

Abstract
Cyclin D1 (CCND1) is a critical regulator of cell proliferation and its overexpression has been linked to the development and progression of several malignancies. CCND1 overexpression is recognized as a major mechanism of therapy resistance in several cancers; tumors that rely on CCND1 overexpression to evade cancer therapy are extremely sensitive to its ablation. Therefore, targeting CCND1 is a promising strategy for preventing tumor progression and combating therapy resistance in cancer patients. Although CCND1 itself is not a druggable target, it can be targeted indirectly by inhibiting its regulators. CCND1 steady-state levels are tightly regulated by ubiquitin-mediated degradation, and defects in CCND1 ubiquitination are associated with increased CCND1 protein levels in cancer. Here, we uncover a novel function of ubiquitin-specific protease 27X (USP27X), a deubiquitinating enzyme (DUB), in regulating CCND1 degradation in cancer. USP27X binds to and stabilizes CCND1 in a catalytically-dependent manner by negatively regulating its ubiquitination. USP27X expression levels correlate with the levels of CCND1 in several HER2-therapy-resistant breast cancer cell lines, and its ablation leads to a severe reduction of CCND1 protein levels, inhibition of tumor growth, and re-sensitization to targeted therapy. Together, the results presented in our study are the first to expose USP27X as a major CCND1 deubiquitinase and provide a mechanistic explanation for how this DUB fosters tumor growth.
Introduction
The cell cycle is a tightly controlled process, coordinated by the temporospatial expression and interaction of cyclins and cyclin-dependent kinases (CDKs). Aberrations in the expression level and activity of these proteins are directly linked to cancer (1). CCND1 has a central role in the transition from G1 to S phase of the cell cycle. This cyclin forms complexes with and regulates the functions of CDK4 and CDK6. Once formed, the CCND1-CDK4/6 complexes phosphorylate members of the retinoblastoma (RB) family of proteins, most notably Rb1, to inhibit its suppressive function and enable the transition to S phase of the cell cycle (2–4).
Despite its redundant role in the cell cycle in normal cells (5), overexpression of CCND1 has been linked to the development of several malignancies including breast cancer (6–9). The functions of CCND1 are essential for HER2-driven tumorigenesis, as Ccnd1-null mice are resistant to breast cancers induced by Neu and Ras oncogenes (10,11). Furthermore, a recent study utilizing patient data and clinically relevant mouse models identified CCND1 overexpression as a major mechanism for HER2 therapy resistance in breast cancer patients (12). This study also demonstrated that therapy resistant cancer cells become “addicted” to CCND1 expression and its ablation can resensitize resistant tumors to HER2 inhibition (12).
CCND1 expression is regulated in two major ways – by the MAP kinase pathway, which activates CCND1 gene transcription (13), and by the PI3K pathway, which regulates CCND1 protein stability (14). While higher CCND1 levels can also be a result of gene amplification (8), overexpression of CCND1 in cancer often reflects defects in protein degradation (15,16). CCND1 is an unstable protein with a short half-life (~20 minutes) and is cleared via proteasomal degradation in a ubiquitin-dependent manner (17).
Protein ubiquitination is a dynamic process. Deubiquitinating enzymes (DUBs) remove ubiquitin moieties from their substrates, which in turn increases the substrate’s stability. Conversely, inhibiting DUB function results in increased substrate ubiquitination and subsequent degradation. Importantly, it has been previously shown that DUBs are centrally involved in regulating steady-state CCND1 protein levels (18–21). Thus, targeting CCND1 deubiquitination, and therefore its stability in cells, holds promise as an option for treating CCND1-dependent cancers.
Ubiquitin-specific protease 27X (USP27X) is one of the ~100 DUBs encoded by the human genome (22). USP27X is an intronless gene located on the X-chromosome in both humans and mice. We initially identified USP27X as an epigenetic modifier that targets histone 2B for deubiquitination and whose functions are important for tumor cell growth (23). Evidence that USP27X fosters tumor growth has now been presented by several groups, defining USP27X as an essential gene for growth and proliferation in glioblastoma (24), liver cancer (25,26), and breast cancer (27). However, the precise mechanisms behind USP27X regulated tumor cell growth remain largely unknown.
Here, we demonstrate that USP27X interacts with and stabilizes the proto-oncogene CCND1. Ablation of USP27X leads to enhanced CCND1 ubiquitination, a drastic reduction in CCND1 protein levels, and abrogated cell growth in several cancer cell lines, including HER2 therapy resistant breast cancer cells and xenograft tumors. USP27X expression correlates with CCND1 levels in several breast cancer cell lines, agreeing with this notion. Moreover, USP27X is overexpressed in invasive breast carcinoma, and its expression levels negatively correlate with patient survival, specifically in HER2-positive disease. Our findings highlight USP27X as a critical regulator of CCND1 and cancer cell proliferation. As CCND1 is undruggable, targeting USP27X may serve as an alternative clinical strategy in CCND1-dependent cancers.
Materials and Methods
Cell lines and in vitro cell culture:
Breast cancer cell lines HCC1954 (CRL-2338), HCC1569 (CRL-2330), HCC1419 (CRL-2326), UACC893 (CRL-1902), BT-474 (HTB-20), SKBR3 (HTB-30), MDA-MB-468 (HTB-132) and the lung cancer cell line H1975 (CRL-5908) were purchased from ATCC (Manassas, VA). The breast cancer cell line JIMT-1 (ACC-589) was purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). JIMT-1, MDA-MB-468, and HEK293T were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), HCC1954 and BT474 in RPMI supplemented with 10% FBS, HCC1419 and HCC1569 in RPMI supplemented with 10% FBS and 2mM L-Glutamine, SKBR3 in McCoy’s 5A culture medium supplemented with 10% FBS, UACC893 in L-15 Leibovitz culture medium supplemented with 10% FBS, and H1975 in RPMI supplemented with 10% FBS and 2mM Sodium Pyruvate. All culture medium was supplemented with 1% penicillin/streptomycin. All cell lines were maintained at 37°C and 5% CO2. All cell lines were authenticated by short tandem repeat (STR) analysis every 6 months. All experiments were performed with cells cultured less than 10 passages to reduce possible genetic drift. Cell lines were routinely screened for mycoplasma contamination using PCR.
Xenografts
6-week-old SCID (C.B-Igh-1b/IcrTac-Prkdcscid) mice were purchased from an in-house colony maintained by the Roswell Park Comprehensive Cancer Center (RPCCC) Laboratory Animal Shared Resource. 1×106 JIMT-1 cells stably expressing pLKO.tet-on-shNC/shUSP27X-1/shUSP27X-2 were mixed at a 1:1 ratio with Matrigel (Cat. #354230, BD Matrigel TM matrix) and injected into the flanks of mice. Once tumors reached 200mm3, mice were placed on a diet containing 625mg/kg doxycycline to induce shRNA expression. Tumor progression was measured with a caliper and tumor volume was determined by the formula (length × width2) × (π/6). At end point, mice were euthanized and tumors harvested. Data represent mean ± SD. Animal experiments were approved by the RPCCC Institutional Animal Care and Use Committee. All mice were kept under standard conditions and diet.
Expression vectors
Lentiviral shRNA plasmids CCND1-1 (TRCN0000295875) and CCND1-2 (TRCN0000312873) were purchased from Sigma-Aldrich. The lentiviral shRNA vector for shRB1 was obtained from the RPCCC pGIPZ library (Clone ID: V2LHS_139). shRNAs targeting USP27X and ATXN7L3 were previously described (23,28). Inducible shRNA vectors were generated by subcloning the USP27X targeting oligos (from the constitutive vectors described above) into the pLKO-Tet-On vector as described in (29). Retroviral vectors were generated by subcloning USP27X and CCND1 into a pBabe-Puro vector (Addgene) and lentiviral USP27X expressing vectors were generated by subcloning USP27X into a pLenti-Puro vector (Addgene).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 9.0. ANOVA or student T-test was used to determine statistical significance compared to control. A p-value < 0.05 was considered statistically significant. All data are reported as the mean ± SD from three independent experiments.
Supplementary materials and methods:
The following are included in the supplementary materials and methods due to space constraints: chemicals, experimental details for generation of knock down cell lines and cells expressing ectopic USP27X and CCND1, cell proliferation assay, colony formation assay, cell line treatment and viability assay, cycloheximide chase assay, RT-qPCR, immunoblotting, antibodies, immunohistochemistry, and immunoprecipitation.
Results:
USP27X regulates cell proliferation independent of H2B deubiquitination
There is a growing body of evidence supporting the notion that USP27X is required for the proliferation of several cancer cell types in vitro and in vivo (23–25,27). In agreement with this, shRNA-mediated depletion of USP27X in the non-small cell lung cancer (NSCLC) cell line H1975 led to a significant decrease in cell proliferation and colony formation abilities (Fig. 1A, B, C). The mechanisms by which USP27X fosters tumor cell growth, however, are not fully elucidated. Our previous work uncovered USP27X as an epigenetic modifier that deubiquitinates histone 2B (H2Bub1) and regulates gene transcription (23). We therefore first reasoned that the cell proliferation defects seen upon USP27X depletion may stem from compromised H2Bub1 deubiquitination. To test this, we used shRNA to deplete ATXN7L3 (Fig. 1D), an activating cofactor protein that is crucial for USP27X activity toward H2Bub1 (23,30). We monitored proliferation and colony formation over time. To our surprise, these experiments revealed that ATXN7L3 depletion has no impact on H1975 proliferation (Fig. 1E) and does not impact colony formation abilities (Fig. 1F). To exclude the possibility that the observed effect of USP27X depletion on cell proliferation is cell line specific, we monitored the impact of USP27X or ATXN7L3 ablation in two different breast cancer cell lines – JIMT-1 and HCC1945. These experiments confirmed that ablation of USP27X strongly impairs cell growth, whereas ATXN7L3 depletion has no impact (Supplementary Fig. 1). Together, these data demonstrate that USP27X regulates cancer cell proliferation through mechanisms independent of H2B deubiquitination.
Figure 1. USP27X functions in cell proliferation are independent of H2B deubiquitination.
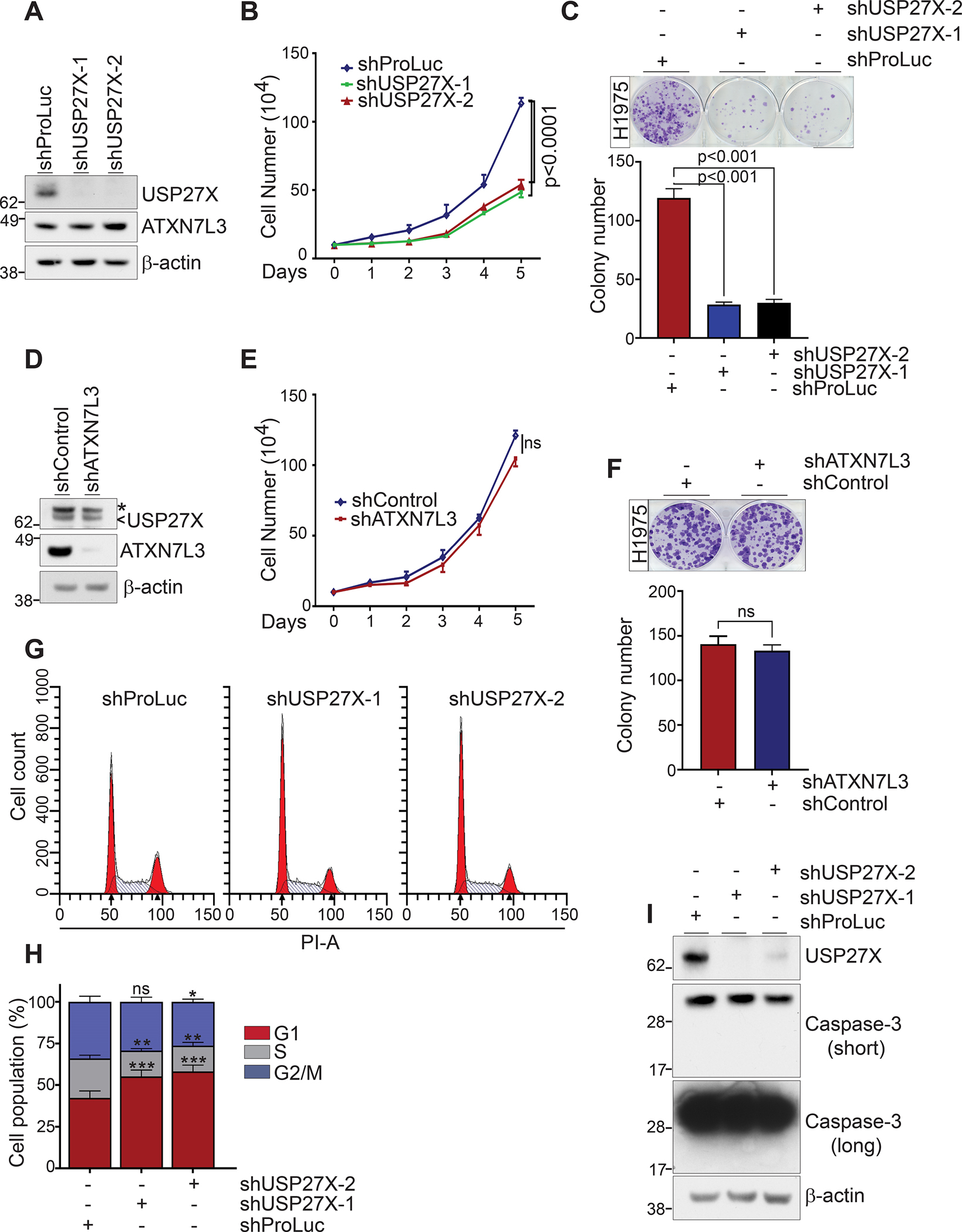
(A) Immunoblots demonstrate efficient silencing of USP27X in the lung cancer cell line H1975. (B) Cell proliferation was monitored by cell counting over the course of 5 days after USP27X depletion in H1975 cells. (C) USP27X depletion impairs the colony formation abilities of H1975 cells. Quantification of C is presented in the bottom panel. (D) Immunoblots demonstrate efficient silencing of ATXN7L3 in H1975 cells. The arrow indicates USP27X while the asterisk indicates a background band. (E) Proliferation of ATXN7L3 depleted H1975 cells over the course of 5 days. (F) ATXN7L3 depletion has no impact on the colony formation abilities of H1975 cells. Quantification of F is presented in the bottom panel. (G) Cell cycle histograms reveal an increased G1 population in USP27X-depleted cells compared to control cells. (H) Quantification of the histograms presented in G. (I) Immunoblots show no caspase-3 activation (cleavage) in USP27X-depleted cells. *p<0.01; **p<0.001; ***p<0.0001. ns = not significant.
Our previous gene expression (RNASeq) and gene ontology analyses in breast cancer cells exposed cyclins and cell cycle regulation as a top pathway affected by USP27X depletion (23). This, in addition to the striking impact USP27X depletion has on proliferation, prompted us to investigate any potential cell cycle defects in these cells. Flow cytometry analyses in USP27X-depleted H1975 cells revealed a substantial increase in cells in G1 phase of the cell cycle (Fig. 1G and H, ~42% vs. 55% and 58%). Interestingly, USP27X ablation did not lead to increased apoptosis, as we did not detect elevated levels of activated (cleaved) caspase 3 in these cells (Fig. 1I). These results suggest that USP27X is required to support cancer cell proliferation as opposed to regulating cell death.
USP27X regulates the steady-state levels of Cyclin D1
To determine how USP27X promotes cancer cell proliferation, we first performed immunoblots comparing the expression of several cell signaling molecules. These experiments revealed that USP27X ablation in H1975 cells leads to a drastic reduction of CCND1 protein levels (Fig. 2A). Notably, cyclin D3 (CCND3), cyclin E1 (CCNE1), CDK4, and CDK6 were not impacted by USP27X depletion; this suggests that the changes seen in CCND1 are specific and not a result of general cell cycle defects upon USP27X depletion. Surprisingly, however, USP27X depletion had no impact on CCND1 transcription as measured by RT-qPCR (Fig. 2B). These experiments were repeated using the JIMT-1 breast cancer cell line. As in the NSCLC cells, USP27X depletion in JIMT-1 cells selectively impacts CCND1 protein levels, but not transcript levels (Fig. 2C, D). It is worth mentioning that the depletion of USP51 (a DUB highly similar to USP27X (23)) had no apparent effect on cell growth or CCND1 levels in these cells (Supplementary Fig. 2).
Figure 2. USP27X regulates the steady-state levels of CCND1.
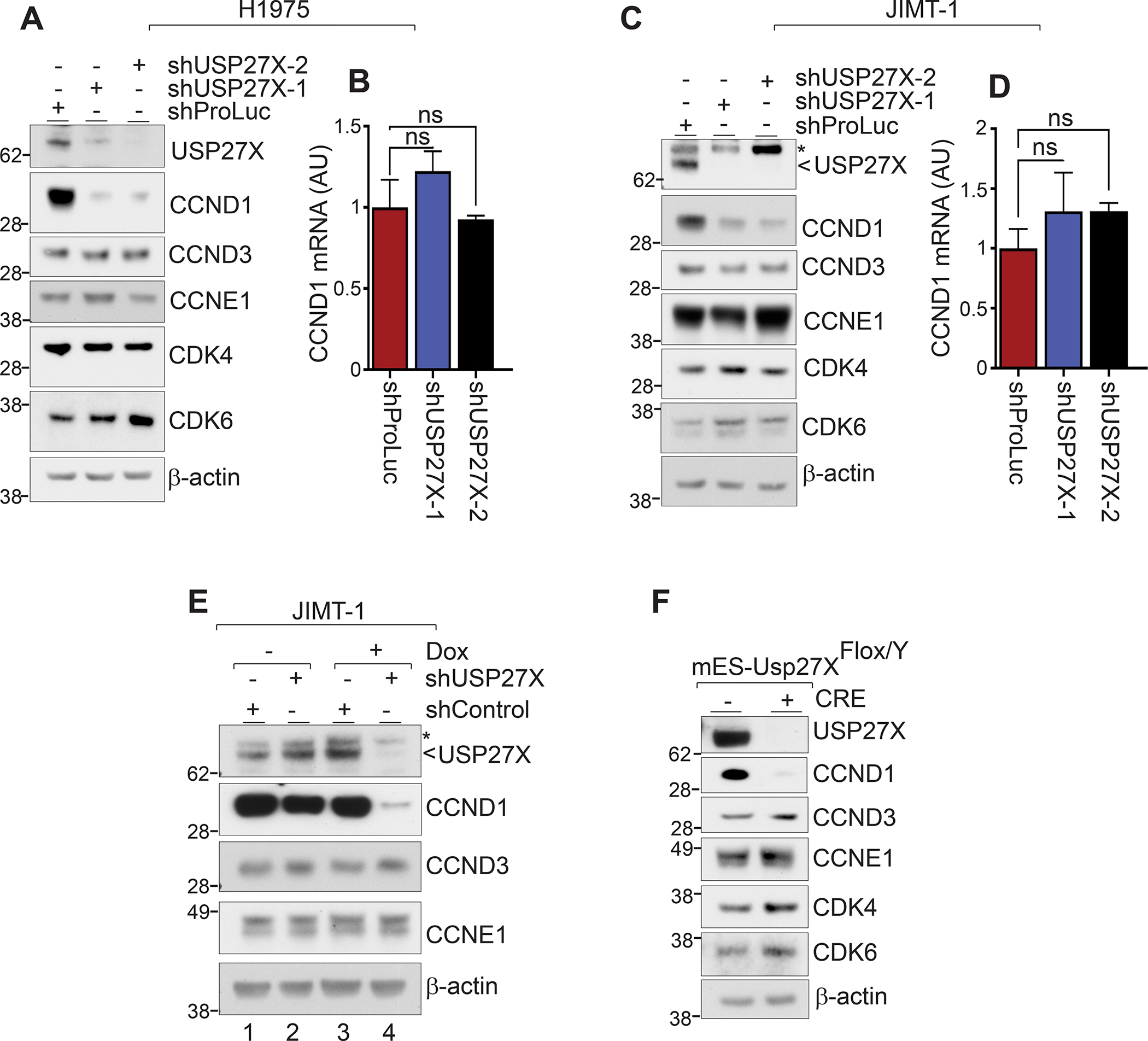
(A) USP2X depletion in H1975 cells leads to a severe reduction in CCND1 levels. No change in CCND3, CCNE1, CDK4, or CDK6 level was detected. CCND2 was not detected in these cells. (B) CCND1 transcript in H1975 USP27X-depleted and control cells measured by RT-qPCR. (C) Immunoblots show reduced CCND1 protein level upon USP27X depletion in JIMT-1 breast cancer cells as well. No changes in CCND3, CCNE1, CDK4, or CDK6 were detected in USP27X-depleted cells. (D) RT-qPCR shows no impact on CCND1 transcription in USP27X-depleted JIMT-1 cells. (E) Doxycycline-induced USP27X depletion in JIMT-1 cells leads to a substantial decrease in CCND1 protein levels. (F) Cre-mediated Usp27X deletion severely impacts the CCND1 protein levels in mESCs.
We next generated doxycycline (Dox) inducible shRNA vectors targeting different parts of USP27X mRNA and stably transduced JIMT-1 cells. We chose this cell line as they express relatively high levels of both USP27X and CCND1. Forty-eight hours after the addition of Dox and therefore shRNA induction, the protein levels of both USP27X and CCND1 were drastically reduced in cells expressing shUSP27X (Fig. 2E, compare lane 4 to lanes 1, 2, and 3). Importantly, we confirmed that USP27X silencing specifically impacts CCND1, as CCND3 and CCNE1 were not affected (CCND2 was not detected in either cell line). Lastly, we utilized a lentiviral vector expressing Cre recombinase to delete Usp27X in mouse embryonic stem cells (mESCs) harboring a ‘floxed’ Usp27X allele (there is only one Usp27X allele in X/Y stem cells). Figure 2F confirms that USP27X loss causes a substantial reduction of CCND1, but not CCND3, CCNE1, or CDK4/6 protein levels in these cells. Taken together, the data described above indicate that USP27X is required for proper maintenance of CCND1 steady-state levels in cells.
USP27X interacts with and stabilizes CCND1
The discrepancy between CCND1 mRNA and protein expression in USP27X-depleted cells was intriguing and led us to investigate how USP27X loss reduces CCND1 protein levels. CCND1 protein degradation is a tightly controlled process mediated by phosphorylation-dependent (14) or phosphorylation-independent (31,32) ubiquitination. We reasoned that USP27X DUB activity may be required for CCND1 deubiquitination, which in turn would prevent CCND1 proteasomal degradation. To test this, we first examined whether inhibition of proteasomal activity would rescue the reduced CCND1 levels in USP27X-depleted cells. Indeed, treatment with the proteasome inhibitor MG132 restored CCND1 levels in USP27X-depleted JIMT-1 cells (Fig. 3A compare lanes 3 and 4 to 7 and 8). Of note, USP27X depletion had no impact on HER2 levels, whose downstream signaling directly regulates CCND1 expression (12). These results were our first indication that USP27X may regulate CCND1 levels through deubiquitination and stabilization of the protein.
Figure 3. USP27X regulates CCND1 ubiquitination and protein degradation in cells.
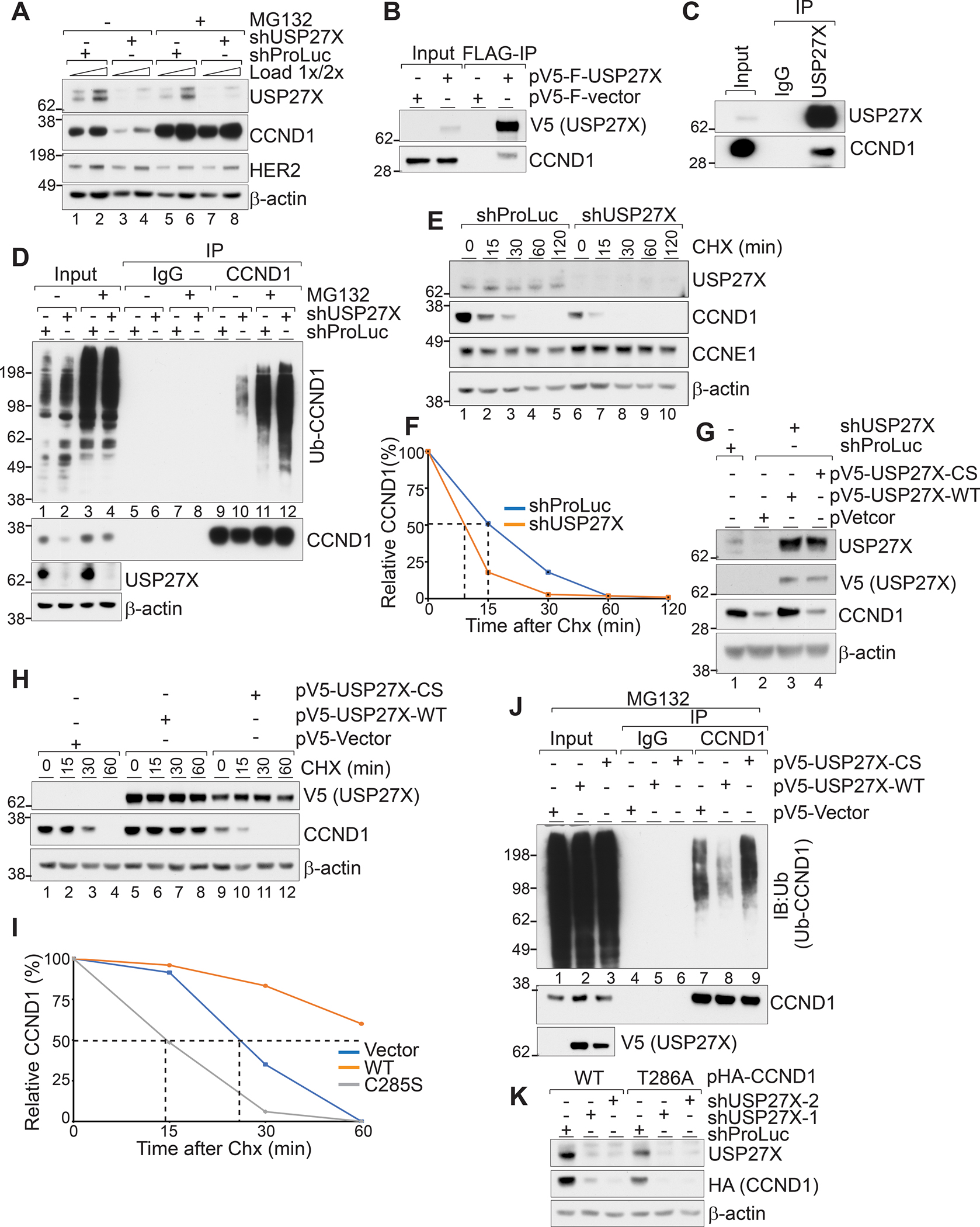
(A) Immunoblots show that proteasomal inhibition by MG132 treatment rescues CCND1 levels in USP27X-depleted cells. (B) Ectopically expressed FLAG and V5-tagged USP27X precipitates endogenous CCND1 form JIMT-1 cells. (C) Endogenous USP27X interacts with endogenous CCND1 in JIMT-1 cells. (D) Ablation of USP27X increases CCND1 ubiquitination. Endogenous CCND1 was precipitated from USP27X-depleted or control JIMT-1 cells. (E) CCND1 protein stability is reduced in USP27X-depleted cells. Immunoblots show enhanced degradation in USP27X-depleted cells after cycloheximide (CHX) treatment. No difference in CCNE1 stability was detected in USP27X-depleted cells under these conditions. (F) Quantification of the changes in E. (G) Expression of WT but not catalytically inactive USP27X rescues CCND1 levels in USP27X-depleted cells. (H) Expression of WT but not catalytically inactive USP27X prolongs the stability of CCND1 in JIMT-1 cells. (I) Quantification of the changes in H. (J) Overexpression of WT but not catalytically inactive USP27X drastically reduces CCND1 ubiquitination levels. Endogenous CCND1 was precipitated from JIMT-1 cells stably expressing USP27X (WT or CS) 4hr after MG132 treatment. (K) Immunoblots show reduced stability of both WT and T286A CCND1 in USP27X-depleted cells.
As USP27X would need to physically interact with CCND1 in order to stabilize it, we next tested whether USP27X binds CCND1. We expressed FLAG and V5-tagged USP27X in JIMT-1 cells, precipitated the tagged protein using anti-FLAG resin, and probed the eluates for endogenous CCND1. Figure 3B confirms that ectopically expressed USP27X interacts with endogenous CCND1. We further established this interaction by precipitating endogenous USP27X from JIMT-1 cell lysates and probing for endogenous CCND1 (Fig. 3C). Next, we sought to test whether USP27X ablation leads to increased CCND1 ubiquitination. To do this, we treated control (expressing shRNA targeting Promega luciferase) and USP27X-depleted cells with MG132 or vehicle. We immunoprecipitated CCND1 from cell lysates and probed the precipitated fractions for ubiquitinated species. As shown in Figure 3D, USP27X depletion led to a substantial increase in CCND1 ubiquitination (Fig. 3D compare lane 9 to 10). This increase in ubiquitination upon USP27X depletion is further highlighted in the presence of MG132, when ubiquitinated CCND1 species are stabilized (Fig. 3D compare lanes 11 and 12). These data demonstrate that ablation of USP27X leads to increased CCND1 ubiquitination in cells.
To further confirm that the stability of CCND1 is compromised upon USP27X loss, we treated control and USP27X-depleted JIMT-1 cells with cycloheximide (CHX) to inhibit protein synthesis and monitored CCND1 decay over time (Fig. 3E, F). As expected, USP27X ablation accelerated the degradation of CCND1 (half-life of ~15 minutes in shProLuc vs. ~8 minutes in shUSP27X), confirming that CCND1 protein stability is compromised upon USP27X loss (Fig. 3E, F). Similar results were obtained in another breast cancer cell line, HCC1945 (Supplementary Fig. 3).
To test whether USP27X catalytic activity is required for stabilization of CCND1, we used shRNA targeting the 3’ UTR to deplete endogenous USP27X and a lentiviral vector to ectopically express either wild type (WT) or catalytically inactive (C285S, hereafter CS) V5-tagged USP27X in JIMT-1 cells. These experiments demonstrated that the expression of WT but not catalytically inactive USP27X can restore the levels of CCND1 in depleted cells (Fig. 3G, compare lanes 2, 3, and 4). The expression of exogenous WT USP27X only increased CCND1 levels slightly above the levels in non-depleted, vector only cells (Fig. 3G, lane1). This is likely due to the fact that JIMT-1 cells have high levels of endogenous USP27X, and the expression of the ectopic protein only moderately increases the steady-state levels of CCND.
Next, we expressed either WT or CS USP27X in JIMT-1 cells and monitored the degradation of endogenous CCND1 over time after CHX treatment. As expected, expression of WT USP27X prolonged CCND1 stability (half-life not detected) while CS USP27X accelerated CCND1 degradation (~18 minutes vs. 15 minutes) (Fig. 3H, compare lanes 3 and 4 to 7, 8,10, 11; Fig 3I). To test if USP27X can deubiquitinate CCND1 in cells, we overexpressed WT or CS USP27X in JIMT-1 cells, treated with MG132 to block proteasomal degradation, and monitored CCND1 ubiquitination after protein immunoprecipitation (Fig. 3J). These experiments revealed that indeed, overexpression of WT but not catalytically inactive USP27X drastically reduces the level of CCND1 ubiquitination (Fig. 3J, compare lanes 7, 8, and 9).
Finally, as CCND1 degradation is mediated through phospho-dependent (triggered by GSK3β mediated phosphorylation on T286) (17) and phospho-independent mechanisms (31,32), we sought to determine which pathway USP27X regulates. We used retroviral vectors to ectopically express WT and phosphorylation-defective (T286A) CCND1 in JIMT-1 cells, used shRNA to deplete USP27X, and probed for levels of the ectopic protein. These experiments revealed that USP27X ablation leads to reduced steady-state levels of both proteins (Fig 3K), indicating that USP27X regulates CCND1 stability independently of its phosphorylation status. Taken together, these data implicate USP27X as a CCND1 DUB. Not only does USP27X bind to CCND1, but USP27X catalytic activity is required for reduction of CCND1 ubiquitination levels and protein stability independent of phosphorylation in cancer cells.
High USP27X expression correlates with higher levels of CCND1 and poor prognosis in HER2 positive (HER2+) breast cancer patients.
Overexpression of CCND1 has been linked to the development and progression of several malignancies including breast cancer, where its role is essential for HER2-driven oncogenesis (10,11). A recent study identified CCND1 overexpression as a major mechanism of HER2-therapy resistance in HER2+ breast cancers (12). Since our previous work identified the importance of UPS27X in breast cancer cell line tumorigenicity (23) and because of the data linking USP27X to CCND1 stability, we next examined online databases to determine if USP27X expression is altered in breast cancer.
We used normalized gene expression datasets from the TCGA breast invasive carcinoma cohort and GTEx breast tissues (generated by RNA-seq) obtained from the TCGA TARGET GTEx study and downloaded from the UCSC Xena Data Portal (33). Identically processed datasets from these databases produce highly reliable comparisons (34). Our analyses showed significantly higher levels of USP27X mRNA in breast invasive carcinoma compared to normal tissue (Fig. 4A). To determine whether USP27X expression levels correlate with patient survival, we used the Kaplan-Meier Plotter. These analyses revealed a statistically significant correlation between high USP27X expression and worse patient survival, specifically in HER2+ (subtype PAM50 HER2+) and lymph node positive disease (Fig. 4B). These data prompted us to check whether levels of USP27X correlate with CCND1. We performed western blot analyses using several breast cancer cell lines, including one triple negative line (MDA-MB-468), two HER2+ and therapy sensitive lines (SKRB-3 and BT-474) and five HER2+ and therapy resistant lines (JIMT-1, UACC893, HCC1954, HCC1569, and HCC1419). As anticipated, CCND1 is highly expressed in HER2 therapy resistant cell lines (Fig 4C). Importantly, this experiment also revealed a strong and significant correlation (r=0.8661, p=0.0054) between USP27X and CCND1 – cells with high CCND1 levels also express more USP27X (Fig. 4C and D). These data suggest that increased expression of USP27X is needed to sustain higher levels of CCND1 in breast cancer cells.
Figure 4. USP27X is highly expressed in breast invasive carcinoma and its expression levels negatively correlate with survival of patients with HER2 and lymph node positive disease.
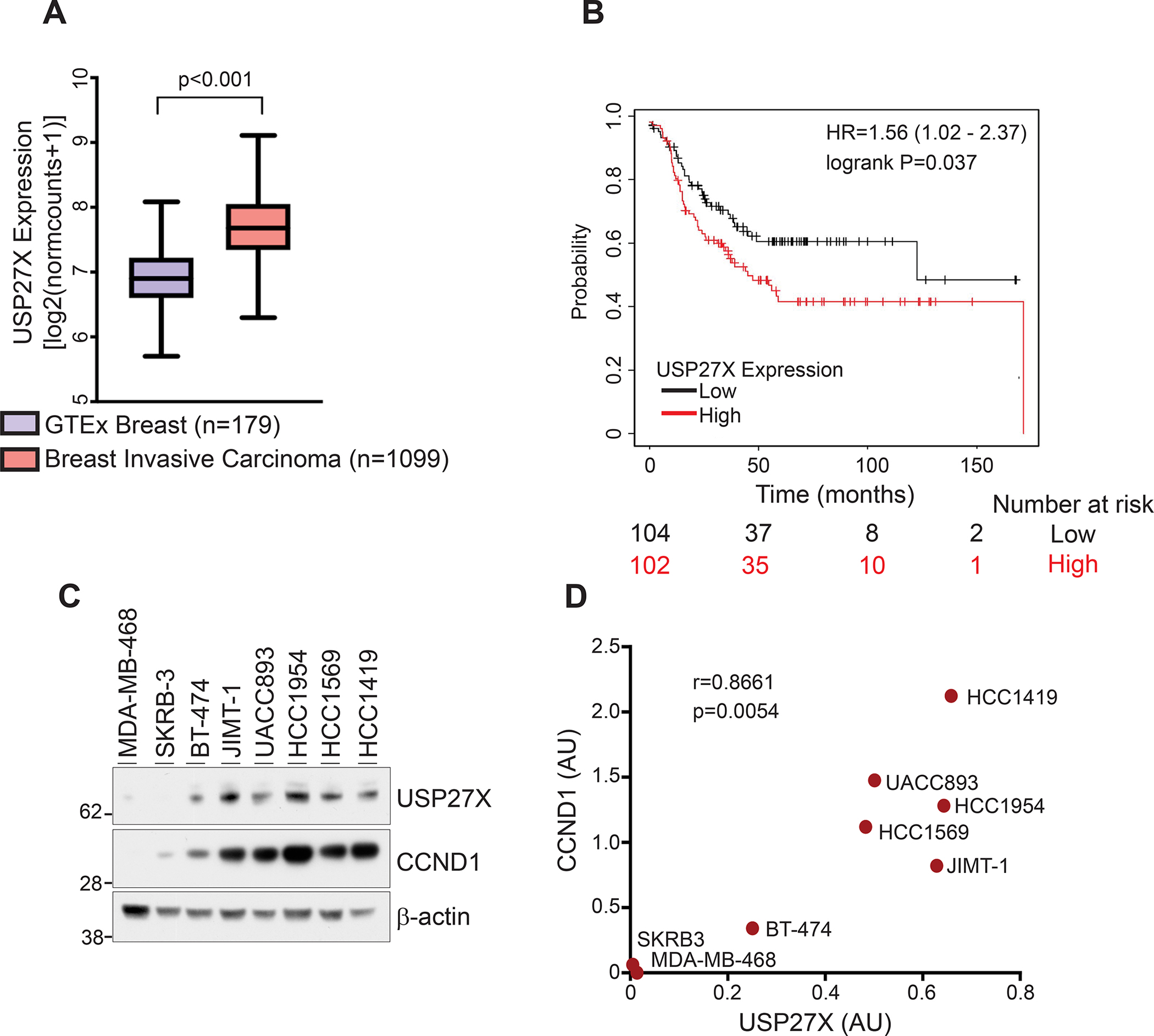
(A) Comparison of normalized gene expression datasets (generated by RNA-seq) demonstrates overexpression of USP27X in invasive breast carcinoma (TCGA breast invasive carcinoma cohort) compared to normal breast tissue (GTEx). (B) Kaplan-Meier analysis of USP27X expression shows a significant correlation between USP27X expression and survival in HER2+ (subtype PAM50 HER2+) and lymph node positive disease in breast cancer patients. (C) USP27X expression correlates with CCND1 expression in several breast cancer cell lines. USP27X and CCND1 are both highly expressed in HER2 therapy resistant breast cancer cells. (D) Pearson correlation analysis of the USP27X and CCND1 expression data presented in C, created using GraphPad Prism 9.0.
USP27X regulates CCND1 steady-state levels and controls cell growth in HER2 therapy-resistant cancer cells lines.
As CCND1 overexpression is a major mechanism by which HER2-therapy resistant tumors diminish their dependence on HER2 signaling (12), we tested whether ablation of CCND1 impaired the growth of HER2 therapy-resistant cells. We used two different shRNAs to deplete CCND1 in the trastuzumab-resistant cell lines JIMT-1 and HCC1954 and monitored cell growth over time. As predicted, CCND1 depletion had a profound effect and almost completely inhibited the growth of both cell lines (Fig. 5A, B), indicating that indeed, HER2 therapy-resistant cells with high expression of CCND1 are extremely sensitive to its ablation. Notably, we obtained similar results when depleting USP27X in these cells (Fig. 5C and D). We speculated these growth defects were due to abrogation of CCND1 in USP27X-depleted cells.
Figure 5. USP27X regulates CCND1 protein levels and cell proliferation in HER2-therapy resistant breast cancer cells.
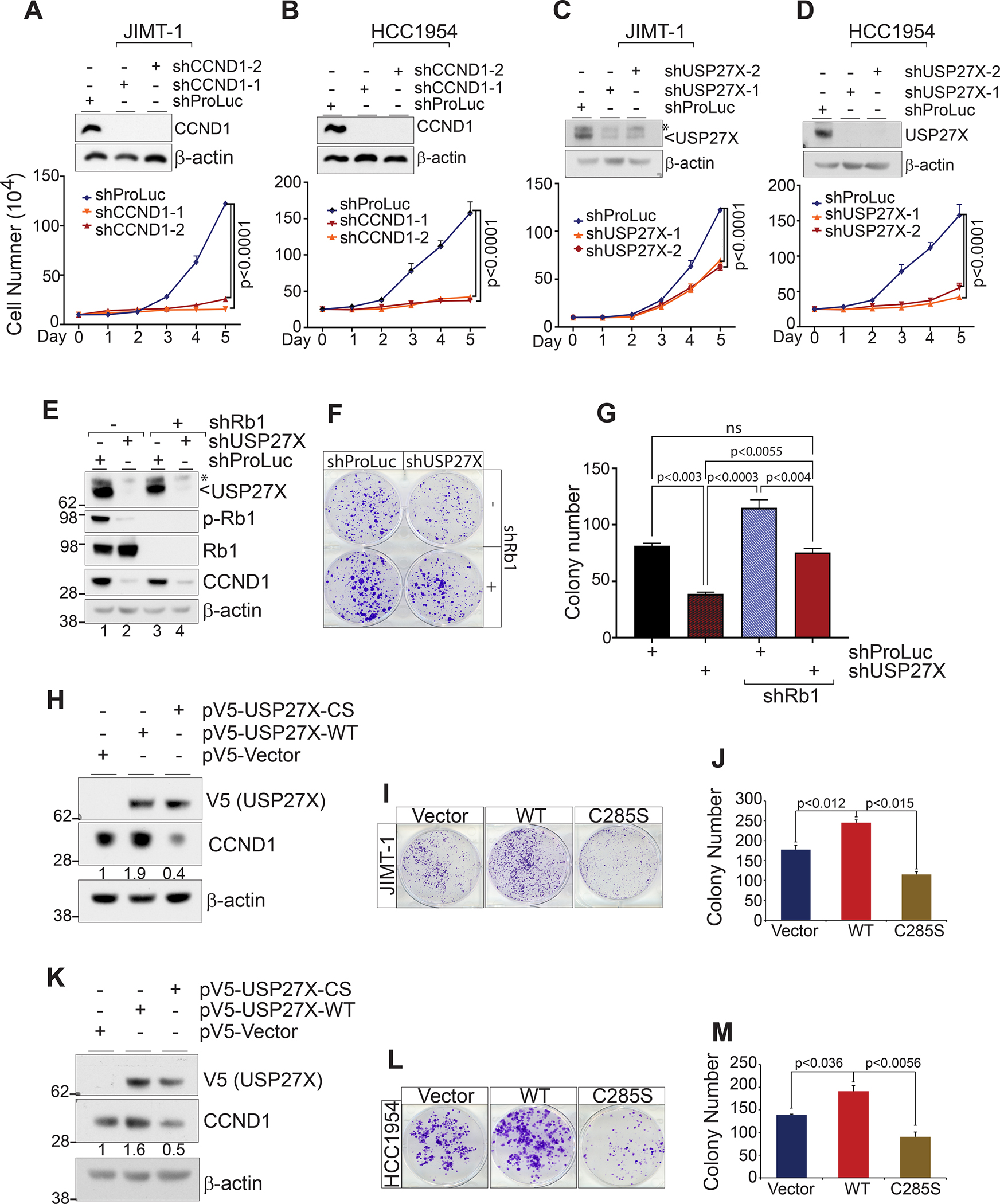
shRNA mediated depletion of CCND1 (A and B) and USP27X (C and D) severely impacts the proliferation of the HER2 therapy resistant breast cancer cell lines JIMT-1 and HCC1954 as measured by cell counting over the course of 5 days. (E) Immunoblots show reduced Rb1 phosphorylation following efficient depletion of USP27X and Rb1 in JIMT-1 cells. (F) Colony formation assay shows USP27X silencing only has a minor impact on cell proliferation in Rb1 depleted cells. (G) Quantification of the data presented in F. (H and K) Immunoblots demonstrating ectopic expression of WT or catalytically inactive (CS) USP27X and its impact on endogenous CCND1 in JIMT-1 and HCC1954 cells, respectively. Expression of USP27X impacts the proliferation of JIMT-1 (I) and HCC1954 (L) in a catalytically-dependent manner. (J and M) Quantification of the data presented in I and L, respectively.
As the main function of the CCND1-CDK4/6 complex is to phosphorylate Rb1 and enable the G1 to S transition, cells lacking Rb1 are largely independent of CCND1 function. As USP27X has several reported substrates (23,25,27,35–37), we wanted to confirm that the growth defects in USP27X-depleted cells are due to abrogation of CCND1 and compromised downstream Rb1 phosphorylation. To do this, we simultaneously depleted USP27X and Rb1 using shRNA in JIMT-1 cells (Fig. 5E). As expected, USP27X depletion substantially reduced both CCND1 levels and Rb1 phosphorylation in these cells (Fig. 5E, compare lane 1 to 2). These data indicated that, indeed, loss of USP27X, which reduces the levels of CCND1, leads to reduction of the CCND1-CDK4/6 complexes and, therefore, loss of Rb1 phosphorylation. Furthermore, our colony formation experiments demonstrate that USP27X depletion has only a minor impact on cell proliferation in Rb1-depleted cells, despite the same impact on CCND1 levels when compared to controls (Fig. 5E, lanes 2 and 4, Fig. 5F and G). Additionally, expression of ectopic CCND1 almost completely rescued the growth defects in USP27X-depleted cells (Supplementary Fig. 5). These data strongly suggest that growth defects in USP27X-depleted cells are largely due to diminished CCND1 levels and the impact on Rb1 phosphorylation and therefore cell cycle progression.
Lastly, we wanted to test whether the growth defects and impact on CCND1 are dependent on USP27X catalytic activity. We used the lentiviral expression vectors from Figure 3G to express either WT or CS V5-tagged USP27X in JIMT-1 and HCC1954 cells. Western blot analyses revealed that overexpression of WT USP27X increased CCND1 levels ~0.9 fold in JIMT-1 and ~0.6 fold in HCC1954 cells. On the other hand, expression of the catalytically inactive DUB reduced CCND1 levels in both cell lines, likely due to dominant-negative effects the mutant DUB exerts on CCND1 (Fig. 5H and K). The cell lines transduced with WT, CS USP27X, or empty vector were used in colony formation assays to measure growth. As shown in Figures 5I, J, L and M, expression of WT USP27X led to a significant increase in colony number, while expression of the catalytically inactive DUB suppressed cell growth in both lines. Taken together, these results indicate that USP27X significantly impacts HER2-therapy resistant cell growth by regulating CCND1 levels. Importantly, these processes rely on USP27X catalytic activity.
Ablation of USP27X in xenograft tumors reduces CCND1 levels and impairs growth.
Our in vitro experiments demonstrated that USP27X ablation severely impacts the growth of HER2 therapy-resistant cells. However, it is not known whether USP27X ablation would have the same impact on CCND1 and tumor growth in vivo, especially after tumors are already established. To test this, we used JIMT-1 cells stably transduced with the dox-inducible USP27X targeting or non-targeting (control) shRNA vectors described above. We subcutaneously injected 1×106 cells into the flanks of SCID mice. After tumors reached 200mm3, mice were placed on a diet containing dox (38) to induce shRNA expression. As shown in Figure 6A, all mice developed similar size tumors before they were placed on the dox diet at day 15. Importantly, dox-induced depletion of USP27X significantly impaired further tumor development. At the experimental end point (Day 40), tumors were harvested. USP27X-depleted tumors weighed ~3 fold less than control tumors (Supplementary Fig. 4). Immunohistochemical (IHC) analyses revealed that USP27X ablation leads to a significant decrease in both CCND1 protein levels (Fig. 6B and C) and proliferation, as measured by Ki67 staining (Fig. 6D and E). Importantly, these analyses confirmed that USP27X ablation has no impact on the expression of Cyclins E1 (CCNE1), A2 (CCNA2), and B1 (CCNB1) (Fig. 6F). These studies recapitulate our in vitro experiments. Taken together, these results demonstrate that functions of USP27X are essential for CCND1 stability and thus cancer cell proliferation and maintenance of tumor growth.
Figure 6. USP27X ablation reduces CCND1 levels in vivo and inhibits tumor growth.
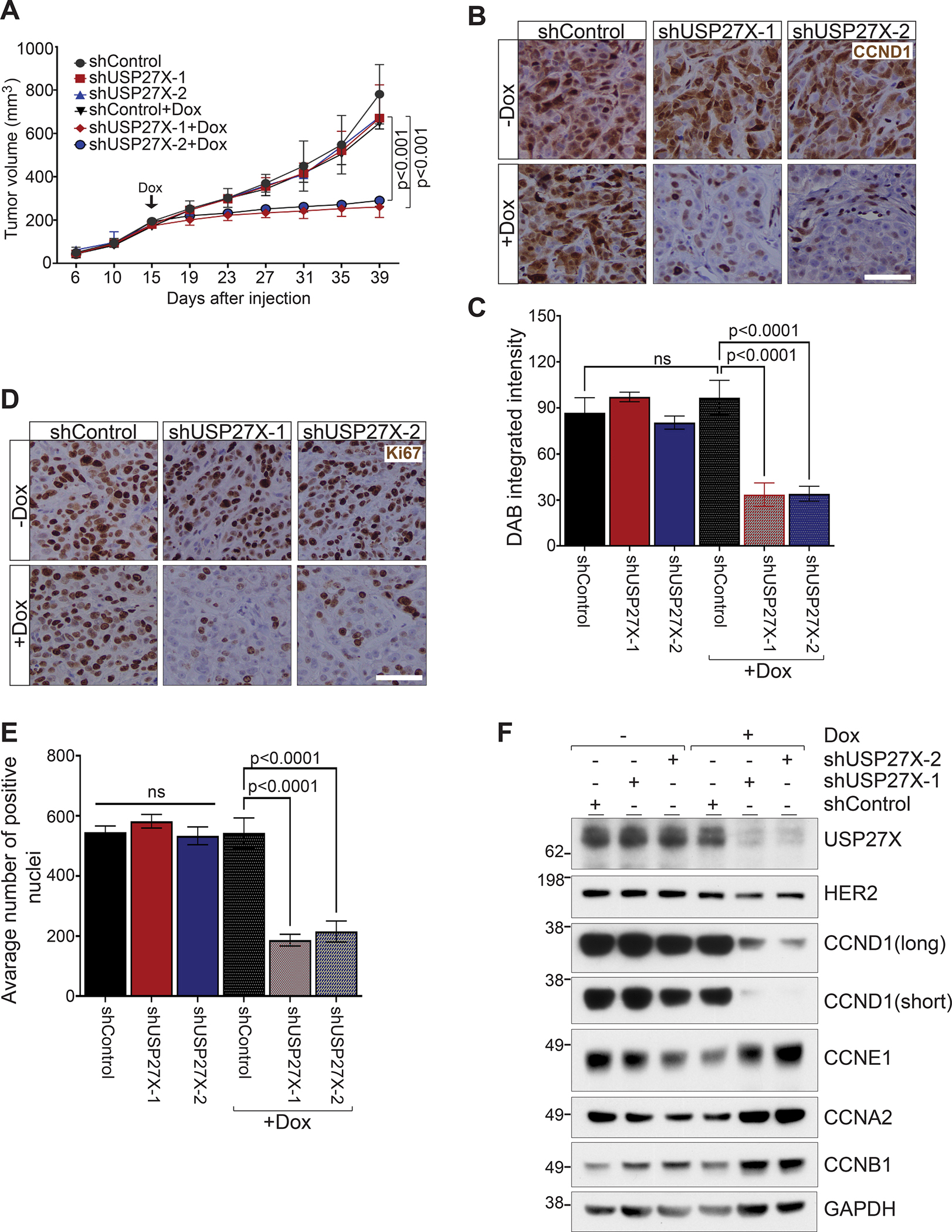
(A) Dox-induced USP27X ablation inhibits tumor progression in a JIMT-1 xenograft tumor model. Once tumors reached 200mm3, mice were placed on a diet containing doxycycline to induce shRNA expression (indicated with Dox and arrow on the graph). Tumor progression was monitored for 40 days. (B) Representative images of IHC analysis of CCND1 protein levels in tumors harvested at the end point of the experiment presented in A. Bar = 50μM. (C) Quantification of the DAB signal intensity of the tumors presented in B. (D) Representative images of IHC analysis of Ki67 protein levels in tumors harvested at the end point of the experiment presented in A. Bar = 50μM. (E) Quantification of the Ki67 positive nuclei presented in D. (F) Immunoblots show severe reduction of CCND1 protein levels in USP27X-depleted tumors. No differences in CCNE1, CCNA2, or CCNB1 were detected in these tumors. HER2 downstream signaling regulates CCND1 expression and HER2 expression was therefore also examined.
USP27X ablation resensitizes HER2-therapy resistant cells to HER2 targeted therapy.
As CCND1-CDK4/6 mediates resistance to HER2 targeted therapy (12), CDK4/6 inhibitors have successfully been used to overcome resistance in preclinical models (12). Such inhibitors have also extended the median progression-free survival in patients previously treated with HER2-targeted therapies by approximately 6 months (39). It has been previously demonstrated that reduction of CCND1 forces therapy resistant cells to renew their dependency on HER2 signaling (12). Hence, we next tested whether USP27X ablation, which reduces CCND1 levels, would resensitize HER2 therapy resistant cells to targeted therapy. To do this, we treated USP27X-depleted or control JIMT-1 and HCC1954 cells with different doses of Lapatinib (10nM to 10μM), a dual HER2/EGFR inhibitor currently used in combination therapy for HER2+ breast cancer patients. We used an MTT assay to assess cell viability after these treatments. USP27X ablation substantially increases the sensitivity of both therapy resistant cell lines to HER2 inhibition, lowering the IC50 ~3 fold in JIMT-1 (Fig. 7A) and HCC1954 cells (Fig. 7B).
Figure 7. USP27X resensitizes HER2+ therapy resistant breast cancer cells to HER2 inhibition.
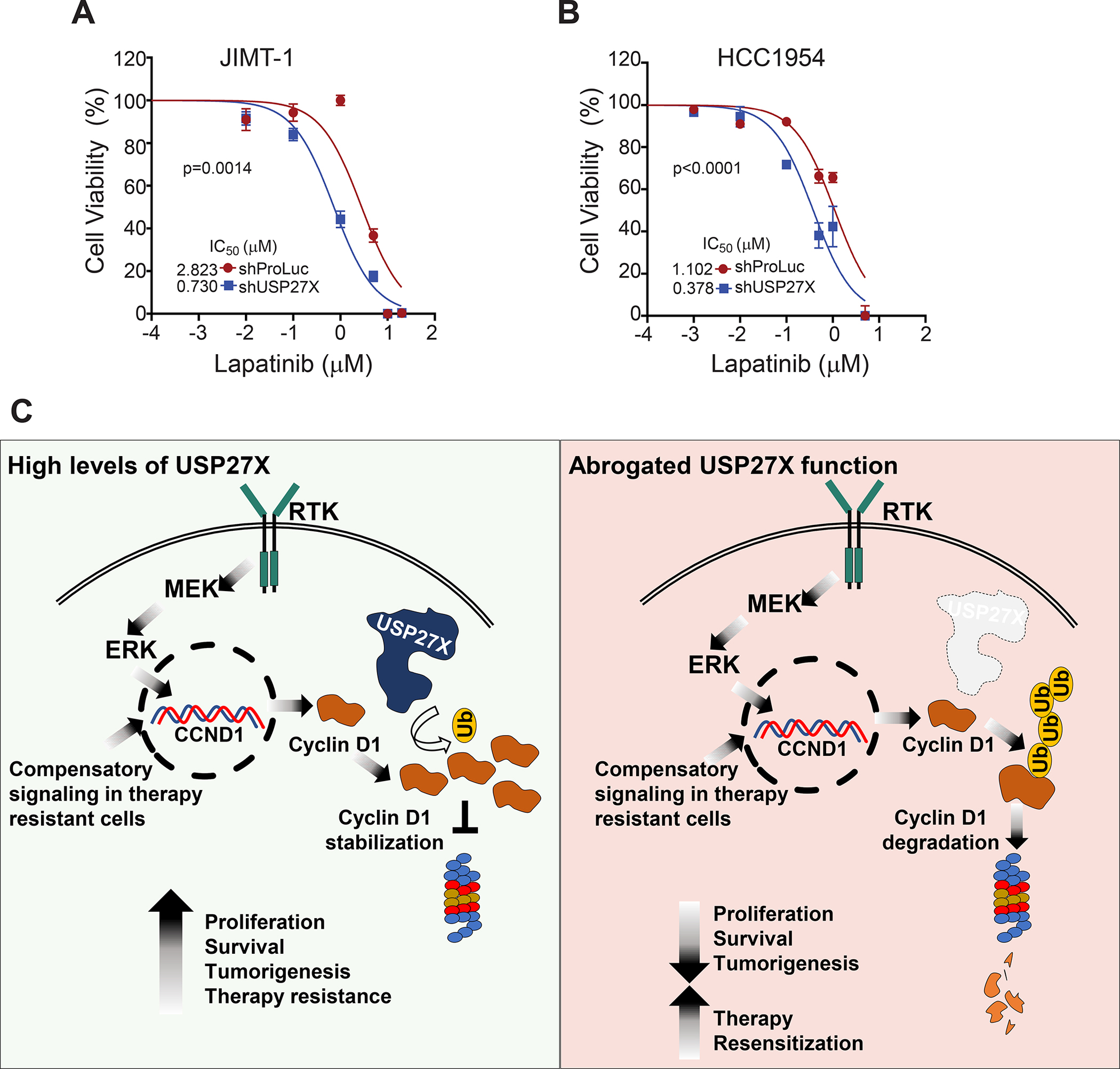
(A) USP27X depletion decreased the IC50 of Lapatinib in JIMT-1 cells and (B) HCC1954 cells, as determined by MTT assays. (C) Model of USP27X mediated regulation of CCND1. In cancer cells, CCND1 transcription is activated by MAP kinase signaling. To maintain tumor growth and evade targeted therapy, cancer cells develop mechanisms to sustain high levels of CCND1 expression. Elevated expression of USP27X is essential to sustain these high levels of CCND1 in tumors (left panel), as CCND1 levels are tightly controlled by ubiquitin-mediated degradation. Ablation of USP27X leads to increased ubiquitination and accelerated CCND1 degradation. This drastic reduction in CCND1 steady-state levels results in impaired tumor growth and resensitizes cancer cells to targeted therapy (right panel).
Discussion
In this study, we provide a mechanism for how USP27X fosters tumor growth (Fig 7C). Our data demonstrate that USP27X functions are required for maintenance of the steady-state levels of CCND1, a proto-oncogene that plays an essential role in cancer cell proliferation. USP27X does not regulate the transcription of CCND1, but rather binds and stabilizes the protein through deubiquitination. Overexpression of USP27X reduces CCND1 ubiquitination and stabilizes the protein, while USP27X ablation reduces CCND1 stability. Inhibition of the ubiquitin-proteasome system rescues this reduced CCND1 stability. Furthermore, our data revealed that USP27X expression levels correlate with CCND1 levels in several HER2+, therapy resistant breast cancer cell lines. Moreover, ablation of USP27X drastically reduces CCND1 levels in these cells and arrests their growth in an Rb1-dependent manner.
USP27X was originally discovered as an epigenetic regulator that targets histone 2B for deubiquitination and thereby regulates transcription of its target loci (23). In recent years, USP27X has emerged as a potential therapeutic target; several studies have demonstrated that USP27X is required for tumor cell growth and proliferation, and its ablation deters cancer progression in different model systems (24–27). Nevertheless, the underlying reasons why loss of this DUB has such a robust impact on tumor progression have not been fully elucidated. The vastly different impact that ATXN7L3 (required for USP27X activity toward H2B) and USP27X depletion have on cell growth indicates that the functions of this DUB in cell proliferation are independent of its role in H2B deubiquitination. Aside from H2B deubiquitination, USP27X has been implicated in several other cellular processes - from antiviral responses (37,40) and regulation of CCNE1 and Snail1 expression (25,27) to regulation of apoptosis via stabilization of the pro apoptotic BH3-only protein Bim (36). The link between USP27X and CCNE1 could provide a plausible explanation for the growth defects observed in USP27X-depleted cells. However, in our work, using several cell lines and USP27X depletion methods, we did not detect any apparent differences in the steady-state levels or decay of CCNE1 (Figs. 2 and 3). Likewise, we did not detect any differences in the steady-state levels of Bim, a proposed USP27X substrate, in USP27X-depleted cells compared to controls (Supplementary Fig. 6). Additionally, Lambies et al. reported Snail1 as USP27X target in breast cancer cells (27). In this study, however, the breast cancer cell growth defects observed upon USP27X ablation were not alleviated by Snail1 expression. While the cause for growth defects in USP27X-depleted cells could not be fully explained by previous studies, our results identify USP27X as a CCND1 DUB and provide a mechanistic explanation for the role of USP27X in cancer progression.
USP27X was not part of the original screen for CCND1 DUBs, which included 76 distinct family members, and identified USP2 as a CCND1-specific DUB (18). In the last several years, additional USPs such as USP22, USP5, and USP10 were reported to also stabilize CCND1 (19–21). CCND1 stability in cancer is likely regulated by several DUBs and CCND1 deubiquitination may be tissue and/or context specific. Stabilization of CCND1 by USP5 was reported to be important in lung cancer (20) whereas CCND1 regulation by USP10 was found in glioblastoma (21). Our data show that USP27X is highly expressed in HER2+ and therapy-resistant breast cancer cells (Fig. 4C) and thus may be the predominant CCND1 DUB in these tumors. Our data also revealed that USP27X regulates CCND1 stability independent of its phosphorylation at T286, suggesting that this DUB regulates a non-canonical pathway of CCND1 proteasomal degradation. Additional studies are needed to determine if different DUBs regulate CCND1 stability in different cancers or through different mechanisms of degradation. Nevertheless, the severely reduced CCND1 levels in USP27X-depleted cells demonstrate that the functions of USP27X in this context are not redundant and cannot be compensated for by other USPs. Interestingly, our previous studies demonstrated that USP27X is inactive in insolation and cannot deubiquitinate histones or digest artificial substrates (such as Ub-AMC) in vitro unless bound by ATXN7L3 (23). However, our current study suggests that USP27X requires different activating cofactor(s) for its activity toward CCND1, as AXTN7L3 depletion had no measurable impact on the steady-state levels of CCND1 (Supplementary Fig. 1E, G). Future studies will elucidate at what stage and in which cellular compartment USP27X interacts with and stabilizes CCND1 and whether additional cofactors or posttranslational modifications are required to facilitate its activity toward this protein.
The importance of CCND1 overexpression for tumor progression is well recognized in several cancers. CCND1 functions are especially important in HER2-driven breast cancers, as its overexpression confers resistance to HER2-targeted therapy (12). Interestingly, our data demonstrate that USP27X expression correlates with CCND1 protein levels in HER2-therapy resistant cells and with poor prognosis in HER2+ invasive breast cancer (Fig. 4). Importantly, ablation of CCND1, either after direct shRNA-mediated gene silencing or through enhanced protein degradation upon USP27X depletion, severely impacts the proliferation of these cells and tumor development in vivo, demonstrating their addiction to CCND1. Although CDK4/6 inhibitors are available to target CCND1-CDK4/6 activity, several studies demonstrate that CCND1 has functions independent of CDK4/6 and that when overexpressed, CCND1 can restart the cell cycle through interaction with CDK2 (41–43). Thus, targeting CCND1 indirectly through USP27X inhibition may offer a superior alternative for the treatment of CCND1-dependent cancers. Furthermore, ablation of USP27X resensitizes HER2-therapy resistant cells to HER2 inhibition (Fig. 7) and highlights a potential mechanism to combat therapy resistance in HER2+ breast cancer. As a deubiquitinating enzyme, USP27X is a druggable target and our studies ultimately illuminate new avenues for therapeutic intervention in CCND1 dependent cancers.
Supplementary Material
Implications:
As a deubiquitinating enzyme, USP27X is a druggable target. Our study illuminates new avenues for therapeutic intervention in CCND1-driven cancers.
Acknowledgments
This work was supported by an award from the Roswell Park Alliance Foundation to B.S.A. and in part by Department of Defense grant W81XWH-21-1-0647 to B.S.A.
This work was also supported in part by the National Cancer Institute (National Institutes of Health) grant P30 CA016056 to the Roswell Park Comprehensive Cancer Center that supports the Pathology Network, Flow and Image Cytometry, Bioinformatics, Biostatistics, Immune Analysis Shared Resources, Genomic Shared Resources and the Onsite Supply Center. We are thankful to Lanni Aquila for critical reading of the manuscript.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153–66 [DOI] [PubMed] [Google Scholar]
- 2.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol 1994;14:2077–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev 1993;7:331–42 [DOI] [PubMed] [Google Scholar]
- 4.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998;18:753–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee YM, Sicinski P. Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle 2006;5:2110–4 [DOI] [PubMed] [Google Scholar]
- 6.Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res 1996;68:67–108 [DOI] [PubMed] [Google Scholar]
- 7.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003;36:131–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillett C, Smith P, Gregory W, Richards M, Millis R, Peters G, et al. Cyclin D1 and prognosis in human breast cancer. Int J Cancer 1996;69:92–9 [DOI] [PubMed] [Google Scholar]
- 9.Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD, et al. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med 1995;1:1257–60 [DOI] [PubMed] [Google Scholar]
- 10.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001;411:1017–21 [DOI] [PubMed] [Google Scholar]
- 11.Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012;22:438–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016;29:255–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004;18:2699–711 [DOI] [PubMed] [Google Scholar]
- 14.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998;12:3499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res 1994;54:1812–7 [PubMed] [Google Scholar]
- 16.Russell A, Thompson MA, Hendley J, Trute L, Armes J, Germain D. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene 1999;18:1983–91 [DOI] [PubMed] [Google Scholar]
- 17.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 1997;11:957–72 [DOI] [PubMed] [Google Scholar]
- 18.Shan J, Zhao W, Gu W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol Cell 2009;36:469–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gennaro VJ, Stanek TJ, Peck AR, Sun Y, Wang F, Qie S, et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc Natl Acad Sci U S A 2018;115:E9298–E307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Cui Z, Xie Z, Li C, Xu C, Guo X, et al. Deubiquitinase USP5 promotes non-small cell lung cancer cell proliferation by stabilizing cyclin D1. Transl Lung Cancer Res 2021;10:3995–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun T, Xu YJ, Jiang SY, Xu Z, Cao BY, Sethi G, et al. Suppression of the USP10/CCND1 axis induces glioblastoma cell apoptosis. Acta Pharmacol Sin 2021;42:1338–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 2012;31:2373–88 [DOI] [PubMed] [Google Scholar]
- 23.Atanassov BS, Mohan RD, Lan X, Kuang X, Lu Y, Lin K, et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol Cell 2016;62:558–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017;547:355–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong L, Yu L, Bai C, Liu L, Long H, Shi L, et al. USP27-mediated Cyclin E stabilization drives cell cycle progression and hepatocellular tumorigenesis. Oncogene 2018;37:2702–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou T, Wang Y, Dong L, Che T, Zhao H, Yan X, et al. Stabilization of SETD3 by deubiquitinase USP27 enhances cell proliferation and hepatocellular carcinoma progression. Cell Mol Life Sci 2022;79:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambies G, Miceli M, Martinez-Guillamon C, Olivera-Salguero R, Pena R, Frias CP, et al. TGFbeta-activated USP27X deubiquitinase regulates cell migration and chemoresistance via stabilization of Snail1. Cancer Res 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, et al. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell 2009;35:352–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009;8:498–504 [DOI] [PubMed] [Google Scholar]
- 30.Morgan M, Ikenoue T, Suga H, Wolberger C. Potent macrocycle inhibitors of the human SAGA deubiquitinating module. Cell Chem Biol 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Jin K, Bunker E, Zhang X, Luo X, Liu X, et al. Structural basis of the phosphorylation-independent recognition of cyclin D1 by the SCF(FBXO31) ubiquitin ligase. Proc Natl Acad Sci U S A 2018;115:319–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Germain D, Russell A, Thompson A, Hendley J. Ubiquitination of free cyclin D1 is independent of phosphorylation on threonine 286. J Biol Chem 2000;275:12074–9 [DOI] [PubMed] [Google Scholar]
- 33.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 2017;35:314–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun 2017;8:1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi T, Iwamoto Y, Takashima K, Isomura A, Kosodo Y, Kawakami K, et al. Deubiquitinating enzymes regulate Hes1 stability and neuronal differentiation. FEBS J 2015;282:2411–23 [DOI] [PubMed] [Google Scholar]
- 36.Weber A, Heinlein M, Dengjel J, Alber C, Singh PK, Hacker G. The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep 2016;17:724–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao X, Chu B, Xin D, Li L, Sun Q. USP27X negatively regulates antiviral signaling by deubiquitinating RIG-I. PLoS Pathog 2020;16:e1008293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cawthorne C, Swindell R, Stratford IJ, Dive C, Welman A. Comparison of doxycycline delivery methods for Tet-inducible gene expression in a subcutaneous xenograft model. J Biomol Tech 2007;18:120–3 [PMC free article] [PubMed] [Google Scholar]
- 39.Tolaney SM, Wardley AM, Zambelli S, Hilton JF, Troso-Sandoval TA, Ricci F, et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncol 2020;21:763–75 [DOI] [PubMed] [Google Scholar]
- 40.Guo Y, Jiang F, Kong L, Li B, Yang Y, Zhang L, et al. Cutting Edge: USP27X Deubiquitinates and Stabilizes the DNA Sensor cGAS to Regulate Cytosolic DNA-Mediated Signaling. J Immunol 2019;203:2049–54 [DOI] [PubMed] [Google Scholar]
- 41.Jahn SC, Law ME, Corsino PE, Rowe TC, Davis BJ, Law BK. Assembly, activation, and substrate specificity of cyclin D1/Cdk2 complexes. Biochemistry 2013;52:3489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Junk DJ, Cipriano R, Stampfer M, Jackson MW. Constitutive CCND1/CDK2 activity substitutes for p53 loss, or MYC or oncogenic RAS expression in the transformation of human mammary epithelial cells. PLoS One 2013;8:e53776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res 2016;76:2301–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.