

Abstract
Current therapies for Duchenne muscular dystrophy (DMD) use phosphorodiamidate morpholino oligomers (PMO) to induce exon skipping in the dystrophin pre-mRNA, enabling the translation of a shortened but functional dystrophin protein. This strategy has been hampered by insufficient delivery of PMO to cardiac and skeletal muscle. To overcome these limitations, we developed the FORCETM platform consisting of an antigen-binding fragment, which binds the transferrin receptor 1, conjugated to an oligonucleotide. We demonstrate that a single dose of the mouse-specific FORCE–M23D conjugate enhances muscle delivery of exon skipping PMO (M23D) in mdx mice, achieving dose-dependent and robust exon skipping and durable dystrophin restoration. FORCE–M23D-induced dystrophin expression reached peaks of 51%, 72%, 62%, 90% and 77%, of wild-type levels in quadriceps, tibialis anterior, gastrocnemius, diaphragm, and heart, respectively, with a single 30 mg/kg PMO-equivalent dose. The shortened dystrophin localized to the sarcolemma, indicating expression of a functional protein. Conversely, a single 30 mg/kg dose of unconjugated M23D displayed poor muscle delivery resulting in marginal levels of exon skipping and dystrophin expression. Importantly, FORCE–M23D treatment resulted in improved functional outcomes compared with administration of unconjugated M23D. Our results suggest that FORCE conjugates are a potentially effective approach for the treatment of DMD.
Graphical Abstract
Graphical Abstract.
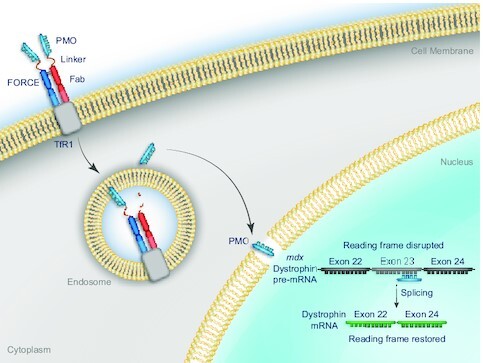
FORCE conjugates deliver an exon skipping PMO via the transferrin receptor 1 and restore dystrophin in muscle of a mouse model of DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a rare, X-linked, progressive neuromuscular disease with an incidence of ∼1 in every 3500–5000 live male births that leads to loss of ambulation before adolescence and premature death in the second to fourth decade of life (1,2). DMD is caused by mutations in the DMD gene encoding dystrophin protein that result in loss of expression. DMD is one of the longest genes in the human genome (∼2.2 million base pairs), making it particularly susceptible to mutations, many of which reduce or abolish the production of dystrophin. The majority (68%) of mutations are large deletions of one or more of the 79 exons that comprise the dystrophin mRNA (3). The remainder are duplications of more than one exon, small deletions or insertions, or point mutations (4). Most of these mutations interrupt the open reading frame resulting in a premature stop codon and the inability to produce functional dystrophin protein (5,6). Dystrophin plays critical roles in stabilizing the sarcolemma in muscle cells by interacting with the dystrophin glycoprotein complex (7,8). Consequently, the absence of dystrophin in patients with DMD leads to damage to the sarcolemma upon contraction, dysregulated cellular function and signaling, and ultimately, muscle cell death. Damaged muscle is progressively replaced with fibrotic and adipose tissue, leading to the clinical manifestations of the disease (9,10).
Exon skipping can restore the open reading frame of the DMD transcript and enable the translation of a shortened but functional dystrophin protein (11,12). This approach has broad clinical applicability as data from the TREAT-NMD DMD global database suggest that up to 55% of all mutations and 80% of deletions in the dystrophin gene in patients with DMD may be amenable to exon skipping therapies (3). Several antisense oligonucleotides (ASOs) that induce exon skipping in the DMD pre-mRNA have been approved as therapies for DMD (11–15). However, the efficacy of currently approved therapeutic approaches using systemic administration of exon skipping ASO with phosphorodiamidate morpholino oligomer (PMO) chemistry is limited by poor muscle delivery due to inefficient cellular uptake and rapid renal clearance (16–18). Thus, there is a need for new strategies to efficiently deliver exon-skipping oligonucleotides to muscle in patients with DMD.
The FORCE™ platform was developed to overcome the limitations of oligonucleotide delivery to muscle. It harnesses properties of the transferrin receptor 1 (TfR1), which is expressed on the surface of skeletal, smooth, and cardiac muscle cells and is critical for iron uptake in these tissues (19–21). TfR1 controls cellular iron homeostasis by mediating endocytosis of iron-bound transferrin (19) and returns to the cell surface with a cycling rate of ∼17 min (22). While divalent antibodies against TfR1 alter recycling to the cell surface, antigen-binding fragments (Fab) do not affect receptor surface expression, internalization, or degradation (23) making them a preferred approach for TfR1 targeting. The TfR1-targeting Fabs at the core of FORCE platform are conjugated to ASOs using a valine-citrulline linker, which is stable in plasma and enzymatically cleaved in the endosomal compartment to enable release of the oligonucleotide payload in the cytosol and subsequent target engagement (24,25).
In the current study, we used the FORCE platform to deliver an exon skipping PMO (26,27) to muscle in mdx mice, an established preclinical model of DMD (28). FORCE–M23D (Figure 1A) is a mouse-specific Fab-PMO conjugate comprised of a Fab fragment, specific to mouse transferrin receptor 1 and known not to interfere with transferrin binding (23,29), conjugated to a PMO payload (M23D) designed to skip exon 23 (26,27). We demonstrate that a single dose of FORCE–M23D achieves robust and durable exon skipping, restoration of functionally competent dystrophin in skeletal and cardiac muscle, and improved locomotor function in mdx mice, whereas a single equimolar dose of unconjugated M23D provides no benefit.
Figure 1.

FORCE–M23D structure, Fab internalization kinetics and FORCE–M23D TfR1 binding affinity in C2C12 mouse myoblasts. (A) Schematic representation of the FORCE–M23D conjugate. (B) Binding to murine TfR1 with the FORCE–M23D conjugate or with a negative control human-TfR1 specific conjugate by ELISA. (C) Uptake of anti-murine TfR1-Cypher5a conjugate and anti-human TfR1-Cypher5e conjugate in C2C12 myoblasts. Data represent mean ± SD. *** P < 0.001. A.U., arbitrary units; ELISA, enzyme-linked immunosorbent assay.
MATERIALS AND METHODS
Reagents
RNAlater (Cat. No. AM7021), iBright FL1000 Imaging System (Cat. No. A32752), Attune NxT Flow Cytometer (Cat. No. AFC2), Rapid Gold BCA bicinchoninic acid assay (Cat. No. A53225), Superscript III Platinum Taq polymerase (Cat. No. 12574018), 20× Tris-buffered Saline with Tween (TBST; Cat. No. J77500-K2), 1× PBS pH 7.4 (Cat. No. 10010023), Zeba desalting column (Cat. No. 89882) goat anti-rat IgG + IgM antibody (Cat. No. A-11006), anti-rabbit Alexa647 (Cat. No. A-21245) and anti-rat Alexa488 (Cat. No. A-21208) antibodies were purchased from ThermoFisher Scientific/Invitrogen, Carlsbad, CA. The Maxwell RSC instrument (Cat. No. AS8500) and Maxwell RSC simplyTissue extraction kit (Cat. No. AS1340) were obtained from Promega, Madison, WI, USA. LabChip GXII Touch HT (CLS138160) and the DNA 5K/RNA/Charge Variant LabChip (Cat. No. 760435) were purchased from Perkin Elmer, Santa Clara, CA, USA. Creatinine kinase (CK) activity assay kit (Colorimetric; Cat. No. ab155901) and the following antibodies were purchased from Abcam (Cambridge, UK): anti-dystrophin (Cat. No. Ab15277), anti-alpha-actinin (Cat. No. Ab9465), anti-Laminin (Cat. No. Ab11576). IRDye 800CW Goat anti-Rabbit IgG (Cat. No. 926-32211) and IRDye 680RD Goat anti-Mouse IgG (Cat. No. 926-68070) antibodies and Odyssey CLX instrument (Cat. No. 9140-09) were purchased from Licor, Lincoln, NE, USA. Cypher5E Mono NHS Ester (Cat. No. PA15401), Sepharose 4 Fast Flow Protein G resin (Cat. No. 17061802) and Butyl HP resin (Cat. No. 28411005) were acquired from Cytiva, Marlborough, MA, USA. Bio-Gel P-4 (Cat. No. 1504124) and ceramic hydroxyapatite (CHT) (Cat. No. 1582000) were purchased from Bio-Rad Laboratories, Hercules, CA, USA. The remaining reagents/instruments were purchased as follows: Vivaflow 50 cassette (Sartorius, Göttingen, Germany; Cat. No. VF05P2); PES Rapidflow (Thermo Scientific Nalgene, Rochester, NY, USA; Cat. No. 09-741-03); Endosafe limulus amebocyte lysate kinetic chromogenic assay (Charles River Laboratories International, Inc., Wilmington, MA, USA; Cat. No. PTS20F); 96-well black flat bottom high bind microplates were purchased from Corning (Corning, NY; Cat. No. 3925); Recombinant mouse TfR1 protein was purchased from R&D Systems (Minneapolis, MN; Cat. No. 9706-TR-050); bovine serum albumin (BSA) was purchased from MP Biomedicals, LLC (Irvine, CA; Cat. No. 840043); phosphorothioate/DNA capture probe (Integrated DNA Technologies Inc., Coralville, IA, USA); Synergy Neo2 Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA, Cat. No. NEO2); Precellys Evolution homogenizer (Bertin, Montigny-le-Bretonneux, France, Cat. No. P000062-PEVO0-A); hard-tissue homogenizer (Omni International, Inc., Kennesaw, GA, USA, Cat. No. TH115); optimal cutting temperature (OCT) mounting medium (Tissue-tek, Torrance, CA, USA, Cat. No. 4583); ZEISS laser scanning 800 confocal microscope (Carl Zeiss Meditec AG, Jena, Germany); Image J software (US National Institutes of Health, Bethesda, MD, USA); M3 multimode plate reader (Molecular Devices, San Jose, CA, USA, Cat. No. M3); Mouse Activity Wheels with dual lickometers (Lafayette Instrument Co., Lafayette, IN, USA, Model 80822); plexiglass square chambers (Med Associates Inc., St Albans, VT, USA). A monoclonal rat anti-mouse IgG2a antibody targeting mouse TfR1 anti-mTfR1 Fab (RI7217-rIgG2a) was purchased from Genescript Biotech Corporation, Piscataway, NJ, USA. A human monoclonal antibody targeting human TfR1 (15G11, Cat. No. TAB-611CT) was acquired from Creative Biolabs, Shirley, NY. The pH-sensitive dye Cypher5E was purchased from Cytiva, Malborough, MA (Cy5E; Cat. No. PA15405). The PMO antisense oligonucleotide for mouse dystrophin exon 23 (M23D; 5′-NH2-GGCCAAACCTCGGCTTACCTGAAAT-3′) (26,27) was purchased from Gene Tools, LLC (Philomath, OR, USA). Nucleic acid-hybridization enzyme-linked immunosorbent assay (hELISA) capture probe, dual labeled with digoxigenin at the 5′ end and biotin at the 3′ end (5′/DigN/ATTTCAGGTAAG CCGAGGTTTGGCC/3Bio/3'; the phosphorothioate ends are highlighted in bold), and PCR primers (forward primer: 5′-CACATCTTTGATGGTGTGAGG-3′; reverse primer: 5′-CAACTTCAGCCATCCATTTCTG-3′) were acquired from Integrated DNA Technologies (Coralville, IA, USA).
Biological resources
Mdx (C57Bl/10ScSn-Dmd <mdx>/J, Jackson Laboratory Stock #1801) and C57Bl/10ScSn WT mice were obtained from The Jackson Laboratory, Bar Harbor, ME, USA. C2C12 myoblast cells (CRL-1772) were acquired from ATCC, Manassas, Virginia. CHO-K1SP cells were purchased from Genescript Biotech Corporation, Piscataway, NJ, USA.
FORCE-Cypher5E conjugate
Anti-TfR1 Fab conjugation to Cypher5E dye
Conjugates were labelled with Cypher5e Mono NHS Ester, a pH-sensitive dye that only fluoresces at acidic pH. Briefly, Fab fragments were diluted to 1 mg/ml in PBS and 0.5 M sodium carbonate (pH 8.3). A 10-molar excess of Cypher5e was added and allowed to incubate in the dark at room temperature for 1 h. After incubation, free dye was removed by passing the mixture through a 0.5 ml, 7 kDa Zeba desalting column. Estimation of final dye/protein (D:P) ratio was calculated as follows:
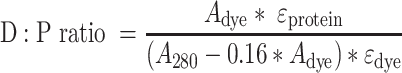 |
A DYE is the absorbance of the labelled antibody conjugate at 500 nm at pH 8.3.
A 280 is the absorbance of the labelled antibody conjugate at 280 nm at pH 8.3.
ϵPROTEIN is the molar extinction coefficient of the antibody (210 000 M−1 cm−1 at 280 nm)
ϵDYE is the extinction coefficient of the dye (40 000 M−1 cm−1 at 500 nm).
FORCE–M23D conjugate
Anti-TfR1 Fab
A monoclonal rat anti-mouse IgG2a antibody Fab targeting mouse TfR1 (RI7-217-rIgG2a; ∼48 kDa) was stably expressed in CHO-K1SP cells. Supernatant was harvested and purified on Protein G resin, eluted, and buffer-exchanged into Dulbecco's phosphate-buffered saline (pH 7.4).
Linker-PMO
To pre-form the linker-PMO, a 25-mer mouse exon 23 PMO antisense oligonucleotide (M23D) was dissolved at 50 mg/ml in dimethyl sulfoxide. The bifunctional valine-citrulline linker generated in house was dissolved in dimethyl sulfoxide at 30 mg/ml and mixed at a 4-molar excess with the PMO for 120 min at room temperature. Reaction completion was measured using the ninhydrin test. Excess reagents were removed using P-4 gel permeation chromatography. The concentration of linker-PMO was measured by ultraviolet–visible spectrophotometry (265 nm, ϵ = 259210 mM–1 cm–1) at 0.1N hydrochloric acid; purity was evaluated by reverse-phase, high-performance liquid chromatography (HPLC).
Fab-linker-PMO conjugate
Before conjugation with linker-PMO, the Fab was diluted with 1/10 (v/v) acetonitrile (HPLC grade). Fab was mixed with a 3-molar excess of linker-PMO for 18 h at room temperature. Reaction was confirmed with sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and analytical size-exclusion chromatography. Unreacted Fab was removed using hydrophilic interaction chromatography (Butyl HP) with an ammonium sulfate gradient. Pooled fractions were diluted with nuclease-free water to reduce conductivity to <8 mS and purified using hydroxyapatite chromatography (CHT) to remove unconjugated linker-PMO. The final Fab-linker-PMO conjugate was eluted directly into formulation buffer (100 mM disodium phosphate, 100 mM sodium chloride, pH 7.2) and concentrated using 10 kDa tangential flow filtration (Vivaflow 50), followed by sterile filtration (0.22 μm, PES Rapidflow). The concentration of the conjugate was measured by bicinchoninic acid assay (Rapid Gold BCA) and the drug antibody ratio was determined by densitometry of an SDS-PAGE gel (iBright FL1000). Endotoxin levels were measured using the Endosafe limulus amebocyte lysate kinetic chromogenic assay.
Anti-TfR1 binding enzyme-linked immunosorbent assay
High binding 96 well plates were coated with 100 μl/well of recombinant TfR1μg/ml in 1× PBS and allowed to incubate overnight at 4°C. Remaining solution was removed and 200 μl of 1% BSA (w/w) in 1× PBS were added. The plates were incubated for 2 hours at room temperature on orbital shaker at 300 rpm. Blocking buffer was removed and plates were washed three times with 300 μl of 1× TBST. An 8-point serial dilution of FORCE–M23D or an unconjugated negative anti-human TfR1 antibody (15G11) was added (concentration range 104–0.05 nM). Plates were incubated at room temperature for 1 hour on an orbital shaker at 300 rpm. Test article solutions were removed, and plates were washed three times with 300 μl of 1× TBST. An anti-rat secondary antibody was diluted 1:500 in 0.5% BSA and 100 μl was added to each well. Secondary antibody was incubated for one hour at room temperature in an orbital shaker at 300 rpm. Secondary antibody was removed, and plates were washed four times with 300 μl of 1× TBST. Signal was then measured on a Synergy Neo2 Multi-Mode Microplate reader at 498 nm excitation, 520 nM emission.
TfR1-mediated in vitro uptake of FORCE conjugates
Anti-TfR1 Fab internalization was measured using flow cytometry. Briefly, C2C12 myoblasts were grown to ∼80% confluency. Cells were then treated with 100 nM of Cy5E-labeled RI7-217 Fab or Cy5E-labeled anti-human TfR1 15G11 Fab for 4 h in standard cell culture conditions. Cells were then released from the cell culture flask and fluorescence was measured at 663 nm using a Attune NxT flow cytometer.
Study designs
Dosing parameters
All mice were administered a single dose of vehicle or test article intravenously (IV) via tail vein injection. All doses of test article are described as the mg/kg equivalent of the M23D payload to allow for comparison of matched doses of the unconjugated M23D. A single dose of 30 mg/kg unconjugated M23D contains an equimolar amount of payload as the 30 mg/kg PMO-equivalent dose of FORCE–M23D. The dose based on the entire conjugate is not described in the text, but the conversions are available in Table 1.
Table 1.
PMO and conjugate dose equivalency for FORCE–M23D
| Reported dose | PMO dose (mg/kg) | Conjugate dose (mg/kg) |
|---|---|---|
| 30 mg/kg unconjugated M23D | 30 | n/a |
| 10 mg/kg PMO-equivalent FORCE–M23D | 10 | 33.8 |
| 30 mg/kg PMO-equivalent FORCE–M23D | 30 | 67.7 |
Kinetics study
To analyze exon skipping and dystrophin restoration, 5-week-old male mdx mice (C57Bl/10ScSn-Dmd <mdx>/J) were injected via tail vein with a single intravenous (IV) dose of vehicle, 30 mg/kg unconjugated M23D, or FORCE–M23D containing the equivalent of 10 or 30 mg/kg PMO (n = 5 animals per treatment). Vehicle-treated mice were necropsied 4-, 8- or 12-week post-dose. Mice treated with 30 mg/kg unconjugated M23D were necropsied 1-, 4- or 8-week post-dose. Mice treated with FORCE–M23D were necropsied at 3-days, 1-, 4- or 8-weeks post-dose for the 10 mg/kg PMO-equivalent cohort and at 3-days, 1-, 4-, 8-, 12-, or 24-weeks post-dose for the 30 mg/kg PMO-equivalent cohort. All mice were anesthetized with a sublethal dose of pentobarbital prior to necropsy. Whole blood was collected using cardiac puncture and processed to produce serum for creatine kinase (CK) analysis. Mice were then perfused with saline, and muscle collected; the left side of each muscle was processed with RNAlater and the right side was snap frozen in liquid nitrogen for subsequent molecular analysis. Samples were removed from RNAlater after ∼48 h, frozen using dry ice, and stored at −80°C.
Functional study
To assess functional performance, 7–8-week-old male mdx mice and C57Bl/10ScSn WT controls were injected via tail vein with a single IV dose of vehicle (n = 10 each mdx and WT mice), 30 mg/kg M23D (n = 10 mdx mice), or FORCE–M23D at a 30 mg/kg PMO-equivalent dose (n = 10 mdx mice). Voluntary running wheel and open-field activity were assessed 2 weeks post-dose (n = 10) and 4 weeks post-dose (n = 5) for each treatment group. At the desired time points, mice were anesthetized with a sublethal dose of pentobarbital. Whole blood was collected using cardiac puncture and processed to produce serum for CK analysis.
Animal husbandry
All mouse studies were conducted at Biomere Biomedical Research Models, Inc. (Worchester, MA, USA) or PsychoGenics (Paramus, NJ, USA) in compliance with local Animal Care and Use Committee guidelines. The environmental temperature was maintained between 20°C and 23°C and the relative humidity at approximately 50%. A light cycle of 12 h light:12 h dark was maintained. Food and water were provided ad libitum. Animals were acclimated to handling and testing procedures prior to functional testing.
Enzyme-linked immunosorbent assay for PMO detection
An hELISA method was adapted from the method of Burki et al. (30) to measure total PMO in the tissue. Briefly, a phosphorothioate/DNA reverse complement capture probe for M23D was dual labeled with digoxigenin at the 5′ end and biotin at the 3′ end. The signal was read on a Synergy Neo2 Multi-Mode Microplate Reader at A450. Concentrations of M23D were calculated using a standard curve of known concentrations ranging from 0 to 500 pM.
Exon skipping analysis
Exon 23 skipping was assessed using end-point PCR followed by capillary electrophoresis (31). Briefly, total RNA was isolated from tissue samples using cryo-pulverization and subsequent RNA extraction using a Maxwell RSC instrument per manufacturer's protocol. Tissue homogenization was performed on tissue fragments from muscle prepared with RNALater. Tissue fragments were weighed and an appropriate amount of 1-thioglycerol/Maxwell homogenization buffer was added to achieve a final tissue concentration of 200 mg/ml buffer. Using the manufacturer's pre-specified ‘hard tissue’ program on a Precellys Evolution, the samples were homogenized in grinding tubes. Tubes were then centrifuged for 1 min at 5000 rpm to reduce foaming; homogenization and centrifuging were repeated twice. Total RNA was extracted using the Maxwell RSC simplyTissue extraction kit per the manufacturer's protocol. Total RNA isolated from tissue samples was used as a template to assay exon 23 skipping. Total RNA (15–75 ng) was used for a 25 μl RT-PCR with Superscript III Platinum Taq. After 35 cycles were completed, the reaction was diluted 1:10 in PCR buffer and was analyzed on a LabChip GXII Touch HT using a DNA 5K/RNA/Charge Variant LabChip according to the manufacturer's protocol. The full-length dystrophin transcript yields a PCR product of 445 base pairs; the exon 23-skipped RNA produces a PCR product of 232 base pairs. Percent exon skipping was quantified using the equation:
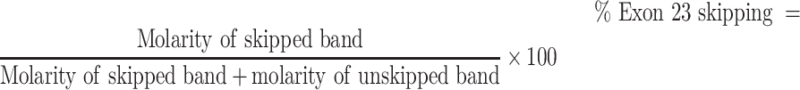 |
Protein extraction and western blot
Muscles were homogenized in a buffer containing 8 M urea, 0.5 M Tris (pH 7.5), 10% sodium dodecyl sulfate, and protease inhibitor in deionized water using a hard-tissue homogenizer at a medium speed for 30 s, four times per sample. Protein concentrations were measured by bicinchoninic acid assay. Total protein (25 μg) was loaded onto a 3–8% Tris-acetate protein gel and run at 60 V for 30 min and 100 V for 1 h. The gel was then transferred to a polyvinylidene difluoride membrane for 120 min at 4°C at 200 mA. The membrane was incubated overnight with anti-dystrophin antibody at a 1:500 dilution and anti-alpha-actinin at a dilution of 1:10 000 at 4°C. The next day, the membrane was washed and incubated with IRDye 800CW Goat anti-Rabbit IgG and IRDye 680RD Goat anti-Mouse IgG secondary antibodies at 1:10 000 dilution to visualize dystrophin and alpha-actinin, respectively. Blots were visualized on an Odyssey CLX with detection of dystrophin at 800 nm and alpha-actinin at 680 nm.
To quantify the amount of dystrophin protein present in each sample, each gel was run with a standard curve of matched muscle matrix from WT mice diluted into muscle matrix from mdx mice. Raw fluorescence was measured in the dystrophin and alpha-actinin channels for each standard and unknown sample. The dystrophin/alpha-actinin ratio of the standards was then generated and plotted against the known percent WT protein in each standard to generate a linear equation. Unknown samples were then interpolated from their dystrophin/alpha-actinin ratio based on the equation of the standard curve. If the raw dystrophin signal of the unknown sample was above the range of the standard curve, the sample was diluted until it fell within the standard curve. A representative western blot image is provided in Supplementary Figure S1A and a representative standard curve calculation is provided in Supplementary Figure S1B, all unedited western blot images can be found in Supplementary Figure S1C and a key to locate specific samples is provided in Supplementary Table S1.
Immunofluorescent microscopy
Quadriceps flash-frozen in OCT mounting medium were sectioned into 10-μm serial sections and mounted on coverslips. Slides were allowed to equilibrate at room temperature before being placed in 1× PBS for 5 min at room temperature. Slides were then fixed in 4% paraformaldehyde and stained with rabbit polyclonal antibody anti-dystrophin primary antibody (1:500) and counter-stained with rat monoclonal antibody anti-laminin (1:1000). Laminin was used as the counterstain because it stains a large glycoprotein just outside of the sarcolemma; this method shows the outline of the muscle fiber without overlapping the membrane, which is relevant when targeting the myofiber outline while avoiding complex image deconvolution of overlapping staining on the membrane. To visualize dystrophin, slides were stained with anti-rabbit Alexa647 (1:500); to visualize laminin, slides were stained with anti-rat Alexa488 (1:500) secondary antibodies prior to imaging. Micrographs were acquired on a ZEISS laser scanning 800 confocal microscope at 20 × magnification. Post-processing of micrographs was completed using ImageJ software (32).
Serum CK assay
Analysis of CK activity was performed using a CK activity assay kit (Colorimetric) according to the manufacturer's protocol. Serum samples were thawed on ice and diluted 1:100 in CK assay buffer (4°C). In a 96-well microplate, diluted nicotinamide adenine dinucleotide standards, positive control, and diluted serum samples were incubated with reaction master mix (CK assay buffer, enzyme mix, developer, ATP, and substrate) at room temperature. Background control reactions using diluted serum samples without the enzyme mix were incubated at room temperature. Readings were obtained at OD450 using an M3 multimode plate reader in kinetic mode every 5 min for a total of 30 min at 37°C. OD450 readings were linear between T1 (time 0) and T2 (10 min). A standard curve was constructed by plotting the OD450 of the standard against the concentration of nicotinamide adenine dinucleotide. Optical density values were used to calculate the CK activity of the serum samples.
Muscle function tests
Running wheel
Voluntary running wheel assessments were conducted on Mouse Activity Wheels with dual lickometers. The polycarbonate chamber measured 35.3 × 23.5 × 20 cm. The aluminum running wheel had an inside diameter of 12.7 cm and an inside width of 5.72 cm for a run distance of 0.40 m/revolution. The run surface consisted of 38 rods each approximately 4.8 mm in diameter on 10.9-mm centers with a 6.14-mm gap. The running wheel was connected to an interface that electronically recorded the animal's activity, including total distance travelled.
Hindlimb fatigue challenge
Locomotor activity, including distance traveled (horizontal activity) was evaluated using an automated open-field test. Activity was measured in a 10-min test session both before and after either a postural hind limb fatigue challenge (HLFC) session of 10 min or placement in the home cage for 10 min. Open field activity was performed in plexiglass square chambers measuring 27.3 × 27.3 × 20.3 cm surrounded by infrared photobeam sources used as open-field chambers. Horizontal activity was measured from consecutive beam breaks. The postural HLFC is a pre-test exercise performed to exacerbate the muscle phenotype of the mdx mouse line. Briefly, the mice are restrained such that they must stand bipedal on their hindlimbs with the majority of their body weight for 10 min. The mice are then subjected to testing as described.
Statistical analysis
A significance level of 0.05 was selected for all inferential statistics. For all molecular analyses, a two-way ANOVA was performed with a Dunnett's Multiple Comparison post-hoc test to evaluate the difference between unconjugated M23D and FORCE–M23D data at the 1-, 4- and 8-weeks post-dose timepoints. For all behavioral assessments, statistical significance was assessed by one-way or two-way analysis of variance with appropriate post hoc comparisons. Outliers were excluded from each of the behavioral assessments. For the running wheel analysis, the total distance traveled during the dark cycle of the first 24 h was assessed. An animal was excluded from the running wheel analysis if its total distance traveled was greater or less than 2 standard deviations (SDs) from the mean compared with the mean performance of all animals in the same treatment group.
For the HLFC, the percent change in total distance traveled in the open field was calculated before and after home cage rest or postural HLFC. For each treatment group, home cage data and HLFC data were analyzed separately. An animal was excluded from either home cage or HLFC analysis if the percent change in its total distance traveled was greater or less than 2 SDs from the mean compared with the mean performance of all animals in the same treatment group.
RESULTS
FORCE–M23D is internalized via the Fab component through TfR1-mediated uptake in C2C12 myoblasts
We designed a mouse TfR1-specific conjugate, FORCE–M23D, to deliver an exon skipping PMO to muscle in mdx mice. FORCE–M23D (Figure 1A) is composed of a Fab fragment specific to mouse transferrin receptor 1 (RI7-217 Fab) conjugated via a val-cit linker to a PMO payload (M23D) designed to skip exon 23. We first confirmed that binding of the RI7-217 Fab component to mouse TfR1 was sufficient for internalization into mouse muscle cells. RI7-217 Fab or a negative control targeting human TfR1 were conjugated to the Cypher5E, a pH-sensitive dye that fluoresces when internalized into the acidified endosomal compartment. We observed >10-fold better uptake of the RI7-217 Fab compared to the human TfR1-targeting Fab in C2C12 myoblasts after 4 h (Figure 1B). An ELISA was then performed to confirm that the FORCE–M23D conjugate retained binding to mouse TfR1. We observed robust binding of FORCE–M23D to the mouse TfR1 with an EC50 of 1.5 nM and observed no binding to mouse TfR1 by a negative control targeting human TfR1 (Figure 1C).(26,27) These data suggest that FORCE–M23D retains mouse TfR1 binding and that FORCE–M23D is internalized via a TfR1-mediated uptake in muscle cells.
FORCE–M23D delivers significantly more exon skipping PMO to skeletal and cardiac muscle of mdx mice than administration of unconjugated M23D
To examine the ability of the FORCE platform to restore dystrophin protein expression in the mdx mouse model of DMD, we conjugated a PMO designed to induce exon 23 skipping in the mouse Dmd pre-mRNA (M23D) to a Fab that binds murine TfR1 (FORCE–M23D). To determine whether FORCE–M23D delivers PMO to muscle, the kinetics of total muscle PMO exposure in mdx mice were measured by hELISA after a single IV administration of FORCE–M23D at doses delivering either 10 or 30 mg/kg PMO, and a parallel cohort injected intravenously with unconjugated M23D at a dose of 30 mg/kg. The hELISA assay measures total PMO present in the tissue and does not differentiate between distinct tissue and cellular compartments. FORCE–M23D led to a dose-dependent distribution of PMO to cardiac and skeletal muscle, and significantly higher levels of exposure than unconjugated M23D alone. Peak PMO levels were detected in quadriceps (Figure 2A) 3 days post-dose and declined steadily thereafter at a similar rate for both dose levels. PMO exposure kinetics in other skeletal muscles, including tibialis anterior (TA; Figure 2B), gastrocnemius (Figure 2C), and diaphragm (Figure 2D) were similar to the kinetics in quadriceps. In the heart, peak PMO levels were achieved between 3 days and 1-week post-dose (Figure 2E). Minimal PMO exposure was detectable in the skeletal and cardiac muscles of mdx mice treated with unconjugated M23D at 1- and 4-weeks post-dose and no payload was detectable at 8 weeks post-dose (Figure 2A-E). These data indicate that the FORCE platform enhances delivery of a PMO payload to muscle.
Figure 2.

A single dose of FORCE–M23D, but not of unconjugated M23D, induces dose-dependent PMO exposure in skeletal and cardiac muscles of mdx mice. Five-week-old mdx mice were injected via tail vein with vehicle, 30 mg/kg unconjugated M23D, or 10 mg/kg or 30 mg/kg PMO-equivalents of FORCE–M23D and sacrificed at the indicated time points. PMO exposure in indicated skeletal and cardiac muscle was determined using hELISA. Data represent mean ± SD. * P < 0.05, ** P < 0.01, **** P < 0.0001. hELISA, hybridization enzyme-linked immunosorbent assay; PMO, phosphorodiamidate morpholino oligomer; WT, wild type.
A single dose of FORCE–M23D, but not of unconjugated M23D, leads to robust and durable exon 23 skipping in muscles of mdx mice
To investigate whether the FORCE–M23D conjugate delivered an active PMO payload to muscle, Dmd exon 23 skipping was assessed using endpoint-RT-PCR and capillary electrophoresis. Dose-dependent exon 23 skipping was detectable 3 days after administration of FORCE–M23D and peaked at 1-week post-dose in all muscles evaluated. Mean maximum levels of exon 23 skipping in quadriceps (Figure 3A), tibialis anterior (Figure 3B), gastrocnemius (Figure 3C), diaphragm (Figure 3D), and heart (Figure 3E) were 24%, 8%, 18%, 18% and 28%, respectively, after a single 10 mg/kg PMO-equivalent dose. Mean maximum levels in the same muscles following treatment with a single 30 mg/kg PMO-equivalent dose of FORCE–M23D were 44%, 30%, 30%, 37% and 40% (Figure 3A–E). Administration of a matched 30 mg/kg dose of M23D alone resulted in significantly lower exon skipping. Exon 23 skipping was only detectable in a single mouse in the 30 mg/kg unconjugated M23D cohort at 1- and 4-weeks post-dose with no detectable skipping evident by 8 weeks post-dose (Figure 3A–E). No exon 23 skipping was detectable in any muscle evaluated in the vehicle-treated control mice at 4-, 8- and 12-weeks post-dose (Figure 3A–E). Importantly, FORCE–M23D-induced skipping was durable and detectable out to 8 weeks in cardiac muscle and diaphragm and up to 12 weeks in quadriceps, tibialis anterior, and gastrocnemius of mice administered a single 30 mg/kg PMO-equivalent dose of FORCE–M23D. These data demonstrate that enhanced PMO delivery by the FORCE platform translates into increased levels of exon skipping in cardiac and skeletal muscle.
Figure 3.

A single dose of FORCE–M23D, but not of unconjugated M23D, induces dose-dependent Dmd exon 23 skipping in skeletal and cardiac muscles of mdx mice. Five-week-old mdx mice were injected via tail vein with vehicle, 30 mg/kg unconjugated M23D, or 10 mg/kg or 30 mg/kg PMO-equivalents of FORCE–M23D and sacrificed at the indicated time points. Exon 23 skipping was measured by RT-PCR and capillary electrophoresis. Percent skipping was calculated as described in Materials and Methods. Data represent mean ± SD. * P < 0.05, ** P < 0.01, **** P < 0.0001. PMO, phosphorodiamidate morpholino oligomer; RT-PCR, reverse transcription polymerase chain reaction; WT, wild type.
A single dose of FORCE–M23D, but not of unconjugated M23D, induces robust and durable dystrophin protein expression in muscles of mdx mice
Consistent with the induction of exon 23 skipping, treatment with a single dose of FORCE–M23D resulted in a durable and dose-dependent restoration of dystrophin protein expression. Representative western blots using undiluted muscle samples of quadriceps, TA, gastrocnemius, diaphragm, and heart of animals treated with 30 mg/kg PMO equivalent of FORCE–M23D show dystrophin restoration that exceeds the upper limit of the WT dystrophin standard curve (Figure 4A; samples were subsequently diluted into the range of the standard curve to generate quantification). Conversely, most vehicle-treated mice had minimal to no detectable dystrophin (Figure 4B–F) consistent with the presence of a low percentage of dystrophin-positive revertant fibers in mdx mice. (33)
Figure 4.

A single dose of FORCE–M23D, but not of unconjugated M23D, induces dose-dependent dystrophin protein expression in skeletal and cardiac muscles of mdx mice. Five-week-old mdx mice were injected via tail vein with vehicle, 30 mg/kg unconjugated M23D, or 10 mg/kg or 30 mg/kg PMO-equivalents of FORCE–M23D and sacrificed at the indicated time points. (A) Representative western blot images of dystrophin expression 28 days after a single 30 mg/kg PMO equivalent dose of FORCE–M23D or a matched 30 mg/kg dose of unconjugated M23D. Dystrophin levels in these samples exceed the upper limit of the standard curve and these western blots were not used for quantification. Additional western blotting (not shown) was performed on samples diluted to within the range of the standard curve and were used for quantification. All bands were quantified based on a standard curve run on the same gel and created using the same muscle tissue matrix. (B–F) Quantification of dystrophin protein levels by fluorimetry analysis of western blot images. Data represent mean ± SD. * P < 0.05, ** P < 0.01, **** P < 0.0001. PMO, phosphorodiamidate morpholino oligomer; WT, wild type.
In the quadriceps (Figure 4B), tibialis anterior (Figure 4C), gastrocnemius (Figure 4D) diaphragm (Figure 4E), and heart (Figure 4F), FORCE–M23D-induced dystrophin expression reached peaks of 20%, 8%, 8%, 11% and 27%, respectively, of WT levels after a 10 mg/kg PMO-equivalent dose. Peak dystrophin expression after a 30 mg/kg PMO-equivalent dose of FORCE–M23D was 51%, 72%, 62%, 90% and 77% of WT levels in the same muscles (Figure 4B–F). Durable dystrophin expression was noted up to 8 weeks in the heart and diaphragm and up to 12 weeks in skeletal muscles of the hindlimb after a single 30 mg/kg PMO-equivalent dose of FORCE–M23D. In contrast, treatment with 30 mg/kg unconjugated M23D led to lower levels of dystrophin production in limb muscles of a minority of mdx mice 1-, 4- and 8-weeks post-dose (Figure 4A, B and D). Dystrophin protein was detected only sporadically in diaphragm and heart of mdx mice 1- and 4-weeks after administration of unconjugated M23D but was no longer measurable after 8 weeks (Figure 4E and F). These data corroborate the observations on Dmd exon skipping and support the superiority of the FORCE as a delivery platform for PMO.
Treatment with FORCE–M23D increases dystrophin-positive myofibers in muscle
To evaluate dystrophin localization to the sarcolemma, its functional niche in skeletal and cardiac muscle, muscle cross-sections were visualized by immunofluorescent micrographs using anti-dystrophin and anti-laminin antibodies. Laminin is a large glycoprotein localized immediately outside the sarcolemma, (34) and laminin staining is commonly used to delineate the muscle myofibers and enable calculation of percent dystrophin-positive myofibers (35). In the same FORCE–M23D-treated mice in which exon skipping and dystrophin restoration were induced, dystrophin was localized to the sarcolemma in quadriceps, with a dose-dependent increase of dystrophin-positive fibers that was durable up to 12 weeks (Figure 5A; Supplementary Figure S2 shows individual fields quantified for 30 mg/kg PMO group at 8 weeks). A single 30 mg/kg PMO-equivalent dose of FORCE–M23D resulted in 68%, 83% and 45% dystrophin-positive fibers in quadriceps at 4, 8, and 12 weeks, respectively (Figure 5B). Similarly, dystrophin was highly localized to the sarcolemma of diaphragm (Figure 5C) and heart (Figure 5D) 4 and 8 weeks after administration of a single 30 mg/kg PMO-equivalent dose of FORCE–M23D. Of note, dystrophin localized to the sarcolemma along the length of the myofibers of the diaphragm, indicating that dystrophin restoration is not a local event but extends to the entirety of the myofiber (Figure 6).
Figure 5.

A single dose of FORCE–M23D is sufficient to restore dystrophin localization to the sarcolemma in skeletal and cardiac muscles of mdx mice. Five-week-old mdx mice were injected via tail vein with vehicle, 30 mg/kg unconjugated M23D, or 10 mg/kg or 30 mg/kg PMO-equivalents of FORCE–M23D and sacrificed at the indicated time points. (A) Representative immunofluorescence images with dystrophin (green) and laminin (red) staining of quadriceps cross-sections isolated 4-, 8- and 12-weeks post-dose from vehicle-treated WT or mdx mice, or from mdx mice treated with 30 mg/kg PMO-equivalent of FORCE–M23D. (B) Quantification of dystrophin positive fibers in quadriceps of mdx mice. (C) Representative immunofluorescence images with dystrophin (green) and laminin (red) staining of diaphragm (C) and heart (D) cross-sections isolated 4-, -8-weeks post-dose from vehicle-treated mdx mice or from mdx mice treated with 30 mg/kg PMO-equivalent of FORCE–M23D. Data represent mean ± SD. **** P < 0.0001. PMO, phosphorodiamidate morpholino oligomer; WT, wild type.
Figure 6.
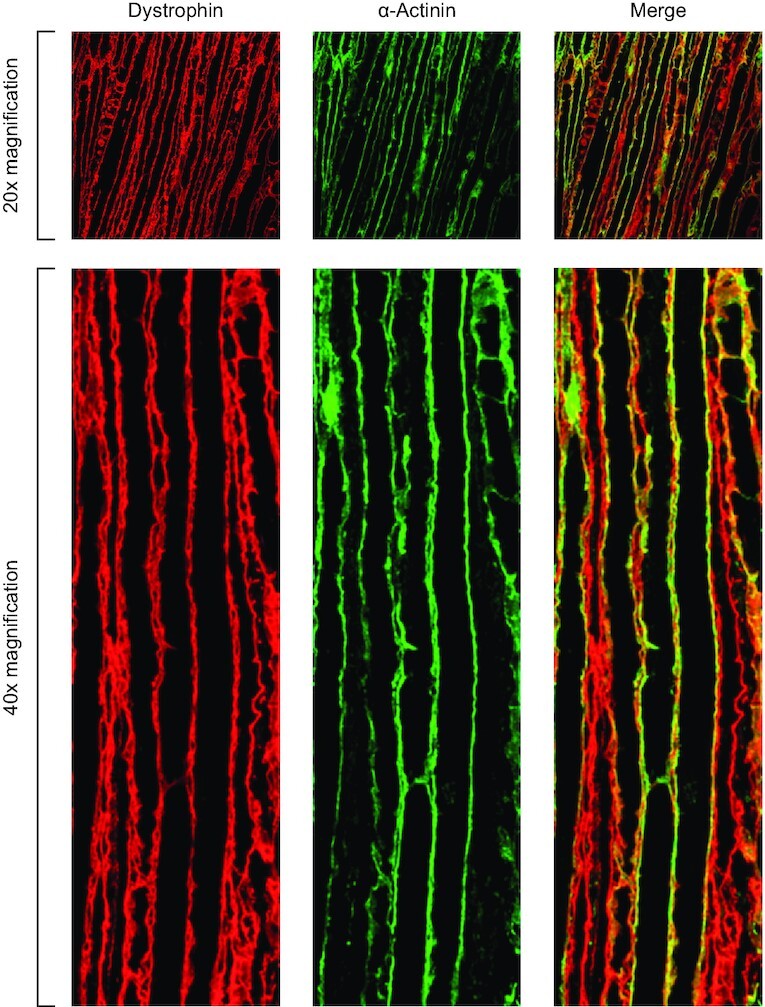
Dystrophin restored by FORCE–M23D is localized to the muscle membranes of mdx mice. Five-week-old mdx mice were injected via tail vein with vehicle or 30 mg/kg PMO-equivalents of FORCE–M23D and sacrificed 4 weeks post-dose. Diaphragm was isolated and longitudinal cross-sections were stained with dystrophin (green) and laminin (red) to image distribution of membrane-localized dystrophin along the entire length of the myofibers. PMO, phosphorodiamidate morpholino oligomer.
A single dose of FORCE–M23D, but not of unconjugated M23D, improves selected functional outcomes in mdx mice
To determine whether FORCE–M23D is superior to its unconjugated exon-skipping PMO in its ability to reduce muscle damage, mdx mice were injected with a single 30 mg/kg PMO equivalent dose of FORCE–M23D or a 30 mg/kg dose of unconjugated M23D. CK levels were measured 2- or 4-weeks after injection and compared with CK levels in WT or mdx mice treated with vehicle. Consistent with the compromised sarcolemma of mdx mice, serum CK levels were higher in these mice compared with their WT counterparts (Figure 7A). A single dose of FORCE–M23D, but not of unconjugated M23D, lowered serum CK in mdx mice at both 2- and 4-weeks post-dose compared with vehicle-treated mdx mice (P < 0.0001). At 4-weeks post-dose, serum CK levels in mdx mice treated with FORCE–M23D were similar to levels in vehicle-treated WT mice, indicating a correction of the mdx phenotype after a single dose of FORCE–M23D. Similar observations at 4- and 8-weeks post-dose confirmed the ability of FORCE–M23D to reduce CK serum levels, and indicated that the effect is dose-dependent (Supplementary Figure S3A and B).
Figure 7.

Treatment with FORCE–M23D, but not with unconjugated M23D, leads to improved functional outcomes in mdx mice. Functional assessments performed 2 weeks following administration of 5-week-old WT or mdx mice with vehicle or mdx mice injected with 30 mg/kg unconjugated M23D or 30 mg/kg PMO-equivalents of FORCE–M23D. Gray bars represent WT vehicle-treated mice, black bars represent mdx vehicle-treated animals, red bars represent mdx mice treated with unconjugated M23D, and blue bars represent mdx mice treated with FORCE–M23D. (A) Serum CK activity. (B) Total distance traveled on a running wheel for an uninterrupted 24-h period. (C) Percent change in total distance traveled in an open field before and after hind limb fatigue challenge. Data are expressed as mean ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. n.s., not significant; PMO, phosphorodiamidate morpholino oligomer; WT, wild type.
To assess whether the ability of FORCE–M23D to ameliorate dystrophy-associated increases in serum CK translates into improved functional outcomes compared with unconjugated M23D, mdx mice (n = 10 per group) received either 30 mg/kg unconjugated M23D or 30 mg/kg PMO equivalent of FORCE–M23D; age-matched control mdx and WT mice (n = 10 per group) were injected with vehicle. Voluntary running wheel and open-field activity after hindlimb fatigue challenge were evaluated 2- (n = 10 mice per treatment group) and 4-weeks (n = 5 mice per treatment group) post-dose.
In a spontaneous running-wheel activity test 2-weeks post-dose, during the dark cycle (active time for mice), mdx mice treated with vehicle traveled a significantly shorter distance than WT mice receiving the same treatment (P = 0.0022) (Figure 7B). Conversely, mdx mice treated with a single dose of FORCE–M23D traveled significantly more than did mdx mice treated with vehicle alone (P = 0.0135). The distance traveled by mdx mice treated with FORCE–M23D was similar to that traveled by WT mice treated with vehicle, indicating a rescue of the mdx phenotype by a single administration of 30 mg/kg PMO-equivalent FORCE–M23D. Furthermore, compared with unconjugated M23D, FORCE–M23D led to increased voluntary running wheel at 4 weeks (Supplementary Figure S4A).
Open-field activity was used to complement the running wheel functional measurements. Without hind-limb fatigue challenge, there was no significant difference in the total distance traveled between WT and mdx mice (Figure 7C). Following hindlimb fatigue challenge which was performed to exacerbate the functional phenotype of the mdx mouse, there was a significant decrease (P < 0.0001) in open-field activity in mdx compared with WT mice analyzed as a percent change in total distance traveled (Figure 7C). A nonsignificant but positive trend toward increased open-field activity after hindlimb challenge was observed in mdx mice treated with a single dose of FORCE–M23D, but not with unconjugated M23D, suggesting that FORCE–M23D increases the muscle delivery and activity of a PMO (Figure 7C, Supplementary Figure S4B).
DISCUSSION
Therapies based on PMO-induced exon skipping have been approved in the United States for the treatment of patients with DMD who have mutations amenable to skipping of exons 51, 53 and 45. (12–15) While exon skipping approaches hold great promise in the treatment of DMD, approved therapies have shown very limited increases in dystrophin protein restoration and marginal, if any, functional benefit. Achieving efficient delivery to muscle has remained an insurmountable obstacle to the development of clinically effective therapies. (17)
ASO delivery has been improved by chemical modifications, use of alternative chemical backbones, and conjugation to delivery moieties such as peptides. (36) PMOs, in which a morpholine ring is substituted for the deoxyribose moiety and a phosphorodiamidate linkage replaces the phosphodiester intersubunit linkage, are more stable against nuclease and protease activity, compared with native DNA or RNA (37,38). Nonetheless, a large body of published work revealed drawbacks of PMOs as therapies for DMD due to the poor uptake into cardiac and skeletal muscle. Consequently, duration of action of a single PMO dose is short, and treatment necessitates high drug dosages and/or repeated administration to achieve a therapeutic effect that is still hampered by inconsistent dystrophin protein restoration. (38)
In the present study, we report that a TfR1-targeting FORCE–M23D conjugate binds to murine TfR1 in vitro with nanomolar affinity and leads to TfR1-mediated uptake into C2C12 mouse myoblasts. We then demonstrate that FORCE–M23D delivers a PMO capable of inducing Dmd exon 23 skipping to skeletal and cardiac muscle on systemic administration to mdx mice. PMO tissue concentrations achieved a peak at the earliest timepoints measured after a single dose, followed closely by peak levels of exon 23 skipping. While both tissue exposure and exon 23 skipping levels declined shortly after achieving maximum levels, high levels of dystrophin protein restoration were observed at 4- and 8-weeks post-dose in cardiac and skeletal muscle, including the diaphragm. This is consistent with the relatively long half-life of dystrophin in skeletal muscle. Administration of FORCE–M23D led to significantly more exposure and a greater pharmacological effect than an equimolar dose of unconjugated M23D. Although not every comparison between the two treatments achieved statistical significance, this is likely due to individual variability in mdx mice. Moreover, in agreement with the known limitations of unconjugated PMO delivery, unconjugated M23D led to an inconsistent, marginal exposure in cardiac and skeletal muscle and molecular as well as functional pharmacologic response. Interestingly, a single animal in the unconjugated M23D group showed exon skipping in the heart at 4 weeks post-dose. This animal showed no detectable dystrophin restoration and none of the other animals in the same cohort showed a detectable level of exon skipping. This finding is inconsistent with the known pharmacology of unconjugated PMO in cardiac muscle and it appears to be an isolated observation.
Immunofluorescence analysis of quadriceps sections complemented the observations on protein expression and revealed that dystrophin localized at the sarcolemma, its physiological site of action, indicating that the shortened dystrophin is functionally sound. Importantly, abundant levels of dystrophin localized to the cell membrane in diaphragm and heart sections, although the observations were qualitative in nature. Interestingly, unlike quadriceps, total dystrophin restoration in the diaphragm and heart appears to decrease rapidly by 8 weeks post-dose. This observation is likely due to the sustained mechanical stress of constant contraction in both the diaphragm and heart. Collectively, these findings indicate that FORCE–M23D delivers substantial amounts of PMO to cardiac and skeletal muscle, and that the dystrophin protein translated from the skipped transcript is likely functionally competent as it localizes to its physiological niche.
The duration of the pharmacodynamic response and the extent of dystrophin localization at the sarcolemma observed after administration of a single dose of FORCE–M23D correlated with improvements in membrane integrity. Additionally, the reduction in serum CK levels combined with the correction of the functional mdx phenotype suggests that treatment with FORCE–M23D restored a meaningful amount of functional dystrophin protein. FORCE–M23D led to a superior molecular and phenotypic correction compared with the modest effect achieved by treatment with unconjugated M23D, confirming the notion that the FORCE platform enhances delivery of an oligonucleotide therapeutic to skeletal and cardiac muscle. Based on these observations, one could speculate that treating DMD patients with a therapy based on the FORCE platform could potentially lead to superior benefit compared with the clinical improvement achieved with infusion of a non-muscle targeting PMO.
A currently available drug for DMD, eteplirsen (EXONDYS51, Sarepta Therapeutics, Cambridge, MA), is a 30-nucleotide PMO that induces the skipping of exon 51 of the DMD transcript. (12) While eteplirsen is approved for use in the United States, a post-marketing confirmatory trial required to verify and describe its anticipated clinical benefit has not yet been completed. Systemic administration of eteplirsen results in non-specific uptake into target tissues, and it is well established from work in animal models that this approach does not restore dystrophin expression in the heart (38). This is of critical importance because cardiovascular complications are a leading cause of morbidity and mortality among patients with DMD (39,40). Considering the magnitude of dystrophin restoration induced by FORCE–M23D in cardiac muscle of mdx mice, the FORCE platform has the potential to deliver levels of PMO to the heart capable of restoring dystrophin expression and potentially reducing mortality secondary to cardiac events in patients with DMD.
To address the limitations of current exon skipping PMO molecules and improve their potential as therapeutics for DMD, conjugation strategies to cell-penetrating peptides (CPP–PMOs) have been implemented. Findings from multiple in vitro and in vivo reports have demonstrated that CPP–PMOs display improved internalization into cells, efficiency in systemic delivery to target tissues, sustained dystrophin production, and enhanced potency at lower doses compared with unconjugated PMOs. (38) Gao et al. (41) reported that mdx mice administered with an exon skipping CPP–PMOs at 25 mg/kg once per week for 3 weeks or a single dose at 75 mg/kg showed effective exon skipping and dystrophin expression in skeletal muscles, but not in the heart, at 2 weeks after the last dose. In a different report, using a PMO-internalizing peptide (Pip6-PMO), Betts et al. (42) reported exon skipping and dystrophin expression in the hearts of mdx mice. Because modest dystrophin expression was seen after a single dose of 10 mg/kg, a longer treatment regimen totaling nine doses of Pip6-PMO was instituted resulting in high levels of exon 23 skipping in the diaphragm (77%) and TA (86%). However, effects on the heart were modest, reaching only 32% skipping and 28% restoration of dystrophin expression. Thus, although peptide conjugation has improved systemic delivery of PMOs, penetration of cardiac muscle remains a challenge. Unlike CPP–PMOs, FORCE–M23D does not contain a membrane destabilizing moiety and relies on cleavage of the linker to release the M23D payload from the conjugate and then spontaneous escape of the PMO from the endosome. Using this strategy, a single 30 mg/kg PMO-equivalent dose of FORCE–M23D in mdx mice produced 40% exon skipping and a 77% dystrophin protein expression peak in the hearts at 4 weeks after administration. These observations suggest that TfR1-mediated uptake is a superior approach for delivery to cardiac tissue compared to CPP–PMOs and possibly other modalities that leverage cell-penetrating strategies for payload uptake.
While CPP–PMO conjugation can increase delivery to muscle, the peptides moieties have known safety liabilities in organs such as the liver and kidneys, potentially leading to toxic side effects that are likely driven by the cationic nature of the peptide (38). A multitude of additional factors such as the duration of treatment, dosage, frequency of systemic administration, and the animal model used to test the CPP–PMOs could also contribute to the observed side effects. Therefore, using lower doses or allowing for longer intervals between dosing could help mitigate some of these risks. (38) At this time, there is an ongoing clinical trial to investigate the use of a CPP–PMO, SRP-5051, for the treatment of DMD patients amenable to exon 51 skipping that may provide clinically relevant safety and pharmacology on the CPP–PMO modality (clinicaltrials.gov, NCT04004065). It is important to note that a single dose of FORCE–M23D achieved similar levels of skipping to those observed in studies with PPMOs in mdx mice, with pharmacological effects in skeletal and cardiac muscles noted up to 12 weeks. The prolonged duration of action combined with lower active dose levels supports the notion that the FORCE platform may potentially have a broader therapeutic index than a delivery mechanism that relies on cell-penetrating peptides.
The mdx mouse is a commonly used animal model for DMD; however, because its dystrophic phenotype is less severe than the manifestations of DMD in patients, it may not represent the best model of human disease, and this is a limitation of the current study. (38) Nevertheless, our findings demonstrate that a single dose of an anti-TfR1 FORCE–M23D conjugate provides superior exon skipping and dystrophin expression, as well as greater improvement in muscle function, compared with approaches that leverage unconjugated oligonucleotides. DMD is a chronic and progressive disease that will require continuous treatment with an exon skipping PMO. While a single dose of FORCE–M23D displays a far superior pharmacodynamic profile compared to the unconjugated payload, additional repeat dose studies will be necessary to understand the ability of FORCE to provide prolonged dystrophin restoration and functional benefit. In conclusion, the FORCE platform combines the pharmacological properties of a biologic drug with the features of an oligonucleotide therapy and has the potential to provide an effective, re-dosable, and titratable treatment for patients with DMD.
DATA AVAILABILITY
All available data have been included in the manuscript.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge Iva Ivanovska Holder, PhD, of Dyne Therapeutics, Inc., for assistance in the preparation of this manuscript, and Amanda Clark, PhD of Biomere. Biomedical Research Models and Sylvie Ramboz, PhD of PsychoGenics, Inc for execution of the mouse studies. Monica Yao was previously employed by Sarepta Therapeutics, Inc., and owns stock options. All remaining authors are either full-time employees of Dyne Therapeutics or were employed by Dyne Therapeutics at the time the work for this study was conducted and may hold stock and/or stock options.
Contributor Information
Cody A Desjardins, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Monica Yao, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
John Hall, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Emma O’Donnell, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Reshmii Venkatesan, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Sean Spring, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Aiyun Wen, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Nelson Hsia, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Peiyi Shen, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Ryan Russo, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Bo Lan, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Tyler Picariello, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Kim Tang, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Timothy Weeden, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Stefano Zanotti, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Romesh Subramanian, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
Oxana Ibraghimov-Beskrovnaya, Research Department, Dyne Therapeutics Inc., Waltham, MA 02451, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Dyne Therapeutics, Inc. Funding for open access charge: Dyne Therapeutics.
Conflict of interest statement. Monica Yao was previously employed by Sarepta Therapeutics, Inc., and owns stock options. All remaining authors are either full-time employees of Dyne Therapeutics or were employed by Dyne Therapeutics at the time the work for this study was conducted and may hold stock and/or stock options.
REFERENCES
- 1. Emery A.E. Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul. Disord. 1991; 1:19–29. [DOI] [PubMed] [Google Scholar]
- 2. Gatheridge M.A., Kwon J.M., Mendell J.M., Scheuerbrandt G., Moat S.J., Eyskens F., Rockman-Greenberg C., Drousiotou A., Griggs R.C.. Identifying non-duchenne muscular dystrophy-positive and false negative results in prior duchenne muscular dystrophy newborn screening programs: a review. JAMA Neurol. 2016; 73:111–116. [DOI] [PubMed] [Google Scholar]
- 3. Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K., Dawkins H., Lamont L., Roy A.J., Chamova T.et al.. The TREAT-NMD DMD global database: analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum. Mutat. 2015; 36:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Flanigan K.M., Dunn D.M., von Niederhausern A., Soltanzadeh P., Gappmaier E., Howard M.T., Sampson J.B., Mendell J.R., Wall C., King W.M.et al.. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009; 30:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koenig M., Beggs A.H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Muller C.R., Lindlof M., Kaariainen H.et al.. The molecular basis for duchenne versus becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 1989; 45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 6. Garcia-Rodriguez R., Hiller M., Jimenez-Gracia L., van der Pal Z., Balog J., Adamzek K., Aartsma-Rus A., Spitali P.. Premature termination codons in the DMD gene cause reduced local mRNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:16456–16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muntoni F., Torelli S., Ferlini A.. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003; 2:731–740. [DOI] [PubMed] [Google Scholar]
- 8. Straub V., Campbell K.P.. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr. Opin. Neurol. 1997; 10:168–175. [DOI] [PubMed] [Google Scholar]
- 9. Duncan C.J. Dystrophin and the integrity of the sarcolemma in duchenne muscular dystrophy. Experientia. 1989; 45:175–177. [DOI] [PubMed] [Google Scholar]
- 10. Brogna C., Lucibello S., Coratti G., Vita G., Sansone V.A., Messina S., Albamonte E., Salmin F., Ferrantini G., Pede E.et al.. Respiratory function and therapeutic expectations in DMD: families experience and perspective. Acta Myol. 2020; 39:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kole R., Krieg A.M.. Exon skipping therapy for duchenne muscular dystrophy. Adv. Drug. Deliv. Rev. 2015; 87:104–107. [DOI] [PubMed] [Google Scholar]
- 12. Lim K.R., Maruyama R., Yokota T.. Eteplirsen in the treatment of duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017; 11:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heo Y.A. Golodirsen: first approval. Drugs. 2020; 80:329–333. [DOI] [PubMed] [Google Scholar]
- 14. Dhillon S. Viltolarsen: first approval. Drugs. 2020; 80:1027–1031. [DOI] [PubMed] [Google Scholar]
- 15. Shirley M. Casimersen: first approval. Drugs. 2021; 81:875–879. [DOI] [PubMed] [Google Scholar]
- 16. Dowdy S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017; 35:222–229. [DOI] [PubMed] [Google Scholar]
- 17. Godfrey C., Desviat L.R., Smedsrod B., Pietri-Rouxel F., Denti M.A., Disterer P., Lorain S., Nogales-Gadea G., Sardone V., Anwar R.et al.. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017; 9:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arechavala-Gomeza V., Anthony K., Morgan J., Muntoni F.. Antisense oligonucleotide-mediated exon skipping for duchenne muscular dystrophy: progress and challenges. Curr. Gene Ther. 2012; 12:152–160. [DOI] [PubMed] [Google Scholar]
- 19. Daniels T.R., Delgado T., Helguera G., Penichet M.L.. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006; 121:159–176. [DOI] [PubMed] [Google Scholar]
- 20. Barrientos T., Laothamatas I., Koves T.R., Soderblom E.J., Bryan M., Moseley M.A., Muoio D.M., Andrews N.C.. Metabolic catastrophe in mice lacking transferrin receptor in muscle. EBioMedicine. 2015; 2:1705–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y., Cheng J.X., Yang H.H., Chen L.P., Liu F.J., Wu Y., Fan M., Wu H.T.. Transferrin receptor 1 plays an important role in muscle development and denervation-induced muscular atrophy. Neural Regen. Res. 2021; 16:1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ciechanover A., Schwartz A.L., Dautry-Varsat A., Lodish H.F.. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J. Biol. Chem. 1983; 258:9681–9689. [PubMed] [Google Scholar]
- 23. Lesley J., Schulte R., Woods J.. Modulation of transferrin receptor expression and function by anti-transferrin receptor antibodies and antibody fragments. Exp. Cell. Res. 1989; 182:215–233. [DOI] [PubMed] [Google Scholar]
- 24. Tsui C.K., Barfield R.M., Fischer C.R., Morgens D.W., Li A., Smith B.A.H., Gray M.A., Bertozzi C.R., Rabuka D., Bassik M.C.. CRISPR-Cas9 screens identify regulators of antibody-drug conjugate toxicity. Nat. Chem. Biol. 2019; 15:949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCombs J.R., Owen S.C.. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J. 2015; 17:339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jearawiriyapaisarn N., Moulton H.M., Buckley B., Roberts J., Sazani P., Fucharoen S., Iversen P.L., Kole R.. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008; 16:1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin H., Moulton H.M., Seow Y., Boyd C., Boutilier J., Iverson P., Wood M.J.. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008; 17:3909–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bulfield G., Siller W.G., Wight P.A., Moore K.J.. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S.et al.. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control Release. 2016; 237:1–13. [DOI] [PubMed] [Google Scholar]
- 30. Burki U., Keane J., Blain A., O’Donovan L., Gait M.J., Laval S.H., Straub V.. Development and application of an ultrasensitive hybridization-based ELISA method for the determination of peptide-conjugated phosphorodiamidate morpholino oligonucleotides. Nucleic Acid Ther. 2015; 25:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spitali P., Heemskerk H., Vossen R.H., Ferlini A., den Dunnen J.T., Hoen P.A., Aartsma-Rus A.. Accurate quantification of dystrophin mRNA and exon skipping levels in duchenne muscular dystrophy. Lab. Invest. 2010; 90:1396–1402. [DOI] [PubMed] [Google Scholar]
- 32. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH image to imagej: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Echigoya Y., Lee J., Rodrigues M., Nagata T., Tanihata J., Nozohourmehrabad A., Panesar D., Miskew B., Aoki Y., Yokota T.. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PLoS One. 2013; 8:e69194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durbeej M. Laminins. Cell Tissue Res. 2010; 339:259–268. [DOI] [PubMed] [Google Scholar]
- 35. Tiger C.F., Champliaud M.F., Pedrosa-Domellof F., Thornell L.E., Ekblom P., Gullberg D.. Presence of laminin alpha5 chain and lack of laminin alpha1 chain during human muscle development and in muscular dystrophies. J. Biol. Chem. 1997; 272:28590–28595. [DOI] [PubMed] [Google Scholar]
- 36. Godfrey C., Desviat L.R., Smedsrød B., Piétri-Rouxel F., Denti M.A., Disterer P., Lorain S., Nogales-Gadea G., Sardone V., Anwar R.et al.. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017; 9:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sazani P., Ness K.P., Weller D.L., Poage D., Nelson K., Shrewsbury A.S.. Chemical and mechanistic toxicology evaluation of exon skipping phosphorodiamidate morpholino oligomers in mdx mice. Int. J. Toxicol. 2011; 30:322–333. [DOI] [PubMed] [Google Scholar]
- 38. Tsoumpra M.K., Fukumoto S., Matsumoto T., Takeda S., Wood M.J.A., Aoki Y.. Peptide-conjugate antisense based splice-correction for duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine. 2019; 45:630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McNally E.M. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu. Rev. Med. 2007; 58:75–88. [DOI] [PubMed] [Google Scholar]
- 40. Ballard E., Grey N., Jungbluth H., Wraige E., Kapetanakis S., Davidson C., Hart N.. Observation cohort study of cause of death in patients with duchenne muscular dystrophy (DMD). Eur. Respir. J. 2012; 40:1720. [Google Scholar]
- 41. Gao X., Zhao J., Han G., Zhang Y., Dong X., Cao L., Wang Q., Moulton H.M., Yin H.. Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 2014; 22:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Betts C.A., Saleh A.F., Carr C.A., Hammond S.M., Coenen-Stass A.M., Godfrey C., McClorey G., Varela M.A., Roberts T.C., Clarke K.et al.. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci. Rep. 2015; 5:8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All available data have been included in the manuscript.