

Abstract
Prostate cancer (PCa) is one of the most heritable human cancers. Genome-wide association studies (GWAS) have identified at least 185 PCa germline risk alleles, most non-coding. We used integrative three-dimensional (3D) spatial genomics to identify the chromatin interaction targets of 45 PCa risk alleles, 31 of which were associated with transcriptional regulation of target genes in 565 localized prostate tumors. To supplement these 31, we verified transcriptional targets for 56 additional risk alleles using linear proximity and linkage disequilibrium (LD) analysis in localized prostate tumors. Some individual risk alleles influenced multiple target genes; others specifically influenced only distal genes while leaving proximal ones unaffected. Several risk alleles exhibited wide-spread germline-somatic interactions in transcriptional regulation, having different effects in tumors with loss of PTEN or RB1 relative to those without. These data clarify functional PCa risk alleles in large linkage blocks and outline a strategy to model multi-dimensional transcriptional regulation.
Introduction
Prostate cancer (PCa) remains a broad health concern because of advancing global population age. It has a heritability of 57%, well above the 33% for all cancers (1). Both rare variants in DNA damage repair genes like BRCA2 and common variants contribute to PCa heritability (2,3). Genome-wide association studies (GWAS) have identified at least 185 common PCa germline risk alleles (4,5). Almost all of these reside in non-coding intergenic (75/185) or intronic regions (99/185).
The mechanisms by which these common non-coding variants influence PCa risk are poorly understood. They are enriched in prostate lineage-specific enhancers and promoters (6–8). Many are thought to contribute to the transcriptional reprogramming of the prostate, akin to the recurrent somatic ERG gene fusions seen in PCa (9). PCa risk alleles have been associated with transcriptional regulation through multiple mechanisms, including DNA methylation, transcription factor binding, and enhancer/promoter activity (5,7,10–18). For example, the PCa risk allele rs684232 and the ERG transcription factor regulate the expression of three neighboring genes in a combinatorial manner in multiple patient cohorts (7,14). Similarly, the PCa risk SNP rs11672691 influences enhancer activity and up-regulates transcription of PCAT19 and CEACAM21 (12,13). Non-coding SNPs influence tumor-specific DNA methylation in the human prostate (19,20). Taken together, these studies suggest that unraveling the genomic and regulatory features of risk alleles can provide novel insights into the biology of PCa.
Identification of the transcriptional targets of risk alleles has been difficult for myriad reasons. First, in many cases it is unclear which of multiple alleles in linkage-disequilibrium (LD) is functional. Second, functional interrogation of risk alleles is complicated by their relatively small effect-sizes and which can compound over decades to subtly influence tumorigenesis (2). Third, individual risk alleles may regulate multiple genes via chromatin interactions, leading to effects in genes distal in the linear genome. Fourth, germline risk alleles occur in the context of an evolving somatic genome and transcriptome, leading to joint germline-somatic influences in regulation of target gene transcription.
To help resolve these problems, we quantified the two-dimensional (2D) and three-dimensional (3D) spatial structure of the PCa genome, and integrated them with germline, somatic and transcriptome data across 565 localized PCa patients. First, we defined the transcriptional targets and chromatin features of PCa risk alleles, including those in LD with tag SNPs. Second, we integrated tumor ChIP-Seq, whole-genome sequencing (WGS), transcriptome and proteome data from PCa patients to quantify germline-somatic interplay in transcriptional regulation between germline risk alleles and somatic mutations. Finally, we demonstrated how PCa risk allele targets divergently influence the major cell lineages of the prostate gland. These studies present a multi-dimensional view of transcriptional dysregulation in PCa.
Results
cis-regulatory targets of germline PCa risk alleles
We examined the DNA sequence and epigenetic features of 185 PCa germline risk alleles (4,17,21) identified by GWAS (Figures 1A and 1B). About one third (63/185) were located outside promoters and gene bodies, while only ~6% (11/185) altered protein coding sequences—implying mechanisms beyond primary protein structure changes (Figure 1C; Table S1). We hypothesized that many PCa risk alleles influence transcription via long-range chromatin interactions. To test this, we focused on the androgen receptor (AR) and FOXA1—master transcription factors in the prostate that are expressed in the LNCaP and VCaP luminal PCa cell lines. We used ChIP-Seq and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) data to evaluate the enrichment of PCa risk loci in various chromatin features, including AR or FOXA1 transcription factor binding sites, insulated neighborhood boundaries marked by the cohesin component RAD21, active enhancers marked by H3K27ac, and RNA Polymerase II (RNA Pol II) binding sites. Enrichment analysis was performed using genome shuffling, controlling for peak number and width (see Supplementary Methods).
Figure 1. Genomic features and cis-regulatory targets of germline PCa risk alleles.
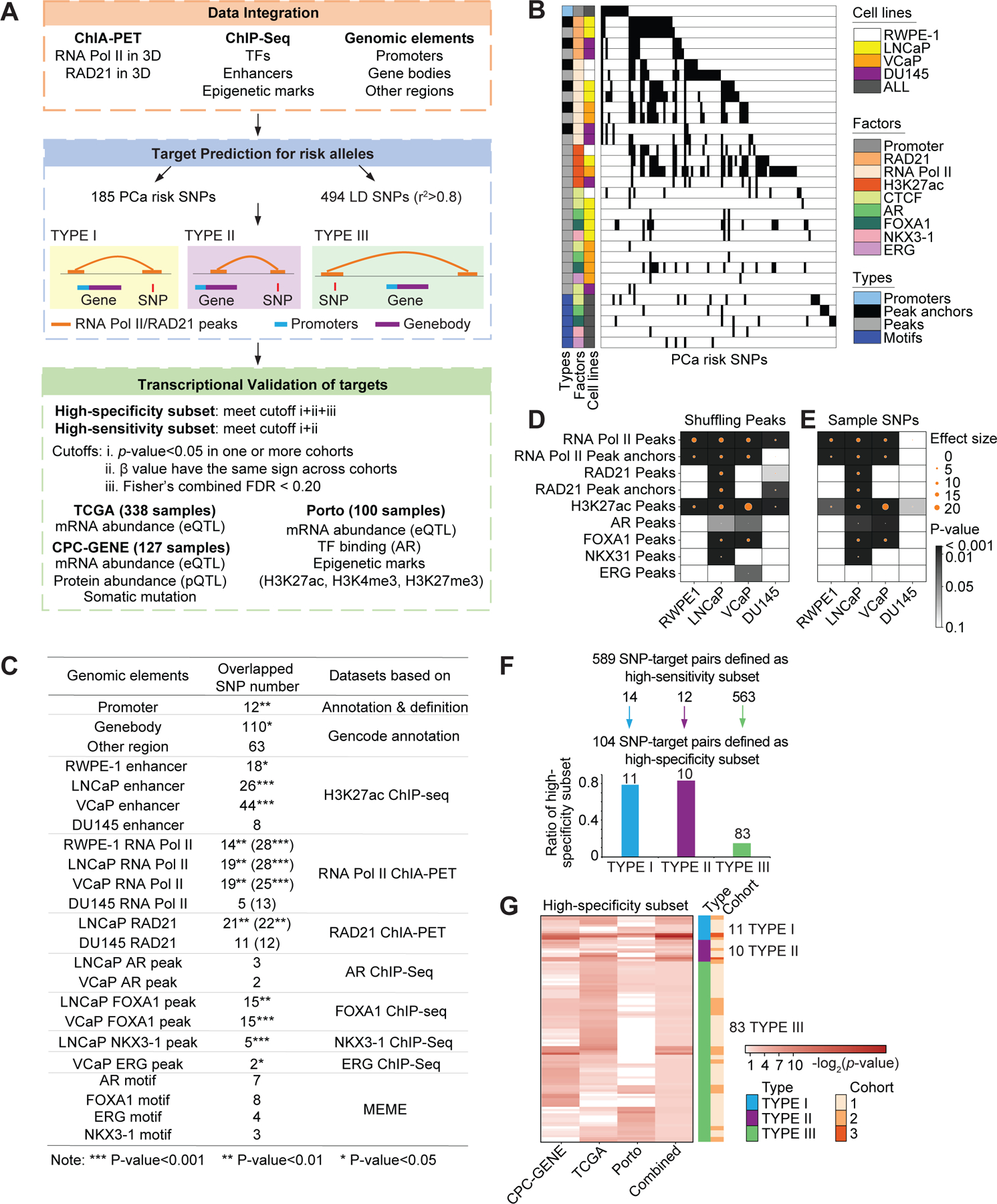
Schematic representation of the overall study design. The 185 PCa risk SNPs were identified from published papers, and 494 LD SNPs were filtered by LD score calculation (r2>0.8). The framework of this study included integrative analysis of multiple omics data, prediction of chromatin interaction targets by ChIA-PET data analysis, and validation of target gene transcription in three clinical cohorts. ChIA-PET data for RNA Pol II and RAD21, and ChIP-Seq for AR, FOXA1, H3K27ac from four cell lines (RWPE-1, LNCaP, VCaP, DU145) were analyzed in this study. eQTL analysis for targets were performed in TCGA, CPC-GENE and Porto cohorts, and chromatin features around the SNP and target gene regions were validated based on ChIP-seq for AR, H3K27ac, H3K4me3 and H3K27me3 in patient tumors from the Porto cohort. (B) Overlap between PCa risk SNPs and genomic features. In the central heatmap, each column represents a SNP, and each row describes a genomic feature. Black color indicates overlap between SNP and genomic feature, while white color indicates no overlap. The individual genomic features are described by the three annotations (types, factors and cell lines). For types, sequences denote the manually defined genomic elements (promoter and motif sequence). Peaks represent antibody bound regions, and anchor peaks represent the sub-set of peaks that interact with other peaks in ChIA-PET analysis. The RWPE-1, LNCaP, VCaP and DU145 cell lines are presented in different colors. (C) Summary table of overlap between PCa risk SNPs and different genomic elements. For the overlap between risk SNPs and ChIA-PET data, the number outside the parentheses is the overlap number with interaction anchor peaks while number in parentheses is the overlap number with all the peaks. P-values were generated by Pearson’s χ2 test. (D and E) Enrichment analysis of GWAS PCa risk SNPs in peak regions versus random regions (D). Enrichment of PCa risk SNPs in RAD21 and RNA Pol II occupied regions (total peaks, anchor peaks), enhancers (H3K27ac marked peaks), AR, FOXA1, NKX3-1 and ERG occupied regions. The circle size indicates effect size between observed and the expected overlap of PCa risk SNPs and the randomly shuffled peaks from 10,000 simulations, and the color indicates the p-value, which was calculated by empirical test. X represents data not available. Enrichment analysis of PCa risk SNPs versus randomly selected SNPs in peak regions (E). The circle size illustrates the effect size between observed (PCa risk SNPs) and expected (random SNPs) overlap of SNPs and the peaks (from sequencing data) from 10,000 simulations. Color intensity indicates the significant differences between the expected and observed values; and p-values were generated by empirical test. (F) 589 high-sensitivity SNP-target pairs with the same β sign across the three cohorts for the subset of SNP-target pairs which were transcriptionally validated in one or more cohorts. Bar chart: 104 high-specificity SNP-target pairs. These are a subset of the high-sensitivity SNP-targets with Fisher’s combined FDR < 0.2. (G) The heatmap represents 104 high-specificity SNP-target pairs. The color intensity in the first three columns denotes the significance in the cohort, which is measured by p-value for β value from linear regression model. The color intensity in the fourth column indicates the combined FDR. The annotation bars in the right for each row describe the interaction context and number of validated cohorts.
The overlap between PCa risk alleles and both AR and FOXA1 peaks was significantly greater than expected by chance (Figures 1D and S1A). PCa risk alleles preferentially occurred in active chromatin marked by H3K27ac or RNA Pol II occupancy. ChIA-PET analysis inferred the subset of peaks that interacted with other peaks associated with the same protein, termed anchor peaks. The enrichment of PCa risk alleles in chromatin landmarks was more pronounced in the benign RWPE-1 cells, and AR positive LNCaP and VCaP PCa cells than in the AR negative DU145 PCa cells. PCa risk alleles were more enriched than random SNPs in 2D and 3D chromatin features in RWPE-1, LNCaP and VCaP cells (Figures 1E and S1B). This enrichment was absent for most chromatin features in DU145 cells. PCa typically begins in a cell that expresses AR; DU145 cells lack AR expression and have undergone a lineage differentiation.
We next sought to identify specific transcriptional targets of PCa risk alleles. Using RNA Pol II and RAD21 ChIA-PET data, we classified targets based on anchor peak location: Type I targets occur when risk loci have 3D chromatin interactions with gene promoters; Type II targets occur when risk loci have 3D chromatin interactions with gene bodies; Type III targets occur when risk loci have 3D chromatin interaction peaks proximal to genes but outside their body or promoter (Figure 1A). A single risk allele can have all of Type I, Type II and Type III targets.
Using this methodology, we identified transcriptional targets of 24.3% (45/185) of the PCa risk alleles. These included 13,384 total SNP-target pairs (107 Type I targets, 111 Type II targets and 13,166 Type III targets). This was significantly more than expected by chance for each target class: 68 additional Type I targets, 72 additional Type II targets and 5,601 additional Type III targets (Figure S1C; Table S2).
To quantify the relationship between risk alleles and transcript abundance, we leveraged three cohorts of localized PCa: the Canadian Prostate Cancer Genome Network (CPC-GENE, 127 samples), The Cancer Genome Atlas (TCGA, 338 samples), and the Porto cohort (100 samples), each with germline genotypes and tumor transcriptomes (22–24). The allele frequencies for risk loci were well correlated across cohorts (Figure S1D). We stratified tumors according to their risk allele genotype to evaluate transcriptional effects of 13,384 SNP-target pairs representing 45 risk alleles. We identified transcriptional changes for 36/45 risk alleles, comprising 589 SNP-target pairs (14 Type I SNP-target pairs, 12 Type II SNP-target pairs, and 563 Type III SNP-target pairs) (Figure 1F; Table S3). These 589 SNP-target pairs met two criteria: (A) p-value < 0.05 in one or more cohorts, and (B) the same direction of effect (β value) in all the three cohorts. The direction of β values (positive and negative β values represent up-regulation and down-regulation, respectively) were more concordant across cohorts for Type I SNP-target pairs, followed by Types II and III. We called these high-sensitivity SNP-target pairs. By applying an FDR <0.2 on the high-sensitivity SNP-target pairs, we retained 272 and 283 SNP-target pairs in the TCGA and CPC-GENE cohorts, respectively, with an overlap of 43 SNP-target pairs (Figures S1E and S1F; Table S4).
We next performed a meta-analysis across the three patient cohorts using Fisher’s method. This prioritized 104 high-specificity SNP-target pairs representing 31 risk alleles (Fisher’s combined FDR < 0.2) (Figures 1F, 1G and S1G; Table S3). Using this criteria, we retained 32 of the 43 shared SNP-target pairs (between TCGA and CPC-GENE) as high-specificity SNP-targets. The presence of an individual risk allele was associated with transcriptional changes in a median of three genes (Figure S1H). Taken together, these data highlight the utility of 3D genome maps in deciphering the transcriptomic targets of PCa risk alleles.
Germline risk alleles shape the transcriptome of primary PCa
Two-dimensional proximity in the linear genome is widely-used to assign transcriptional targets to risk SNPs (25). We therefore applied the same criteria used above for identifying high-specificity targets (Fisher’s combined FDR < 0.2) to the nearest gene in the linear genome for each of the 185 PCa risk alleles. For 48/185, the nearest gene was transcriptionally validated; 13 of these 48 SNP-nearest gene target pairs were rediscovered by our 3D genome approach, 8 of which were intra-genic targets. Intriguingly, 10 out of these 13 SNPs were associated with transcriptional regulation of both proximal and distal genes in primary tumors; only three were associated with transcriptional regulation of proximal genes only (Figure 2A). These results demonstrate that the transcriptional regulation by risk alleles extends beyond its nearest gene, and highlights the complementarity of 2D and 3D approaches.
Figure 2. Transcriptional regulation by the PCa risk alleles in primary tumor cohorts.
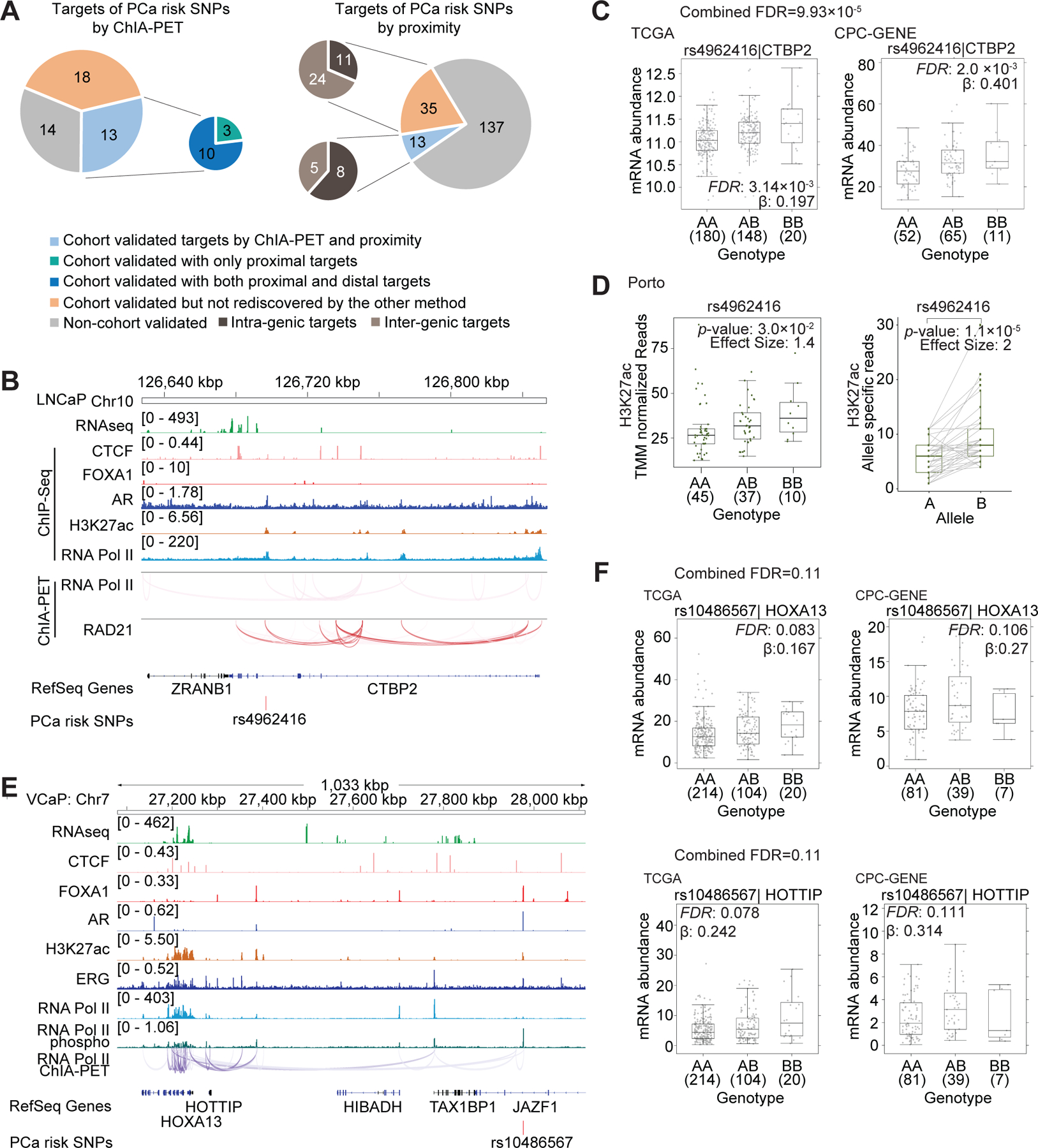
(A) The pie charts represent the number of PCa risk SNPs with high-specificity gene targets determined by ChIA-PET (left pie) and linear proximity (right pie), respectively. The targets defined by linear proximity are further classified as intra-genic (dark-brown) or inter-genic (light-brown) targets based on the location of SNPs. Grey color represents SNPs for which targets were not transcriptionally validated in patient cohorts. Orange color represents PCa risk SNPs for which the transcriptionally validated targets were only predicted by one method but not rediscovered by the other one. Light blue color represents PCa SNPs for which transcriptionally validated targets were defined by both ChIA-PET and linear proximity. For the smaller pie chart: green color represents PCa risk SNPs with only proximal gene targets, while dark blue color represents PCa risk SNPs with both proximal and distal gene targets. (B) Integrated genome view of PCa-risk SNP rs4962416 and its adjacent regions. RNA Pol II and RAD21 ChIA-PET datasets are shown for LNCaP cells. (C) The mRNA abundance of CTBP2 in TCGA and CPC-GENE cohorts. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. mRNA abundance was measured in FPKM. Numbers next to genotypes reflect number of samples in each group. FDR was calculated by “BH” method, and β value are from linear model. (D) Left: H3K27ac binding signal in the PCa SNP rs4962416 locus stratified by genotype in Porto cohort (Mann-Whitney test of recessive model). Y-axis is the number of H3K27ac ChIP-Seq read counts mapped to the SNP rs4962416 locus, which is normalized by TMM method. Right: Allelic imbalance analysis in tumors heterozygous for the risk SNP rs4962416 in Porto cohort. Y-axis denotes the number of read count from allele specific mapping, and the p-value was estimated by paired Wilcoxon test. (E) Integrated genome view of PCa-risk SNP rs10486567 and its adjacent regions. RNA Pol II ChIA-PET, RNA-Seq and CTCF, FOXA1, AR, H3K27ac, ERG, RNA Pol II and phospho–RNA Pol II ChIP-Seq data are shown for VCaP cells. (F) rs10486567 was associated with increased mRNA abundance of HOXA13 and HOTTIP in the CPC-GENE and TCGA cohorts. FDR was calculated by “BH” method, and β value were estimated from linear regression model.
One example of the complex influence of 3D genome architecture on transcriptional regulation by germline PCa risk alleles is the SNP rs4962416, which lies within an intron of CTBP2 (26). The locus harboring the risk SNP rs4962416 exhibited RAD21 and RNA Pol II associated chromatin interactions in LNCaP cells, along with AR occupancy and H3K27ac enrichment (Figure 2B). The presence of the risk allele was associated with increased CTBP2 transcript abundance in primary patient tumors (FDR = 9.93 x 10−5; Figure 2C). The presence of the risk allele was associated with increased H3K27ac (Figure 2D, left). In 37 tumors heterozygous for rs4962416, the risk allele was more associated with H3K27ac than the wildtype allele (Figure 2D, right). This allelic imbalance in an enhancer mark suggests rs4962416 may facilitate enhancer activation in tumors. Consistent with a tumor-promoting role of CTBP2 via modulation of AR signaling (27), our results suggest up-regulation of CTBP2 is a potential mechanism linking the risk SNP with PCa etiology.
While rs4962416 resides in the intron of CTBP2, surprisingly, such regulatory relationships are not generalizable in the absence of 3D genome information. For example, the risk SNP rs636291 is located within an intron of the gene PEX14. DFFA and PEX14 are bidirectional genes with their promoters in a head-to-head orientation. From a 3D genome perspective, the locus harboring the SNP rs636291 is bivalent as it has both the H3K27ac active enhancer mark and the CTCF insulator mark. Consistently, the locus exhibited both RNA Pol II and RAD21 associated chromatin interactions with the promoter of the distal gene APITD1 (Figure S2A). The presence of rs636291 was associated with reduced RNA abundance of APITD1 in patient tumors (FDR = 2.05 x 10−3; Figure S2B). However, we did not observe a relationship between rs636291 and RNA abundance of either the host PEX14 gene or its bidirectional partner gene, DFFA.
A second example of this phenomenon of intronic risk alleles not regulating their host genes is rs10486567, located within an intron of JAZF1 (28). This locus showed AR, FOXA1 and ERG co-occupancy, and the H3K27ac active enhancer mark. We observed RNA Pol II associated long-range chromatin interactions between this locus and the distal HOXA13-HOTTIP bidirectional gene cluster (Figure 2E). rs10486567 genotype was associated with increased RNA abundance of both HOXA13 and HOTTIP expression in patient tumors (FDR = 0.11; Figure 2F). However, we did not observe a relationship between s10486567 and RNA abundance of the host gene JAZF1 (FDR = 0.277; Figure S2C). Taken together, these results indicate that transcriptional targets of risk alleles are not necessarily dictated by linear 2D proximity.
Our strategy elucidated transcription control by both intragenic and intergenic risk alleles. As an example of the latter, the risk SNP rs5759167 is located in an intergenic region between BIK and TTLL1. Its locus exhibited RNA Pol II associated chromatin interaction with the promoter of BIK; but not TTLL1 (Figure S2D). Consistent with this 3D genome data, rs5759167 was associated with increased BIK transcript abundance in patient tumors (FDR = 7.66 x 10−4; Figure S2E), but not TTLL1 transcript abundance (FDR = 0.551; Figure S2F). We replicated several additional expression quantitative trait loci (eQTLs) in patient cohorts (29), including rs10845943 with C1QL4 up-regulation (FDR = 1.30 x 10−4; Figures S2G and S2H), and rs12621278 with ITGA6 down-regulation (FDR = 3.27 x 10−4; Figures S2I and S2J). These results indicate that 3D genome information can improve identification of the transcriptional targets of risk alleles.
Germline-somatic interplay in PCa transcriptional regulation
We hypothesized that somatic driver mutations can modulate the transcriptional effects of germline risk alleles, perhaps enhancing the effect of tumor-promoting alleles and diminishing that of tumor-suppressing ones. To test this, we analyzed the transcriptional effects of a subset of 283 high-sensitivity SNP-target pairs which were also significant in the CPC-GENE cohort (FDR < 0.2) by comparing patient tumors with and without specific somatic driver mutations (somatic mutation status is only available in the CPC-GENE cohort). Because prostate cancer is a C-class disease primarily driven by structural variants (30), we focused on four common driver structural variations that typically occur early and clonally in localized prostate cancer evolution (31): loss of PTEN, loss of RB1, loss of NKX3-1 and ERG gene fusion.
Of these four, PTEN-mutant tumors were systematically associated with larger germline-effects on RNA (Figure 3A). Loss of PTEN potentiated both transcriptional up-regulation and down-regulation by germline risk alleles. Loss of RB1 was also associated with enhanced germline effects on RNA. By contrast, loss of NKX3-1 and ERG gene fusion did not reach statistical significance. RB1 loss potentiated genome-wide transcriptional down-regulation by germline risk alleles, and several individual SNP-target pairs were influenced by NKX3-1 loss or ERG fusion status (Table S5). As an example of these effects, the rs4962416 risk allele was associated with increased CTBP2 transcript abundance in tumors with RB1 loss, but with little or no effect in tumors wild-type for RB1 status (Figure 3B). The same risk allele was associated with increased CTBP2 transcript abundance in ERG negative tumors, but with little or no effect in ERG positive tumors. By contrast, loss of PTEN or NKX3-1 was not associated with changes in the effects of this risk allele on CTBP2 transcript.
Figure 3. The interplay between PCa risk alleles and somatic mutations in transcription control.
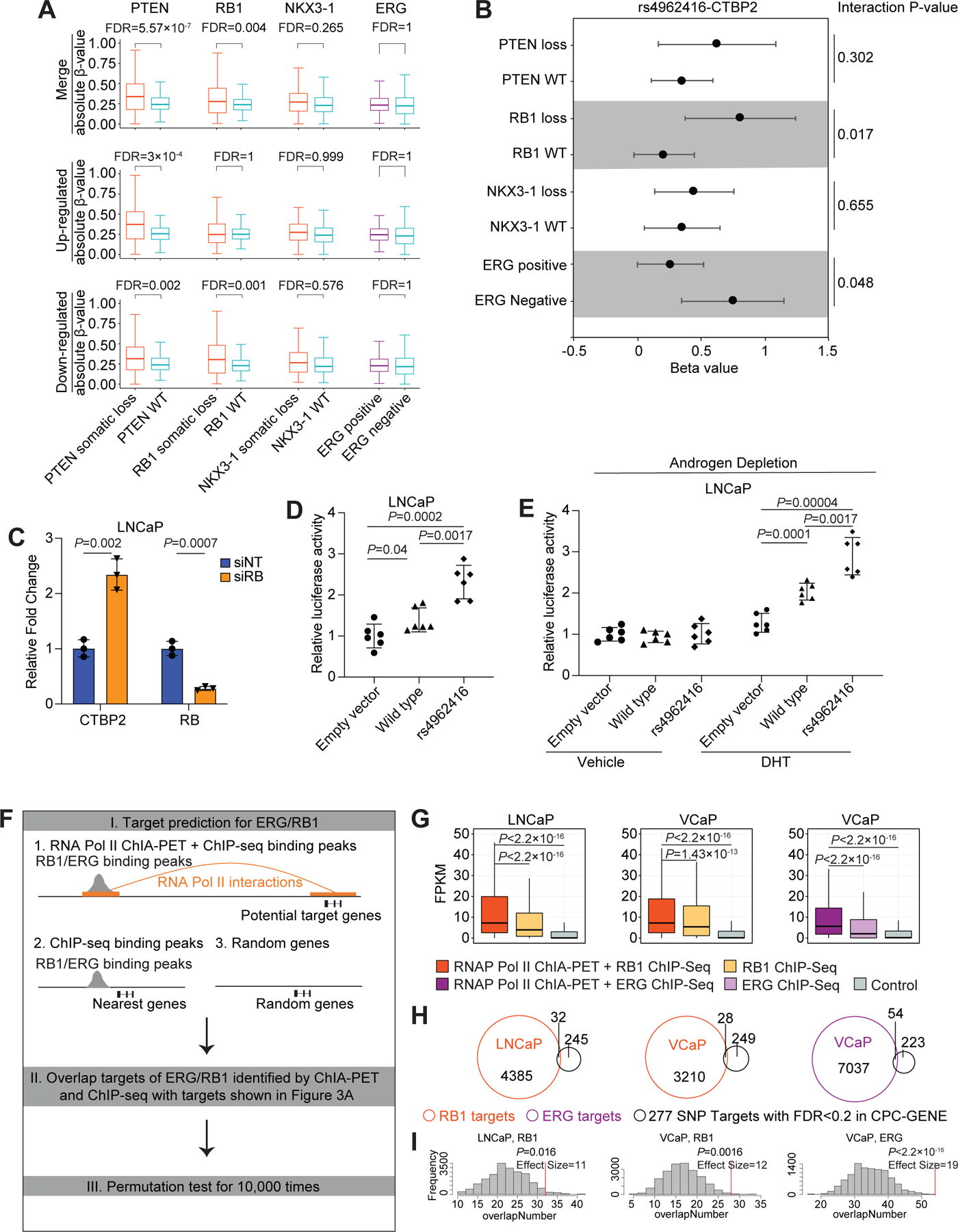
(A) The effect size of transcriptional changes associated with germline risk alleles in tumors stratified by somatic mutations. P-values were generated by Wilcoxon test, and FDR was calculated by “BH” method. (B) Forest plots show eQTL analysis for rs4962416 and the target gene, CTBP2, after patient stratification by PTEN loss, RB1 loss, NKX3-1 loss, and ERG gene-fusion status in the CPC-GENE cohort. Dots and error bars indicate beta value and 95% confidence interval for each group, and p-value from interaction model is shown for each comparison. (C) qPCR validation of RB knockdown and the expression of CTBP2 gene upon treatment of LNCaP cells with RB siRNA. P-value was estimated by 2-tailed Student’s t test. Error bars indicate the SD of 3 technical replicates. (D) Luciferase reporter assays in LNCaP cells. Cells were cotransfected with pSV-Renilla and the luciferase reporter encoding the CTBP2 wild-type or risk allele (rs4962416) and processed 48 hrs after transfection. Firefly Luc/Renilla Luc activity was determined, mean ± SD, n = 6; P-value was estimated by Benjamini-Hochberg Procedure. (E) Luciferase reporter assays in LNCaP cells. Cells were cotransfected with pSV-Renilla and the luciferase reporter encoding the CTBP2 wild-type or risk allele (rs4962416) and processed 48 hrs after transfection. After PBS wash and 10% charcoal stripped FBS media for 2 days, 10 nM of DHT or ethanol was treated for 24 hrs. Firefly Luc/Renilla Luc activity was determined, mean ± SD, n = 6; ; P-value was estimated by Benjamini-Hochberg Procedure. (F) Framework of overlapping targets of ERG/RB1 with targets of SNPs shown in Figure 3A, including three steps: I. Predicting chromatin interaction targets of ERG or RB1 occupied regions by integrating with RNA Pol II ChIA-PET. Here we used the nearest genes to ERG/RB1 binding peaks and randomly sampled genes as control; II. Overlapping targets of ERG/RB1 with targets of SNPs shown in Figure 3A; III. Estimating significance of overlapped targets by conducting permutation test. (G) Expression levels of ERG/RB1 targets identified by ChIA-PET comparing with control gene sets. Gene promoters interacting with RB1 and ERG occupied regions in the RNA Pol II ChIA-PET data are shown in orange and dark purple, respectively. Nearest genes to RB1 and ERG binding peaks are shown in yellow and light purple, respectively. Randomly sampled genes are shown in grey. P was estimated by Wilcoxon test. (H) Scaled venn diagrams show the overlap number between ERG/RB1 targets and SNP targets with FDR < 0.2 in CPC-GENE cohort. (I) The histograms illustrate the results from 10,000 permutation test that assessed the expected overlapped target number, and red line shows the observed overlapped target number. P was estimated by empirical test.
Consistent with the patient tumor data, we observed that siRNA-based knock-down of RB1 resulted in the transcriptional upregulation of CTBP2 in LNCaP cells (Figure 3C). Next, we studied the wild-type (WT) and rs4962416 in transcriptional regulation by conducting dual-luciferase reporter assays in LNCaP cells. In comparison to the vector control, the WT allele increased the reporter luciferase activity, and the magnitude of the effect was further increased in the presence of the risk allele (Figure 3D). Given the observation of AR occupancy in the risk SNP locus (Figure 2B), we tested the effect of androgen signaling on the activity of WT and rs4962416. Depletion of androgens from the media blocked the activity of WT and rs4962416 (Figure 3E). This effect was reversed by the exogenous administration of the AR ligand dihydrotestosterone (DHT) (Figures 3E and S3). Overall, these results indicate that driver somatic mutations influence gene-expression regulation by PCa risk alleles, and thereby contribute to the dynamic transcriptional reprogramming of the prostate epithelium.
We analyzed the chromatin binding profiles of RB1 (in LNCaP and VCaP cells) and ERG (in VCaP cells) (32,33). By overlapping RB1 and ERG binding peaks with RNA Pol II ChIA-PET anchor peaks, we created virtual chromatin contact maps of RB1 and ERG occupancy and traced the target genes. Importantly, the expression of target genes discovered by our new approach was significantly higher than the expression of the nearest neighboring genes as well as control genes in all our comparisons (Figures 3F and 3G). These results suggest that RB1 and ERG regulate transcription via long-range chromatin interactions. Furthermore, the chromatin interaction gene targets (expressed genes; FPKM > 1) of RB1 and ERG significantly overlapped with targets (277 genes corresponding to 283 SNP-target pairs) of PCa risk SNPs (Figures 3H and 3I).
PCa risk SNP rs8102476 and contextual transcriptional regulation
The SNP rs8102476 is an example of simultaneous up-regulation and down-regulation of two adjacent genes by a risk allele. It is located in an intergenic region in chromosome 19q13.2 (34) that exhibited both RNA Pol II associated and RAD21 associated chromatin interactions encompassing the proximal PPP1R14A and distal SPINT2 genes (Figure 4A). These chromatin interactions displayed characteristic CTCF binding and/or the H3K27ac mark in the anchor regions. The presence of the risk allele was associated with down-regulation of PPP1R14A transcript (FDR = 2.85 x 10−8), and up-regulation of SPINT2 transcript (FDR = 0.01) in patient tumors (Figures 4B and 4C). PPP1R14A protein abundance showed a similar effect in a cohort of 63 patients (35) (Figure 4D). Therefore, rs8102476 is both an eQTL and a protein QTL (pQTL) for PPP1R14A. We suggest that changes in the boundaries of the insulated neighborhood and associated changes in enhancer-promoter contacts are likely mechanisms that link the presence of rs8102476 with gene expression changes.
Figure 4. PCa-risk SNP rs8102476 and the transcriptional regulation of PPP1R14A and SPINT2.
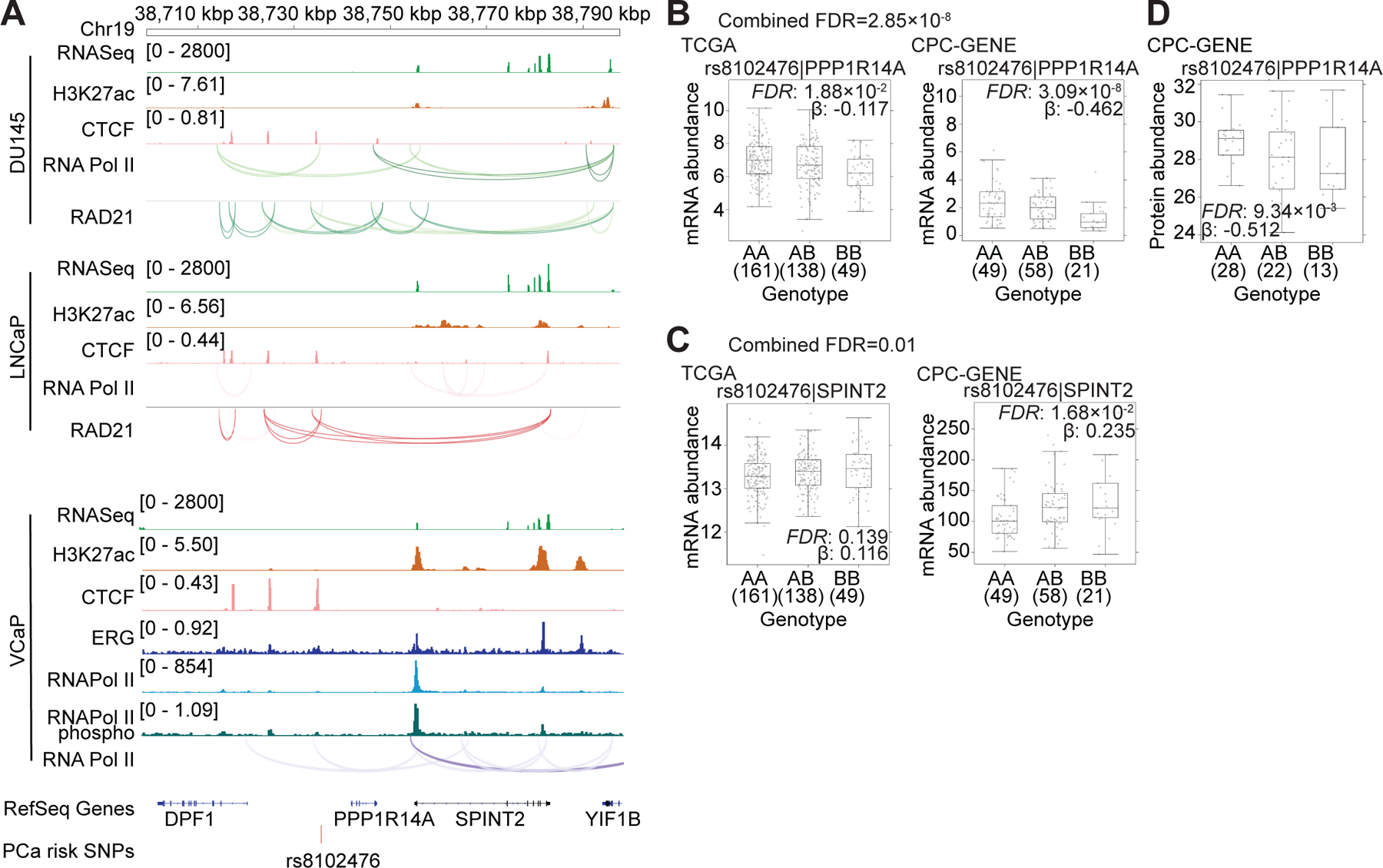
(A) Integrated genome view of PCa-risk SNP rs8102476 and its adjacent regions. ChIA-PET and ChIP-Seq tracks for various factors in LNCaP, VCaP and DU145 cells are shown. (B and C) The mRNA abundance (FPKM) of PPP1R14A and SPINT2 were validated in TCGA and CPC-GENE cohorts. FDR was calculated by “BH” method, and β value were estimated from linear regression model. (D) The protein abundance of PPP1R14A in CPC-GENE cohort.
Cis-regulatory targets of SNPs in LD with the tag risk alleles
LD represents the non-random association of alleles at different loci in the population. We adopted the approach used above to identify the chromatin-interaction targets of alleles in LD to the tag risk SNPs. We identified a total of 494 LD SNPs for the 185 tag SNPs (r2>0.8) (Table S6). The median distance between the tag and LD SNPs was 8,492 bp (Figure S4A). We identified candidate targets for 99 LD SNPs, resulting in 35,836 LD SNP-target pairs (250 Type I SNP-target pairs, 224 Type II SNP-target pairs, and 35,362 Type III SNP-target pairs). We identified 158 additional Type I targets, 133 additional Type II targets and 17,064 additional Type III targets than expected by chance (p-value<0.01) (Figure S4B; Table S7).
We identified transcriptional changes for 82/99 LD SNPs with 3D chromatin linkages, including 70 LD SNPs with high-specificity targets and 12 LD SNPs with high-sensitivity targets (Figure S4C). A total of 247 high-specificity LD SNP-targets were discovered (24 Type I LD SNP-target pairs, 31 Type II LD SNP-target pairs, and 192 Type III LD SNP-target pairs) (Figure 5A; Table S8). The presence of an individual LD SNP was associated with transcriptional changes in a median of two genes (Figure S4D). By applying an FDR <0.2 on the high-sensitivity SNP-target pairs, we retained 691 and 773 SNP-target pairs in the TCGA and CPC-GENE cohorts, respectively, with an overlap of 122 SNP-target pairs (Figure S4E; Table S9). Of the 247 high-specificity LD SNP-target pairs: (A) 45 gene targets were shared between the tag and LD SNPs, and were transcriptionally associated with both, (B) 7 gene targets were common to the tag and LD SNPs, but were transcriptionally associated with LD SNPs only, and (C) 195 gene targets were unique to the LD SNPs only (Figures 5B and S4F). In comparison to the unique targets only identified for LD SNPs, a greater fraction of the common targets was enriched for direct targets (Type I and Type II LD SNP-target pairs) (Figure 5C).
Figure 5. Transcriptional regulation by SNPs in LD with the tag risk alleles.
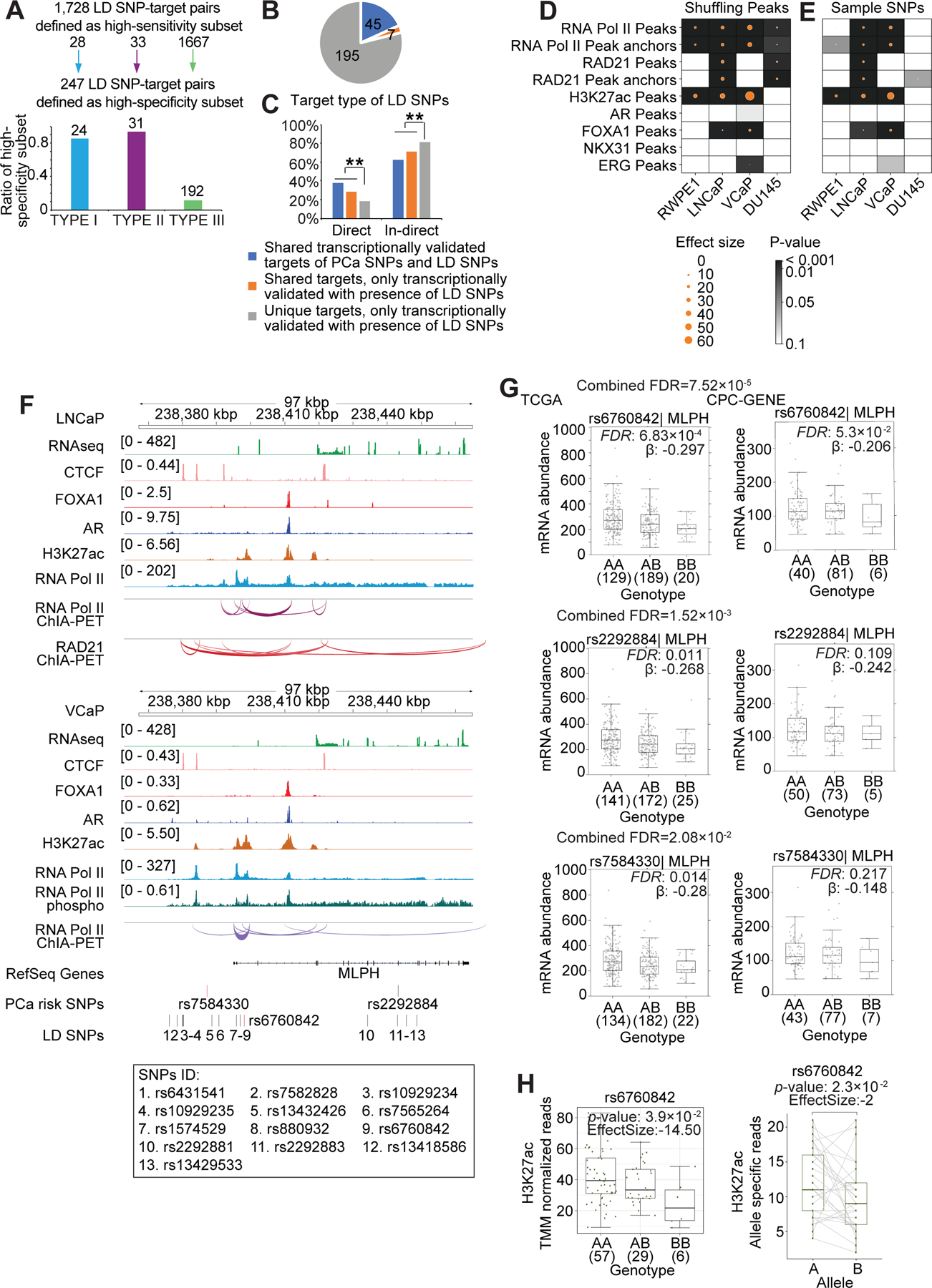
(A) 1,728 high-sensitivity SNP-target pairs with the same β sign across the three cohorts for the subset of SNP-target pairs which were transcriptionally validated in one or more cohorts. Bar chart: 247 high-specificity SNP-target pairs. These are a subset of the high-sensitivity SNP-targets with Fisher’s combined FDR < 0.2. (B) Pie chart shows high-specificity transcriptionally validated LD SNP-target pairs, 45 of which are shared targets of tag and LD SNPs, and transcriptionally validated with the presence of tag SNPs and LD SNPs (blue). 7 SNP-target pairs are shared targets of tag and LD SNPs, but only transcriptionally validated with the presence of LD SNPs (in orange). Others are novel LD SNP-target pairs, unique targets of LD SNPs (grey). (C) Bar chart shows the proportion of direct and in-direct targets for both shared and novel SNP-target pairs (*** P<0.01; Pearson’s χ2 test). (D and E) Enrichment analysis of LD SNPs in peak regions versus random regions (D). Enrichment of LD SNPs in RAD21 and RNA Pol II occupied regions (total peaks, anchor peaks), enhancers (H3K27ac marked peaks), AR and FOXA1 occupied regions. The circle size indicates effect size between observed and the expected overlap of LD SNPs and the randomly shuffled peaks from 10,000 simulations, and the color indicates the p-value, which was calculated by empirical test. Enrichment analysis of LD SNPs versus randomly selected SNPs in peak regions (E). The circle size illustrates the effect size between observed (LD SNPs) and expected (random SNPs) overlap of SNPs and the peaks (from sequencing data) from 10,000 simulations. Color intensity indicates the significant differences between the expected and observed values, and p-values were calculated by empirical test. (F) Integrated genome view of lead SNPs rs7584330, rs2292884, the LD SNP rs6760842 and the adjacent regions. ChIA-PET and ChIP-Seq tracks for various factors in LNCaP and VCaP cells are shown. Additional LD SNP IDs are listed below the tag SNP track. (G) The mRNA abundance of MLPH in TCGA and CPC-GENE cohorts stratified by genotype for tag risk SNPs rs7584330, rs2292884 and the LD SNP rs6760842. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. mRNA abundance was measured in FPKM. Numbers next to genotypes reflect number of samples in each group. FDR was calculated by “BH” method, and β value were estimated from linear regression model. (H) Left: H3K27ac binding signal in the LD SNP rs6760842 locus stratified by genotype in Porto cohort (Mann-Whitney test of recessive model). Y-axis is the number of H3K27ac ChIP-Seq read counts mapped to the LD SNP rs6760842 locus, which is normalized by TMM method. Right: Allelic imbalance analysis in tumors heterozygous for the LD SNP rs6760842 in Porto cohort. Y-axis denotes the number of read count from allele specific mapping, and the p-value was estimated by paired Wilcoxon test.
We identified LD SNPs that were enriched for 2D chromatin features (histone modifications and transcription factor binding) and 3D chromatin interactions (RNA Pol II ChIA-PET and RAD21 ChIA-PET) (Figures 5D, 5E, S4G and S4H). The LD SNPs were enriched in the enhancers of the benign RWPE-1 cells, and AR positive LNCaP and VCaP PCa cells, but not in the AR negative DU145 PCa cells. We studied rs461251, an allele in LD with rs684232, one of the best characterized PCa GWAS risk alleles (7,14,36,37). The two SNPs are located ~200 bp apart. Both the tag SNP rs684232 and the LD SNP rs461251 were associated with loss of enhancer activity (decreased H3K27ac) and down-regulation of VPS53, TLCD3A, and GEMIN4 transcript in patient tumors (Figures S4I–S4M).
Two parallel PCa GWAS reported the tag SNPs rs7584330 and rs2292884 (38,39). Given shared ancestry among study subjects and LD between these two SNPs, the association perhaps reflected the same signal—although rs7584330 gave a stronger signal. Both these lead SNPs had a paucity of 2D chromatin features and 3D chromatin interactions, and lacked clear gene targets. We therefore hypothesized that other LD alleles may be functionally relevant. The SNP rs6760842 is in LD with rs7584330 and its locus exhibited an abundance of chromatin features; it lies in an H3K27ac marked intronic enhancer of MLPH in LNCaP and VCaP cells. This enhancer participated in RNA Pol II associated chromatin interactions with the promoter of MLPH, suggesting MLPH as the target gene (Figure 5F). rs6760842 was associated with down-regulation of MLPH transcript in patient tumors (FDR = 7.52 x 10−5; Figure 5G). rs6760842 was associated with decreased H3K27ac at the locus, indicating a loss of enhancer activity (Figure 5H, left). Tumors heterozygous for rs6760842 exhibited an enrichment of the wild-type allele in the H3K27ac region (Figure 5H, right). Together these data support the idea that rs6760842 is the functional SNP in this GWAS region, influencing PCa etiology through changing MLPH enhancer activity.
3D genomic architecture and transcriptome-wide association study (TWAS) hits
Transcriptome-wide association studies (TWAS) have identified both candidate risk genes at known PCa risk regions, along with novel candidate risk genes outside of them (40,41). To determine the genomic features underlying transcription control of TWAS genes, we considered 222 published SNP-TWAS target pairs (Table S10). Of these, 69 SNP-TWAS target pairs shared the PCa risk SNPs included in this study, and for 27/69 we provide additional 3D genomics support. Six out of these 27 were transcriptionally validated high-specificity SNP-target pairs (Figure 6A).
Figure 6. Transcriptional regulation of TWAS hits.
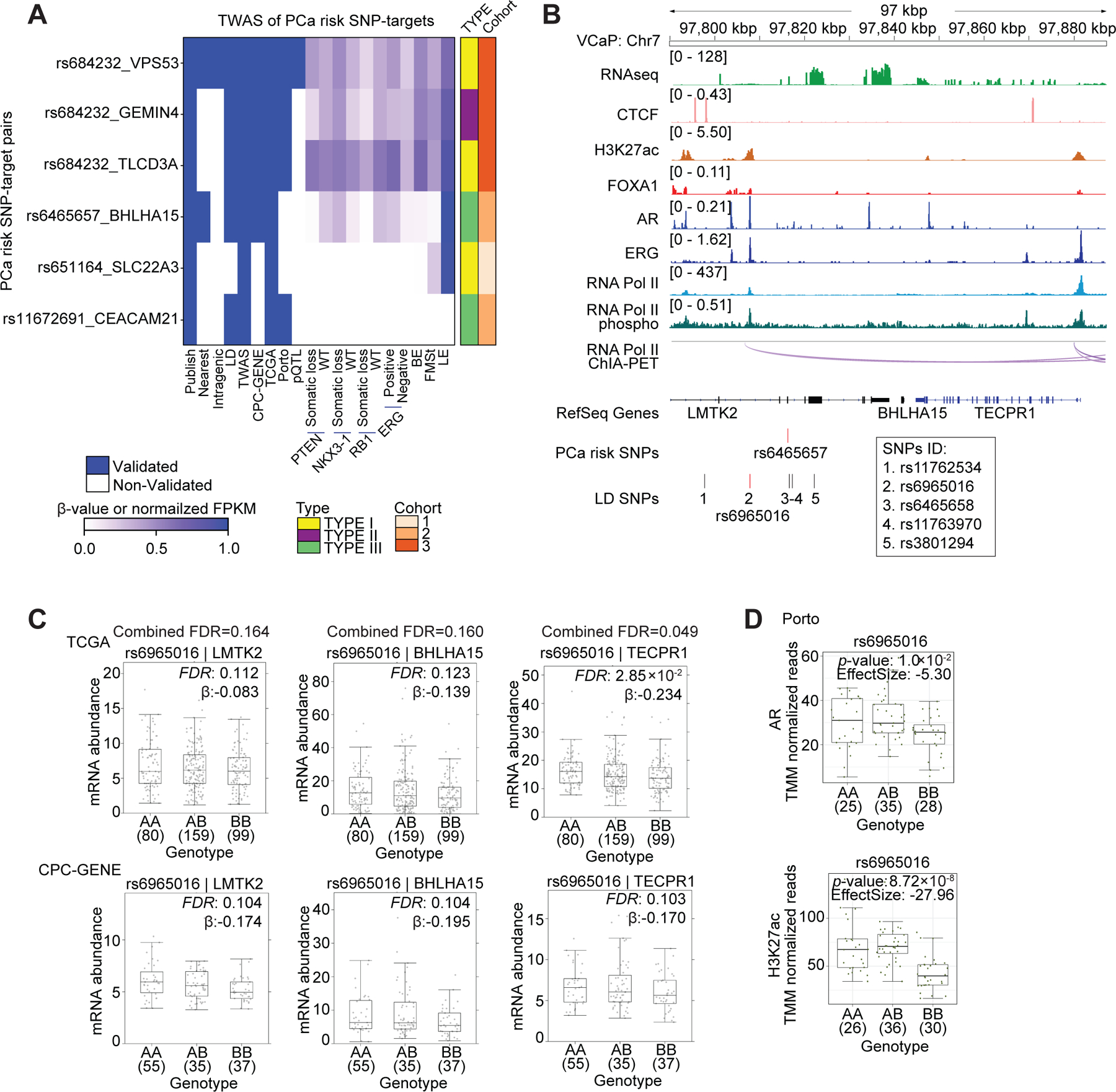
(A) Summary of SNP-TWAS target gene pairs rediscovered by the 3D genomics approach. Each row represents a SNP-target pair, and each column represents a feature. From left to right, the first nine columns describe qualitative features, and the remaining columns describe quantitative features by representing beta value or normalized FPKM in the corresponding dataset. BE: basal epithelia, LE: luminal epithelia, FMSt: fibromuscular stroma. (B) Integrated genome view of the tag risk SNP rs6465657, the LD SNP rs6965016 and the adjacent regions in VCaP cells. RNA Pol II ChIA-PET and ChIP-Seq tracks for various factors in VCaP cells are shown. Additional LD SNPs are listed below the tag SNP track. (C) The mRNA abundance of LMTK2, BHLHA15 and TECPR1 in TCGA and CPC-GENE cohorts stratified by genotype for the LD SNP rs6965016. Boxplot represents median, 0.25 and 0.75 quantiles with whiskers at 1.5x interquartile range. mRNA abundance was measured in FPKM. Numbers next to genotypes reflect number of samples in each group. FDR was calculated by “BH” method, and β value were estimated from linear regression model. (D) Boxplots shows AR and H3K27ac ChIP-Seq signal intensity stratified by genotype for LD SNP rs6965016 in the Porto cohort (Mann-Whitney test of recessive model). Y-axis is the number of ChIP-Seq read counts mapped to LD SNP rs6965016 locus, which is normalized by TMM method.
The 222 published SNP-TWAS target pairs include 86 SNPs from TCGA prostate tumors (40). We extended our analyses pipeline to identify the transcriptional targets of these 86 SNPs, as well as the 371 SNPs in LD with these SNPs. Many TWAS SNPs and LD SNPs were in transcriptionally active regions (Figures S5A and S5B; Table S11). By applying an FDR <0.2, we observed an overlap of 9 TWAS SNP-targets and 80 TWAS LD SNP-targets, respectively, between the CPC-GENE and TCGA cohorts (Figures S5C and S5D; Tables S12 and S13). A total of 33 TWAS SNPs were assigned high-specificity targets by ChIA-PET directly or pairing with LD SNPs followed by ChIA-PET (Figure S5E). The overlap between the 86 TWAS SNPs and 185 PCa risk SNP was 23; seven of these were assigned high-specificity targets (Figure S5F). Eight out of the 12 TWAS SNPs with high-specificity targets identified by ChIA-PET could be rediscovered by TWAS (Figure S5G).
As a representative example, the PCa risk SNP rs6465657 is in an intron of LMTK2 (42) and has been associated with three target genes via TWAS— LMTK2, BHLHA15 and TECPR1 (40). The locus harboring rs6465657 had a paucity of 2D chromatin features and 3D chromatin interactions, and therefore we hypothesized that other LD alleles may be functionally relevant. rs6965016 is in perfect LD with the tag SNP rs6465657—the patient tumors with the AA, AB and BB genotypes were identical for both these SNPs in all cohorts. The locus harboring the LD SNP rs6965016 exhibited an abundance of 2D and 3D genome features, including co-occupancy of AR, FOXA1 and ERG, and the establishment of H3K27ac marked active enhancer (Figure 6B). This locus also exhibited RNA Pol II associated chromatin interactions encompassing the LMTK2, BHLHA15 and TECPR1 genes. rs6965016 was associated with the down-regulation of LMTK2 (FDR = 0.164), BHLHA15 (FDR = 0.160) and TECPR1 (FDR = 0.049) in patient tumors (Figure 6C). rs6965016 was also associated with reduced AR occupancy (Figure 6D, top), and decreased H3K27ac levels—indicating a reduction in enhancer activity—in patient tumors (Figure 6D, bottom). These results suggest that rs6965016 impedes the recruitment of AR, thereby reducing enhancer activity and suppressing the transcription of LMTK2, BHLHA15 and TECPR1.
Cell-lineages associated with the transcriptomic targets of PCa risk alleles
The prostate gland is comprised of diverse cellular lineages (43,44). Although PCa is an epithelial cancer, the microenvironment surrounding the epithelial cells contribute to carcinogenesis (45). To define the expression of the PCa risk allele target genes in the major cellular lineages of the human prostate gland, we examined the mRNA abundance of the transcriptionally validated high-sensitivity target genes of both tag SNPs and LD SNPs in the flow-sorted luminal epithelial, basal epithelial, other epithelial and stromal cells of the normal human prostate gland (43). Unsupervised analysis grouped target genes in to four clusters (Figure 7A; Table S14). Cluster 1 (153 genes) was associated with the stromal cells and was enriched in apoptotic pathways (Figure 7B). Cluster 2 (171 genes) was associated with the luminal epithelial cells and was enriched in membrane tethering processes and transcriptional regulation by RNA Pol II. Cluster 3 (323 genes) was associated with the basal and other epithelial cells, and was predominantly enriched in developmental pathways. The largest of these, Cluster 4 (464 genes) was expressed in all the major cell types of the prostate gland, and was enriched in metabolic processes.
Figure 7. Cell-lineages and molecular pathways associated with the transcriptomic targets of PCa risk alleles.
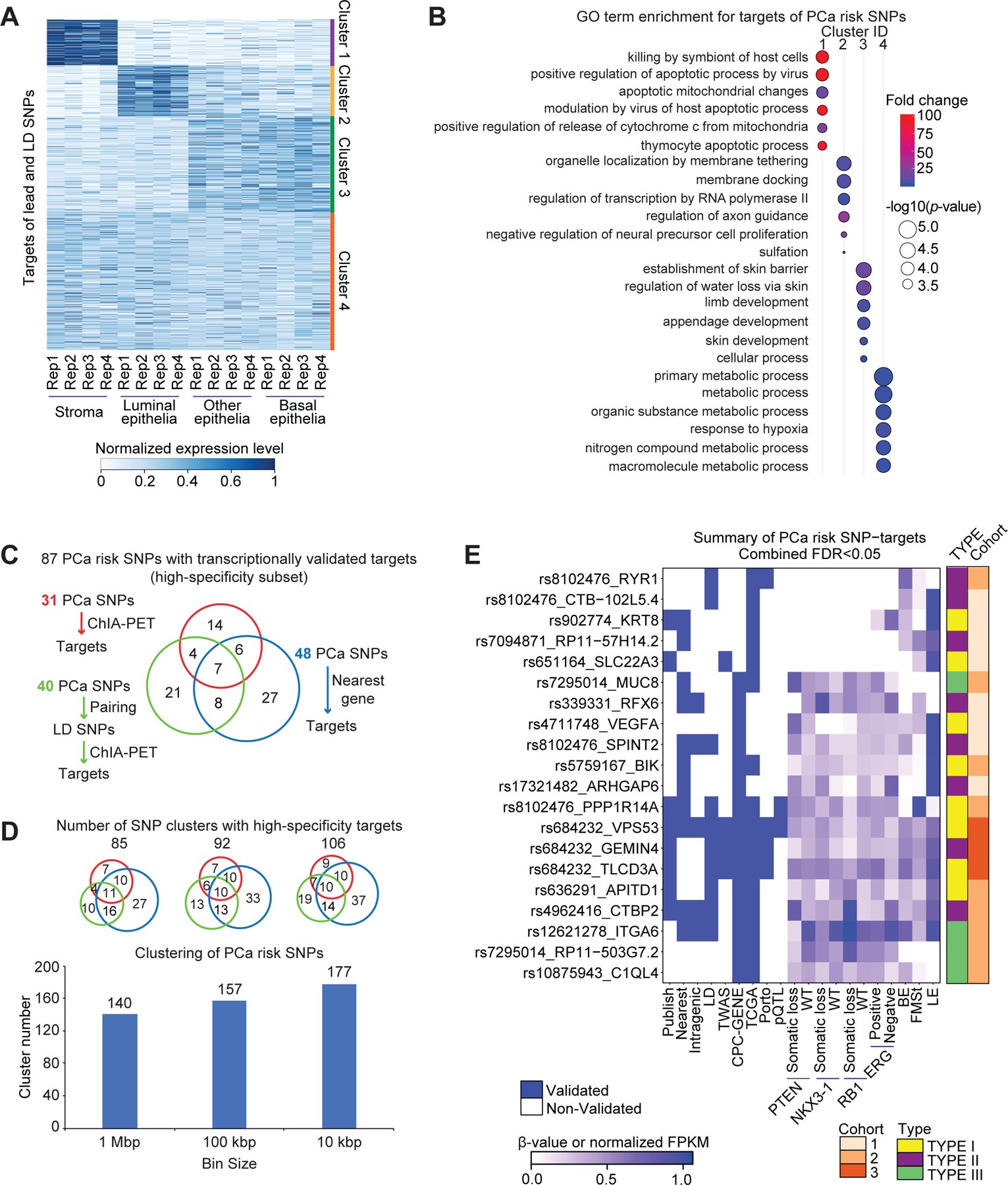
(A) Expression patterns of high-sensitivity target genes of both tag SNPs and LD SNPs in the different cell lineages of the human prostate gland, including stroma, luminal epithelia, basal epithelia, and other epithelia. There are four replicates for each cell type. The expression level is represented by normalized FPKM, and the heatmaps were clustered by Jensen–Shannon divergence (JSD) method. (B) GO term enrichment for targets in each cluster. The p-values transformed by -log10, and fold change are represented by circle size, and color, respectively. (C) Number summary of PCa risk SNPs with transcriptionally validated targets by three strategies. (D) Clustering of PCa risk SNPs by setting different distance cutoffs, including 1 Mbp, 100 kbp and 10 kbp. Scaled Venn diagrams show the number of clusters with transcriptionally validated targets by three strategies. (E) Summary figure for the subset of high-specificity PCa SNP-target pairs (Fisher’s combined FDR < 0.05). Each row represents a SNP-target pair, and each column represents a feature. From left to right, the first nine columns describe qualitative features, and the remaining columns describe quantitative features by representing beta value or normalized FPKM in the corresponding dataset. BE: basal epithelia, LE: luminal epithelia, FMSt: fibromuscular stroma.
We also examined the transcript abundance of a subset of 20 high-specificity target genes (Fisher’s combined FDR < 0.05) in single cell RNA-Seq of the normal human prostate gland (43). We detected transcript abundance for 15 of these 20 genes in the single cell level (Figure S6A; Table S15). Consistent with bulk RNA-Seq analysis, transcriptional targets of PCa risk alleles were expressed in multiple cell lineages of the prostate gland (Figure S6B–S6G). This analysis enabled us to resolve cell-type specific aspects of transcription control by PCa risk alleles. For example, the transcriptional targets of the PCa risk SNP rs8102476—PPP1R14A and SPINT2 genes—were expressed in distinct cellular lineages. SPINT2 expression was restricted to the epithelial cell lineages (luminal, basal, hillock, club and neuroendocrine cells), while PPP1R14A expression was predominantly in smooth muscle cells. By integrating ChIA-PET, single cell RNA-seq, and bulk RNA-Seq data from cell lines, normal prostate, and patient tumors, respectively, we hypothesize that rs8102476 is associated with the down-regulation of PPP1R14A in smooth muscle cells, and the concomitant up-regulation of SPINT2 in epithelial cell lineages (Figures 4 and S6A–S6D).
We associate 87 risk SNPs with high-specificity transcriptionally validated targets in this study: 31 PCa risk SNPs for which targets were defined by ChIA-PET, 48 PCa risk SNPs for which targets were defined by linear proximity, and 40 PCa risk SNPs for which targets were defined by pairing with LD SNPs, followed by ChIA-PET analysis (Figure 7C). We hypothesize that closely spaced risk alleles may perhaps represent the same signal. As described earlier, rs7584330 and rs2292884 are less than 100 kbp apart and are likely to represent the same signal (Figures 5F–5H). Therefore, by setting distance cutoffs of 1 Mbp, 100 kbp and 10 kbp, the 185 risk SNPs analyzed in this study were binned into 140, 157 and 177 clusters, respectively (Figure 7D). Remarkably, regardless of the bin size, we have identified high-specificity SNP-targets in >50% of the clusters (85/140, 1 Mbp clusters; 92/157, 100 kbp clusters; 106/177, 10 kbp clusters) (Figure 7D and Table S16).
We have summarized the molecular and cellular features of the 104 high-specificity SNP-target pairs which were transcriptionally validated in patient cohorts (Table S17). A large majority of high-specificity SNP-target pairs do not involve the nearest genes (91/104). We further filtered the 104 high-specificity SNP-target pairs to identify a subset of 20 SNP-target pairs (Fisher’s combined FDR < 0.05) (Figure 7E; Table S17). Of these 20 SNP-target pairs, (A) 4 SNP-target pairs were previously discovered by TWAS analysis (40), and (B) 9 SNP-target pairs do not involve the nearest genes. The summary heatmaps show how multiple lines of orthogonal evidence converge to illuminate transcription control by PCa germline risk alleles. In conclusion, we have summarized the SNPs that overlapped between published datasets and ours; we have reported the SNP-target pairs that were rediscovered in our dataset; we also report the number of novel SNP-target pairs discovered by our 3D genomics analysis (Table S18–20) (10,14,25,29,40,46–49).
Discussion
It has been long-recognized that germline risk alleles are enriched in gene regulatory elements, yet the specific transcriptional targets of most disease-associated variants have remained elusive. We detect a hitherto unknown pervasive germline-somatic interplay between the PCa risk alleles and somatic loss of PTEN or RB1. These data suggest that the transcriptional effects of PCa risk alleles are not static, but are dynamically tuned by the mutation profile of the cancer. Our discoveries build on prior studies where 3D genome interaction data was used to resolve complex genetic puzzles, and, in so doing, provide a conceptual framework for transcription control by non-coding variants (50,51). We suggest that 3D genome information is critical to deconvolving the relationship between risk SNP location and risk SNP function.
A limitation of our study is the lack of evidence on several PCa risk SNPs. This can be partially addressed by (A) analyses of larger better-powered cohorts, and (B) conducting ChIA-PET analysis of the human prostate and its distinct cell types. Additional mechanisms that are not directly linked to genome architecture or transcription may be in play (some SNPs could be hotspots for DNA breaks or rearrangements). The effect sizes were modest in many cases. This is presumably because of an inherent selection against common strong effect variants. Mendelian variants have high effect size but are present in low frequency in the population. GWAS hits have relatively lower effect size but are present in higher frequency in the population.
While trans-acting proteins, histone modifications and DNA methylation are commonly linked to transcriptional regulation, we suggest that DNA sequence variations can also function as transcriptional activators/repressors by promoting/preventing the recruitment of master transcription factors (tuning enhancer activity) and/or adjusting the boundaries of insulated neighborhoods (tuning enhancer connectivity). We hypothesize that akin to combinatorial binding of transcription factors, such as AR and FOXA1, the combinatorial interaction among cis-regulatory DNA elements can also influence transcription.
Integrating 3D genome data with polygenic risk scores (PRS) can provide insights into genetic pathways associated with an individual’s lifetime PCa risk and can usher the development of intervention strategies to prevent or delay the disease (21). The data from analyses such as these may be used as priors when developing PRS moving forward. In this way, variants with regulatory influences on known prostate cancer pathways may be upweighted while also balancing the fact that not all risk variants may work directly on known pathways and may have biology still to uncover. Studies such as ours may help prioritize variants that fall slightly below typical GWAS significance thresholds but may still inform on risk. Understanding risk SNP function can help distinguish not only cancer risk but provide further insights into the tumor biology and potential vulnerabilities.
Our study has filled-in a key knowledge gap in the field and can stimulate the transition of the GWAS field from association to function. The enhancer-target connectome data emanating from cell line 3D genome studies (representing prostatic lineage) can in principle be used to interpret PCa GWAS signals across all ancestries and will have tremendous value in addressing PCa health disparities. The accuracy of PRS greatly varies across ancestry populations. Mechanistic insights into the function of risk variants along with an increasing understanding of the molecular differences in prostate tumors across diverse populations may help us develop more accurate and generalizable PRS.
Although our study is limited to PCa, our approach can be broadly applied to study other complex diseases with an underlying genetic basis. Integration of GWAS data with population-based LD maps, cell-type-based 3D genome maps and clinical genomics measurements shows significant promise to advance our understanding of the etiology of complex diseases.
Methods
Preprocessing of omics data
The omics data analyzed in this study, include ChIA-PET, ChIP-Seq and RNA-Seq from the benign RWPE-1 cells, the AR positive LNCaP and VCaP PCa cells and the AR negative DU145 PCa cells. ChIA-PET data was processed using ChIAPET2 tool, and all peaks and intra-chromosome interactions were used in the down-stream analysis (52). ChIP-Seq data were processed using bowtie for mapping and MACS2 for peak calling (53). RNA-Seq data were processed by TopHat for mapping (54). All the details about the data preprocessing are described elsewhere (7).
Summary of cohort data
In this study, three cohorts were used for target validation:
TCGA cohort
The TCGA PRAD was used as a validation cohort (22). We included samples with high concordance (>80%) between SNP6 array and whole exome sequencing (WXS) genotypes, and of European descent (338 samples). Genotypes were imputed using the Sanger Imputation Service – pre-phasing using Shapeit2 (55), imputation using PBWT (56) and the Haplotype Reference Consortium (release 1.1) panel (57).
CPC-GENE cohort
We interrogated a previously published cohort of 127 intermediate risk PCa samples that underwent RNA-Seq, described previously (23). Germline SNPs were first identified using GATK (v3.4.0–3.7.0) for each patient individually using HaplotypeCaller followed by VariantRecalibration and ApplyRecalibration. Individual VCFs were merged using bcftools (v.1.8) assuming SNPs not present in an individual VCF were homozygous reference. The minor allele frequency (MAF) of all SNPs within the merged VCF was calculated and filtered to consider only SNPs with MAF > 0.01 (n=10,058,344). Next, all patients were re-genotyped using GATK (v.4.0.2.1) at these sites to produce gVCFs (i.e. with option -ERC GVCF). Individual gVCFs were merged using GenomicsDBImport and joint genotyping was run using GenotypeGVCFs. Finally, SNPs were recalibrated using VariantRecalibrator and ApplyVQSR. Ancestry was determined based on genetic similarity to populations from the 1000 Genomes Project as reported previously (58). Only individuals of European ancestry were included in subsequent analyses (n=127).
Porto cohort
RNA-Seq and ChIP-Seq for AR, H3K27ac, H3K27me3 and H3K4me3 profiles from 100 PCa samples in Porto cohort were integrated in this study (24). RNA-Seq processed data was downloaded from GEO database and raw ChIP-Seq data were downloaded from SRA archive data (GSE120741).
Definition of peak and genomic regions
All RNA Pol II or RAD21 binding peaks and intra-chromosome interactions were defined from the preprocessing results of ChIA-PET. Peaks that interact with other peaks are called anchor peaks or anchor regions. Binding peaks of other TFs and epigenetic marks, e.g., AR, FOXA1, CTCF and H3K27ac were identified by MACS2 with default parameters. The definition of promoters, gene bodies and other regions were based on annotation file Gencode V19. The DNA sequence between the upstream 500 bp and downstream 250 bp from transcription start sites (TSS) were defined as promoters. Gene bodies include both exons and introns. After extracting the coordinates of genes from annotation file, promoter regions were subtracted from whole gene regions, then remaining regions were defined as gene body regions. The regions which were not defined as promoters or gene bodies across the whole genome were defined as other regions. H3K27ac peaks are marks to define enhancer regions, apart from peaks which have overlaps with promoters.
Stratification of patient tumors based on somatic driver mutations
To assess the impact of somatic PTEN loss, RB1 loss, NKX3-1 loss or ERG gene fusions on SNP-target association, the CPC-GENE samples were separated into somatic mutated and matched wild-type groups (30). For each SNP-target pair, eQTL within somatic mutated and corresponding wild-type groups were quantified separately using the same linear regression model described above. Then two p values were calculated for each SNP-target pair, including p value for somatic mutated and matched wild-type groups.
Allelic imbalance analysis of ChIP-Seq data
Allelic imbalance analysis was conducted on ChIP-seq data of H3K27ac and AR from Porto cohort. Firstly, raw reads were mapped to human genome by bwa. Secondly, the mapped bam files were processed by WASP pipeline to remove the allele imbalance mapping biases (59). Thirdly, readergroups were added by AddOrReplaceReadGroups (picard 2.10.3). Finally, allele specific reads were counted by ASEReadCounter (GATK 4.1.4.0). Then, paired wilcox.test was conducted to compare reads counts mapped on reference allele and alternative allele for heterozygous patients on each SNP site.
RNA extraction, cDNA synthesis and quantitative RT-PCR
Total RNA were extracted from LNCaP cells using RNeasy Plus Mini Kit (Qiagen, #74136), and cDNA synthesis was performed using the SuperScript VILO cDNA Synthesis Kit (Invitrogen, #11754050) following the manufacturer’s instructions. Real-time quantitative reverse transcription PCR (qPCR) was carried out on QuantStudio™ 6 Flex Real-Time PCR System (Thermo Fisher) using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher, #A25776) to verify the knockdown efficiency and relative gene expression. The fold change in CTBP2, RB, or PSA mRNA expression levels was calculated by the comparative Ct method, using the formula 2^-(ΔΔCt). The primer sequences for the PCR were RB (forward AGACCCAGAAGCCATTGAAA, reverse CTGGAAAAGGGTCCAGATGA), CTBP2 (forward GTCTGGGACACGATGAACCT, reverse CTTGTTCTTCCTTGGGGTCA), PSA (forward CATCAGGAACAAAAGCGTGA, reverse ATATCGTAGAGCGGGTGTGG), GAPDH (forward TGCACCACCAACTGCTTAGC, reverse GGCATGGACTGTGGTCATGAG).
siRNA transfection
ON-TARGETplus Human RB (#L-003296-02-0005) and negative control small interfering RNAs (siRNA) were purchased from Horizon Discovery. siRNAs were transfected into cells using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher, #13778-150) as described in manufacture’s protocol.
Luciferase reporter assay
The 1 kilobase pair enhancer region containing the wild-type or risk allele rs4962416 was cloned into pGL4 luciferase reporter plasmid (Promega, #E665A) and verified by Sanger sequencing. LNCaP cells were seeded on 12-well plates and transfected with the indicated plasmids. For Dihydrotestosterone (DHT) treatment, 5a-Androstan-17b-OL-3-one was purchased from Sigma Aldrich (#A8380) and dissolved in ethanol. The plated cells were washed with PBS two times, replaced with 10% charcoal stripped FBS media for 2 days, and treated with 10 nM of DHT for 24 h. The Firefly luciferase / Renilla luciferase activities were measured using the Dual-Glo luciferase assay system as described in the manufacturer’s protocol (Promega, #E2940).
Predicting the targets of transcription factors
We integrated ChIP-seq of RB1 and ERG with RNA Pol II ChIA-PET from the same cell line to predict targets of these factors. ChIP-seq peaks were used to define RB1/ERG binding sites, and if the peak was overlapping with one peak anchor from RNA Pol II ChIA-PET, the genes locating in the paired peak anchors would be defined as targets. These genes in the nearest to ChIP-seq binding peaks and randomly selected genes were taken as control groups.
Data Visualization
The track files for multiple factors around different gene loci were visualized by the Integrative Genomics Viewer (IGV) (60). Heatmap was drawn with R package BoutrosLab.plotting.general. Scatterplot and balloon plot were drawn with R package ggpubr. Jensen-Shannon (JS) score was used to measure the expression distance between transcripts in Figure 7A, and then k-means clustering was performed (k=4).
Statistics
Significance of SNP enrichment was estimated by empirical test (Figures 1D and 1E). The significance of mRNA abundance changes was estimated by linear regression model, and p-value and β value were from linear model. The significance of ChIP-Seq binding signal intensity difference was estimated by Mann-Whitney test considering a recessive model. The significance of transcriptional changes after somatic mutation stratification was estimated by Wilcoxon test (Figure 3A). The significance of target position and number of validated cohorts was estimated by χ2 test (Figure 5C). The significance of allele specific binding was estimated by paired Wilcoxon test (Figures 2D and 5H). P-values for qPCR analysis were obtained using two-tailed Student’s t-test. Error bars indicate the SD of 3 technical replicates. P-values for luciferase reporter assay were obtained using Benjamini-Hochberg Procedure. Error bars indicate the SD of 6 technical replicates.
Supplementary Material
Significance.
Many PCa germline risk alleles are enriched in the non-coding regions of the genome and are hypothesized to regulate transcription. We present a 3D genomics framework to unravel risk SNP function and describe widespread germline-somatic interplay in transcription control.
Acknowledgements
We thank James Malter, Diego Castrillon and Ashish Kapoor for insightful comments and discussions. RSM acknowledges funding support from National Cancer Institute (NCI)/NIH grant (R01CA245294), Cancer Prevention and Research Institute of Texas (CPRIT) Individual Investigator Research Award (RP190454), US Department of Defense Prostate Cancer Research Program (PCRP) Impact Award (W81XWH-17-1-0675), and US Department of Defense Breakthrough Award (W81XWH-21-1-0114). KEH was supported by a Vanier Canada Graduate Scholarship. This work was also supported by the NIH/NCI under awards P30CA016042, U01CA214194 and U24CA248265, and by the Prostate Cancer Foundation Special Challenge Award to PCB (Award ID #: 20CHAS01) made possible by the generosity of Mr. Larry Ruvo. The results described here are in part based on data generated by TCGA (http://cancergenome.nih.gov), managed by the NCI and the National Human Genome Research Institute (NHGRI).
Data Availability
The RNA Pol II ChIA-PET data is available in the Gene Expression Omnibus (GEO) under accession number GSE121020, and RAD21 ChIA-PET was downloaded from GEO by accession number GSE127018 and GSE127041. ERG ChIP-Seq data was downloaded from GEO by accession number GSM1328979 and RB1 ChIP-seq data was downloaded from GEO by accession number GSM1974982 and GSM1974981.
References
- 1.Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016;315:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conti DV, Darst BF, Moss LC, Saunders EJ, Sheng X, Chou A, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nature genetics 2021;53:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor RA, Fraser M, Rebello RJ, Boutros PC, Murphy DG, Bristow RG, et al. The influence of BRCA2 mutation on localized prostate cancer. Nat Rev Urol 2019;16:281–90 [DOI] [PubMed] [Google Scholar]
- 4.Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, et al. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol 2014;11:18–31 [DOI] [PubMed] [Google Scholar]
- 5.Farashi S, Kryza T, Clements J, Batra J. Post-GWAS in prostate cancer: from genetic association to biological contribution. Nat Rev Cancer 2019;19:46–59 [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Chng KR, Lingadahalli S, Chen Z, Liu MH, Do HH, et al. An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells. Genome research 2019;29:223–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramanand SG, Chen Y, Yuan J, Daescu K, Lambros MB, Houlahan KE, et al. The landscape of RNA polymerase II-associated chromatin interactions in prostate cancer. The Journal of clinical investigation 2020;130:3987–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nature genetics 2020;52:790–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah YJ, et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nature genetics 2017;49:1336–45 [DOI] [PubMed] [Google Scholar]
- 10.Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nature genetics 2014;46:126–35 [DOI] [PubMed] [Google Scholar]
- 11.Spisak S, Lawrenson K, Fu Y, Csabai I, Cottman RT, Seo JH, et al. CAUSEL: an epigenome- and genome-editing pipeline for establishing function of noncoding GWAS variants. Nat Med 2015;21:1357–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua JT, Ahmed M, Guo H, Zhang Y, Chen S, Soares F, et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 2018;174:564–75 e18 [DOI] [PubMed] [Google Scholar]
- 13.Gao P, Xia JH, Sipeky C, Dong XM, Zhang Q, Yang Y, et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018;174:576–89 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, Yang Y, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nature genetics 2016;48:387–97 [DOI] [PubMed] [Google Scholar]
- 15.Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, et al. Germline mutations in HOXB13 and prostate-cancer risk. The New England journal of medicine 2012;366:141–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Cowper-Sal lari R, Bailey SD, Moore JH, Lupien M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome research 2012;22:1437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo H, Ahmed M, Zhang F, Yao CQ, Li S, Liang Y, et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nature genetics 2016;48:1142–50 [DOI] [PubMed] [Google Scholar]
- 18.Zhang P, Xia JH, Zhu J, Gao P, Tian YJ, Du M, et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat Commun 2018;9:2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houlahan KE, Shiah YJ, Gusev A, Yuan J, Ahmed M, Shetty A, et al. Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med 2019;25:1615–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjostrom M, et al. The DNA methylation landscape of advanced prostate cancer. Nature genetics 2020;52:778–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nature genetics 2018;50:928–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163:1011–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Huang V, Xu X, Livingstone J, Soares F, Jeon J, et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 2019;176:831–43 e22 [DOI] [PubMed] [Google Scholar]
- 24.Stelloo S, Nevedomskaya E, Kim Y, Schuurman K, Valle-Encinas E, Lobo J, et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat Commun 2018;9:4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nature genetics 2014;46:1103–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nature genetics 2008;40:310–5 [DOI] [PubMed] [Google Scholar]
- 27.Takayama K, Suzuki T, Fujimura T, Urano T, Takahashi S, Homma Y, et al. CtBP2 modulates the androgen receptor to promote prostate cancer progression. Cancer research 2014;74:6542–53 [DOI] [PubMed] [Google Scholar]
- 28.Luo Z, Rhie SK, Lay FD, Farnham PJ. A Prostate Cancer Risk Element Functions as a Repressive Loop that Regulates HOXA13. Cell reports 2017;21:1411–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penney KL, Sinnott JA, Tyekucheva S, Gerke T, Shui IM, Kraft P, et al. Association of prostate cancer risk variants with gene expression in normal and tumor tissue. Cancer Epidemiol Biomarkers Prev 2015;24:255–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64 [DOI] [PubMed] [Google Scholar]
- 31.Espiritu SMG, Liu LY, Rubanova Y, Bhandari V, Holgersen EM, Szyca LM, et al. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell 2018;173:1003–13 e15 [DOI] [PubMed] [Google Scholar]
- 32.Gao S, Gao Y, He HH, Han D, Han W, Avery A, et al. Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell reports 2016;17:966–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nature genetics 2009;41:1122–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha A, Huang V, Livingstone J, Wang J, Fox NS, Kurganovs N, et al. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019;35:414–27 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nature genetics 2013;45:385–91, 91e1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larson NB, McDonnell S, French AJ, Fogarty Z, Cheville J, Middha S, et al. Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. American journal of human genetics 2015;96:869–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M, et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nature genetics 2011;43:785–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher FR, Berndt SI, Siddiq A, Jacobs KB, Wang Z, Lindstrom S, et al. Genome-wide association study identifies new prostate cancer susceptibility loci. Human molecular genetics 2011;20:3867–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat Commun 2018;9:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emami NC, Kachuri L, Meyers TJ, Das R, Hoffman JD, Hoffmann TJ, et al. Association of imputed prostate cancer transcriptome with disease risk reveals novel mechanisms. Nat Commun 2019;10:3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harries LW, Perry JR, McCullagh P, Crundwell M. Alterations in LMTK2, MSMB and HNF1B gene expression are associated with the development of prostate cancer. BMC Cancer 2010;10:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry GH, Malewska A, Joseph DB, Malladi VS, Lee J, Torrealba J, et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell reports 2018;25:3530–42 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karthaus WR, Hofree M, Choi D, Linton EL, Turkekul M, Bejnood A, et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science (New York, NY 2020;368:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Bono JS, Guo C, Gurel B, De Marzo AM, Sfanos KS, Mani RS, et al. Prostate carcinogenesis: inflammatory storms. Nature reviews 2020;20:455–69 [DOI] [PubMed] [Google Scholar]
- 46.Grisanzio C, Werner L, Takeda D, Awoyemi BC, Pomerantz MM, Yamada H, et al. Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proceedings of the National Academy of Sciences of the United States of America 2012;109:11252–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q, Stram A, Chen C, Kar S, Gayther S, Pharoah P, et al. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Human molecular genetics 2014;23:5294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thibodeau SN, French AJ, McDonnell SK, Cheville J, Middha S, Tillmans L, et al. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nat Commun 2015;6:8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu X, Hussain WM, Vijai J, Offit K, Rubin MA, Demichelis F, et al. Variants at IRX4 as prostate cancer expression quantitative trait loci. Eur J Hum Genet 2014;22:558–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014;507:371–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. The New England journal of medicine 2015;373:895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li G, Chen Y, Snyder MP, Zhang MQ. ChIA-PET2: a versatile and flexible pipeline for ChIA-PET data analysis. Nucleic acids research 2017;45:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nature methods 2011;9:179–81 [DOI] [PubMed] [Google Scholar]
- 56.Durbin R Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014;30:1266–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics 2016;48:1279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinrich V, Kamphans T, Stange J, Parkhomchuk D, Hecht J, Dickhaus T, et al. Estimating exome genotyping accuracy by comparing to data from large scale sequencing projects. Genome Med 2013;5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Geijn B, McVicker G, Gilad Y, Pritchard JK. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nature methods 2015;12:1061–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology 2011;29:24–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA Pol II ChIA-PET data is available in the Gene Expression Omnibus (GEO) under accession number GSE121020, and RAD21 ChIA-PET was downloaded from GEO by accession number GSE127018 and GSE127041. ERG ChIP-Seq data was downloaded from GEO by accession number GSM1328979 and RB1 ChIP-seq data was downloaded from GEO by accession number GSM1974982 and GSM1974981.