

Abstract
Massively parallel reporter assay (MPRA) is a high-throughput method that enables the study of the regulatory activities of tens of thousands of DNA oligonucleotides in a single experiment. While MPRA experiments have grown in popularity, their small sample sizes compared to the scale of the human genome limits our understanding of the regulatory effects they detect. To address this, we develop a deep learning model, MpraNet, to distinguish potential MPRA targets from the background genome. This model achieves high discriminative performance (AUROC = 0.85) at differentiating MPRA positives from a set of control variants that mimic the background genome when applied to the lymphoblastoid cell line. We observe that existing functional scores represent very distinct functional effects, and most of them fail to characterize the regulatory effect that MPRA detects. Using MpraNet, we predict potential MPRA functional variants across the genome and identify the distributions of MPRA effect relative to other characteristics of genetic variation, including allele frequency, alternative functional annotations specified by FAVOR, and phenome-wide associations. We also observed that the predicted MPRA positives are not uniformly distributed across the genome; instead, they are clumped together in active regions comprising 9.95% of the genome and inactive regions comprising 89.07% of the genome. Furthermore, we propose our model as a screen to filter MPRA experiment candidates at genome-wide scale, enabling future experiments to be more cost-efficient by increasing precision relative to that observed from previous MPRAs.
INTRODUCTION
The decreasing costs and increasing throughput of genomic technologies have propelled the comprehensive characterization of the human genome and identified millions of single-nucleotide variants (SNVs) (1). While the functional effect of variants located in coding regions can be interpreted using our knowledge of the genetic code, the majority of identified SNVs are situated in non-coding regions, and their behavior is less understood (2). Functional non-coding variants located in regulatory elements such as promoters or enhancers may affect binding of transcription factors, while other variants may affect local chromatin structure (3). Non-coding variants have been shown to contribute to disorders such as various cancers (4) and are frequently identified upstream of disease-associated genes (5).
The massively parallel reporter assay (MPRA) allows for high-throughput experimental identification and validation of functional effect in non-coding regions, enabling direct testing of the potential regulatory roles of specific SNVs. Although these experiments can test at the scale of tens of thousands of variants (6), the number identified with significant functional effect is generally in the hundreds, resulting in low precision and cost-effectiveness. As a result, MPRAs usually serve as functional validation of a relatively small set of predefined candidate variants identified by genome-wide association studies (GWAS) or quantitative trait loci (QTL). This prevents a comprehensive understanding of the non-coding variants with detected functional effect under MPRA experiments. Important characterizations that would be of interest include correlations of various epigenetic annotations associated with MPRA functional hits and the location of functional non-coding SNVs such as in enhancer regions of specific gene classes. In addition, the density of functional variants as a function of chromosome position and distribution outside known eQTL regions are still unknown.
Previous studies have developed annotations to predict various forms of functional effect. For example, the Combined Annotation Dependent Deletion (CADD) method integrates multiple annotations into a score that prioritizes variants based on estimated deleterious effect and is trained using a combination of evolutionary conserved and de novo variants (7). The Functional Inference of Regulators of Expression (FIRE) scores SNVs on their potential regulatory effect on nearby genes by distinguishing cis-eQTL from non-eQTL variants (8). Databases such as regBase and Eigen have aggregated multiple such functional scores as input for computing meta-scores (8,9). Furthermore, the Roadmap database contains 25-bp resolution biochemical annotations over a variety of cell and tissue types, including transcription factor binding, chromatin accessibility and modifications, and DNA methylation, which may help identify functional effect in non-coding variants (10). As opposed to predicting the functional consequences of non-coding variants in general, our aim is instead to characterize the variants and effects detected by MPRA experiments for a specific cell line.
In our work, we evaluate existing functional predictions using two of the largest published MPRA datasets (Novaseq (11) and Tewhey (6)) for the identification of causal variants underneath a large number of eQTL and GWAS loci. We observe that existing functional scores represent very distinct functional effects, and most of them fail to characterize the regulatory effect that MPRA detects. To address this, we design a deep learning model to distinguish MPRA-detected functional variants from the background genome. The background genome variants are carefully controlled with respect to allele frequency and proximity to transcription sites in order to isolate MPRA specific effect. Our model, denoted as MpraNet, incorporates local epigenetic annotations across a range of tissues as well as site-specific functional scores from previous studies. We find that a joined convolutional neural network (CNN) and fully connected network (FCN) architecture shows high discriminative performance (AUROC = 0.85).
Using this model, we predict potential MPRA functional variants across the genome and identify the distributions of MPRA effect relative to other characteristics of genetic variation, including allele frequency, alternative functional annotations specified by FAVOR, and phenome-wide associations. We leverage MpraNet to characterize the genome by contrasting a group of significantly positive MPRA variants with a randomly derived background set. MPRA scores for this background set are made possible due to the genome-wide scale of the model which permits inspection of under-studied SNVs such as non-coding and rare variants. Furthermore, we examine the distribution of MpraNet scores genome-wide and discover both MPRA dense and sparse 1kb regions significantly distinct from that of a random uniform spread.
We further show that using our score as a candidate screen for new MPRA experiments can improve cost-efficiency by increasing the number of discovered positives relative to the candidate pool size. MpraNet's genomic precision at finding future positive MPRA candidate SNVs is estimated to surpass that observed in novaSeq and Tewhey, as well as outperform other functional scores. Results after incremental increases to the training set size suggest that the inclusion of future MPRA experiment data will continue to improve AUROC and AUPR performance. These outcomes allow for the possibility of using model-assisted MPRA experiments along with model retraining to enable high-precision regulatory variant detection at a genome-wide scale.
MATERIALS AND METHODS
Train and test partitions
We obtain confirmed MPRA positives from the Tewhey (6) and novaSeq (11) MPRA experiments and reserve 20% of the positives as the validation set. The two MPRA experiments were assessed over the GM12878 lymphoblastoid cell line (LCL). Within the training and validation datasets we match each positive with control SNPs (assumed negatives) from the gnomAD database at a 1:10 ratio. Importantly, since MpraNet predictions are ultimately calculated after a rank-based approach over the whole genome, they are robust to this arbitrarily defined ratio. The control variants are matched by allele frequency percentile to address the MPRA candidates often coming from known high allele-frequency sites. Similarly, they are also controlled by proximity to transcription sites to account for the fact that novaSeq and Tewhey selected cis-eQTL candidate variants. Further details on the matching procedure are given in the Methods section.
Feature extraction
For each candidate SNV we extracted site-specific functional annotations and scores. The annotations comprise eight epigenetic markers including Roadmap histone modification and methylation from 127 different cell and tissue types. The functional scores were obtained from the Eigen and RegBase databases and include multiple previously published methods for predicting various regulatory and functional effects (8,9). Finally, in addition to on-site epigenetic annotations, we collected LCL-specific Roadmap annotations from a 1000 bp window on either side of each SNV to incorporate local sequential epigenetic information. Previous work has shown that prediction performance can be improved by leveraging annotations in nearby base pairs, as was shown for ‘valley scores’ that characterize the local minima of activating histone modifications (H3K4me1, H3K4me3, H3K9ac and H3K27ac) (12,13). We use all these features as inputs for MpraNet, a deep learning model to predict whether a given candidate SNV is MPRA positive.
Model architecture
The method combines a feed-forward neural network (FNN) to learn non-linear functions of site-specific functional scores with a convolutional neural network (CNN) to model Roadmap epigenetic annotations in the local neighborhood of each SNV (Figure 1). The structure of our model can be interpreted as an ensemble combining tissue-specific local annotations with previously developed scores. The resulting prediction scores are then calibrated as a Phred score based on their genome-wide percentile rank in order to increase interpretability. Details on our model and the training procedure are given in the Methods section.
Figure 1.

MpraNet Architecture. The MpraNet model takes as input at-site functional annotations and scores as well as neighboring epigenetic annotations. The neighboring annotations are processed by 1D convolutional layers while at-site scores are processed by fully connected layers. The outputs from these networks are concatenated and passed into the final fully connected layers for classification. The model is run across the genome to produce genome-wide scores, which can be used to prioritize variants for future MPRA. Validated variants can be added to the MpraNet training set to further improve model performance.
RESULTS
MpraNet distinguishes MPRA-positive variants from background genome whereas most existing functional scores represent other functional effects
We evaluate our method on the validation set using AUROC (area under receiver operating characteristic curve) and AUPR (area under precision-recall curve) metrics, shown in Figure 2. As benchmarks we compare against the 44 functional scores available from Eigen and RegBase (median AUROC 0.58, AUPR 0.15), from which we present the seven best-performing scores: GenoCanyon, FunSeq2, FitCons2, FIRE, CADD, LIN-SIGHT and FATHMM-MKL. The remaining functional scores are reported in Supplemental Table S1. Because AUROC and AUPR compare different quantities that are important for assessing classification performance (in particular, sensitivity, specificity, and positive predictive value), a strong method should perform well in both metrics. We observe that MpraNet outperforms the other functional scores in both AUROC (0.85) and AUPR (0.43). For comparison, one of the best-performing benchmarks (FIRE) achieves 0.75 AUROC and 0.20 AUPR. Other scores achieve lower performance, indicating that they measure other forms of functional effects that do not overlap with MPRA effect. For example, the CADD, FitCons2 and LINSIGHT scores estimate various forms of conservation or selection pressure, which may not be strongly associated with assayable regulatory effect. In other cases, FATHMM-MKL uses variants associated with heritable diseases as the target effect, while the GenoCanyon score is unsupervised and may thus reflect a different combination of variant functions.
Figure 2.

Comparison with existing functional scores. (A, B) The performance of our model compared with other functional scores over the validation set (20% of confirmed variants matched with background at 1:10 ratio), as measured by area under the ROC curve (A) And area under precision-recall curve (B). (C, D) The MPRA positive variants from the validation set are embedded along with background using the t-SNE algorithm. (C) We highlight the top 3 percentiles of previously existing functional scores, showing they inhabit different regions of embedding space. (D) We show the location of confirmed MPRA positive variants and the top 3 percentile of our MpraNet score.
The AUROC curve (Figure 2A) shows greater separation between MpraNet and the other methods in minimizing the false positive rate at more selective thresholds. This suggests that MpraNet has greater precision, which is confirmed by the AUPR curve (Figure 2B). In contrast, the FIRE score has strong overall performance in ranking MPRA candidates, as seen by high AUROC, but significantly lower precision than MpraNet among its top ranked candidates. On the other hand, GenoCanyon and FunSeq2 have better precision among their top rankings than FIRE, but they ultimately rank many non-functional variants above functional variants, resulting in worse precision at higher recall levels (Figure 2B).
The AUROC and AUPR show large variation among the top benchmarks, indicating that the annotations may be sensitive to different types of functional effects. To assess the overlap between scores, we take a random sample of SNVs from genomic background and MPRA positives and embed them in two-dimensional space using the t-SNE algorithm, using functional scores and tissue-specific annotations as features. The highest scores, defined by >0.97 quantile, for the seven benchmarks are shown on Figure 2C. The high scores for MpraNet and locations of the MPRA positives are mapped onto the same embedding (Figure 2D).
The high-scoring variants for the functional benchmarks tend to cluster in disjoint regions, although some parts show overlaps. MPRA positives also tend to occur in the regions where FIRE is enriched; however, most benchmarks miss regions of MPRA positives or falsely identify regions without MPRA positives. On the other hand, CADD and FATHMM-MKL scores show concentrations in a region with few positives. The MpraNet scores better track the embedding regions where positives are located, suggesting that MPRA functional effect occurs in a set of variants that partially overlap but are not well-predicted by other scores. Since these other benchmarks are input features for the MpraNet model, this indicates that their characteristics can be combined in a deep learning model to better predict MPRA functional effect. These results reflect the central aim of MpraNet, and validate the subsequent downstream characterizations of MPRA-specific effect in later sections. However, we reference some additional experiments with our model below to provide additional context to the results.
We consider the full list of candidates observed in novaSeq and Tewhey to create an alternative validation set (described further in Methods). Composed of only MPRA positive and negative samples, this validation set contains variants that were all selected by the same sampling scheme implicating cis-eQTLs in the same region. Due to the similarity of variants and the fact that some negatives may contain enriched weaker signals that are currently not passing the significance level due to insufficient power, we expect MpraNet to have a lower discriminative performance which is observed in both AUROC (0.85–0.69) and AUPR (0.43–0.26) in Supplemental Figure S1. However, MpraNet still marginally outperforms other functional scores. For comparison, the best-performing benchmark GenoCanyon achieves 0.66 AUROC and 0.31 AUPR. Although we are more interested in discriminating the background genome than the positive and negative MPRA samples, this test is a valuable control that further indicates MpraNet is capturing MPRA-specific effect.
We also examine an alternate training procedure that instead leverages the MPRA negative samples from novaSeq and Tewhey as training negatives. Since the negative samples were candidate functional variants (eQTLs and/or GWAS hits) in the same region, we hypothesize that a model trained exclusively on MPRA candidates will be inadequate to characterize variants genome-wide. We report the results when implementing this alternate training procedure (labeled MpraNet (+/–) and described further in Methods) in Supplemental Figure S2. We observe that both AUROC (0.85–0.72) and AUPR (0.43–0.33) are reduced when discriminating the background genome with this alternative training procedure. This indicates that the controlled background set is providing valuable information related to MPRA effect and lends itself to our aim of characterizing MpraNet positives genome-wide. In summary, MpraNet successfully discriminates MPRA-positives from the background genome and captures MPRA-specific effect better than other functional metrics.
Genome-wide characterization of MPRA
Here we use the MpraNet predicted scores as a proxy for the distribution of real MPRA signals to identify the distributions of MPRA effect relative to other characteristics of genetic variation, including functional annotations, allele frequency, and phenome-wide associations. In an effort to distinguish high MPRA signals from the background genome, we define MpraNet positive variants as being in the top 95th percentile of scores genome-wide.
Regulatory activities and other functional annotations
First, we assess how MPRA signals are distributed relative to other regulatory signals. To do so, we use regulatory annotations specified by FAVOR (10), an open-access web portal that assembles individual variant functional annotation data including the following categories: Variant Category, Epigenetics, Conservation, Transcription Factors, Chromatin States, Local Nucleotide Diversity, Mutation Density, Mappability and Proximity-To-TSS-TES. For the following analyses, we randomly select 800 variants that are in the top 95th percentile of MpraNet scores as the proxy MPRA signals, referred to as MpraNet positives, and 15 000 randomly selected variants from the background genome (of which ∼5% are expected to be positive). In each group, there is a proportionally equal set of variants from each chromosome. The FAVOR annotations of these two groups are compared with: (i) two-sample independent t-tests where equal population variances are assumed for quantitative annotations and (ii) chi-square tests of independence of variables for qualitative annotations. Significance thresholds are established after Bonferroni correction for the number of tests performed within a graphic.
Promoter and enhancer regions are widely accepted to be integral in the DNA binding of transcription factors and regulation of gene expression (14). Enhancer mediated activation of gene expression is mediated by chromatin looping bringing enhancers close to promoters (14). We thus expect to find that MPRA signals are considerably higher in these regions. GeneHancer (15), SuperHancer (14), and CAGE (16) are databases of human enhancer and promoter locations. Variant scores in these categories are binary depending on a variant's presence within a documented promoter or enhancer. Figure 3a reveals that MpraNet positives are indeed significantly more likely to be found in these regions, with all associated P-values < 1.0e–22 after chi-square tests. This initial result confirms the validity of this nature of analysis and sets the stage for the inspection of other annotations with less certain expectations.
Figure 3.

Genome-wide characterization of MPRA. (A) The prevalence of MpraNet defined positive and background variants are compared for promoter and enhancer regions. (B) The distribution of regulatory regions in which MpraNet positives and the background genome are located is compared. (C) The functional annotation scores of MpraNet positive and background variants are compared. (D, E) The mean MpraNet phred score of common and rare variants are compared and then the allele frequencies of MpraNet positives and the background genome are compared. (F) MpraNet performance is measured between the top hits of a PheWAS and a background set.
We assess the distribution of positives within different types of genomic regions between the positive and background sets in Figure 3B. A chi-square test reveals that the distributions are significantly different from each other (P-value < 1.0e–30), with positives underrepresented in intergenic regions and over-represented in all others. This result is consistent with literature expectations about the location of regulatory activity (17). Intergenic regions reside further from transcription relevant sites than introns, upstream/downstream regions, UTRs and exons (17). Human intergenic prevalence is believed to be ∼50% (18) and intronic prevalence is believed to be ∼35% (19). These numbers are close to the observed proportions in the background genome of 43.2% and 36.3% respectively, further emphasizing the significance of MpraNet positives residing in 16.1% intergenic and 60.1% intronic areas.
These analyses are extended further using the same variants as above to various quantitative functional scores specified by FAVOR. We compare the FAVOR functional scores between the proxy MPRA signals and background genome using two-sample independent t-tests. We present solely the P-values for brevity's sake in Figure 3C. Integrative annotation categories are bolded and summarize their subcategories to the right. For example, the integrative category Proximity-To-TSS-TES is followed by its defining categories: minimum distance to a transcription start site (TSS) and minimum distance to a transcription end site (TES). These subcategories along with the major category have significantly different scores between positives and the background genome. This corroborates literature expectations considering the TSS’s importance in regulating RNAPII (20) and TES’s importance in maintaining the transcriptional machinery(20).
The Transcription Factor major category measures overlap with documented transcription factor sites, and understandably these annotation categories all present higher scores of overlap in MpraNet positives. Transcription factor mediated regulation of expression is one of the primary mechanism by which MPRA experiments aim to assess functionality (21).
MpraNet has a strong relationship with the Epigenetic major category and chromatin states subcategories (no associated major category). The Epigenetic summary category shows a significant effect along with the vast majority of its subcategories. Epigenetic factors, including chromatin states, confer heritable changes to DNA and are known to effect gene expression (3,22). These results are thus unsurprising, but warrant further research into the apparent differences between different epigenetic markers and MPRA signal. For example, the repressive marks (H3K9me3 and H3K27me3) have exceedingly significant P-values. Both the DNA methylation (H3K27ac and H3K4me1) and modifications known to be added as a consequence of transcription (H3K36me3 and H3K79me2) types of epigenetic markers have one category with a much larger degree of significance than the other. The presence of chromatin state structures (such as cHmm E14 and CHmm e3) that are separated by MpraNet positive variants is expected but the reason for varying degrees of significance amongst other chromatin organizations warrants further inspection.
Interestingly, the Conservation annotations are not contrasted heavily by MpraNet positives and the background genome. Conservation has been used as a proxy for regulatory importance in other tools such as CADD (23) and presents a reasonable candidate for MpraNet association. This result may be due to conservation being more important in the context of regulatory coding variants, whereas MPRAs are specialized for non-coding regions (21). This finding warrants further research concerning the effect of conservation on MPRA related regulatory importance.
Local Nucleotide Diversity has a strong relationship with MpraNet score and each of its subcategories are significant. This result is corroborated by previous work which has shown that nucleotide diversity is far higher in intergenic regions and more conserved in gene bodies (24). In fact, nucleotide diversity sharply declines in regulatory rich regions such as 250 bp upstream regions as well as the 3' UTR (24). It is thus expected that MpraNet should differentiate between regions of higher and lower nucleotide diversity.
Mappability is a category which does not have a strong relationship with MpraNet score. Mappability tends to vary significantly between different genes, and thus does not present an overall indicator of functionality (25). There is little evidence to suggest that mappability is correlated with regulatory activity in other studies.
Interestingly, we also find that the Mutation Density major category is significantly distinguished by MpraNet positives from the background genome, even though this is not a very interpretable category in this context. This effect is not as dominant as any of the other significant major categories. Mutation density generally characterizes how often mutations are found within a given range of DNA (26). One of the more likely explanations for this significance is the fact that mutation rates are often determined by chromatin structure, as was shown for cancer genomes (26). Chromatin organization was previously shown to have an MpraNet effect. Should mutation density's relationship to chromatin structure explain its relationship with MpraNet scores, this might explain why the P-values are less significant considering that this is an indirect connection.
We further contrast the same set of random background variants with 1285 novaSeq defined positives (same variants as used during model training). The novaSeq (11) MPRA acts as a ground-truth for MpraNet and we expect similar findings. Replicating Figure 3A and B is not feasible due to the lack of novaSeq positives within different regulatory regions, but the equivalent to Figure 3C (Supplementary Figure S3) shows similar results when comparing functional annotation scores of positive and random background variants. The similarity between MpraNet positives genome-wide and novaSeq positives further suggests that MpraNet is an accurate MPRA proxy signal.
Allele frequency
Most previous MPRA experiments have focused on GWAS-identified common variants (minor allele frequency, MAF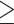 0.01). Rare variants (MAF < 0.01) are believed to be more harmful than common ones but they were understudied by functional experiments (27). For most traits, genetic association studies have shown an inverse relationship between the variant's effect size and its MAF. This relationship is more pronounced for traits most strongly influenced by natural selection, compared with quantitative phenotypes or late-onset diseases. Here we use MpraNet scores to assess the overall difference in regulatory importance between rare and common variants with a new randomly chosen sample of 40k variants (equally distributed amongst each chromosome). We obtained the allele frequency of each variable from the Genome Aggregation Database (gnomAD) (28).
0.01). Rare variants (MAF < 0.01) are believed to be more harmful than common ones but they were understudied by functional experiments (27). For most traits, genetic association studies have shown an inverse relationship between the variant's effect size and its MAF. This relationship is more pronounced for traits most strongly influenced by natural selection, compared with quantitative phenotypes or late-onset diseases. Here we use MpraNet scores to assess the overall difference in regulatory importance between rare and common variants with a new randomly chosen sample of 40k variants (equally distributed amongst each chromosome). We obtained the allele frequency of each variable from the Genome Aggregation Database (gnomAD) (28).
Unlike the effect on disease phenotypes, Figure 3d reveals that common (mean ∼ 4.2) and rare (mean ∼ 4.3) variants do not have a significantly different MpraNet score (P-value > 0.05). Moreover, MpraNet positive and background variants do not have significantly different allele frequencies (P-value > 0.05). This is consistent with our analysis of existing functional scores, where the scores that represent conservation or selection pressure (e.g. CADD, FitCons2 and LINSIGHT) are not predictive of functional effect characterized by MPRAs. Moreover, the conservation scores in FAVOR are not significantly associated with MpraNet score in the previous section. The similarity in behavior between conservation and allele frequency is unsurprising given the intrinsic relationship between these two metrics.
Association with disease phenotypes
We evaluate if predicted MPRA positives are more likely to be associated with disease phenotypes in PheWAS and GWAS. To this end, we assess whether significant variants identified by association studies also have higher MpraNet scores. The UK-BioBank (29) is a large biomedical database from which 20k PheWAS generated top hits are retrieved. Top hits are defined as having a P-value < 10−6 from the PheWAS, of which only the top 2000 for every phenotype are considered. In this case, we select a background set that is percentile matched based on allele frequency and retrieved randomly from each chromosome. Each top hit thus has a corresponding random background variant. Figure 3F separates top hit variants based on their implicated disease major category and compares the MpraNet scores of these top hits vs. the random background set.
Overall, the top hits have a significantly higher mean MpraNet score (mean = 7.00) than allele frequency matched background variants (mean = 6.34) with a P-value < 1e–10 after a t-test. Figure 3F shows the P-values after t-tests for the MpraNet scores of each disease category against their specific background set. Variants implicated in the circulatory system tend to have the most regulatory importance as defined by our screen, however, each disease category assigns a higher regulatory importance to top hits than the background. This version of MpraNet is specific to the lymphoblastoid cell line (LCL) which may affect which phenotypic categories stand out. This may explain why the Circulatory system is distinguished by MpraNet score but the General Symptoms category is not. LCLs are created by modifying peripheral blood lymphocytes (PBL) and are often used as their surrogates for medical discovery (30), which explains why their functional behaviors would be strongly linked.
Distribution of MPRA positives
We aim to evaluate how MPRA positives are distributed across the genome. We hypothesize that MpraNet positives are clumped together due to the presence of regulatory rich and void regions. A large region in Chromosome 1 spanning base pair 70 Mb to 80 Mb according to the GRCh37 reference is chosen as a demonstration. We measure the proportion of MpraNet positives within non-overlapping 1 kb regions to identify especially dense MPRA locations. Figure 4A reveals the continuous set of MpraNet scores in the demonstration region. The y-axis defines different smoothing widths applied to these scores. The presence of high and low density MPRA signal regions can be seen and already it is apparent that MpraNet scores are not evenly distributed.
Figure 4.

Distribution of MpraNet positives. (A) Smoothed MpraNet scores with varying smoothing widths in the Chromosome 1 demonstration region spanning location 70–80 MB in the GrCH37 reference genome. (B) The proportion of MpraNet positive variants in non-overlapping and consecutive 1 kb regions from the demonstration region. Density of MpraNet positives is measured against a uniformly random sequence of scores bearing the same mean. Inactive and active 1kb regions are defined as being outside the uniform distribution's 2.5th percentile and 97.5th percentile scores respectively.
In order to quantify this result, we consider the same demonstration region except we divide it into non-overlapping and continuous 1kb regions absent of smoothing; in total this results in 10 000 1kb regions. In each of these regions, the MpraNet positive proportion is calculated by counting the number of MpraNet positives (defined as being in the top 95th percentile genome-wide) and dividing by 1000. Figure 4b presents the frequencies of MpraNet positive proportions. Among the 10 000 1kb regions, ∼84% contain exactly 0 MpraNet positives and ∼0.48% are 100% positive. The mean MpraNet positive proportion across all 1 kb regions is ∼5% (by the MpraNet positive definition) representing a sum of 50 positives. A clear indication that MPRA signal is clumped together is that when one finds a single positive variant, the mean positive proportion in the surrounding 1 kb region jumps up to 30.20% (3020 positives).
We contrast MpraNet with a Uniform distribution in which positives are defined in the same manner – by scoring in the 95th percentile. With a uniform distribution of scores, 95% of 1kb regions contain MpraNet positive rates within 3.7% and 6.4%. Defining any MpraNet positive proportion outside of this range to be an outlier, 89.07% of MpraNet regions are significantly negative—called inactive regions—while 9.98% are significantly positive—called active regions. We thus find that 99.05% of 1kb regions are either inactive or active. Kolmogorov-Smirnov test for goodness of fit confirms that the two distributions are significantly different with a P-value < 1e–100. We propose that finding the MpraNet positive proportion in 1 kb regions can help the scientific community locate areas of high and low MPRA signal. Queries of this nature can also be localized to genes/areas of interest for researchers. Moreover, active and inactive MPRA regions of different sizes can be found by changing the bp width parameter.
Incremental learning for designing future MPRA experiments
While the current MpraNet scores can be used to assist MPRA experiments to increase precision, future MPRA outputs can also be used to further improve the performance of the deep learning model. We explore the improvement of classification performance when additional MPRA functional variants are added to the training set. In the starting experiment, only 1000 labeled variants are available (100 MPRA positives matched with background at 1:10 ratio). Subsequently, to simulate data becoming available as MPRA experiments are performed, we add 50 MPRA positive variants matched with background at each iteration, ending when all currently available MPRA positives have been added (corresponding to our actual training set). To assess the effect of different data distributions, at first we only add positives from the Tewhey et al. experiment and then the novaSeq experiment. The validation performance at each iteration (Figure 5A) suggests that our method steadily improves as more data becomes available though at a slower rate than in the initial stages. This simulation indicates the potential of leveraging MPRA and deep learning in a feedback loop to accelerate the discovery of functional variants.
Figure 5.

Incremental learning for designing future MPRA experiments. (A, B) MpraNet performance on the validation set increases with the training set size, with MPRA positives and background fixed at a 1:10 ratio. This suggests incorporation of results from future MPRA experiments can further improve MpraNet. (C) MpraNet gives a continuous score from 0 to 1. Choosing a threshold to select top scoring variants gives varying recall on the validation set. In comparison with other benchmark scores, MpraNet achieves higher recall at any threshold. (D) For any fixed threshold and corresponding recall, MpraNet precision is estimated using Bayes’ rule, assuming a value of MPRA function prevalence. Here we estimate precision for four thresholds corresponding to testing the 25 000 to 100000 top scoring variants from the 1000 Genomes Project. For example 100 000 (pink) corresponds to the top 0.011 percentile.
MpraNet can be applied across the genome to score variants using available functional predictors, identifying high-scoring variants as experimental candidates for future MPRA studies. The original output score for a variant ranges from 0 to 1, and we convert it to a Phred score as described in the method section. Then we select a classification threshold above which the variant is predicted as functional. For any given threshold we estimate the recall (the fraction of total functional candidates discovered) of the method and the percent of variants marked functional based on the validation set. We repeat this for each benchmark. For a new candidate variant pool, we can then compute the fraction of top-scoring variants that need to be experimentally assayed in order to detect a certain percent of the true functional variants. As shown in Figure 5B, for any desired recall, MpraNet requires validation of far fewer variants than any other method. Equivalently, at any given experiment size, MpraNet detects a much higher proportion of true MPRA functional variants compared to other methods.
Given a budget to experimentally validate a fixed number of variants, we can further estimate the precision (fraction of tested variants that are functional) of using MpraNet as a candidate screen for MPRA. This uses the estimated recall based on the fraction of top-scoring variants tested (Figure 5B) and also requires making an assumption of the underlying prevalence of MPRA functional SNVs. Specifically, we estimate the precision (or proportion of variants selected for validation that are functional) of our approach using Bayes’ rule:
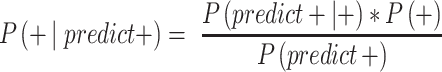 |
where P (+|predict +) is genomic precision; P (predict + |+) is the recall (sensitivity) of our method as estimated from the validation set, P (+) is the (unknown) prevalence of MPRA positive variants across the genome, and P (predict +) is the probability that a given non-coding variant is selected for MPRA. Since our score is continuous, a threshold is chosen based on the number of positive variants desired, thus fixing P (predict +).
We illustrate the resulting precision in Figure 5c at three possible levels of prevalence (0.005, 0.01, 0.02). To give intuition, we assume that the candidate pool of variants are the 9.2 million SNVs from the 1000 Genomes Project and convert the top tested percentile to the corresponding number of tested variants (e.g. in the pink bars, testing 100 000 SNVs is equivalent to testing the top 100 000/9.2 mil = 0.011 percentile). For comparison, the precision of the existing MPRA experiments are indicated with lines: Tewhey et al. with 0.030 (678/22 803) and novaSeq with 0.057 (1684/29 685). We see that for any reasonable prevalence and similar experiment size, MpraNet detects functional variants at much higher precision. The increased ratio of positive to assayed variants translates into more cost-effective experiments.
DISCUSSION
This work shows that our unique feature space comprising epigenetic and functional markers, combined with deep learning techniques, can distinguish MPRA-positive SNPs better than any alternative method in the lymphoblastoid cell line. We expect the scientific community to benefit from the insights gained from characterizing MpraNet positives across the genome, as well as the model's efficacy at prioritizing causal variants for increased precision of future MPRA experiments. The proposed version of MpraNet represents a precursor for future models interested in different cell lines, functional assays, and allelic effects. Moreover, we expect to find that feature spaces similar to our own will be able to characterize other complex genetic traits such as those observed in association studies. It is of great interest to extend the proposed version of MpraNet to the study of different cell lines, allelic effects observed in MPRA, and trans-eQTL ground-truth positives.
Prior work has developed annotations aimed at assessing functional effect, using a wide variety of approaches, but our results show that they do not transfer well to distinguishing MPRA function from background. Another related work, GenoNet, was trained to classify MPRA positive from MPRA negative candidates. Because MPRA candidates are generally from common variant areas (or eQTL regions) and contain many close variants in linkage disequilibrium, the insignificant variants from MPRA are not generally representative of the genomic background. Such prior methods underperform as a genome-scale screening tool as shown in our results. MpraNet achieves an AUROC and AUPR of 0.85 and 0.43, respectively, surpassing all other functional metrics at predicting MPRA specific functional effect.
MpraNet's validation performance will translate to improved cost-efficiency and design of future MPRA experiments. We show that MpraNet's genomic precision exceeds that of Tewhey and novaSeq, as well as other functional scores (Figure 5). This will increase the number of MPRA positive variants found within a set of candidate SNVs. By incrementally supplying more of our dataset to MpraNet, we see continual increases in AUROC and AUPR performance. This suggests that retraining MpraNet after use with future MPRAs will aid the model.
The success of our model permits a variety of analyses concerning functionality on a genome-wide scale. Using MpraNet scores confers several advantages to learning about genomic regulation such as: a genome-wide and continuous set of scores, access to non-coding and rare variants, and access to a set of random control SNVs that aren’t normally present in MPRA experiments. Through the selection of positive MpraNet variants in the top 95th percentile of scores genome-wide, we were able to contrast the background genome with respects to (i) promoter and enhancer prevalence, (ii) intergenic and intronic prevalence (amongst other types of regulatory regions) and (iii) several integration annotation categories specified by FAVOR: Epigenetic, Transcription Factor, Proximity-To-TSS-TES, Mutation Density, Local Nucleotide Diversity and Chromatin States. MpraNet score was also shown to significantly differ between PheWAS generated top hits and the background genome. However, allele frequency did not show a MpraNet dependent effect, and neither did the integrative Conservation or Mappability categories specified by FAVOR. We also examined the distribution of MpraNet positives and found that positives exist in significantly active or inactive 1kb clusters. We hope that researchers continue to expand on these analyses to characterize functional variants in the genome. We propose that MpraNet can serve to not only increase the precision of future MPRAs but also as a tool to undercover underlying mechanisms behind expression regulation.
Although MpraNet has shown success at replicating MPRA results based on model validation, as well as corroborating common findings relating to functional regulation, there are several limitations imposed by the restricted selection criteria observed in novaSeq and Tewhey. Both assays strictly observe cis-eQTLs and are biased towards variants identified by association studies. With a larger feature space, and a larger number of MPRA experiments with which to derive ground truth positives, MpraNet can be further improved by the incremental learning approach. These MPRAs test allelic and expression effects in the lymphoblastoid cell line contributing to a cell-line and site-specific functional effect. We refrain from including further MPRA or STARR-seq based datasets that don’t meet these criteria to prioritize the downstream characterization of MPRA signal genome-wide. The motivation for this inclusion strategy stems from our findings that the prediction of functional effects is sensitive to the training data (Figure 2) and previous work has shown that MPRA predictions are not transferable across cell lines (31). We especially consider the extension of MpraNet to non-site specific effects such as the allelic effects studied within the lymphoblastoid cell line in Griesemer et al. (32) and Kalita et al. (33). With a poor discriminative performance at distinguishing sites containing at least one significant allelic effect (AUROC of 0.57 and 0.56 respectively), preliminary results suggest that the effect captured by MpraNet is too specific for this generalization. It is of great interest to extend MpraNet to other cell types as MPRA labels become increasingly available, and further explore how the proposed methods can be applied to allelic effects.
MATERIALS AND METHODS
MPRA datasets
MPRA positives
We used positive labeled variants from the GM12878 lymphoblastoid cell line (LCL), as identified by two experiments. First, Tewhey et al. identified 842 emVars out of 32 373 variants in 3642 eQTLs and control regions in LCLs. After excluding the emVars that cannot be mapped to a genomic location using the Ensemble database and without available functional scores, we define the remaining 678 emVars out of 22 803 as positive variants.
The second set of positives comes from the novaSeq (11) MPRA which aimed to analyze the distribution of causal variants within eQTL and GWAS loci, with a candidate pool of 32 144 variants in the lymphoblastoid cell line (LCL). After excluding variants without available functional scores, we define the 544 out of 29 685 variants with both expression effect and allelic effect q-value <0.01 as positive variants. Combining the MPRA experiments results in 1222 total positive variants.
Background variants
Each MPRA positive variant was matched with background variants, which are assumed to be negative labels for training a classification model. We noted that MPRA candidates tend to have high allele frequencies since they are generally selected from common variant pools. This can cause a confounding effect when matched with background variants from the genome since random coordinates are usually of low allele frequency. Therefore, we match each positive variant with background SNVs from the gnomAD database by allele frequency at a 1:10 ratio, reflecting the low prevalence of functional variants suggested by existing MPRA experiments. Background SNVs are also matched with respect to proximity to transcription sites to control for this potential confounding effect. This process has an inherent error rate because MPRA functional variants may be randomly selected as the background. However, the low MPRA discovery rates suggests that this occurs with very low probability.
In order to match on allele frequency, the distribution of allele frequencies of the positive set is recorded and the percentile boundaries are calculated. A matching variant must have an allele frequency in the same percentile boundary as the positive it is matched against. A similar procedure is conducted for the proximity to transcription sites metric. This value is extracted from the Phred adjusted Proximity-To-TSS-TES score derived from the Favor (10) database. We use quintile matching to account for the fact that there is low variation of Proximity-To-TSS-TES within the positive candidates and with consideration for the difficulty of finding control variants that are jointly matched between these two effects.
Features
For each variant, 44 functional genomic scores from regBase and Eigen were obtained as predictive features. When available, both raw and PHRED scaled versions of the scores are included. In addition, epigenetic annotations from the Roadmap Epigenomics Project were extracted for each variant in the MPRA datasets, to produce 1016 more features for each non-coding site. The features correspond to eight epigenetic markers assessed in 127 different cells and tissues, including DNase I hypersensitivity and histone methylations. The epigenetic annotations are continuous non-negative values representing P-value scores for each epigenetic marker. In the event of multiplicate entries for a SNV representing allelic variation, scores are averaged across alleles. In addition to the variant-specific annotations described above, we extracted neighboring annotations for the LCL tissue from 1000 bp in both directions for each variant. Since Roadmap is provided at 25bp resolution, this results in a sequence of 81 annotations (including the variant at the center) for each of eight markers.
Functional prediction scores have previously been published for tasks such as predicting regulatory function. These scores were obtained from the regBase database. These are aggregated across the whole genome, and in addition, three additional meta-predictors are developed with XGBoost using the scores as features. We incorporated these meta-predictors as features as well. We also obtained Eigen scores containing non-tissue-specific annotations over the genome, including coding and non-coding variants. Like regBase, a number of different models are compiled and aggregated, and they add their own meta-predictors using the models as features. However, instead of using a supervised method, they model each predictor as conditionally independent given true variant function and apply spectral partitioning.
Alternate validation set comprising only novaSeq and Tewhey candidates
While our primary task is in discriminating MPRA positive variants from their matched backgrounds, we consider an additional validation set where MPRA candidates which do not meet the significance thresholds in the novaSeq and Tewhey experiments are used as the negatives instead. To reduce the chance of including false negatives, we sample from candidates which have adjusted P-value above 0.5. As before we keep a 1:10 ratio of positives to negatives.
Alternate training set comprising only novaSeq and Tewhey candidates
Using the same procedure as above, we also construct an alternative training set by matching positives with MPRA negative candidates. We train a model on this dataset following the same procedure as described in the next section. This model serves to evaluate whether a model that uses the negative candidates is able to generalize at the genomic level. We refer to this model as MpraNet (+/–) and our original primary model as MpraNet (+/bg).
MpraNet model training
Model
Our method aims to distinguish potential MPRA positive variants from genomic background for the LCL (E116) cell line. We model the functional status of a SNV location as a function of (i) E116 annotations at the SNV and around the SNV in a 1000 base-pair window in each direction, (ii) non-E116-specific annotations at the SNV location, and 3) other functional scores at the SNV location. Our model architecture uses convolutional layers on the E116 annotations. Non-LCL annotations and other functional scores are incorporated using a fully connected layer.
Roadmap annotations are at 25 base-pair resolution, giving a sequence of length 81 for eight different annotations. Treating this data as a one-dimensional sequence with eight channels, our CNN architecture consists of two one-dimensional convolutional layers with leaky ReLU activation functions: the first with 32 kernels of width 4, padding 1 and the second with 32 kernels of width 5. Each convolutional layer is followed by an average pooling layer with width 2. Meanwhile the at-site annotations and scores are passed through a fully connected layer with 400 neurons. These outputs are concatenated and passed through a fully connected layer with 256 neurons which is finally fed into a sigmoid classification node. Each fully connected layer uses a sigmoid activation function.
Training
We randomly select 20% MPRA positives and their matched controls as the holdout test data. The remaining data is used for training. The model F(x) was optimized to minimize the binary cross-entropy loss with L2 regularization. The model was trained for 30 epochs at batch size 128 using the Adam optimizer with learning rate 1e-4, with L2 regularization set at 5e-4. These hyperparameters were tuned using 5-fold cross-validation over the training data.
PHRED MpraNet score
Throughout this report, MpraNet scores are transformed in order to increase interpretability. First, raw scores are mapped to a percentile rank resulting in a uniform distribution between 0 and 1. For example, a variant with score 0.95 has a higher score than 95% of variants. Then, they are transformed according to the following equation:
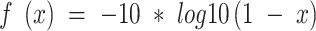 |
For classification applications, we can determine a threshold above which variants are predicted positive. The percentile transform is monotone so thresholds can be converted appropriately without impacting classification performance.
Github
The code supporting our experiments is available at https://github.com/fl16180/MpraScreen.
Online Resources
FAVOR annotations: http://favor.genohub.org/
Allele Frequency Scores from gnomAD: https://gnomad.broadinstitute.org/
UK-BioBank summary statistics: https://pheweb.org/UKB-SAIGE/top_hits
UCSC Genome Browser: https://genome.ucsc.edu/
novaSeq MPRA: https://github.com/nsabell/mpra-v2
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: F.L. and Z.H. developed the concepts for the manuscript and proposed the method. F.L., A.S., Z.H., N.A. and S.M. designed the analyses and applications and discussed results. F.L. and A.S. conducted the main analyses. N.A. and S.M. helped interpret the results. F.L., A.S. and Z.H prepared the manuscript and all authors contributed to editing the paper.
Contributor Information
Fred Lu, Department of Statistics, Stanford University, Stanford, CA 94305, USA.
Aaron Sossin, Department of Biomedical Data Science, Stanford University, Stanford, CA 94305, USA.
Nathan Abell, Department of Genetics, Stanford University, Stanford, CA 94305, USA.
Stephen B Montgomery, Department of Genetics, Stanford University, Stanford, CA 94305, USA; Department of Pathology, Stanford University, Stanford, CA 94305, USA.
Zihuai He, Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA 94305, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH/NIA awards [AG066206 to Z.H.]. Funding for open access charge: NIH/NIA awards [AG066206 to Z.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Siva N. 1000 Genomes project. Nat. Biotechnol. 2008; 26:256. [DOI] [PubMed] [Google Scholar]
- 2. Zappala Z., Montgomery S.B.. Non-Coding Loss-of-Function variation in human genomes. Hum. Hered. 2016; 81:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R.et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mucaki E.J., Caminsky N.G., Perri A.M., Lu R., Laederach A., Halvorsen M., Knoll J.H.M., Rogan P.K.. A unified analytic framework for prioritization of non-coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Med. Genomics. 2016; 9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang F., Lupski J.R.. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015; 24:R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tewhey R., Kotliar D., Park D.S., Liu B., Winnicki S., Reilly S.K., Andersen K.G., Mikkelsen T.S., Lander E.S., Schaffner S.F.et al.. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016; 165:1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ionita-Laza I., McCallum K., Xu B., Buxbaum J.D.. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. gen. 2015; 48:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang S., He Y., Liu H., Zhai H., Huang D., Yi X., Dong X., Wang Z., Zhao K., Zhou Y.et al.. regBase: whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 2019; 47:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X., Li Z., Zhou H., Gaynor S., Liu Y., Chen H., Sun R., Dey R., Arnett D., Aslibekyan S.et al.. {Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet. 2020; 52:969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abell N.S., DeGorter M.K., Gloudemans M., Greenwald E., Smith K.S., He Z., Montgomery S.B.. Multiple causal variants underlie genetic associations in humans. Science. 2022; 375:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Backenroth D., He Z., Kiryluk K., Boeva V., Pethukova L., Khurana E., Christiano A., Buxbaum J.D., Ionita-Laza I.. FUN-LDA: a latent dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: methods and applications. Am. J. Hum. Genet. 2018; 102:920–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramsey S.A., Knijnenburg T.A., Kennedy K.A., Zak D.E., Gilchrist M., Gold E.S., Johnson C.D., Lampano A.E., Litvak V., Navarro G.et al.. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites. Bioinformatics. 2010; 26:2071–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A.. Super-Enhancers in the control of cell identity and disease. 2013; 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M.et al.. GeneHancer: genome-wide integration of enhancers and target genes in genecards. Database. 2017; 2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T.et al.. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farnham P.J. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009; 10:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Francis W.R., Wörheide G.. Similar ratios of introns to intergenic sequence across animal genomes. Genome Biol. Evol. 2017; 9:1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rigau M., Juan D., Valencia A., Rico D.. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019; 15:e1007902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andersson R., Sandelin A.. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020; 21:71–87. [DOI] [PubMed] [Google Scholar]
- 21. Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G., Kinney J.B.et al.. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012; 30:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldberg A.D., Allis C.D., Bernstein E.. Epigenetics: a landscape takes shape. Cell. 2007; 128:635–638. [DOI] [PubMed] [Google Scholar]
- 23. Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M.. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tatarinova T.V., Chekalin E., Nikolsky Y., Bruskin S., Chebotarov D., McNally K.L., Alexandrov N.. Nucleotide diversity analysis highlights functionally important genomic regions. Sci. Rep. 2016; 6:35730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Derrien T., Estellé J., Sola S.M., Knowles D.G., Raineri E., Guigó R., Ribeca P.. Fast computation and applications of genome mappability. PLoS One. 2012; 7:e30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schuster-Böckler B., Lehner B.. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012; 488:504–507. [DOI] [PubMed] [Google Scholar]
- 27. Bodmer W., Bonilla C.. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008; 40:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P.et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen N.E., Sudlow C., Peakman T., Collins R.. UK biobank data: come and get it. Sci. Transl. Med. 2014; 6:224ed4. [DOI] [PubMed] [Google Scholar]
- 30. Hussain T., Mulherkar R.. Lymphoblastoid cell lines: a continuous in vitro source of cells to study carcinogen sensitivity and DNA repair. Int. J. Mol. Cell. Med. 2012; 1:75. [PMC free article] [PubMed] [Google Scholar]
- 31. He Z., Liu L., Wang K., Ionita-Laza I.. A semi-supervised approach for predicting cell-type specific functional consequences of non-coding variation using MPRAs. Nat. Commun. 2018; 9:5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griesemer D., Xue J.R., Reilly S.K., Ulirsch J.C., Kukreja K., Davis J.R., Kanai M., Yang D.K., Butts J.C., Guney M.H.et al.. Genome-wide functional screen of 3′ UTR variants uncovers causal variants for human disease and evolution. Cell. 2021; 184:5247–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalita C.A., Brown C.D., Freiman A., Isherwood J., Wen X., Pique-Regi R., Luca F.. High-throughput characterization of genetic effects on DNA–protein binding and gene transcription. Genome Res. 2018; 28:1701–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.