

Abstract
Neuroblastoma evolution, heterogeneity, and resistance remain inadequately defined, suggesting a role for circulating tumor DNA (ctDNA) sequencing. To define the utility of ctDNA profiling in neuroblastoma, 167 blood samples from 48 high-risk patients were evaluated for ctDNA using comprehensive genomic profiling. At least one pathogenic genomic alteration was identified in 56% of samples and 73% of evaluable patients, including clinically actionable ALK and RAS-MAPK pathway variants. Fifteen patients received ALK inhibition (ALKi) and ctDNA data revealed dynamic genomic evolution under ALKi therapeutic pressure. Serial ctDNA profiling detected disease evolution in 15 of 16 patients with a recurrently identified variant, in some cases confirming disease progression prior to standard surveillance methods. Finally, ctDNA-defined ERRFI1 loss-of-function variants were validated in neuroblastoma cellular models, with the mutant proteins exhibiting loss of wild-type ERRFI1’s tumor suppressive functions. Taken together, ctDNA is prevalent in children with high-risk neuroblastoma and should be followed throughout neuroblastoma treatment.
Introduction
Neuroblastoma is an embryonal tumor that arises from the aberrant growth of neural crest progenitor cells that disproportionately accounts for up to 10% of childhood cancer mortality (1). Overall high-risk neuroblastoma survival rates remain below 50%, and relapsed neuroblastoma is an even more significant clinical challenge (2), providing substantial motivation to better understand genomic evolution and mechanisms of therapeutic resistance to inform the development of new therapeutic strategies for this unrelenting childhood malignancy.
It has been known for decades that cells from human solid tumors such as neuroblastomas, as well as their nucleic acids, are present in the circulation of patients but only recently have technological advances made it possible to consider the clinical application of methodologies to specifically detect this circulating tumor DNA (ctDNA) in patients (3). In turn, multiple adult cancer practitioners have begun to incorporate serial ctDNA profiling into clinical practice to aid in disease surveillance (4), characterize tumor heterogeneity (5), and define mechanisms of therapeutic resistance (6,7). Furthermore, recent targeted analysis of MYCN amplification and anaplastic lymphoma kinase (ALK) gene variants in neuroblastoma suggests potential clinical value of ctDNA profiling for some of these children (8). However, how to optimally utilize comprehensive ctDNA profiling data in the pediatric oncology clinic has not been determined.
While tumors from high-risk neuroblastoma patients at diagnosis are in part characterized by the presence of several clinically prognostic segmental chromosomal aberrations (e.g., MYCN amplification, chromosome 1p or 11q loss, or chromosome 17q gain) and several recurrent structural variants have been identified (e.g., TERT, SHANK2, PTPRD), clonal somatic point mutations in cancer associated genes other than ALK are rare (9–12). However, it is now clear that both primary and relapsed neuroblastomas acquire a significant number of new mutations under the selective pressure of standard neuroblastoma multimodal cytotoxic therapy (13–15), many of which occur in ALK or RAS-MAPK pathway genes (11,13,15). The precise timing for the acquisition of these genomic aberrations is unknown, as is how relapsed neuroblastomas continue to evolve under additional therapeutic pressures, including ALK or other targeted therapies. Furthermore, despite these emerging data, pediatric oncologists are still hesitant to recommend a tissue or bone biopsy for a child with relapsed disease given the specificity of 123I-metaiodobenzylguanidine (MIBG) imaging. Direct neuroblastoma tumor biopsies also have additional challenges, including often being in technically difficult locations (e.g., paraspinal or within bone) and/or requiring procedural sedation. While at some large pediatric academic centers biopsies can be safely achieved via interventional radiological procedures (16), inter- and intratumoral heterogeneity remains a major issue in a cancer that is commonly widely disseminated, perhaps making it an ideal disease to utilize ctDNA profiling.
Current neuroblastoma surveillance strategies rely on cross sectional anatomic imaging and 123I-MIBG scans, along with following urinary vanillylmandelic acid (VMA) and homovanillic acid (HVA) catecholamine levels. However, these methods are clearly insufficient to detect disease progression or relapses early enough to make a difference in clinical outcome for a significant subset of patients. Additionally, imaging evaluations not only expose young children to radiation, but also usually necessitate procedural sedation adding additional clinical risk to these studies. Finally, given that neuroblastomas exist on a spectrum from undifferentiated, clinically aggressive tumors to more benign, differentiated lesions that can harbor significant amounts of ganglioneuroblastoma and/or ganglioneuroma, imaging can often be misleading in deciphering ongoing tumor activity. Thus, utilizing serial ctDNA profiling via a liquid biopsy may directly address many of these common challenges in neuroblastoma clinical care and has the potential for major diagnostic, monitoring, and therapeutic decision-making implications for children with this lethal childhood cancer.
Results
A majority of neuroblastoma patients have ctDNA throughout therapy
Serial ctDNA analysis (Figure 1A) was performed on the peripheral blood of 48 evaluable patients with the FoundationACT™ assay, a previous version of the FoundationOne®Liquid CDx test, profiling a total of 62 genes (27 with complete exon coverage, 34 with partial exon coverage, 6 with intron coverage; Supplementary Table 1). Most of the patients profiled for ctDNA had relapsed neuroblastoma (n=42), but children with newly diagnosed high-risk neuroblastoma (n=4) and children who had disease progression while receiving upfront neuroblastoma therapy (n=2) were also evaluated (Supplementary Table 2). The median age at the time of first ctDNA sample was 89.4 months (7.5 years) and 25% of patients had tumors harboring MYCN amplification. A total of 167 ctDNA samples were sequenced (median of 3 and range 1–10 ctDNA samples/patient) over a median time of 4.4 months (Supplementary Tables 2 and 3).
Figure 1. Circulating tumor DNA is prevalent in children with neuroblastoma.
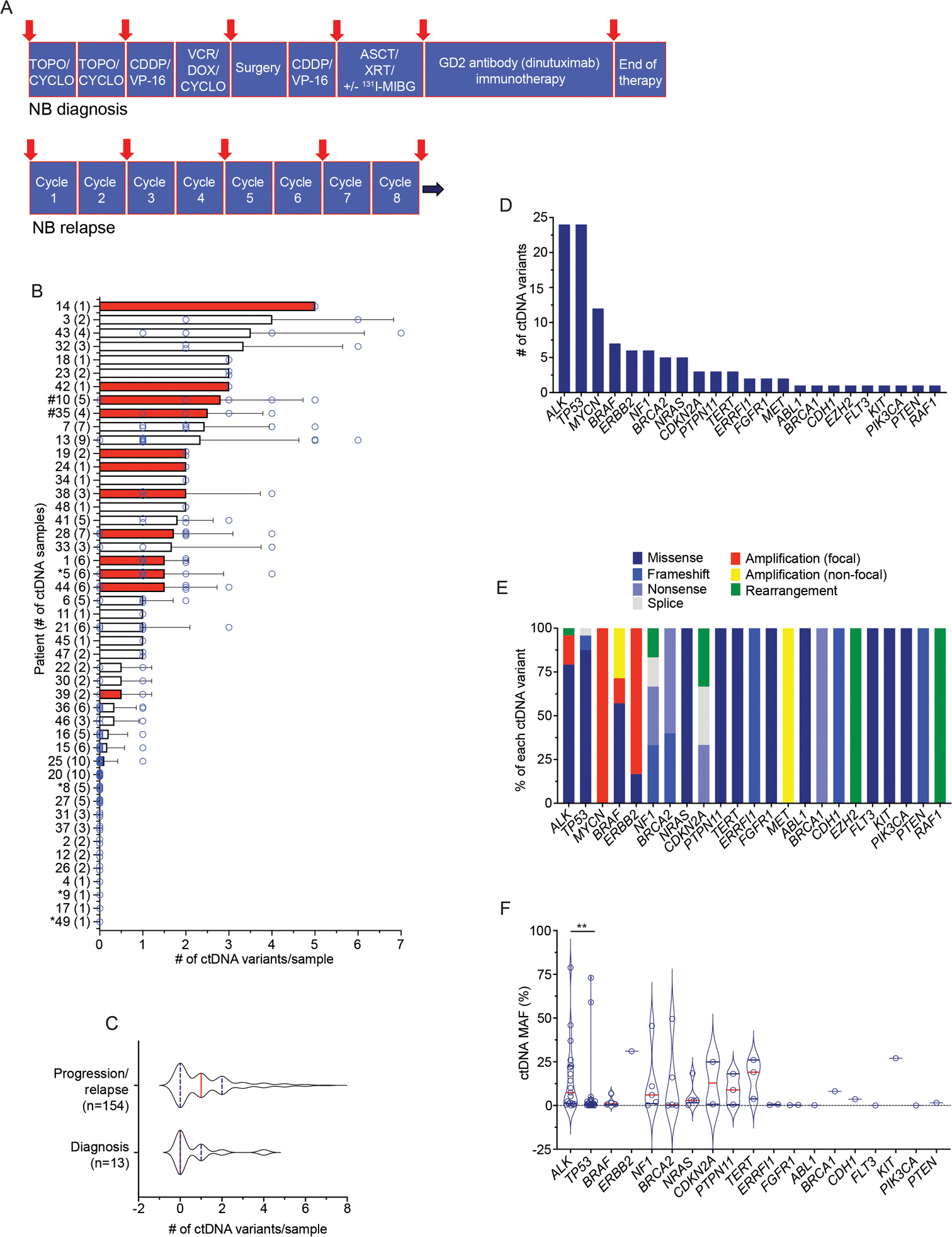
(A) Schema of serial ctDNA profiling sample collection time points for high-risk neuroblastoma patients. Red arrows indicate general ctDNA collection points.
(B) Summary of number of genetic variants identified per ctDNA sample for each patient. The mean ± SEM of ctDNA variants for each patient is shown with each individual ctDNA sample variant number indicated by a circle. The number of ctDNA samples per patient is indicated in parentheses on the y-axis. Red bars indicate patients with MYCN amplified tumors. Newly diagnosed neuroblastoma patients (*) and patients with progressive disease (#) are indicated, with the remaining patients having relapsed neuroblastoma.
(C) Plot of ctDNA variants detected in patients with newly diagnosed neuroblastoma versus those with relapsed disease or disease progression.
(D) Summary of the most altered genes detected in ctDNA.
(E) Summary of the different genetic variants detected per commonly altered gene in ctDNA.
(F) Plot of ctDNA detected genetic alteration MAFs.
The median is indicated with a red line and the quartiles are indicated with blue lines on each violin plot in C and F.
NB, neuroblastoma; TOPO, topotecan; CYCLO, cyclophosphamide; CDDP, cisplatin; VP-16, etoposide; VCR, vincristine; DOX, doxorubicin; XRT, radiation therapy; ASCT, autologous stem-cell transplant; 131I-MIBG, 131I-meta-iodobenzylguanidine, a targeted radiotherapeutic used to treat neuroblastoma. Non-focal amplifications are >20 MB in size with copy numbers ≤ 8 in genes that are recurrently amplified in cancer. Focal amplifications are <20 MB in size with copy numbers > 5 or >20 MB in size with copy numbers > 8 in genes that are recurrently amplified in cancer. **, p < 0.01.
Ninety-eight percent (47/48) of evaluable patients had samples that yielded cell-free DNA (cfDNA) for sequencing (patient 40 had one ctDNA sample that failed quality control assays). ctDNA samples were sequenced to a median exon coverage depth of 9,276x (range 3,168–18,854x). At least one known or likely pathogenic genomic alteration (mutation, amplification, or rearrangement) was found in 56% (93/167) of samples (median of 1 ctDNA variant/sample; range 0–7 variants/sample). Moreover, 73% (35/48) of patients had at least one ctDNA sample with a pathogenic genomic alteration detected (Figure 1B). There was a higher trend but no significant difference between the number of ctDNA variants detected in samples drawn from patients with relapsed disease or disease progression versus newly diagnosed patients with high-risk neuroblastoma (Figure 1C). Missense pathogenic variants in ALK and TP53 were the most common alterations found in ctDNA, followed by MYCN amplification and BRAF alterations (Figure 1D, E). The imputed mutated allele frequency (MAF) varied widely across ctDNA variants, most notably with TP53 variants having lower MAFs when compared to ALK and other genes with point mutations (Figure 1F). Moreover, 21 of the 24 TP53 variants detected occurred only in one ctDNA sample, and 14 of these 21 transient TP53 variants occurred simultaneous with each other with groups of 2–4 TP53 mutations in a given ctDNA sample, collection timepoints that were often preceded by 131I-MIBG radiotherapy (Supplementary Table 4).
ctDNA profiling augments detection of driver mutations throughout neuroblastoma therapy
High-risk neuroblastomas often present with multifocal disease, making it likely impossible to capture all potential driver mutations in a single tumor biopsy. Furthermore, biopsy at the time of neuroblastoma relapse is not always pursued, in part due to the diagnostic specificity of 123I-MIBG scintigraphy, despite recent data that has shown that relapsed neuroblastomas contain significantly more somatic mutations than diagnostic tumors that often occur in potentially clinically targetable genes (11,15). Thus, we next looked to compare ctDNA sequencing findings to those from direct tumor biopsy first in the 28 patients who had temporally paired (within 70 days) blood and tumor sequencing performed (Supplementary Table 5). When considering genes profiled in both tumor and ctDNA assays, 39% (11/28) of patients had discordant ctDNA and tumor sequencing results. Further, 59% (17/29) of detected ctDNA nucleotide variants from these 28 patients were not present in temporally matched tumor samples, including pathogenic alterations in ALK, TERT, TP53, NF1, CDKN2A, FLT3, PIK3CA, and PTPN11 (Figure 2A, B, Supplementary Table 5). However, there was a significantly higher MAF for those variants detected in both ctDNA and tumor sequencing versus variants detected only by ctDNA profiling (median MAF 20.4 vs. 0.7%; Figure 2C). Conversely, there was 100% concordance between the detection of MYCN (9/9) and ALK (3/3) amplification in paired ctDNA/tissue samples (Figure 2A, B). For the 11 patients with ctDNA unique variants detected, we next directly queried the paired tumor sequencing reads and found that in only one case there was any evidence of the ctDNA identified variant in the direct tumor sequencing reads (Supplementary Table 5).
Figure 2. ctDNA profiling enhances the detection of somatic variants at the time of neuroblastoma diagnosis or relapse.
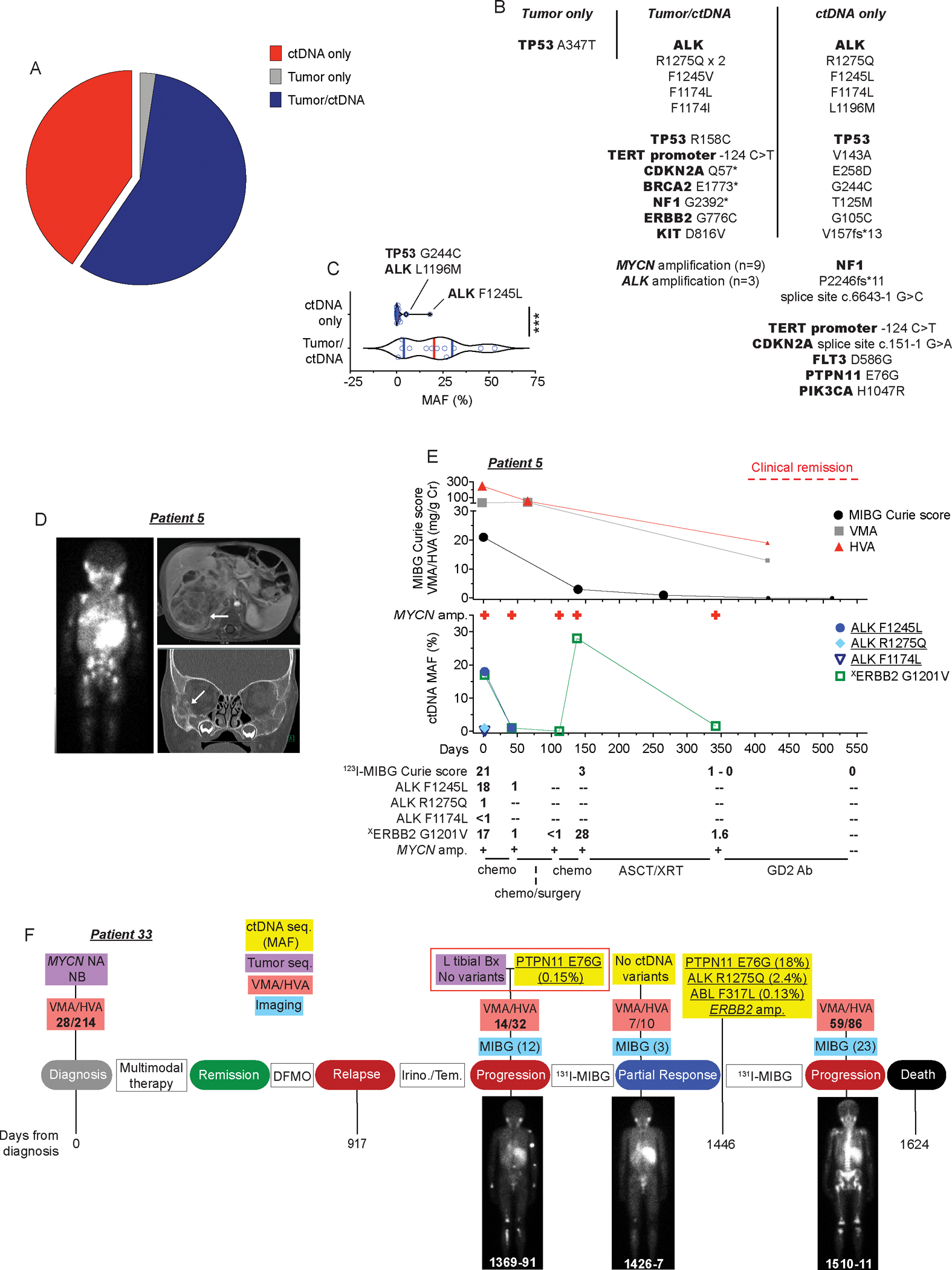
(A) Plot showing concordance between tumor and ctDNA sequencing identified variants from temporally paired samples at the time of diagnosis or relapse (n=28 patients).
(B) Description of gene variants summarized in A.
(C) MAF of gene variants shown in B stratified by those identified in ctDNA only versus those found in both tumor and ctDNA.
(D) 123I-MIBG scan (left), MRI (top right), and CT scan (bottom right) of 1-year-old male (patient 5) who presented with widely metastatic neuroblastoma. White areas of MIBG scan (left) and white arrows on MRI and CT images indicate areas of disease.
(E) Serial ctDNA and disease evaluation correlation plot for patient 5 in D showing relationship between 123I-MIBG Curie scores, a semi-quantitative measure of tumor burden, urine catecholamine levels (VMA/HVA in mg/g Cr; top) and ctDNA profiling data (middle). Larger symbols in plot represent abnormal values. Numbers in bottom table denote MAFs and MIBG Curie scores. General treatment schema shown at bottom of plot.
(F) Serial ctDNA and disease evaluation correlation timeline for patient 33. Images are representative 123I-MIBG images with Curie scores noted in parentheses. VMA/HVA quantification (mg/g Cr) is indicated (bolded values represent abnormal values) and days from neuroblastoma diagnosis is noted on the bottom of plot.
Chemo, chemotherapy; ASCT, autologous stem-cell transplant; XRT, radiation therapy; NB, neuroblastoma; Seq., sequencing; NA, not-amplified; Bx, biopsy; GD2 Ab, GD2-targeting chimeric monoclonal antibody dinutuximab; DFMO, difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase (ODC) used in maintenance neuroblastoma therapy; Irino./Tem., irinotecan/temozolomide, a chemotherapeutic regimen used commonly for relapsed neuroblastoma therapy; 131I-MIBG, 131I-meta-iodobenzylguanidine, a targeted radiotherapeutic used to treat neuroblastoma.
Underlined variants in E and F denote those unique to ctDNA and (X) denotes variant of unknown significance.***p < 0.0001.
One illustrative example of ctDNA/tumor sequencing discordance was with patient 5, a 1-year-old boy who presented to our institution with abdominal pain and proptosis. 123I-MIBG scintigraphy (Figure 2D, left) supported a diagnosis of neuroblastoma revealing widely metastatic, MIBG-avid lesions characteristic of this disease and abdominal magnetic resonance imaging (MRI; Figure 2D, top right) and a computerized tomography (CT) scan (Figure 2D, bottom right) showed significant tumor burden in his abdomen and orbital bones, respectively. An abdominal tumor biopsy confirmed a diagnosis of neuroblastoma and analysis with our institutional next-generation sequencing assay revealed amplification of MYCN and other characteristic neuroblastoma segmental chromosomal aberrations, but no clonal somatic point mutations. Conversely, analysis of this patient’s ctDNA at the same time revealed 3 ALK mutations (F1245L, MAF 18%; R1275Q, MAF 1%; F1174L, MAF <1%), revealing significant tumor heterogeneity. Subsequent re-analysis of the temporally paired-direct tumor sequencing showed one read supporting the highest abundance ALK F1245L ctDNA variant (1/2774 reads) but not the ALK R1275Q or F1174L variants (Supplementary Table 5). Furthermore, the serial ctDNA sequencing data from this patient correlated closely with regular surveillance 123I-MIBG imaging and urine catecholamine (VMA/HVA) levels throughout his disease course (Figure 2E). The ALK variants decreased in frequency after treatment initiation and the ability to detect MYCN amplification and a somatic ERBB2 G1201V variant were only lost from the blood when the patient was concurrently in a clinical remission (Figure 2E, right).
We also observed ctDNA/tumor sequencing discordance in patients profiled with relapsed neuroblastoma. One example is patient 33, who presented to our institution with disease progression after initial neuroblastoma relapse over a year earlier and had a left tibial tumor biopsy performed that did not reveal any genetic variants, yet we identified a PTPN11 E76G variant in the temporally paired ctDNA analysis (Figure 2F, middle red box). Re-analysis of the paired direct tumor sequencing done on the same day did not uncover any evidence of the PTPN11 variant (Supplementary Table 5). Serial ctDNA data for this patient revealed that the PTPN11 E76G variant allele frequency tracked well with their urinary catecholamine (VMA/HVA) values and 123I-MIBG-defined disease burden, all decreasing in parallel after 131I-MIBG radiotherapy (Figure 2F, day 1426). Further, the exponential tumor genomic evolution revealed in this patient’s 3rd ctDNA sample done on day 1446, including identification of a canonical ALK R1275Q hotspot mutation, preceded their overt clinical progression noted 2 months later (123I-MIBG Curie score increased from 3 to 23, significant rise in urinary VMA/HVA; Figure 2F, day 1510) and ultimately their death.
Next, we considered the comparison of ctDNA identified genetic variants (including for patients 28, 42, and 43, ctDNA variants also identified in testing done as part of clinical care during this same period) to all direct tumor or tumor-involved bone marrow sequencing done for each patient over the course of their disease to quantify the additional genomic data that ctDNA profiling provided throughout each patient’s therapy. This analysis showed that 68% (88/129) of the ctDNA variants identified in the 48 evaluable patients were unique to ctDNA and not identified in any direct sequencing performed on tumor tissue or bone marrow involved with metastatic tumor (Figure 3A, Supplementary Table 6). Many of these variants were known driver mutations in ALK or RAS-MAPK pathway genes such as BRAF, NF1, NRAS, PTPN11, and FGFR1 (Figure 3B–D, Supplementary Table 6), further validating the importance of these pathways in neuroblastoma tumorigenesis (11,15). There was also a high prevalence of presumptive loss of function variants in genes that modulate response to DNA damage such as BRCA1, BRCA2, and TP53, many of them also uniquely found in the ctDNA profiling (Figure 3B, C, E, F, Supplementary Table 6). For TP53, while two frameshift variants were identified, most variants identified were missense mutations in the DNA binding domain (DBD), including a R248Q variant that was found in 2 different patients. Two of the TP53 missense variants (R135F and R158C) and one BRCA2 nonsense variant (Y1655*) were found to have MAFs near 50% suggesting that these variants may be of germline origin. These data validated what was known for one patient (patient 14, TP53 C135F) based on prior direct tumor sequencing, but for the other two patients (patient 23, TP53 R158C; patient 45, BRCA2 Y1655*) these presumptive germline mutations had not been defined prior via tumor (or germline) sequencing done as part of their routine clinical care. In turn, for patient 23, the TP53 R158C variant was also observed in a paired tumor biopsy done on the same day as the ctDNA profiling (Supplementary Table 5) as well as a germline sample tested later in their clinical course. However, no tumor or germline sequencing was performed for patient 45 where the BRCA2 Y1655* was identified.
Figure 3. ctDNA unique pathogenic variants in cancer driver genes are commonly identified in relapsed neuroblastoma patients.

(A) Plot of number of ctDNA unique versus ctDNA/tumor common variants for evaluable patients.
(B) Plot of major categories of all ctDNA variants identified.
(C) Plot of major categories of ctDNA unique variants identified.
(D) Charts of ctDNA defined alterations in ALK and RAS-MAPK pathway genes.
(E, F) ctDNA identified genetic variants in BRCA2 (E) and TP53 (F) transposed on their respective protein domains.
Underlined variants denote those unique to ctDNA and (X) denotes variants of unknown significance. In E and F, bolded variants represent those suspected to be of germline origin; red, denotes putative loss-of-function (LOF) variants; purple, denotes variants in splice sites; black, denotes missense variants. (^) denotes splice site variant in TP53 that is predicted to lead to a truncated protein (57). Relative protein domains in E and F (not drawn to scale) (58–60); TAD, transactivation domain; PALB2, PALB2 binding domain; DBD, DNA binding domain; RAD51, RAD51 binding domain; PRD, proline-rich domain; TD, tetramerization domain; BD; basic domain.
*, p<0.05.
Finally, to evaluate the potential therapeutic utility of serial ctDNA analysis, we estimated the number of these ctDNA unique variants that could potentially be clinically targeted by available drugs (Supplementary Table 6). Of the 88 unique ctDNA variants identified across the 48 evaluable patients in this study we estimate approximately 49% (43/88) of these ctDNA unique variants are potentially clinically targetable, primarily with small molecules that inhibit ALK, CDK4/6, or RAS-MAPK pathways, all of which have shown efficacy in neuroblastoma preclinical models and are being tested clinically (Supplementary Table 6) (17–21). Taken together, serial ctDNA profiling significantly enhances the detection of potentially clinically actionable driver mutations in cancer-associated genes throughout neuroblastoma therapy.
Serial ctDNA sequencing identifies dynamic genomic evolution under ALK inhibition in high-risk neuroblastoma
The use of ALK inhibition (ALKi) strategies for children with neuroblastoma harboring kinase activating missense mutations has become increasingly common with the growing evidence of this mutated receptor being a bona fide driver in somatically acquired tumors (9), the major cause of familial neuroblastoma (22), and with the increasing availability of multiple generations of inhibitors of aberrant ALK activation (18,19,23). While ctDNA has been found to be a critical tool in adult cancers in noninvasively defining resistance to ALKi (6,7), much less is known about how neuroblastomas evolve under ALKi therapeutic pressure (19,24–26). Fifteen patients enrolled on this study received an ALKi, including both the first-generation ALKi crizotinib and also several next-generation drugs (Supplementary Table 2), 14 of which had a documented ALK mutation. Nine of these 14 patients with ALK mutant tumors had 3+ serial ctDNA blood draws while receiving ALKi therapy. For 6 of these 9 patients (patients 10, 28, 32, 35, 42, and 43; Figure 4A), serial ctDNA analysis revealed clear genomic evolution of their disease with the acquisition of not only additional mutations in the ALK tyrosine kinase domain (Figure 4B), but also variants in RAS-MAPK pathway genes predicted to activate potential ALK signaling bypass pathways (Figure 4A). Several of these potential ALK bypass pathway mutations were found in the ctDNA from multiple patients, including BRAF V600E (patients 32, 35, and 43), NRAS Q61R (patients 32, 42, and 43), NRAS Q61K (patients 32 and 43), and FGFR1 N546K (patients 32 and 43) variants. Furthermore, in these cases the evolution of ctDNA variant complexity under ALKi therapeutic pressure either immediately preceded or was concurrent with disease progression documented by serial imaging, a rise in urine catecholamine levels, and/or a significant increase in clinical symptoms, including death (Figure 4C, D and Supplementary Figure 1A–C). Thus, serial ctDNA profiling can define clinically relevant genomic evolution in response to ALKi therapy in neuroblastoma patients.
Figure 4. Serial ctDNA sequencing defines dynamic genomic evolution to ALK inhibition in neuroblastoma.

(A) Plot of number of acquired mutations in a subset of patients receiving ALKi therapy (top) with acquisition sequence of specific ctDNA defined variants (bottom). Arrows indicate general timing of ALKi initiation in ctDNA profiling sequence. Only ctDNA samples collected around ALKi therapy shown for clarity (e.g., patient 10 also had MYCN amplification and an ERBB2 G776C mutation identified in their ctDNA and patient 28 had MYCN amplification and a TERT promoter −146 C>T promoter variant identified in their ctDNA, with these variants identified prior and during ALKi).
(B) Diagram of ALK protein structure with location of ALK variants acquired after initiation of ALKi therapy indicated.
(C,D) Plot of patient 32 (C) and 43 (D) treatment courses showing correlation of clinical disease surveillance (top) and serial ctDNA profiling data (bottom) while receiving ALKi therapy. Black triangles indicate general timing of clinical progression including for patient 32 (C) hypertension from tumor mass effect and death and for patient 43 (D) severely increased pain and fatigue and increased skull bone metastases. Representative abdominal MRI images for patient 32 shown at bottom of C.
In B, ALK protein domains (not drawn to scale); SP, signal peptide; MAM, meprin, A5 protein and receptor protein-tyrosine phosphatase mu domain; LDL, low-density lipoprotein receptor class A domain; Tm, transmembrane; TK, tyrosine kinase domain. Underlined variants denote those unique to ctDNA and (^) denotes ctDNA profiling done as part of clinical care.
Serial ctDNA sequencing enriches neuroblastoma surveillance
We next sought to characterize if serial ctDNA sequencing data was comparable to or enhanced standard neuroblastoma clinical surveillance methods. We focused on patients that had at least 3 ctDNA blood draws generally timed with other standard disease evaluations with at least one recurrent ctDNA identified variant. In a majority (15 of 16) of these patients, following serial ctDNA variant MAFs correlated well with clinical disease surveillance data (123I-MIBG Curie score, PET, MRI, or CT scan, and/or urine HVA/VMA levels), some of which have been mentioned earlier due to ctDNA and tumor profiling discordance (patients 5 and 33; Figure 2D–F) or ALKi-induced genomic evolution (patients 28, 32, 42, and 43; Figure 4A–D and Supplementary Figures 1A–C).
Additionally, for three other patients (7, 13, and 21) with prolonged treatment courses and multiple ctDNA samples that revealed genomic evolution of their disease over time, we compared their ctDNA profiling to other disease surveillance data (Figure 5A–F). The rising MAFs of several presumptive driver mutations (NRAS Q61K for patient 7, CDKN2A p14ARF Q57* and PTPN11 G503E for patient 13, and BRCA1 C1225* for patient 21) in these patient’s ctDNA along with overall increased ctDNA variant complexity were both temporally associated with times of disease progression (e.g., increasing 123I-MIBG Curie scores and/or an increase in urine VMA/HVA levels; Figure 5A–F).
Figure 5. ctDNA evolution correlates with disease progression in neuroblastoma patients.
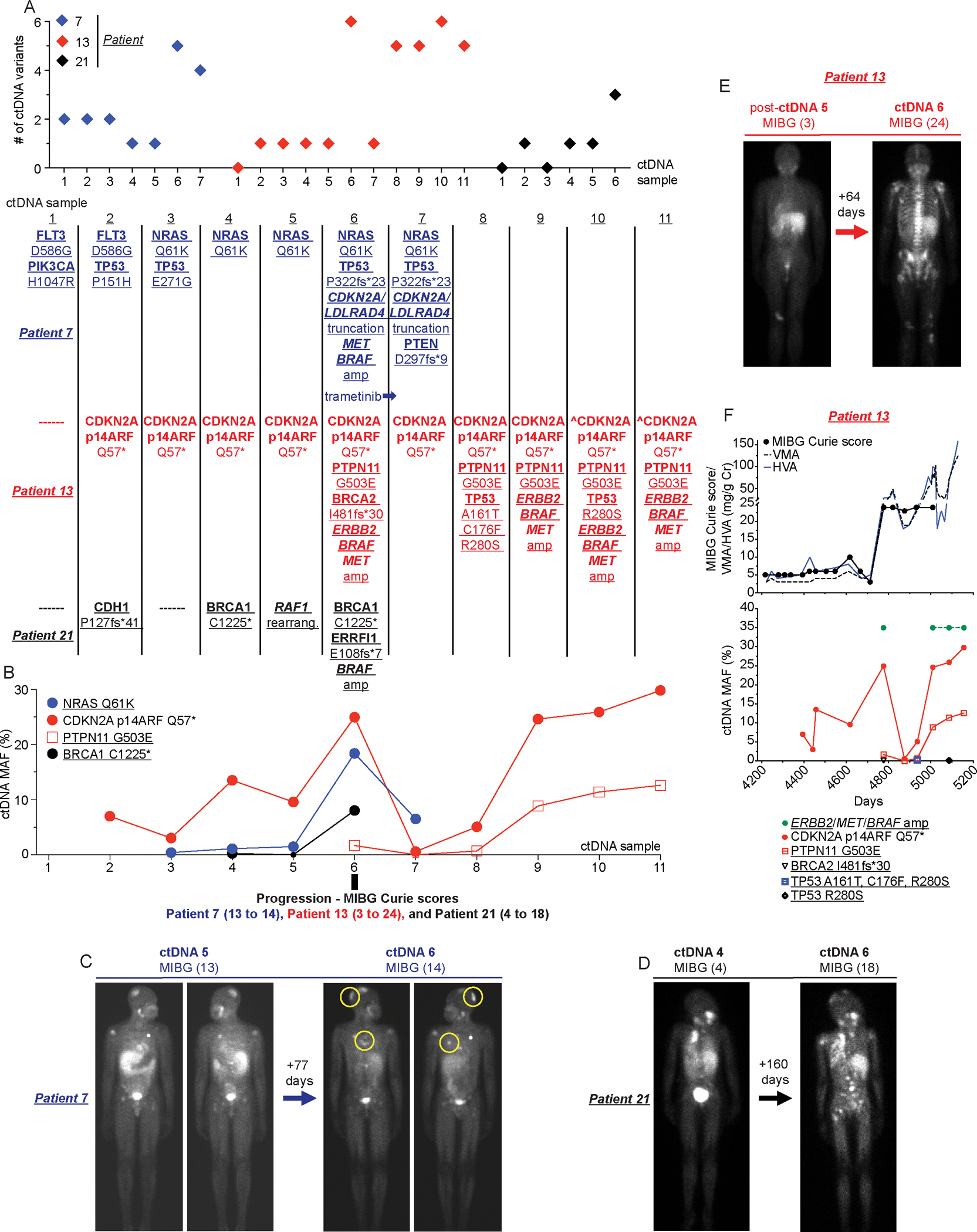
(A) Plot of total number of ctDNA variants identified in serial samples collected from patients 7,13, and 21 (top) with evolution of specific ctDNA variants shown (bottom).
(B) Plot of MAFs for recurrent ctDNA variants summarized in A.
(C-E) Paired 123I-MIBG scans with associated Curie scores for patient 7 (C; new disease noted in yellow circles in right image), patient 21 (D), and patient 13 (E) at the time of rising ctDNA MAFs and increasing ctDNA complexity (ctDNA sample 6 in B as noted).
(F) Serial ctDNA and disease evaluation correlation plot for patient 13 showing relationship between 123I-MIBG Curie scores and urine VMA/HVA levels (top) and ctDNA data (bottom).
Underlined variants denote those unique to ctDNA and (^) denotes ctDNA profiling done as part of clinical care.
For patient 25, a 7-year-old girl with a direct tumor-sequencing identified ALK F1245L mutation and chemotherapy refractory disease, ctDNA profiling also provided a useful adjunct to her neuroblastoma surveillance (Figure 6A). After a partial disease response to 4 cycles of chemoimmunotherapy (irinotecan/temozolomide/dinutuximab), she was started on ALKi. She initially had a partial response to ALKi followed by stable disease for over 16 months (a stable 123I-MIBG Curie score of 7 and normal urine VMA/HVA levels; Figure 6A), with 10 unrevealing ctDNA analyses during this almost 500-day period. However, an ALK F1245L mutation was detected in her 11th ctDNA blood draw on day 875, which correlated temporally with a new MIBG-avid lesion being noted in her left pelvic bone on day 876. Biopsy of this pelvic lesion revealed an ALK F1245L mutated neuroblastoma.
Figure 6. Serial sequencing of ctDNA from neuroblastoma patients can provide enhanced disease surveillance.

(A) Serial ctDNA and clinical disease evaluation correlation plot for patient 25 showing relationship between ctDNA data, 123I-MIBG Curie scores, and urine VMA/HVA levels (top) and representative axial 123I-MIBG images (bottom, 123I-MIBG scan day noted in bottom left).
(B) Serial ctDNA and clinical disease evaluation correlation plot for patient 35 showing relationship between ctDNA data (top) and MRIs, 123I-MIBG Curie scores, urine VMA/HVA levels, and representative axial 123I-MIBG images at the indicated time points (bottom, 123I-MIBG scan day noted in bottom left of image). Clinical remission time frame denoted with red box with ctDNA MAFs and imaging summary during clinical remission noted in bottom table.
(C) Serial ctDNA and clinical disease evaluation correlation plot for patient 6 showing relationship between ctDNA data, 123I-MIBG Curie scores, and urine VMA/HVA levels (top) and representative axial 123I-MIBG images at the indicated time points (bottom, 123I-MIBG scan day noted in bottom right of image).
(D) Serial ctDNA and clinical disease evaluation timeline for patient 39 showing relationship between ctDNA and tumor sequencing data, PET/CT, 123I-MIBG Curie scores, and urine VMA/HVA levels with representative axial PET/CT images at the indicated time points.
Large symbols in plots represent abnormal values and small symbols represent normal values.
Amp, amplification; seq, sequencing. Chemoimmunotherapy, Irinotecan/temozolomide/GD2-targeting chimeric monoclonal antibody dinutuximab, a treatment regimen used commonly for relapsed neuroblastoma.
Underlined variants denote those unique to ctDNA and (X) denotes variants of unknown significance.
Next, for patient 44, a 9-year-old boy with relapsed neuroblastoma who had a KIT D816V variant and MYCN amplification found concurrently in his tumor and ctDNA at the time of relapse, serial ctDNA data revealed KIT D816V MAF and MYCN amplification trends which correlated well with an initial response to chemoimmunotherapy, and ultimately with disease progression after 14 cycles of this therapy (Supplementary Figure 2A). Further, for patient 36, a 2-year-old boy with a BRAF G469A-mutated, relapsed neuroblastoma, loss of detection of this BRAF variant in his ctDNA correlated well with a clinical disease response (Supplementary Figure 2B). This case also highlights that for some neuroblastomas with bulky ganglioneuroblastoma/ganglioneuroma components where imaging is not always an accurate measurement of disease activity (e.g., stable MIBG Curie score), serial ctDNA monitoring may provide an important adjunct to disease surveillance. Finally, for patient 10, whose tumors harbored both an ALK amplification and an ALK F1174L variant, serial ctDNA analyses revealed both correlation with serial surveillance PET scans and also possible spatial tumoral heterogeneity of these genetic alterations, with the ALK amplification likely arising from the neck tumor and the ALK F1174L variant arising from a left-sided abdominal tumor (Supplementary Figure 1C).
In at least 3 cases, serial ctDNA monitoring enriched disease surveillance beyond standard clinical evaluations. One of these cases (patient 33) was previously shown in Figure 2F. Another example was with patient 35, a 2-year-old girl with MYCN amplified high-risk neuroblastoma (Figure 6B). For this child, an ALK R1275Q variant was noted on diagnostic tumor sequencing, and she was enrolled on this serial ctDNA profiling study after a second autologous stem cell transplant (ASCT), from which she suffered extensive clinical morbidity. Although noted to be in clinical remission on a comprehensive set of disease evaluations (no tumor noted on 123I-MIBG or MRI imaging, normal urine HVA/VMA levels) at study enrollment, a low-level of the ALK R1275Q variant was noted in her first ctDNA sample (MAF of 0.17%; day 357). Serial ctDNA evaluations during the following 5 months showed a rising ALK R1275Q MAF (5 to 41%) along with MYCN amplification and other somatic variants, again concurrent with otherwise negative clinical disease evaluations (red box in Figure 6B). Here, the initial ctDNA evidence of recurrent neuroblastoma indicated by a rising ALK R1275Q MAF preceded findings of tumor relapse using standard methods by greater than 100 days. Finally, for patient 6, a 6-year-old male with an ALK R1275Q mutated, relapsed neuroblastoma, rapidly rising ALK R1275Q MAFs were also predictive of his overt clinical progression and ultimately his death (Figure 6C).
In only one of these 16 cases did ctDNA and standard clinical surveillance data not correlate (Supplementary Figure 2C). For patient 41, a 10-year-old boy with relapsed neuroblastoma and multiple TP53 variants (V157fs*13, G334V, and C277Y) identified across serial ctDNA samples, there was little correlation between the TP53 MAFs and other clinical disease evaluations (123I-MIBG Curie score or urine HVA/VMA levels). Here, we hypothesize that the variants identified in cfDNA in this patient may not have been derived from tumor and may be more reflective of clonal hematopoiesis (CH) (27).
Finally, in an unusual case that falls outside of the above filtering parameters (patient 39), ctDNA analysis was supportive of a neuroblastoma relapse approximately 3 months before a confirmative tumor biopsy (Figure 6D). This 8-year-old girl was in a prolonged disease remission after initial treatment for an MIBG-avid, MYCN amplified neuroblastoma when a new right paraspinal lesion was noted by routine PET/CT imaging. Initial ctDNA analysis revealed presence of MYCN amplification, however a subsequent biopsy of the paraspinal tumor showed nodular fasciitis harboring a diagnostic PKM-USP6 fusion. Two months later though, additional PET/CT imaging identified a new left-sided peri-renal lesion which correlated with continued detection of MYCN amplified ctDNA in her peripheral blood. This new mass was biopsied and found to be a MYCN-amplified neuroblastoma prompting initiation of salvage therapy for relapsed disease.
ctDNA-identified ERRFI1 mutations are pathogenic in neuroblastoma cells
Lastly, we looked to understand the functional impact of a subset of ctDNA-defined variants in neuroblastoma cells. For these studies, we focused on two ERRFI1 frameshift putative loss-of-function (LOF) variants (identified in patients 15 and 21) and 2 ERRFI1 missense variants of unknown significance (VUS; identified in patients 15 and 46; Figure 7A, Supplementary Tables 3 and 6). The ERRFI1 gene is encoded on chromosome 1p, a commonly deleted chromosomal arm in neuroblastoma tumors that is associated with a poor prognosis (28). Furthermore, ERRFI1 has been well-documented as a tumor suppressor in other cancers (29–31), in large part by inhibiting EGFR signaling, but little is known about its functional role in neuroblastoma (32). Notably, the 2 LOF ERRFI1 variants we identified in ctDNA are predicted to result in proteins lacking the EGFR/ErbB binding domain (EBD)(31) and the ERRFI1 R247K variant is in a region of the ERRFI1 protein critical to 14–3-3 protein binding and regulation of EGF signaling (Figure 7A) (33,34). Thus, we first sought to better understand ERRFI1’s function in neuroblastoma cells and then how these ctDNA variants may be perturbing these functions. First, consistent with being a putative neuroblastoma tumor suppressor gene, we found that higher ERRFI1 expression was associated with a better prognosis in several larger neuroblastoma tumor data sets (Figure 7B, Supplementary Figure 3A, B). Further, significantly lower ERRFI1 expression was found in neuroblastomas with more clinically aggressive features (Figure 7C, Supplementary Figure 3C, D) and neuroblastoma cell lines had very low ERRFI1 expression (n=39; median ERRFI1 FPKM = 1.3; range 0.3 – 6.3 FPKM) (35) and no measurable ERRFI1 protein (Supplementary Figure 3E). To directly investigate how these ctDNA identified ERRFI1 mutations may be altering ERRFI1’s normal function in neuroblastoma cells, we developed isogenic ERRFI1 wild-type (WT) and ERRFI1 mutant SK-N-AS, SH-SY5Y, and NLF neuroblastoma cell lines. These ERRFI1 isogenic cells showed comparable ERRFI1 expression and ERRFI1 protein was identified in cell lines transduced with one of the LOF variants, along with both missense variants (Figure 7D, E). Remarkably though, exogenous expression of WT ERRFI1 was rapidly lost in isogenic cells generated from lentiviral vectors with several unique exogenous promoters, an effect that could be reversed with 5-azacytidine, suggesting hypermethylation as an additional mechanism of ERRFI1 silencing in neuroblastoma cells (Supplementary Figure 3F–H). Nonetheless, in transient assays WT ERRFI1 displayed characteristics of a tumor suppressor, inhibiting EGF-induced neuroblastoma cell growth and migration, EGFR phosphorylation, as well as EGF-induced PI3K/AKT and MAPK/ERK pathway activation in isogenic SH-SY5Y, SK-N-AS, and NLF neuroblastoma cells (Figure 7F–H, Supplementary Figure 3I, J). However, these ERRFI1 protein inhibitory functions were lost most notably with the ERRF1 putative LOF variants (E108fs*7 and R247fs*16), and with more modest inhibition of EGF-induced effects seen with the ERRFI1 R247K variant (Figure 7F–H, Supplementary Figure 3I, J). Further, while WT ERRFI1 was able to induce EGFR cellular internalization upon EGF stimulation, the mutant ERRFI1 truncated proteins also lost this critical tumor suppressive function (Figure 7I) (36). While the chromosome 1p deletion status was not available for the tumors from these patients, we hypothesize these acquired LOF ERRFI1 mutations are contributing to biallelic loss of the ERRFI1 gene. In fact, for patient 15, the R247K ERRFI1 VUS was found in all 6 ctDNA samples collected at a MAF of ~50%, suggesting a germline origin, and the R247fs*16 was identified in the last sample collected, suggesting a mechanism for the development of biallelic ERRFI1 loss in this patient’s tumor. Thus, for genes such as ERRFI1, serial ctDNA analysis can also highlight likely subclonal events in understudied genes also critically involved in neuroblastoma tumorigenesis.
Figure 7. ctDNA-identified ERRFI1 variants are pathogenic in neuroblastoma cells.
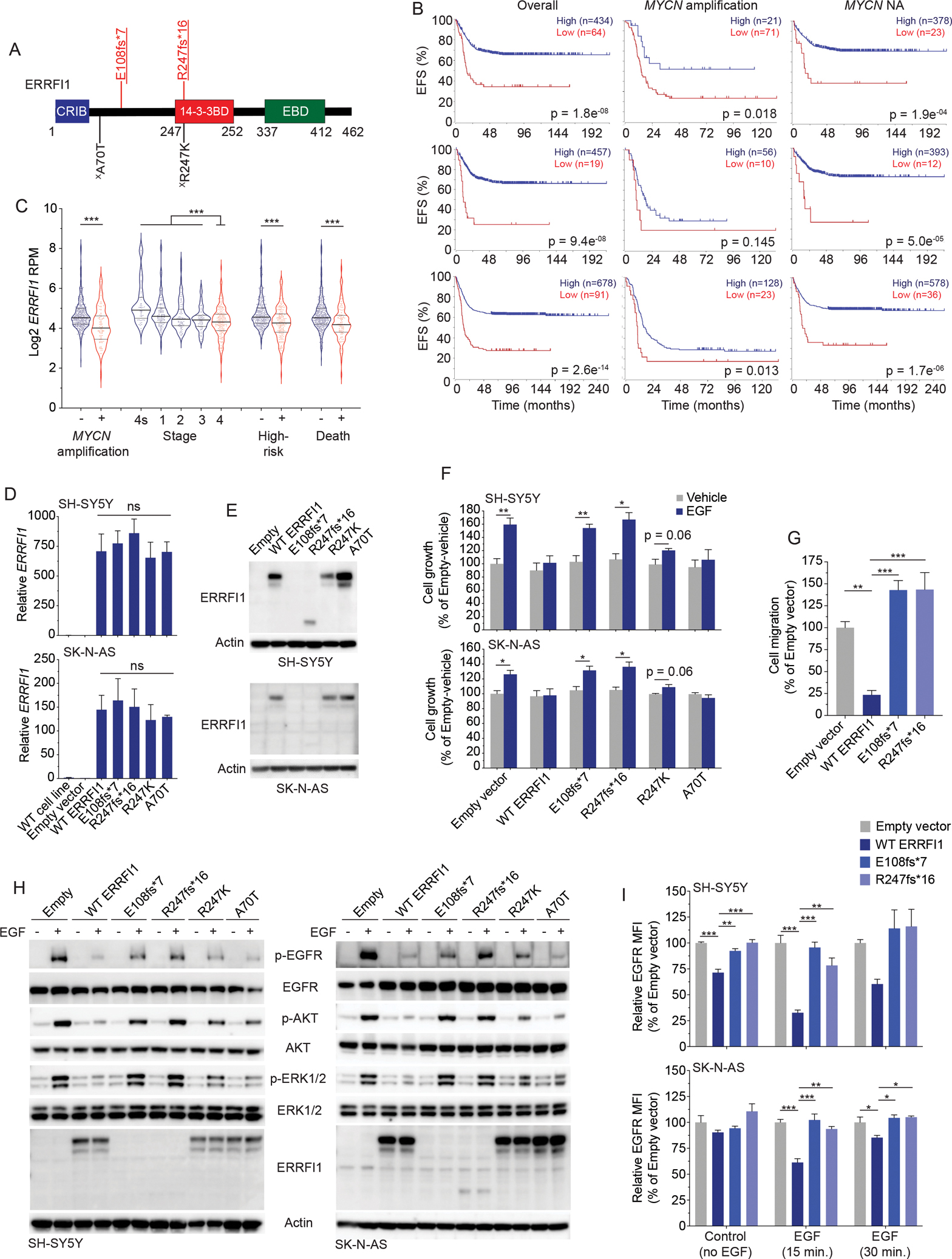
(A) ctDNA-identified variants in ERRFI1 transposed on the ERRFI1 protein domains.
(B) Neuroblastoma event-free survival (EFS) plots stratified by ERRFI1 expression for all patients (left), patients with MYCN amplified tumors (middle), and patients with MYCN non-amplified (NA) tumors (right) in 3 large neuroblastoma data sets, SEQC (n=498, top), Kocak (n=649; middle), and Cangelosi (n=786; bottom). EFS plots were generated in the Genomics Analysis and Visualization Platform (R2; https://hgserver1.amc.nl/cgi-bin/r2/main.cgi).
(C) ERRFI1 expression in neuroblastoma tumors stratified by several clinical covariates (SEQC; n=498 tumors).
(D, E) ERRFI1 expression in ERRFI1 isogenic SH-SY5Y and SK-N-AS neuroblastoma cell lines by RT-PCR (D) and western blot (E).
(F) Relative cell growth plots of ERRFI1 isogenic SH-SY5Y (top) and SK-N-AS (bottom) neuroblastoma cell lines after EGF stimulation (50 ng/mL).
(G) Relative EGF-induced cell migration of ERRFI1 isogenic SK-N-AS neuroblastoma cells.
(H) Western blot of ERRFI1 isogenic SH-SY5Y (left) and SK-N-AS (right) neuroblastoma cell lines after EGF stimulation (50 ng/mL).
(I) Relative EGFR cell surface levels of ERRFI1 isogenic SH-SY5Y (top) and SK-N-AS (bottom) neuroblastoma cell lines with and without EGF stimulation (50 ng/mL).
Underlined variants denote those unique to ctDNA and (X) denotes variants of unknown significance. In A, red denotes putative LOF variant and black denotes missense VUS. Relative protein domains in A (not drawn to scale); CRIB, Cdc42/Rac interactive binding domain; 14–3-3BD, 14–3-3 binding domain, EBD, EGFR or ErbB binding domain.
WT, wild type.
*, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant.
Discussion
The technology now exists to quantify and sequence ctDNA from the plasma of patients. However, how to successfully incorporate comprehensive ctDNA profiling into the clinical care of children with cancer has not yet been realized. Here we provide a broad assessment of the potential clinical utility of serial ctDNA sequencing in children with the commonly lethal pediatric malignancy high-risk neuroblastoma, along with validating the functional relevance of ctDNA defined and novel ERRFI1 variants in neuroblastoma cellular models.
These data support findings from previous studies focused on interrogating ALK (8,37,38) or MYCN amplification or other characteristic neuroblastoma segmental chromosomal aberrations (8,38–42), further validating the ability to commonly detect ctDNA from the blood of children with neuroblastoma. Additionally, these data build upon prior ctDNA gene panel profiling of neuroblastomas from 11 patients at the time of diagnosis (43) and a whole-exome sequencing (WES) ctDNA study that focused on validating clonal evolution with serially sequenced samples in this disease (44). Here we significantly expand upon this work and study the clinical utility of neuroblastoma-focused ctDNA sequencing by using the readily available Foundation Medicine platform to serially profile ctDNA across a panel of cancer driver genes in a diverse cohort of high-risk neuroblastoma patients, both under the therapeutic pressure of conventional neuroblastoma chemoradioimmunotherapy and more molecularly targeted therapies at the time of relapse. Importantly, the Foundation Medicine platform utilizes next-generation sequencing technology that enables sensitive detection of variants even at allele frequencies <1% of the total cfDNA (45) which is more sensitive than whole-exome sequencing approaches previously used in this disease (44). We focus on not only defining the evolution of ctDNA-defined variants in cancer-associated genes, but also how these data compliment or enhance real-world clinical surveillance imaging and biochemical assays and paired-tumor sequencing data. For example, this ctDNA sequencing compared to tumor profiling done throughout each patient’s disease course, exemplified the likely extensive intra- and intertumoral heterogeneity of neuroblastomas as over 50% of variants identified were unique to ctDNA and not found in paired tumor profiling. While many of the ctDNA genetic variants identified were known driver mutations in ALK or RAS-MAPK pathway genes supporting the critical importance of these signaling cascades in neuroblastoma and validating early studies in neuroblastoma focused on ALK variant detection in temporally paired blood and tumor specimens (8,37), some identified variants have not been as commonly associated with neuroblastoma tumorigenesis. The high prevalence of ctDNA variants in DNA damage response genes such as BRCA1, BRCA2, and TP53 was unanticipated, as were variants in the ERRFI1 gene. In fact, ERRFI1 mutations have not been identified in several prior large neuroblastoma tumor sequencing studies (9,46), highlighting their potential subclonality and the perhaps transient nature of these mutations specifically during disease progression. We hypothesize that these lesions may be acquired “second hits” to the ERRFI1 gene in the setting of a somatic chromosomal 1p deletion that were specifically acquired at the time of disease progression. We modeled these ERRFI1 mutations in neuroblastoma cells and indeed found that they induced profound ERRFI1 LOF, consistent with this hypothesis and directly supporting a role for ctDNA identified variants in disease pathogenesis.
Likewise, while TP53 mutations are enriched in relapsed neuroblastomas (11), the number of transient and paired ctDNA TP53 variants with low MAFs identified here was notable. These data, taken together with the increased appreciation for the prevalence of clonal hematopoiesis (CH) (27) that often involves TP53 mutations (47) which can be enriched after radiation or chemotherapy (48), raises the question that some of the TP53 ctDNA mutations identified here may be signs of treatment-induced CH rather than being tumor-derived. A majority (14/21) of the transient TP53 mutations observed in this study were preceded by genotoxic and bone marrow suppressive 131I-MIBG therapy, suggesting that this therapy may have either induced these TP53 mutations or promoted the transient expansion of hematopoietic cells already harboring these TP53 mutations. Most strikingly, for patient 41 with 2 recurrent TP53 mutations identified serially in their ctDNA analyses (V157fs* and G334V), the ctDNA MAFs had no correlation with other disease surveillance data, directly supporting a potential non-tumor origin to these specific TP53 variants in this patient. However, while contamination by CH variants should be contemplated in all ctDNA studies, especially when using ctDNA data to make treatment decisions, most cfDNA identified variants here (and overall in children) are likely tumor-associated. This is particularly true for the ALK or RAS-MAPK pathway variants identified and other variants with a robust connection to cancer tumorigenesis that are not commonly found in CH clones (27,47). Furthermore, specifically considering TP53, even in older patient populations with smoking histories where CH is significantly more prevalent, only a small subset of TP53 variants identified in cfDNA have been proven to be from CH and not tumor-derived (49). Nonetheless, to directly address this issue in future pediatric ctDNA studies, paired sequencing of peripheral blood cells should be routinely implemented to define the origin of cfDNA-identified variants, again a distinction of utmost importance when contemplating utilizing ctDNA data to guide therapeutic decisions.
The putative activation of potential bypass pathways upon ALKi therapy described here, such as RAS-MAPK signaling, is consistent with prior reports in neuroblastoma and lung cancer patients and in model cellular systems (24,26,50). However, this study was not powered to define precise mechanisms of resistance to specific ALK inhibitors, but rather to show the potential clinical utility of ctDNA profiling in this disease under the therapeutic pressure of both targeted and conventional neuroblastoma therapies. The experience in lung cancer has shown that mechanisms of ALKi resistance can vary across drugs (51), which may also be the case in neuroblastoma and thus requires investigation in prospective ALKi-focused trials. Importantly, the serial profiling of ctDNA here also revealed genomic evolution under the pressure of other neuroblastoma therapies, which not only correlated with disease progression, but also nominated other potential therapies for these patients.
Prior studies have shown the potential utility of serial ctDNA profiling in enhancing neuroblastoma surveillance and providing clinically relevant data about ongoing tumor activity (8,44), which is further supported by the data presented here and especially critical in a disease where tumor size does not always correlate directly with tumor activity. Here serial analysis of ctDNA enabled the detection of low levels of neuroblastoma that was below the sensitivity of current clinical surveillance methods, enabling earlier detection of disease progression. Though FoundationACT™ was not designed for disease surveillance, the data generated here provides insight into the future utility of liquid biopsies in disease monitoring. At the time this study was initiated, the FoundationACT™ assay was available for next generation sequencing of ctDNA using a limited panel of 62 cancer-associated genes. In August of 2021, the US Food and Drug Administration approved the FoundationOne®Liquid CDx test that included a 324 gene panel that was analytically and clinically validated across multiple tumor types (52). The availability of this updated liquid biopsy test could allow for the detection of an increased number of potentially targetable gene alterations in pediatric patient samples. However, this, and most other next generation sequencing panels, are focused on those cancer driver genes more commonly aberrant in adult cancers; therefore, it will also be critical to do follow-up studies with pediatric cancer and/or neuroblastoma specific gene panels to fully realize the extent of enhanced genetic data acquired from serial ctDNA profiling in this disease.
Finally, within this rapidly expanding field different approaches to profiling ctDNA have been developed, including droplet-digital PCR (ddPCR) (8,37,38), next-generation sequencing (43), whole-exome-sequencing (WES) (44), and low-passage whole-genome sequencing (WGS) (39,41,42), each with distinct advantages and disadvantages. For example, while ddPCR provides a scalable, cost-effective, and exquisitely sensitive method to detect a single or a few predefined ctDNA variants without the need for complex bioinformatic pipelines, it does not allow for the complex tumor genotyping that can be achieved with NGS-based approaches. Conversely, while WES or NGS approaches have the advantage of being able to detect a large number of ctDNA variants in parallel providing information on tumor evolution and treatment resistance (44), they can also detect often difficult to interpret variants of unknown significance and thus require robust bioinformatic pipelines. Finally, the estimation of ctDNA fraction in total cfDNA may differ dramatically using different sequencing platforms. For example, targeted NGS approaches such as that used here can sensitively detect variants at or below a 1% MAF in cancer associated genes, which is likely identifying subclonal intra- or intertumoral heterogeneity. However other sequencing methods (e.g., WES) can only identify variants with higher MAFs that are often likely somatic passenger mutations. Thus, these differences should be considered when computing total ctDNA fraction using variant MAFs and it is likely the field will rapidly develop more accurate algorithms for quantification of the total ctDNA fraction of cfDNA. Taken together, orthogonal ctDNA detection methods may have complimentary roles in the oncology clinic, such as utilizing highly sensitive ddPCR to serially quantify a few predefined ctDNA variants in a panel of patients harboring tumors with those variants or alternatively using low-passage WGS focused on detecting tumor histotype specific copy number aberrations, allowing for sensitive detection of tumor progression or MRD identification. Alternatively, a WES or an NGS gene panel approach may be utilized at larger academic centers with available analytic pipelines for a smaller number of patients receiving targeted inhibitors to identify clinically actionable variants or relevant treatment resistant clones which could facilitate development of novel therapeutic strategies (3,44).
Based on the observations here, we suggest that serial ctDNA profiling should be integrated into clinical practice for children with high-risk neuroblastoma to provide real-time data on genomic evolution and the development of therapeutic resistance, to identify potentially clinically targetable mutations, and to better enable less invasive and more sensitive tumor surveillance and detection of tumor heterogeneity. In time, ctDNA profiling may allow for decreased use of traditional disease surveillance methods, many of which require procedural sedation along with significant radiation exposure to children already at high risk for secondary malignancies.
Methods
Patient recruitment
Forty-nine (49) high-risk neuroblastoma patients treated at CHOP were enrolled on this study after signing written informed consent between September 2016 and February 2020. The study was approved by the CHOP IRB (#15–012553) and was conducted in accordance with recognized ethical guidelines. Patient 29 was removed from the study when ultimately determined to have intermediate-risk neuroblastoma. Clinical data were collated from CHOP clinical databases, including ctDNA data from the identical Foundation Medicine ctDNA assay. In a few cases, therapy data was generalized (e.g., using “ALKi” versus naming specific drug) or only used in an aggregate fashion to maintain the integrity of ongoing clinical trials.
ctDNA sequencing and analysis
Plasma from neuroblastoma patients was evaluated using the FoundationACT™ hybrid capture-based NGS assay (Foundation Medicine, Cambridge, MA), for which the full validation and methodology have been previously described (45). Cell-free DNA (≥20 ng) was extracted from 5 mL of plasma and subjected to comprehensive genomic profiling using a 62-gene panel with >30,000x raw coverage, 5,000x unique coverage, and approximately 3,000x redundant (e.g., error-corrected) coverage. A set of 12 fragment-level indexed adaptors with variable 6-bp DNA barcodes were used to align read pairs and exclude any sequencing errors. The baitset for this assay targets 0.14 Mb of the human genome including all exons of 27 genes, selected exons of 33 genes, selected introns of 6 genes, and the TERT promoter region that is recurrently mutated in cancer (Supplementary Table 1). Samples were assessed for base substitutions, short insertions/deletions, rearrangements/fusions, and copy number variations as previously described (45). FoundationACT™ has been previously validated to have >99% overall sensitivity (95% CI, 99.1–99.4%) for short variants (base substitutions and short insertions/deletions) at MAF >0.5%, >95% sensitivity (95% CI, 94.2–95.7%) at MAF 0.25% to 0.5%, and 70% sensitivity (95% CI, 68.2–71.5%) at MAF 0.125% to 0.25%.
Tumor sequencing
Tumor sequencing was done as part of routine clinical care, data was collated from the patient medical record, and variants were compared to ctDNA sequencing. Paired tumor sequencing was done either via Foundation Medicine’s FoundationOne® CDx assay or using CHOP’s Comprehensive NGS Solid Tumor Panel, both of which have been comprehensively validated and described previously (53–55), and further described in the supplementary methods. Briefly, the CHOP Comprehensive NGS Solid Tumor Panel interrogates 238 cancer genes for sequence and copy number alterations (CNA) and 110 cancer genes for fusion genes (53). For the FoundationOne CDx assay, ≥50 ng of DNA was extracted from 40 μm of FFPE tumor sections and subjected to whole-genome shotgun library construction and hybridization-based capture. Sequencing was performed on 324 cancer-related genes, including all coding exons from 309 cancer-related genes, one promoter region, one non-coding RNA, and select intronic regions from 34 commonly rearranged genes). Base substitutions, insertions and deletions, rearrangements, and copy number changes were assessed as described previously (https://www.fda.gov/medical-devices/recently-approved-devices/foundationone-cdx-p170019s029) (54,55). Bioinformatic pipelines used for variant calling and filtering for FoundationOne® CDx and FoundationACT are similar but not identical as previously described (45,55), a limitation of these analyses when comparing tumor and ctDNA data.
Cell lines
Human-derived neuroblastoma cell lines (SK-N-AS, RRID:CVCL_1700; SH-SY5Y, RRID:CVCL_0019; and NLF, RRID:CVCL_E217) were obtained from the CHOP cell line bank and cultured in RPMI containing 10% FBS, 2 mM L-Glutamine, and 1% streptomycin/penicillin (RPMI Complete) at 37°C under 5% CO2. Cell lines were genotyped (GenePrint® 24 System; Promega) and mycoplasma tested, both routinely before cells were placed into the cell bank. Cells were typically utilized for the described experiments <5 passages from thaw, genotyping, and mycoplasma testing.
ERRFI1 construct design
The ERRFI1 pLenti CMV puro vector (WT ERRFI1) was constructed from an ERRFI1 pDONR221 vector (DNASU Plasmid Repository, HsCD00044200) cloned into a pLenti CMV puro DEST (w118–1) plasmid, which was a gift from Eric Campeau (56) and was purchased via Addgene (RRID:Addgene_17452), with a Gateway® LR clonase enzyme (Invitrogen) via the manufacturer’s instructions to make a ERRFI1 pLenti CMV puro vector (WT ERRFI1). To achieve the ERRFI1 mutants, the ERRFI1 pLenti CMV puro vector (WT ERRFI1) was mutated utilizing a QuickChange II XL Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer’s instructions. All sequences were verified with Sanger sequencing.
Real-time PCR analysis
Total RNA was isolated utilizing Qiashredder and RNeasy Mini Kits (Qiagen) and mRNAs were converted to cDNA using a SuperScript™ III First-Strand Synthesis System for RT-PCR (Thermo Fisher Scientific). Taqman® gene expression assays were used to quantitate ERRFI1 (Hs01086257_g1) and HPRT1 (Hs02800695_m1). qPCR analysis was performed on an Applied Biosystems 7900HT Sequence Detection System using standard cycling conditions and relative expression values were calculate using the delta-delta Ct method.
Lentiviral preparation and transduction
ERRFI1 (WT, E108fs*7, R247fs*16, R247K, and A70T) pLenti CMV puro vectors were transfected, along with pMD2.G (RRID:Addgene_12259), pMDLg/pRRE (RRID:Addgene_12251), and pRSV-Rev (RRID:Addgene_12253) vectors into HEK293T cells (RRID:CVCL_0063) utilizing Lipofectamine 2000 (Thermo Fisher Scientific). The virus-containing supernatant was collected after 48 and 72 hours, filtered with 0.45 μM nitrocellulose membranes, and concentrated by ultracentrifugation. Virus was added to cells in the presence of 8 μg/ml polybrene (Sigma). Media was changed the next day and puromycin (Sigma) was added for selection for 48 hours prior to further experiments.
EGF stimulation and cell proliferation assays
ERRFI1 isogenic neuroblastoma cells were incubated in RPMI Complete overnight, then serum-starved in 1% FBS RPMI overnight, and then incubated with EGF (50 ng/mL; STEMCELL™ Technologies, #78006.1) for 5 minutes at 37°C. Cells were then harvested for protein lysate preparation. For cell proliferation assays, ERRFI1 isogenic cells were plated in at least triplicate in a 96-well plate in RPMI Complete overnight, serum-starved in 1% FBS RPMI overnight, and then incubated with EGF (50 ng/mL) for 72 hours at 37°C. Luminescence was measured with a CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions in a GloMax plate reader (Promega) and luminescence was quantified relative to vehicle treated cells.
Cell migration assay
Transwell chambers with an 8.0 μm pore polycarbonate membrane insert (Costar) were used to evaluate EGF-induced neuroblastoma cell migration. ERRFI1 isogenic SK-N-AS cells were seeded in the upper chamber in 1% FBS RPMI Complete and 500 uLs of 1% FBS RPMI + EGF (50 ng/mL) was added to the lower chamber. After 24 hours, cells in the lower chamber were fixed with 4% paraformaldehyde, stained with crystal violet, imaged, and counted.
EGFR internalization analysis
ERRFI1 isogenic cells were pretreated with cycloheximide (10 μg/mL) for 1 hour before being treated with EGF (50 ng/mL) for 15–30 minutes. Cells were then washed, fixed for 15 minutes with 4% paraformaldehyde, washed, stained with an anti-EGFR antibody (Biolegend #352904; RRID:AB_10896794) for 30 minutes at 4°C, and subjected to flow cytometric analysis using a CytoFLEX S cytometer.
5-aza-2′-deoxycytidine treatment
ERRFI1 isogenic cells were incubated with 5-aza-2′-deoxycytidine (5-AZA) (2.5 – 5 μM) for 48 hours at 37°C. 5-AZA medium was prepared and replaced daily. Cells were then harvested for whole-cell protein lysate preparation.
Western blotting
Whole-cell protein lysates were prepared with cell lysis buffer [Cell Signaling Technology (CST), #9803], PMSF (Sigma-Aldrich), phosphatase inhibitor cocktail 2 (Sigma-Aldrich, P5726) and 3 (Sigma-Aldrich, P0044), briefly sonicated, rotated for 15 minutes at 4°C, centrifuged for 10 minutes, and then supernatant was removed and protein concentration was quantified by Bradford assays. Lysates were separated on a 4–12% Bis-Tris gels (Life Technologies), transferred to a PVDF membrane, blocked in 5% non-fat milk in Tris-buffered saline and Tween-20 (TBS-T) and blotted using standard protocols. Membranes were typically incubated at 4°C overnight in primary antibody, washed x 3 in TBS-T, then incubated in 1:2,000 diluted HRP-labeled secondary antibody at room temperature for 1 hour, washed an additional x 3 with TBS-T, and then developed with a chemiluminescent reagent (SuperSignal West Femto, Thermo Fisher Scientific). The following primary antibodies were used: β-Actin (1:5,000; CST #4967; RRID:AB_330288), MIG6/ERRFI1 monoclonal antibody (1:1,000; Santa Cruz Biotechnology #sc-137154; RRID:AB_2101524), phospho-p44/42 MAPK (Erk1/2) (T202/Y204) (D13.14.4E) XP (R) Rabbit mAb (1:1,000; CST #4370; RRID:AB_2315112), p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb (1:1,000; CST #4695; RRID:AB_390779), EGF Receptor Rabbit Ab (1:1,000; CST #2232; RRID:AB_331707), Phospho-EGF Receptor (Tyr1173) (53A5) Rabbit mAb (1:1,000; CST #4407; RRID:AB_331795), phospho-AKT (ser473) Rabbit Ab (1:1,000; CST #4060; RRID:AB_2315049), AKT Rabbit Ab (1:1,000; CST #9272; RRID:AB_329827).
Data availability
The ctDNA data generated in this study are available within the article and its supplementary data files. The tumor sequencing and clinical co-variate data collated in this study are not publicly available due to the risk of compromising patient privacy but are included in the article and supplementary data files in a coded and aggregate manner. Any additional data are available upon reasonable request from the corresponding author.
Supplementary Material
Statement of Significance.
Circulating tumor DNA (ctDNA) is prevalent in children with neuroblastoma. Serial ctDNA profiling in neuroblastoma patients improves the detection of potentially clinically actionable and functionally relevant variants in cancer driver genes and delineates dynamic tumor evolution and disease progression beyond that of standard tumor sequencing and clinical surveillance practices.
Financial Support
Kristopher R. Bosse is a Damon Runyon Physician-Scientist supported (in part) by the Damon Runyon Cancer Research Foundation (PST-07–16). This work was also supported by NCI K08 CA230223 (K.Bosse), DOD PRCRP TTSA CA190126P2 (K. Bosse), NCI R35 CA220500 (J. Maris), NCI P01 CA217959 (J. Maris and Y. Mossé), the Giulio D’Angio Endowed Chair (J. Maris), the Patricia Brophy Endowed Chair (Y. Mossé), Alex’s Lemonade Stand Foundation (K. Bosse, G. Pascual-Pasto, and Y. Mossé), the Devaney Family Fund (J. Maris), the Band of Parents Foundation (Y. Mossé), Laney’s Legacy of Hope (K. Bosse), and the EVAN Foundation (K. Bosse).
Footnotes
Conflicts of Interest
M.F., X.L., M.M., M.T., and G.O. are current or past employees of Foundation Medicine. Y.P.M. is a consultant for Pfizer, Inc. No other authors declare any conflict of interest.
References
- 1.Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers 2016;2:16078 doi 10.1038/nrdp.2016.78. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010;362(23):2202–11 doi 362/23/2202 [pii] 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabel L, Proudhon C, Romano E, Girard N, Lantz O, Stern MH, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol 2018;15(10):639–50 doi 10.1038/s41571-018-0074-3. [DOI] [PubMed] [Google Scholar]
- 4.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368(13):1199–209 doi 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 5.Parikh AR, Leshchiner I, Elagina L, Goyal L, Levovitz C, Siravegna G, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019;25(9):1415–21 doi 10.1038/s41591-019-0561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dagogo-Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, et al. Treatment with Next-Generation ALK Inhibitors Fuels Plasma ALK Mutation Diversity. Clin Cancer Res 2019;25(22):6662–70 doi 10.1158/1078-0432.CCR-19-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dagogo-Jack I, Brannon AR, Ferris LA, Campbell CD, Lin JJ, Schultz KR, et al. Tracking the Evolution of Resistance to ALK Tyrosine Kinase Inhibitors through Longitudinal Analysis of Circulating Tumor DNA. JCO Precis Oncol 2018;2018 doi 10.1200/PO.17.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lodrini M, Graef J, Thole-Kliesch TM, Astrahantseff K, Sprussel A, Grimaldi M, et al. Targeted Analysis of Cell-free Circulating Tumor DNA is Suitable for Early Relapse and Actionable Target Detection in Patients with Neuroblastoma. Clin Cancer Res 2022;28(9):1809–20 doi 10.1158/1078-0432.CCR-21-3716. [DOI] [PubMed] [Google Scholar]
- 9.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013;45(3):279–84 doi 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady SW, Liu Y, Ma X, Gout AM, Hagiwara K, Zhou X, et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat Commun 2020;11(1):5183 doi 10.1038/s41467-020-18987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Padovan-Merhar OM, Raman P, Ostrovnaya I, Kalletla K, Rubnitz KR, Sanford EM, et al. Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 2016;12(12):e1006501 doi 10.1371/journal.pgen.1006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellini A, Potschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 2021;39(30):3377–90 doi 10.1200/JCO.21.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 2014;32(25):2727–34 doi JCO.2013.54.0674 [pii] 10.1200/JCO.2013.54.0674. [DOI] [PubMed] [Google Scholar]
- 14.Schmelz K, Toedling J, Huska M, Cwikla MC, Kruetzfeldt LM, Proba J, et al. Spatial and temporal intratumour heterogeneity has potential consequences for single biopsy-based neuroblastoma treatment decisions. Nat Commun 2021;12(1):6804 doi 10.1038/s41467-021-26870-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 2015;47(8):864–71 doi 10.1038/ng.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samoyedny A, Srinivasan A, States L, Mosse YP, Alai E, Pawel B, et al. Image-Guided Biopsy for Relapsed Neuroblastoma: Focus on Safety, Adequacy for Genetic Sequencing, and Correlation of Tumor Cell Percent With Quantitative Lesion MIBG Uptake. JCO Precis Oncol 2021;5 doi 10.1200/PO.20.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin Cancer Res 2017;23(11):2856–68 doi 10.1158/1078-0432.CCR-16-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krytska K, Ryles HT, Sano R, Raman P, Infarinato NR, Hansel TD, et al. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin Cancer Res 2016;22(4):948–60 doi 10.1158/1078-0432.CCR-15-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov 2016;6(1):96–107 doi 10.1158/2159-8290.CD-15-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin Cancer Res 2017;23(7):1785–96 doi 10.1158/1078-0432.CCR-16-1131. [DOI] [PubMed] [Google Scholar]
- 21.Mlakar V, Morel E, Mlakar SJ, Ansari M, Gumy-Pause F. A review of the biological and clinical implications of RAS-MAPK pathway alterations in neuroblastoma. J Exp Clin Cancer Res 2021;40(1):189 doi 10.1186/s13046-021-01967-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455(7215):930–5 doi 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol 2013;14(6):472–80 doi 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res 2018;78(24):6866–80 doi 10.1158/0008-5472.CAN-18-1867. [DOI] [PubMed] [Google Scholar]
- 25.Trigg RM, Lee LC, Prokoph N, Jahangiri L, Reynolds CP, Amos Burke GA, et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat Commun 2019;10(1):5428 doi 10.1038/s41467-019-13315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berlak M, Tucker E, Dorel M, Winkler A, McGearey A, Rodriguez-Fos E, et al. Mutations in ALK signaling pathways conferring resistance to ALK inhibitor treatment lead to collateral vulnerabilities in neuroblastoma cells. Mol Cancer 2022;21(1):126 doi 10.1186/s12943-022-01583-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371(26):2488–98 doi 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 2005;353(21):2243–53 doi 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- 29.Maity TK, Venugopalan A, Linnoila I, Cultraro CM, Giannakou A, Nemati R, et al. Loss of MIG6 Accelerates Initiation and Progression of Mutant Epidermal Growth Factor Receptor-Driven Lung Adenocarcinoma. Cancer Discov 2015;5(5):534–49 doi 10.1158/2159-8290.CD-14-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med 2006;12(5):568–73 doi 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 2007;450(7170):741–4 doi 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caren H, Fransson S, Ejeskar K, Kogner P, Martinsson T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. Br J Cancer 2007;97(10):1416–24 doi 10.1038/sj.bjc.6604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, Takata T, Kawai Y, Ishigaki Y, Kajinami K. Chk1-mediated phosphorylation of receptor-associated late transducer at serine 250 increases its stability by stimulating its interaction with 14–3-3. Genes Cells 2013;18(5):369–86 doi 10.1111/gtc.12043. [DOI] [PubMed] [Google Scholar]
- 34.Xu D, Li C. Gene 33/Mig6/ERRFI1, an Adapter Protein with Complex Functions in Cell Biology and Human Diseases. Cells 2021;10(7) doi 10.3390/cells10071574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harenza JL, Diamond MA, Adams RN, Song MM, Davidson HL, Hart LS, et al. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci Data 2017;4:170033 doi 10.1038/sdata.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C, et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A 2010;107(15):6912–7 doi 10.1073/pnas.0914930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Combaret V, Iacono I, Bellini A, Brejon S, Bernard V, Marabelle A, et al. Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Med 2015;4(4):540–50 doi 10.1002/cam4.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kahana-Edwin S, Cain LE, McCowage G, Darmanian A, Wright D, Mullins A, et al. Neuroblastoma Molecular Risk-Stratification of DNA Copy Number and ALK Genotyping via Cell-Free Circulating Tumor DNA Profiling. Cancers (Basel) 2021;13(13) doi 10.3390/cancers13133365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chicard M, Boyault S, Colmet Daage L, Richer W, Gentien D, Pierron G, et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin Cancer Res 2016;22(22):5564–73 doi 10.1158/1078-0432.CCR-16-0500. [DOI] [PubMed] [Google Scholar]
- 40.Combaret V, Audoynaud C, Iacono I, Favrot MC, Schell M, Bergeron C, et al. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res 2002;62(13):3646–8. [PubMed] [Google Scholar]
- 41.Klega K, Imamovic-Tuco A, Ha G, Clapp AN, Meyer S, Ward A, et al. Detection of Somatic Structural Variants Enables Quantification and Characterization of Circulating Tumor DNA in Children With Solid Tumors. JCO Precis Oncol 2018;2018 doi 10.1200/PO.17.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Roy N, Van Der Linden M, Menten B, Dheedene A, Vandeputte C, Van Dorpe J, et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin Cancer Res 2017;23(20):6305–14 doi 10.1158/1078-0432.CCR-17-0675. [DOI] [PubMed] [Google Scholar]
- 43.Cimmino F, Lasorsa VA, Vetrella S, Iolascon A, Capasso M. A Targeted Gene Panel for Circulating Tumor DNA Sequencing in Neuroblastoma. Front Oncol 2020;10:596191 doi 10.3389/fonc.2020.596191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chicard M, Colmet-Daage L, Clement N, Danzon A, Bohec M, Bernard V, et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin Cancer Res 2018;24(4):939–49 doi 10.1158/1078-0432.CCR-17-1586. [DOI] [PubMed] [Google Scholar]
- 45.Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical Validation of a Hybrid Capture-Based Next-Generation Sequencing Clinical Assay for Genomic Profiling of Cell-Free Circulating Tumor DNA. J Mol Diagn 2018;20(5):686–702 doi 10.1016/j.jmoldx.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012;483(7391):589–93 doi nature10910 [pii] 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 47.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20(12):1472–8 doi 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Gao R, Yao C, Kobayashi M, Liu SZ, Yoder MC, et al. Genotoxic stresses promote clonal expansion of hematopoietic stem cells expressing mutant p53. Leukemia 2018;32(3):850–4 doi 10.1038/leu.2017.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res 2018;24(18):4437–43 doi 10.1158/1078-0432.CCR-18-0143. [DOI] [PubMed] [Google Scholar]
- 50.Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin Cancer Res 2020;26(1):242–55 doi 10.1158/1078-0432.CCR-19-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov 2016;6(10):1118–33 doi 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woodhouse R, Li M, Hughes J, Delfosse D, Skoletsky J, Ma P, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One 2020;15(9):e0237802 doi 10.1371/journal.pone.0237802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med 2019;11(1):32 doi 10.1186/s13073-019-0644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31(11):1023–31 doi 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milbury CA, Creeden J, Yip WK, Smith DL, Pattani V, Maxwell K, et al. Clinical and analytical validation of FoundationOne(R)CDx, a comprehensive genomic profiling assay for solid tumors. PLoS One 2022;17(3):e0264138 doi 10.1371/journal.pone.0264138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 2009;4(8):e6529 doi 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garziera M, Cecchin E, Giorda G, Sorio R, Scalone S, De Mattia E, et al. Clonal Evolution of TP53 c.375+1G>A Mutation in Pre- and Post- Neo-Adjuvant Chemotherapy (NACT) Tumor Samples in High-Grade Serous Ovarian Cancer (HGSOC). Cells 2019;8(10) doi 10.3390/cells8101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andreassen PR, Seo J, Wiek C, Hanenberg H. Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes (Basel) 2021;12(7) doi 10.3390/genes12071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franceschi S, Spugnesi L, Aretini P, Lessi F, Scarpitta R, Galli A, et al. Whole-exome analysis of a Li-Fraumeni family trio with a novel TP53 PRD mutation and anticipation profile. Carcinogenesis 2017;38(9):938–43 doi 10.1093/carcin/bgx069. [DOI] [PubMed] [Google Scholar]
- 60.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8(4):275–83 doi 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ctDNA data generated in this study are available within the article and its supplementary data files. The tumor sequencing and clinical co-variate data collated in this study are not publicly available due to the risk of compromising patient privacy but are included in the article and supplementary data files in a coded and aggregate manner. Any additional data are available upon reasonable request from the corresponding author.