

Abstract
The effects of radiation therapy (RT) on tumor immunity in PDAC are not well understood. To better understand if RT can prime antigen-specific T cell responses, we analyzed human PDAC tissues and mouse models. In both settings, there was little evidence of RT-induced T cell priming. Using in-vitro systems, we found that tumor stromal components, including fibroblasts and collagen, cooperate to blunt RT efficacy and impair RT-induced interferon signaling. Focal Adhesion Kinase (FAK) inhibition rescued RT efficacy in-vitro and in-vivo, leading to tumor regression, T cell priming, and enhanced long-term survival in PDAC mouse models. Based on these data, we initiated a clinical trial of defactinib in combination with SBRT in PDAC patients (NCT04331041). Analysis of PDAC tissues from these patients showed stromal reprogramming mirroring our findings in GEMMs. Finally, the addition of checkpoint immunotherapy to RT and FAKi in animal models led to complete tumor regression and long-term survival.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy with a poor 5-year survival rate(1). Currently, complete surgical resection is the only potentially curative treatment, but 85–90% of patients are diagnosed at unresectable stages(2). While recent advances in diagnosis, chemotherapy, and radiation therapies (RT) have improved patient survival, these treatments have not been proven to be curative(3,4). For many patients with locally advanced PDAC, RT does not achieve resectability, and metastatic PDAC is incurable(2,5–9). Additionally, to date, there are no proven effective immunotherapies for the treatment of PDAC.
Oncogenic mutations in Kras, as well as frequent loss in tumor suppressors like Trp53 (p53) or Cdkn2a (INK4a), drive PDAC initiation, progression, treatment resistance, and metastatic spread(10). Kras accomplishes this through tumor-intrinsic changes in cellular signaling and metabolism, while promoting tumor-permissive interactions with surrounding stroma(10). This fibrotic desmoplastic stroma, composed of dense collagen-rich extracellular matrix and cancer associated fibroblasts (CAFs), likely contributes to PDAC’s resistance to therapy(2,11–18). While checkpoint immunotherapies have been groundbreaking in many solid malignancies(19–21), they have not been effective in PDAC(2,22,23). Similarly, combination treatments of immunotherapies with the standard of care chemotherapy or RT, which are aimed at priming anti-tumor immunity and unlock immunotherapies, have not been tremendously efficacious in gastrointestinal tumors(24–26). Thus, understanding why RT fails to prime anti-tumor immunity in PDAC is important.
The recalcitrant nature of PDAC has been linked in part to its unique tumor microenvironment (TME). In this TME, complex communication between the malignagenetically engineered mouse model nt tumor cells and the non-malignant tumor stroma plays a significant role in dictating PDAC’s resistance to therapy(2,11-13). Further complicating things, PDAC has a very heterogeneous CAF population, which plays diverse roles and contributes to divergent treatment outcomes(27-30). Additionally, PDAC’s fibrotic stroma contributes to poor drug delivery and deprived infiltration of anti-tumor immune cells. These two aspects of the PDAC TME have been confirmed as drivers of PDAC resistance to both chemo- and immunotherapies(31–34). However, it is unclear how PDAC-associated fibrosis and CAFs might impact the efficacy of RT and if this affects RT-induced immune priming.
Historically, studies have focused on RT as a direct tool to damage proliferating tumor cells, which leads to the accumulation of double-strand DNA breaks and cellular death(35). However, recent studies have shown that numerous extrinsic factors, including matrix stiffness, can dampen sensitivity to DNA damaging agents and their subsequent DNA repair mechanisms(36,37). In addition to its direct effect on tumor cells, RT can also prime anti-tumor immunity by releasing tumor-derived antigens and danger signals. This likely plays a critical role in long-term RT efficacy in multiple cancer types(38). Nevertheless, it remains unclear if these immune priming effects of RT are intact in highly fibrotic cancers such as PDAC.
Focal adhesion kinases include Focal Adhesion Kinase 1 (FAK, also known as FAK1 or PTK2) and the closely related Protein Tyrosine Kinase 2 beta (PTK2b, also known as FAK2). Both are non-receptor tyrosine kinases that have been heavily studied and implicated in cancer cell migration, proliferation, and survival. Notably, they also regulate pro-inflammatory pathway activation and cytokine production(39-41). As such, FAK signaling has been shown to be important in wound healing and pathologic fibrosis in various tissues(42,43). FAK signaling is hyperactivated in many cancers, including PDAC, and has been correlated with poor survival(44-46). Our group and others have shown that FAK signaling is an integral driver of the fibrotic and immunosuppressive microenvironment that protects PDAC from immune surveillance and drives resistance to immunotherapy(46-48). In this study, we address the role of stromal-induced RT resistance in immune priming. We show that FAK inhibition can sensitize PDAC to RT and promote both RT-induced immune priming and response to immunotherapy.
RESULTS
Radiation therapy is insufficient to optimally prime robust T cell responses in PDAC
To study the effect of RT on T cell infiltration, we first performed immunohistochemistry (IHC) on human surgical resection samples from patients who received neoadjuvant Chemotherapy in combination with Stereotactic Body RT (Chemo/SBRT) or with conventional RT (Chemo/RT), or no neoadjuvant therapy. Across these cohorts, we found no difference in the number of tumor-infiltrating CD8+ T cells (Fig. 1A, Supplementary Table S1). Even though we did not see T cell changes, we found the total number of CK19+ tumor cells was reduced in the tumor bed of samples from Chemo/SBRT-treated patients (Fig. S1A). In contrast, we observed increased stromal area and collagen deposition in patients who received neoadjuvant RT (Fig. 1B). These data raise the possibility that while RT can lead to temporary disease control in PDAC patients(49,50), it may fail to optimally prime anti-tumor T cell immunity and thus limit long-term benefits. However, due to the heterogeneity of PDAC tumors and patients, these human studies in un-matched patient populations cannot fully answer this question and larger studies with paired samples would be needed to fully address this.
Figure 1: Radiation therapy is insufficient to optimally prime robust T cell responses in PDAC.
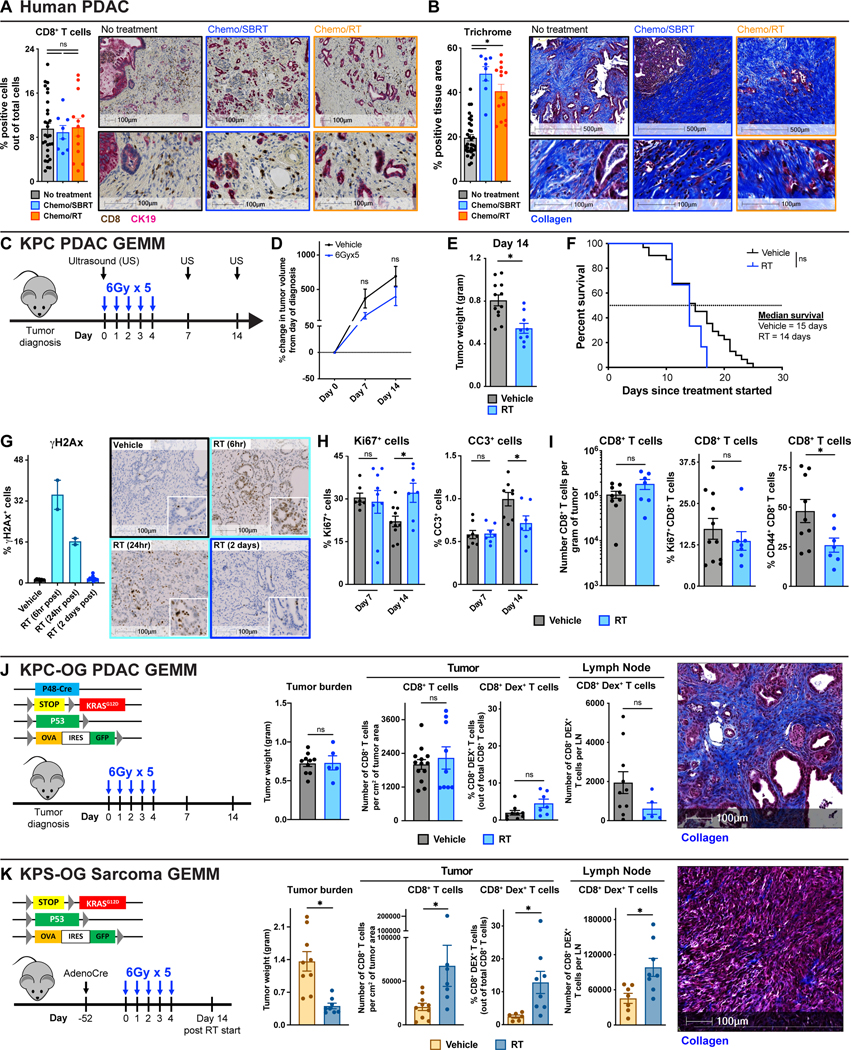
(A-B) Analysis of CD8α T cell number and collagen density in surgical resection samples from patients who received neoadjuvant Chemo/SBRT, Chemo/RT, or no neoadjuvant therapy. Representative CD8α/CK19 IHC and Trichrome images and quantification are depicted. n = 8–32 patients/group. (C) Schematic of RT administration and tumor burden monitoring in KPC GEMMs. KPC mice diagnosed with ultrasound (US) were treated with hypofractionated RT (6Gy x 5) and longitudinally assessed for tumor burden. (D) Tumor growth kinetics of vehicle and RT-treated KPC mice from (C) quantified by US measurements. n = 8–12 mice/group. (E) Day 14 pancreas weight of KPC mice from (C). n = 9–13 mice/group. (F) Kaplan-Meier survival curve of KPC mice from (C). n = 6–31 mice/group. (G) Analysis of DNA damage (γH2Ax) on tissues from KPC vehicle or RT-treated mice at various time points. Representative γH2Ax IHC images are depicted. n = 2–10 mice/group (H) IHC analysis of proliferation (Ki67) and apoptosis (CC3) on tissues from KPC mice at days 7 and 14 post-RT. n = at least 7 mice/group. (I) Flow cytometry quantification of total CD8+ T cells and percentage of Ki67+ or CD44+ CD8+ T cells out of total CD8+ T cells in KPC vehicle or RT-treated mice at day 14. n = at least 7 mice/group. (J) Genetic loci for KPC-OG PDAC GEMMs and schematic of RT administration. Pancreas weight of KPC-OG vehicle or RT-treated mice. IHC analysis of CD8α T cell number from KPC-OG vehicle or RT-treated mice. Flow cytometry quantification of total CD8+ OVA-Dextramer+ tumor-specific T cells from tumor and pancreas draining lymph nodes in KPC-OG vehicle or RT-treated mice. Representative Trichrome image from KPC-OG vehicle-treated mice is depicted. All analyses were done at day 14 post-RT start. n = at least 6 mice/group. (K) Genetic loci for KPS-OG sarcoma GEMMs and schematic of AdenoCre injection and RT administration. Tumor weight of KPS-OG vehicle or RT-treated mice. IHC analysis of CD8α T cell number from KPS-OG vehicle or RT-treated mice. Flow cytometry quantification of total CD8+ OVA-Dextramer+ tumor-specific T cells from tumor and draining lymph nodes in KPS-OG vehicle or RT-treated mice. Representative Trichrome image from KPS-OG vehicle-treated mice is depicted. All analyses were done at day 14 post-RT start. n = at least 7 mice/group.
All graphs depict mean +/− SEM. For comparisons between two groups, “*” denotes p < 0.05 by two-tailed t-test, one-way ANOVA, or Kaplan-Meier as appropriate. “ns” denotes not significant.
To better understand whether RT can directly affect anti-tumor immunity in PDAC, we utilized the p48-Cre/LSL-KrasG12D/p53Flox/Flox (KPC) genetically engineered mouse model (GEMM). The KPC tumors replicate the treatment-resistant nature and abundant stroma of human PDAC(51). Studies on the impact of RT on PDAC TME in pre-clinical models are limited by the lack of methods to precisely deliver targeted RT doses into the pancreata of mice. To overcome this, we employed a Small Animal Radiation Research Platform (SARRP)(52), which incorporates computed tomography (CT) image-guided delivery of high doses of radiation (Fig. S1B). With this instrument, we can safely deliver clinically relevant doses of radiation to the mouse pancreas while limiting off-target delivery to surrounding normal tissues. KPC mice with ultrasound-diagnosed tumors were treated with hypofractionated RT (6Gy x 5) and monitored for tumor progression (Fig. 1C). We found RT could only modestly control tumor growth as measured by longitudinal ultrasound imaging or tumor weights at days 7 and 14 after RT start (Fig. 1D-E, S1C-E). After modest disease control, all KPC mice treated with RT eventually rapidly progressed, which was translated into no benefit in overall long-term survival (Fig. 1F). These data agree with other studies showing, unlike transplantable tumors, this highly fibrotic KPC GEMMs exhibited limited RT response(53,54). To understand if induction of DNA damage was impaired, we analyzed γH2Ax+ foci formation. As expected, at 6 and 24 hours post completion of hypofractionated RT, significant γH2Ax+ foci were observed in most PDAC cells and stroma, but 48 hours post the last RT dose, most γH2Ax+ foci resolved, suggesting DNA repair was intact in this model (Fig. 1G). Analysis of Cleaved Caspase 3+ (CC3+) cells found no induction of apoptosis at 7 or 14 days after RT start. Surprisingly, we also observed no change in the number of Ki67+ proliferating cells on day 7, but by day 14, tumor proliferation in RT-treated tumors was elevated compared to controls (Fig. 1H, S1F). Together, these data show that RT had limited efficacy in killing PDAC cells in stroma-rich KPC GEMMs.
Next, we sought to determine if RT could prime T cell immunity in KPC GEMMs. We found that RT did not lead to increased PDAC-infiltrating CD8+ or CD4+ T cell numbers (Fig. 1I, S1G). We also did not observe any increase in the number of Ki67+ CD8+ T cells, but found CD44+ CD62L− activated T cell fraction was slightly decreased. These data were consistent across 7 and 14 days post-RT timepoints (Fig. 1I, S1G). While there were no major changes in myeloid cell populations, we noted a trend toward increased monocytes, TAMs, granulocytes, and eosinophils at both days 7 and 14 post-RT start (Fig. S1H). To eliminate the possibility that RT did not prime new T cell responses because of the low neoantigen burden characteristic of KPC GEMMs, we studied RT’s ability to induce tumor-specific T cell responses in p48-Cre/LSL-KrasG12D/p53Flox/Flox/LSL-OVA-GFP+ (KPC-OG) PDAC GEMMs(55). KPC-OG mice harbor inducible neoantigens, ovalbumin (OVA) and green fluorescent protein (GFP) expressions, which allow for the tracking of tumor-antigen-specific T cells. Despite neoantigen expression in KPC-OG mice, we found that RT did not lead to an increase in either total or OVA-specific (Dextramer+) CD8+ T cells in PDAC tissues 14 days after treatment start. In keeping with the lack of immune priming effects of RT in this model, we also observed no increase in OVA-specific Dextramer+ T cells in the pancreas draining lymph nodes (Fig. 1J). Furthermore, we found no change in the proximity of CD8+ T cells relative to CK19+ tumor cells in mice treated with RT (Fig. S1I).
To contrast with these KPC-OG GEMMs, we used littermate mice to evaluate RT-induced T cell immunity in sarcoma tumors. These LSL-KrasG12D/p53Flox/Flox/LSL-OVA-GFP+ mice (called KPS-OG here-in) were injected with Adenovirus-Cre into the hindleg muscle to generate sarcoma tumors(56). In contrast to KPC-OG littermates, KPS-OG mice treated with the same RT regimen (6Gy x 5) had a sustained reduction of tumor progression, decreased Ki67+ proliferating tumor cells, and increased tumor-infiltrating CD8+ T cells as well as increased OVA-specific T cells in both the tumor tissues and draining lymph nodes (Fig. 1K, S1J-K). Taken together, these data suggest that in genetically equivalent mice, RT increased antigen-specific T cells in sarcoma GEMMs but failed to prime tumor immunity in PDAC GEMMs. While both models were driven by Kras mutation and p53 loss, one key difference we noted was that the KPC-OG PDAC tissues had higher stromal desmoplasia compared to their KPS-OG sarcoma counterparts (Fig. 1J-K, S1L). These data suggest that the effects of RT on tumor control and T cell priming might be related to stromal differences.
Stromal elements repress RT efficacy and induction of interferons
PDAC tumors are often characterized by dramatic desmoplastic stroma comprised of dense extracellular collagen deposition and diverse CAF populations(57,58). To test the role of PDAC stroma in RT-induced cell death, we used heterotypic organoid cell culture models. These models allow for the co-culture of PDAC organoids with fibroblasts and ECM components. PDAC organoids (KPOG) were derived from KPC-OG mice and growth was tracked by GFP fluorescence (Fig. 2A). We found that while PDAC organoids alone were sensitive to RT, the addition of pancreas-derived fibroblasts rendered tumor cells less sensitive to RT, and the heterotypic organoid co-cultures rapidly grew after RT exposure (Fig. 2A). Similarly, increasing collagen-I density in this culture system also conferred resistance to RT (Fig. S2A-C). The presence of both pancreatic fibroblasts and collagen-I additively protected PDAC organoids from RT-induced growth inhibition (Fig. 2B, S2B-C). To confirm this further, we fluorescence-activated cell sorting (FACS)-sorted PDAC CAFs from KPC tumors and again observed that CAFs and collagen-I worked cooperatively to protect PDAC cells from RT (Fig. S2D). To understand how the PDAC stroma affects RT efficacy, we looked at markers of proliferation, apoptosis, and DNA damage using IHC. First, we found the induction of γH2Ax measured 6 hours post-RT was identical in the presence or absence of fibroblasts, suggesting that DNA damage induction by RT was not impacted, at least in the short-term (Fig. 2C). Similarly, pancreatic fibroblasts did not impact the induction of apoptosis, measured by CC3 at 3 days following RT (Fig. 2D). However, regardless of RT, fibroblasts did significantly increase the number of PDAC cells that entered S-phase after co-culture as measured by BrdU incorporation (Fig. 2E). These data suggest that even though RT could induce apoptosis in some cells, RT was not able to fully blunt proliferation in the presence of PDAC stroma.
Figure 2: Stromal elements repress RT efficacy and induction of interferons.

(A) Experimental schematic of KPOG organoids co-cultured with fibroblasts. Cells were cultured for one day before RT. Representative GFP images taken with a fluorescent microscope at day 6 post co-culture. Tumor growth analysis of KPOG organoids tracked overtime. n = at least 3/group. (B) Experimental schematic of KPOG organoids co-cultured with fibroblasts and collagen-I. Cells were cultured for one day before RT. Representative fluorescent images and quantification of tumor growth inhibition at day 4. n = at least 3/group. (C) IHC analysis and representative images of γH2Ax in KPOG organoid cultures at 6 hours post-RT. n = at least 3 sets/group. (D-E) IHC analysis and representative images of CC3 (D) and 5hr-pulsed BrdU (E) in KPOG organoid cultures at day 3 post-RT. n = at least 3/group. (F) Experimental schematic of KPOG organoids co-cultured with collagen-I. RT-PCR measurements of IFN-related genes measured in KPOG organoid cultures treated with different conditions at the indicated timepoints. n = at least 3/group.
All graphs depict mean +/− SEM. “*” denotes p < 0.05 by two-tailed t-test or one-way ANOVA as appropriate. “ns” denotes not significant.
To understand if there are potential changes in RT-regulated immune signaling due to the collagen-rich stroma, we measured the expression of interferon (IFN)-related genes. We found that when organoids were cultured in basement membrane extract alone, robust increases in IFN-related genes expression were observed 24 to 48 hours after RT (Fig. 2F, S2E). This included increases in genes encoding IFNα, IFNβ, IFNγ, IRF3, IRF7, IRF9, STAT1, CXCL9, CXCL10, and CXCL11 proteins. However, when collagen was added to these cultures, the induction of IFN-related genes was severely dampened (Fig. 2F, S2E). Other inflammatory genes such as IL-1α were not altered, suggesting some specificity for IFN signaling (Fig. 2F). These data suggest that stromal elements may limit not only RT-induced growth suppression, as shown by others(59–61), but also RT-induced IFN signaling, which may relate to RT-induced tumor immunity in-vivo.
To determine if RT alone changes how PDAC cells interact with the TME, we performed RNA sequencing (RNA-seq) and Reverse Phase Protein Array (RPPA) to look at both the gene and protein levels, respectively, after RT. As expected, compared to vehicle-treated cells, RT-treated cells had higher expression of DNA damage and repair proteins, such as γH2Ax, ATM, Rad17, and Rad51 (Fig. S2F) and increases in gene sets related to DNA damage, reactive oxygen species (ROS) and DNA repair (Fig. S2G). However, RT also induced collagen, integrin signaling, and focal adhesion assembly gene signatures (Fig. S2G). Together, these data suggest that RT might also “prime” PDAC cells to engage with the surrounding stroma through integrin and focal adhesion signaling.
Inhibition of Focal Adhesion Kinase overcomes stromal-induced RT resistance
While seldomly mutated, FAK is hyperactivated in >80% of PDAC tumors and plays an integral role in disease progression(45,47). Furthermore, FAK signaling is amplified by stromal density and can be critical to ECM/integrin-induced survival signaling(45,47). To understand whether we can overcome stromal-mediated RT resistance through FAK inhibition, we tested RT in combination with a FAK inhibitor, VS-4718 (FAKi). As above, the presence of fibroblasts and collagen enhanced the growth of PDAC organoids and made them much less responsive to RT (Fig. 3A-C). However, the combination of FAKi and RT led to complete inhibition of PDAC organoid growth, in a FAKi dose-dependent manner (Fig. 3B-C). This was true whether PDAC cells were grown in the presence of CAFs, collagen, or both (Fig. 3C, S3A-D). These findings show that FAKi can be a potential radiosensitizer for PDAC and that FAKi may increase RT efficacy.
Figure 3: Inhibition of Focal Adhesion Kinase overcomes stromal-induced RT resistance.

(A) Tumor growth analysis of KPOG organoids co-cultured with fibroblasts treated with RT +/− FAKi. Cells were cultured as in Fig. 2A. Representative fluorescent images at day 6 post co-culture. n = at least 3/group. (B) Growth analysis of KPOG organoids co-cultured with fibroblasts after treatment with RT +/− FAKi at varying concentrations at day 7 post co-culture. n = at least 3/group. (C) Tumor growth analysis of KPOG organoids co-cultured with fibroblasts and collagen-I treated with RT +/− FAKi. n = at least 3/group. (D) IHC analysis of γH2Ax in KPOG organoid cultures taken 6 hours post-RT. IHC analysis of CC3 and Ki67 in KPOG organoid cultures taken 3 days post-RT. Representative γH2Ax, CC3, and Ki67 IHC images are depicted. n = at least 3/group. (E) RPPA analysis heatmap displaying expression level of proteins related to: (i) DNA damage response/repair and ROS response, (ii) cell cycle checkpoint, (iii) MAPK pathway, (iv) pro-survival, (v) apoptosis/survival, (vi) mTOR/autophagy, and (vii) inflammatory mediators. Proteins were taken from KP2 cells at 24 hours post-RT. n = at least 3/group. (F-G) Bar graphs displaying over-representation analysis of KPOG organoids differentially expressed genes (DEGs) in Molecular Signatures Database (MSigDB)_Hallmark (F) and Gene Ontology (GO) (G) gene sets. All graphs display comparisons of vehicle to RT+FAKi-treated cells. All pathways were filtered with p value < 0.05. (H) Heatmap displaying expression level of genes in MSigDB_Hallmark database for “inflammatory response” and in GO database for “antigen processing and presentation”. (I) mIHC analysis of CK19+ pSTAT1+ cells in tissues of KPC mice from Fig. 4A. Representative CK19 and pSTAT1 fused mIHC images are depicted. n = at least 6 mice/group.
All graphs depict mean +/− SEM. “*” denotes p < 0.05 by two-tailed t-test or one-way ANOVA as appropriate.
To elucidate the effects of adding FAKi to RT, we again looked at markers of proliferation, apoptosis, and DNA damage using IHC. We did not see a difference in the short-term induction of DNA damage as measured by γH2Ax between RT and RT+FAKi in the presence or absence of fibroblasts, suggesting that neither FAK inhibition, nor the presence of stroma, regulated the amount of initial (first 6 hours) DNA damage induced by RT in PDAC cells (Fig. 3D). Next, we found that the combination of RT+FAKi dramatically increased the number of CC3+ apoptotic cells and decreased the number of Ki67+ proliferating cells by 3 days post-treatment and these effects were irrespective of the presence of stroma (Fig. 3D). These data suggest that FAK inhibition can improve RT-induced cell death.
To further understand if the cell death induced by the combination of FAKi and RT would have an immunomodulatory effect, we performed parallel RNA-seq and RPPA in KPOG organoids and the KP2 and KRAS-INK (KI) PDAC cell lines. We found PDAC cells treated with either RT or RT+FAKi had significant upregulation of DNA damage response gene sets and proteins, including DNA damage and ROS-related genes and AMPK, p-ERK, DNA Ligase-IV, TIGAR, and Rad17 proteins (Fig. 3E, S3E). However, unlike RT-treated cells, RT+FAKi-treated cells had downregulation of gene sets related to DNA damage repair and ROS response, as well as downregulation of key DNA damage repair signaling proteins (NQO1, JAB1, KEAP1, Rad23, DDB-1, Fig. 3E, S3E). As expected, RT+FAKi-treated cells also had changes in cell cycle gene sets and cell cycle proteins (CDK1, p27, Cdc2, CyclinD3, and CDK9) corresponding to the observed changes in growth arrest (Fig. 3E-F, S3E). Finally, we observed that RT+FAKi treatment might change key metabolic pathways, including downregulation in genes related to oxidative phosphorylation, glycolysis, cholesterol homeostasis, and DNA replication (Fig. 3E-G, S3E). However, whether these metabolic changes were associated with unique biology or just cell cycle arrest and death is unclear. Taken together, these suggest that while both RT and RT+FAKi can induce DNA damage response, only RT+FAKi inhibits the ability of PDAC cells to repair damage, leading to growth arrest.
Next, we examined the expression of key proliferative signaling mediators for PDAC cells and observed RT+FAKi led to downregulation of numerous MAPK proteins and pro-survival/apoptosis related proteins, including MEK2, ERK5, p38, PLCγ, Notch1, AKT, XIAP and PARP and upregulation of mTOR/autophagy related proteins ATG7, p70-S6K1, mTOR pS2448, Rictor, S6 and ULK1 (Fig. 3E, S3E). Notably, some of these changes were also seen with FAKi monotherapy, suggesting that inhibition of FAK drives these changes in our system.
To understand the potential immunologic impact of RT+FAKi treatment, we analyzed changes in the inflammatory pathways in PDAC cells and organoids. By RNA-seq, we observed upregulation of gene sets involved in TNFα, IFNβ, and IFNγ signaling (Fig. 3F-H). Additionally, RT+FAKi-treated PDAC cells had increased expression level of genes related to antigen processing and presentation (Fig. 3G-H), possibly downstream of increased IFN signaling. Correspondingly, increases in NFκB, JNK, and c-Jun pathways were observed at the protein level with RT+FAKi treatment, while Stat3 and Jak2 protein expressions were decreased (Fig. 3E, S3E). Using orthogonal approaches, we found that STING and phospho-IRF3 were increased by western blot analysis and IFNα, IFNβ, and IFNγ transcripts were upregulated by RT-PCR in KI cells treated with RT+FAKi (Fig. S3F-G). To confirm the observed changes in STAT/IFN signaling in-vivo, we performed multiplex IHC (mIHC) on PDAC tissues from KPC mice and found that RT+FAKi treatment elevated phospho-STAT1 (pSTAT1) expression in CK19+ PDAC cells (Fig. 3I). Taken together, these data suggest that FAK inhibition overcomes stromal-induced RT resistance by mitigating pro-survival and DNA damage repair signaling, which ultimately leads to sustained activation of IFN pathways in PDAC cells.
Combining FAKi with RT leads to immune priming in-vivo
We next sought to determine if FAK inhibition could improve RT efficacy and immune priming in-vivo. To accomplish this, we treated KPC GEMMs with the combination of RT and FAKi (Fig. 4A). Longitudinal ultrasound imaging showed that RT+FAKi led to dramatic PDAC tumor regression in nearly 100% of mice (Fig. 4B-C). These imaging data were verified by absolute pancreas weights from mice sacrificed 7 and 14 days after RT start (Fig. 4D, S4A). Additionally, RT+FAKi led to superior tumor regression than the combination of RT with the chemotherapeutic drug gemcitabine, which has been used to treat PDAC patients (Fig. 4B-C). Similarly, RT+FAKi led to tumor regression and long-term survival in multiple PDAC models representing diverse tumor suppressor alterations, including KPC and KPC-OG GEMMs (p48-Cre/LSL-KrasG12D/p53Flox/Flox/OVA-GFP+), syngeneic KP2 (KrasG12D/p53Flox/+), PDA.69 (KrasG12D/p53R172H/+) and KRAS-INK (KrasG12D/p53+/+/Ink/ArfFlox/Flox) (Fig. 4E-F, S4A-D). Corresponding with the observed efficacy, in KPC GEMMs, RT+FAKi markedly reduced cell proliferation (Ki67+ cells), CK19+ tumor cells, and high-grade PDAC lesions and increased the number of CC3+ apoptotic cells, necrotic tumor, and normal pancreatic tissue areas, compared to tissues from mice treated with RT alone (Fig. 4G-H, S4E-F). Together these data suggest RT+FAKi led to improved PDAC cell killing in-vivo.
Figure 4: Combining FAKi with RT leads to immune priming in-vivo.

(A) Schematic of RT (6Gy x 5) and FAKi (75mg/kg BID) administration and tumor burden monitoring in KPC GEMMs. KPC mice diagnosed with US were treated and longitudinally assessed for tumor burden. (B) Waterfall plot of KPC GEMMs from (A) evaluating tumor growth difference from day 0 to 14 by US measurement. Gemcitabine (GEM) was given at a dose of 75mg/kg every 5 days starting at day 0. n = 6–20 mice/group. (C) Tumor growth kinetics of KPC mice from (B) quantified by US measurements. n = 6–20 mice/group. (D) Day 14 pancreas weight of KPC mice from (A). Representative gross tissue images are depicted. n = 6–20 mice/group. (E) Waterfall plot of KP2 syngeneic tumor-bearing mice treated as depicted in (A) evaluating tumor growth difference from day 0 to 5. n = 5–10 mice/group. (F) Kaplan-Meier survival curve of KP2 syngeneic tumor-bearing mice from (G). n = 5–10 mice/group. (G-H) IHC analysis of Ki67 (G) and CC3 (H) in tissues from KPC mice at days 7 and 14 post-treatment start. Representative Ki67 and CC3 IHC images are depicted. n = at least 7 mice/group. (I) Flow cytometry quantification of total CD8+ OVA-Dextramer+ T cells in tumor and pancreas draining lymph node from KPC-OG mice treated as in (A). n = at least 7 mice/group. (J) Frequency distribution of CD8+ T cell proximity to nearest CK19+ tumor cell in KPC-OG mice treated as indicated. Representative dual stained CD8α and CK19 images are depicted. n = at least 5 mice/group. (K) Flow cytometry quantification of various innate immune infiltrates: (i) cDC1 (CD45+ CD3− CD19− Ly6C− Ly6G− F4/80lo MHC-IIhi CD24hi CD103+), (ii) cDC2 (CD45+ CD3− CD19− Ly6C− Ly6G− F4/80lo MHC-IIhi CD24lo CD11b+), (iii) TAMs (CD45+ CD3− CD19− Ly6C− Ly6G− CD11b+ F4/80+ MHC-II+), (iv) granulocytes (CD45+ CD3− CD19− Ly6C− Ly6G+), and (v) eosinophils (CD45+ CD3− CD19− Ly6C− Ly6G− CD11b+ F4/80− MHC-II−) in KPC mice from (A). n = at least 7 mice/group. (L) Waterfall plot of KP2 syngeneic tumor-bearing mice treated as in (A) evaluating tumor growth difference from day 0 to 5. Depleting αCD4 and αCD8 IgGs were given starting at day −2 every 4 days for a total of 30 days. n = 10 mice/group. (M) Kaplan-Meier survival curve of KP2 syngeneic tumor-bearing mice from (L). n = 10 mice/group.
All graphs depict mean +/− SEM. “*” denotes p < 0.05 by two-tailed t-test or one-way ANOVA or Kaplan-Meier as appropriate. “ns” denotes not significant.
To determine if the observed efficacy of RT+FAKi was also associated with treatment-induced T cell priming, we assessed changes in tumor-antigen-specific T cells in KPC-OG GEMMs. Analysis of these mice at 14 days post dual RT+FAKi treatment demonstrated increases in CD8+ T cell activation (CD44+), increased number of tumor-specific OVA-Dextramer+ CD8+ T cells, and increased CD8+ T cells proximity to CK19+ tumor cells (Fig. 4I-J, S4G). We next analyzed draining lymph nodes from these mice and found that RT+FAKi treatment dramatically increased the total number of activated CD44+ CD8+ T cells and OVA-Dextramer+ CD8+ T cells (Fig. 4I, S4G), suggesting RT+FAKi could lead to treatment-induced immune priming not seen with RT alone. Analysis of other immune cell populations showed that RT+FAKi decreased the total number of CD4+ Foxp3+ T regulatory cells, TAMs, granulocytes, and eosinophils, and increased conventional type 1 dendritic cells (cDC1s) but not type 2 dendritic cells (cDC2s) (Fig. 4K, S4H). In particular, the increase in cDC1, which was >13-fold, and the increase in the ratio of cDC1 to TAMs, which was >24-fold, might be directly contributing to the treatment-induced increase in tumor-specific CD8+ T cells in KPC-OG GEMMs. We next sought to determine whether T cells were critical to initial tumor regression and/or improved long-term disease control. To accomplish this, we depleted CD4+ and CD8+ T cells in mice bearing syngeneic KP2 PDAC tumors and found that loss of T cells did not affect short-term tumor regression (Fig. 4L, S4I), but was critical for long-term disease control (Fig. 4M, S4I). To determine the role of type-I IFNs, we treated mice with αIFNα/β receptor-1 (αIFNAR1) blocking antibody and observed a loss of tumor control by RT+FAKi, suggesting that IFN signaling is important for tumor control (Fig. S4J). Altogether, these data suggest that RT+FAKi remodeled the immune TME to favor T cell priming and that this was critical for long-term disease control.
Tumor-infiltrating immune cells from RT+FAKi treated mice have better anti-tumor signatures
To further understand treatment-induced changes in the immune TME, we performed parallel single cell RNA sequencing (scRNA-seq) and Mass Cytometry (CyTOF). For scRNA-seq, we analyzed CD45+ cells from the PDAC tissues of KPC GEMMs treated with vehicle, RT, FAKi, or RT+FAKi at day 14 post the start of treatment. Using Uniform Manifold Approximation and Projection (UMAP) representation, we could distinguish all major immune cell populations (Fig. 5A, S5A). To elucidate differences in TAM phenotype, we re-clustered these cells and observed five TAM clusters (termed Arg1-TAMs, Ccl7-TAMs, Apoe-TAMs, Ifitm3-TAMs, and Ccl7-TAMs) (Fig. 5B, S5B). Among these, we observed shifts in TAM subsets in RT+FAKi-treated tumors compared to all other treatments. This included decreased frequencies of Arg1-TAMs and increased frequencies of Ifitm3-TAMs and Ccl7-TAMs (Fig. 5C), suggesting possible changes toward T cell supportive and IFN-responsive phenotypes. To further investigate this, we performed Gene Set Enrichment Analysis (GSEA) across all TAMs subsets from vehicle versus RT+FAKi-treated mice and found upregulation of pathways associated with type-I and type-II interferons, T cell activation, phagocytosis, antigen processing and presentation, and ROS production (Fig. 5D-E, S5C-E). In agreement with scRNA-seq, CyTOF analysis of TAMs showed increased CD80, CD86, CD11c, and PDL2 expressions and decreased VISTA in RT+FAKi-treated mice compared to RT-treated mice (Fig. 5F-G, S5F-G). These data suggest that RT+FAKi treatment shifted TAMs away from immunosuppressive phenotypes and toward immune supportive roles.
Figure 5: Tumor-infiltrating immune cells from RT+FAKi treated mice have better anti-tumor signatures.

(A) UMAP dimensionality reduction plot of scRNA-seq data on CD45+ leukocytes from KPC mice at day 14 treated as depicted in Fig. 4A. Annotation shows different cell types. (B) UMAP analysis of TAMs subsets isolated from (A). Five different TAM clusters are depicted. (C) Count ratio of the TAM clusters from (B). (D-E) Bar graph displaying over-representation analysis of DEGs on all the TAMs in (B) to known biological functions in MSigDB_Hallmark (D) and GO databases (E). All graphs display comparisons of vehicle to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (F) UMAP dimensionality reduction plot of CyTOF data on TAMs and cDCs from KP2-OVA-GFP tumor-bearing RT and RT+FAKi-treated mice treated as depicted in Fig. 4A. Annotation shows different cell types. (G) Analysis of median expression levels of various markers on the TAMs, cDC1, and cDC2 in (F). n = at least 4 groups of pooled mice/group. (H) UMAP analysis of cDC subsets isolated from (A). Three different cDC clusters are depicted. (I) UMAP dimensionality reduction plot of scRNA-seq data on the adaptive immune cells from KPC mice. Annotation shows different cell types. (J) Bar graph displaying over-representation analysis of DEGs on cDC1 to known biological functions in GO database. All graphs display comparisons of vehicle to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (K) Dot plot displaying GSEA results from MSigDB_Hallmark database comparing different cDC clusters from (H). All graphs display comparisons of vehicle to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (L) Bar graph displaying over-representation analysis of DEGs on cDC2 to known biological functions in GO database. All graphs display comparisons of vehicle to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (M) Bar graph displaying over-representation analysis of DEGs on CD8+ T cells to known biological functions in GO database for Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). All graphs display comparisons of vehicle to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (N) UMAP dimensionality reduction plot of CyTOF data on CD8+ and CD4+ T cell populations from KP2-OVA-GFP tumor-bearing RT and RT+FAKi-treated mice. Annotation shows different cell types. (O) UMAP dimensionality reduction plot of CyTOF data on CD8+ T cell populations from (N). Annotation shows five different CD8+ T cell subclusters. OVA-Dextramer expression levels projected onto UMAP plots in (N). (P) Analysis of median expression levels of various markers on CD8+ OVA-Dextramer+ T cells in (O). n = at least 4 groups of pooled mice/group. (Q) Quantification of proliferating CD8+ OVA-Dextramer+ T cells out of total CD8+ T cells from mice treated as indicated. n = at least 4 groups of pooled mice/group. (R) CyTOF quantification analysis of exhausted CD8+ T cells in Fig. 5N. n = at least 4 groups of pooled mice/group.
All graphs depict mean +/− SEM. “*” denotes p < 0.05 by two-tailed t-test or one-way ANOVA as appropriate. “ns” denotes not significant.
As cDCs are critical to the induction of T cell priming, we next sought to determine how RT+FAKi altered their phenotype. We first isolated and re-classified cDC populations and adaptive immune cells by UMAP analysis (Fig. 5H-I). In cDCs, we defined three distinct clusters: cDC1, cDC2, and migratory DC (Fig. 5H, S5H). Mirroring our flow cytometry data (Fig. 4K), we found that PDAC tissues from RT+FAKi-treated mice had increased frequency of cDC1s (Fig. S5I). GSEA and over-representation analyses found that RT+FAKi increased expressions of genes in the type-I and type-II interferons, TNFα, and IL-12 pathways, as well as increased gene signatures for phagocytosis, antigen processing and presentation, and T cell activation across multiple cDC populations (Fig. 5J-L, S5J). By contrast, we observed decreases in pathways associated with DNA repair, MYC targets, oxidative phosphorylation, and MTOR signaling (Fig. 5K). Corroborating the changes in antigen presentation, we observed that RT+FAKi upregulated CD80 and CD86 in cDC1s and cDC2s, and decreased Tim3 in the cDC1s by CyTOF analysis (Fig. 5G). Together, these data suggest that cDCs were both numerically increased and phenotypically shifted toward an improved anti-tumor phenotype.
Finally, we utilized parallel scRNA-seq and CyTOF to assess the impact these changes had on T cell phenotype. To accomplish this, we identified CD4 and CD8 α/β T cells by UMAP analysis of the adaptive immune cells in our scRNA-seq data sets (Fig. 5I, S5K). Agreeing with the increase in activated CD8+ T cells found following RT+FAKi by flow cytometry (Fig. S4G), differential gene pathway analyses by scRNA-seq found increases in pathways involved in lymphocyte-mediated cytotoxicity, downstream antigen receptor signaling, lymphocyte activation, diversification of T cell receptor (TCR) genes, lysosome/autophagosome, secretory/zymogen granules, and antigen and cytokine binding, as well as decreases in integrin, VEGF, FGF, and angiogenesis pathways (Fig. 5M, S5L). Heatmap displaying differential gene expression of activation/effector marker also showed that many of these genes were upregulated in the dual RT+FAKi treatment compared to vehicle or monotherapies (Fig. S5M). CyTOF analysis of T cells defined several CD4+ and CD8+ T cell clusters (Fig. 5N-O, S5N-O). OVA-Dextramer+ CD8+ T cells were prominent in the exhausted effector (high for PD1, LAG3 and Tim3, but not Ki67) and the proliferative effector (high for Tbet, Eomes, and Ki67) compartments (Fig. 5O, S5N). We also noted that RT+FAKi-treated mice had increased expression of activation/effector markers, such as Tbet, CD44, and OX40 in OVA-Dextramer+ CD8+ T cells, and increased frequency of Ki67+ OVA-Dextramer+ CD8+ T cells compared with other treatments (Fig. 5P-Q, S5P). However, many OVA-Dextramer+ T cells were in the exhausted cluster with high PD1, LAG3 and Tim3 expression, and RT+FAKi increased the fraction of OVA-Dextramer-negative CD8+ T cells that clustered in the exhausted fraction (Fig. 5O,R, S5N). These data suggest that T cell checkpoint might be engaged during treatment. Analysis of CD4+ effector T cells showed increased OX40 and CD69 expressions in PDAC tissue from mice treated with RT+FAKi, suggesting that, though not as dominant, CD4+ T cells might also play a role (Fig. S5Q). Taken together, our data suggest that both innate and adaptive tumor-infiltrating immune cells from RT+FAKi-treated mice acquired a more anti-tumor phenotype; however, it is unclear if this will be confounded or aided by changes in desmoplastic stromal responses.
FAKi reshapes CAFs to participate in tumor immunity
Like tumor-infiltrating immune cells, CAFs, which are abundant in PDAC, can play a large role in determining treatment response(12,13,17,27,29,62). To further understand how our treatments changed the PDAC CAFs, we performed scRNA-seq from CAF-enriched CD45−, CD31−, EPCAM−, PDPN+ cells isolated by FACS from KPC mice 14 days after treatment start. UMAP analysis of these CAFs generated seven unique subclusters (Fig. 6A, S6A). Using known CAF markers(28,29), we were able to identify the classic myofibroblastic CAFs (myCAFs) marked by Acta2 and Col12a1, inflammatory CAFs (iCAFs) marked by Cxcl12 and Ly6c, and antigen-presenting CAFs (apCAFs) marked by Cd74 and H2-Ab1 (Fig. S6A-C). Interestingly, treatments containing FAK inhibitor generated distinct CAF clusters that did not fall into the 3 classical CAF clusters identified in vehicle or RT samples (Fig. 6A-B, S6C). We named these FAKi-treatment induced CAFs FAK.CAF.1, FAK.CAF.2, FAK.CAF.3, and RTFAK.CAF based on the treatments which induced them (Fig. 6B, S6B). These FAKi-induced phenotypic clusters were also seen with multiple data normalization and integration strategies, suggesting these phenotypic population shifts were not due to batch or computational effects but rather a treatment-induced shift in transcriptional phenotype (Fig. S6D-F). We observed that FAKi-induced CAF subsets became enriched in FAKi-treated samples at the expense of myCAF subsets (Fig. 6B, S6B,D-F). Additionally, we observed a decreased number of αSMA+ CAFs by IHC and collagen density by Sirius Red staining in both FAKi- and RT+FAKi-treated mice compared to controls (Fig. 6C-D). These data suggest that FAKi treatment significantly altered the CAF composition/phenotype.
Figure 6: FAKi reshapes CAFs to participate in tumor immunity.
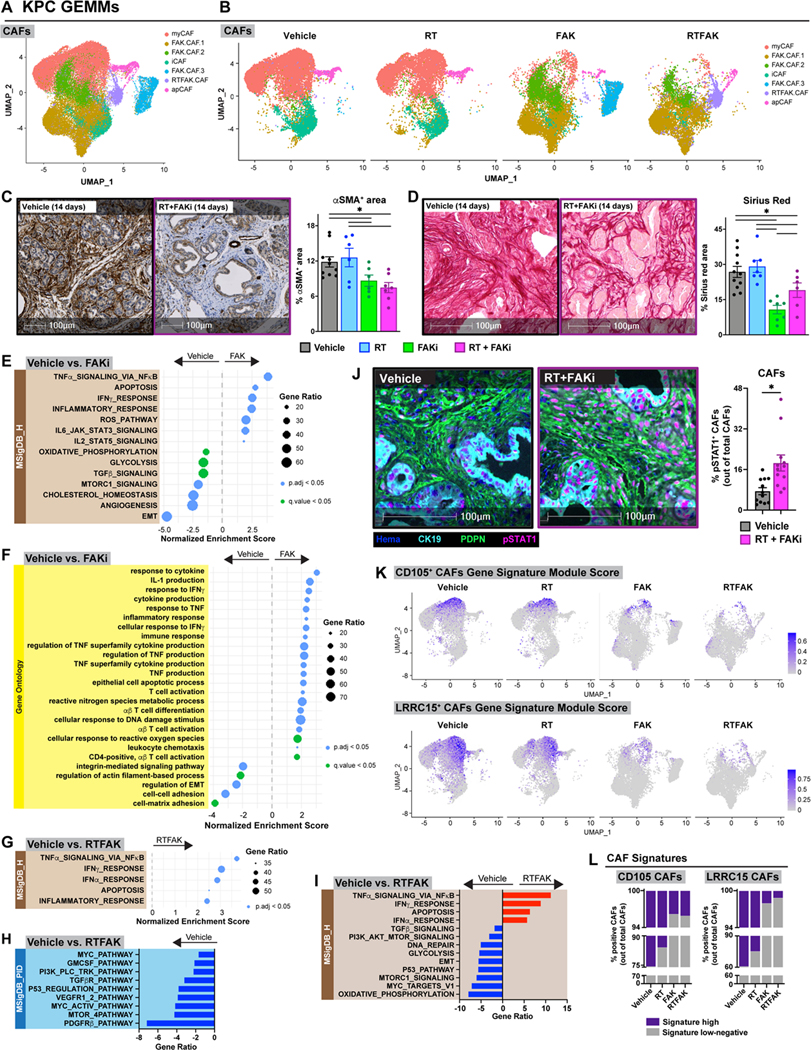
(A) UMAP dimensionality reduction plot of scRNA-seq data on CD45− CD31− Epcam− PDPN+ FACS-sorted CAFs-enriched from KPC mice at day 14 treated as depicted in Fig. 4A. Annotation shows different cell types. (B) UMAP distribution from (A) split by treatment conditions. (C-D) Analysis of αSMA IHC (C) and collagen density (D) on tissues from KPC mice 14 days post-RT start. Representative αSMA IHC and Sirius Red images are depicted. n = at least 7 mice/group. (E-F) Dot plot displaying GSEA results from MSigDB_Hallmark (E) and GO (F) databases. All graphs display comparisons of CAFs from vehicle to FAKi-treated mice. All pathways were filtered with p.adj value or q value < 0.05. (G-I) Dot plot displaying GSEA results from MSigDB_Hallmark database (G) and bar graphs displaying over-representation analysis of DEGs on CAFs in (A) to known biological functions in MSigDB_PID (H) and MSigDB_Hallmark (I) databases. All graphs display comparisons of CAFs from vehicle mice to RT+FAKi-treated mice. All pathways were filtered with p.adj value < 0.05. (J) mIHC analysis of CK19− PDPN+ pSTAT1+ cells in tissues of KPC mice from Fig. 4A. Representative CK19, PDPN, and pSTAT1 fused mIHC images are depicted. n = at least 6 mice/group. (K) UMAP representation of CD105+ CAFs or LRRC15+ CAFs gene signature module scores mapped onto CAFs in (A-B). (L) Quantification of CAFs in (A) for CD105+ CAFs gene signature or LRRC15+ CAFs gene signature.
All graphs depict mean +/− SEM. “*” denotes p < 0.05 by two-tailed t-test or one-way ANOVA as appropriate.
To further elucidate the phenotypic differences in these CAF subsets, we performed GSEA and over-representation analyses comparing CAFs in vehicle-treated mice to CAFs in FAKi-treated mice. In comparison to CAFs from vehicle mice, CAFs from FAKi-treated mice upregulated pathways associated with TNFα, IFNγ, inflammatory response, ROS, IL-6 and IL-2 signaling, and T cell activation while downregulating pathways associated with epithelial-to-mesenchymal transition (EMT), angiogenesis, TGFβ, MTORC1 signaling, oxidative phosphorylation, and glycolysis (Fig. 6E-F, S6G-H). As expected, we also saw downregulation of FAK and integrin signatures in CAFs in FAKi-treated mice (Fig. 6F, S6H).
To confirm that we retained the same anti-tumor phenotypes generated by FAKi treatment, we performed GSEA and over-representation analyses comparing CAFs in vehicle-treated mice to CAFs from RT+FAKi-treated mice. In comparison to vehicle, RT+FAKi-treated mice upregulated pathways associated with TNFα, IFNγ, and inflammatory response, similar to CAFs from FAKi-treated mice. Furthermore, the addition of RT to FAKi resulted in upregulation of pathways associated with IFNα (Fig. 6G-I). We also observed downregulation of TGFβ, MYC, integrins, MAPK, GMCSF, VEGFR, FAK signaling, and DNA repair pathways as well as upregulation of leukocyte migration and chemotaxis (Fig. 6G-I, S6I). To cross-validate the upregulation of IFN signaling in CAFs, we performed mIHC for CK19, PDPN, and pSTAT1 and found an upregulation of pSTAT1+ CK19− PDPN+ CAFs in RT+FAKi-treated mice (Fig. 6J).
Several CAF subsets have been identified in human PDAC with potential immunomodulatory activity and unique origins. These include CD105+(63), LRRC15+(64), and NetrinG1+ CAFs(65), as well as pancreatic stellate cell (PSC)-derived CAFs(66). To understand how our treatments impacted these subsets, we analyzed previously defined signatures for these CAF subsets using an averaged Z-score (Seurat’s “AddModuleScore”) or specific marker genes. Unfortunately, we could not detect sufficient levels of NetrinG1 or PSC-derived transcripts to reach a conclusion for these subsets. However, gene signature-positive or marker genes-positive CD105+ CAFs and LRRC15+ CAFs were decreased in both FAKi and RT+FAKi-treated PDAC tumors (Fig. 6K-L, S6J-K). Taken together, these data suggest that inhibition of FAK signaling remodeled the PDAC CAFs’ phenotype to augment IFN signaling in the stroma which may facilitate anti-tumor immunity.
FAK inhibition in combination with RT in PDAC patients activates interferon signaling
Based on the data above, we initiated a clinical trial of a FAK inhibitor (defactinib, VS-6063) in combination with SBRT in locally advanced PDAC patients (NCT04331041). In the patients from the phase I safety lead-in cohort, we performed scRNA-seq on tissues from pre- and post-treatment biopsies (Fig. 7A). In data from the first five patients, UMAP analysis identified PDAC cells, CAFs, and major immune populations (Fig. 7B, S7A). Mirroring what we found in our pre-clinical studies (Fig. 3), differential gene pathway analysis of the PDAC cells showed that the combination treatment led to upregulation of inflammatory response, interferon, TNFα signaling, T cell activation, and phagocytosis pathways and downregulation of MYC pathways (Fig. 7C, S7B). These data suggest that RT+FAKi might halt PDAC proliferation and augment immunogenicity. To further understand how SBRT + VS-6063 treatment changed the TME in our patients, we re-clustered the CAFs, T cells, and TAMs separately and compared differentially expressed genes and pathways in these pre- and post-treatment biopsies. In CAFs, we observed that SBRT + VS-6063 increased the IFNα, IFNβ, TNFα signaling, and T cell activation pathways, and decreased the TGFβ and integrin signaling pathways (Fig. 7D, S7C-F). Similarly in T cells, we observed increases in IFNα, IFNβ, and TNFα pathways, and in TAMs we observed increases in IFNα, IFNβ, and ROS pathways, and decreases in glycolysis and MTORC1 pathways in SBRT + VS-6063 post-treatment samples (Fig. 7E-G, S7G-I). These results from CAFs, T cells, and TAMs in humans parallel our results obtained in mice, which demonstrate activation of IFN signaling in both the target tumor compartment and the tumor-associated stroma.
Figure 7: FAK inhibition in combination with RT in PDAC patients activates interferon signaling.

(A) Schematic of clinical trial (NCT04331041). Patients received SBRT with a total of 50Gy given in 5 fractions. Defactinib (VS-6063) was given at a dose of 400mg BID starting the end of day 2 post-SBRT and continued up to one year. Tissues were obtained pre-treatment and 12–14 weeks post-SBRT. (B) UMAP analysis of eight pre- and post-treatment biopsies from (A). (C) Bar graph displaying GSEA analysis of DEGs on all the PDAC cells in (B) to known biological functions in MSigDB_Hallmark database. All graphs display comparisons of pre-treatment to post-treatment biopsies. All pathways were filtered with p.adj value < 0.05. (D) Bar graph displaying GSEA analysis of DEGs on all the CAFs in (B) to known biological functions in MSigDB_Hallmark database. All graphs display comparisons of pre-treatment to post-treatment biopsies. All pathways were filtered with p.adj value < 0.05. (E) Bar graph displaying GSEA analysis of DEGs on all the T cells in (B) to known biological functions in MSigDB_Hallmark database. All graphs display comparisons of pre-treatment to post-treatment biopsies. All pathways were filtered with p.adj value < 0.05. (F) Dot plot displaying GSEA analysis of DEGs on all the TAMs in (B) to known biological functions in MSigDB_Hallmark database. All graphs display comparisons of pre-treatment to post-treatment biopsies. All pathways were filtered with p.adj value < 0.05 or q value < 0.05. (G) Bar graph displaying GSEA analysis of DEGs on all the TAMs in (B) to known biological functions in and GO database. All graphs display comparisons of pre-treatment to post-treatment biopsies. All pathways were filtered with p.adj value < 0.05. (H) Schematic of RT (6Gy x 5), FAKi (75mg/kg BID), and ICB (αPD1, 200μg and αCTLA4, 200μg) administration in KP2-OVA-GFP tumor-bearing mice. Mice were treated and longitudinally assessed for tumor burden. (I) Waterfall plot of KP2-OVA-GFP tumor-bearing mice from (H) evaluating tumor growth difference from day 0 to 5. n = 10 mice/group. (J) Kaplan-Meier survival curve of KP2-OVA-GFP mice from (H). n = 10 mice/group. (K) Functional assay of CD8+ and CD4+ T cells by flow cytometry from mice treated as in (H). Tumors were isolated at day 10 post the start of treatment. Bar graphs showed quantification of IFNγ ± TNFα positive CD8+ and CD4+ T cells. n = at least 6 mice/group. (L) Graphical abstract of primary findings.
For comparison between multiple groups, “*” denotes p < 0.05 by two-tailed t-test, one-way ANOVA, or Kaplan-Meier as appropriate.
FAK inhibition in combination with RT renders checkpoint blockade effective
Our data suggest that adding FAK inhibition to RT led to tumor regression and priming of tumor-antigen-specific T cells (Fig. 4B-D,G,I-J, S4A-D,G). However, these effects in turn drove upregulation of T cell exhaustion (Fig. 5N-R, S5N). This suggests that these newly primed CD8+ T cells might become limited by checkpoints. To test this, we treated mice bearing established syngeneic KP2-OVA-GFP PDAC tumors with RT+FAKi in combination with αPD1 and αCTLA4 IgGs (immune checkpoint blockade, ICB, Fig. 7H). Mirroring our T cell depletion studies, short-term tumor regression by RT+FAKi was not impacted by the addition of ICB (Fig. 7I); however, the triple combination of RT+FAKi+ICB led to progressive tumor regression over time and long-term survival in 8 of 9 mice in the treatment group, which was superior to all other treatment groups (Fig. 7J). We next compared the effector phenotype of T cells in these mice. We found that the addition of FAKi to RT+ICB resulted in higher numbers of IFNγ- and TNFα-expressing CD8+ and CD4+ T cells (Fig. 7K). Furthermore, the CD4+ T cells from these RT+FAKi+ICB-treated mice were skewed toward CD4+ T effector cells instead of CD4+ Foxp3+ T regulatory cells (Fig. 7K, S7J). We repeated this in the KRAS-INK PDAC model and observed that the addition of ICB to RT+FAKi also sustained more durable tumor regression (Fig. S7K-L). Our mechanistic mouse models and human data suggest that combining FAK inhibition with RT can reprogram the TME to support IFN signaling and tumor immunity (Fig. 7L). These preclinical checkpoint combination data also suggest that the changes brought by RT+FAKi could be further enhanced with the addition of checkpoint immunotherapy.
DISCUSSION
Many factors contribute to the recalcitrant nature of PDAC(2,15–18). Here, we show that the PDAC stroma can drive RT resistance. Our findings are in agreement with several other studies, which highlight the critical role of β1 integrin(61,67,68) and Caveolin(69) signaling in regulating RT sensitivity in multiple cancer models. ECM can impact RT response by several mechanisms, including directly signaling to tumor cells through integrins and other receptors(59–61,70,71), acting as a reservoir for secreted mitogens(72–74), and serving as a major regulator of oxygen bioavailability(75,76). While these studies showed that the stromal ECM can promote survival of cancer cells following RT, it remains unclear how the PDAC stroma might affect RT-induced immune priming. Interestingly, our data suggest that collagen-rich ECM not only promotes cancer cell survival but also represses RT-induced IFN signaling that may be critical to RT-induced tumor immunity.
In our studies, using KPC GEMMs, we found that although RT resulted in temporary short-term tumor control, it failed to prime T cell responses. This was translated to a failure of long-term tumor control and no survival benefit. This is noteworthy because RT is highly capable of priming T cells and controlling tumor growth in other cancer models(77,78), including the Kras/p53-driven sarcomas we presented here. Even though our PDAC and sarcoma GEMMs carry the same driver mutations in Kras and p53, we noted a major difference in the amount of stromal desmoplasia; the PDAC stroma is highly desmoplastic compared to genetically equivalent sarcomas. In our current study, we found collagen and pancreatic fibroblasts cooperatively mitigated RT efficacy and induction of tumor cell death in-vitro, which was in part mediated through fibroblasts’ support of PDAC cell proliferation and collagen’s ability to blunt RT-induced IFN signaling (Fig. 2). Similar interactions have been reported, in which PSCs’ secreted factors, such as IL-1α and TGFβ, enhance PDAC cell proliferation, migration, invasion, and colony formation, and drive resistance to gemcitabine and RT through MAPK and AKT signaling(79). Furthermore, PSCs can directly interact with PDAC cells to promote radioprotection and stimulate tumor cell proliferation through β1 integrin signaling, which is independent of phosphoinositide 3-kinase but depends on FAK(67,80). The presence of extensive desmoplasia and its role in promoting tumor proliferation in PDAC further explains why, unlike in sarcoma, RT counterintuitively increases PDAC tumor cell proliferation after resistance occurs. This suggests that RT may perform differently in a tumor which is stroma-rich versus stroma-light, and these differences are partly mediated by ECM-receptor FAK signaling.
Retrospective studies of surgical resections have shown that higher levels of fibrosis post-neoadjuvant treatment are correlated with better pathologic response and improved survival(81,82). Conversely, we found that collagen and fibroblasts in treatment-naïve tumors additively blunt RT efficacy, which suggests that treatment-naïve and post-treatment desmoplasia could have distinct characteristics and effects, the earlier which can affect treatment response. It may be more relevant to assess post-treatment PDAC fibrosis/desmoplasia in the context of canonical FAK activation as an indicator of the downstream ECM-receptor signaling effects of collagen deposition.
Prior studies have demonstrated that inhibition of FAK signaling in PDAC changes the phenotype of both tumor cells and stromal cells(41,47,48,83). This prompted us to see if FAKi could overcome stromal-induced protection to result in RT-induced tumor death. To our surprise, we found that FAKi reversed RT resistance in PDAC, and these effects corresponded with changes in DNA damage repair/responses, ROS pathways, cell cycle regulation, and inflammatory signaling (Fig. 3). Similarly, in melanoma and lung cancer, FAKi can resensitize tumor cells to RT by modulating DNA damage responses(84) and NFκB activity(41). These FAKi-specific pathways in tumor cells may play a role in promoting RT efficacy in PDAC. Our in-vitro RNA-seq and Proteomics studies focused on signaling in the cancer cells alone; however, the biologic effects of FAKi and RT are not limited to the cancer cells. The role of these therapies in shaping PDAC stromal cells, particularly fibroblasts, is noteworthy and impactful. Indeed, scRNAseq data from KPC GEMMs and human PDAC patients suggest significant changes in stromal phenotype and potentially tumor-stromal interactions. Further work needs to be done to disentangle the cell type and tissue-specific differences in FAK signaling and their respective contributions to RT resistance.
Aside from tumor cell-directed effects of the combination treatment, we likewise found that in the presence of FAKi, RT could induce immune priming. This was marked by the increase in tumor-infiltrating cDC1s and tumor-antigen-specific T cells, along with shifts in TAMs, cDCs, and T cell phenotypes toward more anti-tumor. We also found an increase in both type-I and type-II IFN responses/pathways in the dual RT+FAKi treatment, but not in either RT or FAKi monotherapy alone. We postulate that these changes in tumor immunity facilitate RT’s ability to prime better anti-tumor T cell responses, both in the targeted tumor tissue and systemically. One possible explanation for the ability of FAK inhibition to rescue RT-induced immune priming is through altering collagen interactions in the stroma and how they contribute to IFN signaling.
CAFs are another major contributor to the stromal modulation of RT treatment response in PDAC(27–30). Similarly, CAFs have been implicated in PDAC resistance to both chemo- and immunotherapies(31–34). However, the distinct roles of CAF subpopulations to therapeutic resistance mechanisms have yet to be fully elucidated(85–87). For example, the extent to which CAF subtypes are interchangeable is unclear, and further study is needed to determine what dictates their numbers, distribution, and function during treatment. Interestingly, we found that RT+FAKi causes significant changes in the composition of CAFs which raised the possibility that these CAFs may participate productively in anti-tumor immunity. Previous studies have shown that CAFs can play a role in tumor immunity through IL-1, TGFβ, and IL-6 signaling(28–30,88), and our study identifies a novel role for CAFs in anti-tumor immunity through IFN signaling. We also found that CAFs in the RT+FAKi-treated mice downregulated TGFβ signaling, which has been shown to be a key signaling pathway in the pro-tumor CD105+ CAFs(63), αSMA+ myCAFs, and LRRC15+ CAFs(64). Accordingly, we found FAKi decreased both CD105 and LRRC15 gene signature-expressing CAFs, which suggest a potential shift toward anti-tumor phenotype. While NetrinG1+ and PSC-derived CAFs have both been identified(65,66), our data on these subsets was inconclusive. It will be interesting to address the role and origin of CAF heterogeneity in FAKi efficacy as more relevant tools are established. Important for this may also be identifying the CAF-intrinsic vs. cancer cell-driven roles of FAK-signaling in shaping CAF heterogeneity.
Overall, this study suggests that stromal modulation by FAKi sensitizes PDAC to RT-induced anti-tumor immunity in both the immune and stromal compartments (Fig. 7L). Furthermore, in combination with FAKi, RT unlocks sustained checkpoint immunotherapy efficacy which is translated into long-term tumor control and/or eradication. Based on our findings, we are currently testing the clinical efficacy of FAK inhibition in combination with SBRT in patients with locally advanced PDAC (NCT04331041), which to this point has mirrored many of the biomarker findings presented in mouse models. Perhaps most significantly, our data strongly supports the addition of immune checkpoint blockade, which may be critical for the prevention of metastatic disease in these PDAC patients.
METHODS
Genetic mice and other models
p48-Cre/LSL-KrasG12D/p53Flox/Flox (KPC) and p48-Cre/LSL-KrasG12D/p53Flox/Flox/OVA-GFP+ (KPC-OG) mice used in these studies were bred to C57Bl/6J background and verified by congenic markers. The generation and studies entailing the details of the KPC-OG mouse were published by our lab(55). KPC and KPC-OG mice were enrolled for treatment studies when the first >0.5 cm tumor was detected by biweekly palpation corroborated by ultrasound measurement. Thereafter, tumor size was assessed weekly by ultrasound (SonoSite m-Turbo). Survival events were scored when mice lost >15% body weight, tumor burden reached >1.8 cm in diameter, moribund appearance, severe cachexia, or per absolute survival. For all studies, care was taken to include negative littermates as well as sex- and age-match in the same experimental setup.
LSL-KrasG12D/p53Flox/Flox/OVA-GFP+ mice, littermates of KPC-OG mice, were injected with Adenoviral-Cre at 8–10 weeks old of age which generated sarcoma GEMMs (KPS-OG) 7–8 weeks post inoculation. Tumor size was assessed by caliper measurement for randomization.
All animal studies were approved by the Washington University IACUC review board.
Human subjects
All human PDAC tissues were obtained under written informed consent from the patients. Washington University Ethics committee approved the study in accordance with recognized ethical guidelines and studies were conducted under institutional review board protocol #201108117 and #201704078 and clinical trial NTC04331041.
Cell lines
3D organoid cell lines (KPOG) were derived from tumor-bearing female KPC-OG mice. KRAS-INK or KI cells used in certain orthotopic transplant experiments were derived from Pdx1-Cre/LSL-KrasG12D/Ink/ArfFlox/Flox mice in Dr. Hanahan’s laboratory (EPFL, Lausanne). KP2 cells were derived from PDAC tumor tissue obtained from 6-month-old p48-Cre/LSL-KrasG12D/p53Flox/+ mice, which were screened for C57BL/6 identity. Cells were grown out on collagen-coated plastic for <12 passages and were tested for CK19, αSMA, Vimentin and CD45 to verify their identity and purity. KP2-OVA-GFP cells were derived from tumor-bearing female KPC-OG, which were used in certain syngeneic subcutaneous experiments. PDA.69 cells were derived from Pdx1-Cre/LSL-KrasG12D/p53R172H/+ mice as previously published(51). A single lot of frozen cells were used for all experiments. These cells were banked and last tested for mycoplasma using two independent commercial kits (Sigma and Lonza) in June 2019.
Three independent WT pancreatic fibroblast primary cell lines were derived from WT C57Bl/6J background mice. Pancreas was harvested (detailed below) and plated on tissue culture flasks under standard antibiotics and the addition of Gentamycin and Amphotericin B. No additional growth factors were supplemented in culture media. After ~3 passages, the culture was composed of pure fibroblasts only. For organoid growth studies, only fibroblasts with passage <10 were used. Fibroblast purity was tested by flow cytometry for PDPN as a pan-marker of fibroblasts. All cell lines were tested negative for mycoplasma.
Two independent CAF lines were derived from end-staged KPC-OG mice. Following tissue digestion, single-cell suspension was resuspended in flow cytometry buffer and stained with fluorophore-conjugated antibodies as detailed below. Cells were sorted for CAFs (GFP− CD45− CD31− Epcam− PDPN+) using FACSAria-II (BD Biosciences), and re-plated in complete medium until stable cell lines were established. CAFs purity was tested by flow cytometry for EPCAM− and PDPN+ as a pan-marker of fibroblasts. All cell lines were tested negative for mycoplasma.
Tumor cell suspension derived after tumor harvest (detailed below) was plated on collagen-coated tissue culture flasks under standard antibiotics for the generation of 2D cell culture. Cell suspension derived after tumor harvest (detailed below) was plated on Matrigel (Cultrex® Organoid Qualified BME, Type 2) dome under standard antibiotics for the generation of 3D organoid cell culture. Protocol for the generation of 3D organoid cell culture was adapted from Dr. Tuveson’s laboratory (Cold Spring Harbor Laboratory)(89). GFP+ tumor cells were sorted on FACSAria-II (BD Biosciences), and re-plated in complete medium until stable cell lines were established. All 2D cell lines were cultured in complete medium (DMEM-F12 with 10% FBS and 1% PenStrep) at 37° C and 5% CO2. All 3D cell lines were cultured in complete medium (Advanced DMEM-F12 with 1% HEPES, 1% L-glutamine, and 1% PenStrep) at 37°C and 5% CO2. All cell lines were passaged <6 times and were tested positive for cytokeratin-19, and negative for smooth muscle actin and vimentin to verify their carcinoma identity and purity. All cell lines were tested negative for mycoplasma.
Tissue harvest
Mice were euthanized by trans-cardiac perfusion using 15 mL of PBS-heparin under isoflurane anesthesia. When taken for histology, tumor tissues were fixed in 10% neutral-buffered formalin overnight at 4°C. When taken for cellular assays, tumor tissues or respective lymph nodes were manually minced and digested in 20 mL of sterile 1X HBSS (Thermo Fisher) containing 2 mg/mL of collagenase A (Roche) and 1X DNase-I (Sigma) for 30 min at 37°C with constant stirring. Digestion was quenched in 5 mL of sterile fetal bovine serum (FBS, Atlanta Biologicals) filtered through 40 μm Nylon mesh, pelleted through centrifugation (2000 RPM for 5 min at 4°C), and resuspended in required media/buffer as single cell suspensions.
Orthotopic and syngeneic implantations
Age-matched 6–8-week-old female C57BL/6 and FVB/NJ mice were used for orthotopic/transplantable mouse models. Syngeneic orthotopic PDAC tumors were established by surgical implantation, as previously described(47). To establish orthotopic models, 100,000 KRAS-INK or KP2 cells in 50 μL of Cultrex (Trevigen) were injected into the pancreas of sex-matched C57BL/6 or FVB/NJ mice as previously described(90). Cohorts of mice were randomized into different treatment groups by gross tumor diameter using thrice-weekly palpation, external caliper measurement, and ultrasound measurement. To establish subcutaneous models, 250,000 KP2 or KP2-OVA-GFP cells or 100,000 PDA.69 cells in 50 μL of Cultrex (Trevigen) were injected into the mammary fat pad of sex-matched C57BL/6 mice as previously described. Cohorts of mice were randomized into different treatment groups by gross tumor diameter using thrice-weekly external caliper measurement.
Pharmacologic inhibitors
FAKi (VS-4718), provided by Verastem, Inc., is a selective bispecific inhibitor with activity against FAK1/PTK2 and Pyk2/PTK2b kinases. Cell-based assays have determined it has biochemical half-maximal inhibitory concentrations (IC50) of 6.0 nM and 20 nM for FAK and Pyk2. For animal experiments, 75mg/kg VS-4718 was formulated in vehicle (0.5% carboxymethyl cellulose and 0.1% Tween-80 (Sigma-Aldrich) in sterile water) and administered by oral gavage twice a day. For in-vitro studies 0.25, 0.5, or 1.0 μM of VS-4718 in DMSO was used.
Radiation therapy
Mice received Radiation Therapy (RT) as five daily hypofractionated doses (6Gy x 5) using the Small Animal Radiation Research Platform (SARRP200, XStrahl Life Sciences). Mice were injected i.p. with an iodine contrast agent (2100mg/kg) before being placed on the irradiation platform one at a time under isoflurane anesthesia. Conebeam computed tomography (CT) imaging was performed for each individual mouse to pinpoint the pancreas, images were imported into Muriplan and used to select an isocenter. The tumor was then irradiated using anterior-posterior-opposed beams using either the 5mm x 5mm or 10mm x 10mm collimator at a dose rate of 3.9 Gy/min. Mice were monitored over 2 weeks for signs of radiation sickness or weight-loss. DietGel recovery gel was provided for 14-day window immediately following radiation therapy in survival studies.
For in-vitro radiation experiments, RS2000 160kV X-ray Irradiator using a 0.3 mm copper filter (Rad Source Technologies) was used.
Immunotherapeutic neutralizing antibodies
For immunotherapy regimen, 250 μg of agonist antibodies (αCTLA4 clone UC10–4F10–11; αPD1 clone RMP1–14; BioXCell) were given by intraperitoneal (i.p.) injection; αPD1 was given every 4 days for a total of 30 days and αCTLA4 was given every 5 days for a total of 4 doses from beginning of treatment. Treatments were discontinued after 30 days to prevent α-rat IgG reaction.
For T cell depletion, CD4 or CD8 neutralizing IgG antibodies (αCD4 clone GK1.5; αCD8 clone 2.43, BioXCell) were administered, with 1st injection (at day −2) containing 400 μg and subsequent injections (every 4 days) containing 200 μg of each IgG. Treatments were discontinued after 30 days. As control, rat IgG2b isotype control (clone LTF-2, BioXCell) was administered.
For type-I IFN neutralization, αIFNAR1 (clone MAR1–5A3, BioXCell) antibody was administered, with 1st injection (at day −2) containing 750 μg and subsequent injections (every 3 days) containing 250 μg of each IgG. Treatments were discontinued after 30 days. As control, mouse IgG1 isotype control (clone MOPC-21, BioXCell) was administered.
Organoid cultures
3D organoid cell lines derived from KPC-OG mice, called KPOG, were passaged in Cultrex® Organoid Qualified BME, Type 2. Pancreatic fibroblasts were derived from C57Bl/6J mice. Organoid cells (~50 structures in a dome of 50μL of Cultrex® Organoid Qualified BME, Type 2) were cultured with or without 100,000 pancreatic fibroblasts or FACS-sorted CAFs per dome. Collagen-I concentrations (1.0 mg/mL) were modulated in the ECM matrix. One day post organoid culture preparation, organoid cultures were treated with FAKi at varying dose concentrations. Two hours post FAKi treatment, culture was treated with radiation.
All organoid experiments were done in 24-well tissue culture treated plates. At least n=3/group was used for statistical power analysis. KPOG organoid growth was monitored using the GFP marker. Fluorescence images were taken using Nikon AZ100 microscope daily and images were analyzed using Metamorph and HALO imaging software for analyses.
Immunohistochemical staining
Tissues were fixed in 10% neutral formalin for 18–24 hours, embedded in paraffin after graded-ethanol dehydration, and sectioned into 5-μm sections using a microtome. Where applicable, formalin-fixed, paraffin embedded (FFPE) tissue sections were stained for Hematoxylin & Eosin (Thermo Fisher), Picro-Sirius Red (Sigma-Aldrich) and Masson’s Trichrome (Diagnostic Biosystems) according to manufacturer’s instructions. Automated staining of tissues was carried out on the Bond RXm (Leica Biosystems) following dewaxing and appropriate antigen retrieval. Immunostaining was chromogenically visualized using the Bond Polymer Refine Detection alone or in conjunction with Bond Intense R Detection Systems (DS9263, Leica Biosystems). Slides were dehydrated through graded ethanol, followed by xylene, then mounted using Xylene-based Cytoseal (Thermo Fisher) or Vectamount (Vector Labs) as appropriate.
Staining was performed with the following antibodies: αSMA (Abcam ab5694), Podoplanin (Biolegend 127402), Ki67 (Abcam ab15580), BrdU (Abcam ab2284), CC3 (Cell Signaling 9661S), Cytokeratin 19 (DSHB TROMA-III), Cytokeratin 17/19 (Cell Signaling 12434S), CD8α (Cell Signaling 98941), pSTAT1 (Cell Signaling 8826s), CD4 (Abcam ab183685), Foxp3 (eBioscience 14–5773-82), γH2Ax (Cell Signaling 9718S).
Flow cytometry
Following tissue digestion, single cell suspensions were resuspended in flow cytometry buffer (PBS containing 1% BSA and 5 mM EDTA), FcR blocked with rat α-mouse CD16/CD32 antibodies (EBioscience) for 10 min and pelleted by centrifugation. Where applicable, CD8+ T cells-specific for antigen OVA were labeled by incubating cell suspension with H2Kb::SIINFEKL-specific MHCI dextramer (1:5; Immudex protocol) for 10 min at room temperature prior to extracellular staining. Cells were consequently labeled with 100 μL of fluorophore-conjugated α-mouse extracellular antibodies at recommended dilutions for 25 min on ice. Intracellular staining for intracellular markers was conducted subsequently using the EBioscience Transcription Factor Staining buffer set, according to manufacturer’s instructions. All antibodies are listed below. FCS Data were acquired on BD Fortessa X-20 (BD Biosciences) within 3–4 days and analyzed using FlowJo software (v10).
For T cell functional (cytokine release) assay, following tissue digestion, primary tumor cell suspension containing 100,000 cells were incubated in 96-well plate with 1μM Brefeldin A (Biolegend) and 2μM Monensin Solution (Biolegend) and 1X Stimulation Cocktail (eBioscience) for 5 hours at 37C and 5% CO2. After incubation, cells were then labeled with fluorophore-conjugated anti-mouse antibodies as above.
Staining was performed with the following antibodies: CD45 (eBioscience 25–0451), CD45 (BD Biosciences 564225), CD3ε (eBioscience 17–0031-82), CD11b (eBioscience 56–0112-82), MHC-II (eBioscience 48–5321-82), Ly6G (Biolegend 127608), Ly6C (eBioscience 45–5932), F4/80 (eBioscience 15–4801-82), CD11c (eBioscience 47–0114-82), CD24 (eBioscience 11–0241-82), CD24 (eBioscience 11–0242-82), CD103 (Biolegend 121423), CD8α (BD Biosciences 563786), CD8α (BD Biosciences 562283), CD4 (eBioscience 11–0043-82), CD62L (Biolegend 104437), CD44 (eBioscience 48–0441-82), PD1 (eBioscience 12–9985-82), Tim3 (Biolegend (119715), Ki67 (eBioscience 50–5698-82), Foxp3 (eBioscience 35–5773-82), MHC-I (eBioscience 11–5998-82), B220 (Biolegend 103212), SIINFEKL Dextramer (Immudex JD2163), IFNγ (Biolegend; clone XMG1.2), TNFα (Invitrogen; clone MP6-XT22).
Mass cytometry
Tumors taken from KP2-OVA-GFP tumor bearing mice were taken at day 14 post-treatment start. Two to three tumors were pooled as one sample. Tumor samples were digested in HBSS supplemented with 2mg/ml collagenase A (Roche), 2.5U/ml hyaluronidase and DNase I at 37C for 30 min with agitation to generate single-cell suspensions. Cell suspensions were counted and stained in 5 μM cisplatin per million cells for exactly 3 min on ice and washed with Cy-FACS buffer (PBS, 0.1% BSA, 0.02% NaN3, 2 mM EDTA) twice. Cells were incubated with FcR blocking reagent plus surface-antibody cocktail for 40 min on ice. After incubation, surface-marker stained cells were washed twice with Cy-FACS buffer. Cells were then fixed with 4% PFA for 10 min on ice and permeabilized with permeabilization buffer (Invitrogen) for 40 min containing the intracellular stain cocktail. All antibodies are listed below. Cells were then washed twice with PBS and stained with 200 μl of DNA intercalator per million cells. Cells were acquired on a CyTOF2 mass cytometer (Fluidigm) and data were uploaded to Cytobank for further analysis. Events were gated on singlets, live, and CD45+. A maximum of 100,000 events were then visualized using standard t-SNE algorithm. Populations of interest were manually gated and verified based on lineage marker-expression.
Staining was performed with the following antibodies: CD45 (Fluidigm 3089005B), CD90 (Biolegend 105202), CD11c (Fluidigm 3142003B), CD68 (Biolegend 137001), MHC-I (Fluidigm 3144016B), CD206 (Biolegend 141702), F4/80 (Fluidigm 3146008B), MHC-II (Biolegend 107602), CD11b (Fluidigm 3148003B), CD172α/SIRPα (Biolegend 144002), Ly6C (Fluidigm 3150010B), Ly6G (Fluidigm 3151010B), CD64 (Biolegend 139301), XCR1 (Biolegend 148202), CD103 (Biolegend 121402), NK1.1 (BioXcell BE0036), Bst2 (Novous/imgenx DDX0390P-100), IRF4 (Biolegend 646402), CD83 (Thermofisher Scientific 14–0831-82), CD40 (Fluidigm 124601), Ox40L (Biolegend 108802), CCR2 (RnD systems MAB55381–100), Cx3CR1 (Fluidigm 3164023B), CCR7 (Biolegend 120101), PDL2 (BioXCell BE0112), VISTA (Biolegend 150202), Tim3 (BioXcell BE0115), PDL1 (BioXCell BE0101), CD80 (Biolegend 104702), CD135/FLT3 (Thermofisher Scientific 14–1351-82), CD86 (Fluidigm 3172016B), Tim4 (Biolegend 130002), B220 (Fluidigm 3144011B), CD44 (Leinco C382), GITR (BioXcell BE0063), CD25 (Leinco C1194), CD38 (eBioscience 14–0381-82), CD90 (Biolegend 105202), Lag3 (Leinco L306), CD27 (eBioscience 50–124-94), KLRG1 (Bioxcell BE0201), CD103 (Biolegend 121402), CD4 (BioXcell BE0003–1), CD45 (Fluidigm 3089005B), CD62L (Leinco C2118), ICOS (eBioscience 14–9949-82), OX-40 (BioXcell BE0031), PD-1 (eBioscience 14–9981-82), TIGIT (BioXcell BE0274), CD69 (eBioscience 14–0691-82), TCRβ (BioXcell BE0102), CD127 (BioXcell BE0065), CD39 (Biolegend 143802), NK1.1 (BioXcell BE0036), CD8α (Leinco C375), TCRγδ (eBioscience 14–5711-82), Tim3 (BioXcell BE0115), H2-Kb OVA (WUSTL CHiiPs core), Ki67 (Novus NBP2–22112), Foxp3 (eBioscience 14–5773-82), GATA3 (eBioscience 14–9966-82), Granzyme B (eBioscience MA1–80734), CTLA-4 (eBioscience 50–129-16), TCF1 (R&D MAB8224), ROR-γt (eBioscience 14–6988-82), Eomes (eBioscience 50–245-556), T-bet (Biolegend 644802).
Western immunoblot
Cell lysates were harvested using radioimmunoprecipitation assay (RIPA) lysis buffer [25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS] supplemented with protease and phosphatase inhibitors (Roche). Cell lysates were resolved in Tris-glycine sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) gels and transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen). After blocking in 1X TBST buffer with 5% w/v BSA, membranes were probed with primary antibodies overnight at 4°C. Membranes were washed thrice in 1X TBST and probed with HRP-conjugated secondary antibody for 1 hour at RT. Membranes were developed with Pierce ECL Western Blotting Substrates and detected using a ChemiDoc XRS+ system (Bio-Rad).
Staining was performed with the following antibodies: β-actin (Cell Signaling 4970), p-IRF3 (Cell Signaling 29047S), IRF3 (Cell Signaling 4302S), STING (Cell Signaling 13647S), γH2Ax (Cell Signaling 9718S).
RNA and cDNA isolation
Total RNA was extracted from live cultured cells using an E.Z.N.A.® Total RNA kit (OMEGA) according to manufacturer’s instructions and banked in −80° C until use. cDNA for downstream applications were synthesized using qScript cDNA SuperMix kit (QuantaBio) according to manufacturer’s instructions.
RNA sequencing (RNA-seq) and analysis
RNA samples from treated KPOG organoid cultures were prepared according to kit manufacturer’s protocol, ribo-depleted using RiboZero protocol and subsequently indexed, pooled, and sequenced on HiSeq 3000 (Illumina) at the Genome Technology Access Center (GTAC), Washington University. Differential expression analysis of normalized counts (after standard base calling and demultiplexing) was performed to analyze for differences between conditions and the results were filtered for only those genes with Benjamini-Hochberg FDR adjusted p values ≤ 0.05. For each contrast extracted with Limma, global perturbations in known Gene Ontology (GO) terms and KEGG pathways were detected using the R/Bioconductor package GAGE to test for changes in expression of the reported log2 fold-changes reported for each term versus the background log2 fold-changes of all genes found outside the respective term.
Mouse tissue single cell RNA (scRNA) sequencing data analysis
PDAC tissues were taken from vehicle-treated, RT-treated, FAKi-treated, or RT+FAKi-treated KPC pancreatic tumor 14 days post-treatment start. Cells were sorted into two samples: Immune cells (CD45+) and CAFs (CD45− CD31− Epcam− PDPN+) using Aria-II cell sorter (BD Biosciences). Each sample was generated from a pooled of 2–3 mice/treatment group. For a total of 4 treatment groups and 2 different cell types, we made 8 total libraries to be sequenced.
Sorted cells from each sample were encapsulated into droplets and libraries were prepared using Chromium Single Cell 3’v3 Reagent kits according to the manufacturer’s protocol (10x Genomics, Pleasanton, CA, USA). The generated libraries were sequenced by a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA) to an average of 50,000 mean reads per cell. Cellranger mkfastq pipeline (10X Genomics) was used to demultiplex IIllumina base call files to FASTQ files. Files from the KPC tumors were demultiplexed with > 97% valid barcodes, and > 94% q30 reads. Afterwards, fastq files from each sample were processed with Cellranger counts and aligned to the mm10 reference (v.3.1.0, 10X Genomics, mouse reference mm10–2020-A from https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz) to generate feature barcode matrix.
The filtered feature barcode matrices from were loaded into Seurat (v4.1.2). For each Seurat object, genes that were expressed in less than 3 cells and cells that expressed less than 1000 or more than 6000 genes, were excluded. Cells with greater than 10% mitochondrial RNA content were also excluded. SCTransform with default parameters was used on each individual sample to normalize and scale the expression matrix against the sequence depths and percentages of mitochondrial genes(91). Cell cycle scores and the corresponding cell cycle phase for each cell were calculated and assigned after SCTransform based on the expression signatures for S and G2/M genes (CellCycleScoring). The differences between the S phase score and G2/M score were regressed-out by SCTransform on individual samples. Variable features were calculated for each sample independently and ranked, based on the number of samples they were independently identified (SelectIntegrationFeatures). The top 3,000 shared variable features were used for multi-set canonical correlation analysis to reduce dimensions and identify projection vectors that defined shared biological states among samples and maximized overall correlations across datasets. Principle component analysis (PCA) was performed on the 3000 variable genes calculated earlier (function RunPCA). Multiple datasets were then integrated using Harmony Integration (RunHarmony(92)) using the calculated PCA values. Mouse immune cells datasets were integrated using Harmony (Fig. 5), mouse CAFs datasets (Fig. 6) were not integrated, and mouse CAFs Harmony integrated datasets were shown in Fig. S6. A UMAP dimensional reduction was performed using the Harmony-corrected values to obtain a two-dimensional representation. Then, these defined 30 dimensionalities were used to refine the edge weights between any two cells based on Jaccard similarity (FindNeighbors), and were used to cluster cells through FindClusters functions, which implemented shared nearest neighbor modularity optimization. To characterize clusters, the FindAllMarkers function with log2-fold threshold = 0.25 and minimum 0.25-fold difference and MAST test was used to identify signatures alone with each cluster. Then, the TAMs, cDCs, and adaptive immune (B cells, T cells, NK cells, γδT cells) were selected and top 3000 variable features were recalculated to re-cluster using the above described methodology. Differently expressed genes (DEGs) between the two groups were calculated for each dataset with min.pct of 0.1 and logfc.threshold of -Inf and MAST test (PrepSCTFindMarkers then FindMarkers). Then the DEG lists from each dataset were filtered with p.value < 0.05 and ranked based on foldchange. These ranked gene sets were fed into Gene Set Enrichment Analysis (GSEA) to test for GO terms, KEGG pathways, Reactome and Molecular Signatures Database (MSigDB) gene sets with FDR < 0.05 in ClusterProfiler(93).
Human sample single cell RNA (scRNA) sequencing data analysis
PDAC tissues were taken from patients using ultrasound-guided biopsies and details of the trial design are available on clincaltrials.gov (NCT04331041) and briefly here. Patients received SBRT with a total of 50Gy given in 5 fractions. FAKi (VS-6063) was given at a dose of 400mg BID starting the end of day 2 post-SBRT and continued up to one year. Tissues were obtained pre-treatment and after 12–14 weeks post-SBRT. Biopsy tissues were processed and digested as described above. Following tissue digestion, processed cells from each sample were encapsulated into droplets and libraries using BD Rhapsody Single-Cell Capture and cDNA Synthesis Analysis System (BD Biosciences) according to the manufacturer’s protocol. For a total of 6 pre- and post-treatment groups, we made 10 total libraries to be sequenced.
The generated libraries were sequenced by a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA) to an average of 50,000 mean reads per cell. FASTQ files from each sample were processed and aligned with BD Rhapsody WTA Analysis Pipeline (SevenBridges) using reference genome (GRCh38-PhiX-gencodev29.tar.gz) to generate feature barcode matrix.
The filtered feature barcode matrices were loaded into Seurat as Seurat objects (Seurat v4). For each Seurat object, genes that were expressed in less than 3 cells and cells that expressed less than 1000 or more than 6000 genes, were excluded. Cells with greater than 25% mitochondrial RNA content were also excluded. Seurat NormalizeData and ScaleData with default parameters was used on each individual sample to normalize and scale the expression matrix against sequence depth and percentage of mitochondrial genes. Variable features were calculated for each sample independently and ranked based on the number of samples they were independently identified (FindVariableFeatures). The top 3000 shared variable features were used for multi-set canonical correlation analysis to reduce dimension and identify projection vectors that define shared biological states among samples and maximize overall correlation across datasets. Principle component analysis (PCA) was performed on the 3000 variable genes calculated earlier (function RunPCA). Multiple datasets were then integrated based using Harmony Integration (RunHarmony). A UMAP dimensional reduction was performed on the scaled matrix using the first 30 PCA components to obtain a two-dimensional representation of cell states. Then, these defined 30 dimensionalities were used to refine the edge weights between any two cells based on Jaccard similarity (FindNeighbors), and were used to cluster cells through FindClusters functions, which implemented shared nearest neighbor modularity optimization. To characterize clusters, the FindAllMarkers function with logfold threshold = 0.25 and minimum 0.25-fold difference and MAST test was used to identify signatures alone with each cluster. Differently expressed genes (DEGs) between the two groups were calculated for each dataset with min.pct of 0.1 and logfc.threshold of 0.01 and MAST test (FindMarkers). Then the DEG lists from each dataset were filtered with p.value <0.05 and ranked based on foldchange. These ranked gene sets were fed into Gene Set Enrichment Analysis (GSEA) to test for GO terms, KEGG pathways, Reactome and Molecular Signatures Database (MSigDB) gene sets with FDR < 0.05 in ClusterProfiler.
Reverse Phase Protein Array (RPPA)
Cell extracts were lysed using (RIPA) lysis buffer [25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS] supplemented with protease and phosphatase inhibitors (Roche). Samples were then submitted to the MD Anderson Cancer Center for the RPPA assay. The Functional Proteomics RPPA Core is supported by MD Anderson Cancer Center Support Grant # 5 P30 CA016672-40.
Image analysis
Whole-tissue scans at 10X or 20X magnification were obtained on a Zeiss Axio Scan Z1 brightfield/fluorescence Slide Scanner. Whole-tissue scans were analyzed with HALO software (Indica Labs) using Area quantification, Cytonuclear, or HighPlex modules. Where noted, grading was conducted by a trained pathologist in a blinded fashion and verified by principal investigator post-hoc.
Multiplex IHC (mIHC)
Serial staining was performed with multiple markers as indicated, adapted from a previously published study(94). FFPE tissue sections were loaded onto the BOND RXm autostainer (Leica Biosystems) for iterative staining cycles for markers including CK19, CD8, CD4, Foxp3, PDPN, and pSTAT1. Slides were baked for 60 minutes and deparaffinized prior to the first cycle. Based on antibody host species, default manufacturer protocols and reagents were used up to labeling with HRP. Antigen labeling was chromogenically visualized with a ready-to-use AEC substrate (Abcam). Post-staining, the slides were manually counterstained for hematoxylin, coverslipped using VectaMount AQ Aqueous mounting media (Vector), then scanned using an Axio Scan Z1 (Zeiss). The slides were then decoverslipped and destained using an ethanol gradient, including a 1% hydrochloric acid wash and blocked, as needed, with avidin/biotin blocking kit (Vector Laboratories) and anti-rabbit or anti-rat Fab fragments to eliminate carryover signal from previous cycles, prior to starting another staining cycle. Citrate based antigen retrieval was performed before each staining cycle.
Images of the same specimen but different stains were cropped into multiple segments using ZEN software (Zeiss). Using HALO image analysis software (Indica Labs), each marker was then converted into pseudocolors using the Deconvolution algorithm and compiled into composite multi-marker images. Markers of interest were pseudo-colored and quantified through the HighPlex FL algorithm in HALO software.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software v9, with input from a Biostatistics core expert at Washington University. All data are representative of at least two independent experiments, unless specifically noted. Sample size was pre-calculated to satisfy power-requirements (with >85% confidence) in most experiments. To accomplish randomization for orthotopic or syngeneic tumor experiments, animals were sorted by a blinded investigator with tumor sizes in ascending order and then groups were assigned in descending order. Each group was checked post-hoc to verify no statistical difference in average starting tumor size. Data are shown as mean ± SEM, unless otherwise noted. Statistical tests such as unpaired parametric Student’s t-test, ANOVA analysis (Bonferroni multiple comparison), or unpaired non-parametric Mann–Whitney U test were used appropriately based on normality of data. For survival analyses, Log-Rank (Mantel-Cox) test was used. p < 0.05 was considered statistically significant for all studies; ns denotes not significant.
Supplementary Material
SIGNIFICANCE.
Checkpoint immunotherapeutics have not been effective in PDAC, even when combined with RT. One possible explanation is that RT fails to prime T cell responses in PDAC. Here, we show that FAK inhibition allows RT to prime tumor immunity and unlock responsiveness to checkpoint immunotherapy.
Acknowledgements:
VEL was supported by the NCI F30CA243233. JIB was supported by the Canadian Institutes of Health Research Doctoral Foreign Study Award. GDH was supported by the NCI F30CA254087. LIK was supported by the NIH 5T32EB021955. JMB was supported by NIH F31DK122633.
JKS was supported by NIH R01CA248917, Siteman Investment Program, and AACR/Bristol Meyers Squibb Female Investigator Award. HK was supported by NIH R01CA248917.
DGD and study costs were supported by NCI R01CA273190, R01CA177670, R01CA262506, R01CA248917, P30CA09184215, P50CA196510, and the BJC Cancer Frontier Fund.
We would like to thank Dr. Gregory D. Longmore, Dr. Robert D. Schreiber, and Dr. Sheila A. Stewart for providing reagents and feedback while drafting the manuscript.
We would like to thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital and the Institute of Clinical and Translational Sciences (ICTS) at Washington University in St. Louis, MO. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA091842 and the ICTS is funded by the National Institutes of Health’s NCATS Clinical and Translational Science Award (CTSA) program grant #UL1 TR002345.
We would like to thank the Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine for imaging experiments, the CHiiPs Immunomonitoring Laboratory for CyTOF experiments, the Flow Cytometry & Fluorescence Activated Cell Sorting Core for sorting and flow cytometry experiments, and the Genome Technology Access Center for scRNA-seq and RNA-seq experiments. We would also like to thank the RPPA Core at MDACC which is supported by NCI Grant # CA16672 and Dr. Yiling Lu’s NIH R50 Grant # R50CA221675: Functional Proteomics by Reverse Phase Protein Array in Cancer.
Footnotes
The authors declare no potential conflicts of interest.
Data availability
Bulk-RNA and scRNA sequencing data from KPC pancreatic lesions and human PDAC biopsies (NCT04331041) can be found at the Gene Expression Omnibus Repository (GEO) accession number GSE207718 and GSE207536. All software packages used are publicly available through commercial vendors.
References
- 1.Cid-Arregui A, Juarez V. Perspectives in the treatment of pancreatic adenocarcinoma. World J Gastroenterol 2015;21(31):9297–316 doi 10.3748/wjg.v21.i31.9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarantis P, Koustas E, Papadimitropoulou A, Papavassiliou AG, Karamouzis MV. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J Gastrointest Oncol 2020;12(2):173–81 doi 10.4251/wjgo.v12.i2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol 2018;24(43):4846–61 doi 10.3748/wjg.v24.i43.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hull A, Li Y, Bartholomeusz D, Hsieh W, Allen B, Bezak E. Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature. Cancers 2020;12(2) doi 10.3390/cancers12020481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazard L. The role of radiation therapy in pancreas cancer. Gastrointest Cancer Res 2009;3(1):20–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman KA, Hajj C. Role of radiation therapy in the management of pancreatic cancer. J Surg Oncol 2013;107(1):86–96 doi 10.1002/jso.23137. [DOI] [PubMed] [Google Scholar]
- 7.Yao W, Maitra A, Ying H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020;53:102655 doi 10.1016/j.ebiom.2020.102655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017;66(1):124–36 doi 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63(11):1769–81 doi 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nature Reviews Gastroenterology & Hepatology 2020;17(3):153–68 doi 10.1038/s41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- 11.Valkenburg KC, De Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nature Reviews Clinical Oncology 2018;15(6):366–81 doi 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krisnawan VE, Stanley JA, Schwarz JK, DeNardo DG. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers (Basel) 2020;12(10) doi 10.3390/cancers12102916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nature Reviews Clinical Oncology 2021;18(12):792–804 doi 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214(3):579–96 doi 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohlund D, Lundin C, Ardnor B, Oman M, Naredi P, Sund M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. British journal of cancer 2009;101(1):91–7 doi 10.1038/sj.bjc.6605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res 2015;21(15):3561–8 doi 10.1158/1078-0432.Ccr-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18(16):4266–76 doi 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. The Journal of Clinical Investigation 2016;126(11):4140–56 doi 10.1172/JCI87734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. New England Journal of Medicine 2018;378(22):2078–92 doi 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 20.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. New England Journal of Medicine 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. New England Journal of Medicine 2015;372(26):2521–32 doi 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 22.Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. Journal for immunotherapy of cancer 2016;4:14- doi 10.1186/s40425-016-0119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Reilly EM, Oh D-Y, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncology 2019;5(10):1431–8 doi 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, et al. A Randomized Trial of Chemoradiotherapy and Chemotherapy after Resection of Pancreatic Cancer. New England Journal of Medicine 2004;350(12):1200–10 doi 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 25.Wainberg ZA, Hochster HS, Kim EJ, George B, Kaylan A, Chiorean EG, et al. Open-label, Phase I Study of Nivolumab Combined with <em>nab</em>-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clinical Cancer Research 2020;26(18):4814 doi 10.1158/1078-0432.CCR-20-0099. [DOI] [PubMed] [Google Scholar]
- 26.Gajiwala S, Torgeson A, Garrido-Laguna I, Kinsey C, Lloyd S. Combination immunotherapy and radiation therapy strategies for pancreatic cancer—targeting multiple steps in the cancer immunity cycle. Journal of Gastrointestinal Oncology 2018;9(6):1014–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biffi G, Tuveson DA. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev 2021;101(1):147–76 doi 10.1152/physrev.00048.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discovery 2019;9(2):282 doi 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery 2019;9(8):1102–23 doi 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214(3):579–96 doi 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieskoski MD, Marra K, Gunn JR, Hoopes PJ, Doyley MM, Hasan T, et al. Collagen Complexity Spatially Defines Microregions of Total Tissue Pressure in Pancreatic Cancer. Scientific Reports 2017;7(1):10093 doi 10.1038/s41598-017-10671-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007;99(19):1441–54 doi 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 33.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 2000;60(9):2497–503. [PubMed] [Google Scholar]
- 34.DuFort CC, DelGiorno KE, Carlson MA, Osgood RJ, Zhao C, Huang Z, et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys J 2016;110(9):2106–19 doi 10.1016/j.bpj.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baskar R, Dai J, Wenlong N, Yeo R, Yeoh K-W. Biological response of cancer cells to radiation treatment. Frontiers in molecular biosciences 2014;1:24- doi 10.3389/fmolb.2014.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng M, Lin J, Nowsheen S, Liu T, Zhao Y, Villalta Peter W, et al. Extracellular matrix stiffness determines DNA repair efficiency and cellular sensitivity to genotoxic agents. Science Advances;6(37):eabb2630 doi 10.1126/sciadv.abb2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deville SS, Cordes N. The Extracellular, Cellular, and Nuclear Stiffness, a Trinity in the Cancer Resistome—A Review. Frontiers in Oncology 2019;9:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaur P, Asea A. Radiation-induced effects and the immune system in cancer. Front Oncol 2012;2:191 doi 10.3389/fonc.2012.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrels A, Lund T, Serrels B, Byron A, McPherson Rhoanne C, von Kriegsheim A, et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015;163(1):160–73 doi 10.1016/j.cell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 2014;14(9):598–610 doi 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014;514(7520):112–6 doi 10.1038/nature13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao X-K, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Scientific Reports 2016;6(1):19276 doi 10.1038/srep19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 2011;10(11):2135–45 doi 10.1158/1535-7163.Mct-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlaepfer DD, Hunter T. Signal transduction from the extracellular matrix--a role for the focal adhesion protein-tyrosine kinase FAK. Cell Struct Funct 1996;21(5):445–50 doi 10.1247/csf.21.445. [DOI] [PubMed] [Google Scholar]
- 45.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005;6(1):56–68 doi 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 46.Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunology, Immunotherapy 2017;66(8):1037–48 doi 10.1007/s00262-017-2003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nature Medicine 2016;22(8):851–60 doi 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature medicine 2016;22(5):497–505 doi 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venkatesulu BP, Hsieh C-E, Sanders KL, Krishnan S. Recent advances in radiation therapy of pancreatic cancer. F1000Res 2018;7:F1000 Faculty Rev-931 doi 10.12688/f1000research.16272.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. The Lancet 2004;363(9414):1049–57 doi 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 51.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7(5):469–83 doi 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 52.Wong J, Armour E, Kazanzides P, Iordachita I, Tryggestad E, Deng H, et al. High-resolution, small animal radiation research platform with x-ray tomographic guidance capabilities. International journal of radiation oncology, biology, physics 2008;71(5):1591–9 doi 10.1016/j.ijrobp.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016;150(7):1659–72.e5 doi 10.1053/j.gastro.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mills BN, Connolly KA, Ye J, Murphy JD, Uccello TP, Han BJ, et al. Stereotactic Body Radiation and Interleukin-12 Combination Therapy Eradicates Pancreatic Tumors by Repolarizing the Immune Microenvironment. Cell Rep 2019;29(2):406–21.e5 doi 10.1016/j.celrep.2019.08.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020;37(3):289–307.e9 doi 10.1016/j.ccell.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med 2007;13(8):992–7 doi 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 57.Thomas D, Radhakrishnan P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer 2019;18(1):14- doi 10.1186/s12943-018-0927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 2004;29(3):179–87 doi 10.1097/00006676-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 59.Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, et al. Β1 Integrin Inhibitory Antibody Induces Apoptosis of Breast Cancer Cells, Inhibits Growth, and Distinguishes Malignant From Normal Phenotype in Three Dimensional Cultures and in Vivo. Cancer Research 2006;66(3):1526–35 doi 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park CC, Zhang HJ, Yao ES, Park CJ, Bissell MJ. Β1 Integrin Inhibition Dramatically Enhances Radiotherapy Efficacy in Human Breast Cancer Xenografts. Cancer Research 2008;68(11):4398–405 doi 10.1158/0008-5472.CAN-07-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. Β1-Integrin-Mediated Signaling Essentially Contributes To Cell Survival After Radiation-Induced Genotoxic Injury. Oncogene 2006;25(9):1378–90 doi 10.1038/sj.onc.1209164. [DOI] [PubMed] [Google Scholar]
- 62.Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proceedings of the National Academy of Sciences 2013;110(50):20212 doi 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021;39(9):1227–44.e20 doi 10.1016/j.ccell.2021.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 2020;10(2):232–53 doi 10.1158/2159-8290.Cd-19-0644. [DOI] [PubMed] [Google Scholar]
- 65.Francescone R, Barbosa Vendramini-Costa D, Franco-Barraza J, Wagner J, Muir A, Lau AN, et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast-Driven Nutritional Support and Immunosuppression. Cancer Discov 2021;11(2):446–79 doi 10.1158/2159-8290.Cd-20-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discovery 2022;12(2):484–501 doi 10.1158/2159-8290.CD-21-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cordes N. Integrin-mediated cell-matrix interactions for prosurvival and antiapoptotic signaling after genotoxic injury. Cancer Letters 2006;242(1):11–9 doi 10.1016/j.canlet.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 68.Goel HL, Sayeed A, Breen M, Zarif MJ, Garlick DS, Leav I, et al. β1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-Jun amino terminal kinase 1. J Cell Physiol 2013;228(7):1601–9 doi 10.1002/jcp.24323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klein D, Schmitz T, Verhelst V, Panic A, Schenck M, Reis H, et al. Endothelial Caveolin-1 regulates the radiation response of epithelial prostate tumors. Oncogenesis 2015;4(5):e148-e doi 10.1038/oncsis.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DSA, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proceedings of the National Academy of Sciences of the United States of America 2008;105(47):18490–5 doi 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, et al. Inhibition of integrin αvβ6, an activator of latent transforming growth factor-β, prevents radiation-induced lung fibrosis. American Journal of Respiratory and Critical Care Medicine 2008;177(1):82–90 doi 10.1164/rccm.200706-806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chetty C, Bhoopathi P, Rao JS, Lakka SS. Inhibition of matrix metalloproteinase-2 enhances radiosensitivity by abrogating radiation-induced FoxM1-mediated G2/M arrest in A549 lung cancer cells. International Journal of Cancer 2009;124(10):2468–77 doi 10.1002/ijc.24209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biswas S, Freeman ML, Arteaga CL, Biswas S, Guix M, Rinehart C, et al. Inhibition of TGF Beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression Find the latest version : Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic. Journal of Clinical Investigation 2007;117(5):1305–13 doi 10.1172/JCI30740.Some. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: The importance of dose and fractionation. Frontiers in Oncology 2014;4 JAN(January):1–12 doi 10.3389/fonc.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ansiaux R, Baudelet C, Jordan BF, Beghein N, Sonveaux P, De Wever J, et al. Thalidomide radiosensitizes tumors through early changes in the tumor microenvironment. Clinical Cancer Research 2005;11(2 I):743–50. [PubMed] [Google Scholar]
- 76.Crokart N, Jordan BF, Baudelet C, Ansiaux R, Sonveaux P, Grégoire V, et al. Early reoxygenation in tumors after irradiation: determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys 2005;63(3):901–10 doi 10.1016/j.ijrobp.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 77.Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy Promotes Tumor-Specific Effector CD8 + T Cells via Dendritic Cell Activation. The Journal of Immunology 2012;189(2):558–66 doi 10.4049/jimmunol.1200563. [DOI] [PubMed] [Google Scholar]
- 78.Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 2009;114(3):589–95 doi 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Research 2008;68(3):918–26 doi 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Research 2011;71(10):3453–8 doi 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sasson AR, Wetherington RW, Hoffman JP, Ross EA, Cooper H, Meropol NJ, et al. Neoadjuvant chemoradiotherapy for adenocarcinoma of the pancreas. International Journal of Gastrointestinal Cancer 2003;34(2):121–7 doi 10.1385/IJGC:34:2-3:121. [DOI] [PubMed] [Google Scholar]
- 82.Chun YS, Cooper HS, Cohen SJ, Konski A, Burtness B, Denlinger CS, et al. Significance of Pathologic Response to Preoperative Therapy in Pancreatic Cancer. Annals of Surgical Oncology 2011;18(13):3601–7 doi 10.1245/s10434-011-2086-4. [DOI] [PubMed] [Google Scholar]
- 83.Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC. Targeting FAK in anticancer combination therapies. Nature Reviews Cancer 2021;21(5):313–24 doi 10.1038/s41568-021-00340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang K-J, Constanzo JD, Venkateswaran N, Melegari M, Ilcheva M, Morales JC, et al. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(23):5851–63 doi 10.1158/1078-0432.CCR-15-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25(6):719–34 doi 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, et al. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021;39(4):548–65.e6 doi 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lo A, Li C-P, Buza EL, Blomberg R, Govindaraju P, Avery D, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017;2(19):e92232 doi 10.1172/jci.insight.92232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McAndrews KM, Chen Y, Darpolor JK, Zheng X, Yang S, Carstens JL, et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL-6 Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discovery 2022;12(6):1580–97 doi 10.1158/2159-8290.CD-20-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160(1–2):324–38 doi 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 2009;4(11):1670–80 doi 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 2019;20(1):296 doi 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 2019;16(12):1289–96 doi 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2(3):100141 doi 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 2017;19(1):203–17 doi 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk-RNA and scRNA sequencing data from KPC pancreatic lesions and human PDAC biopsies (NCT04331041) can be found at the Gene Expression Omnibus Repository (GEO) accession number GSE207718 and GSE207536. All software packages used are publicly available through commercial vendors.