

Abstract
The spliceosome undergoes extensive rearrangements as it assembles onto precursor messenger RNAs. In the earliest assembly step, U1snRNA identifies the 5′ splice site. However, U1snRNA leaves the spliceosome relatively early in assembly, and 5′ splice site identity is subsequently maintained through interactions with U6snRNA, protein factor PRP8, and other components during the rearrangements that build the catalytic site. Using a forward genetic screen in Caenorhabditis elegans, we have identified suppressors of a locomotion defect caused by a 5′ss mutation. Here we report three new suppressor alleles from this screen, two in PRP8 and one in SNRNP200/BRR2. mRNASeq studies of these suppressor strains indicate that they also affect specific native alternative 5′ss, especially for suppressor PRP8 D1549N. A strong suppressor at the unstructured N-terminus of SNRNP200, N18K, indicates a novel role for this region. By examining distinct changes in the splicing of native genes, examining double mutants between suppressors, comparing these new suppressors to previously identified splicing suppressors from yeast, and mapping conserved suppressor residues onto cryoEM structural models of assembling human spliceosomes, we conclude that there are multiple interactions at multiple stages in spliceosome assembly responsible for maintaining the initial 5′ss identified by U1snRNA for entry into the catalytic core.
INTRODUCTION
Precursor messenger RNA (pre-mRNA) splicing occurs in a large ribonucleoprotein complex called the spliceosome (1). The spliceosome is a dynamic series of complexes that assemble onto the pre-mRNA from small nuclear ribonucleoprotein complexes (snRNPs) and transiently associated factors. The splicing reactions require the action of 5 UsnRNAs and over 100 proteins. Through a complex series of ordered binding and rearrangement events, many involving ATP-dependent helicases (2,3), the catalytic core is built to promote the two trans-esterification reactions of splicing. In recent years, there has been a revolution in our understanding of the spliceosome based on advancements in cryo-electron microscopy (4,5). For the human spliceosome, there now exist a series of snapshots of the spliceosome assembly and reaction process; these provide new insights into how the splicing machinery functions (6). Even with this series of still pictures of the spliceosome, it is evident that there are dramatic structural changes between the stages, and these transitions are not well understood (7).
The definition of sites on the pre-mRNA for splicing occurs during the initiation of spliceosome assembly (1). The 5′ splice site (ss) is defined by its interaction with 5′ end of U1snRNA, the 3′ss by its interaction with the U2 auxiliary factor (U2AF), and the branch point by its interaction with U2snRNA in A complex. In the case of the 5′ss, extensive work has been done to identify the importance of base-pairing interactions between the pre-mRNA and U1snRNA in 5′ss selection and how point mutations at the splice site affect this identification step (8). However, as the U4/U6•U5 tri-snRNP assembles into the spliceosome to form pre-B complex, extensive rearrangements occur and U1snRNP leaves the spliceosome in the transition to the B complex (9,10). The 5′ss is then held in complex with the U6snRNA ACAGAGA region, and cryoEM studies have shown that multiple protein factors help to escort the 5′ss region as the spliceosome goes through assembly steps B (11), pre-Bact1, pre-Bact2 (12), Bact (13) and B* (14) to build towards the first catalytic transesterification reaction of splicing. Some examples of protein interactions with the 5′ss region that have been identified in cryoEM studies include parts of PRP8 in all steps, RNF113A in Bact (13), and Yju1/Isy1 heterodimer at the first catalytic transition step (14). This list is not complete, and the changes in the interactions between the cryoEM ‘snapshots’ of spliceosome assembly steps, and the identification of unique transient protein interactions at pre-Bact1, pre-Bact2, Bact and B* complexes indicate that there are dramatic changes in the interactions surrounding the 5′ss region. Through all these dynamic changes, the identity of the intron start must be maintained to ensure that it is properly loaded into the active site.
We have been utilizing a genetic screen in Caenorhabditis elegans to identify amino acid residues in spliceosome components that are required for maintaining 5′ss identity. In this screen, a mutation of G to T at the first position of the 16th intron of the unc-73 gene (the e936 allele) leads to an uncoordinated phenotype (15). By performing a screen for phenotypic suppression of uncoordination, we are able to identify factors important for identifying and maintaining the proper 5′ss (16–18). UNC-73 encodes a rac guanine nucleotide exchange factor important for promoting proper axon guidance. The G to T mutation to the +1 position of the 16th intron disrupts the nearly invariant GU dinucleotide that begins introns and leads to activation of two new cryptic 5′ss; cryptic splice sites are sites that are used only upon mutation of the original 5′ss (Figure 1A). In the presence of this mutation, 73.5% of splicing occurs at an out-of-frame cryptic site defining an intron that starts GU, at a position 23nt into the intron (the +23 site). Another cryptic splice site, 1nt upstream of the original site (the −1 site) also defines an intron starting with GU that is created by mutation to the +1 position of the intron; the out-of-frame −1 site is used 12.9% of the time. We do detect 13.6% of splicing at the original wt site, which leads to a wildtype mRNA even though the intron begins UU. We have previously demonstrated that the out-of-frame UNC-73 mRNAs are not subject to nonsense-mediated decay, indicating that the ratios of cryptic mRNAs are a readout of the relative use of the splice sites by the spliceosome (19). The e936 mutation of unc-73 provides the spliceosome with ambiguous choices of suboptimal sites, and we have identified extragenic suppressors that influence how the splice site is identified and its identity maintained. Essentially, any change in splicing that increases the amount of in-frame message from ∼14% in a wildtype background to ∼20% or higher will yield sufficient amounts of full-length UNC-73 protein to provide phenotypic suppression. This allows for a sensitive screen for alleles that change splice site usage.
Figure 1.

New extragenic splicing suppressors of unc-73(e936). (A) Schematic of the unc-73 exon 16/intron 16 boundary. The top line shows the wildtype sequence and the bottom line shows the e936 G→T point mutation at the start of intron 16 in bold. Positions of the −1, wt and +23 cryptic 5′ splice site intron starts are indicated by arrows below. (B) A thrash test assay to demonstrate phenotypic suppression of unc-73(e936) uncoordination by the new suppressor allele mimics generated by CRISPR. The number of movements across the body axis per minute in liquid were measured for 20 different L4s for each indicated strain. (C) Reverse transcription and polymerase chain reaction (RT-PCR) analysis of unc-73(e936) splicing in the presence of different extragenic suppressors. Genotype of each strain is indicated above each lane (N2 is the wild isolate Bristol N2 strain), and the bands corresponding to the different cryptic splice sites are shown on the left. The reverse PCR primer was 5′ end labeled with Cy3, and the products separated on a 6% acrylamide denaturing gel and visualized using a Typhoon scanner. The three new suppressor alleles described in this manuscript are snrp-200(N18K), prp-8(R540K) and prp-8(D1549N). Previously identified class II suppressors snrp-27(M141T) (16), prp-8(G654E) and prp-8(T524S) (17) are shown here for comparison. (D) Quantitation of RT-PCR products for unc-73(e936) for the indicated strains. The mean value and standard deviation of three separate experiments are shown (see Supplementary Table S1 for experimental values and P-values relative to the unsuppressed strain). Note that in these class II suppressors, +23 splicing is decreased and −1 and wt site usage are both increased.
We have previously reported eight different unc-73(e936) extragenic suppressor alleles that fall into three distinct classes. The one member of Class I is a compensatory mutation in one of the 12 U1 snRNA genes, at the position that base pairs with the +1G of the intron, known as sup-39. This U1 snRNA mutation allows for initial recognition of splice sites beginning with UU. For the unc-73(e936) mutant, this suppressor is hypothesized to increase initial identification of the in-frame wt splice site that begins UU, but we also detect an equal increase in usage of the −1 site (18). This result indicated that the discrepancy between the overlapping wt and −1 site usage occurs after U1snRNA’s initial splice site identification. Class II suppressors are in protein factors that enter the spliceosome after U1 snRNA and promote the use of both the −1 and wt site over the use of the +23 site. We have previously identified three alleles in this class. We have identified a key residue in the protein SNRP-27, M141, in the worm homolog of the human tri-snRNP 27K protein, providing the first functional evidence of a role for this protein in splice site identity (16,20). Cryo-EM structural models of the tri-snRNP and pre-B complexes show a role for this factor in stabilizing the U4/U6 quasi-pseudoknot at the base of the ACAGAGA box loop in U6 snRNA (9), and we hypothesize that the normal function of the M141 residue of tri-snRNP 27K is to help prevent pre-mature availability of the ACAGAGA region for interaction with the pre-mRNA. We have also identified two residues of the largest and most highly conserved spliceosomal protein, PRP8, which when mutated give a class II suppressor splicing phenotype (17). One suppressor residue, G654, appears to be involved in helping to stabilize the U6 internal stem loop (ISL) that holds the Mg2+ ions important for catalysis. A second suppressor allele at T524 interacts with the 5′ss at the +2 U residue of the intron. These PRP-8 suppressors appear to be important for stabilizing the active site and maintaining the identity of the 5′ss through assembly of the catalytic core. We recently published the discovery of a third class of suppressors that function specifically to alter that ratio of adjacent −1/wt splice site usage, specifically promoting the use of the UU splice site. These class III suppressors consist of two alleles each in KIN17 and PRCC; this screen led to the first demonstration of a role for both of these proteins in splicing (21).
With one exception (the class III suppressor PRCC) these suppressor alleles identified in this screen are not null mutants and in our screens we have yet to identify the same suppressor twice. Because of this, we have continued to screen for extragenic suppressors of the e936 uncoordination and splicing phenotypes with a total of 1 000 000 mutagenized genomes now screened. In this manuscript we report the identity of three new extragenic suppressors that belong to class II. Two of these new suppressors are in PRP-8. One suppressor is at R540, a region involved in intra-PRP-8 interactions in the spliceosome, which while suppressing the e936 phenotype does not lead to global changes in splicing. A suppressor at D1549 of PRP-8 is one of the strongest suppressors we have found in this screen, and it leads to dozens of changes in both 5′ss and 3′ss usage globally. CryoEM models of spliceosome assembly indicate that this residue has important interactions around the 5′ss at multiple steps. A third suppressor is in the N-terminus of the spliceosomal helicase SNRNP200/BRR2 at N18, in a region of the protein that is poorly characterized both structurally and mechanistically. In this case, our data show an important role for the very N-terminal region of BRR2 in maintaining 5′ss identity.
MATERIALS AND METHODS
Strains used in this study
| Strain | Genotype | Allele description |
|---|---|---|
| N2 | Wildtype strain (22) | |
| SZ181 | unc-73(e936)I | unc-73 intron16 G + 1T substitution |
| SZ118 | unc-73(e936) snrp-27(az26)I | SNRP-27(M141T) (16) |
| SZ155 | unc-73(e936)I; prp-8(az29)III | PRP-8(G654E)* (17) |
| SZ197 | unc-73(e936)I; prp-8(az50)III | PRP-8(T524S)* (17) |
| SZ211 | snrp-27(az56) | SNRP-27(M141T)* (20) |
| SZ299 | unc-73(e936)I; prp-8(az117)III | PRP-8(R540K)* |
| SZ304 | unc-73(e936)I; prp-8(az119)III | PRP-8(D1549N)* |
| SZ305 | unc-73(e936)I; snrp-200(az123)II | SNRP-200(N18K)* |
| SZ306 | unc-73(e936)I; snrp-200(az124)II | SNRP-200(E103K)* |
| SZ328 | snrp-200(az140)II | SNRP-200(N18V)* |
| SZ329 | snrp-200(az141)II | SNRP-200(N18P)* |
| SZ330 | snrp-200(az142)II | SNRP-200(N18G)* |
| SZ331 | snrp-200(az143)II | SNRP-200(N18N)* |
| SZ332 | snrp-200(az144)II | SNRP-200(N18S)* |
| SZ333 | snrp-200(az145)II | SNRP-200(N18C)* |
| SZ334 | snrp-200(az146)II | SNRP-200(N18R)* |
| SZ335 | snrp-200(az147)II | SNRP-200(N18F)* |
| SZ336 | snrp-200(az148)II | SNRP-200(N18A)* |
| SZ337 | snrp-200(az149)II | SNRP-200(N18I)* |
| SZ338 | snrp-200(az150)II | SNRP-200(N18T)* |
| SZ339 | snrp-200(az151)II | SNRP-200(N18E)* |
| SZ342 | snrp-200(az154)II | SNRP-200(Δ1–47)* |
| SZ343 | snrp-200(az155)II | SNRP-200(E12D and Δ13–22)* |
| SZ372 | prp-8(az164)III | PRP-8(D1549D)* |
| SZ373 | prp-8(az165)III | PRP-8(D1549I)* |
| SZ374 | prp-8(az166)III | PRP-8(D1549V)* |
| SZ396 | prp-8(az179)III | PRP-8(K528R)* |
| SZ398 | prp-8(az180)III | PRP-8(T1561K)* |
| SZ400 | prp-8(az181)III | PRP-8(R143G)* |
| SZ404 | prp-8(az182)III | PRP-8(L1478F)* |
| SZ406 | prp-8(az183)III | PRP-8(R1674K)* |
| SZ408 | snrp-200((az178)II | SNRP-200(R267I)* |
| SZ410 | prp-8(az184)III | PRP-8(E1881K)* |
| SZ413 | unc-73(e936)I; prp-8(az179)III | PRP-8(K528R)* |
| SZ414 | unc-73(e936)I; prp-8(az180)III | PRP-8(T1561K)* |
| SZ415 | unc-73(e936)I; prp-8(az182)III | PRP-8(L1478F)* |
| SZ417 | unc-73(e936)I; prp-8(az181)III | PRP-8(R143G)* |
| SZ419 | unc-73(e936)I; prp-8(az184)III | PRP-8(E1881K)* |
| SZ420 | unc-73(e936)I; snrp-200(az178)II | SNRP-200(R267I)* |
| SZ421 | unc-73(e936)I; prp-8(az183)III | PRP-8(R1674K)* |
| SZ423 | unc-73(e936) snrp-27(az185)I; snrp-200(az123)II | SNRP-27(M141T)* SNRP-200(N18K)* |
| SZ429 | unc-73(e936) snrp-27(az185)I; prp-8(az119)III | SNRP-27(M141T)* PRP-8(D1549N)* |
| *Indicates an allele generated using CRISPR engineering. |
Mutagenesis and identification of putative suppressed strains
High throughput screens for genetic suppression of the unc-73(e936) uncoordination phenotype are straightforward (23). Larval stage L4 SZ181 (unc-73(e936)) hermaphrodites were exposed to 0.5 mM N-nitroso-N-ethylurea (ENU) for 4 hours as described previously (16). After extensive washing, four animals were placed at the edge of a 10c m NGM-agar plate with a lawn of OP50 Escherichia coli, for 500 plates, and allowed to self-propagate. This represents 500 000 mutagenized genomes (4 worms per plate, 125 progeny per worm, 2 genomes in each progeny and 500 plates). NGM plates were maintained at 20°C. Whereas the unc-73(e936) animals’ movement defects confine them in place, after 8 days, suppressed F2 animals were able to crawl away from the crowded pile of uncoordinated animals, and were identified by their improved locomotion on the far side of the plate that still contained food.
Thrashing assay
A thrash test was performed to measure how well the suppressed worms move in comparison to the uncoordinated mutant unc-73(e936) worms (16). Live L4 worms were transferred to a drop of M9 solution on an NGM agar plate (usually seven individual drops of M9 per plate) and observed at 20°C. For a period of 60 s, worms were observed and the number of times that they bent across their body axis was recorded. Twenty L4 animals were assayed for each strain.
Identification of extragenic splicing suppressors
The unc-73 gene in each of the suppressed lines from this screen was sequenced to distinguish between extragenic and intragenic suppressors. Extragenic suppressor alleles were mapped to chromosomes using a snip-SNP strategy (24,25). Briefly, each extragenic suppressor strain identified in the genetic screen was crossed against a polymorphic Hawaiian isolate, CB4856, and uncoordinated F2 animals that continued to have only uncoordinated offspring were recovered. These new Unc strains were then screened for chromosomal regions that are homozygous for CB4856 snip-SNP markers. Approximately 20 uncoordinated strains derived from each extragenic suppressor strain outcrossed to the Hawaiian strain were recovered and DNA extracted and combined. For each chromosomal region, we expected to see a mix of Hawaiian and Bristol N2 single nucleotide polymorphisms (SNPs), except in the region of the suppressor mutation, where we expect to see 100% Hawaiian SNPs (loss of the suppressor allele) and in the unc-73 region of chrI, where we expected to see 100% N2 SNPs (as these are uncoordinated animals). Using this approach, we were able to narrow down the suppressors to approximately one third of the length of a chromosome.
At the same time as the Hawaiian mapping, we performed high throughput genomic sequencing of the non-outcrossed suppressor strains. We used STAR (26) to map those sequences back to the C. elegans genome. Diploid SNPs relative to the original SZ181 strain were identified using GATK (27), and the snpEff tool (28) was used to identify SNPs within genes in the chromosomal region identified by the Hawaiian strain mapping. That list of putative suppressors was cross-referenced to the Spliceosome Database (29) (http://spliceosomedb.ucsc.edu/), and SNP-containing spliceosome-associated genes and RNA binding proteins in the delimited genomic region were chosen for further analysis. The suppressor allele identity was verified by de novo re-creation of each putative suppressor allele using CRISPR/Cas9 genome editing, and those resulting in both suppression of the movement defect and the molecular splicing phenotype as assayed by reverse-transcription/polymerase chain reaction were confirmed as bona fide suppressors.
CRISPR/Cas9 genome editing
Cas9 guides were chosen from the CRISPR guide track on the UCSC Genome Browser C. elegans reference assembly (WS220/ce10) (30,31) and crRNAs were synthesized by Integrated DNA Technologies (www.idtdna.com). Cas9 CRISPR RNA guides were assembled with a standard tracrRNA; these RNAs were heated to 95°C and incubated at room temperature to allow for annealing. The full guides were then incubated with Cas9 protein to allow for assembly of the CRISPR RNA complex (32). That mix, along with a single stranded repair guide oligonucleotide was then micro-injected into the syncytial gonad of young adult hermaphrodite animals. A dpy-10(cn64) or unc-58(e665) co-CRISPR strategy was used to identify F1 animals showing homology-directed CRISPR repair in their genomes (33). Silent restriction sites were incorporated into or deleted from the repair oligo design so that mutations could be easily tracked by restriction digestion of PCR products from DNA extracted from single worms. Injected animals were moved to plates using recovery buffer, allowed to recover for 4 hours, and surviving worms were plated individually. F1 offspring were screened for the dpy-10(cn64) dominant roller (Rol) co-injection marker phenotype or the unc-58(e665) dominant Unc phenotype. F1 animals expressing the co-CRISPR marker were plated individually, allowed to lay eggs, and then the adult was removed and checked for allele of interest by PCR and restriction enzyme digestion followed by gel electrophoresis. If an F1 worm showed the presence of a heterozygous DNA fragment matching the programmed restriction site, non-roll or non-unc animals in the F2 progeny were screened by electrophoresis of digested PCR products. Individuals that had lost the co-injection marker but were homozygous for the allele of interest were retained and sequenced at the gene of interest to verify error-free insertion of sequences guided by the repair oligo.
CRISPR crRNA guide RNA sequences (entered as DNA into IDTdna.com crRNA order form).
| SNRP-200 N18 and Δ13–22 | GTCGACCGACAGAACCAAGT |
| SNRP-200 E103 | AGCCTACAAGCCCAGAACCC |
| PRP-8 R540 | GAAGGATTTCTCTGCACAAA |
| PRP-8 D1549 | CAGTCAAATCGAGTTGTACT |
| SNRP-27 M141 | CCGCCAATACATGAACCGAA |
| PRP-8 K528 | AGTTAAAACATTGACAACAA |
| PRP-8 T1561 | ATGAGAGAAATCTTGAGCGT |
| SNRP-200 R267 | AGTACTTGGCGAGCGATCTT |
| PRP-8 R143 | TGGCACAATGGGGAACGATG |
| PRP-8 L1478 | TGGAGGAGTAGAAGGAATTT |
| PRP-8 R1674 | TATAGTCAAGGAATTTTGCA |
| PRP-8 E1881 | TTTCCCGAATATTGTCATCA |
CRISPR repair oligonucleotides (lower case differ from wt)
| SNRP-200 N18K | GGCCGACGAGCTAGCCCGAATCCAGCAGTATGAGTACCGGCAGAAtTCgAAaTTGGTTCTGTCGGTCGACTACAATCTGACGGATCGGCGTGGCAGA |
Creates silent EcoRI site GAATTC
Silent PAM disruption
Changes N18 (AAC) to K18 (AAA)
| SNRP-200 E103K | TTGTCGACAATAATGAGCTCATGGGAGCCTACAAGCCCAGAACCCAGaAGACGAAGCAGACTTATGAAGTGATTTTGTCTTTCATTTTGGA |
Changes E103 (GAG) to K103 (AAG) and disrupts an ScrFI restriction site CCNGG
| SNRP-200 N18 Randomize | GGCCGACGAGCTAGCCCGAATCCAGCAGTATGAGTACCGGCAGAAtTCgnnnTTGGTTCTGTCGGTCGACTACAATCTGACGGATCGGCGTGGCAGA |
Creates silent EcoRI site GAATTC
Silent PAM disruption
Randomizes N18 (AAC) to NNN
| SNRP-200 Δ13–22 | ATGGCCGACGAGCTAGCCCGAATCCAGCAGTATGAC…GTCGACTACAATCTGACGGATCGGCGTGGCAGAGAG |
36nt flank on each side of deletion
Created a G→C change at the 5′ end of the deletion junction to create an AatII site GACGTC
Changed E12 (GAG) to D12 (GAC)
Deleted 30nt coding amino acids 13 to 22
| PRP-8 R540K | ACAACAAAGGAAAGAAAGAAATCTCGTTTCGGAAATGCATTtCATTTGTGCAaAGAgATCCTTCGTCTCACAAAACTTGTTGTTGATGCCCATGTTCAGTA |
Silent PAM disruption
Changes R540 (AGA) to K540 (AAA)
Creates silent MboI site GATC
| PRP-8 D1549N | CTCCAACTATCAACCGTGCTAATGTCTACGTCGGTTTtCAAGTACAgCTgaATTTGACTGGAATTTTCATGCACGGAAAGATCCCGACGCTCAAGATTTCT |
Silent PAM disruption
Creates silent PvuII site CAGCTG
Changes D1549 (GAT) to N1549 (AAT)
| PRP-8 D1549 Randomize | CTCCAACTATCAACCGTGCTAATGTCTACGTCGGTTTtCAAGTACAgCTgnnnTTGACTGGAATTTTCATGCACGGAAAGATCCCGACGCTCAAGATTTCT |
Silent PAM disruption
Creates silent PvuII site CAGCTG
Randomizes D1549 (GAT) to NNN
| SNRP-27 M141T | CGTGGATGGCTGCGTGAACATCAAGAAACCTCGCAGATACCGCCAgTAtAcGAACCGAAAaGGAGGATTCAATCGTCCACTTGATTTTATGGGATAATTTA |
Silent PAM disruption
Silent creation of BstZ17I restriction site GTATAC
Changes M141 (ATG) to T (ACG)
| PRP-8 K528R | Mimic of S.cerevisiae U4-cs1 suppressor |
| TTACAACTTCAATTTGAAACCAGTTAAAACATTGACAACAAAaGAAcggcgcAAATCTCGTTTCGGAAATGCATTCCATTTGTGCAGAGAAATCCTTCGT | |
Silent PAM disruption
Creates HhaI restriction site GCGC
Changes K528 (AAG) to R (CGC)
| PRP-8 T1561K | Mimic of S.cerevisiae U4-cs1 suppressor |
| AGTACAACTCGATTTGACTGGAATTTTCATGCACGGAAAGATCCCGAaGCTtAAGATTTCTCTCATTCAAATCTTCCGTGCTCACTTGTGGCAAAAGAT | |
Changes T1561 (ACG) to K (AAG)
Creates a new HindIII restriction site AAGCTT
| SNRP-200 R267I | Mimic of S.cerevisiae U4-cs1 suppressor |
| CACGAGGAATTCTTCATCCACGTGACATCGACGCTCATTGGATCCAgAtcTCGCTCGCCAAGTACTTCAAAGATCCTCTGATCGCCCAACAGAAGCAAAC | |
Changes R267 (AGA) to I (ATC)
Combines with a silent mutation in Q266 to create a new BglII site AGATCT
| PRP-8 R143G | Mimic of S.cerevisiae Prp28-1 suppressor |
| ATTCCGCGTGTCATCGAGCCGGTCTACATGGCACAATGGGGtACcATGTGGATTATGATGcGcgGAGAAAAACGTGATCGTCGTCATTTCAAGCGAATGAGATTCCCT | |
R142 has a silent change AGA to CGC
Creates a new HhaI restriction site GCGC
Changes R143 (AGA) to G (GGA)
| PRP-8 L1478F | Mimic of S.cerevisiae first step suppressor |
| AACTATAGAACAGACATGATTCAAGCACTTGGAGGAGTAGAAGGAATTTTcGAACATACATTATTCAGGGTAAGTGTTAACAGATATAATTTGCTTTCTT | |
Changes L1478 (TTG) to F (TTC)
Single point mutation also disrupts PAM site and creates a new BstBI restriction site (TTCGAA)
| PRP-8 R1674K | Mimic of S.cerevisiae first step suppressor |
| CAGCTTCGTTGGGGAGATTATGATTCTCACGACGTTGAACGtTACGCCaagGCAAAATTCCTTGACTATACCACTGATAACATGTCGATTTATCCTTCAC | |
R1674 (CGT) to K (AAG) – also disrupts PAM
Silent mutation in R1671 creates a new AclI restriction site AACGTT
| PRP-8 E1881K | Mimic of S.cerevisiae first step suppressor |
| CATTGGAGGTGCATCTTCTCGATTTCCCGAATATTGTCATCAAaGGATCcaAGCTCATGTTACCATTCCAAGCTATCATGAAGGTTGAAAAATTCGGTGA | |
Changes E1881 (GAG) to K (AAG)
Silent mutation at S1880 helps to create a new BamHI restriction site GGATCC
Oligonucleotides for genomic PCR amplification
| Region | Forward oligo | Reverse oligo |
|---|---|---|
| SNRP-200N18 | GGA TAT TCG ACT ATC CCG AG | CAG GCT TAT TCT CCT CTT CG |
| PRP-8 R540 | AAT GCC CGT GAA AGT TCG TG | TCC ACG CAA GAA GAA CAA CC |
| PRP-8 D1549 | GAA CCT ACT TCC CAA CAT GG | CTG TCT GCC ATC AAA GAA GG |
| SNRP-27M141 | GAG TCG TTA CAA AGT GGA GC | TTC GCC ATG GTC AAA TTC CC |
| PRP-8 K528 | AAT GCC CGT GAA AGT TCG TG | CAA AAA CCA CAT CCA GGT CC |
| PRP-8 T1561 | TCA AGC ACT TGG AGG AGT AG | CTG TCT GCC ATC AAA GAA GG |
| SNRP-200R267 | CAT CTA CGA GAA TGA GGG TG | GTG CAA GAA TCG GAT GAA GC |
| PRP-8 R143 | AAG GAA AGC GGT ATT CGG AG | GAA CCA TTC AGC AAC AGC TC |
| PRP-8 L1478 | TTC CGG AAT GAG TCA CGA TG | GTT CCC AGA ACA AAC CTT CC |
| PRP-8 R1674 | GTC TAG GCC TTC TTT GAT GG | GTC TTG TGA ATG GTG ACA CG |
| PRP-8 E1881 | TCG AAG GTA ACC TAA CGA CC | ATC GTC GTC AGA AAG TGT GG |
Oligonucleotides for reverse transcription—polymerase chain reactions
| Gene | Forward oligo | Reverse oligo |
|---|---|---|
| UNC-73 | GCA GTG TGC CTA GAA AAG TG | TCG TCC CTT AAA GTA GGC TC |
| MAB-10 | TTT GAG TCA GCT GTC GTC AG | CAG ATG AGG TGG ATG ATG AC |
| T01D3.6 | ACA AGT TCC TGA TCC TTC GC | TTT CCA AGT CCC TGA CAA GC |
| HIL-1 | TCC GCA TGG GAA AAG AAT GC | CTT ATT GGT GGA CAT GGA GC |
| NTL-4 | CAT AAC TCA ATG TTG GAT GGT AGG | CTG TGT GCT TTC CAA GAT GC |
| MDT-15 | TCT AGA CGT CGC TTT CAA CG | ATC CAT TCC ACG AAG ACT CG |
| TOFU-7 | CGT TCT AAG ATC GTA CAC ACC | TGC GCG AAC CAG TAG ATA AG |
| AKAP-1 | AAC TCC ACT CCT CAC AAT GC | CTC CAT GAG AAT GAA GCT GG |
| Y71H2AM.2 | ACA AGA TCT ACA TGG TGC CG | ATT TGG ATT TCA GCC GGG TG |
| E01B7.2 | ACG TGA GAA GCT TGA AGA CG | TCG AAT ATC GAG CCA TCA CG |
| B0001.7 | TGG TGG AAA ATT CGC AGA GC | AAT AAT CTC GAC GCT GCT CC |
RNA extraction, cDNA production and PCR amplification
RNA from indicated strains was extracted from mixed stage populations of animals using TRIzol reagent (Invitrogen), then phase-separated with CHCl3 and the aqueous phase precipitated by isopropanol. Total RNA was reverse transcribed with gene-specific primers using SuperScript III (ThermoFisher) or AMV reverse transcriptase (Promega). cDNA was PCR-amplified for 25 cycles with 5′-Cy3-labeled reverse primers (IDT) and unlabeled forward primers using either Taq polymerase or Phusion high-fidelity polymerase (NEB). PCR products were separated on 40 cm tall 6% polyacrylamide urea denaturing gels and then visualized using a Molecular Dynamics Typhoon Scanner. Band intensity quantitation was performed using ImageJ software (https://imagej.nih.gov/ij/). For quantitation, a box of the same size was drawn around each alternative splicing product on a gel in ImageJ, and a control background box of the same size was drawn between them in each lane (or just above the two if the bands were too close together). The background volume value was subtracted from each band's value within a lane and then the relative usage of the splice sites was calculated. Relative splice site usage data for each sample in each repeated trial are included in Supplementary Table S1.
RNASeq
Total RNA isolations from three biological replicates were done for strains SZ181, SZ299, SZ304 and SZ305. mRNA isolation and sequencing library preparation for each RNA isolation were performed by RealSeq Biosciences (Santa Cruz, CA). 75nt x 75nt paired-end reads were obtained on a Novaseq 6000 sequencer, with nine libraries combined in a lane. RNA-seq results were trimmed, subjected to quality control, and two-pass aligned to UCSC Genome Browser C. elegans reference assembly (WS220/ce10) (this earlier assembly release was used to facilitate comparison to previous RNA-seq datasets obtained by our lab) using STAR (26).
High stringency ΔPSI analysis
Alternative 5′ (A5) and alternative 3′ (A3) splicing events found in the STAR mappings of all of the libraries were identified and filtered for those introns with at least 5 reads of support (total across all samples) and a maximum of 50 nucleotides between the alternative ends (either 5′ or 3′ respectively). In addition, alternative first exon (AF), alternative last exon (AL), skipped exon (SE), retained intron (RI), mutually exclusive exon (MX) and multiple skipped exon (MS) events were derived from the Ensembl gene predictions Archive 65 of WS220/ce10 (EnsArch65) using junctionCounts ‘infer pairwise events’ function (https://github.com/ajw2329/junctionCounts). The percent spliced in (PSI) for each event in each sample was derived using junctionCounts with the option suppress_eij_use for A3 and A5 events. Each strain had 3 biological replicates, therefore between any two strains, there are a total of nine possible pairwise comparisons for each event. For each suppressor strain, only alternative splicing events with a minimum of 15 junction counts that showed a change in the same direction >15% ΔPSI compared to the SZ181 control in all nine pairwise comparisons (pairSum = 9) were considered further. Those events with a mean ΔPSI >20% across the nine comparisons were chosen for examination. The reads supporting that alternative splice site choice event were then examined individually on the UCSC Genome Browser C. elegans reference assembly (WS220/ce10) to ensure that the algorithmically flagged events looked like real examples of alternative splice site choice. Supplementary Table S2 has the chromosomal location, ΔPSI measurements, distance between alternative splice sites, alternative splice sequences and notes for all alternative splicing events that fit these criteria.
Consensus motifs
Consensus motifs were created using WebLogo (34); https://weblogo.berkeley.edu/logo.cgi.
Multiple sequence alignments
Multiple sequence alignments were generated using Multalin (35) at the on-line web interface; http://multalin.toulouse.inra.fr/multalin/.
RESULTS
Identification of new unc-73(e936) extragenic suppressor alleles
To expand the range of potential cryptic splicing suppressors, we performed the genetic screen for suppression of the uncoordination phenotype of unc-73(e936) on an additional 500 000 ENU-mutagenized genomes. Here, we characterize three new extragenic suppressor alleles from this screen that belong to class II. Suppressors were mapped to a chromosomal region using Hawaiian SNP mapping (25), and then putative suppressor mutations were identified after high-throughput genomic sequencing; spliceosome-associated factors and RNA binding proteins within the mapped region that had mutations in their coding regions were flagged for additional analysis. CRISPR technology and homology-directed recombination (32) were then used to create these putative suppressor alleles de novo and these were tested for uncoordination suppression using a thrash assay (Figure 1B) and cryptic splicing changes in unc-73(e936) using reverse transcription and PCR (Figure 1C and D).
Using this mapping and high throughput sequencing approach, we identified the three new putative suppressor alleles as prp-8(R540K), prp-8(D1549N) and either snrp-200(N18K) and/or (E103K). When we sequenced the original suppressed strain containing the mutation that mapped to snrp-200, that gene had two point mutations resulting in the two amino acid substitutions in the original suppressed strain. We made CRISPR mimics for the two unique new prp-8 suppressor alleles and for both snrp-200 mutations individually, N18K and E103K. We used genetic crosses to pair these mutations with unc-73(e936) (worm strains homozygous for both the unc-73 and suppressor mutant alleles) and tested whether these four new CRISPR alleles had suppressor activity. We used a thrash assay (16) to compare the movement of these putative suppressor strains against the unsuppressed unc-73(e936) strain and to previously identified class II suppressors snrp-27(M141T) (16), prp-8(G654E) and prp-8(T524S) (17)) along with a wildtype N2 control. Figure 1B shows that the new alleles snrp-200(N18K), prp-8(R540K) and prp-8(D1549N) all show strong suppression of the unc-73(e936) uncoordination phenotype. In contrast, snrp-200(E103K) showed no change in movement relative to unc-73(e936) and is therefore not considered a suppressor and not followed further. Of this group of six suppressors tested in Figure 1B, snrp-27(M141T), snrp-200(N18K) and prp-8(D1549N) appear to have the highest level of movement suppression, similar to wildtype N2 worms. During the process of doing crosses with CRISPR mimic strains to generate unc-73(e936); suppressor homozygous double mutants for the new suppressors prp-8(D1549N), prp-8(R540K) and snrp-200(N18K), we noted that worms homozygous for unc-73(e936) and heterozygous for the suppressor were ‘unc-ish’; they moved better than the unc-73(e936) starting strain, but still showed somewhat uncoordinated movement. This is consistent with the possibility that these new suppressor alleles are semi-dominant, as had been previously reported for class I and class II suppressors (16,17,23). However, we have not formally tested for suppressor allele semi-dominance using balancers in strains harboring the unc-73(e936) allele.
We next tested these new suppressors for changes in unc-73(e936) cryptic splice site usage (Figure 1C and D) and compared them against the previously reported class II suppressors snrp-27(M141T) (16) and prp-8(G654E) and (T524S) (17). The new snrp-200(N18K) and prp-8(D1549N) alleles are the strongest protein-coding extragenic suppressors that we have isolated to date in terms of their ability to promote in frame messages for unc-73(e936). They increase wt cryptic 5′ss usage from 13.6% in the unsuppressed strain to 37.2% and 38.6%, respectively in the suppressed strains. The three new suppressors all decrease the +23 site usage while increasing both -1 and wt site usage, so we also consider these as class II suppressors.
Determining global effects on splicing for the new suppressor alleles
To get an understanding of the role in splicing of the residues we found mutated in these new prp-8 and snrp-200 suppressor alleles, we used high throughput mRNASeq to identify native genes whose splicing changes in the presence of these suppressors. Libraries were made from strains containing unc-73(e936) alone as a control, or unc-73(e936) in combination with each suppressor allele, prp-8(R540K), prp-8(D1549N) or snrp-200(N18K). Three biological replicate samples from each strain were made into libraries (12 libraries in total) and between 50M and 79M raw paired-end 75nt reads were obtained from each library, of which between 20M and 35M reads mapped uniquely to the C. elegans genome. We ran an alternative splicing analysis which looked at both annotated and unannotated alternative 5′ and 3′ splicing events, as well as Ensembl-annotated skipped exon, mutually exclusive exon, multiple skipped exons, intron inclusion, alternative first exon and alternative last exon events (see Methods). For each alternative splicing event with a minimum of 15 junction spanning counts in each library, we quantified relative usage of each junction in each of the 12 libraries as the Percent Spliced In (PSI). We performed pairwise comparisons between each of the three biological replicates of a suppressor strain against each of the three biological replicates of the control strain, for a total of nine comparisons for each suppressor for each alternative splicing event, and asked how many of those nine comparisons generated a ΔPSI of >15%. Those events for which all nine pairwise comparisons had a ΔPSI >15% (pairSum = 9) were then analyzed individually on the UCSC Genome Browser with the RNASeq tracks to confirm the alternative splicing event. We then filtered these confirmed pairSum = 9 events for those with a >20% average ΔPSI over the nine pairwise comparisons. Table 1 summarizes the number of visually confirmed alternative splicing events meeting these criteria for each suppressor strain. Supplementary Table S2 contains a spreadsheet with chromosome location, ΔPSI values, splice junction sequences and notes on each event in Table 1. Supplementary Table S3 shows the junction counts for the three alternative cryptic splicing events for unc-73(e936) in each library. These counts of alternative 5′ splice junctions are consistent with class II suppression by increasing the usage of –1 and wt cryptic splicing while decreasing +23 cryptic splicing. However, because only one of the 12 libraries has more than 15 reads spanning this alternative region, it would not have been flagged by our stringent analysis pipeline.
Table 1.
Changes in global splicing in new suppressor alleles
| Events\Allele | prp-8(R540K) | prp-8(D1549N) | snrp-200(N18K) |
|---|---|---|---|
| Alt 3′ss | 30 | 1 | |
| Alt 5′ss | 1 | 73 | 19 |
| Skipped exon | 3 | ||
| Retained intron | 10 | 5 | |
| Multi-skip exon | 1 | ||
| Mut. Excl. exon | |||
| Alt first exon | 4 | 1 | |
| Alt last exon |
For each suppressor allele, we identify the number of events of each type with changes in splicing relative to an unsuppressed strain as determined by mRNASeq analysis. The number of events that fit the criteria of having a minimum of 15 junction spanning reads in each library, a ΔPSI > 0.15 for all nine pairwise comparisons (pairSum = 9), and a mean ΔPSI > 0.20 for the nine pairwise comparisons are indicated.
Alternative 5′ splice site changes in native substrates in the presence of suppressors
From this mRNASeq analysis, there are clear differences between the suppressors in the global changes in splicing that they promote. For the PRP-8(R540K) suppressor, we can only detect a single alternative splicing event in a native gene that changes. It involves an alternative 5′ss within an alternative first exon in the crh-1 gene, a splicing change that is shared with the PRP-8(D1549N) strain. This indicates that PRP-8(R540K), while having a measurable role in changing cryptic splicing for e936, does not affect splicing overall. In comparison, both PRP-8(D1549N) and SNRP-200(N18K) showed 73 and 19 alternative 5′ss events in native genes affected by the suppressors respectively. Of these alternative 5′ss events flagged in this analysis, in only three cases, all in PRP-8(D1549N), does the suppressor activate a 5′ss whose usage cannot be detected in the mRNA-Seq analysis in the control strain. This indicates that vast majority of the changes in the alternative 5′ss usage that we see are in introns that contain true alternative 5′ss events, and that the suppressors very rarely activate new cryptic alternative 5′ss. It should be noted that these experiments were done in strains containing an intact nonsense-mediated decay (NMD) pathway (36), so potential new splice sites that switch reading frame and are subject to NMD might not have been detected.
We chose to confirm some of the alternative 5′ss events determined by the mRNA-Seq using an RT-PCR assay. Several alternative 5′ splice sites were chosen for RT-PCR testing based on strong changes in the PRP-8(D1549N) and SNRP-200(N18K) strains, a range of intersplice site distances, and some representative overlap with SNRP-27(M141T) from our previous study (20). Figure 2A shows the results for all class II suppressor strains on 6 of the 73 introns that our mRNA-Seq analysis indicated should show changes in alternative 5′ss usage in the presence of the PRP-8(D1549N) mutation. The stars indicate lanes in which the average ΔPSI measured by RT-PCR in a suppressed strain compared to the unc-73 mutant control strain (the lane on the left) is >20%. mab-10 and T01D3.6 alternative splicing were previously described as changing in the presence of SNRP-27(M141T) (20), while the hil-1 splicing change was not identified in the previous SNRP-27(M141T) analysis; the change in splicing is less dramatic for SNRP-27(M141T) compared to PRP-8(D1549N). Three other substrates shown only had alternative splicing changes in the presence of PRP-8(D1549N). Figure 2B shows the results for three of the 19 native introns whose alternative 5′ss usage is affected in the presence of the SNRP-200(N18K) suppressor. In two of these cases, AKAP-1 and E01B7.2, the SNRP-200(N18K) suppressor alone among the suppressors we tested changes the usage of these native alternative 5′ss. For one event, an intron in Y71H2AM.2, we see an interesting phenomenon. In an unsuppressed background, the two alternative 5′ss are used relatively evenly. In the presence of both the PRP-8(D1549N) and SNRP-200(N18K) suppressors, there is a strong shift to usage of the downstream 5′ss. In the presence of the SNRP-27(M141T) suppressor, there is a strong and opposite effect where there is a shift towards almost exclusive usage of the upstream 5′ss which was noted previously (20). In addition to the two major splice products, we do see weak usage of a downstream 5′ss defining an intron beginning with GU, and another that defines an intron beginning with UU (usage of both is detected in the sequenced libraries) - the usage of these sites is not affected greatly by these suppressor alleles. The ability to identify alternative 5′ splicing events whose changes are unique to a particular suppressor, even when all these suppressors have similar effects on unc-73(e936) cryptic splicing, indicates that these class II suppressors may be working at different and unique stages of spliceosome assembly and/or employing unique mechanisms to maintain the identity of 5′ splice sites.
Figure 2.

Reverse transcription and polymerase chain reaction (RT-PCR) analysis of RNA from unc-73(e936) suppressor strains on selected alternatively spliced introns. Sequence of the 5′ss (A and B) or 3′ss (C) region of the alternative spliced gene is indicated at the right and filled and clear arrows indicate alternative splice sites and correspond to the bands indicated on the scanned gels. Samples with a mean ΔPSI >20% compared to the unc-73(e936) unsuppressed control strain are indicated with a blue star at the top of the lane; the number of independent trials used to calculate mean ΔPSI (n) is indicated. Quantitation data for these experiments is provided in Supplementary Table S1. (A) Sample alternative 5′ splicing events with changes in the prp-8(D1549N) mutant strain. Note in the first three examples that the snrp-27(M141T) mutant strain has similar effects on alternative splicing while in the next three examples the effect is specific to D1549N. (B) Sample alternative 5′ splicing events from the snrp-200(N18K) mutant strains. Note that for the Y71H2AM.2 intron shown, the prp-8(D1549N) mutation has a similar effect on alternative 5′ss choice, while the snrp-27(M141T) mutation has the dramatic opposite effect. Two minor confirmed alternative 5′ss for Y71H2AM.2 are indicated above the sequence with small arrows. (C) An example of an alternative 3′ splicing event uncovered in the prp-8(D1549N) RNASeq analysis. Note that the change in alternative 3′ss usage seen for this intron in the prp-8(T524S) suppressor strain was reported previously (17).
We analyzed the information at the 5′ss at the beginning of introns for these native alternative 5′ splicing events. Alternative splice sites whose usage is increased in the presence of these suppressors are almost evenly split between upstream and downstream location relative to the alternative 5′ss in a pair (Figure 3A and B). Figure 3C shows sequence logos for random intron 5′ss and for the pairs of 5′ss whose usage is reduced or promoted in the presence of the PRP-8(D1549N) and SNRP-200(N18K) suppressors. The 5′ss in the alternative pair whose usage is reduced in the presence of PRP-8(D1549N) resembles the consensus 5′ss for C. elegans, while the 5′ss whose usage is promoted in the presence of this suppressor only shows a strong signal for the GT dinucleotide at the start of the intron. For the SNRP-200(N18K) suppressor, the 5′ss in the alternative pair whose usage is reduced in the presence of the suppressor has a striking lack of a preference for G at the fifth intron position; perhaps this is a clue as to why these alternative 5′ss are sensitive to the SNRP-200(N18K) mutation. The 5′ss that is activated in the presence of the SNRP-200(N18K) suppressor has only a strong signal for the GT dinucleotide at the start of the intron. These differences offer some indications of features of alternative 5′ss that may lead to altered splicing by these suppressors. Here it should be noted that, in contrast to the trend in native alternative splicing events affected by PRP-8(D1549N) and SNRP-200(N18K), for unc-73(e936) cryptic splicing the +1 and wt cryptic splice sites promoted by these suppressors both have G at their fifth intronic positions (Figure 1A). As almost all these alternative 5′ss pairs have the usage of both isoforms detected in a wildtype background, these two suppressors appear to allow for the increased usage of a weaker-consensus authentic alternative 5′ss positioned nearby.
Figure 3.

Analysis of global 5′ss changes for prp-8(D1549N) and snrp-200(N18K) and 3′ss changes for prp-8(D1549N). (A) Histogram showing relative distances of alternative 5′ss for prp-8(D1549N) (n = 73) (Table 1 and Supplementary Table S2). (B) Histogram showing relative distances of alternative 5′ss for snrp-200(N18K) (n = 19) (Table 1 and Supplementary Table S2). The arrow at zero in (A) and (B) is the position of the 5′ss whose usage is reduced in the presence of the mutant allele. The distances of the sites whose usage is increased in the presence of the mutants are shown in the histogram. Note that there is no apparent bias for use of an upstream or downstream 5′ss with either suppressor allele. (C) The consensus of the first 7nt of 10 000 randomly chosen C. elegans intron starts is shown at the top. The second and third logos compare the 73 alternative 5′ss from the prp-8(D1549N) RNAseq library analysis identified in Table 1. The alternative 5′ss whose usage is lessened in the presence of prp-8(D1549N) is shown first, followed by the alternative 5′ss whose usage is increased in the presence of the prp-8(D1549N) allele. The fourth and fifth logos compare the 19 alternative 5′ss from the snrp-200(N18K) mutants. The alternative 5′ss whose usage is lessened in the presence of snrp-200(N18K) is shown first, followed by the logo for the alternative 5′ss in the pair whose usage is increased in the presence of the snrp-200(N18K) allele. Consensus sequence logos were generated using WebLogo (34). (D) Histogram showing the positions of the 30 alternative 3′ss promoted by prp-8(D1549N) relative to the partner alternative 3′ss whose usage is reduced. Note that all but one of the mutant-promoted sites is downstream. (E) The consensus logo of the last 7nt of 10 000 randomly chosen introns is shown at the top, matching the TTTTCAG consensus. The second and third logos compare the 30 alternative 3′ss from the prp-8(D1549N) RNAseq analysis from Table 1 and Supplementary Table S2. The sequence logo of the alternative 3′ss whose usage is lessened in the presence of prp-8(D1549N) is shown first, followed by the alternative 3′ss whose usage is increased.
We analyzed the overlap between the native alternative splice sites affected by PRP-8(D1549N) and SNRP-200(N18K), along with SNRP-27(M141T) alternative splicing events that we reported previously (20). For SNRP-27 we reported 26 native alternative 5′ splicing events that were changed in the mutant strain. Between PRP-8(D1549N) and SNRP-27(M141T), 9 alternative splicing events are shared, and 8/9 change splicing in the same direction. The one exception is the event in Y71H2AM.2, whose splicing is shown in figure 2B. Between SNRP-27(M141T) and SNRP-200(N18K), three alternative 5′ splicing events are shared, and surprisingly all three are changed in opposite directions between the strains. Between PRP-8(D1549N) and SNRP-200(N18K), seven alternative 5′ splicing events are shared between the two, and they all change splicing in the same direction. Overlapping events are noted in Supplementary Table S2. The limited overlap in native 5′ splicing targets between the suppressor alleles is consistent with these suppressors affecting different sites in the spliceosome with distinct roles in the maintenance of 5′ss identity as the spliceosome is assembled.
Alternative 3′ splicing changes in the presence of the PRP-8(D1549N) suppressor
The PRP-8(D1549N) suppressor also shows global changes in alternative 3′ss usage. There are 30 events identified in Table 1 (detailed in Supplementary Table S2) and all but one promote the usage of a downstream 3′ss in the presence of the suppressor allele (Figure 3D). In all 30 cases, we can detect the usage of both splice sites in the pair in the wildtype control background, indicating that these are native alternative splicing events. Figure 2C shows a confirmation of one of these events, an intron in B0001.7, by RT-PCR. We had previously reported that the PRP-8(T524S) suppressor had effects on alternative 3′ss usage on 10 native introns, including this one, which is confirmed in this figure. What is surprising is that of the 10 reported alternative 3′ splicing events affected by PRP-8(T524S), there was a 5:5 split as to whether the suppressor mutation promoted the upstream or downstream alternative 3′ss (17). This is in sharp contrast to PRP-8(D1549N) in which 29 of the 30 alternative 3′ss promoted by the suppressor mutation promoted the downstream 3′ss (Figure 3D). This unidirectional change in splicing is similar to a phenomenon that we reported previously for developmentally-regulated alternative 3′ss usage (37). In that study we reported 203 examples of alternative 3′ss pairs in which germline cells increased the usage of the upstream 3′ss relative to somatic cells. A feature of those alternative splicing pairs is that the upstream 3′ss only had an AG dinucleotide at the end of the intron as the distinguishing feature, while the somatic-preferred downstream 3′ss was a strong match to the C. elegans 3′ss consensus sequence. The majority of those alternative 3′ss were within 12nt of each other. Figure 3E shows that for the 30 alternative splicing pairs affected by PRP-8(D1549N), similar consensus features are observed; the downstream 3′ss promoted by the suppressor matches the 3′ss consensus sequence for the animal, while the upstream 3′ss only has an AG dinucleotide at the end of the intron as its distinguishing feature. Of these 30, 8 overlap with the 203 alternative 3′ splicing events identified by Ragle et al. as germline regulated (37). These eight events are indicated in Supplementary Table S2.
Double suppressor strains allow insight into the order in which suppressor mutations function
To better understand when in spliceosome assembly class II suppressors exert their influence, we attempted to generate double mutants for the class II suppressors with the highest number of changes in native gene splicing; PRP-8(D1549N), SNRP-200(N18K) and SNRP-27(M141T). Because of the challenges of doing crosses with strains containing unc-73(e936) and two suppressors without individual phenotypes, we instead opted to use CRISPR to insert a second suppressor allele into an already suppressed strain. Using this approach, we attempted to insert prp-8(D1549N) using CRISPR into strain SZ305 which has the genotype unc-73(e936); snrp-200(N18K). While we could identify fertile animals that were heterozygous for a CRISPR allele of prp-8(D1549N) (containing the genotype unc-73(e936)I; snrp-200(N18K)II; prp-8(D1549N)/+III) and those animals had the expected 25% progeny that were homozygous for prp-8(D1549N), the animals homozygous for D1549N were completely sterile and we could not maintain that strain. We confirmed the homozygous D1549N mutation in the sterile adults by sequencing. This result suggests that spliceosomes containing both PRP-8(D1549N) and SNRP-200(N18K) mutant proteins have too many complications in splicing to support the ability to produce embryos.
We did successfully use CRISPR to create a snrp-27(M141T) allele in suppressed strains to generate strains with the genotypes unc-73(e936)snrp-27(M141T)I; prp-8(D1549N)III and unc-73(e936)snrp-27(M141T)I; snrp-200(N18K)II. These double class II suppressor strains are viable and have wildtype movement. We performed RT-PCR analysis to measure the splicing of unc-73(e936) in the double class II suppressor strains (shown in Figure 1D for ease of comparison with single suppressor strains). We find that the snrp-27(M141T);prp-8(D1549N) strains used the wt e936 splice site in 36.7% of the messages, which is similar to prp-8(D1549N) on its own (38.6%) and greater than snrp-27(M141T) on its own (23.0%) (Figure 1D). A two-tailed students t-test of the prp-8(D1549N) single suppressor vs. the snrp-27(M141T);prp-8(D1549N) double suppressors on e936 wt splice site usage (38.6% vs. 36.7% average respectively) yields a P-value of 0.253, indicating that there is no significant difference between these mutant strains. The snrp-27(M141T);snrp-200(N18K) double mutant showed 43.0% of the e936 messages splicing to the wt site, which is greater than the snrp-200(N18K) suppressor on its own (37.2%) and the highest wt splice site usage we have measured for protein suppressors on e936. The 5.8% average increase in snrp-27(M141T); snrp-200(N18K) double mutant wt splice site usage relative to the snrp-200(N18K) single mutant has a P-value of 0.0017, indicating that this change is significant. All three of the suppressors used in these double-mutant strains are expected to move e936 splicing in the same direction, toward the –1 and wt sites. Only in the case of the snrp-27(M141T);snrp-200(N18K) double mutant is there evidence for an additive effect.
We next tested these two double suppressor strains on three additional native alternative 5′ splicing events that we showed in Figure 2 to be targets of these suppressors (Figure 4A and B). The Y71H2AM.2 alternative splicing event is interesting because of the differential response to these class II suppressors. There is roughly even usage of the two main alternative splice sites in a wildtype background, while the snrp-27(M141T) allele promotes use of the upstream splice site while the prp-8(D1549N) and snrp-200(N18K) suppressors promote use of the downstream 5′ss. The snrp-27(M141T);prp-8(D1549N) double mutant splices this intron similar to the prp-8(D1549N) suppressor alone and very differently than the snrp-27(M141T) alone, indicating that the prp-8(D1549N) allele overrides the alternative splicing change promoted by the snrp-27(M141T) allele. For the snrp-27(M141T);snrp-200(N18K) double mutant, splicing essentially resembles wild type, indicating that these two mutant alleles independently counteract their opposite effects on this substrate, leading to no change in splicing from wildtype.
Figure 4.

Analysis of class II suppressor double mutant alternative splicing on select introns. (A) Reverse transcription and PCR reactions for alternative splicing. Strains are indicated at top and alternative splicing products and sequences of 5′ss regions are indicated at right. The results of three biological replicates of the double suppressor strains are shown. (B) Quantitation of alternative splicing results of the indicated suppressor strains. Percent usage of the upstream 5′ss for each intron in each strain is shown, along with the standard deviation and number of trials. Quantitation of the splice products are found in Supplementary Table S1.
For the E01B7.2 alternatively spliced intron, we find that the snrp-27(M141T) mutant increases upstream splice site usage, while the prp-8(D1549N) and snrp-200(N18K) mutants increase downstream 5′ss usage, with the biggest percent change coming from snrp-200(N18K). Here we observe again that the snrp-27(M141T);prp-8(D1549N) double mutant splicing resembles the prp-8(D1549N) mutant alone. In contrast, the snrp-27(M141T);snrp-200(N18K) double mutant resembles the snrp-27(M141T) alone. This indicates that snrp-27(M141T) may be acting downstream of snrp-200(N18K) to override the splicing change promoted by it. For the mab-10 alternative 5′ss intron, snrp-27(M141T) and prp-8(D1549N) both promote upstream 5′ss usage while snrp-200(N18K) shows a 7% average decrease in upstream 5′ss usage (P-value 0.0011). The snrp-27(M141T);prp-8(D1549N) double mutant promotes upstream 5′ss usage, and the snrp-27(M141T);snrp-200(N18K) resembles the snrp-27(M141T) single mutant in promoting upstream 5′ss usage. Taken together, analyzing the effects of these two double mutant strains on targets of alternative splicing suggests that snrp-200(N18K) mutation affects events at the same time or before snrp-27(M141T), and that prp-8(D1549N) suppressor allele exerts its influence on alternative splicing after the other two suppressors.
Isolation of two additional PRP-8(D1549) substitution alleles
In order to test a range of different amino acid substitutions of PRP-8 at position D1549, we used a CRISPR approach to randomize that position and asked what substitutions we could recover and their effects on splicing. While not an exhaustive screen, we were able to recover viable substitutions of D1549I and D1549V. We also identified substitutions D1549R, D1549F and D1549Y that were detectable as heterozygotes after CRISPR but were homozygous sterile. We also recovered a D1549D allele in which silent mutations to disrupt the PAM sequence and create a restriction site were recovered but the amino acid identity of D served as a nice control strain. Figure 5 shows a test for these three new alleles on three different native substrates whose splicing changes in the presence of the D1549N substitution. For the mab-10 and tofu-7 introns, the D1549I and D1549V substitutions perform similarly to D1549N. In contrast, on the Y71H2AM.2 intron, the D1549I and D1549V substitutions had no effects on alternative splicing. The fact that not all substitutions are viable as homozygotes indicates the importance of this residue of PRP-8. The substrate specificity for the substitutions in splicing changes indicates that some alternative splicing events are differentially sensitive to substitutions at D1549.
Figure 5.
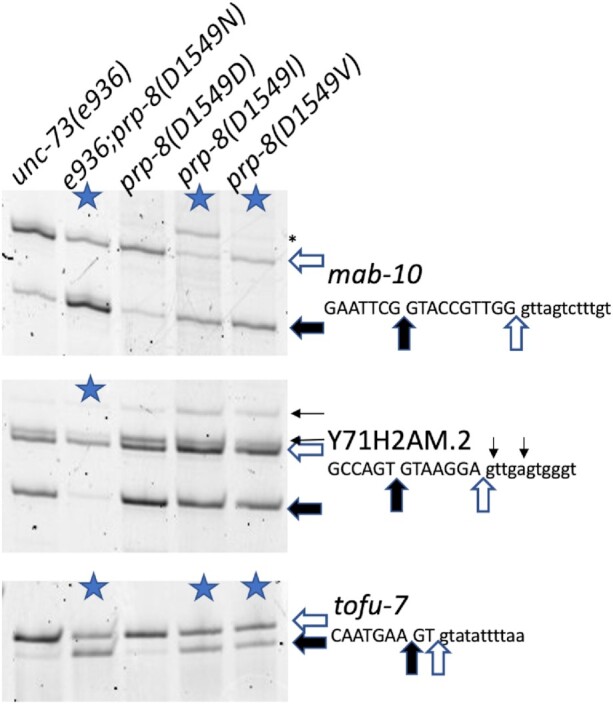
Analysis of alternative splicing of two additional prp-8(D1549) mutant alleles. New strains tested include a D1549D control allele from the CRISPR experiment, and the new viable alleles D1549I and D1549V. RNA was isolated from the indicated strains and the alternative 5′ splicing events in mab-10, Y71H2AM.2 and tofu-7 analyzed. Those strains with >20% ΔPSI in this experiment compared to the control on the left are indicated with a blue star (average of two independent experiments, see Supplementary Table S1 for quantitation data). An asterisk on the right of the mab-10 gel denotes a spurious background band that is not reproducible for prp-8(D1549I).
Analysis of additional SNRP-200 (BRR2) mutants
SNRNP200/BRR2 is a member of the ski2-like subfamily of RNA helicases and is one of the 8 RNA helicases required for the spliceosome assembly/disassembly cycle (3). During catalytic activation of the spliceosome, BRR2 helicase activity is required to unwind the U4/U6 snRNA duplex (38). Mutations within the ratchet helix of the RNA binding channel in the human homolog SNRNP200 are associated with the degenerative vision disease retinitis pigmentosa (39,40). Genetic experiments have shown that yeast with a deletion of the N-terminal 112 amino acids (41) or 120 amino acids (42) of BRR2 have growth defects associated with a lower spliceosome assembly efficiency. Additionally, a defect in unwinding stabilized U1snRNA-pre-mRNA helices has been observed in the N-terminal deletion (42). Crystal structure models of the BRR2 protein implicate the 500aa N-terminal domain in autoinhibition of the BRR2 activity through substrate competition and conformational clamping (43), however the first 110 amino acids of the protein are absent from that structure.
The N-terminal 120 amino acids of BRR2 are poorly conserved between Humans, C. elegans Arabidopsis thaliana, S. pombe and S. cerevisiae, but the N18K suppressor allele of SNRP-200 is in a short region of amino acids, S(N)(L)V(L) that is conserved between the five species (those amino acids in parentheses are identical in 4/5 species; the worm and human protein sequences are identical in this short stretch) (Figure 6A). To further characterize this region, we generated a series of 11 additional single amino acid substitutions at N18 using a CRISPR homologous repair approach to generate random sequences at that residue. In addition, we used CRISPR and homology-directed repair to generate a deletion of SNRP-200 amino acids 13–22 to encompass the conserved region with the N18K suppressor (Δ13–22). In the process generating that allele, we also recovered a larger deletion allele generated by non-homologous end joining, az154, that deletes 131nt from the genome. The deletion begins 16nt before the start codon of SNRP-200, within the annotated 5′ UTR of the first exon and removes the first 39 amino acids of the coding region. Downstream of the deletion, the next three ATG triplets that could serve as start codons are all in-frame at amino acid positions 48, 51 and 53 of the annotated region. While we have not confirmed the actual start codon used, we have been referring to this allele as Δ1–47. Animals homozygous for either the Δ13–22 or Δ1–47 are viable, providing metazoan evidence that is consistent with the viability of S. cerevisiae deletions of the very N-terminal region of BRR2 (41,42).
Figure 6.

Alternative 5′ splicing tests on a range of SNRP-200(N18) substitutions. (A) Alignment of the N-terminal region of SNRNP200/BRR2 from C. elegans, human, Arabidopsis thaliana, the fission yeast Schizosaccharomyces pombe and S. cerevisiae. The large yellow arrow indicates the N18 residue in C. elegans. (B) RT-PCR analysis of mutants on specific introns. The first lane on the left is from RNA from wild type strain N2. The second lane uses RNA derived from a strain with a mutation in snrp-27(M141T). The next 13 lanes use RNA derived from strains generated by CRISPR mutagenesis at the N18 position; the amino acid substitutions at position N18 are indicated. Note that we recovered an N18N substitution by CRISPR, and that is a useful control in the seventh lane. The last two lanes use RNA derived from strains with N-terminal deletions of SNRP-200, amino acids 1–47 or amino acids 13–22 respectively. The 5′ splice site regions of the three introns, from Y71H2AM.2, E01B7.2 and akap-1 are indicated below each gel image. Lanes in which the products show ΔPSI > 0.2 compared to wild type in two biological replicates are indicated with a blue star (see Supplementary Table S1 for quantitation).
We tested these new SNRP-200 alleles for effects on alternative 5′ splice site choice in three different native introns that show splicing changes in the presence of the N18K allele (Figure 6B). For the Y71H2AM.2 and E01B7.2 introns tested, only the N18R substitution showed a >20% ΔPSI as assayed by RT-PCR (average of two independent experiments for each strain, see Supplementary Table S1 for quantitation). This result indicates that the positively charged N18K and N18R substitutions represent specific gain-of-function alleles in splicing. Also, note that for Y71H2AM.2, the N18P allele only can promote an increase in a downstream intron start site (indicated by short arrows in the figure). We see this reproducibly for N18P and not in any other suppressor we have tested on Y71H2AM.2. In contrast to the Y71H2AM.2 and E01B7.2 results, when we look at the akap-1 intron alternative splicing, substitutions at N18 of K, V, F, A, I and E, along with Δ13–22 and Δ1–47 all give >20% ΔPSI compared to strains with wildtype SNRP-200. Other N18 substitutions, P, G, S, C, R and T fail to meet this 0.20 threshold for ΔPSI (see Supplementary Table S1). It therefore appears that the role of the N-terminal domain in regulating alternative 5′ss choice is dependent on the both the nature of the mutation in SNRP-200 as well as features of the particular alternative splicing event examined.
Comparison of yeast splicing suppressors of known mechanism to type class II splicing suppressors
To better understand the potential mechanism of class II suppression, we performed an experiment to compare the role of suppressors in S. cerevisiae that may also affect 5′ splice site maintenance. We chose three classes of S. cerevisiae splicing suppressors to study based on hypotheses as to how class II suppressors may function in worms. One hypothesis for unc-73(e936) class II suppressors is that they may enhance or bypass the unwinding of U4/U6 to promote nearby alternative 5′ss usage. The first class of yeast suppressors that we test are U4-cs1 suppressors (44). These suppress a cold-sensitive mutation in U4 that stabilizes its base pairing with U6. The U4-cs1 suppressor alleles we chose to test are in BRR2 (R295I) (the S. cerevisiae homolog of SNRP-200) and PRP8 (K611R and T1640K) (44,45). Another hypothesis for our unc-73(e936) suppressor function is that they work at the level of U1/5′ss unwinding to promote alternative U6/5′ss interactions. The second type of suppressor we tested addresses this hypothesis by suppressing a cold-sensitive defect in Prp28 called prp28-1 (46,47). PRP28 is a DEAD-box helicase responsible for unwinding the U1/5′splice site interaction which then allows for U6 to interact with the 5′ss. The suppressors isolated in the prp28-1 cold sensitive suppressor screen localize to the bromodomain-like region in the N-terminal domain of PRP8 (48). A third hypothesis for class II suppressor mechanism is that they accelerate the first step of splicing thus allowing sub-optimal 5′ss to load into the active site without proper proofreading. This can be tested by a third class of suppressors that suppress first step splicing defects in yeast. These suppressors in PRP8, L1557F(syf77), R1753K and E1960K(prp8-101) (49–51) accelerate the first catalytic step of splicing (52). To test these hypotheses, we used CRISPR to generate seven new C. elegans alleles (six in prp-8 and one in snrp-200) representing these three classes of yeast suppressors (Figure 7A). We chose these candidates based on being in regions of high conservation with the C. elegans homolog (Supplementary Figure S1A) and for proximity to class II unc-73(e936) suppressors. Figure 7A summarizes the yeast suppressor alleles chosen for study and their homologous C. elegans positions that were targeted by CRISPR.
Figure 7.
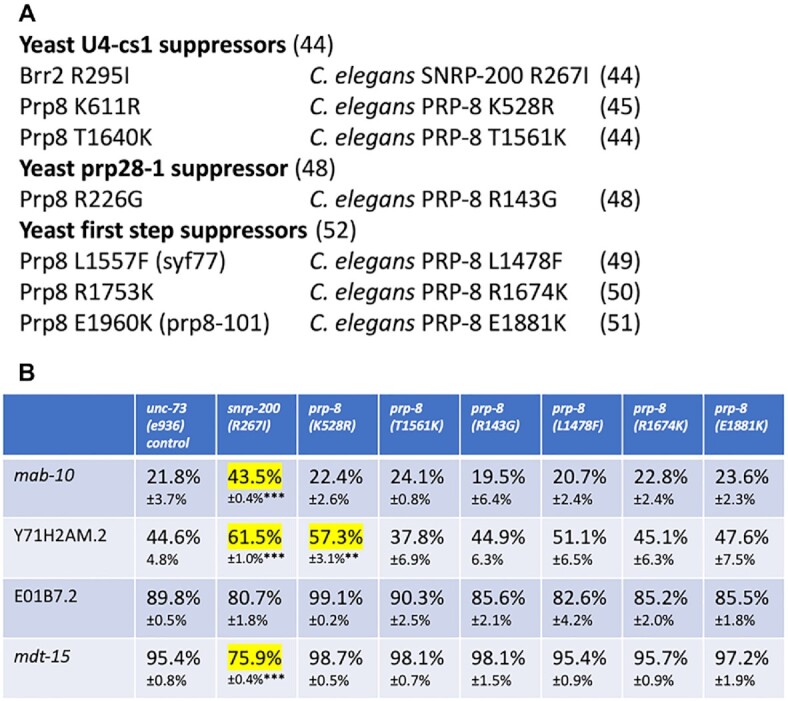
Generation and testing of yeast splicing suppressors of known classes in C. elegans. (A) The three different classes of yeast suppressor tested, the location of the seven individual alleles tested (with their location in S. cerevisiae BRR2 and PRP8 on the left and the homologous position of the suppressor in C. elegans on the right. Corresponding citations for the individual alleles and classes are indicated in parentheses. (B) Quantitation of alternative splicing of several introns regulated by class II suppressors in C. elegans strains bearing mimics of the yeast suppressors compared to an unsuppressed control in the left column. Percent usage of the upstream 5′ss and standard deviation (n = 3) are indicated. Events with >10% changes in upstream 5′ss usage relative to the control strain are highlighted in yellow. For the ones highlighted we determined the P-value for the change relative to the control. ** indicates P-value from student's t-test <0.01 and *** indicates P-value <0.001.
We first tested these new mimics of yeast suppressor alleles for the ability to suppress movement defect in unc-73(e936) by using crosses to generate strains homozygous for each new suppressor and unc-73(e936). Using the thrash assay, we found that none of these new suppressor alleles could suppress the uncoordination defect caused by unc-73(e936) (Supplementary Figure S1B). This result is quite different than the suppression seen for the six class II suppressors that we isolated in forward genetic screens (Figure 1B).
We next tested whether the seven new yeast suppressor mimic alleles could have any effects on splicing of four native introns identified by RNA-seq as having alternative 5′ss choice affected by the class II suppressors. Triplicate biological replicates from each strain were tested for splicing of the alternative 5′ss intron in mab-10, Y71H2AM.2, E01B7.2 and mdt-15. These test alternative 5′ splicing events were chosen for their differential specificity for the class II suppressors snrp-27(M141T), snrp-200(N18K) and prp-8(D1549N) (Figure 2A and B). Quantitation of the alternative splicing of these new strains on these alternative events is shown in Figure 7B and in Supplementary Table S1. For the prp28-1 suppressor and the three first step suppressors tested, we saw no changes in upstream 5′ss usage vs. the control strain greater than 10% on any of these four substrates. One of the U4-cs1 suppressor mimics, snrp-200(R267I) did allow for significant changes in upstream 5′ss usage greater than 10% on three of the test alternative introns (Figure 7B). While these three changes were consistent in the direction of splicing change as found for snrp-27(M141T) (Figure 2), none of these changes approached the level of the more dramatic changes seen in the class II suppressors (Figures 2A, B and 4A, B). The inability of these yeast suppressor mimics to suppress the unc-73(e936) uncoordination phenotype and the inability of these suppressor mimics to recapitulate the level of alternative splicing changes promoted by class II suppressors on native genes, suggests that class II suppression may occur through unique mechanisms from the three classes of yeast suppressors tested.
DISCUSSION
Using a forward genetic screen for suppressors of an uncoordination defect caused by a mutation to a splice site, we identified three new suppressor mutations that affect cryptic splice site choice. These new suppressors, PRP-8(R540K), PRP-8(D1549N) and SNRP-200 (N18K) join three previously identified suppressors SNRP-27(M141T) (16), PRP-8(G654E) and PRP-8(T524S) (17) as class II suppressors. These have a specific effect on unc-73(e936) cryptic splicing, resulting in less splicing at the +23 cryptic splice site and a concomitant increase in splicing at both the −1 and wt cryptic sites (Figure 1D). We previously identified a different suppressor allele, now referred to as a class I suppressor, that consisted of a compensatory mutation in one of the 12 U1snRNA genes, sup-39, that allowed for U1snRNA to base pair with a 5′ end of an intron that begins UU (18). This suppressor also resulted in less splicing at the +23 site with a concomitant increase in splicing at both the −1 and wt cryptic sites (19). Class I suppression must act before class II suppression, since SNRP-27, PRP-8 and SNRP-200 enter the spliceosome with the U4/U6-U5 tri-snRNP after U1 snRNA initially identifies the 5′ss. We assume that wildtype U1snRNA initially identifies and promotes the +23 site ∼74% of the time (we assume this since a mutation in one of the 12 U1 snRNA genes can alter this choice). Therefore, the changes in splicing that we observe in class II suppressors are consistent with a decrease in the ability to maintain the 5′ss initially identified by U1snRNA base pairing through to loading into the catalytic site of the spliceosome. The 5′ss is initially identified by U1snRNA, but U1snRNA leaves the assembling spliceosome during the preB to B transition. After that, the identity of the 5′ss must be maintained through multiple structural rearrangements from B to preBact1 to preBact2 to Bact to B* complex, where catalysis occurs (4,12).
One hypothesis to explain changes that class II suppressors promote in unc-73(e936) cryptic splicing, along with the changes in alternative 5′ss usage on specific native substrates, is that these suppressors allow the spliceosome to slip from the site identified by U1 snRNA to use a nearby 5′ss if one is available. This slippage must occur in the period after U1snRNA has initially identified a 5′ss and prior to catalysis. In Figure 8, we look to the published cryoEM structures of human spliceosomal assembly intermediates to see if we can gain some understanding of suppressor function by looking at the modeled interactions of the corresponding wild type residues. Figure 8 shows human spliceosomal structures from four different stages of spliceosome assembly and on that we can map some of the suppressors; we use the worm protein numbering system in these images for the class II suppressors, but they are mapped onto their human conserved counterparts; human SNRNP27K M141(C. elegans SNRP-27 M141), human PRPF8 T532 (C. elegans T524), PRPF8 R548 (C. elegans R540), PRPF8 G662 (C. elegans G654), and PRPF8 D1556 (C. elegans D1549). The extreme N terminus of SNRNP200/BRR2 is not modeled in any of the human or yeast spliceosome structures, so we cannot model human N18 which corresponds to C. elegans SNRP-200 N18. ChimeraX .csx session files for the four images shown in Figure 8 are available as Supplementary Material.
Figure 8.

CryoEM modeling of prp-8 suppressors in the context of the assembling human spliceosome. (A) Human spliceosome preB complex 6QX9 (9). The unstructured ACAGA box, anchored by the quasi-pseudoknot above and the U4/U6 helix below is represented by the bold dashed line. The 5′ss paired with U1snRNA is located just off the image to the lower right corner. (B) Human spliceosome B complex 6AHD (11). (C) Human spliceosome Bact complex 6FF4 (13). (D) Human spliceosome catalytic step 1 complex 5YZG (14). The coloring scheme for snu66, 27K, U4snRNA, U6snRNA, pre-mRNA, PRP8, RNF113A, YJU2, and ISY1 used in the images is indicated above the figures. PRP8 residues T524, D1549, and H1573 are labeled and shown as stick models, as are snu66 H765 and 27K M141 (we are using the C. elegans numbering imposed on their homologous positions in the human spliceosome). The G at the +1 and the U at the +2 positions of the intron, and the branchpoint A (bpA) are also labeled.
In human pre-B complex (9) (Figure 8A), the SNRNP27K M141 residue is modeled to interact with H765 of SNU66 (H734 in humans) and H1573 of PRP8 (H1580 in humans). These appear to stabilize the U4/U6 quasi-pseudoknot that forms one end of the unstructured ACAGAGA box region of U6 (shown as an orange dotted line in 8A) (9). The ACAGAGA sequence will interact with the 5′ end of the intron when U1snRNA becomes displaced from it by PRP28 activity (Figure 8B). In Figure 8A, the 5′ end of the intron paired with the 5′ end of U1snRNA is just off the lower right corner of the image. One hypothesis to explain 5′ss slippage by SNRP-27(M141) mutants is that this disruption can destabilize the U4/U6 quasi-pseudoknot, allowing for the ACAGAGA region to pair with a second 5′ss while the primary 5′ss identified by U1snRNA is still interacting with U1snRNA. Normally SNRNP200/BRR2 unwinds the U4/U6 interaction to make the ACAGAGA box available; although the N terminal region of SNRP200 is not modeled in these cryoEM structures, we hypothesize that the N18K mutation may cause premature unwinding of the U4/U6 interaction, leading to a similar result. This is consistent with our observation of alternative 5′ss usage in the snrp-27(M141T); snrp-200(N18K) double mutant that indicate that these alleles act at a similar time, with snrp-200(N18K) acting earlier on some substrates (Figure 4). PRP8 D1549 is in proximity to SNRNP27K M141 in this model (Figure 8A), so perhaps some of the overlap between native alternative 5′ss activated by SNRP-27(M141T) and PRP-8(D1549N) can be explained by similar destabilization of a key region in preB complex. However, in a snrp-27(M141T); prp-8(D1549N) double mutant, an alternative splicing events regulated in opposite directions by both suppressors (Y71H2AM.2) has a splicing pattern expected for prp-8(D1549N); this indicates that prp-8(D1549N) exerts its effects after snrp-27(M141T) (Figure 4). Consistent with this observation, PRP-8(D1549) has additional important interactions with the 5′ss in later assembly steps (discussed below).
In B complex (11) (Figure 8B), the 5′ss of the pre-mRNA forms interactions with the ACAGAGA region of U6 snRNA. In this complex, PRP8 T524 has direct interactions with the backbone of the intron between the G and the U of the GU dinucleotide at the intron start. At the same time, D1549 interacts with the A at the 3rd position of the intron. These two PRP8 positions appear to stabilize the 5′ss within the assembling spliceosome after U1snRNA has left the complex. We hypothesize that mutation to T524S or D1549N may allow the 5′ss to be held less tightly allowing for slippage to a nearby 5′ss. The interaction of T524 with the GU dinucleotide region of the 5′ss is also maintained in both Bact (13) and catalytic step 1 C complex (14) (Figure 8C and D). In contrast to T524, D1549 is displaced from the 5′ss in Bact by RNF113A with which it interacts. In B*/C complex, YJU2 and ISY1 occupy the space that RNF113A had occupied in Bact, and they also interact with the branchpoint A region of the intron as it is building a transition state for the first step of catalysis; D1549 interacts with YJU2 in this complex. For D1549, there appear to be important distinct interactions at each of these four snapshots of spliceosome assembly, so it is possible that our observed changes in both 5′ and 3′ splice site choice could occur because of several different disruptions caused by this suppressor position.
Suppressor PRP-8 R540 is in an alpha helix that interacts with other PRP-8 alpha helices in these structures (not shown). A potential function for this suppressor is hard to pin down from the cryoEM models, and only a single alternative native 5′ss change was noted in our mRNASeq study of the R540K mutant strain. In the case of PRP-8 R540K, we hypothesize that this minor change to the spliceosome may accommodate the unique nature of the e936 5′ss region leading to the changes in splicing and uncoordination that we detect. PRP8 G654 is in a region that is unstructured until Bact complex, when it is one end of a structured protein loop of PRP8 that forms across the U6 ISL. We previously referred to this region of PRP8 as the 740 loop (based on the numbering of this region in the S. cerevisiae homolog) (17). The U6 ISL is a part of U6 that holds the catalytic Mg2+ ions, and we hypothesized that the PRP8 740 loop forms a structure across the ISL that stabilizes the catalytic core of the spliceosome, and that mutations at G654 would leave this region more open and potentially allow for an alternative 5′ss to slip into the catalytic site. This hypothesis is supported by the work of Eysmont et al. who showed that the U6-ISL within the catalytic core toggles between catalytic and non-catalytic conformations due to flexibility within the lower ISL segment (53). They demonstrated that changes of relative stabilities of these conformations affect splicing fidelity.
The ability to perform suppressor genetic screens to identify changes in spliceosome specificity is a powerful approach, especially when combined with the ability to use cryoEM models to map these suppressors. This combined suppressor screening/cryoEM mapping approach has been used in S. cerevisiae to map onto the spliceosome suppressors of a cold sensitive phenotype caused by an extended U4/U6 basepairing interaction (44). We tested whether mimics of S.cerevisiae U4-cs1 suppressors (these allow splicing to proceed when the U4–U6 interaction is stabilized by additional base pairing), a prp28-1 cs suppressor (that allows splicing to proceed when PRP28 is inhibited in unwinding U1/5′ss pairing under cold conditions), and first step suppressors that allow the first catalytic step of splicing to proceed. None of the mimics we created (Figure 7A) could suppress unc-73(e936) uncoordination (Supplementary Figure S1B). In addition, these suppressor mimics did not behave like the class II suppressors on four native alternative 5′ss events (Figure 7B). The potential exception is the U4-cs1 suppressor mimic SNRP-200(R267I), which changes splicing on some native substrates in the same direction as SNRP-27(M141T), but to a much lesser extent (Figure 7B). These negative results suggest that the class II suppressors represent a new type of suppressor with a mode of action that is distinct from the three classes of yeast suppressors we explored. Alternatively, there may exist enough differences between S. cerevisiae and metazoan splicing mechanisms that transferring the yeast suppressors to animals would not fully replicate the mechanism of suppressor function; this alternative interpretation supports our approach of using suppressor genetics in animals to explore splicing. The advantage of our screen is that we employed forward genetics in a metazoan system, where 5′ss consensus is less strict and in which there are distinct factors not found in S. cerevisiae (for example SNRNP27K). Combined with the ability to map these suppressors onto cryoEM structures of human spliceosomes, we can begin to explore how the 5′ss is handed off from U1snRNA to the spliceosome's catalytic core. Although these genetic screens are lower throughput than a yeast genetic selection, we have identified six class II alleles in spliceosomal proteins that are important for ensuring the accurate maintenance of 5′ss identity during spliceosome assembly. The unique global effects on splicing of these six different class II suppressors, and the mapping of the suppressors onto the cryoEM models at different stages of spliceosome assembly, is consistent with the hypothesis that these suppressors function at several distinct stages of spliceosome assembly to chaperone the 5′ss into the active site. Our double suppressor analysis with snrp-27(M141T) paired with snrp-200(N18K) or prp-8(D1549N) suggests that snrp-200(N18K) acts before or at the same time as snrp-27(M141T), while prp-8(D1549N) acts downstream of these two alleles. Our analysis has revealed that multiple parts of the spliceosome, acting at distinct steps downstream from the initial 5′ss identification by U1snRNA, are important for guiding accurate 5′ss loading into the catalytic core.
DATA AVAILABILITY
Raw mRNA sequencing data for 12 libraries in fastq format, along with files in gtf format that indicate the PSI and (1-PSI) values for the two alternatives of all analyzed splicing events, are available at the NCBI Gene Expression Omnibus (GEO - https://www.ncbi.nlm.nih.gov/geo/) accession GSE189437. Fastq format files of raw genomic DNA sequencing of the original ENU-mutagenized suppressed strains from the screen (not outcrossed) can be accessed from the Sequence Read Archive at NCBI project PRJNA77886 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA778860/).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Joshua Arribere for help with genomic DNA library preparation and sequence alignments to help map suppressors, Melissa Jurica for help with visualizing mutants in published cryoEM structures, Noel Ng for early help with some of the genetic crosses and Eliana Duran with help in some CRISPR screening and RNA preparation.
Contributor Information
Catiana H Cartwright-Acar, Department of MCD Biology and The Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
Kenneth Osterhoudt, Department of MCD Biology and The Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
Jessie M N G L Suzuki, Department of MCD Biology and The Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
Destiny R Gomez, Department of MCD Biology and The Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
Sol Katzman, Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
Alan M Zahler, Department of MCD Biology and The Center for Molecular Biology of RNA, University of California Santa Cruz, Santa Cruz, CA 95060, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1613867 to A.M.Z.]; National Institutes of Health [5R01GM135221 to A.M.Z.]; D.G. was supported by the UCSC MARC Program [T34GM007910]. Funding for open access charge: National Institutes of Health [5R01GM135221 to A.M.Z.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Will C.L., Luhrmann R.. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011; 3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Semlow D.R., Blanco M.R., Walter N.G., Staley J.P.. Spliceosomal DEAH-Box ATPases remodel Pre-mRNA to activate alternative splice sites. Cell. 2016; 164:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Bortoli F., Espinosa S., Zhao R.. DEAH-Box RNA helicases in Pre-mRNA splicing. Trends Biochem. Sci. 2021; 46:225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilkinson M.E., Charenton C., Nagai K.. RNA splicing by the spliceosome. Annu. Rev. Biochem. 2020; 89:359–388. [DOI] [PubMed] [Google Scholar]
- 5. Wan R., Bai R., Zhan X., Shi Y.. How is precursor messenger RNA spliced by the spliceosome. Annu. Rev. Biochem. 2020; 89:333–358. [DOI] [PubMed] [Google Scholar]
- 6. Fica S.M. Cryo-EM snapshots of the human spliceosome reveal structural adaptions for splicing regulation. Curr. Opin. Struct. Biol. 2020; 65:139–148. [DOI] [PubMed] [Google Scholar]
- 7. Mayerle M., Guthrie C.. Genetics and biochemistry remain essential in the structural era of the spliceosome. Methods. 2017; 125:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong M.S., Kinney J.B., Krainer A.R.. Quantitative activity profile and context dependence of all human 5′ splice sites. Mol. Cell. 2018; 71:1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charenton C., Wilkinson M.E., Nagai K.. Mechanism of 5′ splice site transfer for human spliceosome activation. Science. 2019; 364:362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Staley J.P., Guthrie C.. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein prp28p. Mol. Cell. 1999; 3:55–64. [DOI] [PubMed] [Google Scholar]
- 11. Zhan X., Yan C., Zhang X., Lei J., Shi Y.. Structures of the human pre-catalytic spliceosome and its precursor spliceosome. Cell Res. 2018; 28:1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Townsend C., Leelaram M.N., Agafonov D.E., Dybkov O., Will C.L., Bertram K., Urlaub H., Kastner B., Stark H., Luhrmann R.. Mechanism of protein-guided folding of the active site U2/U6 RNA during spliceosome activation. Science. 2020; 370:eabc3753. [DOI] [PubMed] [Google Scholar]
- 13. Haselbach D., Komarov I., Agafonov D.E., Hartmuth K., Graf B., Dybkov O., Urlaub H., Kastner B., Luhrmann R., Stark H.. Structure and conformational dynamics of the human spliceosomal B(act) complex. Cell. 2018; 172:454–464. [DOI] [PubMed] [Google Scholar]
- 14. Zhan X., Yan C., Zhang X., Lei J., Shi Y.. Structure of a human catalytic step I spliceosome. Science. 2018; 359:537–545. [DOI] [PubMed] [Google Scholar]
- 15. Steven R., Kubiseski T.J., Zheng H., Kulkarni S., Mancillas J., Ruiz Morales A., Hogue C.W., Pawson T., Culotti J.. UNC-73 activates the rac GTPase and is required for cell and growth cone migrations in C.elegans. Cell. 1998; 92:785–795. [DOI] [PubMed] [Google Scholar]
- 16. Dassah M., Patzek S., Hunt V.M., Medina P.E., Zahler A.M.. A genetic screen for suppressors of a mutated 5′ splice site identifies factors associated with later steps of spliceosome assembly. Genetics. 2009; 182:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayerle M., Yitiz S., Soulette C., Rogel L.E., Ramirez A., Ragle J.M., Katzman S., Guthrie C., Zahler A.M.. Prp8 impacts cryptic but not alternative splicing frequency. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:2193–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zahler A.M., Tuttle J.D., Chisholm A.D.. Genetic suppression of intronic +1G mutations by compensatory U1 snRNA changes in Caenorhabditis elegans. Genetics. 2004; 167:1689–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roller A.B., Hoffman D.C., Zahler A.M.. The allele-specific suppressor sup-39 alters use of cryptic splice sites in Caenorhabditis elegans. Genetics. 2000; 154:1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zahler A.M., Rogel L.E., Glover M.L., Yitiz S., Ragle J.M., Katzman S.. SNRP-27, the C. elegans homolog of the tri-snRNP 27K protein, has a role in 5′ splice site positioning in the spliceosome. RNA. 2018; 24:1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki J.M., Osterhoudt K., Cartwright-Acar C.H., Gomez D.R., Katzman S., Zahler A.M.. A genetic screen in C. elegans reveals roles for KIN17 and PRCC in maintaining 5′ splice site identity. PLoS Genet. 2022; 18:e1010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brenner S. The genetics of caenorhabditis elegans. Genetics. 1974; 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Run J.Q., Steven R., Hung M.S., van Weeghel R., Culotti J.G., Way J.C.. Suppressors of the unc-73 gene of Caenorhabditis elegans. Genetics. 1996; 143:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis M.W., Hammarlund M., Harrach T., Hullett P., Olsen S., Jorgensen E.M.. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005; 6:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wicks S.R., Yeh R.T., Gish W.R., Waterston R.H., Plasterk R.H.. Rapid gene mapping in caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 2001; 28:160–164. [DOI] [PubMed] [Google Scholar]
- 26. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.et al.. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X.Y., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: SNPs in the genome of drosophila melanogaster strain w(1118); iso-2; iso-3. Fly. 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cvitkovic I., Jurica M.S.. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013; 41:D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J.et al.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paix A., Folkmann A., Rasoloson D., Seydoux G.. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics. 2015; 201:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arribere J.A., Bell R.T., Fu B.X., Artiles K.L., Hartman P.S., Fire A.Z.. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014; 198:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988; 16:10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arribere J.A., Kuroyanagi H., Hundley H.A.. mRNA editing, processing and quality control in Caenorhabditis elegans. Genetics. 2020; 215:531–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ragle J.M., Katzman S., Akers T.F., Barberan-Soler S., Zahler A.M.. Coordinated tissue-specific regulation of adjacent alternative 3′ splice sites in C. elegans. Genome Res. 2015; 25:982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raghunathan P.L., Guthrie C.. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998; 8:847–855. [DOI] [PubMed] [Google Scholar]
- 39. Cvackova Z., Mateju D., Stanek D. Retinitis pigmentosa mutations of SNRNP200 enhance cryptic splice-site recognition. Hum. Mutat. 2014; 35:308–317. [DOI] [PubMed] [Google Scholar]
- 40. Ledoux S., Guthrie C.. Retinitis pigmentosa mutations in bad response to refrigeration 2 (Brr2) impair ATPase and helicase activity. J. Biol. Chem. 2016; 291:11954–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Absmeier E., Wollenhaupt J., Mozaffari-Jovin S., Becke C., Lee C.T., Preussner M., Heyd F., Urlaub H., Luhrmann R., Santos K.F.et al.. The large N-terminal region of the Brr2 RNA helicase guides productive spliceosome activation. Genes Dev. 2015; 29:2576–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang L., Li X., Hill R.C., Qiu Y., Zhang W., Hansen K.C., Zhao R.. Brr2 plays a role in spliceosomal activation in addition to U4/U6 unwinding. Nucleic Acids Res. 2015; 43:3286–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Absmeier E., Becke C., Wollenhaupt J., Santos K.F., Wahl M.C.. Interplay of cis- and trans-regulatory mechanisms in the spliceosomal RNA helicase Brr2. Cell Cycle. 2017; 16:100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brow D.A. An allosteric network for spliceosome activation revealed by high-throughput suppressor analysis in Saccharomyces cerevisiae. Genetics. 2019; 212:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kuhn A.N., Brow D.A.. Suppressors of a cold-sensitive mutation in yeast U4 RNA define five domains in the splicing factor Prp8 that influence spliceosome activation. Genetics. 2000; 155:1667–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Strauss E.J., Guthrie C.. A cold-sensitive mRNA splicing mutant is a member of the RNA helicase gene family. Genes Dev. 1991; 5:629–641. [DOI] [PubMed] [Google Scholar]
- 47. Strauss E.J., Guthrie C.. PRP28, a ‘DEAD-box’ protein, is required for the first step of mRNA splicing in vitro. Nucleic Acids Res. 1994; 22:3187–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Price A.M., Gornemann J., Guthrie C., Brow D.A.. An unanticipated early function of DEAD-box ATPase prp28 during commitment to splicing is modulated by U5 snRNP protein Prp8. RNA. 2014; 20:46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ben-Yehuda S., Dix I., Russell C.S., McGarvey M., Beggs J.D., Kupiec M.. Genetic and physical interactions between factors involved in both cell cycle progression and pre-mRNA splicing in Saccharomyces cerevisiae. Genetics. 2000; 156:1503–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schneider S., Campodonico E., Schwer B.. Motifs IV and v in the DEAH box splicing factor Prp22 are important for RNA unwinding, and helicase-defective Prp22 mutants are suppressed by Prp8. J. Biol. Chem. 2004; 279:8617–8626. [DOI] [PubMed] [Google Scholar]
- 51. Umen J.G., Guthrie C.. A novel role for a U5 snrnp protein in 3′-splice-site selection. Genes Dev. 1995; 9:855–868. [DOI] [PubMed] [Google Scholar]
- 52. Liu L., Query C.C., Konarska M.M.. Opposing classes of Prp8 alleles modulate the transition between the catalytic steps of pre-mRNA splicing. Nat. Struct. Mol. Biol. 2007; 14:519–526. [DOI] [PubMed] [Google Scholar]
- 53. Eysmont K., Matylla-Kulinska K., Jaskulska A., Magnus M., Konarska M.M.. Rearrangements within the U6 snRNA core during the transition between the two catalytic steps of splicing. Mol. Cell. 2019; 75:538–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mRNA sequencing data for 12 libraries in fastq format, along with files in gtf format that indicate the PSI and (1-PSI) values for the two alternatives of all analyzed splicing events, are available at the NCBI Gene Expression Omnibus (GEO - https://www.ncbi.nlm.nih.gov/geo/) accession GSE189437. Fastq format files of raw genomic DNA sequencing of the original ENU-mutagenized suppressed strains from the screen (not outcrossed) can be accessed from the Sequence Read Archive at NCBI project PRJNA77886 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA778860/).