

Abstract
The many microbial communities around us form interactive and dynamic ecosystems called microbiomes. Though concealed from the naked eye, microbiomes govern and influence macroscopic systems including human health, plant resilience, and biogeochemical cycling. Such feats have attracted interest from the scientific community, which has recently turned to machine learning and deep learning methods to interrogate the microbiome and elucidate the relationships between its composition and function. Here, we provide an overview of how the latest microbiome studies harness the inductive prowess of artificial intelligence methods. We start by highlighting that microbiome data – being compositional, sparse, and high-dimensional – necessitates special treatment. We then introduce traditional and novel methods and discuss their strengths and applications. Finally, we discuss the outlook of machine and deep learning pipelines, focusing on bottlenecks and considerations to address them.
Subject terms: Microbiome, Metagenomics
Introduction
All around us, microbial communities are at work. These communities contribute to biogeochemical cycles [1], augment or buffer environmental shifts [2], and are essential to understand disease and health of humans and other organisms [3–6]. Characteristic microbial communities and their metabolites form a dynamic and interactive micro-ecosystem that we call the microbiome [7]. Insights into the workings and relations in these networks hold promise for sustainable agriculture [4, 8], disease prevention and treatment [9], and anthropogenic impact evaluation [10]. A frontier in microbiome research is microbiome engineering to establish a microbiome that supports a desired outcome, be it better health or a higher crop yield [11]. Nevertheless, successful engineering requires knowledge about what constitutes the functioning of a given microbial community, whether certain species within the microbiome are more important than others, and how and to what degree composition and function can be manipulated.
To untangle the complexity of the microbiome, researchers have turned to artificial intelligence. Owing to their powerful predictive and informative potential, machine learning and deep learning have emerged as key tools to advance microbiome research. In this review, we present an overview of how these novel techniques can be used to study the interplay of the microbiome constituents and its links to phenotype.
Microbiome data types
Even though only a fraction of microbial species can be described through traditional isolation and cultivation approaches [12], advances in omics and high-throughput sequencing have opened the door to a comprehensive description of the microbiome and the generation of large-scale microbiome datasets [13, 14]. The most commonly used methods to analyze the microbiome are amplicon and metagenomic sequencing. In the amplicon methodology, samples are characterized using the reads of specific taxonomic marker genes like the evolutionarily conserved 16 S rRNA gene [15] or the ITS region [16]. Typically, a predefined identity threshold roughly delineates prokaryotic taxa and creates clusters known as operational taxonomic units (OTUs) [17]. Amplicon sequence variants (ASVs) are a newer analog to OTUs. ASVs are generated by a denoising approach and do without an arbitrary dissimilarity threshold, thus allowing resolution of even rare members of the community [18]. In contrast, shotgun metagenomics comprehensively catalogs the totality of genomes within a sample by non-specific sequencing [19]. Through different algorithms, shotgun metagenomic reads can be aligned to curated databases for functional or taxonomic annotation [14]. Furthermore, shotgun metagenomics enables the recovery of metagenome-assembled genomes (MAGs) from the communities using binning strategies such as MetaBAT2 [20] and VAMB that resolve genomes by contig-clustering [21]. Latest advances have even made it possible to characterize the virome, allowing a more comprehensive characterization of the microbiome using shotgun data [22].
These approaches produce feature tables, in which each cell represents the abundance or presence of a specific taxon or function per sample. Whether taxonomic or functional profiles provide a better discriminatory power in downstream analysis is subject to debate [23–25]. In any case, it is due to acknowledge the particularities and challenges related to this data type. Firstly, feature tables are compositional. Compositional data describes relationships between its components, so its parts are not independent and their sum is arbitrary [26, 27]. In addition, feature tables are usually sparse, having excessive zero counts [28], and are high-dimensional, with a larger number of features per sample. This subjects downstream analysis to the curse of dimensionality. The curse is two-fold: a high number of features inflates the computational cost, while a relatively low number of samples impoverishes generalization to other datasets [29].
Different strategies are used to deal with microbiome data. Since common distance and association measures are invalid for compositional data, statistical methods such as log-ratio transformations [26], staying-in-the-simplex approach [30], and calculating component ratios [31] have been established. Traditional log-ratio transformation methods cannot deal with sparsity, so the data is oftentimes imputed; commonly zeros are replaced with pseudo-counts [32]. On the other hand, feature selection and extraction techniques can help overcome the curse of dimensionality. Feature selection entails selecting an optimal subspace of relevant and non-redundant features [33, 34]. In contrast, feature extraction attempts to reduce the dimensionality of a dataset by building a compressed representation of the input features (see examples in further sections). Altogether, the nature of microbiome data demands pre-processing steps that have profound implications on differential feature analysis; arguably, this is bound to affect the performance of machine learning methods [35, 36].
Machine learning
Machine learning (ML) is a subset of artificial intelligence (AI) methods, which leverage large datasets to recognize, classify, and predict patterns [37]. In microbiome research, ML has been applied to tackle tasks such as phenotyping (namely, predicting an environmental or host phenotype), microbial feature classification (i.e., determining the abundance, diversity, or distribution of the microbiota), studying the complex physical and chemical interactions between the microbiome’s components, and monitoring for changes in the composition of the microbiome [9, 10]. In Table 1, we enumerate select examples of each of these tasks.
Table 1.
Examples of common tasks and ML methods used in microbiome research.
| Task | Predictive goal | Method | Reference |
|---|---|---|---|
| Phenotyping | Sponge bacterial density | Random forests | Moitinho-Silva et al. [42] |
| Phenotyping | Crop productivity | Random forests | Chang et al. [43] |
| Phenotyping | Food allergy | Recurrent neural network (LSTM) | Metwally et al. [56] |
| Phenotyping | Disease (inflammatory bowel disease) | Random forests, lasso, elastic nets | Wirbel et al. [40] |
| Phenotyping | Disease (e.g., cirrhosis, type 2 diabetes, inflammatory bowel disease) | Convolutional neural networks | Sharma et al. [53], Reiman et al. [54, 55] |
| Microbial feature classification | Microbiome composition | Autoencoder | García-Jiménez et al. [93] |
| Microbial feature classification | Metabolic profile | Autoencoder | Le et al. [73] |
| Interaction analysis | Microbe-metabolite interactions | Embedding | Morton et al. [65] |
| Interaction analysis | Microbe co-ocurrence patterns | Embedding | Tataru and David [66] |
| Monitoring composition | Response to diet change | Autoencoder | Reiman and Dai [61] |
Classical methods
Among the classical ML methods, linear regression models, random forests (RFs), and support vector machines (SVMs) have been found to perform well on microbiome data [38, 39]. However, the latter has fallen into disuse in recent studies, relegated to benchmarking. Linear regression methods like lasso and elastic nets model an output, such as a phenotype, as a linear combination of inputs making the interpretation of these methods straightforward. These methods have been recently used in host dysbiosis prediction studies, with comparable results to other methods such as RF [40]. RFs aggregate decision trees, flowchart-like structures constructed by making decisions on how to split a dataset into similar groups. By growing multiple trees from randomly-sampled feature subsets, one can assemble an RF, which has an improved performance over a singular tree [41]. Using microbiome census data, RFs have resolved the symbiont density of sponges [42], predicted maize productivity [43], and differentiated between individuals with or without a substance use disorder [44].
Dimensionality reduction techniques
Unsupervised ordination methods reduce dimensionality and simplify data for human interpretation. These algorithms are apt for creating visualizations or so-called projections. By computing a linear or non-linear combination of the existing features, these methods generate a compressed representation of the input data. Linear methods, like principal component analysis (PCA) and principal coordinate analysis (PCoA), are popular tools to visualize and contrast microbial communities, such as identifying the habitat or geographic origin of microbiota samples [45, 46]. Methods like t-stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP) faithfully capture and reveal local and non-linear relationships in complex microbiome datasets, but their tuning is finicky [47–49].
Deep learning
Deep learning (DL) is a class of ML algorithms that involves various artificial neural network architectures. DL models rely on nodes (also called neurons or units), which are functions that transform inputs and forward the outputs to other nodes. The connections between nodes result in a network consisting of multiple layers (hence the name deep neural networks), which can be connected and organized in different layouts, or architectures.
The most basic neural network architecture is the fully-connected neural network (FCNN), in which the nodes from one layer are fully connected to every node from the subsequent layer. Lo and Marculescu [50] employed this architecture to predict host phenotype from raw metagenomic count data, obtaining better classification accuracy over traditional methods across different datasets. While the FCNN is an effective standalone model, it is most often the basic building block of more complex architectures.
Picturing microbiomes
Researchers have found creative ways to enrich OTU abundance matrices with spatial information (such as that inherent in phylogenetic trees). By doing so, they can leverage the inductive capabilities of convolutional neural networks (CNNs). CNNs excel at summarizing local structure in their input; thus, they are well-suited to handle data conveying spatial information, such as images. Nguyen et al. [51, 52] rendered an OTU table into an image by reshaping each sample into a square, where each pixel was colored based on the abundance or presence of microbial taxa (Fig. 1A). taxoNN rearranges an OTU table based on its inherent phylogenetic information [53], whereas PopPhy-CNN [54, 55] populates a phylogenetic tree with OTU abundances, and then transforms the tree into a two-dimensional matrix (Fig. 1B). Generally, these approaches have outperformed their benchmarks (both traditional ML methods and FCNNs) in the task of host phenotype prediction.
Fig. 1. Examples of CNN image inputs generated from OTU tables.
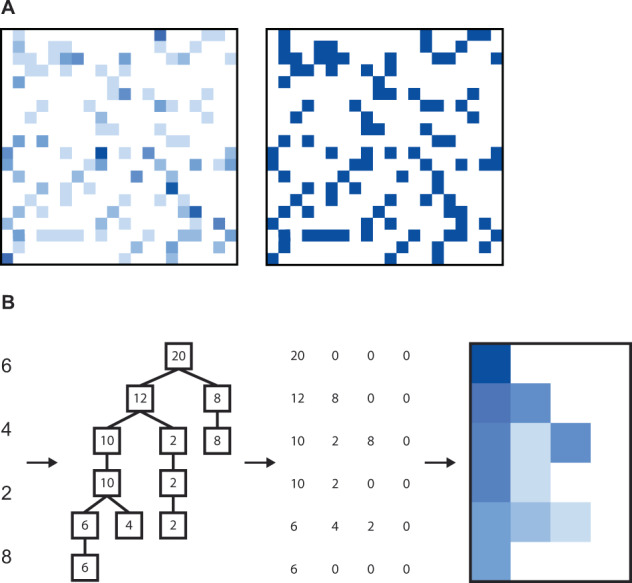
A The image is filled with species abundances (left) or presences (right). B For a single sample, the phylogenetic tree is constructed, populated with species abundances, and rearranged into a matrix.
Examining patterns in temporal data
Recurrent neural networks are mostly used to explore sequential or historical patterns. These architectures are oftentimes chain-like, consisting of loops that pass the information from one point in time to the next. In microbiome studies, RNNs allow the prediction of temporal dependencies and dynamic patterns. Metwally et al. [56] were one of the first to build a predictive model based on longitudinal microbiome profiles. Based on data from a study tracking infants’ allergic phenotype over three years, their model was built to predict food allergy, outperforming traditional ML models and FCNNs, but not reaching a performance suitable for clinical utilization. phyLoLSTM [57], an RNN-based framework, improves on previous classification accuracy by using taxoNN for feature extraction. Around the same time, Chen et al. [58] proposed a different time-aware framework, combining imputation of inconsistent temporal data and feature engineering to enrich the input tables with phylogenetic information. Their method was tested on multiple longitudinal microbiome datasets, with the task to predict different host statuses: such as type of diet, nationality, food allergy, disease, and drug use.
Unveiling latent information
For the sake of computational cost and efficiency, it is often beneficial to reduce the dimensions of microbiome feature tables. In DL, this low-dimensional latent representation is called an embedding, and it is often created with an autoencoder [59]. The autoencoder architecture consists of an encoder network that learns a latent representation of the supplied input and a decoder network that tries to reconstruct the input from this representation. By minimizing the difference between original and reconstructed data, the network learns to faithfully compress information. DeepMicro [60] presents multiple autoencoder variations and how each different latent representation improves prediction of irritable bowel syndrome and type 2 diabetes.
The modularity of autoencoders enables multimodal-data integration, holding promise for better and more comprehensive models. As presented by Reiman and Dai [61], a bimodal autoencoder can integrate diet and microbial composition to predict the microbial dynamics response to dietary change. Grazioli et al. [62] introduce a disease prediction model that relies on the product-of-experts approach to integrate the information from two autoencoders, each expert on a different modality: abundance (species-level) and presence (strain-level) features, respectively.
Other algorithms that produce embeddings draw inspiration from word processing methods, such as word2vec [63] and GloVe [64]. These methods can create dense embeddings that capture co-occurrence patterns [65, 66]. Such representations summarize the relations in the microbiome samples (e.g., microbe-metabolite interactions) and are useful for host-phenotype classification tasks.
Outlook
Bottlenecks for further applications
Even though ML was promised as a powerful predictive tool in microbiome research, it is challenged by various obstacles that limit its wide and readily application [67]. Common limitations have to do with interpretability, data hungriness, and model evaluation and selection. Plainly, ML empirically establishes a link between an input and a response without any mechanistic understanding of the underlying logic behind such a relationship. This has led to ML models being generally regarded as black boxes with inexplicable innards. The issue becomes evident, for instance, in clinical decision-making, where mechanistic insight is instrumental to trust causal inference [67]. Although the concept of interpretability is ill-defined, there is growing interest in interpretable ML [68]. For instance, the deep forest algorithm ranks features by importance and has already been explored in microbiome-wide association studies [69, 70]. Zhu et al. also proposed an approach to embed a microbial interaction network into an FCNN, thus constraining the learning process with a priori knowledge [71]. Other frameworks, such as DeepCoDA [72], prioritize feature attribution by relying on linear transformations, whereas SparseNED, an encoder-decoder model, has been used to capture microbe-metabolite relationships associated with inflammatory bowel disease through a sparse and interpretable latent space [73]. More generally-applicable ways to open the black box are thoroughly reviewed by Guidotti et al. [74].
The second hurdle is the dearth of voluminous, high-quality, and correctly-labeled data required to reliably train ML models [75–78]. Adadi [78] highlights strategies to tackle the issue of data availability of ML, including data augmentation, non-supervised learning, transfer learning, and hybrid models. Data augmentation comprises a set of practices to create synthetic samples. Lo and Marculescu [50] modeled and sampled microbiome profiles from a negative binomial distribution to enlarge their training dataset and improve the host phenotype classification performance of their FCNN model. Sayyari et al. [79] addressed the pervasive limitation of low-sample numbers and under-represented classes by introducing a tree-based associative data augmentation (TADA) approach to generate new OTU samples from an inferred phylogenetic tree. The non-supervised learning paradigm encompasses semi- and unsupervised learning approaches (think autoencoders), which are less reliant on labeled samples. Transfer learning and hybrid learners are yet to be explored in the context of microbiome research.
A paramount consideration is data quality, and, as such, our advice is to be aware of the source, deficiencies, and biases of the microbiome dataset [80]. Techniques to curb this obstacle include deduplication, class balancing, outlier removal, and imputation. These techniques influence a model’s performance, as noted by Chen et al. [58], who assay the effect of different imputation techniques on longitudinal microbiome data. Even though the collection of large and properly-annotated sample sizes is difficult to overcome in the microbiome setting, researchers can (after ensuring samples are collected and processed under the same regime) aggregate data from multiple studies, allowing the study of cohort-dependent effects [40, 81]. In any case, we stress that ML models are tightly dependent on their training dataset, so special attention should be paid to the data that feeds them.
An additional challenge microbial ecologists face has to do with the evaluation, selection, and tuning of the appropriate ML model for a given task. While choosing among the many models and fishing for a set of suitable hyperparameters seems like a daunting task, we encourage aspiring ML partakers to take advantage of the fertile ML ecosystem. Implementation has been facilitated by continuous development of Python and R libraries, such as scikit [82], PyTorch [83], Tensorflow [84], and mlr3 [85]. Moreover, high-level frameworks, like FastAI [86], PyTorch Lightning [87], and Keras [88], make implementation even more approachable. Tuning and developing ML models should also take advantage of existing frameworks for generating synthetic microbiome datasets like those provided by the CAMI consortium [89]. Not only will synthetic and pre-labeled microbiomes help guide the choice of hyperparameters and model design, but it also provides a basis for benchmarking and comparison. Comparison across multiple datasets enables assessing the robustness of ML methods, but, as remarked in neutral benchmarking studies [90], the selection of a reference dataset is critical to ensure fair comparisons.
Lastly, we summarize the key steps of ML-assisted microbiome analysis in Fig. 2, and provide the following quick tips and heuristics:
Get familiar with the dataset. An early inspection of the input data can help gauge the size of the feature space, identify whether the dataset contains unbalanced classes, or determine if imputation or feature engineering is an option.
Set up a model selection and benchmarking strategy. Either split the dataset into training, validation, and test subsets (in the case of a large dataset) or plan for cross-validation (for smaller datasets). Select appropriate metrics to compare models and estimate their performance.
Choose the appropriate method. Although the choice is data- and task-dependent, traditional ML algorithms are good starting points, as they require minimum tuning and are relatively easy to implement. If large-scale or multi-modal data is available, consider a DL approach like an autoencoder to incorporate all data facets into informative embeddings. In the case of sequential data with a longitudinally-profiled microbiome, try an RNN framework that is suitable for capturing temporal dependencies. If spatial information can be embedded into the input such as a phylogenetic tree that can be decomposed into a 2D matrix, consider CNNs.
Fig. 2. Key steps of ML-assisted microbiome analysis.

Generally, analysis begins with a feature table describing the functional or taxonomic profile of a microbiome. As part of the pre-processing step, this table can be transformed, imputed, or augmented, among other processes. The outcome of pre-processing can be tabular data or a set of image-like representations or embeddings per sample. The next step entails training and tuning ML or DL models, such as random forests, fully-connected neural networks, convolutional neural networks, recurrent neural networks, and autoencoders. Finally, the results help to elucidate the link between the microbiome composition and a continuously- (regression) or discretely-described (classification, clustering, and visualization) phenotype.
Novel techniques to keep on the watchlist
A comprehensive evaluation of DL models by LaPierre et al. suggests that it is likely that the upper limits on predictive accuracy from only metagenomic data have been reached [91]. Nonetheless, previous research has demonstrated improved predictive power can be attained by marrying different data modalities, such as microbiome, genetic, and environmental data [92]. For instance, García-Jiménez et al. [93] implemented a concept of multimodal embedding by minimizing the distance between the two latent spaces created by the separate encoders of two modalities (environmental variables and microbial composition). A lineage of work on multimodal variational autoencoders investigates the most suitable way of combining the latent spaces of individual modalities depending on the dataset properties [94–99]. Although multimodal VAEs [96] have been used to analyze single-cell multi-omics data [100], to the best of our knowledge, this kind of learner has not yet been applied to multi-omics microbiome data.
Conclusions
The study of microbial communities is lush. Amplicon and metagenomic sequencing produce feature tables that taxonomically or functionally describe a microbiome, and that, with appropriate labels, can fuel ML and DL-based methods. DL models are powerful tools with a wide array of applications in the field of microbiome research. Notably, these methods enable linking specific taxa to a host phenotype or monitoring the dynamics and host response to changes in the composition of the microbiome. Although different configurations of ML and DL models exist, the choice is task and input-dependent. In this review, we have not only provided examples of applications of AI in the realm of microbiome research but also presented a list of considerations to heed when using these models. Further research into the current bottlenecks of data availability and model interpretability will further propel the use of DL in microbiome studies and expand our understanding of the microbial interactions that shape our world.
Glossary
- Architecture
Organization and size of the layers of a neural network.
- Autoencoder
A neural network consisting of an encoder-decoder pair. The encoder reduces the dimensionality of the input, thus creating a so-called latent representation; whereas, the decoder is tasked with generating a reconstruction of the original input from such latent space.
- Benchmarking
The practice of comparing the performance of different approaches using a reference dataset.
- Compositional data
Data with the following characteristics: the total sum of each component is defined by the sampling technique (such as the capacity of a high-throughput sequencing instrument), and the difference between values is only meaningful proportionally (that is, values are relative to an arbitrary total and not independent).
- Convolution
A mathematical operation between an input matrix and filter matrix of the same rank, consisting of multiplication between a slice of the input and a filter and subsequent summation of the resulting product.
- Convolutional layer
A layer that uses the convolution operation. They are widely used to capture spatial relationships within a sample’s features and are suited for image classification.
- Elastic net
A linear regression method with regularization that shrinks both irrelevant and outlier coefficients towards zero. Such shrinkage prevents overfitting, making the model more generalizable to other datasets.
- Embedding
Typically, a low-dimensional continuous-valued representation of the feature space.
- Feature
An input variable of a model.
- Fully-connected layer
An abstraction of a matrix multiplication between an input matrix and a weight matrix, representing a connection between every input and output node.
- Label
A categorical or binary output of a model.
- Lasso
A linear regression method with regularization to shrink irrelevant coefficients towards zero. This regularization is useful to handle highly-sparse datasets.
- Layer
An abstraction of a numerical transformation.
- Machine learning paradigm
A machine learning pattern. There are two main paradigms used in microbiome research: unsupervised learning (learning from unlabelled data) and supervised learning (learning from labeled data). Other paradigms include semi-supervised learning and transfer learning.
- Random forest
An aggregated collection of independently-trained decision trees, where each decision tree is trained on a randomly-sampled subset of the training dataset.
- Recurrent layer
An extension of the fully connected layer that is looped multiple times. As each run feeds from a previous state, this layer excels at capturing time dependencies present in sequential or longitudinal data.
Author contributions
SR and MN conceptualized the study. RHM and SK wrote the first draft of the manuscript. KNN, JJ, LHH, MN, and SR contributed to ideas and manuscript editing. All authors read and approved the final version of the manuscript.
Funding
RHM was supported financially by the Novo Nordisk Foundation (grant NNF20SA0035590). SR and JJ were supported by the Novo Nordisk Foundation (grant NNF14CC0001). RHM, SK, KNN, LHH, MN, and SR were supported by the Novo Nordisk Foundation (grant NNF19SA0059348).
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mads Nielsen, Email: madsn@di.ku.dk.
Simon Rasmussen, Email: simon.rasmussen@cpr.ku.dk.
References
- 1.Toju H, Peay KG, Yamamichi M, Narisawa K, Hiruma K, Naito K, et al. Core microbiomes for sustainable agroecosystems. Nat Plants. 2018;4:247–57. doi: 10.1038/s41477-018-0139-4. [DOI] [PubMed] [Google Scholar]
- 2.Labbate M, Seymour JR, Lauro F, Brown MV. Editorial: anthropogenic impacts on the microbial ecology and function of aquatic environments. Front Microbiol. 2016;7:1044. doi: 10.3389/fmicb.2016.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller DB, Vogel C, Bai Y, Vorholt JA. The plant microbiota: systems-level insights and perspectives. Annu Rev Genet. 2016;50:211–34. doi: 10.1146/annurev-genet-120215-034952. [DOI] [PubMed] [Google Scholar]
- 5.Pita L, Rix L, Slaby BM, Franke A, Hentschel U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome. 2018;6:46. doi: 10.1186/s40168-018-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberdi A, Andersen SB, Limborg MT, Dunn RR, Gilbert MTP. Disentangling host–microbiota complexity through hologenomics. Nat Rev Genet. 2021;1–17. [DOI] [PubMed]
- 7.Berg G, Rybakova D, Fischer D, Cernava T, Vergès M-CC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8:1–22. doi: 10.1186/s40168-020-00875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sessitsch A, Mitter B. 21st century agriculture: integration of plant microbiomes for improved crop production and food security. Microb Biotechnol. 2015;8:32–3. doi: 10.1111/1751-7915.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcos-Zambrano LJ, Karaduzovic-Hadziabdic K, Loncar Turukalo T, Przymus P, Trajkovik V, Aasmets O, et al. Applications of machine learning in human microbiome studies: a review on feature selection, biomarker identification, disease prediction and treatment. Front Microbiol. 2021;0:313. doi: 10.3389/fmicb.2021.634511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta MM, Gupta A. Survey of artificial intelligence approaches in the study of anthropogenic impacts on symbiotic organisms – a holistic view. Symbiosis. 2021;84:271–83. doi: 10.1007/s13199-021-00778-0. [DOI] [Google Scholar]
- 11.Albright MBN, Louca S, Winkler DE, Feeser KL, Haig S-J, Whiteson KL, et al. Solutions in microbiome engineering: prioritizing barriers to organism establishment. ISME J. 2022;16:331–8. doi: 10.1038/s41396-021-01088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis WH, Tahon G, Geesink P, Sousa DZ, Ettema TJG. Innovations to culturing the uncultured microbial majority. Nat Rev Microbiol. 2021;19:225–40. doi: 10.1038/s41579-020-00458-8. [DOI] [PubMed] [Google Scholar]
- 13.Jiang X, Li X, Yang L, Liu C, Wang Q, Chi W, et al. How microbes shape their communities? A microbial community model based on functional genes. Genomics Proteomics Bioinformatics. 2019;17:91–105. doi: 10.1016/j.gpb.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y-X, Qin Y, Chen T, Lu M, Qian X, Guo X, et al. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 2021;12:315–30. doi: 10.1007/s13238-020-00724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012;109:6241–6. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005;71:1501–6. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci. 2011;3:347–71. doi: 10.1146/annurev-marine-120709-142811. [DOI] [PubMed] [Google Scholar]
- 20.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nissen JN, Johansen J, Allesøe RL, Sønderby CK, Armenteros JJA, Grønbech CH, et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat Biotechnol. 2021;39:555–60. doi: 10.1038/s41587-020-00777-4. [DOI] [PubMed] [Google Scholar]
- 22.Johansen J, Plichta DR, Nissen JN, Jespersen ML, Shah SA, Deng L, et al. Genome binning of viral entities from bulk metagenomics data. Nat Commun. 2022;13:965. doi: 10.1038/s41467-022-28581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z, Malmer D, Gi Langille M, Way SF, Knight R. Which is more important for classifying microbial communities: who’s there or what they can do? ISME J. 2014;8:2357–9. doi: 10.1038/ismej.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ning J, Beiko RG. Phylogenetic approaches to microbial community classification. Microbiome. 2015;3:47. doi: 10.1186/s40168-015-0114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aitchison J. The statistical analysis of compositional data. J R Stat Soc. 1982;44:139–60. [Google Scholar]
- 27.Quinn TP, Erb I, Richardson MF, Crowley TM. Understanding sequencing data as compositions: an outlook and review. Bioinformatics. 2018;34:2870–8. doi: 10.1093/bioinformatics/bty175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu T, Gallins P, Zhou Y-H. A zero-inflated Beta-binomial model for microbiome data analysis. Stat. 2018;7:e185. doi: 10.1002/sta4.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu K, Bellet A. Escaping the curse of dimensionality in similarity learning: Efficient Frank-Wolfe algorithm and generalization bounds. Neurocomputing. 2019;333:185–99. doi: 10.1016/j.neucom.2018.12.060. [DOI] [Google Scholar]
- 30.Mateu-Figueras G, Pawlowsky-Glahn V, Egozcue JJ. Compositional Data Analysis. Chichester, UK: John Wiley & Sons, Ltd; 2011. The principle of working on coordinates; pp. 29–42. [Google Scholar]
- 31.Greenacre M. Towards a pragmatic approach to compositional data analysis. Economics Working Papers. 2017.
- 32.Costea PI, Zeller G, Sunagawa S, Bork P. A fair comparison. Nat Methods. 2014;11:359. doi: 10.1038/nmeth.2897. [DOI] [PubMed] [Google Scholar]
- 33.Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans Pattern Anal Mach Intell. 2005;27:1226–38. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 34.Ditzler G, Morrison JC, Lan Y, Rosen GL. Fizzy: feature subset selection for metagenomics. BMC Bioinformatics. 2015;16:358. doi: 10.1186/s12859-015-0793-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13:342. doi: 10.1038/s41467-022-28034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarca AL, Carey VJ, Chen X-W, Romero R, Drăghici S. Machine learning and its applications to biology. PLoS Comput Biol. 2007;3:e116. doi: 10.1371/journal.pcbi.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Statnikov A, Henaff M, Narendra V, Konganti K, Li Z, Yang L, et al. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome. 2013;1:11. doi: 10.1186/2049-2618-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasolli E, Truong DT, Malik F, Waldron L, Segata N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput Biol. 2016;12:e1004977. doi: 10.1371/journal.pcbi.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wirbel J, Zych K, Essex M, Karcher N, Kartal E, Salazar G, et al. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021;22:93. doi: 10.1186/s13059-021-02306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho TK. Random decision forests. Proceedings of 3rd International Conference on Document Analysis and Recognition. vol. 1, 1995, pp 278–282.
- 42.Moitinho-Silva L, Steinert G, Nielsen S, Hardoim CCP, Wu YC, McCormack GP, et al. Predicting the HMA-LMA status in marine sponges by machine learning. Front Microbiol. 2017;8:752. doi: 10.3389/fmicb.2017.00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang H-X, Haudenshield JS, Bowen CR, Hartman GL. Metagenome-Wide Association Study and Machine Learning Prediction of Bulk Soil Microbiome and Crop Productivity. Front Microbiol. 2017;8:519. doi: 10.3389/fmicb.2017.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kosciolek T, Victor TA, Kuplicki R, Rossi M, Estaki M, Ackermann G, et al. Individuals with substance use disorders have a distinct oral microbiome pattern. Brain Behav Immun Health. 2021;15:100271. doi: 10.1016/j.bbih.2021.100271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Porras AM, Shi Q, Zhou H, Callahan R, Montenegro-Bethancourt G, Solomons N, et al. Geographic differences in gut microbiota composition impact susceptibility to enteric infection. Cell Rep. 2021;36:109457. doi: 10.1016/j.celrep.2021.109457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17:260–73. doi: 10.1016/j.chom.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X, Xie Z, Yang Z, Li D, Xu X. A t-SNE based classification approach to compositional microbiome data. Front Genet. 2020;11:620143. doi: 10.3389/fgene.2020.620143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armstrong G, Martino C, Rahman G, Gonzalez A, Vázquez-Baeza Y, Mishne G, et al. Uniform manifold approximation and projection (UMAP) reveals composite patterns and resolves visualization artifacts in microbiome data. mSystems. 2021;6:e0069121. doi: 10.1128/mSystems.00691-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo C, Marculescu R. MetaNN: accurate classification of host phenotypes from metagenomic data using neural networks. BMC Bioinformatics. 2019;20:314. doi: 10.1186/s12859-019-2833-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen TH, Chevaleyre Y, Prifti E, Sokolovska N, Zucker J-D. Deep Learning for Metagenomic Data: using 2D Embeddings and Convolutional Neural Networks. arXiv [csCV]. 2017. Available from: https://arxiv.org/abs/1712.00244.
- 52.Nguyen TH, Prifti E, Chevaleyre Y, Sokolovska N, Zucker J-D. Disease Classification in Metagenomics with 2D Embeddings and Deep Learning. arXiv [csCV]. 2018. Available from: https://arxiv.org/abs/1806.09046.
- 53.Sharma D, Paterson AD, Xu W. TaxoNN: ensemble of neural networks on stratified microbiome data for disease prediction. Bioinformatics. 2020;36:4544–50. doi: 10.1093/bioinformatics/btaa542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiman D, Metwally AA, Dai Y. Using convolutional neural networks to explore the microbiome. Conf Proc IEEE Eng Med Biol Soc. 2017;2017:4269–72. doi: 10.1109/EMBC.2017.8037799. [DOI] [PubMed] [Google Scholar]
- 55.Reiman D, Metwally AA, Sun J, Dai Y. PopPhy-CNN: A Phylogenetic Tree Embedded Architecture for Convolutional Neural Networks to Predict Host Phenotype From Metagenomic Data. IEEE J Biomed Health Inform. 2020;24:2993–3001. doi: 10.1109/JBHI.2020.2993761. [DOI] [PubMed] [Google Scholar]
- 56.Metwally AA, Yu PS, Reiman D, Dai Y, Finn PW, Perkins DL. Utilizing longitudinal microbiome taxonomic profiles to predict food allergy via Long Short-Term Memory networks. PLoS Comput Biol. 2019;15:e1006693. doi: 10.1371/journal.pcbi.1006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma D, Xu W. phyLoSTM: a novel deep learning model on disease prediction from longitudinal microbiome data. Bioinformatics. 2021;37:3707–14. doi: 10.1093/bioinformatics/btab482. [DOI] [PubMed] [Google Scholar]
- 58.Chen X, Liu L, Zhang W, Yang J, Wong K-C. Human host status inference from temporal microbiome changes via recurrent neural networks. Brief Bioinform. 2021;22:bbab223. doi: 10.1093/bib/bbab223. [DOI] [PubMed] [Google Scholar]
- 59.Kingma DP, Welling M. Auto-encoding variational Bayes. International Conference on Learning Representations (ICLR). 2014.
- 60.Oh M, Zhang L. DeepMicro: deep representation learning for disease prediction based on microbiome data. Sci Rep. 2020;10:6026. doi: 10.1038/s41598-020-63159-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reiman D, Dai Y. Using Autoencoders for Predicting Latent Microbiome Community Shifts Responding to Dietary Changes. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2019. pp 1884–91.
- 62.Grazioli F, Siarheyeu R, Alqassem I, Henschel A, Pileggi G, Meiser A. Microbiome-based disease prediction with multimodal variational information bottlenecks. PLoS Comput Biol. 2022;18:e1010050. doi: 10.1371/journal.pcbi.1010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mikolov T, Chen K, Corrado G, Dean J. Efficient Estimation of Word Representations in Vector Space. arXiv [csCL]. 2013. Available from: https://arxiv.org/abs/1301.3781.
- 64.Pennington J, Socher R, Manning C. Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP) Stroudsburg, PA, USA: Association for Computational Linguistics; 2014. Glove: Global vectors for word representation. [Google Scholar]
- 65.Morton JT, Aksenov AA, Nothias LF, Foulds JR, Quinn RA, Badri MH, et al. Learning representations of microbe-metabolite interactions. Nat Methods. 2019;16:1306–14. doi: 10.1038/s41592-019-0616-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tataru CA, David MM. Decoding the language of microbiomes using word-embedding techniques, and applications in inflammatory bowel disease. PLoS Comput Biol. 2020;16:e1007859. doi: 10.1371/journal.pcbi.1007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ching T, Himmelstein DS, Beaulieu-Jones BK, Kalinin AA, Do BT, Way GP, et al. Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface 2018;15:20170387. [DOI] [PMC free article] [PubMed]
- 68.Murdoch WJ, Singh C, Kumbier K, Abbasi-Asl R, Yu B. Definitions, methods, and applications in interpretable machine learning. Proc Natl Acad Sci USA. 2019;116:22071–80. doi: 10.1073/pnas.1900654116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu Q, Pan M, Liu L, Li B, He T, Jiang X, et al. An Ensemble Feature Selection Method Based on Deep Forest for Microbiome-Wide Association Studies. 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2018. pp 248–53.
- 70.Zhu Q, Li B, He T, Li G, Jiang X. Robust biomarker discovery for microbiome-wide association studies. Methods. 2020;173:44–51. doi: 10.1016/j.ymeth.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 71.Zhu Q, Jiang X, Zhu Q, Pan M, He T. Graph embedding deep learning guides microbial biomarkers’ identification. Front Genet. 2019;10:1182. doi: 10.3389/fgene.2019.01182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quinn TP, Nguyen D, Rana S, Gupta S, Venkatesh S. DeepCoDA: personalized interpretability for compositional health data. arXiv [csLG]. 2020. Available from: https://arxiv.org/abs/2006.01392.
- 73.Le V, Quinn TP, Tran T, Venkatesh S. Deep in the Bowel: Highly Interpretable Neural Encoder-Decoder Networks Predict Gut Metabolites from Gut Microbiome. BMC Genomics. 2020;21:256. doi: 10.1186/s12864-020-6652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guidotti R, Monreale A, Ruggieri S, Turini F, Giannotti F, Pedreschi D. A Survey of Methods for Explaining Black Box Models. ACM Comput Surv. 2018;51:1–42. doi: 10.1145/3236009. [DOI] [Google Scholar]
- 75.Raudys SJ, Jain AK. Small sample size effects in statistical pattern recognition: recommendations for practitioners. IEEE Trans Pattern Anal Mach Intell. 1991;13:252–64. doi: 10.1109/34.75512. [DOI] [Google Scholar]
- 76.van der Ploeg T, Austin PC, Steyerberg EW. Modern modelling techniques are data hungry: a simulation study for predicting dichotomous endpoints. BMC Med Res Methodol. 2014;14:137. doi: 10.1186/1471-2288-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alwosheel A, van Cranenburgh S, Chorus CG. Is your dataset big enough? Sample size requirements when using artificial neural networks for discrete choice analysis. J Choice Model. 2018;28:167–82. doi: 10.1016/j.jocm.2018.07.002. [DOI] [Google Scholar]
- 78.Adadi A. A survey on data‐efficient algorithms in big data era. J Big Data. 2021;8:1–54.. doi: 10.1186/s40537-021-00419-9. [DOI] [Google Scholar]
- 79.Sayyari E, Kawas B, Mirarab S. TADA: phylogenetic augmentation of microbiome samples enhances phenotype classification. Bioinformatics. 2019;35:i31–40. doi: 10.1093/bioinformatics/btz394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCoubrey LE, Elbadawi M, Orlu M, Gaisford S, Basit AW. Harnessing machine learning for development of microbiome therapeutics. Gut Microbes. 2021;13:1–20. doi: 10.1080/19490976.2021.1872323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee KA, Thomas AM, Bolte LA, Björk JR, de Ruijter LK, Armanini F, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med. 2022;28:535–44. doi: 10.1038/s41591-022-01695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30. [Google Scholar]
- 83.Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, et al. Proceedings of the 33rd International Conference on Neural Information Processing Systems. Red Hook, NY, USA: Curran Associates Inc.; 2019. PyTorch: an imperative style, high-performance deep learning library; pp. 8026–37. [Google Scholar]
- 84.Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv [csDC]. 2016. Available from: https://arxiv.org/abs/1603.04467.
- 85.Lang M, Binder M, Richter J, Schratz P, Pfisterer F, Coors S, et al. mlr3: A modern object-oriented machine learning framework in R. J Open Source Softw. 2019;4:1903. doi: 10.21105/joss.01903. [DOI] [Google Scholar]
- 86.Howard J. fastai. 2018. https://docs.fast.ai/.
- 87.Falcon W, The PyTorch Lightning Team. PyTorch Lightning. 2019. https://www.pytorchlightning.ai.
- 88.Chollet F, Keras Team. Keras. 2015. https://keras.io/.
- 89.Fritz A, Hofmann P, Majda S, Dahms E, Dröge J, Fiedler J, et al. CAMISIM: simulating metagenomes and microbial communities. Microbiome. 2019;7:17. doi: 10.1186/s40168-019-0633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sidorczuk K, Gagat P, Pietluch F, Kała J, Rafacz D, Bąkała L, et al. Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief Bioinform 2022;23:bbac343. [DOI] [PMC free article] [PubMed]
- 91.LaPierre N, Ju CJ-T, Zhou G, Wang W. MetaPheno: a critical evaluation of deep learning and machine learning in metagenome-based disease prediction. Methods. 2019;166:74–82. doi: 10.1016/j.ymeth.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–5. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 93.García-Jiménez B, Muñoz J, Cabello S, Medina J, Wilkinson MD. Predicting microbiomes through a deep latent space. Bioinformatics. 2021;37:1444–51. doi: 10.1093/bioinformatics/btaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suzuki M, Nakayama K, Matsuo Y. Joint Multimodal Learning with Deep Generative Models. International Conference on Learning Representations Workshop (ICLR) Workshop Track. 2017.
- 95.Wu M, Goodman N Multimodal Generative Models for Scalable Weakly-Supervised Learning. Advances in Neural Information Processing Systems 31 (NIPS). 2018. pp 5575–85.
- 96.Shi Y, N S, Paige B, Torr P. Variational Mixture-of-Experts Autoencoders for Multi-Modal Deep Generative Models. Advances in Neural Information Processing Systems (NeurIPS). 2019. pp 15718–29.
- 97.Wu M, Goodman N. Multimodal Generative Models for Compositional Representation Learning. arXiv [csLG]. 2019. Available from: https://arxiv.org/abs/1912.05075.
- 98.Kutuzova S, Krause O, McCloskey D, Nielsen M, Igel C. Multimodal Variational Autoencoders for Semi-Supervised Learning: In Defense of Product-of-Experts. arXiv [csLG]. 2021. Available from: https://arxiv.org/abs/2101.07240.
- 99.Sutter TM, Daunhawer I, Vogt JE. Generalized Multimodal ELBO. arXiv [csLG]. 2021. Available from: https://arxiv.org/abs/2105.02470.
- 100.Minoura K, Abe K, Nam H, Nishikawa H, Shimamura T. A mixture-of-experts deep generative model for integrated analysis of single-cell multiomics data. Cell Rep Methods. 2021;1:100071. doi: 10.1016/j.crmeth.2021.100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.