

Summary
CDK11 is a cyclin-dependent kinase that controls proliferation by regulating transcription, RNA splicing, and the cell cycle. As its activity is increasingly associated with cancer, CDK11 is an attractive target for the development of small molecule inhibitors. However, the development of CDK11 inhibitors with limited off-target effects against other CDKs poses a challenge based on the high conservation of sequence across family members. OTS964 is notable as it displays a measure of specificity for CDK11 in cells. To understand the basis for OTS964’s specificity for CDK11, we solved a 2.6 Å crystal structure of the CDK11 kinase domain bound to OTS964. Despite the absence of cyclin, CDK11 adopts an active like conformation when bound to OTS964. We identified amino acids likely to contribute to the specificity of OTS964 for CDK11 and assessed their contribution to OTS964 binding by ITC in vitro and by resistance to OTS964 in cells.
eTOC Blurb
Kelso et al. report the crystal structure of the CDK11 kinase domain bound to the small molecule inhibitor OTS964. Mutational analysis explores the contribution of specific contact residues towards OTS964 binding and specificity.
Graphical Abstract
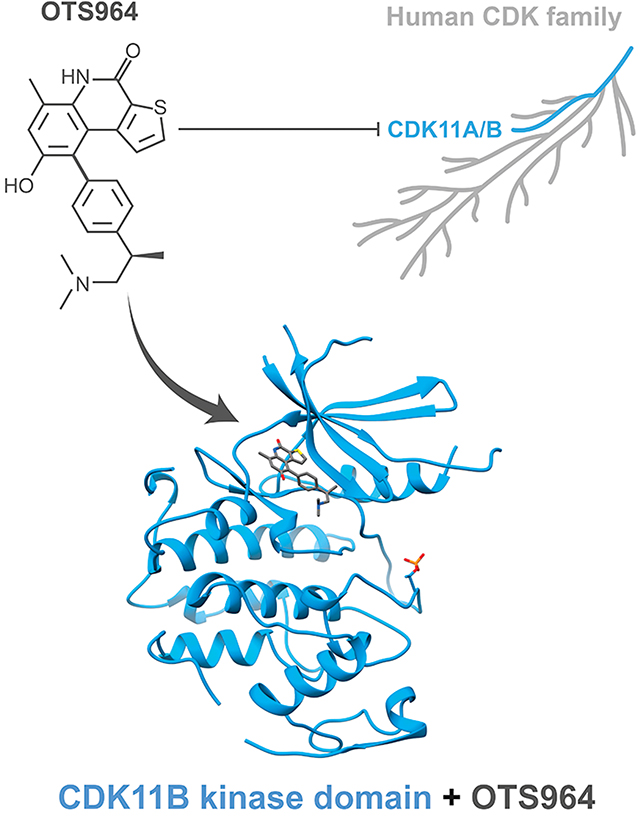
Introduction
The cyclin-dependent kinases (CDKs) are a large family of 20 serine/threonine protein kinases that regulate diverse cellular processes ranging from transcription to progression through the cell cycle (Malumbres, 2014). CDK kinase activity is activated by hetero-dimerization with another large family of proteins termed cyclins and by phosphorylation events within their activation segments. Some CDKs are also inhibited by interaction with inhibitor proteins of the Kip/Cip and INK4 families and by inhibitory phosphorylation events elsewhere on the kinase domain such as within the G-loop (Wood and Endicott, 2018). Due to their important roles in regulating cell growth and division and the druggable nature of the protein kinase fold, the CDKs have garnered attention as targets for the development of small molecule cancer therapeutics (Ferguson and Gray, 2018).
Early generations of CDK inhibitors display broad cross reactivity across many CDKs and exhibited limited therapeutic utility in clinical trials (Asghar et al., 2015). For this reason, efforts have focused on developing inhibitors that target a single CDK or a small subset of CDKs sharing common cellular functions. A major impediment to these efforts is the high degree of conservation in the kinase active site across CDK family members (Martin et al., 2017). Despite this challenge, some success has been reported. Most notably, Palbociclib, Ribociclib, and Abemaciclib which target both CDK4 and CDK6 have been approved for the treatment of hormone-receptor positive advanced breast cancer (Braal et al., 2021). All three inhibitors, which show little cross reactivity towards other CDK family members, function to prevent cell cycle entry by blocking phosphorylation of the tumour suppressor retinoblastoma protein by CDK4/CDK6-cyclin D complexes. Some success at inhibitor development has also been achieved for transcriptional CDKs such as CDK7, CDK8/CDK19, CDK9, and CDK12/CDK13 (Zhang et al., 2021) with inhibitors of CDK7 and of CDK9 now in Phase I clinical trials. Many of these CDK specific inhibitors exploit sequence features of the kinase active site that are unique to particular CDKs including surfaces just outside of the direct ATP binding site (Martin et al., 2017).
Still, some CDKs continue to present a major challenge for drug development. CDK2 has long been the focus of drug discovery efforts and is a target of many potent existing pan-CDK inhibitors. However, identifying small molecules that selectively bind to CDK2 has proven difficult. In particular, CDK2 inhibitors frequently exhibit off-target reactivity against the closely related kinase CDK1. The two enzymes play very different biological roles in cell cycle regulation; while CDK2 regulates entry into S-phase, CDK1 regulates aspects of mitosis. Despite their different functional roles, CDK1 and CDK2 have near identical amino acid compositions in their active sites and adopt similar structures in their active cyclin-bound states. For this reason, most ATP competitive inhibitors of CDK2 also inhibit CDK1 with comparable potencies (Tadesse et al., 2019). ATP competitive inhibitors that show some measure of specificity for CDK2-cyclin A over CDK1-cyclin B often show even greater specificity for CDK2 over CDK1 when no cyclins are present (Wood et al., 2019). This increased specificity has been attributed to differences in the dynamics of the two kinase domains in solution as crystal structures of CDK2 and CDK1 bound to the same inhibitor show strikingly similar binding modes despite large differences in affinity (~20X for Dinaciclib and ~170X for AZD5438; Wood et al., 2019). Furthermore, many small molecule inhibitors described as dual CDK2/CDK1 inhibitors in vitro have been shown to engage an even wider range of CDKs in cells (Wells et al., 2020). While some inhibitors have begun to explore binding outside the CDK2 ATP binding pocket (Alexander et al., 2015; Betzi et al., 2011), more efforts are needed to determine if this will be a viable strategy for achieving selectivity.
CDK11 is an emerging therapeutic target in numerous cancers, including liposarcoma, osteosarcoma, multiple myeloma, acute myeloid leukemia, breast cancer, colon cancer, cervical cancer, ovarian cancer, melanoma, and esophageal squamous cell carcinoma (Zhou et al., 2016; Liu et al., 2016; Ahmed et al., 2019; Du et al., 2019). CDK11 is encoded by two genes CDK11A and CDK11B that are 99% identical in protein sequence (Gururajan et al., 1998; Malumbres et al., 2009). CDK11 plays a regulatory role in both RNA transcription and splicing (Trembley et al., 2004) as well as in the progression through the G2/M phase of the cell cycle (Renshaw et al., 2019). These activities are mediated by distinct CDK11 isoforms, CDK11p110 and CDK11p58, respectively. The p110 isoform is expressed in a non-cell cycle dependent manner and comprises the full-length version of CDK11 containing the central RE (Arg/Glu) domain, poly-E (Glu) domain, and C-terminal kinase domain. In contrast, the p58 isoform is generated from an internal ribosome-entry site upstream of the same kinase domain during the G2/M phase transition only (Cornelis et al., 2000). Both isoforms are activated by binding to the L-type cyclins, L1 and L2 (Berke et al., 2001; Dickinson et al., 2002), while the p58 isoform has also been reported to bind cyclin D3 (Zhang et al., 2002). Increased expression of CDK11 has been noted for several cancer cell lines, and knockdown effects of CDK11 in breast cancer, myeloma, osteoscarcoma, and liposarcoma cells implicates CDK11 as important for cell survival (Zhou et al., 2016).
To date, only one inhibitor has been disclosed that was prospectively developed to target CDK11, namely ZNL-05–044 (Li et al., 2022). Although it displays promising on-target function in cells, ZNL-05–044 appears equally cross reactive towards CDKs 4, 6, and 7 in NanoBRET and KINOMEscan assays (Li et al., 2022). Despite being developed specifically to target CDK11, ZNL-05–044 is less potent at targeting CDK11 than the pre-existing kinase inhibitor OTS964. In 2019, Lin et al. discovered that the kinase inhibitor OTS964, developed to target TOPK (Matsuo et al., 2014) (T-lymphokine-activated killer cell-Originated Protein Kinase) aka PBK (PDZ-Binding Kinase), has significant off-target activity against CDK11 and that this activity is primarily responsible for the inhibitor’s toxic effect on cancer cells. A KINOMEscan assay for OTS964 against a panel of CDKs demonstrated that OTS964 binding to CDK11 is surprisingly specific (10X more potent than against CDK7, and no apparent binding to other CDKs tested; Lin et al., 2019). Interestingly, mutation of a single amino acid in CDK11 (specifically Gly223Ser - amino acid numbering from CDK11B p58) caused resistance to OTS964 (Lin et al., 2019). The identity of the residue at this position is also partially responsible for the specificity of OTS964 across CDK family members as all CDKs except CDK11 have an alanine at this position rather than glycine. Currently no structures are available for OTS964 bound to CDK11 or bound to its original target kinase TOPK. Hence the basis for how OTS964 binds CDK11 and the role of Gly223 and other potential features of the CDK11 kinase domain in mediating OTS964 specificity and sensitivity remain in question.
Recently, O’Brien et al. (2022) showed that cancer cell lines harbouring inactivating mutations in the tumour suppressor gene FBXW7 are dependent on CDK11-cyclin L1 activity for survival. With the knowledge that OTS964 is a potent cross-reactive inhibitor of CDK11 in cells, they showed that tumor cell lines harbouring FBXW7−/− mutations are more sensitive to OTS964 treatment than those harbouring the FBXW7 wild-type gene. This finding suggests that the selective inhibition of CDK11 may be a viable strategy for treating cancers driven by mutations in FBXW7. With this observation in mind, we determined the 2.6 Å X-ray crystal structure of OTS964 bound to the kinase domain of CDK11. The structure provides insight into how OTS964 selectively binds CDK11 versus other CDK family members and how the Gly223Ser mutation imparts resistance to OTS964. Interestingly, OTS964 binds to CDK11 in an active-like conformation of the kinase domain that manifests in the crystal environment even in the absence of cyclin binding. As expected, OTS964 engages the ATP binding pocket of CDK11 along the hinge region and makes contacts with many amino acids conserved between CDK11 and other CDKs. Gly223 lies in close proximity to OTS964 explaining why its mutation to a larger serine residue may impact on binding. Further analysis reveals other residues that contribute to binding affinity and specificity in vitro, although their contributions to binding in vivo context appear negligible. Structural insight gained from studies of OTS964 together with the pioneering drug development efforts of Li et al. (2022), may provide a starting point for developing other more effective and selective inhibitors of CDK11.
Results
Structure determination of CDK11 bound to OTS964
We expressed the kinase domain of CDK11 (residues 72–380 of CDK11B p58) with an N-terminal, TEV (tobacco etch virus) protease cleavable His-tag in Sf9 cells. Our multiple attempts to purify the cyclin domain of the CDK11 activator cyclin L1 from either Sf9 or E. coli cell culture proved unsuccessful. While we achieved some success producing cyclin D3, we could not demonstrate binding to CDK11 in vitro. Thus, we focused our crystallization efforts on CDK11 in the absence of an activating cyclin. We co-crystallized CDK11 with OTS964 yielding well diffracting crystals that enabled collection of a near complete 2.6 Å resolution data set. We determined the structure of CDK11 by molecular replacement using a homology model of CDK11 derived from the CDK2 kinase domain bound to cyclin A (PDB 2IW8, Pratt et al., 2006) lacking N-terminal residues 72–78, β3-αC loop residues 115 to 123, the activation segment residues 229 to 244, and C-terminal residues 368 to 380. We observed six highly similar copies of the CDK11 kinase domain in the asymmetric unit (cα superimpose with average RMSD 0.486Å, CDK11 residues modelled in all six copies of CDK11 are indicated in Figure 1). Data collection and refinement statistics are located in Table 1. See Sup. Figure 1 for representative electron density maps.
Figure 1. Protein sequence alignment of CDKs 1–12 and TOPK.

Multiple sequence alignment showing the conservation of amino acids between human CDK11B, TOPK, and human CDKs 1–12. Protein sequences of human kinases were aligned using Clustal Omega and visualized using Jalview. Boxed residues of CDK11 were modelled in all six copies of CDK11 in the asymmetric unit. Yellow circles indicate amino acids located within 5.0 Å of OTS964 and red circles indicate amino acids located within 5.0 Å of OTS964 that were mutated as a part of this study.
Table 1.
Diffraction data collection and refinement statistics.
| CDK1172-380-OTS964 | |
|---|---|
|
| |
| Data collection | |
| Space group | P 65 |
| Cell dimensions | |
| a, b, c (Å) | 146.68, 146.68, 230.94 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| Resolution (Å) | 200.00–2.60 (2.65–2.60) |
| Rmeas | 11.4 (280.1) |
| Rpim | 4.1 (95.5) |
| Rmerge | 11.4 (262.9) |
| I/σI | 13.2 (0.8) |
| CC1/2 | 99.9 (18.8) |
| Completeness (%) | 99.9 (97.2) |
| Reflections (unique) | 86336 |
| Redundancy | 8.6 (8.3) |
| Refinement | |
| Resolution (Å) | 127.03–2.60 (2.64–2.60) |
| No. reflections | 86184 (3761) |
| Rwork | 0.222 (0.308) |
| Rfree | 0.236 (0.331) |
| No. atoms | |
| Protein | 13231 |
| Ligands | 198 |
| B-factors | |
| Protein | 73.8 |
| Ligands | 70.9 |
| R.M.S. deviations | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.503 |
| Ramachandran Plot | |
| Most favored (%) | 96.8 |
| Allowed (%) | 2.8 |
| Disallowed (%) | 0.3 |
| Clashscore | 6.80 |
| MolProbity score | 1.57 |
| PDB ID code | 7UKZ |
Highest resolution shell is shown in parenthesis.
OTS964 binds CDK11 in the active conformation
CDK11 bound to OTS964 (Figure 2A) adopts an active-like kinase conformation defined by productive position of helix αC, a contiguous regulatory spine, and a productive conformation of the activation segment harbouring phosphorylation on the Thr239 positive regulatory site. Despite the absence of cyclin binding, helix αC adopts an inwardly rotated conformation more similar to CDK2 bound to its activator cyclin A (Figure 2B) than to other CDK kinases in the absence of a cyclin. This allows the subdomain III side chain of Glu129 to point inward and form a salt bridge with subdomain II sidechain of Lys111 within the β3 strand. The inward position of helix αC also orients the hydrophobic side chain of Ile133 towards the kinase core to complete the regulatory spine (R-spine), a network of contiguous hydrophobic amino acids observed in all active kinase conformations (Figure 2C). Finally, the activation segment is ordered and adopts a productive conformation supported by near stochiometric phosphorylation of Thr239 phospho-regulatory site. The phosphate on Thr239 is coordinated by three arginine residues Arg128, Arg205, and Arg229 (Figure 2D). We cannot rule crystal packing interactions as contributing to the unexpected productive conformation of helix αC (Sup. Figure 2).
Figure 2. OTS964 binds CDK11 in an active-like conformation.
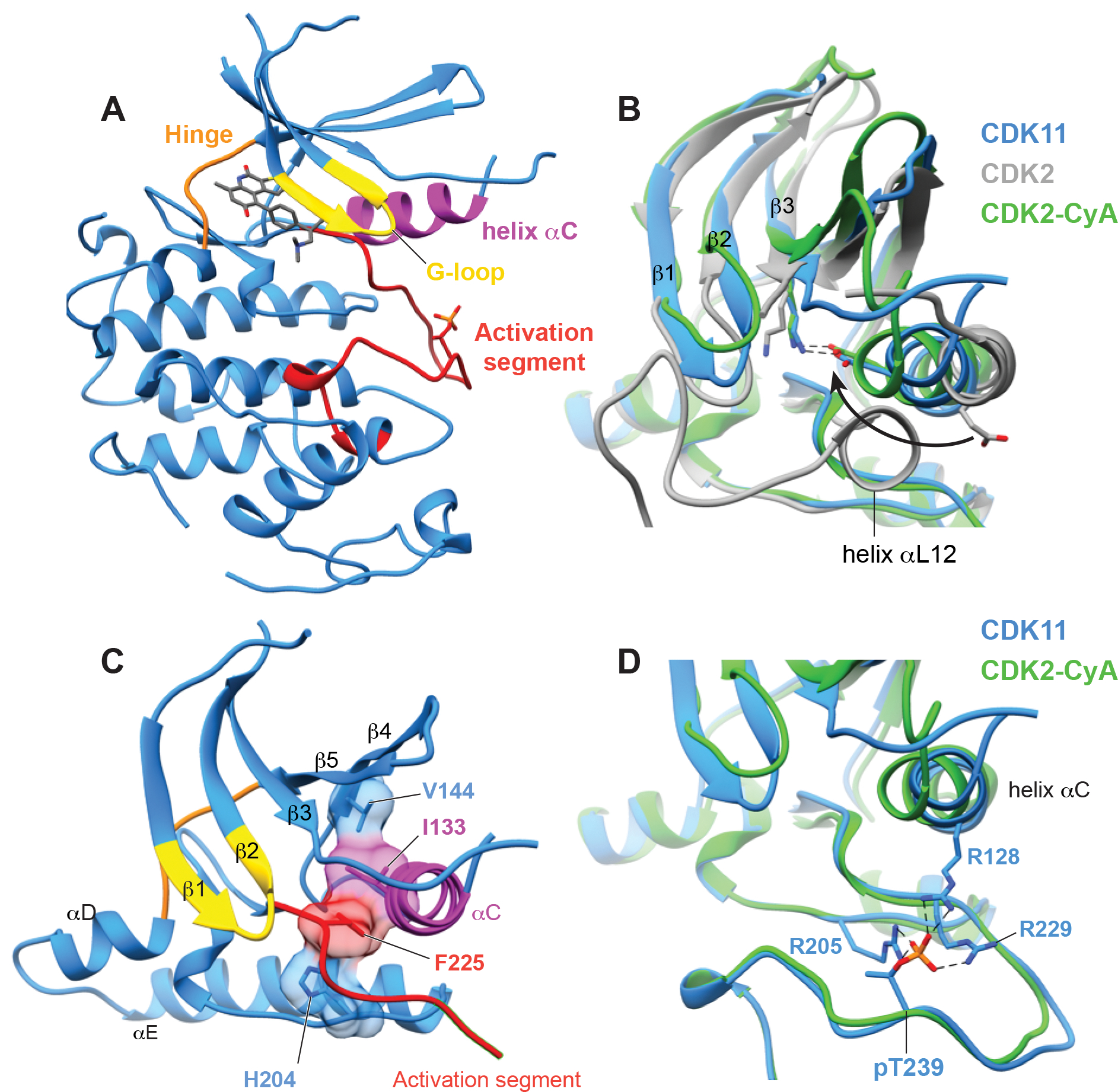
(A) The overall structure of CDK11 with OTS964 bound in the active site, highlighting the position of important kinase substructures the G-loop (yellow), helix αC (magenta), the hinge between the N and C lobes (orange), and the activation segment (red).
(B) View of helix αC of CDK11 comparing the overall position of the helix and direction of conserved glutamate (CDK11 E129, CDK2 E51) to structures of CDK2-cyclin A (PDB 1QMZ, Brown et al., 1999) and CDK2 without a cyclin (PDB 1HCK, Schulze-Gahmen et al., 1996).
(C) View of the R-spine residues of CDK11 which are aligned within the kinase domain.
(D) View of on the conformation of the phosphorylated activation segment of CDK11 compared to the phosphorylated activation segment CDK2 and showing the position of phospho-Thr229 which is coordinated by indicated arginine residues.
The binding mode of OTS964 to CDK11
Consistent with its designation as a type-I (i.e., ATP competitive) kinase inhibitor, OTS964 occupies the ATP binding pocket of the CDK11 kinase domain (Figure 3A). After refining the structure of CDK11, we observed electron density for OTS964 in the active site of all six copies of CDK11 in the asymmetric unit (Sup. Figure 1A). Based on the size and shape of the density, we could reasonably dock OTS964 into the active site in two possible orientations (Sup. Figure 3A, left). In both, the fused ring portion of OTS964 sits along the kinase hinge in the nucleotide binding pocket, while the tail portion of OTS964 extends towards the opening of the active site cleft. Contouring the electron density map prior to docking and refinement of OTS964 coordinates at 5 σ or greater revealed a single electron density peak near the gate keeper residue Met160, which we reasoned corresponded to the position of the electron rich sulfur atom of the thiophene ring (Sup. Figure 3A, right). This specific feature was conserved for all six copies of the CDK11-OTS964 complex in the asymmetric unit, providing confidence in the correctness of binding mode 1. In further support, binding mode 1 allowed for the formation of three hydrogen bonds between CDK11 and OTS964 whereas the alternate binding mode allowed for only a single hydrogen bond (Figure 3B, Sup. Figure 3B,C). Specifically, the amine and carbonyl groups on the central hydroxypyridine ring of OTS964 form hydrogen bonds to the backbone of CDK11 hinge residue Val96, while the hydroxyl group of the benzene ring forms a hydrogen bond to the Asp166 sidechain. Hydrophobic side chains of Ile88, Val96, Ala109, Val142, Tyr162, and Leu213 form complementary interaction with hydrophobic features of OTS964 (Figure 3B). We concluded that OTS964 binds within the ATP binding pocket of CDK11 in orientation 1.
Figure 3. The binding mode of OTS964 to CDK11.
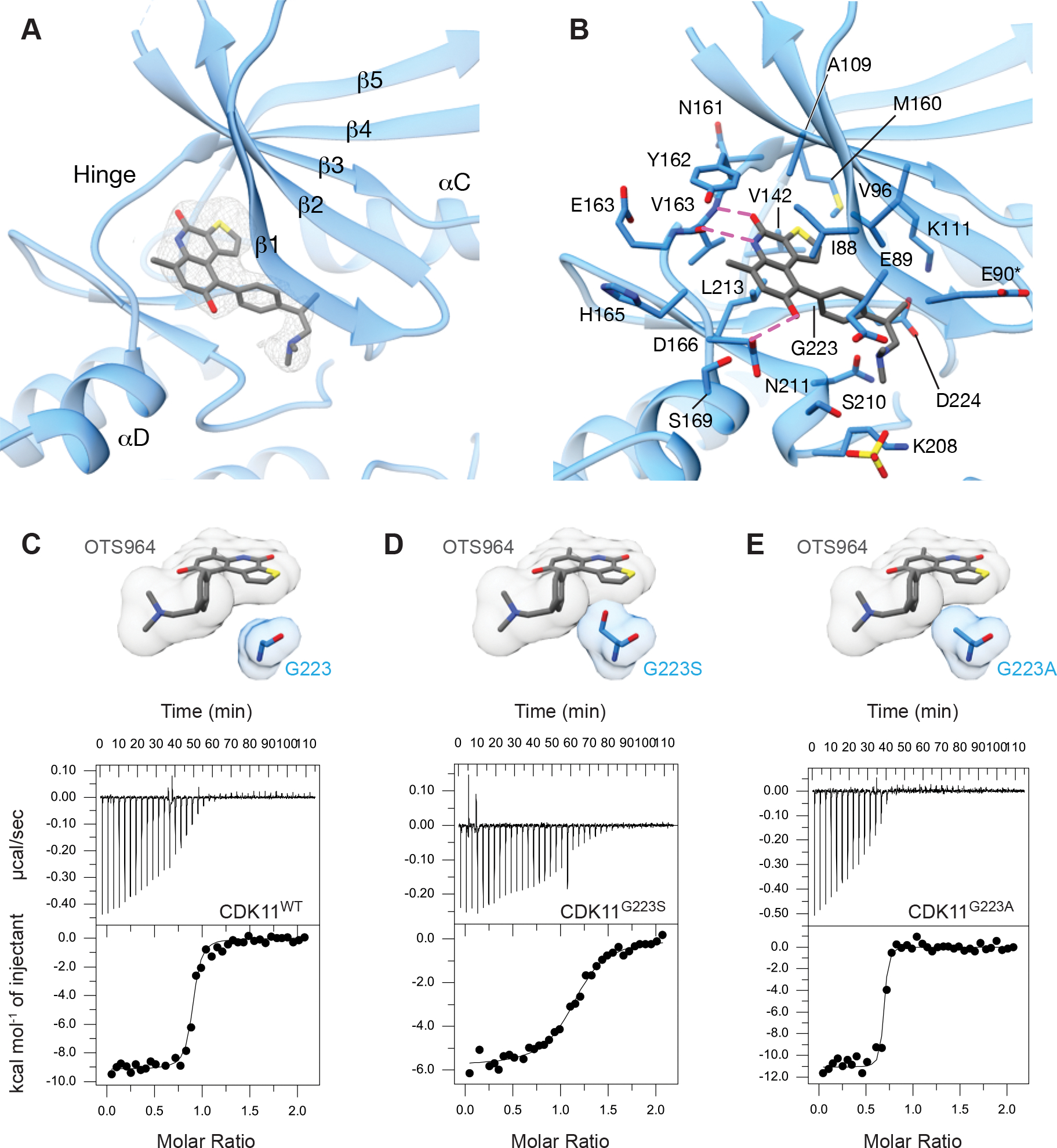
(A) View of OTS964 bound to the ATP binding pocket of CDK11 showing |Fo-Fc| electron density at 2.5 σ from prior to docking OTS964.
(B) View of OTS964 bound to the ATP binding pocket of CDK11 highlighting the position of nearby amino acids and possible hydrogen bonds in magenta. Side chains of residues marked with an asterisk were modeled for clarity.
(C) View of the position of the G223 side chain relative to OTS964, accompanied by a representative ITC (isothermal calorimetry) trace of binding between CDK11 wild-type and OTS964.
(D) View of the G223 side chain modeled as a serine (G223S), highlighting the position of the side chain relative to OTS964. Representative ITC (isothermal calorimetry) trace of binding between CDK11 G223S and OTS964.
(E) View of the G223 side chain modelled as an alanine (G223A), highlighting the position of the side chain relative to OTS964. Representative ITC (isothermal calorimetry) trace of binding between CDK11 G223A and OTS964.
The identity of the xDFG residue is important for the binding of OTS964 to CDK11
Simultaneous to the discovery that OTS964 binds CDK11 in cells, Lin et al. (2019) determined that mutation of Gly223 of CDK11 confers resistance to cell killing by OTS964. In CDK11, Gly223 occupies the position preceding the DFG (Asp-Phe-Gly) motif that demarks the start of the activation segment (denoted xDFG). Mutation of Gly223 to serine caused strong resistance to OTS964, while mutation to alanine caused an intermediate effect (Lin et al., 2019). Interestingly, CDK11 is the only member of the CDK family that has a glycine in the xDFG position, while all other members have an alanine at this position (Figure 1). This specific difference correlates with the finding that OTS964 binds CDK11 with at least 75-fold higher affinity than other CDKs (KD 40 nM for CDK11 and >3 μM for other CDKs, Lin et al., 2019).
To understand how the identity of the xDFG residue effects OTS964 binding to CDK11 we examined the structure of CDK11 bound to OTS964. Due to the close proximity of Gly223 to the thiophene ring (~5 Å) (Figure 3C,D), we predict that mutation to serine (or any residue larger) would be expected to cause steric crowding with OTS964 binding. The finding that mutation of the xDFG position to alanine imparts resistance to OTS964 in cells is more difficult to rationalize since the alanine substitution appears to be accommodated sterically (Figure 3E).
To determine the precise effect of xDFG mutation on OTS964 binding CDK11 in vitro, we made both Gly223Ser and Gly223Ala mutants and tested binding by ITC (Figure 3C–E, Table 2). Compared to wild-type CDK11 (KD mean ± SD 65.4 ± 18.0 nM), the Gly223Ser mutant displayed ~13-fold impairment in OTS964 binding (KD mean ± SD 853 ± 56 nM). However, the Gly223Ala mutant displayed enhanced binding to OTS964 (KD mean ± SD 31.1 ± 13.8 nM). While this is surprising based on the previous experiments performed in cells, this is consistent with observation within our crystal structure. We reason that other factors beyond the xDFG residue must contribute to the selectivity of OTS964 for CDK11.
Table 2.
ITC binding constants for OTS964 binding CDKs
| CDK | N (sites) | KD (nM) | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS (cal/mol/K) |
|---|---|---|---|---|---|
|
| |||||
| CDK11 | |||||
| Wild-type | 0.882 | 65.4 ± 18.0 | −9.82 ± 0.16 | −9.33 ± 0.27 | 1.62 ± 0.35 |
| Gly223Ser | 1.12 | 853 ± 56 | −8.28 ± 0.04 | −5.93 ± 0.16 | 7.89 ± 0.42 |
| Gly223Ala (G223A) | 0.668 | 31.7 ± 13.8 | −10.4 ± 0.3 | −11.4 ± 0.4 | −3.65 ± 2.14 |
| Glu89Gly (E89G) | 0.882 | 50.9 ± 3.3 | −9.95 ± 0.79 | −9.82 ± 0.42 | 0.455 ± 1.27 |
| Met160Phe (M160F) | 0.794 | 556 ± 26 | −8.29 ± 0.32 | −8.13 ± 0.26 | 0.540 ± 1.94 |
| G223A/E89G/M160F | 0.616 | 320 ± 66 | −8.87 ± 0.12 | −9.89 ± 0.24 | −3.42 ± 1.21 |
| CDK2 | |||||
| Wild-type | 0.704 | 16300 ± 1700 | −6.54 ± 0.06 | −9.12 ± 0.60 | −8.66 ± 1.80 |
| Gly11Glu (G11E) | 0.866 | 6820 ± 530 | −7.05 ± 0.05 | −9.45 ± 0.32 | −8.05 ± 1.23 |
| Phe80Met (F80M) | 0.804 | 32400 ± 1900 | 17.5 ± 0.6 | 11.8 ± 0.3 | −19.1 ± 1.1 |
| Ala144Gly (A144G) | 0.554 | 16000 ± 4600 | −6.56 ± 0.17 | −10.6 ± 1.2 | −13.4 ± 4.5 |
| CDK2-cyclin AWT | |||||
| Wild-type | 1.00 | 1820 ± 500 | −7.85 ± 0.18 | −8.64 ± 1.06 | −2.64 ± 2.99 |
| Gly11Glu (G11E) | 1.16 | 621 ± 16 | −8.47 ± 0.02 | −7.13 ± 0.21 | 4.48 ± 0.76 |
| Phe80Met (F80M) | 1.28 | 4980 ± 440 | −7.24 ± 0.05 | −9.69 ± 0.08 | −8.23 ± 0.44 |
| Ala144Gly (A144G) | 1.04 | 3030 ± 150 | −7.53 ± 0.03 | −6.92 ± 0.01 | 2.04 ± 0.11 |
Data represents the mean ± standard deviation (SD) of two replicate experiments.
The Gatekeeper residue is a binding determinant for OTS964
We next focused our attention on Glu89 and Met160, both of which reside within 5 Å of OTS964 (Figure 1). Glu89 is the first amino acid of the G-loop that closes over the ATP binding pocket of the kinase. In nearly all kinases other than CDK11, including TOPK and all other CDKs, this position is occupied by a glycine residue rather than glutamic acid. Although the conformation of the Glu89 sidechain varies between the six copies of CDK11 in the asymmetric unit (Sup. Figure 1B), it is nonetheless well placed to interact with the phenyl group of the OTS964 tail. Met160 in CDK11 (and also TOPK) is the gatekeeper residue, located at the back of the ATP binding pocket, close to the thiophene ring of OTS964. Both CDK11 and CDK10 have methionine at the gatekeeper position, while all other CDKs have a phenylalanine. Notably, the Met160 side chain is clearly visible in the OTS964-CDK11 structure and makes direct contact with the thiophene ring (Sup. Figure 1C).
To test whether Glu89 and Met160 contribute favourably and selectively to OTS964 binding, we mutated them individually to the corresponding amino acids in most other CDKs (Glu89Gly and Met160Phe). As assessed by ITC (Table 2, isotherms shown in Sup. Figure 4), the Glu89Gly mutation had no effect on OTS964 binding CDK11 (KD mean ± SD 50.9 ± 3.3 nM for Glu89Gly compared to 65.4 ± 18.0 nM for wild-type) while the Met160Phe gate keeper mutation, caused a near 9-fold impairment of binding (KD mean ± SD 556 ± 26 nM).
We next asked whether the combined mutation of the three CDK11 specific residues (Glu89, Met160, and Gly223) in the ADP binding pocket could confer a more exaggerated loss of binding to OTS964 (as would be expected if the three together were responsible for the majority of why OTS964 is selective for CDK11 over other CDKs in cells). As assessed by ITC, OTS964 bound to the triple (Glu89Gly/Met160Phe/Gly223Ala) CDK11 mutant with a KD mean ± SD 320 ± 66 nM, only ~5-fold less tightly than wild-type CDK11. This result is consistent with an additive rather than a synergistic effect of the individual mutants on OTS964 binding (Table 2, isotherms shown in Sup. Figure 4) and suggests there may be other factors at play that contribute to the selectivity of OTS964 on CDK11 versus other CDKs.
Effect of CDK11 mutations on sensitivity to cell killing by OTS964
We next investigated the effect of CDK11 mutations on the ability of OTS964 to kill cells. For this we expressed either wild-type or mutant CDK11 (p58 isoform) stably in the human melanoma cell line A375 (Figure 4A, full western blot images in Sup. Figure 5A). We first verified that each CDK11 mutant was catalytically active in cells by demonstrating that cells harboring the CDK11 mutants could survive after CRISPR knock-out of the endogenous CDK11B gene (Sup. Figure 5B). Importantly, to perform this experiment we used a gRNA selectively targeting CDK11B-p110 to knock-out the endogenous CDK11B alleles while leaving the ectopically expressed CDK11B-p58 cDNA intact. We then compared the sensitivity of each of the A375 cell lines expressing the individual CDK11 mutants with wild-type A375 cells upon treatment with 100 nM OTS964 for 12 days before quantifying cell confluence (Figure 4B,C). Using this system, we could recapitulate the findings of Lin et al. (2019) showing the Gly223Ser CDK11 mutant confers resistance to cell killing by OTS964, while the Gly223Ala mutant confers weaker resistance (Figure 4B,C). While the former is fully consistent with our in vitro binding studies, the latter is not.
Figure 4. Mutations in CDK11 cause resistance to OTS964.
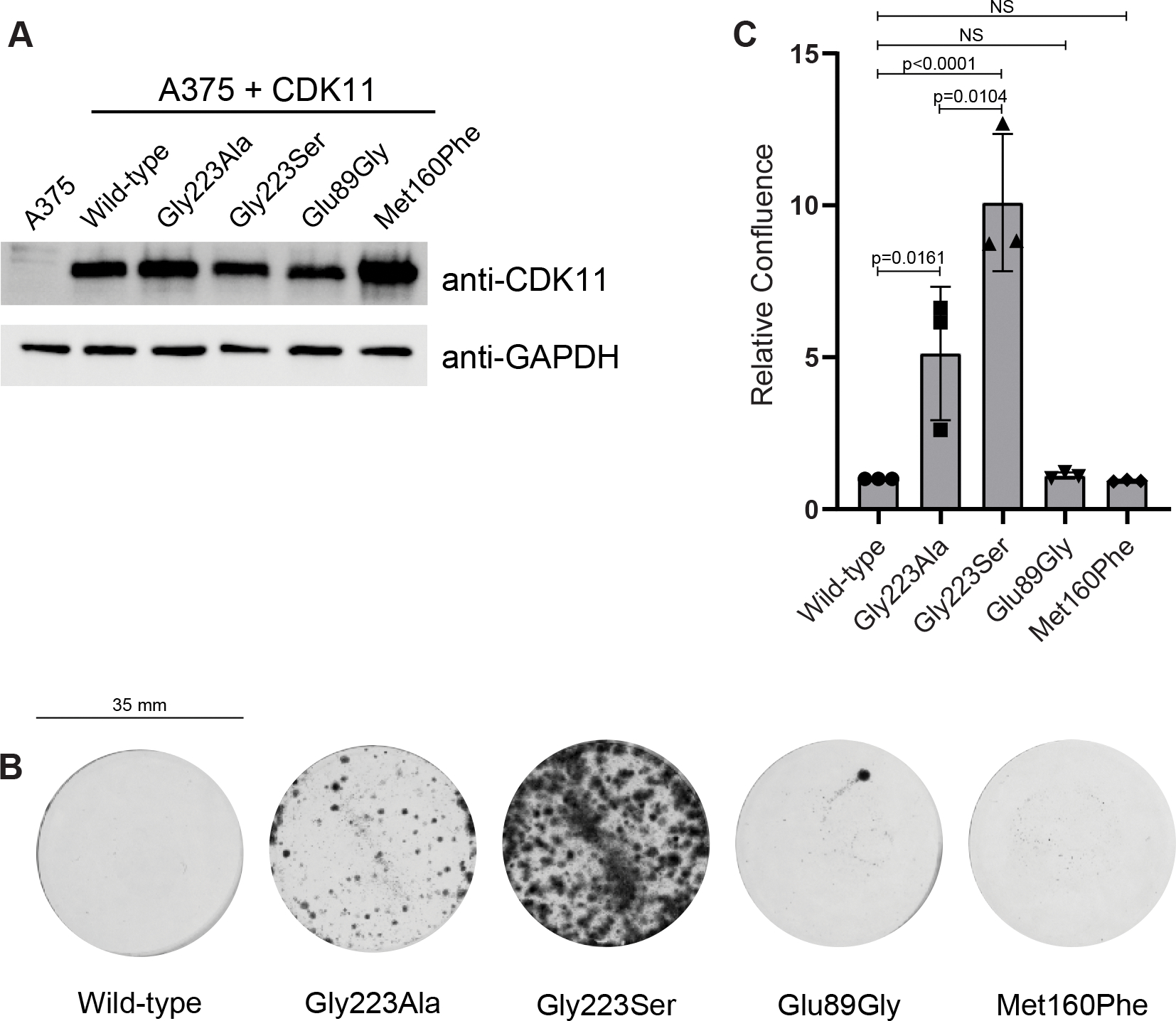
(A) Western-blot demonstrating level of CDK11 expression in A375 cell lines stably expressing either CDK11 wild-type or mutants.
(B) Representative clonogenic assay of A375 cells stably expressing either CDK11 wild-type or the indicated CDK11 mutants in 6-well tissue culture plates (diameter 35 mm) after 12 days of treatment with 100 nM OTS964. Representative images of three independent replicates.
(C) Relative confluence of A375 cell lines stably expressing either CDK11 wild-type or the indicated CDK11 mutants after 12 days of treatment with 100 nM OTS964. Quantification of three independent biological replicates, mean ± standard error (SEM).
We next tested the effect of expressing the Met160Phe and Glu89Gly CDK11 mutants (Figure 4B,C). The Glu89Gly mutant did not confer any resistance to cell killing, consistent with our observation that this mutant displayed equal or better binding to OTS964 in vitro and the disorder of the Glu89 side chain in the crystal structure. Surprisingly, the Met160Phe mutant also did not confer resistance to OTS964 in cells, despite displaying a 9-fold impaired binding to CDK11 in vitro. We conclude that the effect of CDK11 mutations on the binding of CDK11 in isolation to OTS964 is not sufficient to predict the sensitivity of cells to OTS964. We conclude that other factors must be at play.
Cyclin binding improves binding of OTS964 to CDKs
The unexpected behaviour of CDK11 mutants in cells led us to consider what other factors might impact OTS964 binding to CDK11. We first examined the potential role of cyclin binding. The structure of CDK11 bound to OTS964 suggests that OTS964 favors binding to the active conformation of the kinase domain. In the absence of cyclin binding, CDKs typically adopt an inactive conformation characterized by a laterally displaced helix αC and broken regulatory spine. Binding of a cyclin causes a transition to an active conformation defined by the inward movement of helix αC and the formation of a contiguous regulatory spine (depicted in Figure 2B,C). As noted above, active-like features are maintained for CDK11 bound to OTS964, despite the absence of cyclin binding. This finding suggests that the binding of a cyclin to CDK11 could contribute favourably to OTS964 binding by reinforcing a productive conformation of the kinase domain.
As we were not able to express and purify soluble CDK11-cyclin L1 or CDK11-cyclin D3 complexes to allow for direct affinity measurements, we used CDK2 as a surrogate to examine the effect of cyclin binding on CDK binding to OTS964. CDK2 in the absence of a cyclin bound OTS964 with a KD of 16.3 ± 1.7 μM, (mean ± SD; ~250-fold weaker than monomeric CDK11). However, in the presence of cyclin A, binding of CDK2 to OTS964 was enhanced 10-fold (KD mean ± SD 1.8 ± 0.5 μM; Table 2, isotherms shown in Sup. Figure 6). We also generated and tested the effect of the CDK2 mutations Gly11Glu, Phe80Met, and Ala144Gly, that are reciprocal to the CDK11 mutations Glu89Gly, Met160Phe, and Gly223Ala, for their effect on binding to OTS964. As observed for CDK2 wild-type, the presence of cyclin A enhanced binding of OTS964 to all three mutants (KD values mean ± SD in Table 2, isotherms shown in Sup. Figure 6).
Making CDK2 more CDK11-like by introduction of the Gly11Glu mutation enhanced binding affinity of OTS964 to CDK2 compared to wild-type both in the presence and absence of cyclin A (KD values mean ± SD in Table 2, isotherms shown in Sup. Figure 6). This suggests that the specific residue at the equivalent position in CDK11 (Glu89) may contribute to the preferential binding of OTS964 to CDK11 over other CDKs. However, the enhancement of affinity was not expected since the Glu89Gly mutation in CDK11 had almost no effect on OTS964 binding. Making CDK2 more CDK11-like by introduction of the Phe80Met mutation (or to a lesser extent the Ala144Gly mutation) decreased binding affinity of OTS964 to CDK2 compared to wild-type in the presence and absence of cyclin A (KD values mean ± SD in Table 2, isotherms shown in Sup. Figure 6). This was unexpected as it suggests that the identity of the CDK2 residue at this position is preferred for OTS964 binding over the residue that naturally occurs in CDK11. However, our experiments show that mutation of the equivalent residue in CDK11, Met160Phe, also decreased binding affinity for OTS964.
Together, these results support the notion that factors both within (such as identity of inhibitor contacting residues) and beyond the kinase domain (such as cyclin binding) affect the binding affinity and potentially the selectivity of OTS964 binding to CDKs. The fact that single site substitutions of OTS964 contacting residues between CDK11 and CDK2 do not give consistent increases or decreases in binding affinity based on sequence conservation alone, suggests that binding affinity and specificity is achieved by a combination of many interdependent substitutions, including residues beyond the direct inhibitor contact surface.
Discussion
Inhibitor specificity is a challenging goal for protein kinases considering the conservation of active site features across the 500 plus members in the human kinome. This is an especially daunting task within subfamilies of protein kinases such as the CDKs which share even greater similarity in structure and regulation. Here, our goal was to understand how OTS964 inhibits CDK11 with great selectivity over other members of the CDK family. Previous studies identified the xDFG residue Gly223 within the kinase active site as a critical determinant for OTS964 inhibition of CDK11 in cells (Lin et al., 2019). To better understand the role for the xDFG position and other structural features of the kinase domain on selective inhibition by OTS964, we determined the co-crystal structure of the CDK11-OTS964 complex. In addition to demonstrating the close proximity of the xDFG position to OTS964, which can partly explain its contribution to CDK11 binding, the crystal structure revealed Met160 (at the gatekeeper position) and Glu89 (in the kinase G-loop) as being well positioned near OTS964 to influence binding. The identity of both of these residues is unique to CDK11 as compared to other CDKs.
We showed that mutation of Gly223 to Ser (but not Ala) and Met160 to Phe had a pronounced detrimental effect on binding to CDK11 in vitro whereas mutation of Gly223 to Ala actually enhanced binding. The Glu89Gly mutation had no effect on OTS964 binding. These results were readily rationalized by the crystal structure of OTS964 bound to CDK11 and suggested that the identity of the residue at both the gatekeeper position (Met160) and the xDFG position would contribute to the sensitivity of CDKs to OTS964 in vivo. However, in vitro binding of CDK11 to OTS964 did not fully correlate with effects on OTS964 resistance observed in cells. Consistent with expectations, the Gly223Ser substitution, which weakened binding in vitro, caused strong resistance to OTS964, and the Glu89Gly substitution, which had no effect on binding in vitro, had no effect on cell resistance to OTS964. Contrary to expectations however, the Met160Phe mutation, which weakened binding in vitro did not impart resistance to OTS964, while the Gly223Ala mutation, which enhanced binding weakly in vitro, caused resistance to OTS964. We reason that these discrepancies may be due in part to differences between the in vitro and in cellulo test systems.
One difference between the two test systems relates to the presence in cellulo, or absence in vitro, of interacting proteins. Indeed, we showed using cyclin A-CDK2 as a surrogate system that cyclin A binding to CDK2 played a strong role in enhancing binding to OTS964 in vitro. A second difference between the two test systems relates to the presence of ATP, which is expected to compete with OTS964 for binding to the kinase domain of CDK11. In the in vitro system, we monitored the effect of CDK11 mutations on OTS964 binding while in the in cellulo system we monitored the effect of mutations on kinase activity, measured as resistance to OTS964. To inhibit CDK11, OTS964 must compete with ATP for CDK11 binding. If a mutation in question adversely affects ATP binding to a greater extent than OTS964 binding, then this could explain why cells bearing the CDK11 mutation would counterintuitively display continued sensitivity to killing by OTS964. Whatever the cause for discrepancies, our results highlight a limitation of studying inhibitors on CDK kinases in isolation.
If the identity of Met160 (at the gatekeeper position), Glu89 (in the kinase G-loop), and Gly223 (the xDFG residue) together cannot account for the degree of specificity OTS964 exhibits toward CDK11 over other CDKs, then where does the remaining specificity come from? Excluding the three explored positions, 20 residues are located within 5 Å of OTS964 with 9 being fully conserved in all 20 CDK family members. Of the nine remaining variable residues, 5 (Ile88, Glu90, Gly94, Val142, and Tyr162) show no clear patterns of conservation that suggest an obvious role in conferring sensitivity to OTS964 binding. Notably CDK11 has the same residue as half of all CDKs at these positions, making them unlikely candidates for imparting the broad specificity observed for OTS964. Three of the 9 positions, Asn161, Ser169, and Ser210 display some bias for CDK11 versus other CDKs, making them better candidates for contributing to OTS964 selectivity, but are located in positions unlikely to impact OTS964 binding. Asn161 is unique to CDK11, but the side chain points to solvent, in the opposite direction of the inhibitor. Ser169 and Ser210 are conserved between CDKs 10 and 11 and 10, 11, 12, and 13, respectively, but reside at the outer edge of the catalytic cleft facing solvent where there is room for a larger side chain that is not expected to perturb OTS964 binding. Finally, for the three of 9 remaining residues (Val163, Glu164, and His165), amino acids at these positions vary widely among CDKs and all reside within the kinase hinge with side chains facing solvent.
Aside from residues within 5 Å proximity to OTS964 there are an additional 250 out of 280 residues that diverge between CDK11 and other CDKs. While hard to rationalise possible mechanisms of action based on a static X-ray crystal structure, we reason that these residues could act through allostery or effects on protein dynamics to favour OTS964 binding to CDK11. A more extensive genetic screen for resistance mutations in CDK11 in cells, like that used to identify a role for Gly223, would be a good path to exploring the issue of inhibitor specificity further.
Our crystal structure of CDK11 bound to OTS964 represents an atomic picture of the CDK11 kinase domain. Interestingly, the structure of CDK11 in isolation more closely resembles the kinase domain of CDKs in their active state bound to cyclin rather than inactive states typified in the absence of cyclin. While this observation may be due in part to crystal packing (Sup. Figure 6), this suggests that the OTS964 favours and promotes the active conformation of the CDK11. Conversely, the binding of cyclin to CDK11 which should also promote an active conformation of kinase domain, should also promote CDK11 binding to OTS964. This inference is consistent with the 7-fold enhancement of binding we see for OTS964 to CDK2 in the presence versus the absence of cyclin A. While our results provide a compelling case for cyclin binding promoting OTS964 binding to CDKs, whether cyclin L1 or L2 binding to CDK11 contributes to the selectivity of OTS964 over other CDKs remains an open question that will require the development of a suitable expression system for these cyclins to allow further biophysical characterizations.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Frank Sicheri (sicheri@lunenfeld.ca).
Materials availability
Materials generated in this study can be made available upon request from the Lead contact.
Data and code availability
The accession number for the CDK1172-380-OTS964 coordinates and structure factors reported in this paper is Protein Data Bank: 7UKZ. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial cultures
Escherichia coli DH5alpha cells (for cloning) and BL21 (DE3) cells (for protein expression) were grown in LB media containing 100 mg/mL ampicillin. Cultures were incubated at 37°C with 200 rpm shaking. For protein expression, IPTG was added to a final concentration of 0.5 mM and the temperature was changed to 18°C overnight.
Insect cell culture
Sf9 cells were cultured in I-max culture media (Wisent Bio) supplemented with 5% fetal bovine serum (Wisent Bio); 100 U and 100 μg/mL of penicillin and streptomycin, respectively (Wisent Bio); and 2 mM L-glutamine (Wisent Bio). Cultures were incubated at 27°C with 110 rpm shaking.
Human cell line culture
A375 and HEK293T cells (ATCC) were cultured in DMEM (Gibco) with 10% fetal bovine serum (Gibco) and 1X antibiotic-antimycotic (Gibco) at 37°C and 5% CO2.
METHOD DETAILS
Cloning and plasmid construction
Baculovirus for 6XHis-CDK11 expression was generated by inserting the gene encoding CDK11B residues 72–380 (p58 isoform, Uniprot Identifier P21127–12) into a pFastBac HTb plasmid by restriction cloning. Bacmid was isolated from DH10 cells transformed with the pFastBac HTb-CDK11 plasmid. Sf9 cells were transfected with isolated CDK11 bacmid using Qiagen transfection reagent, and baculovirus was subsequently amplified for protein expression. Mutations were introduced in the pFastBac HT-CDK11 plasmid via site directed mutagenesis (primers listed in Table S1) and baculovirus was generated for each mutant. These mutants were also generated in full-length CDK11p58 and cloned into pLentiPuro (Addgene #39481) through restriction cloning for lentiviral production. The pGEX 4T-3 plasmid co-expressing human CDK2 residues 1–298 (GST-tagged, PreScission protease cleavable, sequence verified) and Saccharomyces cerevisiae CAK1 (GST-tagged, non-cleavable) was a gift from Tanja Mittag (St. Jude Children’s Research Hospital). The 6XHis-cyclin A plasmid was constructed by inserting the gene encoding cyclin A residues 173–432 pProEX HTb by restriction cloning.
Protein expression and purification
CDK11B (residues 72–380) was expressed as a TEV protease cleavable N-terminal 6XHis fusion protein in Sf9 cells. Insect cell pellets were lysed in buffer containing 25 mM HEPES pH 7.5, 250 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM TCEP, and 2 mM PMSF. Cleared lysates were passed over a 5 mL HiTrap Ni-chelation column (GE LifeScience Inc.) and were eluted using a gradient from 20 mM to 500 mM imidazole. Fractions containing 6XHis tagged CDK11 were pooled and dialyzed against a buffer of 25 mM HEPES pH 7.5, 250 mM NaCl and 2 mM β-mercaptoethanol overnight at 4°C in the presence of 500 μg non-cleavable 6XHis tagged TEV protease. Dialyzed protein was then passed over a 5 mL HiTrap Ni-chelation column to remove TEV protease and the cleaved 6XHis-tag. Finally, CDK11 was concentrated and loaded onto a Superdex S75 sizing column equilibrated in buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl, and 1 mM TCEP.
CDK2 (residues 1–298) was expressed as a PreScission protease cleavable N-terminal GST fusion protein. Bacterial pellets were lysed in buffer containing 25 mM HEPES pH 7.5, 250 mM NaCl, 10% glycerol, 2 mM β-mercaptoethanol and 2 mM PMSF. Cleared lysates were then passed over glutathione Sepharose resin (GE LifeScience Inc.) and eluted by incubation with 1 mg non-cleavable GST-PreScission protease overnight at 4°C. Eluted CDK2 was concentrated and loaded onto a Superdex S75 column equilibrated in buffer containing 25 mM HEPES pH 7.5, 250 mM NaCl, and 1 mM DTT.
CDK2-cyclin A complexes were formed by combining bacteria pellets expressing either GST-CDK2 or 6XHis-cyclin A. Pellets were combined in a ratio of 1:1 and resuspended in lysis buffer containing 25 mM HEPES pH 7.5, 400 mM NaCl, 20 mM imidazole, 10% glycerol, 2 mM β-mercaptoethanol, and 2 mM PMSF. Like cyclin A above, cleared lysates were passed over a 5 mL HiTrap Ni-chelation column (GE LifeScience Inc.) and eluted using a gradient from 20 mM to 500 mM imidazole. Fractions containing CDK2-cyclin A were combined and then passed over glutathione Sepharose resin (GE LifeScience Inc.). CDK2-cyclin A complexes were then released by incubation with 1 mg GST-PreScission protease and 500 μg 6XHis-TEV protease overnight at 4°C. Following tag cleavage, the complex was passed over a 5 mL HiTrap Ni-chelation column to removed 6XHis-TEV protease and the cleaved 6XHis-tag. After concentrating, the 1:1 complex was loaded onto a Superdex S75 column equilibrated in a buffer containing 25 mM HEPES pH 7.5, 400 mM NaCl, and 1 mM DTT.
Crystallography
OTS964 and CDK11 were mixed prior to crystallization with OTS964 in 2 molar excess (CDK11 120 μM, and OTS964 240 μM). Crystals of OTS964-CDK1172-380 were grown in hanging drop 24-well plates by mixing 1 μL of protein with 1 μL well solution (1.75 M NH4SO4 and 0.1M Tris-HCl pH 7.5) at 20°C. For cryoprotection, crystals were soaked in well solution supplemented with 25% ethylene glycol. Diffraction data was collected from a single frozen crystal at 0.97911 Å wavelength on beamline NE-CAT (APS, Chicago, Il) and processed with XDS (Kabsch, 2010). Molecular replacement was performed using Phaser (McCoy et al., 2007) with a structural model of CDK11 as a search model that was generated using SWISS-MODEL (Waterhouse et al., 2018) from a previous structure of CDK2 bound to cyclin A (PDB: 2IW8, Pratt et al., 2006). PHENIX Xtriage indicated that the data was twinned, so the twin operator H, -H -K,-L was used during structure refinement. The refined twin fractions are 35.0% for H, K, L and 65.0% for H, -H -K, -L. Refinement was performed using PHENIX with optimizing target weights on and Ramachandran restraints on (Adams et al., 2011). Model building was done in Coot (Emsley et al., 2010). Software used in this project was curated by SBGrid (Morin et al., 2013). Final structure consists of six OTS964-CDK1172-380 complexes in the asymmetric unit.
Isothermal Calorimetry
Calorimetric titrations were performed on a Malvern MicroCal Auto-iTC200 (SBC Facility at The Hospital for Sick Children) at 25°C. CDK11 samples were prepared in a buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, and 2.0% DMSO, and CDK2 or CDK2-cyclin A samples were prepared in a buffer containing 25 mM HEPES pH 7.5, 250 mM NaCl, 1 mM DTT, and 2.0% DMSO. OTS964 samples were prepared in the same buffer as the CDK for each experiment. To measure binding between OTS964 CDK11 or the CDK2-cyclin complex, 370 μM OTS964 in the syringe was titrated into 37 μM protein in the cell. To measure binding between OTS964 and CDK2, 2250 μM OTS964 in the syringe was titrated into 150 μM CDK2 in the cell. A total of 38 1μL injections were performed with 180 seconds between injections for all experiments. Data was processed using Origin 7 (v7.0552).
OTS964 resistance experiments
Lentivirus was prepared as follows: HEK293T cells were seeded to 60% confluence, and the following day transfected with 6 μg target plasmid, 6 μg psPAX2 (Addgene #12260) and 1 μg pMD2.G (Addgene #12259) in 60 μg polyethylenimine (Sigma-Aldrich) and Opti-MEM (Gibco). 24 hours post-transfection, media was replaced. Lentivirus was harvested 48 hours post-transfection, filtered through a 0.45 μm filter, and aliquoted and stored at −80°C prior to use. Following collection of lentiviruses, A375 cells were infected in the presence of 8 μg/ml polybrene (Sigma) for 24h. The following day, infected cells were selected by treatment with 1 μg/ml puromycin for 48 hours. Following puromycin selection, cells were expanded and expression of CDK11 constructs was verified by western blotting.
Clonogenic growth assay: A375 cells expressing indicated CDK11-p58 constructs were seeded to 6-well plates at 100k cells/well. Cells were treated with 100 nM OTS964 (Selleck Chemicals) for 12 days. Cells were fixed in 100% methanol (BioShop) at −20°C for 40 minutes. Cells were washed and stained in 0.5% crystal violet for 15 minutes. Plates were dried and imaged on a Chemidoc-MP (BioRad). Crystal violet was destained from cells using 10% acetic acid, and absorbance read at A595 on an Envision MultiLabel plate reader to quantify cell confluence.
Western blot: Samples were harvested in 4X Laemmli buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue) and stored at −20°C until processing. Samples were sonicated, boiled at 95°C for 5 minutes, and centrifuged. Next, approximately 10 μg of protein was loaded into BioRad Precast 4–15% SDS-PAGE TGX Stain-Free gels and run at 150V for 60 minutes. Gels were transferred to methanol activated PVDF membrane at 90V for 2h. Blots were incubated in 1:1000 dilution of primary antibody (Mouse anti-GAPDH; ThermoFisher AM4300, Rabbit anti-CDK11: Abcam ab19393) overnight at 4°C. The following day, blots were washed and incubated in secondary antibody (Donkey anti-Mouse-HRP; Cedarlane 715–035-150, Donkey-anti-Rabbit-HRP; Cedarlane 711–035-152) for 1h at room temperature. Blots were washed extensively and imaged using chemiluminescent substrate (ThermoFisher 34577) and a Chemi-doc MP imager (BioRad).
Assessing survival after CDK11B CRISPR knock-out
Two independent gRNAs targeting CDK11B-p110 were cloned into the Lenticrispr_V2 plasmid that harbors a blasticidin resistance cassette. A375 parental cells and A375 cells stably expressing CDK11B-p58 mutants were transduced with lentivirus coding for gRNA targeting CDK11B-p110 and were selected in 8 μg/ml blasticidin for 5 days. Following selection, cells were seeded at low density and growth was tracked over 6 days in an incucyte (Sartorius). Confluence was measured in the incucyte and normalized to Day 0. Confluence was further normalized to A375 parental cell growth following CDK11B-p110 knock-out. Cell pellets were collected for editing analysis by TIDE. Statistical tests were performed on three independent replicates, one t-test per row.
TIDE analysis: gDNA from indicated cells was isolated using the Lucigen QuickExtract solution (Lucigen). Sites surrounding the gRNAs were amplified by PCR and sequenced using Sanger Sequencing at The Centre for Applied Genomics at SickKids hospital. Sequencing chromatograms were assessed for editing using the TIDE algorithm (Brinkman et al. 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
X-ray data collection and refinement statistics are shown in Table 1.
The number of replicate experiments (n) represented by the data can be found in the individual figure legends or table footnotes. P-values for statistical tests can be found in the figures.
Each ITC binding experiment was performed n=2 times. N, KD, ΔH, and ΔS values were calculated for each replicate using Origin 7 (v7.0552). ΔG for each replicate was calculated in Microsoft Excel (version 16.64) using the formula: ΔG = ΔH − TΔS. Mean and standard deviation (SD) for N, KD, ΔH, ΔS, and ΔG were calculated from both replicates in Excel and are reported in Table 2.
Clonogenic growth assays were performed n=3 times. For each replicate, the confluence of the mutants was quantified relative to cells stably expressing wild-type CDK11. For each CDK11 variant, the mean and standard error of the mean (SEM) of all three replicates were calculated using GraphPad Prism (version 8.2.1) and are shown in Figure 4C. We compared the relative confluence each mutant to wild-type CDK11 using a one-way ANOVA test with a Tukey’s multiple corrections. P-values for these comparisons were calculated using GraphPad Prism and are reported in Figure 4C.
Survival after CDK11B knock-out experiments were performed n=3 times. For each replicate, the confluence of the cell lines expressing wild-type or mutants was quantified relative to the parental A375 cell line. For each cell line, the mean and standard error of the mean (SEM) of all three replicates were calculated using GraphPad Prism and are shown in Sup. Figure 5B. The relative confluence of each cell line stably expressing a CDK11 variant was compared the parental cell line using multiple t-tests. P-values for these comparisons were calculated using GraphPad Prism and are reported in Sup. Figure 5B.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GAPDH | ThermoFisher | Cat# AM4300; RRID: AB_2536381 |
| Rabbit polyclonal anti-CDK11 | Abcam | Cat# Ab19393; RRID: AB_444893 |
| Donkey polyclonal anti-Mouse-HRP | Cedarlane | Cat# 715-035-150; RRID: AB_2340770 |
| Donkey polyclonal anti-Rabbit-HRP | Cedarlane | Cat# 711-035-152; RRID: AB_10015282 |
| Bacterial and virus strains | ||
| BL21(DE3) chemically competent E. coli | ThermoFisher | Cat# C600003 |
| DH5alpha chemically competent E. coli | ThermoFisher | Cat# EC0112 |
| Chemicals, peptides, and recombinant proteins | ||
| OTS964 | Selleck Chemicals | Cat# S7648 |
| Western blot chemiluminescent substrate | ThermoFisher | Cat# 34577 |
| Polyethylenimine | Sigma-Aldrich | Cat# 408727 |
| Polybrene | Sigma-Aldrich | Cat# 28728-55-4 |
| Lucigen QuickExtract | Lucigen | Cat# LGN-QE09050 |
| Deposited data | ||
| CDK11B72-380 + OTS964 | This paper | PDB:7UKZ |
| CDK2 | Schulze-Gahmen et al., 1996 | PDB:1HCK |
| CDK2-cyclin A | Pratt et al., 2006 | PDB:2IW8 |
| CDK-cyclin A-substrate peptide | Brown et al., 1999 | PDB:1QMZ |
| Experimental models: Cell lines | ||
| A375 cells | ATCC | Cat# CRL-1619; RRID: CVCL_0132 |
| HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Sf9 cells | ThermoFisher | Cat# 11496015 |
| Oligonucleotides | ||
| gRNA1 CDK11B p110: AAAAGAGAAAAGAGAAACGT | ThermoFisher | N/A |
| gRNA2 CDK11B p110: TCCTTATTGTGATCTCCATG | ThermoFisher | N/A |
| Additional oligonucleotides listed in Supplementary Table 1 | This paper | N/A |
| Recombinant DNA | ||
| pFastBac CDK11B p58 (human 72–380) | This paper | N/A |
| pGEX 4T-3 CDK2 (human 1–298) | Gift from Tanja Mittag (St. Jude Children’s Research Hospital) | N/A |
| pProEX Cyclin A (human 173–432) | This paper | N/A |
| pLentiPuro CDK11B p58 (human 1–439) | This paper | N/A |
| lenticrispr_V2 plasmid CDK11B p110 gRNA | This paper | N/A |
| psPAX2 | Gift from Didier Trono (no citation) | RRID: Addgene_12260 |
| pMD2.G | Gift from Didier Trono (no citation) | RRID: Addgene_12259 |
| Software and algorithms | ||
| Adobe Illustrator (version 25.4.1) | Adobe | www.adobe.com |
| Chimera (version 1.15) | Pettersen et al., 2004 | www.cgl.ucsf.edu/chimera/ |
| Clustal Omega | Sievers et al., 2011 | www.ebi.ac.uk/Tools/msa/clustalo/ |
| Coot (version 0.9) | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Excel (version 16.64) | Microsoft | www.microsoft.com |
| GraphPad Prism (version 8.3.0) | GraphPad | www.graphpad.com |
| Jalview (version 2.11.1.4) | Waterhouse et al., 2009 | https://www.jalview.org |
| Origin 7 (version 7.0552) | Malvern Panalytical | www.malvernpanalytical.com |
| Phaser (version 2.8.3) | McCoy et al., 2007 | https://www-structmed.cimr.cam.ac.uk/phaser_obsolete/ |
| PHENIX (version 1.20.1) | Adams et al., 2011 | https://www.phenix-online.org/download/ |
| TIDE algorithm | Brinkman et al. 2014 | https://tide.nki.nl/ |
| XDS (version 20210322) | Kabsch, 2010 | https://xds.mr.mpg.de |
Highlights:
Determined the crystal structure of CDK11 bound to the kinase inhibitor OTS964
The CDK11 kinase domain adopts an active-like conformation
The G223S mutation causes OTS964 resistance by interfering with OTS964 binding
Acknowledgements
This work was supported by a CIHR foundation grant (FDN-143277) to F.S., CIHR grant (PJT-148691) to S.A. and an NSERC PGS D to S.K. and was based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. We thank and acknowledge The Hospital for Sick Children’s Structural & Biophysical Core Facility staff and infrastructure.
Footnotes
Declaration of Interests
F.S. is a founder and consultant of Repare Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed RL, Shaughnessy DP, Knutson TP, Vogel RI, Ahmed K, Kren BT, and Trembley JH (2019). CDK11 loss induces cell cycle dysfunction and death of BRAF and NRAS Melanoma Cells. Pharmaceuticals 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander LT, Möbitz H, Drueckes P, Savitsky P, Fedorov O, Elkins JM, Deane CM, Cowan-Jacob SW, and Knapp S (2015). Type II Inhibitors Targeting CDK2. ACS Chem. Biol. 10, 2116–2125. [DOI] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, and Knudsen ES (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD, Ronique Sgambato V, Zhu P-P, Lavoie B, Vincent M, Krause M, and Hyman SE (2001). Dopamine and Glutamate Induce Distinct Striatal Splice Forms of Ania-6, an RNA Polymerase II-Associated Cyclin dence that some genes are induced as homeostatic responses to excessive dopaminergic stimulation and that such neuronal adaptations contribute t. Neuron 32, 277–287. [DOI] [PubMed] [Google Scholar]
- Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, and Schönbrunn E (2011). Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol. 6, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, and Jager A (2021). Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 81, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and Van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NR, Noble MEM, Endicott JA, and Johnson LN (1999). The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, and Beyaert R (2000). Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol. Cell 5, 597–605. [DOI] [PubMed] [Google Scholar]
- Dickinson LA, Edgar AJ, Ehley J, and Gottesfeld JM (2002). Cyclin L is an RS domain protein involved in pre-mRNA splicing. J. Biol. Chem. 277, 25465–25473. [DOI] [PubMed] [Google Scholar]
- Du Y, Yan D, Yuan Y, Xu J, Wang S, Yang Z, Cheng W, Tian X, and Kan Q (2019). CDK11p110 plays a critical role in the tumorigenicity of esophageal squamous cell carcinoma cells and is a potential drug target. Cell Cycle 18, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 17, 353–376. [DOI] [PubMed] [Google Scholar]
- Gururajan R, Lahti JM, Grenet J, Easton J, Gruber I, Ambros PF, and Kidd VJ (1998). Duplication of a genomic region containing the Cdc2L1 −2 and MMP21–22 genes on human chromosome 1p36.3 and their linkage to D1Z2. Genome Res. 8, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan M. Lou, Sausville EL, Lukow DA, Liu L, Chait AR, et al. (2019). Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gao Y, Shen J, Yang W, Choy E, Mankin H, Hornicek FJ, and Duan Z (2016). Cyclin-dependent kinase 11 (CDK11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol. Cancer Ther. 15, 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome Biol. 15, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai L, and Wolgemuth DJ (2009). Cyclin-dependent kinases : a family portrait. 11, 1275–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MP, Endicott JA, and Noble MEM (2017). Structure-based discovery of cyclin-dependent protein kinase inhibitors. Essays Biochem. 61, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Park JH, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, and Nakamura Y (2014). TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci. Transl. Med. 6, 259ra145. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. Elife 2013, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien S, Kelso S, Steinhart Z, Orlicky S, Mis M, Kim Y, Lin S, Sicheri F, Angers S (2022). SCF-FBXW7 regulates G2-M progression through control of CCNL1 ubiquitination. BioRxiv. DOI: 10.1101/2022.09.26.509608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pratt DJ, Bentley J, Jewsbury P, Boyle FT, Endicott JA, and Noble MEM (2006). Dissecting the determinants of cyclin-dependent kinase 2 and cyclin-dependent kinase 4 inhibitor selectivity. J. Med. Chem. 49, 5470–5477. [DOI] [PubMed] [Google Scholar]
- Renshaw MJ, Panagiotou TC, Lavoie BD, and Wilde A (2019). CDK11p58–cyclin L1β regulates abscission site assembly. J. Biol. Chem. 294, 18639–18649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Gahmen U, De Bondt HL, and Kim S-H (1996). High-Resolution Crystal Structures of Human Cyclin-Dependent Kinase 2 with and without ATP: Bound Waters and Natural Ligand as Guides for Inhibitor Design. J. Med. Chem. 39, 4540–4546. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse S, Caldon EC, Tilley W, and Wang S (2019). Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 62, 4233–4251. [DOI] [PubMed] [Google Scholar]
- Trembley JH, Loyer P, Hu D, Li T, Grenet J, Lahti JM, and Kidd VJ (2004). Cyclin Dependent Kinase 11 in RNA Transcription and Splicing. Prog. Nucleic Acid Res. Mol. Biol. 77, 263–288. [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, De Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CI, Vasta JD, Corona CR, Wilkinson J, Zimprich CA, Ingold MR, Pickett JE, Drewry DH, Pugh KM, Schwinn MK, et al. (2020). Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DJ, and Endicott JA (2018). Structural insights into the functional diversity of the CDK–cyclin family. Open Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DJ, Korolchuk S, Tatum NJ, Wang LZ, Endicott JA, Noble MEM, and Martin MP (2019). Differences in the Conformational Energy Landscape of CDK1 and CDK2 Suggest a Mechanism for Achieving Selective CDK Inhibition. Cell Chem. Biol. 26, 121–130.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, Zheng Q, and Cai C (2021). CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res 11, 1913–1935. [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Cai M, Zhang S, Xu S, Chen S, Chen X, Chen C, and Gu J (2002). Interaction of p58PITSLRE, a G2/M-specific protein kinase, with cyclin D3. J. Biol. Chem. 277, 35314–35322. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Shen JK, Hornicek FJ, Kan Q, and Duan Z (2016). The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget 7, 40846–40859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the CDK1172-380-OTS964 coordinates and structure factors reported in this paper is Protein Data Bank: 7UKZ. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.