


Abstract
Mitochondrial translation is of high significance for cellular energy homeostasis. Aminoacyl-tRNA synthetases (aaRSs) are crucial translational components. Mitochondrial aaRS variants cause various human diseases. However, the pathogenesis of the vast majority of these diseases remains unknown. Here, we identified two novel SARS2 (encoding mitochondrial seryl-tRNA synthetase) variants that cause a multisystem disorder. c.654–14T > A mutation induced mRNA mis-splicing, generating a peptide insertion in the active site; c.1519dupC swapped a critical tRNA-binding motif in the C-terminus due to stop codon readthrough. Both mutants exhibited severely diminished tRNA binding and aminoacylation capacities. A marked reduction in mitochondrial tRNASer(AGY) was observed due to RNA degradation in patient-derived induced pluripotent stem cells (iPSCs), causing impaired translation and comprehensive mitochondrial function deficiencies. These impairments were efficiently rescued by wild-type SARS2 overexpression. Either mutation caused early embryonic fatality in mice. Heterozygous mice displayed reduced muscle tissue-specific levels of tRNASers. Our findings elucidated the biochemical and cellular consequences of impaired translation mediated by SARS2, suggesting that reduced abundance of tRNASer(AGY) is a key determinant for development of SARS2-related diseases.
Graphical Abstract
Graphical Abstract.
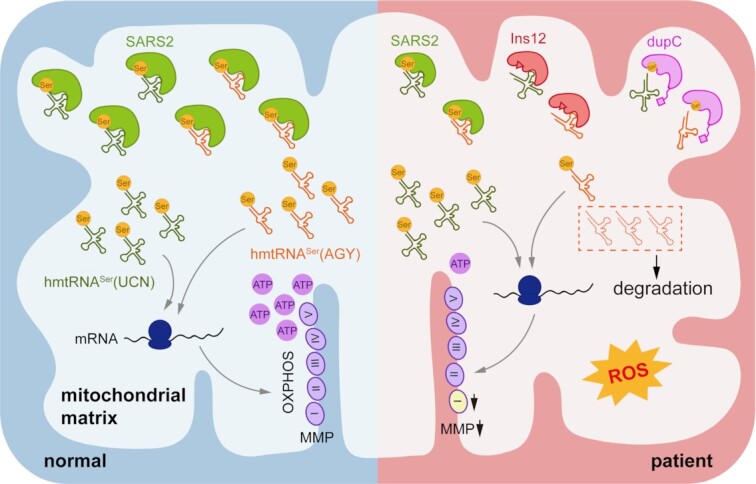
A proposed pathogenic mechanism of two novel disease-causing SARS2 mutations. Selective degradation of the D-armless tRNASer(AGY) led to comprehensive mitochondrial function deficiencies.
INTRODUCTION
Mitochondria provide ATP to cells via oxidative phosphorylation (OXPHOS). Mitochondria are dynamic organelles that undergo constant fusion and fission (1,2); they are also the main source of ROS (3). Several quality control mechanisms, including mitochondrial autophagy (mitophagy) (4,5), ensure functional mitochondria in cells.
Mitochondrial translation is pivotal for proper mitochondrial and cellular function by generating 13 essential subunits for respiratory chain complexes, while other subunits are encoded by nuclear DNA (nDNA) (6–8). Aminoacyl-tRNA synthetases (aaRSs), divided into two classes (9), charge tRNAs with cognate amino acids to generate aminoacyl-tRNAs for mRNA translation (10). tRNA charging is performed via two reactions for most aaRSs: amino acid activation and aminoacylation (10,11).
Two sets of nDNA-encoded aaRSs are required to meet cytoplasmic and mitochondrial translation in human cells (12,13). Mutations in aaRS genes cause many tissue-specific human diseases, primarily affecting the central and peripheral nervous system, heart and muscle (12,14–17). The comprehensive effect of most mitochondrial aaRS (mito-aaRS) mutations on protein architecture, protein function, mitochondrial translation, and mitochondrial function remains poorly understood. Elucidation of etiology of individual aaRS disease–causing mutation is pivotal for patient diagnosis, treatment and prognosis.
SARS2 encodes class II mitochondrial seryl-tRNA synthetase (SerRS) (18), which aminoacylates two tRNAs, human mitochondrial tRNASer(AGY) (hmtRNASer(AGY)) and hmtRNASer(UCN) (Figure 1A). hmtRNASer(AGY) is a highly unique tRNA in humans, lacking D-stem and D-loop structures; hmtRNASer(UCN) maintains canonical tRNA domains (6,18–20). Several mutations in SARS2, including c.1169A > G (p.D390G), c.667G > A (p.V223M), c.1031G > A (p.R344Q), c.1205G > A (p.R402H), c.1347G > A (causing mRNA mis-splicing), and c.1343A > T (p.H448L), have been associated with conditions such as HUPRA (hyperuricemia, pulmonary hypertension, renal failure, and alkalosis) syndrome (OMIM #613845) or progressive spastic paresis (21–26). These studies reinforce that SARS2 is highly associated with human diseases; however, detailed information regarding pathogenesis of SARS2-related diseases remains elusive.
Figure 1.

Two SARS2 mutations identified in a Chinese pedigree with a multisystem disorder. (A) Secondary structures of human mitochondrial tRNASers. (B) Pedigree of the Chinese family. Half-solid symbols represent individuals carrying heterozygous SARS2 mutations. Solid symbols represent the affected individual. The slash-through symbol represents a deceased individual. The proband is indicated by the arrow. (C) Cerebral MRI results of the patient showing normal findings at 19 months, diffuse edema at 3.5 years, and atrophy at 4.5 years of age. (D) DNA sequence identifying c.654–14T > A and c.1519dupC mutations in SARS2. (E) Scheme showing the impact of the c.654–14T > A and c.1519dupC mutations on SARS2 transcription. (F) Primary sequence alignment showing the impact of c.654–14T > A and c.1519dupC mutations on the SARS2 amino acid sequence. Hs cyto, human SARS1; Sc, Saccharomyces cerevisiae cytoplasmic ThrRS; other sequences represent SARS2 from various species (Hs, Homo sapiens; Bt, Bos taurus; Gg, Gallus gallus; Pt, Pan troglodytes; Mm, Mus musculus; Ss, Sus scrofa; Rn, Rattus norvegicus). (G) Ectopic expression of wild-type and mutant SARS2 in HEK293T cells. The band intensities were measured and normalized to those of GAPDH (lower panel). (H) HEK293T cells were treated with CHX at the indicated time points. Western blot was performed on cell extracts using anti-Myc and anti-GAPDH antibodies. The band intensity was measured, normalized to GAPDH, and plotted as a percentage of the initial band intensity (right panel). (I) Detection of dimerization of wild-type and mutant SARS2 by Co-IP analysis. The red arrow indicates decreased homodimerization of Ins12-FLAG/Ins12-Myc. The coprecipitated FLAG-tagged proteins were measured and normalized to enriched Myc-tagged proteins (WT/WT dimer was defined as 1). (J) Tm values of purified mature SARS2 and mutants determined by TSA. Results in (G), (H) and (I) represent mean ± SD (n= 3–4), and P values were determined by unpaired two-tailed t-tests. **P< 0.01, ***P< 0.001, ****P< 0.0001, ns: not significant.
In this study, we examined a Chinese family with multisystem SARS2-related disorders, with both overlapping and distinct phenotypes of HUPRA syndrome and spastic paresis, and investigated the biochemical and cellular pathogenesis. Selective degradation of hmtRNASer(AGY) stood out as a primary and key determinant in inducing profound cellular deficiencies. We suggest that hmtRNASer(AGY) is particularly susceptible to RNA degradation if uncharged or unprotected by SARS2, likely because of its unstable structure. Our data provide a valuable basis for the onset, development, and diagnosis of SARS2-related diseases.
MATERIALS AND METHODS
Whole exome sequencing (WES) and variant verification
Genomic DNA was extracted from peripheral blood leukocytes using a Gentra Puregene Blood Kit (Qiagen, Hilden, Germany). DNA samples (3 μg of patient DNA) were subjected to WES using a SureSelect XT Human All Exon V6 reagent kit (Agilent Technologies, Santa Clara, CA, USA). The original sequencing data were assessed using FastQC (version0.11.2) for quality control. Sequence reads were aligned to the reference human genome (GRCh37/hg19) using NextGENe (SoftGenetics, StateCollege, PA, USA). All single-nucleotide variants and indels were saved in VCF format and uploaded to the Translational Genomics Expert platform (Flash Interpretation Biological Technology Company, Shanghai, China) for annotation, analysis, and prioritization (27). Variations in SARS2 detected by WES were further verified in the pedigree by PCR amplification and Sanger sequencing.
Generation and identification of induced pluripotent stem cells (iPSCs)
Nucleofection and generation of iPSCs were performed with peripheral blood mononuclear cells (PBMCs) as previously described (28). Briefly, pEV SFFV-OCT4-E2A-SOX2 (OS), pEV SFFV-MYC-E2A-KLF4 (MK) and pEV SFFV-BCL-XL (Bcl-XL) episomal vectors were mixed and transferred to the cell pellet (1 × 106 cells). After nucleofection, the cells were transferred to a culture plate pre-seeded with feeder cells. The cells were cultured in reprogramming medium composed of knockout DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% KnockOut™ Serum Replacement (Invitrogen), 1% l-glutamine (Invitrogen), 2 mM nonessential amino acids (Invitrogen), 1% penicillin/streptomycin (Invitrogen), 1% 2-mercaptoethanol (Invitrogen), and 20 ng/ml FGF2 (PeproTech, Cranbury, NJ, USA) for 7 days. The cells were then cultured in PSCeasy medium (Cellapy, Beijing, China) until iPSCs were generated. The iPSCs were characterized by karyotyping, analysis of the expression of embryonic stem cell markers, teratoma formation, and SARS2 sequencing.
Cell culture
PBMCs collected by Ficoll-Paque density gradient centrifugation were expanded for 6–10 days in serum-free erythroid culture medium (ECM) as described previously (28). Human iPSCs were cultured on Matrigel-coated plates (BD Biosciences, San Diego, CA, USA) in PSCeasy medium (Cellapy), which was changed daily. The iPSCs were passaged with EDTA (5 × 10−4 M) when reaching 60% confluence. Human embryonic kidney 239T (HEK293T) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 atmosphere at 37°C.
Mitochondrial respiratory complex (MRC) activity
Mitochondria were isolated from iPSCs as previously described (29). A Bradford Protein Assay Kit (Beyotime, Shanghai, China) was used to quantify the mitochondrial samples. The activity of the MRC complexes was assessed by colorimetry using commercial kits (Abbkine, Wuhan, China) with a SynergyNEO Multi-Mode Reader (BioTek, Winooski, VT, USA) following manufacturer instructions.
Pulse-chase experiments
Pulse-chase experiments were conducted as previously described (30). For pulse labeling, iPSCs were pre-incubated in methionine- and cysteine-free medium and treated with 100 μg/mL emetine to block nDNA translation. The iPSCs were then incubated in 200 μCi/ml PRO-MIX ([35S]-methionine and [35S]-cysteine, PerkinElmer, Waltham, MA, US) to label the mitochondrial translation products. For chase labeling, 40 μg/ml chloramphenicol was added to the cell culture in advance. The labeling was carried out as described in the pulse labeling section, except that 100 μg/ml cycloheximide (CHX) was substituted for emetine. After labeling for 2 h, iPSCs were cultured in PSCeasy medium for 18 h (Chase). Protein samples were separated on 12–20% gradient SDS-polyacrylamide gels. Radioactive signals were detected by phosphor screens using the FLA-9000 fluorescent and radioisotope science imaging systems (Fijifilm, Tokyo, Japan) and quantified using ImageQuant v5.0 software (GE Healthcare, Chicago, IL, USA).
In vitro transcription of hmtRNASer(AGY) and hmtRNASer(UCN)
The T7 promoter sequence and the hmtRNASer(AGY) and hmtRNASer(UCN) genes were directly synthesized and cloned into the pTrc99b vector. The pTrc99b recombinant vector was used to amplify transcription template by PCR. tRNAs were transcribed as described previously (31). The obtained hmtRNASer (AGY) and hmtRNASer (UCN) were incubated at 85°C for 10 min, and then naturally cooled to room temperature. Biotin-labelled tRNAs were obtained as described previously (32).
Gene expression and protein purification
A construct expressing the full-length mature form of SARS2 with a C-terminal His6-tag was developed according to a previous report (33). Constructs expressing Ins12 and dupC were generated using mutagenesis kits (TOYOBO, Osaka, Japan). Escherichia coli BL21-Codon Plus (DE3)-RIL cells were transfected with plasmids encoding wild-type and mutant human SARS2. A single clone was selected and cultured in Luria Broth at 37°C. When the cells reached the mid-log phase (OD value 0.6–0.8), isopropyl β-D-thiogalactoside was added at a final concentration of 100 μM to induce expression at 18°C for ∼16 h. Cells were collected by centrifugation at 5000 rpm and 4°C. Protein purification was performed as described previously (34). For crystallization purpose, the coding sequence of N-terminal Met1-Val180-truncated SARS2 (WT or Ins12) was inserted in a pET28a vector which expressed a His6-tag at the N-terminal end of the SARS2 protein. The truncated proteins were initially purified using NTA affinity chromatography (Qiagen) and then further purified with an ion-exchange column (Cytiva, Uppsala, Sweden), followed by gel filtration using a Superdex 200 column (GE Healthcare) equilibrated with 20 mM Tris·HCl (pH 8.0), 150 mM NaCl, and 5 mM 2-mercaptoethanol. The peak fractions were concentrated for crystallization. All purification steps were performed at 4°C.
Structure determination
Crystal diffraction data were obtained from the National Facility for Protein Science in Shanghai at the Shanghai Synchrotron Radiation Facility. The dataset was processed using the HKL2000 program (35). The structures were determined by molecular replacement using Molrep (36). The bovine SARS2 structure (PDB No. 1WLE) was used as the search model. Iterative model building and refinement were performed using Coot (37) and Phenix (38). The data collection and refinement statistics were presented in Supplementary Table S2.
Study approval
The study was approved by the ethics committee of the Shanghai Children's Medical Center (Shanghai, China). Informed consent was obtained from the parents of the child included in this study. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Shanghai Children's Medical Center.
Further information about the methodology is included in Supplemental Information.
RESULTS
Clinical presentation
The patient was a female aged 8.5 years, born to a nonconsanguineous Chinese couple (Figure 1B). A series of clinical phenotypes began to emerge in her first year of life, including brain atrophy observed by cerebral magnetic resonance imaging (MRI) at 4.5 years of age (Figure 1C). Detailed clinical manifestations were presented in the ‘Clinical manifestation’ subsection of the Supplemental Information.
Compound heterozygous SARS2 mutations were identified
To identify the causal gene, we performed WES and identified sixteen candidate single nucleotide variants in 14 genes (Supplementary Table S1). Among these, SARS2 was recognized as the most likely causal gene because of its functional and clinical relevance. Two heterozygous variants of SARS2, (NM_017827.4) c.654–14T > A and c.1519dupC (p.R507Pfs*41), were detected in the patient. Sanger sequencing of family members revealed that c.654–14T > A and c.1519dupC were paternally and maternally inherited, respectively (Figure 1B, D). The SARS2 variants (c.654–14T > A and c.1519dupC) had not been previously reported in public databases, such as the Human Genome Mutation Database (http://www.hgmd.cf.ac.uk), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), or the Genome Aggregation Database (gnomAD, http://gnomad-sg.org/), and were also absent from our local database (for individuals of the same ethnicity).
The two mutations caused mRNA mis-splicing and frameshifting
The c.654–14T > A variant was located at the 3′ end of intron 6 (Figure 1E). This paternal variant may potentially cause mRNA mis-splicing due to the presence of a 3′ splice site, AG, after mutation. Analyses of cDNA from peripheral leukocytes showed that wild-type mRNA accounted for approximately 60% of paternal RNA (Figure 1E). Considering the wild-type SARS2 allele in paternal cells, it suggested that the allele with the c.654–14T > A mutation still generated ∼10% normal transcripts. In addition, 36.5% mRNA (SV1) contained a 12-bp intron retention, showing that the mutation indeed led to mRNA mis-splicing (Figure 1E). We also detected two other mRNA isoforms, one derived from the activation of an upstream 3′ splice site in intron 6 causing a 39-bp intron retention (SV2), and the other causing 12-bp intron retention and a 39-bp truncation involving partial exons 7 and 8 (c.757-c.795) (SV3) (Figure 1E). SV2 and SV3 only accounted for the 4% of total mRNAs (Figure 1E). Aberrant SARS2 transcripts were not detected in a healthy control sample. Therefore, the c.654–14T > A variant caused aberrant mRNA splicing, and SV1 was further investigated because of its overwhelming abundance. Translation of SV1 resulted in the SARS2 K218R mutation plus an insertion of four amino acids (NLSP) between positions Q217 and R218 (Figure 1F, Supplementary Figure S1A). The SV1-encoded mutant was named Ins12. Class II SARS2 has three class-defining motifs in its active site (9,39) (Figure 1F, Supplementary Figure S1A). In the bovine SARS2 structure with serine adenylate (Ser-AMS) (PDB No. 1WLE) (19), a highly flexible loop in motif II (designated loop A), ranging from 329YRAETDTGKEPWGLYRVH346, extensively interacts with the adenosine moiety (Supplementary Figure S1A). Of note, a glycine at the tip of this loop, G341, uses its main chains to interact with side chain of Q217, which is in a spatially opposite loop (ranging from 217QKRLSHVSGHRSY229, designated loop B) (Figure 1F, Supplementary Figure S1A). In the Ins12 mutant, a four-amino acid peptide (NLSP) was inserted between Q217 and R218 (Figure 1F, Supplementary Figure S1A), likely causing loss of Q217-G341 interaction. The maternal SARS2 c.1519dupC mutation led to a frameshifting and stop codon readthrough, replacing of the last 12 residues with an extended peptide of 40 amino acids (Figure 1E, F). The c.1519dupC-encoded mutant was named dupC.
Structural determination suggested Ins12 and dupC exhibited defects in dimerization and tRNA binding
To understand the potential effects of mutations on the structure, we crystallized the N-terminal Met1-Val180-truncated SARS2 and Ins12 (Supplementary Table S2). Full-length proteins could not be crystallized, nor could full-length or truncated dupC. In wild-type and Ins12 crystal structures, the electron densities of a few regions, including 218–227, 263–301, 331–340 and 495–518 residues, were untraceable (Supplementary Figure S1B). Where the crystal structures were defined, Ins12 showed a very similar structure to the wild-type, with a root-mean-square deviation (RMSD) as low as 0.177 Å (over 209 Cα atoms). In addition, both wild-type and Ins12 showed high similarity to the bovine SARS2 structure (PDB No. 1WLE) (19), with RMSD values of 0.442 Å (over 203 Cα atoms) and 0.383 Å (over 198 Cα atoms), respectively (Supplementary Figure S1B). These results indicate that the Ins12 mutation altered the local, rather than global, structure of SARS2.
Because the Ins12 mutation occurs in loop B, which lacks electron density, we could not directly compare it with the wild-type protein from the determined structures. Therefore, we developed structural models of SARS2, Ins12, and dupC using AlphaFold (40,41). The calculated models of wild-type and Ins12 fit very well onto the determined crystal structures, with RMSD values of 0.470 Å (over 200 Cα atoms) and 0.421 Å (over 210 Cα atoms) (Supplementary Figure S1C, D), respectively, indicating high confidence of the models. The AlphaFold-calculated structures reveals that the Ins12 mutation is located at the dimer interface. The peptide insertion enlarged the original loop B and may introduce an extra steric hindrance for SARS2 dimerization (Supplementary Figure S1E). In addition, loop A is located on the opposite side of the mutation site and is essential for ATP binding (Supplementary Figure S1E). To further study the effect of the Ins12 mutation on tRNA binding, we superimposed AlphaFold models onto the Thermus thermophilus SerRS-tRNA structure (PDB No. 1SER) (39). In wild-type SARS2, K218 was close to the tRNA base 67. The substitution of K218 with the extended loop ‘NLSPR’ may cause a clash with tRNA at bases 67 and 68 (Supplementary Figure S1F). Therefore, the Ins12 mutation may have affected dimerization and substrate-binding.
Similarly, we superimposed the dupC model onto the SARS2 dimer model and T. thermophilus SerRS-tRNA structure (PDB No. 1SER) (39) (Supplementary Figure S1G). The region with replacement and extension of the C-terminus in dupC was predicted to be a long loop, likely oscillating over a large space in solution and potentially interfering with SARS2 dimerization and tRNA binding (Supplementary Figure S1G).
Ins12 was defective in homodimerization
We ectopically expressed genes encoding C-terminal Myc-tagged SARS2 (SARS2-Myc), Ins12 (Ins12-Myc) and dupC (dupC-Myc) in HEK293T cells. The steady-state protein abundance of both Ins12-Myc and dupC-Myc was significantly lower than that of wild-type SARS2-Myc (Figure 1G). No obvious protein aggregates were observed for Ins12-Myc or dupC-Myc, indicating their relatively stable structures in vivo. Moreover, the CHX chase assay revealed that the in vivo stability of both mutants was comparable with that of SARS2, suggesting that decreased amounts of Ins12-Myc and dupC-Myc were not attributable to accelerated protein degradation (Figure 1H). Immunofluorescence analysis using anti-Myc suggesting that the two mutations did not influence proper cellular localization (Supplementary Figure S2).
Dimerization of class II SARS2 is required for tRNA aminoacylation (9,10). The capacity of Ins12 and dupC to form homodimers or heterodimers in vivo was also determined. Three proteins, dupC, Ins12, and wild-type SARS2 (encoded by the paternal c.654–14T > A allele), co-existed in the patient cells. Genes encoding C-terminal FLAG-tagged SARS2 (SARS2-FLAG), Ins12 (Ins12-FLAG), or dupC (dupC-FLAG) were co-expressed with genes encoding SARS2-Myc, Ins12-Myc, or dupC-Myc in HEK293T cells. Co-immunoprecipitation (Co-IP) analysis clearly showed that homo- or heterodimerization between SARS2-FLAG/dupC-Myc, SARS2-FLAG/Ins12-Myc, and dupC-FLAG/Ins12-Myc was not obviously impaired while homodimerization between dupC-FLAG/dupC-Myc was in fact even higher; however, homodimerization of Ins12-FLAG/Ins12-Myc was significantly decreased (Figure 1I). This observation was consistent with the results of AlphaFold-based Ins12 model (Supplementary Figure S1E) and the fact that loop B directly mediates dimer formation (19).
We have successfully purified mature SARS2 from E. coli (32,33). We obtained mature forms of wild-type, Ins12, and dupC, and then performed thermal shift assays (TSA) to determine the stability of purified proteins in vitro (42). The apparent melting temperature (Tm) value of dupC (54.0°C) was comparable with that of SARS2 (54.2°C); however, that of Ins12 (52.1°C) was lower, suggesting a more relaxed conformation in solution (Figure 1J).
The above data showed that the two mutants displayed a decreased steady-state abundance when overexpressed, without impairment of in vivo stability or mitochondrial localization. Furthermore, homodimerization of Ins12 was impaired, implying a reduction in tRNA aminoacylation.
SARS2 mutants exhibited weakened tRNA binding capacities and abolished or impaired tRNA charging activities at the aminoacylation level
We determined the tRNA-binding affinity of SARS2 and the two mutants by measuring KD values. Ins12 and SARS2 exhibited the same KD value (0.08 μM) for hmtRNASer(UCN); however, that of dupC was evidently elevated (0.28 μM) (Table 1). For tRNASer(AGY), the KD value of Ins12 increased approximately 3.7-fold, and that of dupC increased approximately 11-fold. These data suggest that Ins12 exhibited an impaired tRNA binding affinity for tRNASer(AGY), whereas dupC displayed dramatically impaired binding for both tRNAs, highlighting the crucial role of the C-terminus in tRNA binding (19). Alteration in KD values was mainly derived from kon constants. Furthermore, the different effects of Ins12 on binding between the two tRNAs suggested that their acceptor ends were differently accommodated into the active site.
Table 1.
Binding affinities of SARS2, Ins12 and dupC for hmtRNASer(AGY) and hmtRNASer(UCN)
| enzyme | tRNA | K D (μM) | k on*102 (μM−1s−1) | k off *104 (s−1) | Fold/SARS2 |
|---|---|---|---|---|---|
| SARS2 | hmtRNASer(AGY) | 0.03 ± 0.01 | 2.55 ± 0.03 | 7.77 ± 0.08 | 1 |
| Ins12 | 0.11 ± 0.01 | 0.82 ± 0.01 | 8.79 ± 0.05 | ∼3.7 | |
| dupC | 0.33 ± 0.03 | 0.16 ± 0.01 | 5.29 ± 0.04 | 11 | |
| SARS2 | hmtRNASer(UCN) | 0.08 ± 0.01 | 1.22 ± 0.01 | 9.43 ± 0.04 | 1 |
| Ins12 | 0.08 ± 0.01 | 1.08 ± 0.01 | 8.69 ± 0.05 | 1 | |
| dupC | 0.28 ± 0.02 | 0.22 ± 0.01 | 6.17 ± 0.04 | 3.5 |
Mean ± SD, n = 6.
SARS2 activates Ser and transfers it to tRNASers. Amino acid activation kinetics in ATP-PPi exchange reactions (Table 2) showed that both mutants exhibited simultaneous decreases in Km and kcat values for ATP and Ser, suggesting that local structural rearrangement altered ATP and Ser binding. The resultant catalytic efficiency (kcat/ Km) of dupC for both substrates was not negatively impaired; however, that of Ins12 was slightly decreased. The aminoacylation kinetics (Table 3) showed that those of Ins12 for both hmtRNASer(AGY) and hmtRNASer(UCN) were too low to be determined accurately. Meanwhile, For hmtRNASer(AGY), dupC exhibited a nearly 6-fold increase in Km and 3-fold decrease in kcat, leading to only ∼5% activity when compared with that of SARS2. For hmtRNASer(UCN), the catalytic efficiency of dupC decreased by ∼60% due to an increase in Km and a decrease in kcat, although the changes were less pronounced than those for hmtRNASer(AGY).
Table 2.
Kinetics of SARS2, Ins12 and dupC for Ser and ATP in ATP-PPi exchange reaction
| Enzyme | Substrate | K m [mM (Ser), μM (ATP)] | k cat (s−1) | k cat/ Km (μM−1min−1) | Relative kcat/ Km (%) |
|---|---|---|---|---|---|
| SARS2 | Ser | 2.40 ± 0.04 | 2.11 ± 0.03 | 0.88 | 100 |
| Ins12 | 1.66 ± 0.03 | 0.98 ± 0.02 | 0.59 | 67 | |
| dupC | 1.28 ± 0.08 | 1.88 ± 0.07 | 1.47 | 167 | |
| SARS2 | ATP | 112.83 ± 10.54 | 1.87 ± 0.05 | 16.57 | 100 |
| Ins12 | 64.69 ± 4.48 | 0.91 ± 0.01 | 14.07 | 84.9 | |
| dupC | 61.37 ± 2.20 | 1.50 ± 0.03 | 24.44 | 147.5 |
Mean ± SD, n = 3.
Table 3.
Kinetics of SARS2, Ins12 and dupC for hmtRNASer(AGY) and hmtRNASer(UCN) in aminoacylation
| Enzyme | tRNA | K m (μM) | k cat (min−1) | k cat/Km (μM−1min−1) | Relative kcat/Km(%) |
|---|---|---|---|---|---|
| SARS2 | hmtRNASer(AGY) | 1.03 ± 0.07 | 1.71 ± 0.06 | 1.66 | 100 |
| Ins12 | ND | ND | |||
| dupC | 5.76 ± 1.12 | 0.51 ± 0.04 | 0.09 | 5.4 | |
| SARS2 | hmtRNASer(UCN) | 0.82 ± 0.27 | 0.87 ± 0.04 | 1.06 | 100 |
| Ins12 | ND | ND | |||
| dupC | 1.27 ± 0.37 | 0.53 ± 0.10 | 0.42 | 39.6 |
ND, not accurately determined due to low activity. Mean ± SD, n= 3.
Together, these data showed that both Ins12 and dupC, particularly the latter, exhibited impaired tRNA binding capacities. In addition, both mutants displayed abolished or impaired tRNA aminoacylation activity in vitro.
A significant decrease in hmtRNASer(AGY) due to selective degradation impaired mitochondrial mRNA translation in patient cells
To understand whether these two mutations could affect mitochondrial translation in vivo, we constructed four lines of iPSCs (Supplementary Figure S3) using peripheral blood samples from the patient, her parents, and a healthy control (28).
We raised an antibody against SARS2 using purified SARS2 as the antigen. The steady-state level of SARS2 was detected, showing that dupC, with its large molecular mass, could be readily detected in both patient and maternal samples in comparable levels. The abundance of Ins12 and/or SARS2 (indistinguishable in size) was dramatically reduced in patient cells when compared with SARS2 from other three cells (Figure 2A). Maternal cells harbored a wild-type or a mutant allele in each chromosome; however, the level of dupC was only ∼6.3% that of wild-type SARS2 (Figure 2A).
Figure 2.

SARS2 mutations led to decreased hmtRNASer(AGY) due to selective degradation and a reduction of specific OXPHOS components. (A) Expression of SARS2 in four iPSCs. dupC was indicated with a red arrow; a non-specific band was indicated by an asterisk. The band intensities were measured and normalized to those of GAPDH (right panel). (B) Steady-state abundance of mitochondrial tRNAs detected by northern blot; 5S rRNA was used as the loading control. The data were normalized to those of normal control iPSCs. (C) iPSCs were treated with EB for the indicated time points. Northern blot was performed to detect steady-state abundance of hmtRNASer(AGY). The band intensity was measured, normalized to 5S rRNA, and plotted as a percentage of the initial band intensity (lower panel). The asterisks showed statistical differences between patient and normal control iPSCs. (D) Aminoacylation level of mitochondrial tRNAs determined by acidic northern blot. The proportion of aminoacylated hmtRNASers is quantified (right panels). (E) Western blot analysis of mtDNA-encoded mitochondrial proteins. HSP60 was used as a loading control. The expression of mtDNA-encoded mitochondrial proteins was normalized to that of controls (right panel). (F) Pulse-chase analysis showing synthesis (pulse) and stability (chase) of mtDNA-encoded proteins. The lane of the patient is indicated with a red asterisk. The loaded volume was normalized by G250 staining. (G) Western blot analysis of nDNA-encoded mitochondrial proteins. The band intensities were measured and normalized to those of GAPDH (lower panel). Values are expressed as mean ± SD (n= 2–6). P indicates significance according to the unpaired two-tailed t-test. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001.
We then compared the steady-state abundance of hmtRNASer(AGY) and hmtRNASer(UCN) in various cell lines using northern blot and found that level of hmtRNASer(AGY) was obviously decreased to approximately 25% in patient cells when compared with that in control cells (Figure 2B). However, the level of hmtRNASer(UCN) was not significantly different between samples. Furthermore, the amounts of noncognate hmtRNAThr and hmtRNALeu(UUR), transcribed from the same H-strand as hmtRNASer(AGY), were similar between patient and control cells, suggesting that hmtRNASer(AGY) in patient cells was rapidly degraded (Figure 2B). The steady-state of hmtRNASer(AGY) in four iPSCs was monitored in the presence of ethidium bromide (EB), a mitochondrial transcription inhibitor (43,44). The results showed a significantly shortened lifespan of hmtRNASer(AGY) in patient-derived iPSCs compared to that in other samples (Figure 2C). The half-life of patient hmtRNASer(AGY) was estimated to be approximately 5.5 h, whereas that in other three cells was over 20 h. We therefore concluded that the abnormal decrease in patient hmtRNASer(AGY) was due to the high degradation rate.
Aminoacylation levels of hmtRNASer(AGY) and hmtRNASer(UCN) were determined under acidic conditions to preserve the labile bond between Ser and A76. Results showed that hmtRNASer(UCN) charging was not markedly influenced, whereas charging of residual hmtRNASer(AGY) in patient cells was slightly but significantly lower than that in other cell lines (Figure 2D). Furthermore, aminoacylation of the noncognate hmtRNAThr and hmtRNALeu(UUR) was consistent across samples (Figure 2D). Both the reduced amount and aminoacylation level of hmtRNASer(AGY) suggested impaired mitochondrial translation. Indeed, the levels of both mitochondrial DNA (mtDNA)-encoded MT-ND3 (complex I (CI) component) and MT-ATP8 (CV component) were significantly lower in patient cells than in other samples (Figure 2E). However, the levels of MT-ND1 (CI component), MT-CYB (CIII component), MT-CO1 (CIV component), and MT-ATP6 (CV component) were comparable. Considering no AGY codon in MT-ATP8, we suggested that its lower amount was an indirect effect of dysfunctional mitochondrial translation in patient cells. We further performed quantitative reverse transcription PCR (RT-PCR) to detect the transcript level of the mtDNA-encoded mRNAs, revealing comparable amounts of various mRNAs and suggesting impaired mRNA translation, but not transcription (Supplementary Figure S4).
To investigate potential defects in mitochondrial translation caused by SARS2 variants, iPSCs were pulse-labeled with [35S]-methionine and [35S]-cysteine and nDNA translation was blocked with emetine. The overall rate of mitochondrial translation in patient cells was apparently higher than that in other cells (Figure 2F). The iPSCs were then pulse-labeled and chased for 18 h to detect the degradation rate of mitochondrial translation products. The results showed that remaining signals of [35S]-labelled proteins in patient cells were weaker than those in other samples, indicating a shorter half-life of mitochondrial translation products (Figure 2F). This suggests that, although the mitochondrial translation rate was higher in patient cells, possibly due to a compensatory effect, its protein products were unstable and prone to degradation.
The above data suggest that, in patient cells, it had a dramatically decreased abundance of SARS2 (or its mutants) and hmtRNASer(AGY) due to rapid selective degradation. In combination with the impaired tRNA aminoacylation activity of the mutants, Ser-hmtRNASer(AGY) was sharply reduced and mitochondrial translation was impaired.
Patient cells exhibited reduced complex I activity and respiratory capacity
Comparable levels of mtDNA-replication and transcription-related POLG2 and TFAM (45), other mito-aaRSs (AARS2, IARS2, KARS, NARS2 and TARS2) (12) and protein trafficking-associated TOM40 and TIM23 (46), were observed among four cells (Supplementary Figure S5), indicating two mutations had little effect on mitochondrial biogenesis.
OXPHOS complexes are of dual origin (47). In patient cells, a reduced abundance of NDUFS1 (in CI) and UQCRC2 (in CIII) was observed; however, that of SDHA and SDHB in CII, which harbors only nDNA-encoded members, was consistent. In addition, normal level of NDUFS2 (CI component) or ATP5A1 (CV component) was observed in patient cells, suggesting selective downregulation of OXPHOS components (Figure 2G).
Activities of CI-CIV determination revealed that CI activity in patient was significantly lower than that in all other samples, whereas the activities of CII-CIV in patient were not consistently lower than those of other cell lines (Figure 3A).
Figure 3.

Multiple mitochondrial dysfunctions caused by SARS2 mutations. (A) Activities of CI-CIV from mitochondria isolated from various iPSCs. (B) OCR, as determined with inhibitors including oligomycin, FCCP, antimycin A, and rotenone. The results of non-mitochondrial, basal, maximal, proton leak, ATP production respiration, and spare respiratory capacity OCR were normalized by cell count. (C) ECAR, as determined with glucose, oligomycin, and 2-Deoxy-D-glucose (2-DG). The results of non-glycolysis, glycolysis, glycolytic capacity, and glycolytic reserve ECAR were normalized by cell count. (D) MMP, measured using the fluorescence probe JC-10 assay system. (E) ROS levels detected using MitoSOX with or without H2O2 stimulation. (F) Fluorescent micrographs showing intracellular ROS formation, as indicated by DCFH-DA staining. (G) Western blot analysis of OGDH and SOD2. The band intensities were measured and normalized to those of GAPDH (right panel). (H) Level of protein carbonylation in various iPSCs. (I) Apoptosis levels of various iPSCs. Values are expressed as mean ± SD (n= 3–5). P values were determined by unpaired two-tailed t-tests. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001; ns, not significant. The results are representative of at least two independent experiments.
Subsequently, to evaluate whether the mutations affected energy production, we examined the oxygen consumption rates (OCR) of all four iPSC lines. The basal OCR, ATP production OCR, proton leak OCR, maximal respiration OCR, spare respiratory capacity, and nonmitochondrial oxygen consumption in patient were significantly lower (Figure 3B). Collectively, these results showed that patient OXPHOS function and energy production were affected by the two mutations.
We also measured the extracellular acidification rate (ECAR) of the iPSCs. The non-glycolytic acid levels were similar across cell types. Lifting of ECAR was observed after the addition of glucose. The increase in extracellular acidification levels in patient cells was higher than that in the control cells. The glycolytic capacity of patient cells was close to that of paternal and maternal cells, but reached 3-fold that of control cells in the presence of oligomycin. Glycolysis in patient cells was similar with that in parent cells, but stronger than that of the control cells (Figure 3C). These results reflected the damaged glycolytic capacity and reserve in patient cells.
A normal membrane potential (MMP) is a prerequisite for producing ATP (48). Flow cytometry results indicated that the MMP of parent-derived cells was ∼40% lower than that of the control, whereas MMP of patient-derived cells was 87.5% lower than that of the control (Figure 3D). This suggests that parental cells exhibited a certain degree of mitochondrial depolarization.
High ROS levels were generated in patient-derived iPSC cells
We analyzed the level of superoxide, a representative ROS, in various iPSCs using the mitochondrial superoxide indicator mitoSOX. The results showed that, in a basic environment, levels of superoxide free radicals among the four cell lines were almost identical, with only slightly high levels in patient- and mother-derived cells (Figure 3E). After treatment with H2O2, the superoxide content in patient cells obviously increased (Figure 3E). Superoxide levels in maternal cells also increased noticeably in the presence of H2O2. ROS production, as detected by the fluorescence intensity, in patient and maternal cells was slightly higher than other two cells (Figure 3F). Western blot results showed that levels of the ROS scavenging–related protein, SOD2 (49), was significantly elevated in patient cells. However, no significant difference in the level of ROS production-related proteins, such as OGDH (50), was observed among the four cells (Figure 3G), implying that ROS were mainly produced by perturbed electron transport. Analysis of protein carbonylation, mediated by excessive ROS (51), showed that protein carbonylation level in patient-derived iPSCs was apparently higher than that in other cell lines (Figure 3H).
Based on higher ROS and lower MMP in the patient cells, we further investigated the apoptosis level. Both the early apoptotic and late apoptotic or necrotic cell populations increased significantly, whereas the level of viable cells was lower in the patient-derived iPSCs. A slight increase in the number of late apoptotic or necrotic cells and a slight decrease in the number of viable cells were also observed in maternally-derived cells (Figure 3I).
The two mutations led to change in mitochondrial dynamics and activated mitophagy
We further compared the amount of several key regulators during mitochondrial dynamics, including DRP1 (for mitochondrial division) (2), MFN1, and MFN2 (for mitochondrial fusion) (1). Abundance of both DRP1 and MFN2 was notably decreased in patient cells; MFN1 levels were comparable between iPSCs from the patient, maternal, and control cells, but higher in paternal cells (Figure 4A). Mitochondria in various iPSCs were further observed via transmission electron microscopy (TEM) (Figure 4B). In the cytoplasm of paternal, maternal, and control cells, mitochondria presented a clear and dense double-membrane structure with visible, inwardly folded cristae structures and a round, oval, or elongated stick-shaped appearance. In patient cells, however, many mitochondria were swallowed likely by autophagosomes or autolysosomes, and the inner membrane structure was destroyed, with disordered vacuoles (Figure 4B). These differences indicate that mitochondrial morphology was affected by impairments in fusion and/or fission processes.
Figure 4.

Aberrant mitochondrial dynamics and activated mitophagy and in patient cells. (A) Protein levels of DRP1, MFN1, MFN2 and OPA1 (two forms indicated with one or two asterisks) in four iPSCs. GAPDH was used as a loading control. (B) Mitochondria in various iPSCs were observed by TEM. Red arrowheads indicate autophagolysosomes in patient iPSCs. Yellow arrowheads indicate normal mitochondria. (C, D) Western blot analysis of LC3 and p62 with or without Baf A1. GAPDH was used as a loading control. The LC3 II/I ratio and the levels of p62 and LC3 II were calculated and normalized to those of the normal control (right panels). (E) Western blot analysis of BCL2L13, PINK1, and Parkin. The band intensities were measured and normalized to those of GAPDH (right panel). Values are expressed as mean ± SD (n= 3–4). P values were determined by unpaired two-tailed t-tests. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001; ns, not significant.
Due to the observed potential autophagosomes, we further detected the mitophagy (52). No significant difference was observed in LC3 II/I levels between the four cell lines, but patient cells exhibited significantly lower p62 levels than other samples (Figure 4C). Considering that LC3II may be depleted by autophagy-lysosomal degradation, the levels of LC3II/I and p62 were further analyzed after the cells were treated with an autophagy inhibitor bafilomycin A1 (Baf A1) (53). The results of the autophagy flux assay revealed significantly increased LC3II and p62 levels in patient iPSCs, suggesting a high abundance of autophagosomes and an active autophagic flux in patient cells (Figure 4D). Examination of mitochondrial autophagy-related protein levels among cells showed that both Parkin and BCL2L13 levels in patient cells were significantly higher than other cells (Figure 4E), indicating that both Parkin-dependent (4) and-independent (5) mitophagy were activated.
Overexpression of wild-type SARS2 rescued the comprehensive defects in patient-derived iPSCs
To understand whether SARS2 malfunction is the predominant driver for the above pleiotropic effects, we stably expressed the wild-type SARS2 in patient-derived iPSCs and obtained a cell line referred to as P-WT. SARS2 was more expressed in P-WT cells than in the other four iPSCs (Figure 5A).
Figure 5.

Overexpression of the wild-type SARS2 rescued hmtRNASer(AGY) levels and corrected mitochondrial translational defects. (A) Expression of SARS2 in iPSCs, as detected by western blot assay using anti-SARS2 antibody. GAPDH was used as a loading control. (B) Steady-state abundance of mitochondrial tRNAs detected by northern blot; 5S rRNA was used as the loading control. Data were normalized to those of normal control iPSCs (right panel). (C) iPSCs were treated with EB for the indicated time points. Northern blot was performed to detect steady-state abundance of hmtRNASer(AGY). The band intensity was measured, normalized to 5S rRNA, and plotted as a percentage of the initial band intensity (lower panel). The asterisks showed statistical differences between P-WT and patient iPSCs. (D) Western blot analysis of mtDNA-encoded MT-ND3 and MT-CO1. The band intensities were measured and normalized to those of HSP60 (right panel). (E) Synthesis and stability of mtDNA-encoded proteins, as determined by pulse-chase analysis. The lane of the patient is indicated with a red asterisk. The lane of P-WT is denoted by a blue asterisk. The loaded volume was normalized by G250 staining. (F) Expression of NDUFS1, UQCRC2, and DRP1, as detected by western blot. GAPDH was used as a loading control. Data were normalized to those of normal control iPSCs (right panel). Values are expressed as mean ± SD (n= 3–6). P indicates significance according to the unpaired two-tailed t-test. *P< 0.05, **P< 0.01, ***P< 0.001, ****P< 0.0001.
We investigated whether the sharp decrease in patient hmtRNASer(AGY) levels could be reversed. A remarkable increase (3–4-fold) in the steady-state level of hmtRNASer(AGY) was observed in P-WT, reaching a level comparable to that in other cells (Figure 5B). No significant changes were seen in the steady-state levels of hmtRNASer(UCN), hmtRNAThr, or hmtRNALeu(UUR), suggesting a specific effect of SARS2 variants on hmtRNASer(AGY) abundance. The steady-state level of hmtRNASer(AGY) in P-WT remained consistent over 24 h and was more stable than that in maternal cells, indicating that SARS2 overexpression protected against and effectively reversed the physiological degradation of hmtRNASer(AGY) in patient cells (Figure 5C). hmtRNASer(AGY) in P-WT was efficiently aminoacylated. Because the hmtRNASer(AGY) in patient-derived iPSCs was highly aminoacylated, the difference in aminoacylation level between patient and P-WT cells was not significant (Supplementary Figure S6). These data clearly show that the replenished hmtRNASer(AGY) in P-WT was readily charged. Aminoacylation levels of hmtRNASer(UCN), hmtRNAThr, or hmtRNALeu(UUR) between samples were comparable (Supplementary Figure S6).
MT-ND3 level was recovered in P-WT (Figure 5D), suggestive of restored mitochondrial mRNA translation. Pulse-chase experiments showed that the mitochondrial translation rate in P-WT was comparable to that in parental and control cells. Moreover, mtDNA-encoded protein stability in P-WT was similar with that in parental and control cells, as evidenced by a higher proportion of products remaining stable than that in patient cells (Figure 5E). These data suggest that SARS2 replenishment rescued the mitochondrial translation dysfunction in patient cells.
NDUFS1 (in CI) was significantly reduced in patient cells, but increased by ∼2 fold in P-WT to a level comparable to that in other cells (Figure 5F). In addition, steady-state levels of UQCRC2 (in CIII) and DRP1 were evidently increased in P-WT after SARS2 replenishment (Figure 5F). The above results suggest that abnormal levels of nDNA-encoded mitochondria-related proteins in patient cells were associated with SARS2 variants.
The CI activity of P-WT reached a level close to that of the healthy control and was more than 1.5-fold that in patient iPSCs (Figure 6A). Meanwhile, the activity of CII-CIV in P-WT was also greatly improved (Figure 6A), despite a lack of obvious impairment between the patient and control cells. Consistently, the respiratory activity of P-WT was also improved compared to that of the patient cells. The basal, proton leak, maximal respiration, non-mitochondrial, and ATP production OCR values in P-WT were comparable to control levels (Figure 6B). However, the spare respiratory capacity of P-WT was close to that of patient cells. Rising glycolysis and impaired glycolytic reserve could be apparently improved by expressing SARS2. The ECAR in P-WT decreased considerably after SARS2 overexpression (Figure 6C), resembling that in healthy controls. Remarkably, the damaged glycolytic reserve seen in patient cells was efficiently repaired in P-WT. The protein carbonylation level in P-WT dropped significantly to a level close to that of the control cells (Figure 6D).
Figure 6.

Mitochondrial malfunctions were rescued by overexpression of wild-type SARS2. (A) Activities of CI-CIV of various cell lines. (B) OCR determination of various cell lines. The results of non-mitochondrial, basal, maximal, proton leak, ATP production respiration, and spare respiratory capacity OCR were normalized by cell count. (C) ECAR determination of various cell lines. The results of non-glycolysis, glycolysis, glycolytic capacity, and glycolytic reserve ECAR were normalized by cell count. (D) The level of protein carbonylation was determined by western blot. GAPDH was used as a loading control. (E) Western blot analysis of LC3 and p62 with Baf A1. The band intensities were measured and normalized to those of GAPDH (lower panels). (F) Mitochondria in various iPSCs were observed by TEM. The red arrow indicates autophagolysosomes. The yellow arrow indicates morphologically normal mitochondria. Values are expressed as mean ± SD (n= 3–4). P indicates significance according to the unpaired two-tailed t-test. *P< 0.05, **P< 0.01, ***P< 0.001; ns, not significant.
Autophagy flux experiments showed that, after Baf A1 treatment, the LC3II and p62 levels of P-WT were significantly lower than those in patient cells, reaching only ∼20% and ∼30% of the previously recorded levels and approaching the levels in parental and control cells (Figure 6E). These results indicate that SARS2 replenishment successfully rescued active autophagy in patient cells.
The morphology of the mitochondria in P-WT and patient cells was compared using TEM. Clear and dense mitochondrial double-layer membrane structures were observed in P-WT, and the number of vesicle structures, autophagosomes, and autolysosomes in P-WT cells was lower than that in patient cells (Figure 6F), suggesting that SARS2 effectively rescued abnormal mitochondrial morphology.
Ins12 and dupC mutations caused embryonic lethality in mice
To further understand whether either mutation impairs mitochondrial translation in model animals, we assessed the potential influence of these two mutations on mitochondrial tRNASer(AGY) and translation in mice.
Due to the conservation of SARS2 mRNAs between humans and mice, an allele carrying a dupC mutation was constructed by inserting a cytosine before c.1519C into the donor DNA fragment in the CRISPR/Cas9 system to yield heterozygous dupC mice (Sars2dupC/+). Although c.654–13T (human c.654–14T counterpart) was present in the corresponding site in mouse Sars2, only 11 nucleotides (TCTGTCTCCAG) were immediately upstream of the splicing acceptor site of intron 6 (Supplementary Figure S7A), in contrast to the 12 nucleotides (TCTGTCTCCCAG) in human SARS2. If we directly constructed a c.654–13T > A knock-in mutation, we are not sure whether the introduced AG in Sars2 could function as a splicing acceptor site as in human cells; the efficiency during splicing, even as an acceptor site, is unknown. Lastly and most importantly, after intron retention, the inclusion of 11 nucleotides would lead to frameshifting of the mutated mRNA and premature termination during translation. Based on these considerations, we constructed an Ins12 knock-in mouse (Sars2Ins12/+) using CRISPR/Cas9 (Supplementary Figure S7B). RNA transcribed from this allele completely contained the mutant mRNA with 12 nucleotides (TCTGTCTCCCAG) between exons 6 and 7, without the wild-type version, and thus only translated the Ins12 mutant.
Mating between Sars2Ins12/+ or Sars2dupC/+ mice did not yield the corresponding homozygous offspring (Sars2Ins12/Ins12 or Sars2dupC/dupC) (Supplementary Table S3). Furthermore, we failed to obtain Sars2Ins12/dupC mice and Sars2Ins12/dupC embryos during the embryonic stage at E13.5 by mating Sars2Ins12/+ and Sars2dupC/+ mice (Figure 7A, Supplementary Table S3). This phenomenon demonstrated the embryonic lethality of Sars2Ins12/dupC, Sars2Ins12/Ins12 and Sars2dupC/dupC genotypes, suggesting that the survival of the human patient was related to the production of wild-type SARS2 mRNA from the allele with c.654–14T > A variation.
Figure 7.

Sars2 mutations caused embryonic lethality in mice and led to impaired mitochondrial translation and morphology in the skeletal muscle of heterozygous mice. (A) Mouse embryos at E13.5, showing fetal arrest in Sars2Ins12/dupC mice. (B) Steady-state levels of Sars2 in skeletal muscle and heart tissues. Gapdh was used as a loading control. (C) Steady-state abundance of mitochondrial tRNAs detected in heart, brain, and skeletal muscle tissues; 5S rRNA was used as the loading control. Data were normalized to those of the wild-type mice (lower panels). Values are expressed as mean ± SD (n= 4–5). P values were determined using the unpaired two-tailed t-test. *P< 0.05, **P< 0.01, ***P< 0.001; ns, not significant. (D) Western blot analysis of mtDNA-encoded proteins in the skeletal muscle and heart tissues. Hsp60 was used as a loading control. (E) Western blot analysis of nDNA-encoded proteins in the skeletal muscle and heart tissues. Gapdh was used as the loading control. (F) Mitochondria in skeletal muscle tissues were observed by TEM. Yellow arrowhead indicates morphologically normal mitochondria; red arrowheads indicate abnormal mitochondria. (G) Model of the molecular etiology underlying Ins12 and dupC-induced human disease.
Taken together, these observations demonstrated that Ins12 and dupC mutations impaired Sars2 function and were lethal to normal embryogenesis in mice in vivo.
Skeletal muscle from heterozygous mice exhibited lower mtRNASer(AGY) and mtRNASer(UCN) levels and impaired mitochondrial translation and morphology
We further analyzed mitochondrial translation and function in Sars2Ins12/+ and Sars2dupC/+ mice. Weight, length, and organ size of Sars2Ins12/+ and Sars2dupC/+ mice were not notably different from those of wild-type mice from birth to adulthood. Among energy-consuming tissues, western blot revealed obvious lower Sars2 levels in the heart and skeletal muscle (bilateral hindlimb) tissues of Sars2Ins12/+ and Sars2dupC/+ mice than those in wild-type mice, at one month of age. No obvious dupC was detected for Sars2dupC/+ mice (Figure 7B). Sars2 levels in the brain tissue among wild-type, Sars2Ins12/+ and Sars2dupC/+ mice showed no obvious difference (Supplementary Figure S7C).
Northern blot showed that the steady-state levels of both mouse mitochondrial tRNASer(AGY) (mmtRNASer(AGY)) and mmtRNASer(UCN) were comparable between the hearts and brains of various mice (Figure 7C). mmtRNASer(AGY) level in skeletal muscle from Sars2dupC/+ mice was 50% lower than that from wild-type mice, but remained unaltered in Sars2Ins12/+ mice (Figure 7C). In addition, mmtRNASer(UCN) levels in Sars2Ins12/+ and Sars2dupC/+ mice were significantly lower to 75% and 40%, respectively, when compared with that in wild-type mice (Figure 7C). These results suggest that the stability of mmtRNASer(AGY) and mmtRNASer(UCN) were highly sensitive to impairment of Sars2 function in skeletal muscle; however, despite noticeably lower levels of Sars2 in the heart, little influence on mmtRNASer(AGY) and mmtRNASer(UCN) was observed, suggesting tissue-specific dependence and vulnerability of mitochondrial tRNASers to Sars2. The charging levels of mmtRNASer(AGY) and mmtRNASer(UCN) in the heart, brain, and skeletal muscle were comparable between heterozygous and wild-type mice (Supplementary Figure S7D), which was consistent with the results in human iPSCs that (the remaining) mitochondrial tRNASers were nearly fully aminoacylated.
As seen in patient iPSCs, mt-Nd3 level in skeletal muscle from Sars2dupC/+ and Sars2Ins12/+ mice was obviously reduced compared with that in wild-type mice, whereas mt-Nd1 abundance was not affected (Figure 7D). However, no alteration in the expression of nDNA-encoded OXPHOS subunits, including Ndufs1 and Uqcrc2, was observed in the skeletal muscle (Figure 7D). In line with the lack of decrease in both tRNASer abundance and tRNA charging levels, mitochondrial translation in the mouse heart was not affected, as evidenced by comparable levels of mtDNA-encoded mt-Nd1 and mt-Atp6 among groups (Figure 7D). No changes in Ndufs1 or Uqcrc2 expression were observed in mouse heart tissue (Figure 7E). Moreover, no abnormal protein levels were observed in the brains of heterozygous mice (Supplementary Figure S7C). These results suggest that the muscle tissue–specific downregulation of mitochondrial mRNA translation occurred in heterozygotes.
Western blot revealed that Drp1 and Mfn2 levels were lower in muscle tissue of Sars2Ins12/+ (Drp1: 71.6%; Mfn2: 78.6% of wild-type) and Sars2dupC/+ (Drp1: 92.5%; Mfn2: 77.9% of wild-type) mice, but not heart tissue of heterozygous mice (Figure 7E). Mitochondrial morphology in the mouse heart, brain, and skeletal muscle tissues was observed by TEM, which showed that the number and structure of mitochondria in the heart and brain tissues were not apparently different between heterozygous and wild-type mice (Supplementary Figure S7E). However, mitochondria in the skeletal muscle of heterozygous mice were fewer in number and smaller in size than those in wild-type mice (Figure 7F).
We further explored whether mitochondrial translation defects in the muscle yielded phenotypic consequences in male mice at 13–16 weeks of age. No obvious histological abnormalities were found in the rectus femoris muscle sections of Sars2dupC/+ or Sars2Ins12/+ mice. Quantification of the myofiber cross-sectional area also showed no significant difference in myofiber size distribution among wild-type and mutant mice (Supplementary Figure S7F). In the treadmill test, three groups of mice showed no significant difference in running time (Supplementary Figure S7G). No significant difference was observed between groups in the treadmill speed when mice were exhausted (Supplementary Figure S7G).
Altogether, the above evidence indicates that mmtRNASer(UCN) and/or mmtRNASer(AGY) levels were reduced in muscle from both Sars2dupC/+ and Sars2Ins12/+ mice, which subsequently led to impaired mitochondrial translation and morphology in heterozygotes.
DISCUSSION
SARS2 is highly associated with HUPRA syndrome and progressive spastic paresis (21–26). The clinical features of our patient overlapped with the phenotypes of both previously reported HUPRA syndrome and progressive spastic paresis (Supplementary Table S4). However, compared with HUPRA syndrome, the condition of the patient progressed more slowly, and the disease severity was relatively moderate. Metabolic alkalosis and premature birth, which are common in patients with HUPRA syndrome, were absent in this patient. The nervous system, which only affects progressive spastic paresis, was more seriously damaged in our patient. Hypertonia and varying degrees of paralysis observed in patients with SARS2-related progressive spastic paresis were also present in our patient. Moreover, frequent seizures, which have not been reported in SARS2-related patients, were one of the most striking features in our patient. Thus, our findings expanded the spectrum of the SARS2-related diseases.
The aminoacylation kinetics of Ins12 for both tRNASers could not be accurately quantified. Considering that Ins12 could activate Ser and bind tRNASers, its inability to aminoacylate tRNAs suggested that the enlarged loop B failed to orient the CCA terminus to synthetic active site. dupC exhibited a nearly 6-fold Km value for hmtRNASer(AGY) but that for hmtRNASer(UCN) was only marginally increased. Moreover, The KD determination echoed the aminoacylation kinetic data that the affinity of dupC for hmtRNASer(AGY) was more significantly impaired. These data suggest a more critical role of the C-terminal tail for binding hmtRNASer(AGY) in catalysis, in accordance with the previously reported results (19). Indeed, a very recent study has reported the cyro-EM structure of SARS2-tRNASer(AGY), which clearly shows that the very C-terminus provides an important interaction site for binding the T-stem sugar-phosphate backbone of tRNASer(AGY) (54).
The binding affinity and charging level of both mutants for hmtRNASer(AGY) were dramatically impaired in vitro. Accordingly, in vivo, hmtRNASer(AGY) abundance in patient cells was only 25% of that in control cells. Consistently, a reduced amount of hmtRNASer(AGY) was also observed in HUPRA patients with a SARS2 c.1169A > G mutation, and in patients carrying the SARS2 c.1347G > A mutation with progressive spastic paresis (21,25). We therefore suggest that SARS2 mutations weakened the binding affinity for hmtRNASer(AGY), leading to defects in the aminoacylation of hmtRNASer(AGY) by both mutants. Subsequently, impaired aminoacylation of hmtRNASer(AGY) led to the naked hmtRNASer(AGY), which is more prone to degradation by the mitochondrial RNA degradosome (55,56) and thus could not be detected by northern blot analysis. However, the residual aminoacylated hmtRNASer(AGY) remained intact; therefore, we visualized seemingly unchanged aminoacylation levels of hmtRNASer(AGY) when compared with maternal, paternal, and control cell lines, suggesting hmtRNASer(AGY) integrity was protected from degradation by tRNA aminoacylation. Besides, we cannot absolutely exclude the possibility that SARS2 was able to serve as a tRNA ‘chaperone’ to stabilize, in particular, the unusual tRNASer(AGY). With a reduced abundance of SARS2 and its mutants in patient cells, tRNASer(AGY) was more prone to be degraded. Such a chaperone role has been observed for LARS2 and its C-terminal domain (57,58). The residual pool of Ser-hmtRNASer(AGY) subsequently caused an abnormal rate of mitochondrial translation. Common observations of reduced hmtRNASer(AGY) in different patients (21,25), albeit with distinct clinical manifestations, suggest that a decreased level of hmtRNASer(AGY) due to rapid degradation of uncharged hmtRNASer(AGY) is the primary etiology of SARS2-related diseases reported to date.
Several studies with other mitochondrial aaRS-related diseases have also described similar downregulation of cognate tRNA levels. The level of mitochondrial tRNAAsn was markedly reduced while its aminoacylation level seems to be unaltered in a patient with NARS2 mutations (59). An intronic mutation in RARS2 causes a truncated ArgRS and a lower amount of mitochondrial tRNAArg, while the remaining transcript is fully aminoacylated (60). In addition, the steady-state level of mitochondrial tRNATyr is reduced in the cell lines derived from patients with Leber's hereditary optic neuropathy (LHON) phenotypes due to a YARS2 c.572G > T mutation (61). All these examples suggested that aminoacylation status of some mitochondrial tRNAs determines its cellular abundance. In line with this suggestion, we found that re-expression of wild-type SARS2 was able to restore hmtRNASer(AGY) amount to a comparable level with control cell lines. In addition, the restored hmtRNASer(AGY) was fully charged. Consistently, overexpression of LARS2 could improve the aminoacylation efficiency and stability of mitochondrial tRNALeu(UUR) A3243G mutant, which leads to tRNALeu(UUR) instability and Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes (MELAS) syndrome (62); likewise, HARS2 overexpression in cybrids carrying the m. 12201 T > C mutation elevates the steady-state level of the mitochondrial tRNAHis mutant (63).
Previous studies have uncovered that aberrant mitochondrial tRNA species are prone to be spuriously polyadenylated following the 3-terminal A76 by mitochondrial poly(A)polymerase (mtPAP) (64) and subsequently degraded by the mitochondrial degradosome, composed of polynucleotide phosphorylase (PNPase) and the RNA helicase SUV3 (56). On other hand, 2′,5′-phosphodiesterase PDE12 functions as an important tRNA surveillance factor to remove such spurious poly(A) tails (64,65). The balance of polyadenylation and de-adenylation likely provides a strategy to regulate tRNA abundance and to retain a functional tRNA pool in human mitochondria. Of note, in most cases, the poly(A) modification and degradation occur at the structurally aberrant tRNAs (7,56,64,65). Here, we suggested that naked A76 of hmtRNASer(AGY) is easily polyadenylated by mtPAP despite that hmtRNASer(AGY) is native without structural variation and the competition of de-adenylation by PDE12 likely insufficiently compromised the destructive effect on hmtRNASer(AGY) stability. Considering decreased level of hmtRNASer(AGY) seems to underlie all SARS2-related human mitochondrial disease, the hmtRNASer(AGY) metabolism needs to be further explored. Elevating abundance of hmtRNASer(AGY) based on its metabolism pathway might provide therapeutic potential for treatment of SARS2-related diseases. Beside overexpression of SARS2 explored here, manipulation of processes in hmtRNASer(AGY) lifecycle, including t6A modification by YRDC-OSGEPL1 (32,66), m3C biogenesis by NSUN2 (67,68), poly(A) addition by mtPAP (64), de-adenylation by PDE12 (65), degradation by degradosome (56) etc, if elevating its abundance, poses promising potentials to improve SARS2-related mitochondrial diseases. In addition, mitochondria from both lower and higher eukaryotes are able to import nuclear-expressed tRNAs (69,70). It is worthy to explore any possibility to overexpress and import nuclear-expressed hmtRNASer(AGY) to increase its organelle concentration as a treatment choice for SARS2-related mitochondrial diseases.
The mechanisms of tissue-specific involvement in mito-aaRS-related diseases remain unclear at present (71). Various sensitivities of mitochondrial tRNASers to SARS2 in different organs may contribute to its tissue specificity, albeit why tRNASers are selectively decreased in muscle tissue is still unclear.
In summary, we identified two novel SARS2 variants in a Chinese pedigree. These two mutations led to reduced SARS2 abundance and remodeling of the SARS2 local structure. The weakened tRNA binding capacity, which was particularly pronounced for tRNASer(AGY), caused impaired or abolished tRNASer(AGY) aminoacylation, which subsequently resulted in its rapid degradation and thus insufficient aminoacylation of tRNASer(AGY) in patient cells, leading to pleiotropic defects in mitochondrial biogenesis, homeostasis, and function (Figure 7G). The destructive effect of either mutation on tRNASer(AGY) stability, mitochondrial function, and animal viability was also observed in mouse models. Our findings expand the SARS2-related disease spectrum and clarify the molecular etiology of SARS2 mutations.
DATA AVAILABILITY
Structures of WT SARS2 and Ins12 have been deposited to PDB database with accession codes 7YDF and 7YDG. All other data supporting the findings of this study are available within the article and its Supplementary Information.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Prof. En-Duo Wang (Shanghai Institute of Biochemistry and Cell Biology, CAS) for consistent support and encouragement. We thank Prof. Yanxin Li (Shanghai Children's Medical Center) for the assistance in iPSCs generation.
Author contributions: X.Z. conceptualized, designed and coordinated the study, performed in vitro biochemical studies and drafted and revised the manuscript. J.W. conceptualized and coordinated the study, and revised the manuscript. T.Y. and Y.Z. carried out most of the experiments, and drafted and revised the manuscript. W.Z., Y.Z. and G.L. contributed to several experiments and helped with data analysis. J.W., R.Y. and N.L. performed the genetic tests, interpreted the variations and evaluated the phenotype. W.S. and P.F. performed protein structure analysis.
Contributor Information
Tingting Yu, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China.
Yi Zhang, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China; State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Wen-Qiang Zheng, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Siqi Wu, State Key Laboratory of Bioorganic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China.
Guoqiang Li, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China.
Yong Zhang, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Niu Li, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China.
Ruen Yao, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China.
Pengfei Fang, State Key Laboratory of Bioorganic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China.
Jian Wang, Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics, Shanghai Children's Medical Center, School of Medicine, Shanghai Jiao Tong University, 1678 Dong Fang Road, Shanghai 200127, China.
Xiao-Long Zhou, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2021YFA1300800, 2021YFC2700903 to X.Z. and 2020YFA0804000 to J.W.]; Natural Science Foundation of China [82171849 to T.Y. and 31822015, 81870896, 32271300 to X.Z.]; Committee of Science and Technology in Shanghai [22ZR1481300, 22JC1400503 to X.Z.]; CAS Project for Young Scientists in Basic Research [YSBR-075 to X.Z.]; State Key Laboratory of Bioorganic and Natural Products Chemistry (to P.F.). Funding for open access charge: the National Key Research and Development Program of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen Y., Liu Y., Dorn G.W. 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011; 109:1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ikeda Y., Shirakabe A., Maejima Y., Zhai P., Sciarretta S., Toli J., Nomura M., Mihara K., Egashira K., Ohishi M.et al.. Endogenous drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015; 116:264–278. [DOI] [PubMed] [Google Scholar]
- 3. Simon H.U., Haj-Yehia A., Levi-Schaffer F.. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000; 5:415–418. [DOI] [PubMed] [Google Scholar]
- 4. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T.et al.. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014; 510:162–166. [DOI] [PubMed] [Google Scholar]
- 5. Murakawa T., Yamaguchi O., Hashimoto A., Hikoso S., Takeda T., Oka T., Yasui H., Ueda H., Akazawa Y., Nakayama H.et al.. Bcl-2-like protein 13 is a mammalian atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015; 6:7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Suzuki T., Nagao A., Suzuki T.. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 2011; 45:299–329. [DOI] [PubMed] [Google Scholar]
- 7. Kummer E., Ban N.. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021; 22:307–325. [DOI] [PubMed] [Google Scholar]
- 8. Hallberg B.M., Larsson N.G.. Making proteins in the powerhouse. Cell Metab. 2014; 20:226–240. [DOI] [PubMed] [Google Scholar]
- 9. Eriani G., Delarue M., Poch O., Gangloff J., Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990; 347:203–206. [DOI] [PubMed] [Google Scholar]
- 10. Ibba M., Soll D.. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000; 69:617–650. [DOI] [PubMed] [Google Scholar]
- 11. Guo M., Yang X.L., Schimmel P.. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010; 11:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antonellis A., Green E.D.. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008; 9:87–107. [DOI] [PubMed] [Google Scholar]
- 13. Bonnefond L., Fender A., Rudinger-Thirion J., Giege R., Florentz C., Sissler M.. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005; 44:4805–4816. [DOI] [PubMed] [Google Scholar]
- 14. Moulinier L., Ripp R., Castillo G., Poch O., Sissler M.. MiSynPat: an integrated knowledge base linking clinical, genetic, and structural data for disease-causing mutations in human mitochondrial aminoacyl-tRNA synthetases. Hum. Mutat. 2017; 38:1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sissler M., Gonzalez-Serrano L.E., Westhof E.. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med. 2017; 23:693–708. [DOI] [PubMed] [Google Scholar]
- 16. Yao P., Fox P.L.. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013; 5:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lightowlers R.N., Taylor R.W., Turnbull D.M.. Mutations causing mitochondrial disease: what is new and what challenges remain. Science. 2015; 349:1494–1499. [DOI] [PubMed] [Google Scholar]
- 18. Yokogawa T., Shimada N., Takeuchi N., Benkowski L., Suzuki T., Omori A., Ueda T., Nishikawa K., Spremulli L.L., Watanabe K.. Characterization and tRNA recognition of mammalian mitochondrial seryl-tRNA synthetase. J. Biol. Chem. 2000; 275:19913–19920. [DOI] [PubMed] [Google Scholar]
- 19. Chimnaronk S., Gravers Jeppesen M., Suzuki T., Nyborg J., Watanabe K.. Dual-mode recognition of noncanonical tRNAs(Ser) by seryl-tRNA synthetase in mammalian mitochondria. EMBO J. 2005; 24:3369–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belostotsky R., Ben-Shalom E., Rinat C., Becker-Cohen R., Feinstein S., Zeligson S., Segel R., Elpeleg O., Nassar S., Frishberg Y.. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011; 88:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rivera H., Martin-Hernandez E., Delmiro A., Garcia-Silva M.T., Quijada-Fraile P., Muley R., Arenas J., Martin M.A., Martinez-Azorin F.. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013; 14:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y., Zhong C., Yang Q., Zhang G., Yang H., Li Q., Wang M.. Novel SARS2 variants identified in a chinese girl with HUPRA syndrome. Mol. Genet. Genomic Med. 2021; 9:e1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Colin E., Courtois G., Brouzes C., Pulman J., Rabant M., Rotig A., Taffin H., Lion-Lambert M., Fabrega S., Da Costa L.et al.. Biallelic mutations in the SARS2 gene presenting as congenital sideroblastic anemia. Haematologica. 2021; 106:3202–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linnankivi T., Neupane N., Richter U., Isohanni P., Tyynismaa H.. Splicing defect in mitochondrial seryl-tRNA synthetase gene causes progressive spastic paresis instead of HUPRA syndrome. Hum. Mutat. 2016; 37:884–888. [DOI] [PubMed] [Google Scholar]
- 26. Souza P.V.S., Bortholin T., Dias R.B., Chieia M.A.T., Burlin S., Naylor F.G.M., Pinto W., Oliveira A.S.B.. New genetic causes for complex hereditary spastic paraplegia. J. Neurol. Sci. 2017; 379:283–292. [DOI] [PubMed] [Google Scholar]
- 27. Dahary D., Golan Y., Mazor Y., Zelig O., Barshir R., Twik M., Iny Stein T., Rosner G., Kariv R., Chen F.et al.. Genome analysis and knowledge-driven variant interpretation with TGex. BMC Med. Genomics. 2019; 12:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu H., Huang X., Xu J., Song L., Liu S., Zhang X.B., Yuan W., Li Y.. Optimizing the method for generation of integration-free induced pluripotent stem cells from human peripheral blood. Stem Cell Res. Ther. 2018; 9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandez-Vizarra E., Ferrin G., Perez-Martos A., Fernandez-Silva P., Zeviani M., Enriquez J.A.. Isolation of mitochondria for biogenetical studies: an update. Mitochondrion. 2010; 10:253–262. [DOI] [PubMed] [Google Scholar]
- 30. Sasarman F., Antonicka H., Shoubridge E.A.. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008; 17:3697–3707. [DOI] [PubMed] [Google Scholar]
- 31. Mao X.L., Li Z.H., Huang M.H., Wang J.T., Zhou J.B., Li Q.R., Xu H., Wang X.J., Zhou X.L.. Mutually exclusive substrate selection strategy by human m3C RNA transferases METTL2A and METTL6. Nucleic Acids Res. 2021; 49:8309–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou J.B., Wang Y., Zeng Q.Y., Meng S.X., Wang E.D., Zhou X.L.. Molecular basis for t6A modification in human mitochondria. Nucleic Acids Res. 2020; 48:3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang M.H., Peng G.X., Mao X.L., Wang J.T., Zhou J.B., Zhang J.H., Chen M., Wang E.D., Zhou X.L.. Molecular basis for human mitochondrial tRNA m3C modification by alternatively spliced METTL8. Nucleic Acids Res. 2022; 50:4012–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou X.L., Zhu B., Wang E.D.. The CP2 domain of leucyl-tRNA synthetase is crucial for amino acid activation and post-transfer editing. J. Biol. Chem. 2008; 283:36608–36616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 36. Vagin A., Teplyakov A.. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:22–25. [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Biou V., Yaremchuk A., Tukalo M., Cusack S.. The 2.9 a crystal structure of t. thermophilus seryl-tRNA synthetase complexed with tRNA(Ser). Science. 1994; 263:1404–1410. [DOI] [PubMed] [Google Scholar]
- 40. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A.et al.. Highly accurate protein structure prediction with alphafold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A.et al.. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blocquel D., Li S., Wei N., Daub H., Sajish M., Erfurth M.L., Kooi G., Zhou J., Bai G., Schimmel P.et al.. Alternative stable conformation capable of protein misinteraction links tRNA synthetase to peripheral neuropathy. Nucleic Acids Res. 2017; 45:8091–8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fukuhara H., Kujawa C.. Selective inhibition of the in vivo transcription of mitochondrial DNA by ethidium bromide and by acriflavin. Biochem. Biophys. Res. Commun. 1970; 41:1002–1008. [DOI] [PubMed] [Google Scholar]
- 44. Hayakawa T., Noda M., Yasuda K., Yorifuji H., Taniguchi S., Miwa I., Sakura H., Terauchi Y., Hayashi J., Sharp G.W.et al.. Ethidium bromide-induced inhibition of mitochondrial gene transcription suppresses glucose-stimulated insulin release in the mouse pancreatic beta-cell line betaHC9. J. Biol. Chem. 1998; 273:20300–20307. [DOI] [PubMed] [Google Scholar]
- 45. Gustafsson C.M., Falkenberg M., Larsson N.G.. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016; 85:133–160. [DOI] [PubMed] [Google Scholar]
- 46. Wiedemann N., Pfanner N.. Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 2017; 86:685–714. [DOI] [PubMed] [Google Scholar]
- 47. Vercellino I., Sazanov L.A.. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022; 23:141–161. [DOI] [PubMed] [Google Scholar]
- 48. Zorova L.D., Popkov V.A., Plotnikov E.Y., Silachev D.N., Pevzner I.B., Jankauskas S.S., Babenko V.A., Zorov S.D., Balakireva A.V., Juhaszova M.et al.. Mitochondrial membrane potential. Anal. Biochem. 2018; 552:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palma F.R., He C., Danes J.M., Paviani V., Coelho D.R., Gantner B.N., Bonini M.G.. Mitochondrial superoxide dismutase: what the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid. Redox Signal. 2020; 32:701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tretter L., Adam-Vizi V.. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 2004; 24:7771–7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cattaruzza M., Hecker M.. Protein carbonylation and decarboylation: a new twist to the complex response of vascular cells to oxidative stress. Circ. Res. 2008; 102:273–274. [DOI] [PubMed] [Google Scholar]
- 52. Sciarretta S., Maejima Y., Zablocki D., Sadoshima J.. The role of autophagy in the heart. Annu. Rev. Physiol. 2018; 80:1–26. [DOI] [PubMed] [Google Scholar]
- 53. Mauvezin C., Neufeld T.P.. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015; 11:1437–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuhle B., Hirschi M., Doerfel L.K., Lander G.C., Schimmel P.. Structural basis for shape-selective recognition and aminoacylation of a D-armless human mitochondrial tRNA. Nat. Commun. 2022; 13:5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Borowski L.S., Dziembowski A., Hejnowicz M.S., Stepien P.P., Szczesny R.J.. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013; 41:1223–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Toompuu M., Tuomela T., Laine P., Paulin L., Dufour E., Jacobs H.T.. Polyadenylation and degradation of structurally abnormal mitochondrial tRNAs in human cells. Nucleic Acids Res. 2018; 46:5209–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Perli E., Giordano C., Pisano A., Montanari A., Campese A.F., Reyes A., Ghezzi D., Nasca A., Tuppen H.A., Orlandi M.et al.. The isolated carboxy-terminal domain of human mitochondrial leucyl-tRNA synthetase rescues the pathological phenotype of mitochondrial tRNA mutations in human cells. EMBO Mol. Med. 2014; 6:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hornig-Do H.T., Montanari A., Rozanska A., Tuppen H.A., Almalki A.A., Abg-Kamaludin D.P., Frontali L., Francisci S., Lightowlers R.N., Chrzanowska-Lightowlers Z.M.. Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol. Med. 2014; 6:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Simon M., Richard E.M., Wang X., Shahzad M., Huang V.H., Qaiser T.A., Potluri P., Mahl S.E., Davila A., Nazli S.et al.. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and leigh syndrome. PLos Genet. 2015; 11:e1005097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O.. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007; 81:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giege R.et al.. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am. J. Hum. Genet. 2010; 87:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li R., Guan M.X.. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell Biol. 2010; 30:2147–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gong S., Wang X., Meng F., Cui L., Yi Q., Zhao Q., Cang X., Cai Z., Mo J.Q., Liang Y.et al.. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNA(His) mutation. J. Biol. Chem. 2020; 295:940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fiedler M., Rossmanith W., Wahle E., Rammelt C.. Mitochondrial poly(A) polymerase is involved in tRNA repair. Nucleic Acids Res. 2015; 43:9937–9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pearce S.F., Rorbach J., Van Haute L., D'Souza A.R., Rebelo-Guiomar P., Powell C.A., Brierley I., Firth A.E., Minczuk M. Maturation of selected human mitochondrial tRNAs requires deadenylation. Elife. 2017; 6:e27596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lin H., Miyauchi K., Harada T., Okita R., Takeshita E., Komaki H., Fujioka K., Yagasaki H., Goto Y.I., Yanaka K.et al.. co2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018; 9:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shinoda S., Kitagawa S., Nakagawa S., Wei F.Y., Tomizawa K., Araki K., Araki M., Suzuki T., Suzuki T.. Mammalian NSUN2 introduces 5-methylcytidines into mitochondrial tRNAs. Nucleic Acids Res. 2019; 47:8734–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Van Haute L., Lee S.Y., McCann B.J., Powell C.A., Bansal D., Vasiliauskaite L., Garone C., Shin S., Kim J.S., Frye M.et al.. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019; 47:8720–8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Duchene A.M., Pujol C., Marechal-Drouard L.. Import of tRNAs and aminoacyl-tRNA synthetases into mitochondria. Curr. Genet. 2009; 55:1–18. [DOI] [PubMed] [Google Scholar]
- 70. Mercer T.R., Neph S., Dinger M.E., Crawford J., Smith M.A., Shearwood A.M., Haugen E., Bracken C.P., Rackham O., Stamatoyannopoulos J.A.et al.. The human mitochondrial transcriptome. Cell. 2011; 146:645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gonzalez-Serrano L.E., Chihade J.W., Sissler M.. When a common biological role does not imply common disease outcomes: disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019; 294:5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structures of WT SARS2 and Ins12 have been deposited to PDB database with accession codes 7YDF and 7YDG. All other data supporting the findings of this study are available within the article and its Supplementary Information.