recorded following previous DTP, history of any serious adverse reactions following previous DTP vaccination, history of administration of immunosuppressive agents, immunoglobulin or blood products within the previous 3 months or during the trial, major congenital defects, neurological including seizure disorders and acute febrile illness | |
| Interventions | Booster (aP versus wP)
Number studied: 165 aP, 165 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | All symptoms (solicited or unsolicited) were classified by the investigators as not related, unlikely, suspected or probably related. But not a clear temporal definition of this criterion (before or after data collection) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Lewis 1986.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 18 to 24 months; healthy; completed DTwP primary series Excluded: contraindication to DTwP as specified in ref CID 1982 or in the Wyeth [W] pack insert | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 40 aP, 20 wP Dose schedule: 1 dose (18 to 24 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Marcinak 1993.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 15 to 20 months; completed DTwP primary series Excluded: routine contraindications to DTP vaccine; immunodeficiency; immunosuppressive therapy; major congenital malformation; serious chronic disease; personal or immediate family history of developmental delay or neurological disorder; antibiotic therapy within 7 days before enrolment | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 164 aP, 82 wP Dose schedule: 1 dose (15 to 20 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Study report states that only adverse events for which there was a significant difference between vaccines were reported. Not clear whether adverse event types were omitted or just certain time points for individual adverse events Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation but no statement regarding file locking |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | Low risk | Vomiting is the only target adverse event without data. Redness reported only for moderate/severe category |
Miller 1991.
| Methods | Site: UK Design: parallel‐group RCT Parents and study nurses recorded adverse events on forms |
|
| Participants | Included: infants attending clinics for 1st dose DTP (due at age 3 months) Excluded: history of neurological disorder, serious chronic disease or confirmed pertussis; immediate family history of idiopathic epilepsy | |
| Interventions | Primary series (aP versus wP)
Number randomised: 94 aP, 94 wP Dose schedule: 3 doses (3, 5, 8 to 10 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Minor adverse event data for fever only. Anorexia, drowsiness, irritability, prolonged crying and vomiting combined as "any systemic symptom" No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data for redness, swelling collected but not reported (stated not to differ in frequency) |
Morgan 1990.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 4 to 6 years; healthy; completed DTwP primary series and 15‐ to 24‐month booster Excluded: not stated | |
| Interventions | Booster (wP.wP.aP versus wP.wP.wP)
Number randomised: 41 aP, 42 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Pichichero 1992.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on questionnaires and measured their child's rectal temperature |
|
| Participants | Included: infants due to receive 1st dose DTP (mean age = 2 months), 2nd dose (mean age = 4 months) and 3rd dose (mean age = 6 months) Excluded: contraindication to DTP vaccine as specified in ref CID 1988 | |
| Interventions | Primary series (aP versus wP)
Number randomised:
Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | The pattern of crying is not clear, considered unusual, or high‐pitched cries. 7 children left the study for severe reactions not described and 2 experienced hypotonic/hyporesponsive episodes following the second vaccination. Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Pichichero 1993.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: infants due to receive 1st dose DTP (mean age = 2 months) Excluded: contraindication to DTP vaccine as specified in ref CID 1988 | |
| Interventions | Primary series (aP versus wP)
Number randomised: 88 aP, 22 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | The choice of reported adverse event types was not limited to those showing a significant difference between vaccines but was based on the preliminary results of Decker 1995 (reported in Pichichero 1995), which identified the chosen adverse events as sufficient to differentiate between DTP vaccines in regard to reactogenicity. Irritability and pain were reported only for the moderate/severe category. Reactive antipyretic/analgesic use allowed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | High risk | Data on anorexia, drowsiness and vomiting were collected but not reported |
Pichichero 1994.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 2 months (6 to 12 weeks); healthy Excluded: contraindication to DTP vaccine as specified in ref CID 1988 | |
| Interventions | Primary series (aP versus wP)
Number randomised: 62 aP, 18 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Redness and pain were reported only for the moderate/severe category Antipyretic/analgesic use discouraged but allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Parents and investigators are unaware of the type of vaccination |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
Pichichero 1996.
| Methods | Site: USA
Design: parallel‐group RCT. Subjects had been randomised and received primary series in Pichichero 1993 and Pichichero 1994 (latter excluded due to discrepancies in reported data). 158 out of 190 (83%) parents elected to remain blinded and allow child to participate in booster study Parents recorded adverse events on forms |
|
| Participants | Included: age 15 to 20 months; primary series DTaP or DTwP given in Pichichero 1993 or Pichichero 1994 Excluded: not stated | |
| Interventions | Booster (aP.aP versus wP.wP)
Number studied: 124 aP, 34 wP Dose schedule: 1 dose (15 to 20 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Possible bias due to parental self selection of 83% subset who continued in the study. Number of subjects studied for redness and pain in the DTwP group is unclear due to discrepancies in the data table. Irritability and pain were reported only for the moderate/severe category Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Pichichero 1997.
| Methods | Site: USA
Design: parallel‐group RCT. Subjects had been randomised and received primary series in Decker 1995. 1374 of 2264 (61%) parents elected to remain blinded and allow child to participate in booster study Parents recorded adverse events on forms |
|
| Participants | Included: completed DTaP or DTwP primary series in Decker 1995 Excluded: see Decker 1995; contraindications to DTP vaccine as per ref CID 1994 | |
| Interventions | Booster (aP.aP versus wP.aP versus wP.wP) Children who had received DTaP primary series received the same DTaP as a booster, except for those who had received Lederle[3P] (no longer available) who were boosted with Lederle/Takeda[4F2]. Children who had received DTwP primary series were re‐randomised to receive 1 of the 12 DTaP vaccines or DTwP
Number studied: 1079 aP.aP, 187 wP.aP, 16 wP.wP Dose schedule: 1 dose (15 to 20 months) Concurrent vaccine: OPV |
|
| Outcomes |
|
|
| Notes | Most failures to proceed from primary series to booster study were due to prior receipt of booster dose or reluctance regarding venipuncture. Possible selection bias exists due to loss of 39% of subjects after primary series, although authors state that DTwP primary series recipients with severe reactions were not less likely to proceed to the booster study. Safety results are combined for all acellular vaccines. Data for drowsiness and vomiting were collected but not reported (stated to be not significantly different between groups) Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Vaccine vials labelled with letter codes instead of type/manufacturer details but unable to ascertain from study report whether vaccinators remained unaware of what each code represented |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Vaccinators took no further part in the study and did not participate in follow‐up data collection, so outcome assessment was double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
| Selective reporting (reporting bias) | High risk | Data for drowsiness and vomiting were collected but not reported (stated to be not significantly different between groups) |
Pichichero 2000.
| Methods | Site: USA Design: parallel‐group RCT. Subjects had been randomised and received primary series in Decker 1995 and Pichichero 1997. Details of allocation concealment in Decker 1995 and Pichichero 1997. Parents recorded adverse events on forms. Parents elected to remain blinded and allow child to participate in booster study | |
| Participants | Included: age 4 to 6 years of age who had completed earlier National Institute of Allergy and Infectious Diseases (NIAID) multicentre acellular pertussis vaccine trials in Decker 1995, Pichichero 1997 Excluded: subjects with contraindications or precautions to immunisations as specified in the Report of the Committee on Infectious Diseases of the American Academy of Pediatrics |
|
| Interventions | Booster (same aPaPaP, mixed aPaPaP, wPaPaP versus wPwPwP)
Number of studies: 316 aP, 10 wP Dose schedule: 1 dose (4 to 6 years) Concurrent vaccine: OPV |
|
| Outcomes |
|
|
| Notes | Possible bias due to parental self selection of 83% subset who continued in the study Reactive antipyretic/analgesic use allowed |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Podda 1994.
| Methods | Site: Italy Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 2 months; healthy Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number randomised: 240 aP, 240 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
PVSG 1998.
| Methods | Site: Germany
Design: parallel‐group RCT Active case ascertainment (bi‐weekly telephone). Case incidence adjusted for follow‐up duration by use of incidence density. Parents recorded adverse events in diary |
|
| Participants | Included: age 2 to 4 months; healthy Excluded: known or suspected neurological disorder, immunological dysfunction or immunosuppressive therapy; birth weight < 2 kg; significant congenital abnormality or chronic illness; history of convulsions; hereditary disease in the family with an increased risk of neurological manifestations after vaccination (for example, tuberous sclerosis); immunoglobulins in previous 4 weeks | |
| Interventions | Primary series + booster (aP.aP versus wP.wP)
A third non‐randomised group received DT (n = 1739) Number randomised: 4273 aP, 4259 wP Dose schedule: 4 doses (dose 1: 2 to 4 months; dose 2: at least 6 weeks after dose 1; dose 3: at least 6 weeks after dose 2 but before 1st birthday; dose 4: at least 6 months after dose 3, at 15 to 18 months of age). DT vaccinees received doses at times corresponding to doses 1, 2 and 4 of the randomised schedule Concurrent vaccines: not stated |
|
| Outcomes |
|
|
| Notes | Macrolide prophylaxis was not documented During the total period of the study (until at least 6 months after 3rd dose) convulsions occurred in 46 (1.1%) DTaP, 56 (1.3%) DTwP and 19 (1.1%) DT recipients Redness and induration were reported only for the moderate/severe category No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Rothstein 1993.
| Methods | Site: USA
Design: parallel‐group RCT Parents recorded adverse events on forms |
|
| Participants | Included: age 15 to 16 months; healthy; completed DTwP primary series Excluded: past pertussis, mumps, measles or rubella; previous DTaP; contraindication to DTP, OPV or MMR vaccine | |
| Interventions | Booster (wP.aP versus wP.wP)
Number randomised: 48 aP, 49 wP Dose schedule: 1 dose (age 15 to 16 months) Concurrent vaccine: OPV and MMR vaccine |
|
| Outcomes |
|
|
| Notes | No "severe" or contraindicating adverse events but these not defined in the study report. DTaP or DTwP administered at same time as OPV and MMR (but MMR at a different injection site) Reactive antipyretic/analgesic use allowed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Simondon 1996.
| Methods | Site: Senegal
Design: parallel‐group RCT Study physicians visited homes, recorded adverse events and measured temperature at 2 to 3 days after each dose. Parents interviewed regarding deaths |
|
| Participants | Included: age 2 months Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number randomised: 141 aP, 145 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: Bacille Calmette‐Guérin (BCG) (2 months); IPV (2, 4, 6 months), measles and yellow fever (6 months) |
|
| Outcomes |
|
|
| Notes | Study conducted in Senegal in an area with high background infant mortality. High loss to follow‐up (see text). Data included for 1st dose only. Second and third doses excluded because follow‐up for those doses was less than 80% in both vaccine groups. Irritability and pain were reported only for the moderate/severe category No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Simondon 1997.
| Methods | Site: Senegal
Design: parallel‐group RCT Active case ascertainment (weekly visit by field workers). Case incidence adjusted for follow‐up duration by use of incidence density. Field workers recorded adverse events during 2 weekly visits after each dose |
|
| Participants | Included: age 2 months Excluded: serious congenital defect; serious chronic illness manifested as failure to thrive or cardiac failure; history of seizure or neurological disorder; history of pertussis | |
| Interventions | Primary series (aP versus wP)
Number studied (efficacy): 1847 aP, 1772 wP Number studied (safety): 2396 aP, 2379 wP Dose schedule: 3 doses (2, 4, 6 months) Concurrent vaccine: BCG (2 months); IPV (2, 4, 6 months) |
|
| Outcomes |
|
|
| Notes | Number randomised not stated. A total of 4821 were studied for safety. No statement on macrolide prophylaxis Study conducted in Senegal in an area with high background infant mortality. Deaths recorded are due to any cause and may include some due to injury. No breakdown by vaccine group of the number of deaths due to infection A household contact substudy compared pertussis attack rates with non‐randomised unvaccinated controls No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Tian 1993.
| Methods | Site: China
Design: parallel‐group RCT Adverse event recording method not stated |
|
| Participants | Included: age 3 to 6 months Excluded: not stated | |
| Interventions | Primary series (aP versus wP versus P)
Number studied: 105 aP, 101 wP, 100 P Dose schedule: 3 doses at 4‐week intervals Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | Number randomised not stated Data for redness and tenderness excluded (combined as 1 outcome in study report) No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Trollfors 1995.
| Methods | Site: Sweden
Design: DB parallel‐group RCT Passive and active case ascertainment (parent report and monthly telephone). Case incidence adjusted for follow‐up duration by use of incidence density. Parents recorded adverse events in diary |
|
| Participants | Included: full‐term, healthy infants Excluded: none stated | |
| Interventions | Primary series (aP versus DT)
Number randomised: 1724 aP, 1726 DT Dose schedule: 3 doses (3, 5, 12 months) Concurrent vaccine: nil |
|
| Outcomes |
|
|
| Notes | Macrolide prophylaxis not used (not recommended in Sweden for age > 6 months and main efficacy follow‐up in this study started at age 10 months) Target adverse events assessed in the trial included anorexia, irritability, fever, redness and induration. Data for anorexia and irritability were collected but not reported (stated to be equally frequent with both vaccines). Data for other events are not included in the review because the number of children experiencing the event could not be determined with sufficient confidence (fever) and because data were reported only for the moderate/severe category (redness and induration) No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | Vaccines indistinguishable |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
| Selective reporting (reporting bias) | High risk | Data for anorexia and irritability were collected but not reported (stated to be equally frequent with both vaccines) |
Vanura 1994.
| Methods | Site: Austria and Switzerland
Design: parallel‐group RCT Parents recorded adverse events in diary |
|
| Participants | Included: age 10 to 16 weeks; healthy Excluded: not stated | |
| Interventions | Primary series (aP versus wP)
Number studied: 200 aP, 101 wP Dose schedule: 3 doses (3, 4, 5 months) Concurrent vaccine: not stated |
|
| Outcomes |
|
|
| Notes | The 2 acellular vaccine formulations contained differing amounts of PT. In this review, data for the 2 acellular vaccine formulations are combined Total 308 enrolled but cannot determine number randomised to each vaccine. Denominator for primary series non‐completion is the number with adverse event data at the first dose (200 DTaP, 101 DTwP) No statement on antipyretic/analgesic use | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
Interventions Primary series: primary series pertussis immunisation performed in the study. Booster: pertussis booster immunisation performed in the study.
The pertussis immunisation history of study participants (including doses received in the study under consideration) is indicated in the form (X.Y.Z). The first element (X) indicates the type of pertussis vaccine received in the primary series (aP = acellular, wP = whole‐cell, P = placebo, DT = diphtheria‐tetanus toxoids). The second element (Y) indicates the vaccine type received at the 15‐ to 24‐month booster and the third element (Z) indicates the vaccine type received at the 4‐ to 6‐year booster. For example: aP = acellular primary series; wP.wP.aP = acellular 4‐ to 6‐year booster after whole‐cell primary series and whole‐cell 18‐ to 24‐month booster.
The vaccines used in each study are further identified by type, manufacturer and number, and type of pertussis components, as follows:
Vaccine types: aP: acellular pertussis vaccine DTaP: diphtheria‐tetanus‐acellular pertussis vaccine wP: whole‐cell pertussis vaccine DTwP: diphtheria‐tetanus‐whole‐cell pertussis vaccine DT: diptheria‐tetanus (toxoids) vaccine DTP: diptheria‐tetanus (toxoids) pertussis vaccine P: placebo
Manufacturer abbreviations: CAMR: Centre for Applied Microbiology and Research Canton: Canton Department of Health JNIH: Japanese National Institute of Health Massachusetts: Massachusetts Public Health Laboratory Michigan: Michigan Department of Health Porton: Porton Products (later Speywood Pharmaceuticals) SKB: SmithKline Beecham SSVI: Swedish Serum and Vaccine Institute Wuhan: Wuhan Department of Health
Pertussis components (in square brackets after manufacturer: [1] = PT (inactivated pertussis toxin); [2] = PT+FH (filamentous haemagglutinin); [3] = PT+FH+Prn (pertactin); [4] = PT+FH+Fim2&3 (fimbrial antigen serotypes 2 and 3) or PT+FH+Prn+Fim2 or PT+FH+Prn+Fim (serotype unspecified); [5] = PT+FH+Prn+Fim2&3; [W] = killed whole Bordetella pertussis organisms.
Outcomes Non‐completion of the primary series due to adverse events, defined as withdrawal from the study due to an adverse event or events before completion of all scheduled doses of the primary series. Deaths, encephalopathy, convulsions, hypotonic‐hyporesponsive episodes (HT‐HR): these events generally led to withdrawal from the study and were reported and analysed across all doses in a study. Minor adverse events: fever, irritability, drowsiness, anorexia, vomiting, redness, pain/tenderness and swelling/induration. These events usually did not lead to withdrawal and data were reported and analysed separately for each dose within a study. Data for these events were not combined across doses (see 'Methods' section of the review for reasons).
Other abbreviations BCG: Bacille Calmette‐Guérin DB: double‐blind (claim made in study report) Hib: Haemophilus influenzae type B vaccine IPV: inactivated polio vaccine MMR: measles‐mumps‐rubella vaccine OPV: oral polio vaccine RCT: randomised controlled trial SIDS: sudden infant death syndrome
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Annunziato 1994 | Booster Efficacy: not studied Primary series non‐completion: booster Deaths: booster, no data Encephalopathy: booster, no data Convulsions: no data Hypotonic‐hyporesponsive episodes: no data Minor AEs: data expressed as percentages in graph form only. Could not be converted to numeric values with sufficient accuracy |
| Bernstein 1993 | Booster Efficacy: not studied Primary series non‐completion: booster Deaths: booster, no data Encephalopathy: booster, no data Convulsions: no data Hypotonic‐hyporesponsive episodes: no data Minor AEs: data expressed as percentages in graph form only. Could not be converted to numeric values with sufficient accuracy |
| Bernstein 1995 | Primary series Efficacy: not studied Primary series non‐completion: cannot determine number studied for each vaccine Deaths: cannot determine number studied for each vaccine Encephalopathy: cannot determine number studied for each vaccine Convulsions: cannot determine number studied for each vaccine Hypotonic‐hyporesponsive episodes: cannot determine number studied for each vaccine Minor AEs: data combined across 3 doses. Cannot determine number receiving each vaccine or experiencing AEs at each dose |
| Hori 1994 | Primary series Efficacy: not studied Primary series non‐completion: no data Deaths: no data Encephalopathy: no data Convulsions: no data Hypotonic‐hyporesponsive episodes: no data Minor AEs: unable to extract data separately for each event |
| Hori 1995 | Booster Efficacy: not studied Safety: not reported (immunogenicity only) |
| Just 1991 | Primary series Report of 2 studies comparing acellular versus whole‐cell primary series immunisation (1 study in Turkey, 1 in Switzerland); Swiss study possibly reported in Vanura 1994 Efficacy: not studied Primary series non‐completion: no data Deaths: 1 death (pneumonia) in acellular arm in Turkey but cannot determine number receiving each vaccine Encephalopathy: cannot determine number studied for each vaccine Convulsions: cannot determine number studied for each vaccine Hypotonic‐hyporesponsive episodes: cannot determine number studied for each vaccine Minor AEs: unable to determine number studied for each vaccine (Turkey). Swiss results were combined across doses or across different reaction types |
| Miller 1997 | Primary series Efficacy: not studied Primary series non‐completion: no data Deaths: no data Encephalopathy: no data Convulsions: no data Hypotonic‐hyporesponsive episodes: no data Minor AEs: data combined across 3 doses. Unable to extract data separately for each dose This report examines 2‐4‐6 and 3‐5‐10 month immunisation schedules. It includes some data previously reported in Miller 1991 |
| Murphy 1983 | Primary series Efficacy: not studied Primary series non‐completion: no data Deaths: cannot determine number studied for each vaccine Encephalopathy: cannot determine number studied for each vaccine Convulsions: cannot determine number studied for each vaccine Hypotonic‐hyporesponsive episodes: cannot determine number studied for each vaccine Minor AEs: cannot determine number studied for each vaccine |
| Pichichero 1987 | Booster Efficacy: not studied Primary series non‐completion: booster Deaths: follow‐up of only 65% of participants Encephalopathy: follow‐up of only 65% of participants Convulsions: follow‐up of only 65% of participants Hypotonic‐hyporesponsive episodes: follow‐up of only 65% of participants Minor AEs: follow‐up of only 65% of participants |
| Shek 2003 | Primary series Efficacy: not studied Primary series non‐completion: no data Deaths: no data Encephalopathy: no data Convulsions: no data Hypotonic‐hyporesponsive episodes: no data Minor AEs: data combined across 3 doses |
AEs: adverse events
Contributions of authors
Inge Axelsson (IA) contributed to updating the Background section and the section "Agreements and disagreements with other studies or reviews" in the Discussion section. Silvio Prietsch (SP) was responsible for updating the Background section. He participated in study selection, quality assessment and data collection for the updated review. He also provided input to updating the section Risk of bias in included studies. Linjie Zhang (LZ) was responsible for study selection, quality assessment, data collection and data analysis for the updated review. He was also responsible for updating the Abstract, Methods, Results, Discussion and References. He contributed to updating the Background section. Scott Halperin (SH) performed a critical review of the manuscript and provided input to updating the Discussion section. The final version of the updated review was approved by all review authors.
Sources of support
Internal sources
No sources of support supplied
External sources
-
UK Department of Health Cochrane Incentive Scheme 2008, UK.
This updated review received an award from the UK Department of Health Cochrane Incentive Scheme 2008
Declarations of interest
Inge Axelsson was in part paid by the Research and Development Unit, Jämtland County Council, Östersund, Sweden. "In the last 10 years, I have been a Clinical Investigator in a trial of an infant combination vaccine containing acellular pertussis (aP) vaccine from Sanofi Pasteur MSD but I have not received any fee from any pharmaceutical company".
Scott A Halperin has undertaken clinical trials of acellular pertussis vaccines for most manufacturers of these products including GlaxoSmithKline, Sanofi Pasteur, Novartis and Wyeth. He has given Continuing Medical Education talks (not as part of a manufacturers' speakers bureau) for which an honorarium has been paid. He has served on occasional ad hoc advisory boards of vaccine manufacturers but does not have any ongoing consultancies.
Linjie Zhang: none known. Sílvio OM Prietsch: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Afari 1996 {published data only}
- Afari EA, Kamiya Y, Nkrumah FK, Dunyo SK, Akpedonu P, Kamiya H, et al. Randomized controlled trial of acellular diphtheria, pertussis and tetanus vaccines in southern Ghana. Annals of Tropical Paediatrics 1996;16(1):39‐48. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
AHGSPV 1988 {published data only}
- Ad Hoc Group for the Study of Pertussis Vaccines. Placebo‐controlled trial of two acellular pertussis vaccines in Sweden‐protective efficacy and adverse events. Lancet 1988;1(8592):955‐60. [PubMed] [Google Scholar]
- Blackwelder WC, Storsaeter J, Olin P, Hallander HO. Acellular pertussis vaccines. Efficacy and evaluation of clinical case definitions. American Journal of Diseases of Children 1991;145:1285‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Kallings LO, Olin P, Storsaeter J. Protective effect of 2 acellular pertussis vaccines in a double‐blind placebo controlled clinical trial [Skyddseffekten av tva acellulara kikhostevacciner i en dubbelblind placebokontrollerad klinisk provning]. Lakartidningen 1988;85:1994‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Olin P, Romanus V, Storsaeter J. Invasive bacterial infections during an efficacy trial of acellular pertussis vaccines ‐ implications for future surveillance in pertussis vaccine programmes. Tokai Journal of Experimental and Clinical Medicine 1988;13:143‐4. [MEDLINE: ] [PubMed] [Google Scholar]
- Olin P, Storsaeter J, Linder T, Winter C, Sönstenby C. A clinical trial of acellular pertussis vaccines in Sweden. Technical Report 1988. [MEDLINE: ]
- Storsaeter J, Hallander H, Farrington CP, Olin P, Mollby R, Miller E. Secondary analyses of the efficacy of two acellular pertussis vaccines evaluated in a Swedish phase III trial. Vaccine 1990;8:457‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Storsaeter J, Olin P, Renemar B, Lagergård T, Norberg R, Romanus V, et al. Mortality and morbidity from invasive bacterial infections during a clinical trial of acellular pertussis vaccines in Sweden. Pediatric Infectious Disease Journal 1988;7(9):637‐45. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
AHGSPV 1997 {published data only}
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Heijbel H, Rasmussen F, Olin P. Safety evaluation of one whole‐cell and three acellular pertussis vaccines in Stockholm trial II. Developments in Biological Standardization 1997;89:99‐100. [MEDLINE: ] [PubMed] [Google Scholar]
- Olin P, Gustafsson L, Rasmussen H, Hallander H, Heijbel H, Gottfarb P. Efficacy trial of acellular pertussis vaccines. Technical report trial II with preplanned analysis of efficacy, immunogenicity and safety. Swedish Institute for Infectious Disease Control 1997.
- Olin P, Rasmussen F, Gustafsson L, Hallander HO, Heijbel H, Ad Hoc Group for the Study of Pertussis Vaccines. Randomised controlled trial of two‐component, three‐component, and five‐component acellular pertussis vaccines compared with whole‐cell pertussis vaccine. Lancet 1997;350:1569‐77. [DOI] [PubMed] [Google Scholar]
Anderson 1988 {published data only}
- Anderson EL, Belshe RB, Bartram J. Differences in reactogenicity and antigenicity of acellular and standard pertussis vaccines combined with diphtheria and tetanus in infants. Journal of Infectious Diseases 1988;157(4):731‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bernstein 1992 {published data only}
- Bernstein HH, Rothstein EP, Pichichero ME, Francis AB, Kovel AJ, Disney FA, et al. Clinical reactions and immunogenicity of the BIKEN acellular diphtheria and tetanus toxoids and pertussis vaccine in 4‐ through 6‐year‐old US children. American Journal of Diseases of Children 1992;146(5):556‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bernstein 1994 {published data only}
- Bernstein HH, Rothstein EP, Reisinger KS, Blatter MM, Arbeter AM, Fontana ME, et al. Comparison of a three‐component acellular pertussis vaccine with a whole‐cell pertussis vaccine in 15‐ through 20‐month‐old infants. Pediatrics 1994;93(4):656‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Black 1997 {published data only}
- Black SB, Shinefield HR, Bergen R, Hart C, Kremers R, Lavetter A, et al. Safety and immunogenicity of Chiron/Biocine recombinant acellular pertussis‐diphtheria‐tetanus vaccine in infants and toddlers. Pediatric Infectious Disease Journal 1997;16(1):53‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Blennow 1988 {published data only}
- Blennow M, Granstrom M, Jaatmaa E, Olin P. Primary immunization of infants with an acellular pertussis vaccine in a double‐blind randomized clinical trial. Pediatrics 1988;82(3):293‐9. [MEDLINE: ] [PubMed] [Google Scholar]
- Blennow M, Granstrom M, Olin P, Tiru M, Jäätmaa E, Askelöf P, et al. Preliminary data from a clinical trial (phase 2) of an acellular pertussis vaccine, J‐NIH‐6. Developments in Biological Standardization 1986;65:185‐90. [MEDLINE: ] [PubMed] [Google Scholar]
- Blennow M, Hedenskog S, Granstrom M. Protective effect of acellular pertussis vaccines. European Journal of Clinical Microbiology and Infectious Diseases 1988;7(3):381‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Blumberg 1990 {published data only}
- Blumberg DA, Mink CM, Cherry JD, Reisinger KS, Blatter MM, Congeni BL, et al. Comparison of an acellular pertussis‐component diphtheria‐tetanus‐pertussis (DTP) vaccine with a whole‐cell pertussis‐component DTP vaccine in 17‐ to 24‐month‐old children, with measurement of 69‐kilodalton outer membrane protein antibody. Journal of Pediatrics 1990;117(1 Pt 1):46‐51. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Cherry JD, Mortimer EA, Hackell JG, Scott JV, Multicenter APDT Vaccine Study Groups. Clinical trials in the United States and Japan with the Lederle‐Takeda and Takeda acellular pertussis‐diphtheria‐tetanus (APDT) vaccines. Developments in Biological Standardization 1991;73:51‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Blumberg 1991 {published data only}
- Blumberg DA, Mink CM, Cherry JD, Johnson C, Garber R, Plotkin SA, et al. Comparison of acellular and whole‐cell pertussis‐component diphtheria‐tetanus‐pertussis vaccines in infants. The APDT Vaccine Study Group. Journal of Pediatrics 1991;119(2):194‐204. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Decker 1995 {published data only}
- Decker MD, Edwards KM. The multicenter acellular pertussis trial: an overview. Journal of Infectious Diseases 1996;174(Suppl 3):270‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Decker MD, Edwards KM, Steinhoff MC, Rennels MB, Pichichero ME, Englund JA, et al. Comparison of 13 acellular pertussis vaccines: adverse reactions. Pediatrics 1995;96(3 Pt 2):557‐66. [MEDLINE: ] [PubMed] [Google Scholar]
- Deloria MA, Blackwelder WC, Decker MD, Englund JA, Steinhoff MC, Pichichero ME, et al. Association of reactions after consecutive acellular or whole‐cell pertussis vaccine immunizations. Pediatrics 1995;96(3 pt 2):592‐4. [PubMed] [Google Scholar]
- Edwards KM, Meade BD, Decker MD, Reed GF, Rennels MB, Steinhoff MC, et al. Comparison of 13 acellular pertussis vaccines: overview and serologic response. Pediatrics 1995;96(3 Pt 2):548‐57. [MEDLINE: ] [PubMed] [Google Scholar]
Edwards 1986a {published data only}
- Edwards KM, Lawrence E, Wright PF. Diphtheria, tetanus, and pertussis vaccine. A comparison of the immune response and adverse reactions to conventional and acellular pertussis components. American Journal of Diseases of Children 1986;140(9):867‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Edwards 1986b {published data only}
- Edwards KM, Lawrence E, Wright PF. Diphtheria, tetanus, and pertussis vaccine. A comparison of the immune response and adverse reactions to conventional and acellular pertussis components. American Journal of Diseases of Children 1986;140(9):867‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Edwards 1989a {published data only}
- Edwards KM, Bradley RB, Decker MD, Palmer PS, Savage J, Taylor JC, et al. Evaluation of a new highly purified pertussis vaccine in infants and children. Journal of Infectious Diseases 1989;160(5):832‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Edwards 1989b {published data only}
- Edwards KM, Bradley RB, Decker MD, Palmer PS, Savage J, Taylor JC, et al. Evaluation of a new highly purified pertussis vaccine in infants and children. Journal of Infectious Diseases 1989;160(5):832‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Edwards 1989c {published data only}
- Edwards KM, Bradley RB, Decker MD, Palmer PS, Savage J, Taylor JC, et al. Evaluation of a new highly purified pertussis vaccine in infants and children. Journal of Infectious Diseases 1989;160(5):832‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Englund 1992 {published data only}
- Englund JA, Glezen WP, Barreto L. Controlled study of a new five‐component acellular pertussis vaccine in adults and young children. Journal of Infectious Diseases 1992;166(6):1436‐41. [DOI] [PubMed] [Google Scholar]
Englund 1994a {published data only}
- Englund JA, Decker MD, Edwards KM, Pichichero ME, Steinhoff MC, Anderson EL. Acellular and whole‐cell pertussis vaccines as booster doses: a multicenter study. Pediatrics 1994;93(1):37‐43. [MEDLINE: ] [PubMed] [Google Scholar]
Englund 1994b {published data only}
- Englund JA, Decker MD, Edwards KM, Pichichero ME, Steinhoff MC, Anderson EL. Acellular and whole‐cell pertussis vaccines as booster doses: a multicenter study. Pediatrics 1994;93(1):37‐43. [MEDLINE: ] [PubMed] [Google Scholar]
Feldman 1992 {published data only}
- Feldman S, Perry S, Andrew M, Jones L, Moffitt JE. Comparison of acellular (B type) and whole‐cell pertussis‐component diphtheria‐tetanus‐pertussis vaccines as the first booster immunization in 15‐ to 24‐month‐old children. Journal of Pediatrics 1992;121(6):857‐61. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Feldman 1993 {published data only}
- Feldman S, Perry CS, Andrew M, Jones L, Moffitt JE, Lamb D, et al. Primary immunization series for infants: comparison of two‐component acellular and standard whole‐cell pertussis vaccines combined with diphtheria‐tetanus toxoids. Southern Medical Journal 1993;86(3):269‐75, 284. [MEDLINE: ] [PubMed] [Google Scholar]
Glode 1992 {published data only}
- Glode M, Joffe L, Reisinger K, Blatter M, Plotkin S, Watson B, et al. Safety and immunogenicity of acellular pertussis vaccine combined with diphtheria and tetanus toxoids in 17‐ to 24‐month‐old children. Pediatric Infectious Disease Journal 1992;11(7):530‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Greco 1996 {published data only}
- Ciofi degli Atti ML, Olin P. Severe adverse events in the Italian and Stockholm I pertussis vaccine clinical trials. Developments in Biological Standardization 1997;89:77‐81. [MEDLINE: ] [PubMed] [Google Scholar]
- Greco D. Italian trial on acellular pertussis vaccines. Developments in Biological Standardization 1997;89:55‐7. [MEDLINE: ] [PubMed] [Google Scholar]
- Greco D, Salmaso S, Mastrantonio P, Giuliano M, Tozzi AE, Anemona A, et al. Progetto Pertosse Working Group. A controlled trial of two acellular vaccines and one whole‐cell vaccine against pertussis. New England Journal of Medicine 1996;334(6):341‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Tozzi AE, Olin P. Common side effects in the Italian and Stockholm I trials. Developments in Biological Standardization 1997;89:105‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Gustafsson 1996 {published data only}
- Ciofi degli Atti ML, Olin P. Severe adverse events in the Italian and Stockholm I pertussis vaccine clinical trials. Developments in Biological Standardization 1997;89:77‐81. [MEDLINE: ] [PubMed] [Google Scholar]
- Gustaffson L, Hallander H, Olin P, Reizenstein E, Storsaeter J. Efficacy trial of acellular pertussis vaccines. Technical report Trial I with results of preplanned analysis of safety, efficacy and immunogenicity. Swedish Institute for Infectious Disease Control 1995.
- Gustafsson L, Hallander HO, Olin P, Reizenstein E, Storsaeter J. A controlled trial of a two‐component acellular, a five‐component acellular, and a whole‐cell pertussis vaccine. New England Journal of Medicine 1996;334(6):349‐55. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Olin P. Efficacy trial of acellular pertussis vaccines; trial I. Developments in Biological Standardization 1997;89:52‐4. [MEDLINE: ] [PubMed] [Google Scholar]
- Tozzi AE, Olin P. Common side effects in the Italian and Stockholm I trials. Developments in Biological Standardization 1997;89:105‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Halperin 1994a {published data only}
- Halperin SA, Barreto L, Eastwood BJ, Law B, Roberts EA. Safety and immunogenicity of a five‐component acellular pertussis vaccine with varying antigen quantities. Archives of Pediatrics and Adolescent Medicine 1994;148(11):1220‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Halperin 1994b {published data only}
- Halperin SA, Barreto L, Eastwood BJ, Law B, Roberts EA. Safety and immunogenicity of a five‐component acellular pertussis vaccine with varying antigen quantities. Archives of Pediatrics and Adolescent Medicine 1994;148(11):1220‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Halperin 1995 {published data only}
- Halperin SA, Mills E, Barreto L, Pim C, Eastwood BJ. Acellular pertussis vaccine as a booster dose for seventeen‐ to nineteen‐month‐old children immunized with either whole‐cell or acellular pertussis vaccine at two, four and six months of age. Pediatric Infectious Disease Journal 1995;14(9):792‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Halperin 1996 {published data only}
- Halperin SA, Eastwood BJ, Barreto L, Friesen B, Medd L, Meekison W, et al. Adverse reactions and antibody response to four doses of acellular or whole‐cell pertussis vaccine combined with diphtheria and tetanus toxoids in the first 19 months of life. Vaccine 1996;14(8):767‐72. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Halperin 1999 {published data only}
- Halperin SA, Scheifele D, Barreto L, Pim C, Guasparini R, Medd L, et al. Comparison of a fifth dose of a five‐component acellular or a whole‐cell pertussis vaccine in children four to six years of age. Pediatric Infectious Disease Journal 1999;18(9):772‐9. [DOI] [PubMed] [Google Scholar]
Halperin 2003 {published data only}
- Halperin SA, Scheifele D, Mills E, Guasparini R, Humphreys G, Barreto L, et al. Nature, evolution, and appraisal of adverse events and antibody response associated with the fifth consecutive dose of a five‐component acellular pertussis‐based combination vaccine. Vaccine 2003;21(19‐20):2298‐306. [DOI] [PubMed] [Google Scholar]
Heininger 1994 {published data only}
- Heininger U, Cherry JD, Christenson PD, Eckhardt T, Göering U, Jakob P, et al. Comparative study of Lederle/Takeda acellular and Lederle whole‐cell pertussis‐component diphtheria‐tetanus‐pertussis vaccines in infants in Germany. Vaccine 1994;12(1):81‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kanra 1993a {published data only}
- Kanra G, Ceyhan M, Vandevoorde D, Bogaerts H. Acellular pertussis diphtheria‐tetanus‐pertussis vaccine containing separately purified pertussis toxoid, filamentous haemagglutinin and 69 kDa outer membrane protein as a booster in children. European Journal of Pediatrics 1993;152(6):478‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kanra 1993b {published data only}
- Kanra G, Ceyhan M, Vandevoorde D, Bogaerts H. Acellular pertussis diphtheria‐tetanus‐pertussis vaccine containing separately purified pertussis toxoid, filamentous haemagglutinin and 69 kDa outer membrane protein as a booster in children. European Journal of Pediatrics 1993;152(6):478‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kosuwon 2003 {published data only}
- Kosuwon P, Warachit B, Hutagalung Y, Borkird T, Kosalaraksa P, Bock HL, et al. Reactogenicity and immunogenicity of reduced antigen content diphtheria‐tetanus‐acellular pertussis vaccine (dTpa) administered as a booster to 4‐6 year‐old children primed with four doses of whole‐cell pertussis vaccine. Vaccine 2003;21(27‐30):4194‐200. [DOI] [PubMed] [Google Scholar]
Lewis 1986 {published data only}
- Lewis K, Cherry JD, Holroyd HJ, Baker LR, Dudenhoeffer FE, Robinson RG. A double‐blind study comparing an acellular pertussis‐component DTP vaccine with a whole‐cell pertussis‐component DTP vaccine in 18‐month‐old children. American Journal of Diseases of Children 1986;140(9):872‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Marcinak 1993 {published data only}
- Marcinak JF, Ward M, Frank AL, Boyer KM, Froeschle JE, Hosbach PH IV. Comparison of the safety and immunogenicity of acellular (BIKEN) and whole‐cell pertussis vaccines in 15‐ to 20‐month‐old children. American Journal of Diseases of Children 1993;147(3):290‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Miller 1991 {published data only}
- Miller E, Ashworth LA, Robinson A, Waight PA, Irons LI. Phase II trial of whole‐cell pertussis vaccine versus an acellular vaccine containing agglutinogens. Lancet 1991;337(8733):70‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Morgan 1990 {published data only}
- Cherry JD, Mortimer EA, Hackell JG, Scott JV, Multicenter APDT Vaccine Study Groups. Clinical trials in the United States and Japan with the Lederle‐Takeda and Takeda acellular pertussis‐diphtheria‐tetanus (APDT) vaccines. Developments in Biological Standardization 1991;73:51‐8. [MEDLINE: ] [PubMed] [Google Scholar]
- Mink CM, Uhari M, Blumberg DA, Knip M, Lewis K, Christenson PD, et al. Metabolic and hematologic effects and immune complex formation related to pertussis immunization. Pediatric Research 1990;27(4 Pt 1):353‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Morgan CM, Blumberg DA, Cherry JD, Reisinger KS, Blatter MM, Blumer JL, et al. Comparison of acellular and whole‐cell pertussis‐component DTP vaccines. A multicenter double‐blind study in 4‐ to 6‐year‐old children. American Journal of Diseases of Children 1990;144(1):41‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pichichero 1992 {published data only}
- Pichichero ME, Francis AB, Blatter MM, Reisinger KS, Green JL, Marsocci SM, et al. Acellular pertussis vaccination of 2‐month‐old infants in the United States. Pediatrics 1992;89(5 Pt 1):882‐7. [PubMed] [Google Scholar]
Pichichero 1993 {published data only}
- Pichichero ME, Francis AB, Marsocci SM, Green JL, Disney FA. Comparison of a diphtheria and tetanus toxoids and bicomponent acellular pertussis vaccine with diphtheria and tetanus toxoids and whole‐cell pertussis vaccine in infants. American Journal of Diseases of Children 1993;147(3):295‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pichichero 1994 {published data only}
- Pichichero ME, Green JL, Francis AB, Marsocci SM, Lynd AM, Litteer TTI. Comparison of a three‐component acellular pertussis vaccine with whole‐cell pertussis vaccine in two‐month‐old children. Pediatric Infectious Disease Journal 1994;13(3):193‐6. [DOI] [PubMed] [Google Scholar]
Pichichero 1996 {published data only}
- Pichichero ME, Green JL, Francis AB, Marsocci SM, Murphy AM, Buscarino C. Antibody response and reactions to completion of a four‐dose series with a two‐ or three‐component acellular pertussis vaccine compared to whole‐cell pertussis vaccine. Scandinavian Journal of Infectious Diseases 1996;28(2):159‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pichichero 1997 {published data only}
- Pichichero ME, Deloria MA, Rennels MB, Anderson EL, Edwards KM, Decker MD, et al. A safety and immunogenicity comparison of 12 acellular pertussis vaccines and one whole‐cell pertussis vaccine given as a fourth dose in 15‐ to 20‐month‐old children. Pediatrics 1997;100(5):772‐88. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pichichero 2000 {published data only}
- Pichichero ME, Edwards KM, Anderson EL, Rennels MB, Englund JA, Yerg DE, et al. Safety and immunogenicity of six acellular pertussis vaccines and one whole‐cell pertussis vaccine given as a fifth dose in four‐ to six‐year‐old children. Pediatrics 2000;105(1):e11. [DOI] [PubMed] [Google Scholar]
Podda 1994 {published data only}
- Podda A, Luca EC, Contu B, Furlan R, Maida A, Moiraghi A, et al. Comparative study of a whole‐cell pertussis vaccine and a recombinant acellular pertussis vaccine. The Italian Multicenter Group for the Study of Recombinant Acellular Pertussis Vaccine. Journal of Pediatrics 1994;124(6):921‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
PVSG 1998 {published data only}
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Schmitt‐Grohe S, Stehr K, Cherry JD, Heininger U, Uberall MA, Laussucq S, et al. Pertussis Vaccine Study Group. Minor adverse events in a comparative efficacy trial in Germany in infants receiving either the Lederle/Takeda acellular pertussis component DTP (DTaP) vaccine, the Lederle whole‐cell component DTP (DTP) or DT vaccine. Developments in Biological Standardization 1997;89:113‐8. [MEDLINE: ] [PubMed] [Google Scholar]
- Stehr K, Cherry JD. A comparative efficacy trial in Germany in which infants received either the Lederle/Takeda acellular pertussis component DTP (DTaP) vaccine, the Lederle whole‐cell component DTP (DTP) vaccine or DT vaccine. Developments in Biological Standardization 1997;89:58‐62. [MEDLINE: ] [PubMed] [Google Scholar]
- Stehr K, Cherry JD, Heininger U, Schmitt‐Grohé S, Überall M, Laussucq S, et al. A comparative efficacy trial in Germany in infants who received either the Lederle/Takeda acellular pertussis component DTP (DTaP) vaccine, the Lederle whole‐cell component DTP vaccine, or DT vaccine. Pediatrics 1998;101(1):1‐11. [DOI] [PubMed] [Google Scholar]
- Uberall MA, Stehr K, Cherry JD, Heininger U, Uberall MA, Laussucq S, et al. Pertussis Vaccine Study Group. Severe adverse events in a comparative efficacy trial in Germany in infants receiving either the Lederle/Takeda acellular pertussis component DTP (DTaP) vaccine, the Lederle whole‐cell component DTP (DTP) or DT vaccine. Developments in Biological Standardization 1997;89:83‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Rothstein 1993 {published data only}
- Rothstein EP, Bernstein HH, Glode MP, Laussucq S, Nonenmacher J, Long SS, et al. Simultaneous administration of a diphtheria and tetanus toxoids and acellular pertussis vaccine with measles‐mumps‐rubella and oral poliovirus vaccines. American Journal of Diseases of Children 1993;147(8):854‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Simondon 1996 {published data only}
- Simondon F, Yam A, Gagnepain JY, Wassilak S, Danve B, Cadoz M. Comparative safety and immunogenicity of an acellular versus whole‐cell pertussis component of diphtheria‐tetanus‐pertussis vaccines in Senegalese infants. European Journal of Clinical Microbiology and Infectious Diseases 1996;15(12):927‐32. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Simondon 1997 {published data only}
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Preziosi M‐P, Ndiaye M, Coll‐Seck A, Simondon F. The Senegal pertussis trial: safety and surveillance of adverse reactions. Developments in Biological Standardization 1997;89:91‐7. [MEDLINE: ] [PubMed] [Google Scholar]
- Simondon F. Senegal pertussis trial. Developments in Biological Standardization 1997;89:63‐6. [MEDLINE: ] [PubMed] [Google Scholar]
- Simondon F, Preziosi MP, Yam A, Kane CT, Chabirand L, Iteman I, et al. A randomized double‐blind trial comparing a two‐component acellular to a whole‐cell pertussis vaccine in Senegal. Vaccine 1997;15(15):1606‐12. [DOI] [PubMed] [Google Scholar]
Tian 1993 {published data only}
- Tian X. An observation on immunization reaction and serological effect of adsorbed purified pertussis vaccine. Chung Hua Liu Hsing Ping Hsueh Tsa Chih 1993;14(3):155‐9. [MEDLINE: ] [PubMed] [Google Scholar]
Trollfors 1995 {published data only}
- Heijbel H, Ciofi degli Atti MC, Harzer E, Liese J, Preziosi MP, Rasmussen F, et al. Hypotonic hyporesponsive episodes in eight pertussis vaccine studies. Developments in Biological Standardization 1997;89:101‐3. [MEDLINE: ] [PubMed] [Google Scholar]
- Taranger J, Trollfors B, Knutsson N. Adverse reactions of a pertussis toxoid vaccine in a double‐blind placebo‐controlled trial. Developments in Biological Standardization 1997;89:109‐12. [MEDLINE: ] [PubMed] [Google Scholar]
- Trollfors B, Taranger J. The Gothenburg pertussis vaccine study. Developments in Biological Standardization 1997;89:49‐51. [MEDLINE: ] [PubMed] [Google Scholar]
- Trollfors B, Taranger J, Lagergard T, Lind L, Sundh V, Zackrisson G, et al. A placebo‐controlled trial of a pertussis‐toxoid vaccine. New England Journal of Medicine 1995;333(16):1045‐50. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vanura 1994 {published data only}
- Vanura H, Just M, Ambrosch F, Berger RM, Bogaerts H, Wynen J, et al. Study of pertussis vaccines in infants: comparison of response to acellular pertussis DTP vaccines containing 25 micrograms of FHA and either 25 or 8 micrograms of PT with response to whole‐cell pertussis DTP vaccine. Vaccine 1994;12(3):210‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Annunziato 1994 {published data only}
- Annunziato PW, Rothstein EP, Bernstein HH, Blatter MM, Reisinger KS, Pichichero ME. Comparison of a three‐component acellular pertussis vaccine with a whole‐cell pertussis vaccine in 4‐ through 6‐year‐old children. Elmwood Pediatric Associates, Pennridge Pediatric Associates. Archives of Pediatrics and Adolescent Medicine 1994;148(5):503‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bernstein 1993 {published data only}
- Bernstein DI, Smith VE, Schift GM, Rathfon HM, Boscia JA. Comparison of acellular pertussis vaccine with whole‐cell vaccine as a booster in children 15 to 18 months and 4 to 6 years of age. Pediatric Infectious Disease Journal 1993;12:131‐5. [DOI] [PubMed] [Google Scholar]
Bernstein 1995 {published data only}
- Bernstein HH, Rothstein EP, Pichichero ME, Green JL, Reisinger KS, Blatter MM, et al. Reactogenicity and immunogenicity of a three‐component acellular pertussis vaccine administered as the primary series to 2, 4 and 6 month old infants in the United States. Vaccine 1995;13(17):1631‐5. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hori 1994 {published data only}
- Hori H, Afari EA, Akanmori BD, Kamiya Y, Sakatoku H, Nkrumah FK, at al. A randomized controlled trial of two acellular pertussis‐diphtheria‐tetanus vaccine in primary immunization in Ghana: antibody responses and adverse reactions. Annals of Tropical Paediatrics 1994;14(2):91‐6. [DOI] [PubMed] [Google Scholar]
Hori 1995 {published data only}
- Hori H, Afari A, Akanmori BD, Kamiya Y, Sakatoku H, Nkrumah FK, et al. Pertussis immunization with acellular vaccines in Ghanaian children. Annals of Tropical Paediatrics 1995;15(2):141‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Just 1991 {published data only}
- Just M, Kanra G, Bogaerts H, Berger R, Ceyhan M, Petre J. Two trials of an acellular DTP vaccine in comparison with a whole‐cell DTP vaccine in infants: evaluation of two PT doses and two vaccination schedules. Developments in Biological Standardization 1991;73:275‐83. [MEDLINE: ] [PubMed] [Google Scholar]
Miller 1997 {published data only}
- Miller E, Ashworth LA, Redhead K, Thornton C, Waight PA, Coleman T. Effect of schedule on reactogenicity and antibody persistence of acellular and whole‐cell pertussis vaccines: value of laboratory tests as predictors of clinical performance. Vaccine 1997;15(1):51‐60. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Murphy 1983 {published data only}
- Murphy MD, Rasnack J, Dickson HD, Dietch M, Brunell PA. Evaluation of the pertussis components of diphtheria‐tetanus‐pertussis vaccine. Pediatrics 1983;71(2):200‐5. [MEDLINE: ] [PubMed] [Google Scholar]
Pichichero 1987 {published data only}
- Pichichero ME, Badgett JT, Rodgers GC Jr, McLinn S, Trevino Scatterday B, Nelson JD. Acellular pertussis vaccine: immunogenicity and safety of an acellular pertussis versus a whole‐cell pertussis vaccine combined with diphtheria and tetanus toxoids as a booster in 18‐ to 24‐month old children. Pediatric Infectious Disease Journal 1987;6(4):352‐63. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Shek 2003 {published data only}
- Shek LP, Khor ES, Tan GH, Low KT, Ong D, Roy J, et al. Comparative study of the reactogenicity of a three‐component acellular pertussis vaccine and whole‐cell pertussis vaccine administered to healthy Singaporean infants. Southeast Asian Journal of Tropical Medicine and Public Health 2003;34(4):863‐8. [PubMed] [Google Scholar]
Additional references
Bamberger 2008
- Bamberger ES, Srugo I. What is new in pertussis?. European Journal of Pediatrics 2008;167:133‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berlin 1997
- Berlin JA, University of Pennsylvania Meta‐analysis Blinding Study Group. Does blinding of readers affect the results of meta‐analyses?. Lancet 1997;350:185‐6. [DOI] [PubMed] [Google Scholar]
Bettinger 2007
- Bettinger JA, Halperin SA, Serres G, Scheifele DW, Tam T. The effect of changing from whole‐cell to acellular pertussis vaccine on the epidemiology of hospitalized children with pertussis in Canada. Pediatric Infectious Disease 2007;26(1):31‐5. [DOI] [PubMed] [Google Scholar]
Bisgard 2005
- Bisgard KM, Rhodes P, Connelly BL, Bi D, Hahn C, Patrick S, et al. Pertussis vaccine effectiveness among children 6 to 59 months of age in the United States, 1998‐2001. Pediatrics 2005;116(2):e285‐94. [DOI] [PubMed] [Google Scholar]
Blackwelder 1991
- Blackwelder WC, Storsaeter J, Olin P, Hallander HO. Acellular pertussis vaccines. Efficacy and evaluation of clinical case definitions. American Journal of Diseases of Children 1991;145(11):1285‐9. [DOI] [PubMed] [Google Scholar]
Carlsson 2009
- Carlsson RM, Trollfors B. Control of pertussis ‐ lessons learnt from a 10‐year surveillance programme in Sweden. Vaccine 2009;27(42):5709‐18. [DOI] [PubMed] [Google Scholar]
Casey 2005
- Casey JR, Pichichero ME. Acellular pertussis vaccine safety and efficacy in children, adolescents and adults. Drug 2005;65(10):1367‐89. [DOI] [PubMed] [Google Scholar]
CDC 2009
- Centers for Disease Control. Recommendations and Guidelines: 2009 Child and Adolescent Immunization Schedules. www.cdc.gov/vaccines/recs/schedules/child‐schedule.htm 2009.
CDC 2011
- Centers for Disease Control. Local health department costs associated with response to a school‐based pertussis outbreak ‐ Omaha, Nebraska, September–November 2008. Morbidity and Mortality Weekly Report 2011;60:4‐5. [PubMed] [Google Scholar]
Cherry 1996
- Cherry JD. Historical review of pertussis and the classical vaccine. Journal of Infectious Diseases 1996;174(Suppl 3):8259‐63. [DOI] [PubMed] [Google Scholar]
Cherry 1998
- Cherry JD, Gornbein J, Heininger U, Stehr K. A search for serologic correlates of immunity to Bordetella pertussis cough illnesses. Vaccine 1998;16:1901–6. [DOI] [PubMed] [Google Scholar]
Cherry 2012
- Cherry JD. Epidemic pertussis in 2012 ‐ the resurgence of a vaccine‐preventable disease. New England Journal of Medicine 2012;367(9):785‐7. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Zellweger‐Zähner T, Schneider M, Junker C, Lengeler C, Antes G. Language bias in randomised controlled trials published in English and German. Lancet 1997;350:326‐9. [DOI] [PubMed] [Google Scholar]
Feunou 2010
- Feunou PF, Bertout J, Locht C. T‐ and B‐cell‐mediated protection induced by novel, live attenuated pertussis vaccine in mice. Cross protection against parapertussis. PLoS One 2010;5(4):e10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fine 1997
- Fine PE. Implications of different study designs for the evaluation of acellular pertussis vaccines. Developments in Biological Standardization 1997;89:123‐33. [PubMed] [Google Scholar]
Forsyth 2004
- Forsyth KD, Campins‐Marti M, Caro J, Cherry JD, Greenberg D, Guiso N, et al. New pertussis vaccination strategies beyond infancy: recommendations by the global pertussis initiative. Clinical Infectious Diseases 2004;39(12):1802‐9. [DOI] [PubMed] [Google Scholar]
Galanis 2006
- Galanis E, King AS, Varughese P, Halperin SA. Changing epidemiology and emerging risk groups for pertussis. Canadian Medical Association Journal 2006;174(4):451‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Granoff 1997
- Granoff DM, Rappuoli R. Are serological responses to acellular pertussis antigens sufficient criteria to ensure that new combination vaccines are effective for prevention of disease?. Developments in Biological Standardization 1997;89:379‐89. [PubMed] [Google Scholar]
Gustafsson 2006
- Gustafsson L, Hessel L, Storsaeter J, Olin P. Long‐term follow‐up of Swedish children vaccinated with acellular pertussis vaccines at 3, 5, and 12 months of age indicates the need for a booster dose at 5 to 7 years of age. Pediatrics 2006;118(3):978‐84. [DOI] [PubMed] [Google Scholar]
Harnden 2009
- Harnden A. Practice: Easily missed? Whooping cough. BMJ 2009;338:b1772. [DOI] [PubMed] [Google Scholar]
Hedenskog 1987
- Hedenskog S, Granström M, Olin P, Tiru M, Sato Y. A clinical trial of a monocomponent pertussis toxoid vaccine. American Journal of Diseases of Children 1987;141(8):844‐7. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Chichester: Wiley‐Blackwell, 2011. [Google Scholar]
Hviid 2004
- Hviid A, Stellfeld M, Andersen PH, Wohlfahrt J, Melbye M. Impact of routine vaccination with a pertussis toxoid vaccine in Denmark. Vaccine 2004;22(27‐8):3530‐4. [DOI] [PubMed] [Google Scholar]
Jacquet 2006
- Jacquet JM, Bégué P, Grimprel E, Reinert P, Sandbu S, Silfverdal SA, et al. Safety and immunogenicity of a combined DTPa‐IPV vaccine administered as a booster from 4 years of age: a review. Vaccine 2006;24(13):2440‐8. [DOI] [PubMed] [Google Scholar]
Jefferson 2003
- Jefferson T, Rudin M, DiPietrantonj C. Systematic review of the effects of pertussis vaccines in children. Vaccine 2003;21(17‐18):2003‐14. [DOI] [PubMed] [Google Scholar]
Kerr 2000
- Kerr JR, Matthews RC. Bordetella pertussis infection: pathogenesis, diagnosis, management, and the role of protective immunity. European Journal of Clinical Microbiology and Infectious Diseases 2000;19:77‐88. [DOI] [PubMed] [Google Scholar]
Kuno‐Sakai 2004
- Kuno‐Sakai H, Kimura M. Safety and efficacy of acellular pertussis vaccine in Japan, evaluated by 23 years of its use for routine immunization. Pediatrics International 2004;46(6):650‐5. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Liese 1997
- Liese JG, Meschievitz CK, Harzer E, Froeschle J, Hosbach P, Hoppe JE, et al. Efficacy of a two‐component acellular pertussis vaccine in infants. Pediatric Infectious Disease Journal 1997;16(11):1038‐44. [DOI] [PubMed] [Google Scholar]
Liko 2013
- Liko J, Robison SG, Cieslak PR. Priming with whole‐cell versus acellular pertussis vaccine. New England Journal of Medicine 2013;368(6):581‐2. [DOI] [PubMed] [Google Scholar]
Mahid 2008
- Mahid SS, Qadan M, Hornung CA, Galandiuk S. Assessment of publication bias for the surgeon scientist. British Journal of Surgery 2008;95(8):943‐9. [DOI] [PubMed] [Google Scholar]
Millis 1998
- Mills KHG, Ryan M, Ryan E, Mahon BP. A murine model in which protection correlates with pertussis vaccine efficacy in children reveals complementary roles for humoral and cell‐mediated immunity in protection against Bordetella pertussis. Infection and Immunity 1998;66:594‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mills 1999
- Mills KHG, Ryan M, McGuirk P, Griffin F, Murphy G, Mahon B. The immunology of Bordetella pertussis infection. Biologicals 1999;27(2):77. [DOI] [PubMed] [Google Scholar]
Petitti 1994
- Petitti DB. Meta‐analysis, Decision Analysis and Cost‐Effectiveness Analysis: Methods for Quantitative Synthesis in Medicine. Oxford: Oxford University Press, 1989. [Google Scholar]
Pichichero 2005
- Pichichero ME, Casey JR. Acellular pertussis vaccines for adolescents. Pediatric Infectious Disease Journal 2005;24(Suppl 6):117‐26. [DOI] [PubMed] [Google Scholar]
Pichichero 2006
- Pichichero ME, Casey JR, Francis AB, Murphy M, Hoeger W, Cleary C. Acellular pertussis vaccine boosters combined with diphtheria and tetanus toxoid boosters for adolescents: safety and immunogenicity assessment when preceded by different 5‐dose DTaP/DTwP schedules. Clinical Pediatrics 2006;45(7):613‐20. [DOI] [PubMed] [Google Scholar]
Plotkin 2008
- Plotkin SA. Correlates of vaccine‐induced immunity. Clinical Infectious Diseases 2008;47:401–9. [DOI] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Rodríguez‐Cobo 2008
- Rodríguez‐Cobo I, Chen YF, Olowokure B, Litchfield I. Clinical and economic assessment of different general population strategies of pertussis vaccine booster regarding number of doses and age of application for reducing whooping cough disease burden: a systematic review. Vaccine 2008;26:6768‐76. [DOI] [PubMed] [Google Scholar]
Sato 1984
- Sato Y, Kimura M, Fukumi H. Development of a pertussis component vaccine in Japan. Lancet 1984;1(8369):122‐6. [DOI] [PubMed] [Google Scholar]
Schmitt 1996
- Schmitt HJ, König CH, Neiss A, Bogaerts H, Bock HL, Schulte‐Wissermann H, et al. Efficacy of acellular pertussis vaccine in early childhood after household exposure. JAMA 1996;275(1):37‐41. [PubMed] [Google Scholar]
Sheridan 2012
- Sheridan SL, Ware RS, Grimwood K, Lambert SB. Number and order of whole cell pertussis vaccines in infancy and disease protection. JAMA 2012;308(5):454‐6. [DOI] [PubMed] [Google Scholar]
Singh 2006
- Singh M, Lingappan K. Whooping cough. Chest 2006;130:1547‐53. [DOI] [PubMed] [Google Scholar]
Stephens 2008
- Stephens DS. Vaccines for the unvaccinated: protecting the herd. Journal of Infectious Diseases 2008;197:643‐5. [DOI] [PubMed] [Google Scholar]
Storsaeter 1998
- Storsaeter J, Hallander HO, Gustafsson L, Olin P. Levels of anti‐pertussis antibodies related to protection after household exposure to Bordetella pertussis. Vaccine 1998;16:1907–16. [DOI] [PubMed] [Google Scholar]
Taranger 1997
- Taranger J, Trollfors B, Lagergård, Lind L, Sund VH, Zackrisson G, et al. Unchanged efficacy of a pertussis toxoid vaccine throughout the two years after the third vaccination of infants. Pediatric Infectious Disease Journal 1997;16:180‐4. [DOI] [PubMed] [Google Scholar]
Taranger 2000
- Taranger J, Trollfors B, Lagergård T, Sundh V, Bryla DA, Schneerson R, et al. Correlation between pertussis toxin IgG antibodies in postvaccination sera and subsequent protection against pertussis. Journal of Infectious Diseases 2000;181:1010‐3. [DOI] [PubMed] [Google Scholar]
Tran Minh 1998
- Tran Minh NN, Edelman K, He Q, Viljanen MK, Arvilommi H, Mertsola J. Antibody and cell‐mediated immune responses to booster immunisation with a new acellular pertussis vaccine in school children. Vaccine 1998;16:1604‐10. [DOI] [PubMed] [Google Scholar]
Watanabe 2005
- Watanabe M, Nagai M. Acellular pertussis vaccines in Japan: past, present and future. Expert Review of Vaccines 2005;4(2):173‐84. [DOI] [PubMed] [Google Scholar]
WHO 1991
- World Health Organization. WHO meeting on case definitions of pertussis. MIN/EPI/PERT/91 1991:4‐5.
WHO 2010
- World Health Organization. Pertussis vaccines: WHO position paper. Weekly Epidemiological Record, WHO 2010;No. 40, 85:385–400. [Google Scholar]
References to other published versions of this review
Tinnion 1999
- Tinnion ON. Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 1999, Issue 2. [DOI: 10.1002/14651858.CD001478.pub2] [DOI] [PubMed] [Google Scholar]
Tinnion 2001
- Tinnion ON, Hanlon M. Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 2001, Issue 4. [DOI: 10.1002/14651858.CD001478.pub2] [DOI] [PubMed] [Google Scholar]
Zhang 2009
- Zhang L, Prietsch SOM, Axelsson I, Halperin SA. Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD001478.pub4] [DOI] [Google Scholar]
Zhang 2012
- Zhang L, Prietsch SOM, Axelsson I, Halperin SA. Acellular vaccines for preventing whooping cough in children. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD001478.pub5] [DOI] [PubMed] [Google Scholar]