

Abstract
Caesarean section delivery (CSD) disrupts mother-to-neonate transmission of specific microbial strains and functional repertoires as well as linked immune system priming. Here we investigate whether differences in microbiome composition and impacts on host physiology persist at 1 year of age. We perform high-resolution, quantitative metagenomic analyses of the gut microbiomes of infants born by vaginal delivery (VD) or by CSD, from immediately after birth through to 1 year of life. Several microbial populations show distinct enrichments in CSD-born infants at 1 year of age including strains of Bacteroides caccae, Bifidobacterium bifidum and Ruminococcus gnavus, whereas others are present at higher levels in the VD group including Faecalibacterium prausnitizii, Bifidobacterium breve and Bifidobacterium kashiwanohense. The stimulation of healthy donor-derived primary human immune cells with LPS isolated from neonatal stool samples results in higher levels of tumour necrosis factor alpha (TNF-α) in the case of CSD extracts over time, compared to extracts from VD infants for which no such changes were observed during the first year of life. Functional analyses of the VD metagenomes at 1 year of age demonstrate a significant increase in the biosynthesis of the natural antibiotics, carbapenem and phenazine. Concurrently, we find antimicrobial resistance (AMR) genes against several classes of antibiotics in both VD and CSD. The abundance of AMR genes against synthetic (including semi-synthetic) agents such as phenicol, pleuromutilin and diaminopyrimidine are increased in CSD children at day 5 after birth. In addition, we find that mobile genetic elements, including phages, encode AMR genes such as glycopeptide, diaminopyrimidine and multidrug resistance genes. Our results demonstrate persistent effects at 1 year of life resulting from birth mode-dependent differences in earliest gut microbiome colonisation.
Subject terms: Metagenomics, Microbiome, Antibiotics
Introduction
The rate of caesarean section delivery is constantly increasing worldwide, which is partly driven by increases in overall income and access to health facilities.1 According to a 2015 report, 29.7 million births occurred via CSD in that year accounting for ~18% of the births in 169 countries.1 At 25% in Europe, this number is higher than the global average.2 The short-term risks of CSD include delayed or altered development of the immune system,3 reduced gut microbiome diversity,4 limited transmission of bacterial strains from mother to neonate5,6 and microbiome-borne functional deficiencies.7–10 Although few studies associate CSD with metabolic disorders11,12 and allergies,13,14 the long-term effects of birth mode are not well understood. Shao et al. reported that CSD may predispose individuals to colonisation by opportunistic pathogens including those carrying antimicrobial resistance (AMR) genes.15 On the one hand, several reports including our previously published study8 addressed questions concerning the very early development of the neonate’s gut microbiomes14,16 and immune system priming3 in relation to disease development.17,18 On the other hand, only few reports19–22 follow the effects of birth mode during the first year of life especially in relation to immune system priming, development and evolution of AMR, and the contribution of mobile genetic elements to the persistence of AMR genes.
Factors including environmental exposure,14 breast feeding and diet19,23,24 and genetics25 play crucial roles in the development of an infant. Aside from this, it is now generally accepted that birth mode, i.e. vaginal delivery (VD) or CSD, has a pronounced impact on early microbiome structure.3,8,11,26,27 While the majority of these studies focus on overall microbiome structure, analyses of the functional contribution of the microbiome has attracted attention due to its sensitivity to perturbation.28 For example, we previously reported that the microbiome in VD-born babies was enriched in bacterial genes encoding for lipopolysaccharide (LPS) biosynthesis, cationic antimicrobial peptide resistance as well as two-component systems.8 Interestingly, higher levels of LPS biosynthesis genes were associated with increased immune responses in VD neonates, whereas CSD neonates had reduced levels of TNF-α and IL-18 immediately after birth. Noteworthy in this context is previous work by Vatanen et al. which showed that differing LPS immunogenicity contributes to autoimmunity thereby affecting the long-term health outcomes of infants exposed to different antigens.29 Furthermore, others have hypothesized and reported3,30 a similar phenomenon, whereby the gut microbiome contributes to the development of the immune system during a “critical window” of development.3,30–36 In a neonatal cohort at risk for the development of asthma, bacterial metabolites were shown to specifically impede immune tolerance.37 However, some of the reports described above do not elaborate on the continuous effect of early immune system priming in the context of the birth mode and especially over the course of the first year of life, including whether these effects normalize over time.
Aside from the well-studied factors and consequences of development described above, the role of commensal microbiota in the emergence and spread of AMR is not well understood. Recent studies have reported that antibiotic exposure in infancy affects microbial diversity, and enriches AMR genes. Interestingly, Ravi et al. have suggested that the infant gut microbiome acts as a reservoir for multidrug resistance that persists throughout infancy up to 2 years of age.38 They reported that integrons (int1 gene) in the gut could potentially be responsible for this phenomenon. Nevertheless, the effect of birth mode, CSD or VD, on the transmission and occurrence of AMR remains unresolved.
Here, we address the aforementioned gaps in knowledge concerning the effect of birth mode on the persistence of the gut microbiota over the first year of life including their inherent functions, immunogenic properties and their role in conferring AMR. Our results highlight birth mode-dependent differences in gut microbiome structure and their association with immune function. We found that the gut microbiota becomes similar between CSD and VD babies at 1 year of age, with the exception of an immunostimulatory commensal, Faecalibacterium prausnitzii, which was enriched in the VD group. In addition, we identified an increased abundance in AMR genes directed against synthetic and semi-synthetic antibiotics in CSD as early as 5 days postpartum. Strikingly, we found that mobile genetic elements (MGEs) including plasmids and bacteriophages are key contributors to the establishment and persistence of AMR, irrespective of birth mode. Collectively, our findings suggest that birth mode-dependent effects persist through the first year of life including the delayed immunostimulation of CSD infants likely affecting tolerance mechanisms as well as the apparent role of bacteriophages in conferring AMR.
Results
Birth mode-dependent gut microbiota differences during the first year
We previously described the initial seeding and colonisation processes within the human gut microbiome and identified differences in microbiome structure and function as well as linked immunogenicity and immune system priming, which stratified according to birth mode.8,39 Building on this work, we aimed to understand the long-term effects in relation to the observed differences, especially through the first year of life which represents a “critical window” of development including physiological growth and immune system maturation. To achieve this, we followed VD and CSD neonates in our cohort and collected faecal samples at crucial intervals after birth, including 5 days, 1 month, 6 months and at 1 year of age (Fig. 1a). In one of our previous studies,8 a multivariate analysis was performed to compare the profiles of CSD (±SGA) to VD neonates. The results of these analyses demonstrated that delivery mode was the strongest determining factor in the microbial profile and predicted functions, irrespective of the infants were born SGA or not.8 In light of these analyses, we included the SGA samples within the CSD group. We reconstructed microbial genomes and identified differentially abundant taxa and functions between the groups using metagenomic sequencing data. Based on metagenomic operational taxonomic units (mOTUs), we calculated the Jensen–Shannon divergence index and found that the intra-group variability within CSD or VD was minimal while the inter-group variability between CSD and VD groups was significantly different (Supplementary Fig. 1). At the genus level, our data also recapitulated previously described8 significantly increased levels of Bacteroides (FDR-adjusted p < 0.05, Wilcoxon rank-sum test) in the VD neonates compared to CSD at the early timepoints (day 5 after birth and at 1 month). B. caccae also showed an increasing trend in the CSD group at 6 months and after 1 year of age, while B. caecimuris was significantly increased in CSD at 5 days after birth (Fig. 1b; FDR-adjusted p < 0.05, Wilcoxon rank-sum test) and showed an increasing trend at 1 year of age. However, at 1 year of age, the abundance of this genus in samples from CSD neonates was comparable to the levels in the VD group. In contrast, the levels of Bifidobacterium were increased in VD after 6 months, while Faecalibacterium prausnitzii, a commensal associated with healthy human microbiomes,40 was found to be significantly increased in the VD group at 1 year of age (Fig. 1b, FDR-adjusted p < 0.05, Wilcoxon rank-sum test). We further found that both birth mode and the neonatal age affect the gut microbiome community structure, whereby the latter contributes highly to variation within and between the groups (Fig. 1c). The taxonomic profiles at 1 year of age were distinct when compared to day 5, 1 month and 6 months from both groups.
Fig. 1. Gut microbiome profiles throughout the first year of life.

a Workflow representation of DNA and LPS isolation from faecal samples for metagenomic, immune and functional AMR analyses. b Relative abundances of metagenomic operational taxonomic units (mOTUs) >1% abundance at day 5 after birth, 1 month, 6 months, and at 1 year of age. c Canonical correlation analyses (CCA) resolving the stratification of taxonomic profiles based on two covariates, i.e. birth mode and time when samples were sequenced.
Assessment of differences in metagenomic functional potential at 1 year of age
Taxonomic differences within the gut microbiome populations may not always manifest as differences in functional diversity due to the redundancy in the latter. To address this, we assigned KEGG41 orthology identifiers (KOs) to each gene identified from both groups. We found 84 differentially abundant KOs between VD and CSD samples at 1 year of age (Fig. 2a). In addition, we linked all identified KOs (n = 7103) to their corresponding KEGG orthology pathways (Fig. 2b) and performed differential pathway analyses. We found that the VD group showed an increase in the gene copy numbers of pathways involved in carbapenem and phenazine biosyntheses (Fig. 2c). We found that 21 unique genera were associated with carbapenem biosynthesis across both groups (Supplementary Data 1) spanning all major phyla found within the gut.
Fig. 2. Functional differences at 1 year of age.
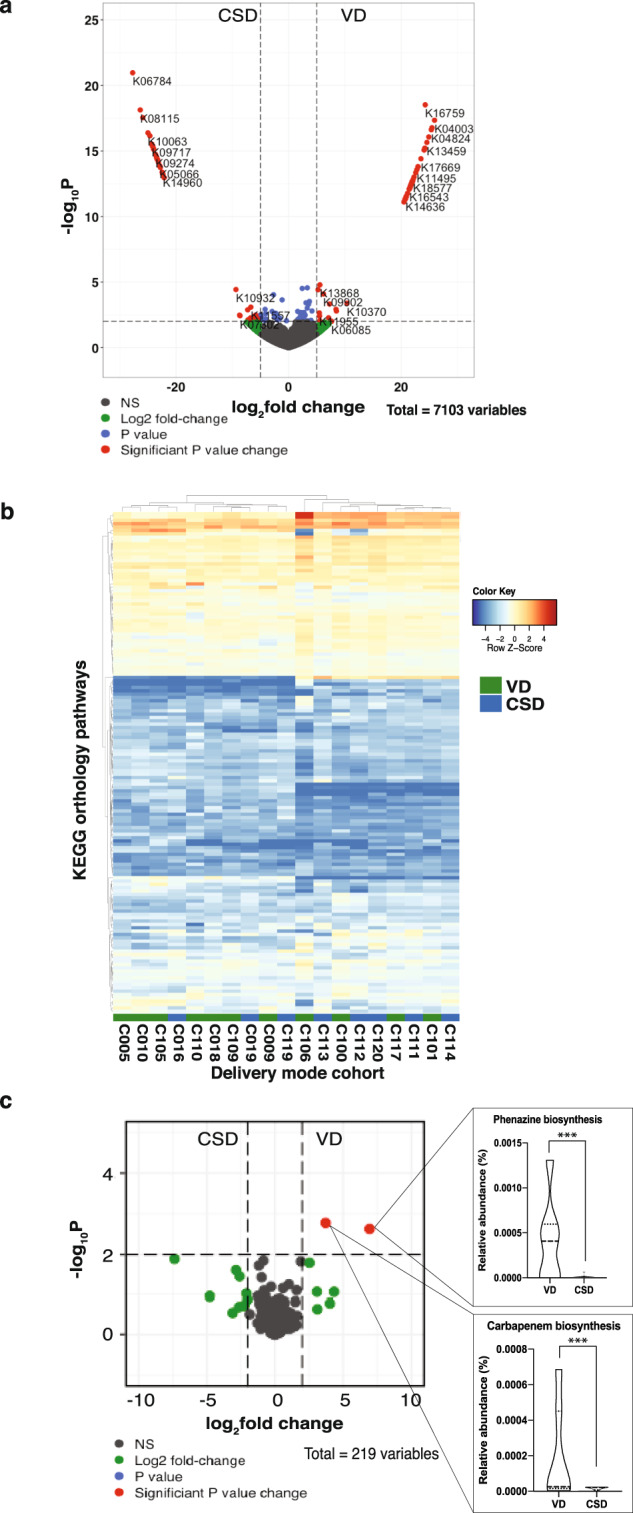
a Volcano plot depicting the statistically significantly different KEGG orthologs found in both CSD and VD groups at 1 year of age. A total of 6413 variables were tested, with −log10(p-value) shown on the y-axis. Green dots indicate KOs with a fold change >2. Red dots indicate KOs with a significant fold-change cut-off of 2, and with a false-discovery rate-adjusted p-value cut-off of 0.01. b Heatmap based on the KEGG pathways found in both CSD and VD samples at 1 year of age. Each row denotes a pathway represented by the KO genes, with the hierarchical clustering being based on Euclidean distances using Ward’s clustering algorithm. c Volcano plot of the 219 KEGG pathways to which the KOs were mapped, tested for significance with a fold-change cut-off of 2, and with a false-discovery rate-adjusted p-value cut-off of 0.01. The insets show carbapenem and phenazine biosynthesis pathways that were statically significantly different. ***p-value < 0.001.
Pro-inflammatory immune responses elevated in CSD after 1 year of life
In the early stages of neonatal development, we found that the immune activation potential of LPS was significantly increased in samples from VD neonates,8 whereby the isolated LPS triggered the secretion of TNF-α and IL-18 by monocyte-derived dendritic cells (MoDCs) from four healthy adult donors. To determine if the immunostimulatory potential persisted at 1 year of age, we stimulated the MoDCs (obtained from four healthy adult donors) with LPS isolated from the faecal samples of the CSD and VD groups. In addition to TNF-α and IL-18, we also tested the potential of LPS to stimulate secretion of pro- and anti-inflammatory cytokines such as IL-1ß, IL-12, IL-8 and IL-10 (Fig. 3a). Interestingly, at 1 year of age, IL-18 was below the detection limits. We did not find any significant differences between the CSD and VD groups with respect to the levels of secreted TNF-α at 1 year of age. However, contrary to the patterns observed at 5 days after birth, we found that the levels of TNF-α stimulated by LPS were significantly increased at 1 year of age within the CSD group (p < 3.5 × 10−5, paired two-way ANOVA; Fig. 3b). Interestingly, the increase in stimulated TNF-α levels in CSD at 1 year of age was similar to the level of the cytokine stimulated by LPS from the day 5 VD samples (Fig. 3b). In addition, we found that the stimulated TNF-α levels at 1 year of age were positively correlated with the abundance of several mOTUs, including Bacteroides caecimuris and Haemophilus influenzae (Fig. 3c). Previous reports42 suggest that Enterobacteriaceae levels correlate with inflammatory levels. However, we did not find a correlation of these taxa with LPS levels in our study (Supplementary Fig. 2). Our data also indicate an increase in the number of Gram-negative (G-ve) bacteria at 1 year of age compared to day 5 after birth in the CSD group (Supplementary Fig. 3). The increase in Shannon diversity at 1 year of age compared to day 5 coupled with the increase in G-ve bacteria provides a mechanistic explanation why the LPS stimulation of donor cells from faecal samples of CSD resulted in similar levels of TNF-α (Fig. 3b), as observed with faecal samples from the VD group at 1 year of age.
Fig. 3. Immunostimulatory potential at 1 year of age.
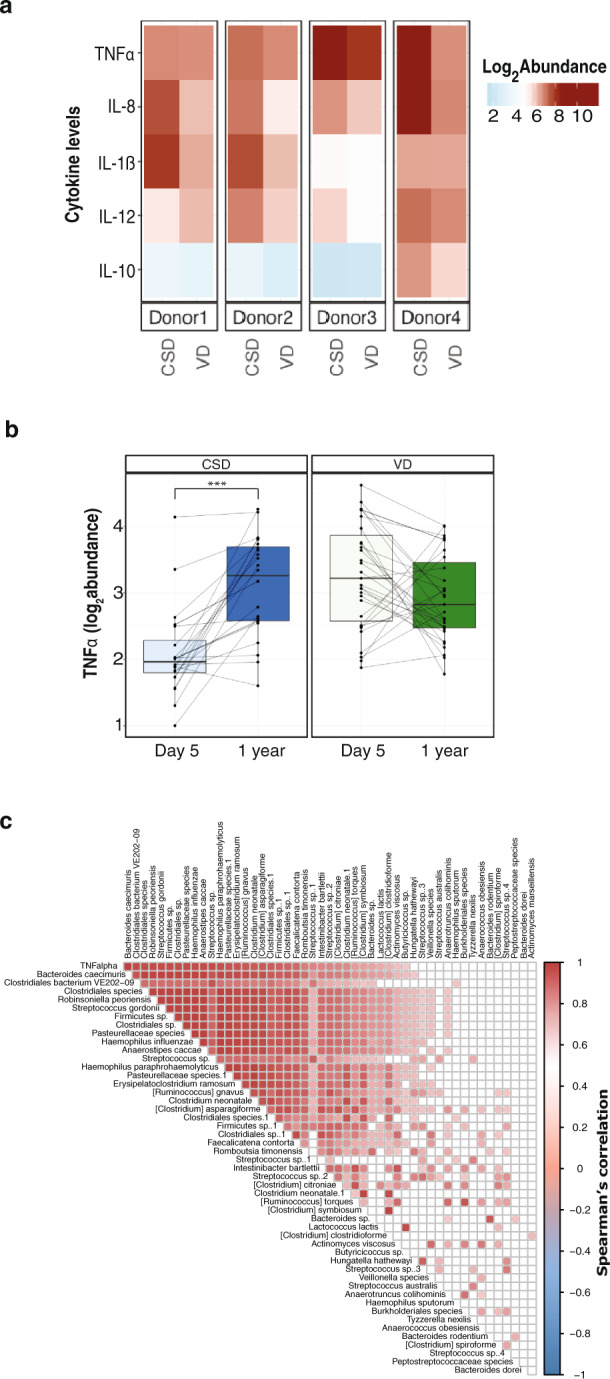
a Heatmap depicting the abundance (log2) of pro- and anti-inflammatory cytokines at 1 year of age. Cytokine levels were measured by stimulating MoDCs from healthy donors (Donor 1–4) with LPS isolated from faecal samples of CSD and VD neonates. b Boxplots depicting the TNF-α levels in both groups (CSD and VD) at day 5 after birth and 1 year of age. Paired two-way ANOVA (analysis of variance) p-values are listed in the plot to depict significant differences. ***p-value < 0.001. c Correlation of TNF-α levels (row 1) with the relative abundance of metagenomic OTUs based on canonical correlation analysis. Filled squares indicate significantly correlated taxa, whereas colour indicates positive (red) or negative (blue) correlation.
Antimicrobial resistance modulated by birth mode
The analyses of the functional potential based on KEGG orthology revealed a stratification of antibiotic biosynthesis pathways based on whether an infant was born by CSD or VD (Fig. 2c). To assess and validate the impact of birth mode on the presence and persistence of AMR, we used a deep-learning approach43 to annotate antibiotic resistance genes in our metagenomic data.44 We determined the presence and relative abundance of AMR genes in samples collected from both CSD and VD at day 5 after birth, 1 month, 6 months and at 1 year of age. The samples collected from CSD neonates exhibited an increased abundance in AMR genes at the earliest time point (day 5) compared to the VD group. In addition, we found that the number of AMR genes detected in CSD infants at 1 year of age was significantly reduced in comparison to the CSD samples at day 5 (FDR-adjusted p < 0.0021, Wilcoxon rank-sum test; Fig. 4a, Supplementary Fig. 4a). To corroborate our observations on the levels of AMR genes, we assessed the abundance of AMR genes using a random subset of samples from the resistome study by Gasparrini et al.45 We found that the overall levels of AMR genes starting at 1 month through to 1 year of age were similar in their study to those observed in our own cohort (Supplementary Fig. 4b). Meanwhile in our study, at 1 year of age, we found several genes that were differentially abundant between the CSD and VD groups (FDR-adjusted p < 0.05, Wilcoxon rank-sum test; Fig. 4b). Since various genes can confer resistance to the same antibiotic, we regrouped the genes into their respective categories such as multidrug, tetracycline resistance etc. We found that genes conferring glycopeptide, phenicol, pleuromutilin, bacitracin, sulfonamide and diaminopyrimidine resistance were significantly increased in CSD compared to VD at day 5 after birth (Fig. 4c, Supplementary Fig. 5; FDR-adjusted p < 0.05, Wilcoxon rank-sum test). Interestingly, diaminopyrimidine, phenicol, pleuromutilin, and sulfonamide are synthetic or semi-synthetic antibiotics and, likely prevalent in the hospital environment.46–49 However, these differences did not persist over time. In addition, the mothers in our cohort across both groups (CSD:6 and VD:1) received prophylactic treatment against group B Streptococcus (Supplementary Data 2) in the form of cephalosporin. However, we did not find any distinguishing patterns within the resistance categories corresponding to this treatment regimen. Albeit a limited sample size, we also tracked the diet including feeding method (bottle- or breast-fed), antibiotic regimen and physical characteristics through the first year and did not find any significant correlations with functional pathways including AMR (Supplementary Fig. 6 and Supplementary Data 3).
Fig. 4. Antimicrobial resistance gene abundances over time.
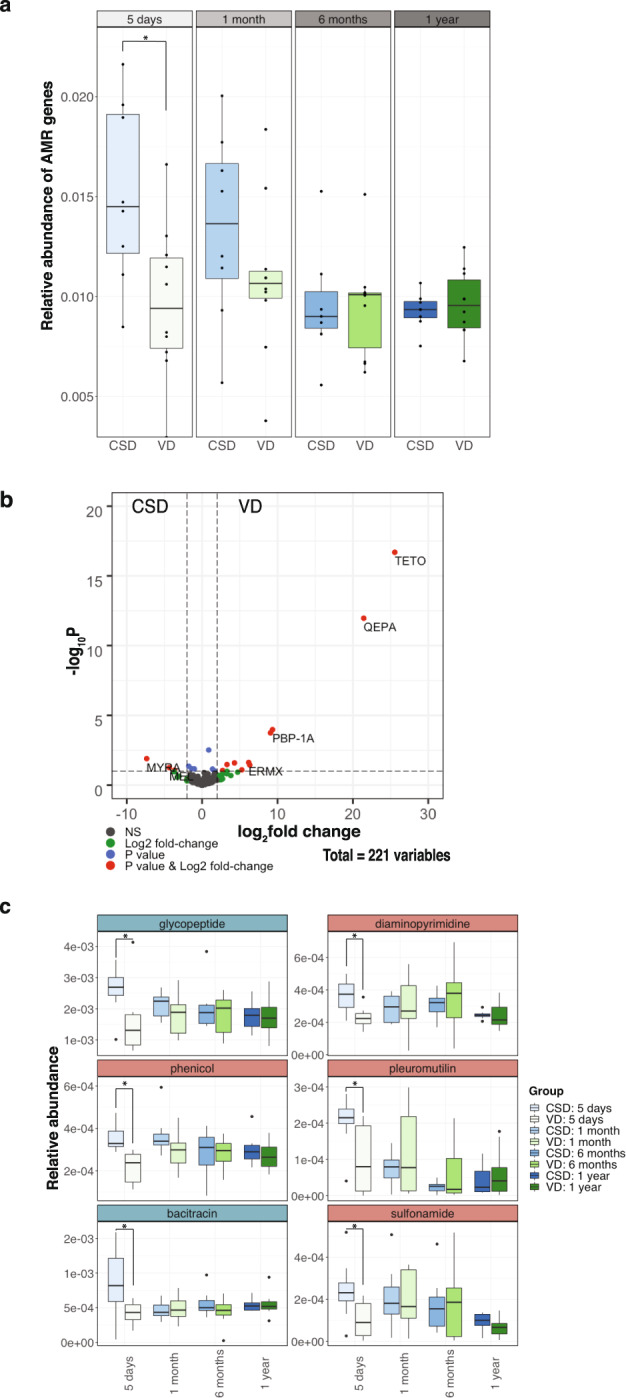
a Boxplots of the overall AMR gene abundance in CSD and VD samples at different timepoints including 5 days after birth and at 1 year of age. Wilcoxon rank-sum tests were used to test for significance. *p < 0.05. b Volcano plot depicting the significantly enriched genes in either CSD or VD samples at 1 year of age. c AMR categories which are significantly different between the groups at any of the timepoints are shown. Wilcoxon rank-sum tests were used to test for significance. *p < 0.05.
Taxa associated with antimicrobial resistance
Since AMR genes were found in the metagenomic data, we investigated which taxa carried these genes by reconstructing metagenome-assembled genomes (MAGs), classifying them taxonomically based on the GTDB database,50 and linking AMR genes to individual MAGs. We then compared taxa contributing to AMR between birth mode at day 5 after birth and 1 year of age as well as the change over time within the individual groups (Fig. 5a).
Fig. 5. Taxa and mobile genetic elements associated with antimicrobial resistance.
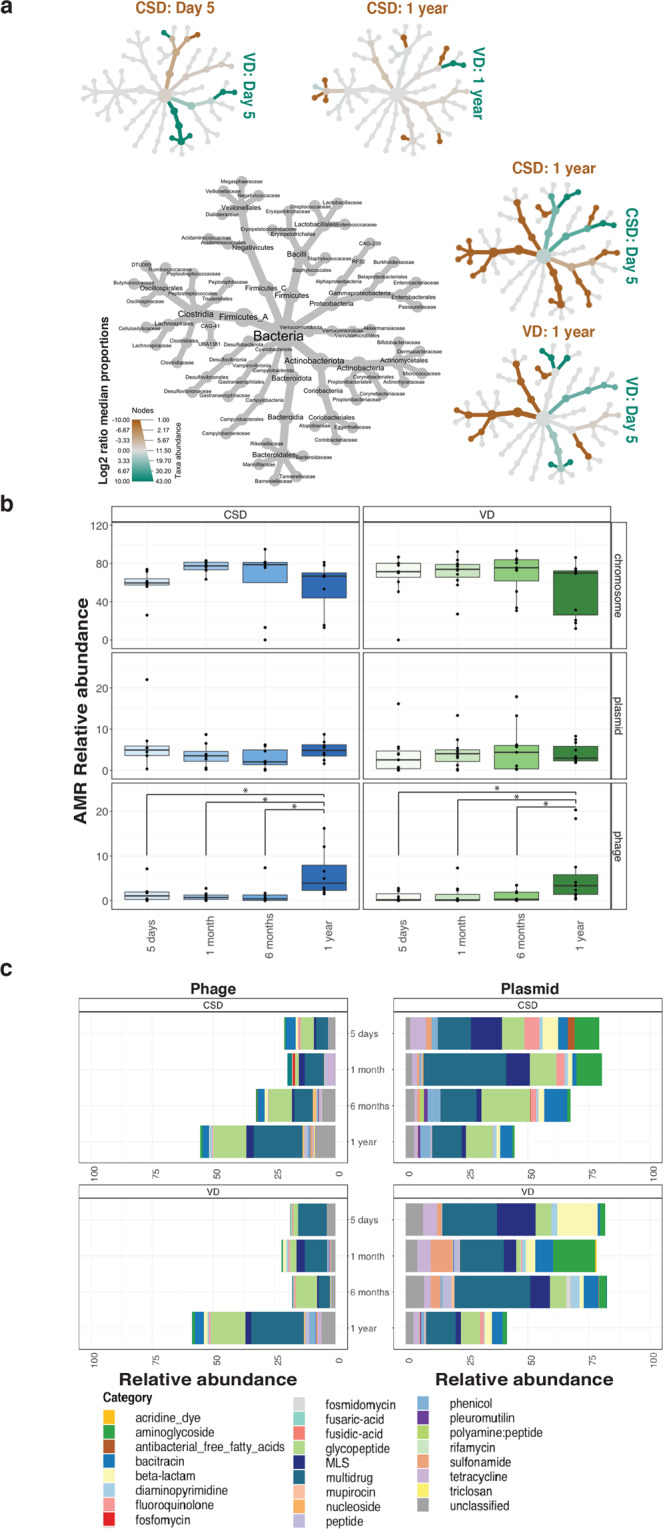
a Tree plots depicting the median proportion of AMR-associated taxa at day 5 after birth and at 1 year of age comparing the CSD and VD groups. Values used for plotting the trees are an average of all the samples within the group. b The relative abundances of AMR genes found on the bacterial chromosome, plasmids or phages at the different timepoints, ranging from day 5 after birth through to 1 year of age. CSD = blue (left panel), VD = green (right panel). Paired two-way ANOVA was used to assess significant differences; adjusted p-values are shown in the plot to depict significant differences. *p-value < 0.05. c Stacked bar plot depicting the AMR categories transmitted via phages and plasmids at all timepoints. Each colour in the plot is associated with a category listed in the legend on the right. The plot represents mean values for all samples in each group.
Comparing CSD with VD, we found that shortly after birth, i.e. at day 5, Bacilli contributing to AMR were enriched in CSD, while Bifidobacteriaceae, Bacteroidales, Eggerthellaceae, and Actinobacteria linked to AMR were more abundant in VD samples. At 1 year of age, other taxa including Burkholderiaceae, Ruminococcaceae and Bacteroidales were prevalent in CSD samples (Fig. 5a). In contrast, taxa including Enterobacteriales were enriched in VD compared to CSD at 1 year of age.
We compared samples from day 5 after birth versus 1 year of age within each birth mode group independently to differentiate between taxonomic groups contributing to AMR. Within CSD samples, we found that Enterobacteriales and Staphylococcaceae were enriched at day 5 after birth while major AMR contributors at 1 year of age were Lachnospiraceae, Bacteroidaceae, Actinobacteria and Oscillospirales. Conversely, within VD samples early AMR resistance was mainly attributed to the abundance of Bacteroidales, Lactobacillales, Propionibacteraceae and Enterobacteriaceae at day 5 after birth. Meanwhile, at 1 year of age, VD samples were enriched in taxa including Lachnospiraceae, Ruminococcaceae, Veillonellales and Eggerthellaceae with respect to contribution of AMR genes to the resistome (Fig. 5a). These data suggest that AMR genes are also encoded by commensals apart from pathogens which, in the context of the present study, sustain their presence throughout the first year of life.
Role of mobile genetic elements in antimicrobial resistance
Bacterial genomes have been fine-tuned over evolutionary timescales,51 potentially refining their defence mechanisms against various biocidal agents including chemicals. Aside from these, bacterial components such as MGEs are known to be potent factors in the spread of AMR52 and can transfer genes across distinct taxonomic clades. An example of such MGEs are plasmids as well as viruses including bacteriophages, which actively drive the transfer of genetic material.53 To determine the role of MGEs in conferring AMR in our neonate cohort, we analysed the genomic context of the AMR genes. The contigs were classified as chromosomal, plasmid, phage, ambiguous (those that could not be resolved) and unclassified. In this study, chromosomal sequences refer to the bacterial genome excluding plasmids, in accordance with the PlasFlow54 methodology. These criteria were used to assess the role of MGEs at all timepoints. The majority (average of ~75%) of the AMR genes were encoded on the bacterial chromosome (Fig. 5b). This phenomenon was prominent in the VD samples irrespective of sampling time point. On the other hand, the mean relative abundance of AMR genes encoded on plasmids (~5%) was marginally increased in the CSD group at both 5 days after birth and at 1 year of age. Overall, we found that phages encoded lower levels (1–3%) of AMR genes compared to the plasmids. However, we found that the relative abundances of phages encoding AMR were significantly increased after 1 year of age (Fig. 5b). Interestingly, we did not find any significant differences between the birth modes in relation to the virome profiles at any of the timepoints (FDR-adjusted p > 0.05, two-way ANOVA, Supplementary Fig. 7). However, a large proportion of the contigs were either ambiguous or unclassified but demonstrated an even distribution across all timepoints (Supplementary Fig. 8a). When ambiguous sequences mapping to both the bacterial chromosome and phages are included in the phage abundance metrics, it results in a higher abundance of AMR genes conferred by phage compared to plasmids (Supplementary Fig. 8b).
Distribution of AMR categories encoded by mobile genetic elements
Assessing AMR conferred by MGEs, we found that both plasmids and phages encoded genes conferring resistance to several classes of antibiotics (Fig. 5c). Though significant differences were not apparent, we found that phage-encoded AMR genes against vancomycin (glycopeptide) and numerous other antimicrobials were dominant in both birth mode groups. In addition, plasmids conferred resistance to diaminopyrimidine and bacitracin, as well as β-lactams, phenicol, MLS and tetracyclines (Fig. 5c). Strikingly, these data suggest that MGE-mediated AMR, encoded by phages, is a potential factor in conferring AMR or serve as a reservoir for antimicrobial resistance throughout the first years of life.
Phage-mediated horizontal gene transfer (HGT)
To understand phage-mediated horizontal gene transfer of AMR we analysed, in detail, phage contigs encoding AMR genes. We identified several genes that were horizontally transferred within the CSD and VD groups (Supplementary Data 4 and Supplementary Fig. 9). CSD samples (n = 3; C112, C113 and C119), exhibited HGT involving AMR genes including resistance to glycopeptide and multidrug (Fig. 6a–d). The majority (~88%) mapped to the bacterial chromosome. However, two genes encoding multidrug resistance were encoded by both chromosome and phage (C119: contig 2568, Supplementary Data 4). The contig was found to be a candidate prophage based on detailed inspection and was found to encode several genomic regions with prophage signatures flanking the multidrug resistance genes (Fig. 6f). In addition, the coverage of the contig across its entire length was more variable in the genomic regions where the prophage and AMR gene sequences were identified. Resolution of the taxa involved indicated HGT between the Intestinimonas butyriciproducens (GCA 003096335) and Clostridium bolteae (ATCC BAA 613 GCA 000154365) strains belonging to the Oscillospirallales and Lachnospirallales orders, respectively (Supplementary Fig. 10).
Fig. 6. Horizontal gene transfer (HGT) events.

Schematics depicting the a diaminopyrimidine resistance gene transfer between Blautia and Dorea in sample C112 (CSD). The lighter blue and green arrows represent genes localized on the forward and reverse strands, respectively. Red bars indicate matching regions between contigs with darker shades representing higher similarity. b HGT event transferring glycopeptide resistance between Clostridium_P and Clostridium in C113 (CSD). c–e Multidrug resistance genes were transferred horizontally between the following genera: Intestimonas and Clostridium, Ruminococcus and Roseburia, Enterococcus and Clostridium, in sample C119 (CSD). The timepoints at which the genomes were recovered and to which the HGT event corresponds are indicated next to the genera nomenclature. f Linear representation of ‘contig_2568’, which mapped to both bacterial chromosome and phage. Area plot (grey) and dot plot (red) indicating the coverage of the genomic regions. Triangles (red) indicate the prophage sequences (Supplementary Data 4) and their localisation coordinates along the contig, while the dark-blue triangle indicates the location of the multidrug AMR genes (n = 2) on the contig.
Discussion
Birth mode is postulated to represent a major factor in shaping earliest gut microbiome colonisation and the linked development of neonates especially in relation to the priming of the neonates’ immune system.3 Apart from birth mode, additional aspects such as diet and medical factors have been described to have a significant effect on neonate colonisation and succession.19 Whether or not such effects persist during the first year of life remains an essential question. Here, we performed an in-depth longitudinal analysis of the gut microbiome using high-resolution metagenomics on samples collected during the first few days postpartum through to the first year of age. We specifically assessed the pervasive effect of birth mode-dependent microbiome differences in relation to immune system priming and AMR. In addition, we analysed the contribution of mobile genetic elements and the role of horizontal gene transfer in conferring AMR.
Our previous findings8 along with the longer-term trends of the present study underpin the notion that persistent structural and functional differences exist in the gut microbiomes of neonates born by CSD. More specifically, our results agree with other studies which have highlighted a reduced abundance and colonisation by taxa such as Bifidobacterium and Bacteroides in CSD neonates.6,8,24,55,56 In addition, we found that the levels of Faecalibacterium prausnitzii were significantly elevated in VD infants after 1 year of age. F. prausnitzii is a highly abundant commensal in the human gut including those with higher levels of diversity and richness when compared to individuals following a Western lifestyle.57 Concurrently, this taxon has been reported to be reduced in the gut of patients with ulcerative colitis and Crohn’s disease,58 which in turn may be linked to it being a keystone taxon conferring anti-inflammatory properties in humans.59,60 Further studies are necessary to effectively understand the longer-term consequences of the differential abundance of F. prausnitzii in humans beyond the first year of age.
Our previously published study8 highlighted a higher potential for LPS-mediated immune priming in VD compared to CSD at day 5 after birth. Conversely, we found that LPS extracts from the CSD samples taken at 1 year of age resulted in significantly higher TNF-α levels compared to 5 days after birth. Our results indicate that a reduction in the earliest immune system priming through key immunogenic molecules occurs in CSD neonates. This might lead to persistent effects throughout the first year of life which, in turn, may explain the higher rates of immune system-linked diseases observed in CSD infants in later life including metabolic disorders11,55 and allergies.12,13 Along these lines, Jakobsson et al. previously showed that children born via CSD have reduced Th1 responses.4 Furthermore, other groups have reported that early-life immune system stimulation impacts immune-disorders including asthma,61 allergies,62 diabetes and IBD.63 In this context, our findings indicate that birth mode-dependent gut microbiota alterations affect the status of the immune system throughout the first year of life, and likely beyond. This in turn may explain immunological deficits linked to numerous chronic diseases for which a higher propensity is observed in individuals born by CSD.64
Birth mode-associated alterations of the gut microbiota may facilitate colonisation by opportunistic pathogens, including those encoding antimicrobial resistance.15 Functional analyses of our metagenomic data highlighted enrichments in carbapenem and phenazine biosynthesis genes in the VD group after the first year of life, potentially a consequence of endogenous gut bacteria-mediated resistance mechanisms against opportunistic pathogens in the gut. Both carbapenem and phenazine are known to be bacterial compounds that are used clinically in fighting Gram-positive and Gram-negative pathogens.65,66 This data suggests that the indigenous gut microbiota plays a crucial, early role in conferring colonisation resistance against pathogens. In addition, we found that CSD is associated with resistance against semi- and synthetic antibiotics as early as 5 days after birth. It is well established that mothers undergoing CSD are administered antibiotics to prevent nosocomial infections, as a prophylactic policy.42,67,68 Interestingly, we did not find resistance towards the antibiotic treatment administered to mothers in our cohort. It, however, remains plausible that the enrichment in AMR genes especially against phenicol, pleuromutilin and diaminopyrimidine classes at day 5 after birth in CSD neonatal samples is linked to the hospital environment including the actual caesarean section.
In conjunction with the observed differences in AMR between CSD and VD our study also highlights the potential mode of AMR transmission via mobile genetic elements including via plasmids and/or bacteriophages.69–71 Parnanen et al. reported the presence of AMR genes and MGEs in infant faecal samples at 1 and 6 months of age.72 Our findings agree with their results and expand on these by additionally providing data on the abundance of AMR genes and MGEs at 5 days after birth. Furthermore, we identified 27 categories of AMR genes and linked these to both bacterial taxonomy and MGEs. We found that both plasmids and phages encoded genes which confer resistance to several classes of antibiotics. Of all MGEs, plasmids conferred resistance to a variety of antimicrobial compounds. Furthermore, we found that glycopeptide and multidrug resistance were transferred via phages, in accordance with previous reports.73–75 We also found that horizontal gene transfer plays a critical role in the continued transmission of AMR during the first year of life. While ~88% of HGT occurred via canonical methods involving the bacterial chromosome and plasmids, we found that prophages contributed to multidrug resistance in one CSD sample (C119). We detected prophage signature sequences flanking two multidrug resistance genes, horizontally transferred between bacteria from two distinct orders. Our findings thereby highlight the role of prophages, typically thought to mediate AMR in humans pathogens including Staphylococcus aureus,76 Salmonella and shiga toxin-producing Escherichia coli,77 as mediators of HGT even among commensals. Intriguingly, HGT events in the VD samples did not indicate any AMR gene transmission. On the other hand, considering the smaller sample size, further studies with an increased power are needed to clarify the role of phage-mediated AMR resistance especially during the first few days of life.
The persistence of differences in early-life exposure is an important but challenging research question, not least because of the paucity of long-term, longitudinal studies ranging from immediately after birth until early childhood. Our findings imply that birth mode leads to persistent gut microbiota structural and functional differences. We acknowledge that the limited sample size and the lack of detailed dietary information cannot rule out other confounding factors such as the in utero and postpartum environments of the infants. However, and importantly, our data suggest that gut microbiota structural and functional effects may predispose infants delivered by CSD to delayed immune priming resulting in a deficiency in tolerance. Our results pave the way for future, rational interventions aimed at restoring key functional features of the microbiota. In this context, further studies including following the children over extended periods of time are needed to understand birth mode-mediated manifestations of disease. Concurrently, an important research direction which arises from our study centres on the role of the gut mobilome in conferring AMR and how this affects microbiome trajectories and linked phenotypic outcomes in humans. Considering current global efforts directed at limiting the emergence of antibiotic resistance,78 appreciation of the role of phages as an additional source of resistance may be necessary for success in reducing the overall burden of AMR in the future.
Methods
Ethics statement
All aspects concerning the recruitment and collection of mother-neonate pairs including handling, processing and storing of samples as well as data were approved by the Luxembourg Comité national d’éthique de recherche, under reference number 201110/06 and by the Luxembourg National Commission for Data Protection under reference number A005335/R000058. Prior to specimen collection, following a detailed consultation; written and informed consent was obtained from all mothers enrolled in the study.
Sample collection
Based on our previous study,8 the present study design aimed at testing the hypothesis that birth mode elicits longer-term functional microbiome changes which may impact neonatal health and development (with particular foci on antimicrobial resistance and lipopolysaccharide biosynthesis) and we performed the corresponding power analyses using data from our previous study. Founded on the increase in fold-change [caesarean section delivery (CSD) versus vaginal delivery (VD)] in antimicrobial resistance genes, a sample size calculation revealed a minimum number of four individual mother–infant pairs per group to achieve a power of 80% with a significance threshold of 5%. For the LPS-mediated functional cytokine measurements, we estimated a minimum sample size per group of six pairs based on a fold-change of 1.40x in TNF-α, i.e., a 40% difference of means between the samples (Supplementary Fig. 11). As previously published,8,39 we found that the functional microbiome differences provide clearer delineations when comparing groups than the typically reported taxonomic profiles. Based on our hypothesis, we focused on functional endpoints, in particular on the emergence and acquisition of AMR genes and the LPS-mediated immune stimulation. As per the results of the power analyses highlighting a minimum requirement of 6 mother–infant pairs per group, we further inflated the per-group sample size by 50% leading to a minimum of 9 mother–infant pairs per group. In the present study, babies delivered via caesarean (CSD, n = 11) and vaginal (VD, n = 9) deliveries were sampled during the first days of life and were followed-up at 1 month, 6 months and at 1 year of age (Supplementary Data 2). Samples were collected during follow-up visits into sterile plastic vials and immediately flash-frozen in liquid nitrogen. Faecal samples were stored until further processing at −80 °C.
Faecal processing and nucleic acid extraction
Genomic DNA was isolated from 50 mg of frozen stool samples aseptically weighed into sterile vials, prior to processing with the DNeasy PowerSoil Kit (Qiagen, Luxembourg) including an additional incubation step at 65 °C and milling, as described previously.8 All the study samples yielded sufficient DNA for metagenomic sequencing including artefact-curated metagenomic data as described previously8 for subsequent analyses. DNA extracted from all timepoints was thereafter stored at −80 °C until further use.
DNA sequencing
All DNA samples were subjected to random shotgun sequencing. Briefly, 250 ng of DNA was sheared using Bioruptor NGS (Diagenode, UCD300) with 30-s ON and 30-s OFF for 15 cycles. The sequencing libraries were prepared using TruSeq Nano DNA library preparation kit (Illumina, FC-121-4002) using the protocol provided with the kit. The libraries were prepared considering 350 bp average insert size. Prepared libraries were quantified using Qubit (Invitrogen) and the quality was checked on a Bioanalyzer (Agilient). Sequencing was performed on the NextSeq500 (Illumina) instrument using 2 × 150 bp read length at the LCSB Sequencing Platform.
Data processing for metagenomics, including genome reconstruction
Paired forward and reverse sequences were processed using the metagenomic workflow of the Integrated Meta-omic Pipeline79 (IMP). The metagenomic processing workflow includes pre-processing, assembly, genome reconstruction and functional annotation of genes based on custom databases in reproducible manner. Briefly, the adapter sequences were trimmed in the pre-processing step including the removal of human reads. Thereafter the de novo assembly was performed using the MEGAHIT (version 2.0) assembler.80 Default IMP parameters were retained for all samples. Subsequently, we used MetaBAT281 and MaxBin282 for binning in addition to an in-house binning methodology previously described.83 This involved ignoring ribosomal RNA sequences in kmer profiles, clustering from VizBin embeddings,84 using density-based non-hierarchical clustering algorithms and depth of coverage for genome reconstructions. The reconstructed genomes are hereafter referred to as bins or metagenome-assembled genomes (MAGs). We obtained a non-redundant set of MAGs using DASTool85 with a score threshold of 0.7 for downstream analyses.
Metagenomic taxonomic classification, virome and functional analyses
Trimmed and pre-processed read pairs were used as input to determine the microbial abundance and population genomic profiles based on the mOTUs86 (version 2) tool. Based on the marker genes in the mOTU2 database taxonomic profiling was performed. The relative abundances of the mOTUs were estimated using a minimum alignment length of 125 basepairs (bp), where the read counts were normalized to the gene length while also accounting for base coverage of the genes. This was done using the motus profile option with the built-in option (-c) for relative abundance values per samples. Simultaneously, to improve specificity and minimise false positives, a cut-off of seven genes that deviated from the median was used as an additional parameter to improve both sensitivity and precision. For the reconstructed MAGs, completeness and contamination were determined using CheckM,87 while the taxonomy for each MAG was assigned using the GTDB (Genome Taxonomy Database) toolkit (gtdb-tk)50 using the lineage_wf option and by using the fasta files as inputs for the MAGs.
For the analyses of functional potential from the assembled contigs, open-reading frames were predicted from the assembled contigs using a modified version of Prokka88 that includes Prodigal89 gene predictions for complete and incomplete open-reading frames. The identified genes were annotated with a hidden Markov models90 (HMM) approach, trained using an in-house database84 including all KO,41 TIGRFAM and SWISS-PROT91 groups and using hmmsearch from HMMER 3.1.92 Where multiple functional groups were assigned to genes, the best hits based on bit scores were selected. FeatureCounts93 was used to extract the number of reads per functional category, using the arguments -p and -O, thus yielding counts for each functional category. After the LPS-cytokine analysis, insufficient faecal sample for one of the CSD samples (C118) remained for metagenomic sequencing. Therefore, the sample was removed from subsequent metagenomic analyses. For the virome analyses, we used an iterative annotation method to recover microbial (bacterial and archaeal) viruses,94 and subsequently taxonomically annotated using a network-based classification protocol defined by Bolduc et al.95 Samples C109 was not included in the virome analyses due to viral contigs being below detection confidence thresholds.
Identification of antimicrobial resistance genes and association with mobile genetic elements
We used a deep-learning approach, DeepARG,43 to predict and identify AMR genes within our metagenomic data. The output from Prokka, i.e. the translated fasta sequence files for all open-reading frames, was used as input for the AMR analyses. AMR genes were collapsed into categories based on the Comprehensive Antibiotic Resistance Database (CARD)44 and identified using DeepARG. Thereafter, the relative abundance of the AMR genes was calculated using the Rnum_Gi method described by Hu et al.96
Identified AMR genes and their categories were consecutively linked to associated bacterial taxonomy using the metagenomic bin classification. Furthermore, AMR genes were linked to predicted mobile genetic elements (MGEs; phages and plasmids) to identify probable transmission of AMR between taxa. For the identification of plasmids in the metagenomic data, PlasFlow54 was used with a threshold for filtering set to 0.7. Simultaneously, DeepVirFinder97 and VirSorter98 were used to identify phage sequences within the VD and CSD groups. Predictions from both these tools were subsequently merged to obtain a comprehensive catalogue of phage sequences. For the prediction of phage sequences, the DeepVirFinder thresholds for filtering were set at a p-value of < 0.05 and a score of 0.7, while for VirSorter the category 1 and 2 predictions were used for downstream analyses. To link both the MGEs and the taxonomy to the AMR genes, we mapped the genes to assembled contigs, followed by identifying the corresponding bins (MAGs) to which the contigs belonged. By considering all different predictions of MGEs, a final classification was made based on the genomic contexts of the AMR genes encoded on plasmids, phages or chromosomes, including classification of those that could not be resolved (ambiguous). Those AMR genes that could not be assigned to either the MGEs or bacterial chromosomes were further referred to as unclassified genomic signatures. Certain AMR genes were encoded on both the bacterial chromosome and phage genomes (Supplementary Data 4). In such cases, we recorded the encoded AMR gene as being ambiguous. The confirmation of AMR genes and their associated mode of transfer was performed manually alongside the mapping of identical 1 Kbp flanking regions, via the MetaCHIP analyses pipeline.99 Briefly, groups of genes among all input MAGs with maximum average identity were considered putative HGT genes. To validate the predicted candidates, a pairwise BLASTN was used to assess each pair of flanking regions of 10 Kbp. Visual representations of the genomic regions were extracted alongside the results for visual interpretation and inspection. Coverage of the genomic regions was additionally assessed through the IGV viewer using the bam file, and manually plotted based on per base coverage statistics for the latter.
LPS isolation and in vitro immunostimulation for cytokine profiling
From the 1 year of age time point, 150 mg faecal samples were weighed aseptically and lipopolysaccharide (LPS) was extracted alongside an extraction blank to serve as a negative control. We also used an in-house pure culture of E. coli, from which extracted LPS was used as a positive control. To maximise yields, the samples were divided into triplicates, i.e. 50 mg per vial, prior to LPS extraction using the hot phenol-water protocol as previously described.8 After extractions the triplicates from each sample were pooled and quantified using an endotoxin-detection assay (Endolisa, #609033, Hyglos GmbH, Germany). All samples produced sufficient quantities of LPS. The purified LPS was used to stimulate monocyte-derived dendritic cells (MoDCs). Briefly, primary human monocytes were derived from blood samples from four healthy donors obtained through the Luxembourg Red Cross. The monocytes were further differentiated into MoDCs, in RPMI 1640 medium (ThermoFisher Scientific) supplemented with 10% foetal bovine serum (ThermoFisher Scientific), 20 ng ml−1 of granulocyte-macrophage colony-stimulating factor (Peprotech, London, UK), 20 ng ml−1 IL-4 (Peprotech) and 1% penicillin–streptomycin (Invitrogen). Subsequently, the immunostimulatory potential of the LPS fractions isolated from the 1 year of age faecal samples was determined. For this, MoDCs were treated with LPS extracts from VD and CSD samples. The amount of LPS from each sample that was used to stimulate the MoDCs was adjusted as described by Wampach et al.8 Briefly, the MoDCs were stimulated with 7.5 µl/well of LPS while a positive control was established using 15 EU/well LPS isolated from E. coli, and a negative control was set up by incubating MoDCs with 7.5 µl/well of the LPS extraction blank. For the in vitro stimulation, the amount of MoDCs was 1 × 105 cells/well. Treatments were performed on cells from all the healthy donor-derived samples, and analysed for the presence of pro- and anti-inflammatory cytokines (TNF-α, IL-8, IL-18, IL-1β, IL-12 and IL-10) using both Human Instant and uncoated ELISA kits (ThermoFisher Scientific).
Data analysis
All figures for the study including visualizations derived from the taxonomic, functional and cytokine profiling were created using version 3.6 of the R statistical software package.100 DESeq2101 and Wilcoxon rank-sum tests with FDR-adjustments for multiple testing were used to assess significant differences for the AMR and taxonomic analyses whereas a paired two-way ANOVA (analysis of variance) within the nlme package was used for identifying statistically significant differences in the cytokine profiles. Volcano plots were generated using the EnhancedVolcano package.102 Corrplots were generated using the corrgram package developed for R.103 The metacoder104 package was used to visualize the AMR-linked taxonomy in R.
Supplementary information
Acknowledgements
We are grateful to all the parents and neonates who participated in the study. We thank the dedicated clinical staff and neonatologists of the paediatric clinic and gynaecologists at the CHL for participant recruitment and sample collection, especially Alain Noirhomme and all involved study nurses of the Clinical and Epidemiological Investigation Centre (CIEC) who performed sample and data collection at the CHL and at home as well as the scientific staff of the IBBL for sample storage. The experiments presented in this paper were carried out using the HPC facilities of the University of Luxembourg. We are thankful for the insightful conversations about AMR assays and immune profiling with Dr. Camille-Martin Gallausiaux, and for the assistance of Audrey Frachet, Lea Grandmougin, Annegrat Daujemont and Laura Lebrun (LCSB) for laboratory support. We are also grateful for the feedback on the manuscript by Dr. Cedric Christian Laczny. The present work was partially financed by the Fondation André et Henriette Losch. It was further supported by an ATTRACT programme grant (ATTRACT/A09/03) and CORE programme grants (CORE/15/BM/104040 and CORE/C15/SR/10404839) to P.W. and (CORE Junior/14/BM/8066232) to J.V.F., Aide à la Formation Recherche grants to L.W. (AFR PHD-2013-5824125) and S.N. (AFR PHD-2014-1/7934898), all funded by the Luxembourg National Research Fund (FNR). P.W. acknowledges the European Research Council (ERC-CoG 863664). L.dN., P.W. and P.M. were supported by the Luxembourg National Research Fund PRIDE17/11823097. S.B.B. was supported by the Synergia grant (CRSII5_180241) through the Swiss National Science Foundation (in collaboration with Dr. Tom Battin at EPFL, Switzerland). A.H.-B. was funded by the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig of the German Research Foundation (FZT 118 - 202548816). Sample collection, processing and storage were co-funded by the Integrated BioBank of Luxembourg under the Personalised Medicine Consortium Diabetes programme.
Author contributions
S.B.B. and L.dN. participated in the study design and performed the biomolecular extractions, and analyses including cytokine, metagenomic, including taxonomy, functional, AMR and horizontal gene transfer. J.H. along with L.dN. prepared the monocyte-derived dendritic cells, while J.H. and S.B.B. performed the cytokine ELISAs and subsequent data analyses. L.W. was instrumental in identifying all samples and previously sequenced metagenomic data. J.F. set up immune profiling protocols. P.M. and A.H.-B. were involved in the curation, trimming, assembly and annotation of the metagenomic data. R.H. was instrumental in sequencing the samples for metagenomics. C.dB. and P.W. conceived the study, participated in its design, were involved in data interpretation and coordinated the study. S.B.B., L.dN. and P.W. wrote the manuscript. All authors read and approved the final manuscript.
Data availability
The sequencing data and the MAGs generated during the current study are available from NCBI under bioproject accession number PRJNA595749. Supplementary Data 1 lists the taxonomic classifications of the carbapenem biosynthesis KEGG pathway identified within the MAGs. A reporting summary for this article is available as a Supplementary Information file (Supplementary Data 2). Supplementary Data 3 provides the clinical characteristics of all babies including details on diet, antibiotic and growth tracking. A description of the HGT events between reconstructed genomes and the manual validation of phage-conferred AMR is available in Supplementary Data 4, while the accession numbers for the Gasparrini et al.45 sequence data are listed in Supplementary Data 5. Legends for the supplementary figures are provided in the Supplementary material file. A description of the AMR and HGT analyses including pre-processing steps along with the scripts and config files can be found at GitLab: https://git-r3lab.uni.lu/susheel.busi/cosmic2.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Susheel Bhanu Busi, Laura de Nies.
Supplementary information
The online version contains supplementary material available at 10.1038/s43705-021-00003-5.
References
- 1.Boerma T, et al. Global epidemiology of use of and disparities in caesarean sections. The Lancet. 2018;392:1341–1348. doi: 10.1016/S0140-6736(18)31928-7. [DOI] [PubMed] [Google Scholar]
- 2.Betrán AP, et al. The increasing trend in Caesarean section rates: global, regional and national estimates: 1990–2014. PLoS ONE. 2016;11:e0148343. doi: 10.1371/journal.pone.0148343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gensollen T, et al. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–533. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakobsson HE, et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by Caesarean section. Gut. 2014;63:559–566. doi: 10.1136/gutjnl-2012-303249. [DOI] [PubMed] [Google Scholar]
- 5.Wang S, et al. Maternal vertical transmission affecting early-life microbiota development. Trends Microbiol. 2019;28:28–45. doi: 10.1016/j.tim.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Guittar J, et al. Trait-based community assembly and succession of the infant gut microbiome. Nat. Commun. 2019;10:1–11. doi: 10.1038/s41467-019-08377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandall J, et al. Short-term and long-term effects of caesarean section on the health of women and children. The Lancet. 2018;392:1349–1357. doi: 10.1016/S0140-6736(18)31930-5. [DOI] [PubMed] [Google Scholar]
- 8.Wampach L, et al. Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nat. Commun. 2018;9:5091. doi: 10.1038/s41467-018-07631-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korpela K, et al. Fucosylated oligosaccharides in mother’s milk alleviate the effects of caesarean birth on infant gut microbiota. Sci. Rep. 2018;8:1–7. doi: 10.1038/s41598-018-32037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asnicar, F. et al. Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems2, e00164-16 (2017). [DOI] [PMC free article] [PubMed]
- 11.Bouhanick, B. et al. Mode of delivery at birth and the metabolic syndrome in midlife: the role of the birth environment in a prospective birth cohort study. BMJ Open4, e005031 (2014). [DOI] [PMC free article] [PubMed]
- 12.Magne, F. et al. The Elevated Rate of Cesarean Section and Its Contribution to Non-Communicable Chronic Diseases in Latin America: The Growing Involvement of the Microbiota. Front Pediatr.5, e192 (2017). [DOI] [PMC free article] [PubMed]
- 13.Loo EXL, et al. Associations between caesarean delivery and allergic outcomes: Results from the GUSTO study. Ann. Allergy Asthma Immunol. 2017;118:636–638. doi: 10.1016/j.anai.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamburini S, et al. The microbiome in early life: implications for health outcomes. Nat. Med. 2016;22:713–722. doi: 10.1038/nm.4142. [DOI] [PubMed] [Google Scholar]
- 15.Shao Y, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. 2019;574:117–121. doi: 10.1038/s41586-019-1560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stearns JC, et al. Intrapartum antibiotics for GBS prophylaxis alter colonization patterns in the early infant gut microbiome of low risk infants. Sci. Rep. 2017;7:1–9. doi: 10.1038/s41598-017-16606-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivas MN, et al. The microbiome in asthma. Curr. Opin. Pediatr. 2016;28:764–771. doi: 10.1097/MOP.0000000000000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez KA, et al. Increased weight gain by C-section: functional significance of the primordial microbiome. Sci. Adv. 2017;3:eaao1874. doi: 10.1126/sciadv.aao1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäckhed F, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Muinck EJde, Trosvik P. Individuality and convergence of the infant gut microbiota during the first year of life. Nat. Commun. 2018;9:1–8. doi: 10.1038/s41467-018-04641-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferretti P, et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe. 2018;24:133–145.e5. doi: 10.1016/j.chom.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wopereis H, et al. The first thousand days—intestinal microbiology of early life: establishing a symbiosis. Pediatr. Allergy Immunol. 2014;25:428–438. doi: 10.1111/pai.12232. [DOI] [PubMed] [Google Scholar]
- 23.Baumann-Dudenhoeffer AM, et al. Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nat. Med. 2018;24:1822–1829. doi: 10.1038/s41591-018-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bokulich NA, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016;8:343ra82. doi: 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller NT, et al. The infant microbiome development: mom matters. Trends Mol. Med. 2015;21:109–117. doi: 10.1016/j.molmed.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stokholm J, et al. Cesarean section changes neonatal gut colonization. J. Allergy Clin. Immunol. 2016;138:881–889.e2. doi: 10.1016/j.jaci.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 27.Keoning JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl Acad. Sci. USA. 2011;15:4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heintz-Buschart A, Wilmes P. Human gut microbiome: function matters. Trends Microbiol. 2018;26:563–574. doi: 10.1016/j.tim.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Vatanen T, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–853. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jennewein MF, et al. Neonate-omics: charting the unknown immune response in early life. Cell. 2018;174:1051–1053. doi: 10.1016/j.cell.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Romano-Keeler J, Weitkamp J-H. Maternal influences on fetal microbial colonization and immune development. Pediatr. Res. 2015;77:189–195. doi: 10.1038/pr.2014.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spencer SJ, et al. Early-life immune challenge: defining a critical window for effects on adult responses to immune challenge. Neuropsychopharmacology. 2006;31:1910–1918. doi: 10.1038/sj.npp.1301004. [DOI] [PubMed] [Google Scholar]
- 33.Torow N, Hornef MW. The neonatal window of opportunity: setting the stage for life-long host-microbial interaction and immune homeostasis. J. Immunol. 2017;198:557–563. doi: 10.4049/jimmunol.1601253. [DOI] [PubMed] [Google Scholar]
- 34.Gopalakrishna KP, et al. Maternal IgA protects against the development of necrotizing enterocolitis in preterm infants. Nat. Med. 2019;25:1110–1115. doi: 10.1038/s41591-019-0480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Muñoz ME, et al. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. 2017;5:48. doi: 10.1186/s40168-017-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olin A, et al. Stereotypic immune system development in newborn children. Cell. 2018;174:1277–1292.e14. doi: 10.1016/j.cell.2018.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levan SR, et al. Elevated faecal 12,13-diHOME concentration in neonates at high risk for asthma is produced by gut bacteria and impedes immune tolerance. Nat. Microbiol. 2019;4:1851–1861. doi: 10.1038/s41564-019-0498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravi A, et al. The commensal infant gut meta-mobilome as a potential reservoir for persistent multidrug resistance integrons. Sci. Rep. 2015;5:1–11. doi: 10.1038/srep15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wampach, L. et al. Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Front. Microbiol. 10.3389/fmicb.2017.00738 (2017). [DOI] [PMC free article] [PubMed]
- 40.Miquel S, et al. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013;16:255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salguero MV, Al-Obaide MAI, Singh R, Siepmann T, Vasylyeva TL. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019;18:3461–3469. doi: 10.3892/etm.2019.7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arango-Argoty G, et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 2018;6:23. doi: 10.1186/s40168-018-0401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alcock, B. P. et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 10.1093/nar/gkz935 (2020). [DOI] [PMC free article] [PubMed]
- 45.Gasparrini A, et al. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019;4:2285–2297. doi: 10.1038/s41564-019-0550-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fong, I. W. et al. Antimicrobial Resistance in the 21st Century (Springer, 2018).
- 47.Greenwood, D. Antibiotic and Chemotherapy 9th edn (Elsevier, 2010).
- 48.Paukner, S. & Riedl, R. Pleuromutilins: potent drugs for resistant bugs—mode of action and resistance. Cold Spring Harb. Perspect. Med. 7, a027110 (2017). [DOI] [PMC free article] [PubMed]
- 49.Wright PM, et al. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. Engl. 2014;53:8840–8869. doi: 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaumeil, P.-A. et al. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020). [DOI] [PMC free article] [PubMed]
- 51.Martínez-Cano, D. J. et al. Evolution of small prokaryotic genomes. Front. Microbiol. 5, e742 (2015). [DOI] [PMC free article] [PubMed]
- 52.Woodford N, et al. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011;35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 53.Gao, N. L. et al. Prokaryotic genome expansion is facilitated by phages and plasmids but impaired by CRISPR. Front. Microbiol. 10, e2254 (2019). [DOI] [PMC free article] [PubMed]
- 54.Krawczyk PS, et al. PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018;46:e35. doi: 10.1093/nar/gkx1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reyman M, et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl Acad. Sci. USA. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rinninella, E. et al. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms7, e14 (2019). [DOI] [PMC free article] [PubMed]
- 58.Machiels K, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 59.Sokol H, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rossi O, et al. Faecalibacterium prausnitzii A2-165 has a high capacity to induce IL-10 in human and murine dendritic cells and modulates T cell responses. Sci. Rep. 2016;6:18507. doi: 10.1038/srep18507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lloyd CM, Hawrylowicz CM. Regulatory T cells in asthma. Immunity. 2009;31:438–449. doi: 10.1016/j.immuni.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vuillermin PJ, et al. Microbial exposure, interferon gamma gene demethylation in naïve T-cells, and the risk of allergic disease. Allergy. 2009;64:348–353. doi: 10.1111/j.1398-9995.2009.01970.x. [DOI] [PubMed] [Google Scholar]
- 63.Zhuang L, et al. Intestinal microbiota in early life and its implications on childhood health. Genomics Proteomics Bioinformatics. 2019;17:13–25. doi: 10.1016/j.gpb.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keag OE, et al. Long-term risks and benefits associated with cesarean delivery for mother, baby, and subsequent pregnancies: Systematic review and meta-analysis. PLoS Med. 2018;15:e1002494. doi: 10.1371/journal.pmed.1002494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coulthurst SJ, et al. Regulation and biosynthesis of carbapenem antibiotics in bacteria. Nat. Rev. Microbiol. 2005;3:295–306. doi: 10.1038/nrmicro1128. [DOI] [PubMed] [Google Scholar]
- 66.Pierson LS, Pierson EA. Metabolism and function of phenazines in bacteria: impacts on the behavior of bacteria in the environment and biotechnological processes. Appl. Microbiol. Biotechnol. 2010;86:1659–1670. doi: 10.1007/s00253-010-2509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vangay P, et al. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. 2015;17:553–564. doi: 10.1016/j.chom.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stokholm J, et al. Prevalence and predictors of antibiotic administration during pregnancy and birth. PLoS ONE. 2013;8:e82932. doi: 10.1371/journal.pone.0082932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gómez-Gómez C, et al. Infectious phage particles packaging antibiotic resistance genes found in meat products and chicken feces. Sci. Rep. 2019;9:1–11. doi: 10.1038/s41598-019-49898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown-Jaque M, et al. Transfer of antibiotic-resistance genes via phage-related mobile elements. Plasmid. 2015;79:1–7. doi: 10.1016/j.plasmid.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Cruz Fdela, Davies J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 2000;8:128–133. doi: 10.1016/S0966-842X(00)01703-0. [DOI] [PubMed] [Google Scholar]
- 72.Pärnänen K, et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-06393-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang, M. et al. Metagenomic insights into the contribution of phages to antibiotic resistance in water samples related to swine feedlot wastewater treatment. Front. Microbiol. 9, e2474 (2018). [DOI] [PMC free article] [PubMed]
- 74.Bearson, B. L. et al. The agricultural antibiotic carbadox induces phage-mediated gene transfer in Salmonella. Front. Microbiol. 5, e52 (2014). [DOI] [PMC free article] [PubMed]
- 75.Torres-Barceló, C. The disparate effects of bacteriophages on antibiotic-resistant bacteria. Emerg. Microbes Infect. 7, e168 (2018). [DOI] [PMC free article] [PubMed]
- 76.Haaber J, et al. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat. Commun. 2016;7:1–8. doi: 10.1038/ncomms13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Colavecchio, A. et al. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family—a review. Front. Microbiol. 8, e1108 (2017). [DOI] [PMC free article] [PubMed]
- 78.Hoffman SJ, et al. Strategies for achieving global collective action on antimicrobial resistance. Bull. World Health Organ. 2015;93:867–876. doi: 10.2471/BLT.15.153171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Narayanasamy S, et al. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016;17:260. doi: 10.1186/s13059-016-1116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li D, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinforma. Oxf. Engl. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 81.Kang DD, et al. MetaBAT2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu YW, et al. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 83.Heintz-Buschart A, et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2017;2:16180. doi: 10.1038/nmicrobiol.2016.180. [DOI] [PubMed] [Google Scholar]
- 84.Laczny CC, et al. VizBin—an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome. 2015;3:1. doi: 10.1186/s40168-014-0066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sieber MKC, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018;3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Milanese A, et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019;10:1–11. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parks DH, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinforma. Oxf. Engl. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 89.Hyatt D, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoon B-J. Hidden Markov models and their applications in biological sequence analysis. Curr. Genomics. 2009;10:402–415. doi: 10.2174/138920209789177575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eddy SR. Accelerated profile HMM searches. PLOS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liao Y, et al. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 94.Kieft K, Zhou Z, Anantharaman K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome. 2020;8:90. doi: 10.1186/s40168-020-00867-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolduc B, et al. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ. 2017;5:e3243. doi: 10.7717/peerj.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu Y, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013;4:1–7. doi: 10.1038/ncomms3151. [DOI] [PubMed] [Google Scholar]
- 97.Ren, J. et al. Identifying viruses from metagenomic data using deep learning. Quant Biol. 8, 64–77 (2020). [DOI] [PMC free article] [PubMed]
- 98.Roux S, et al. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015;3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song W, et al. MetaCHIP: community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches. Microbiome. 2019;7:36. doi: 10.1186/s40168-019-0649-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Team, R. C. R.: A Language and Environment for Statistical Computing (2013).
- 101.Love, M. I. et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, e550 (2014). [DOI] [PMC free article] [PubMed]
- 102.Blighe, K. et al. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. GitHub Repository, https://github.com/kevinblighe/EnhancedVolcano (2019).
- 103.Chambers, J. M. Graphical Methods for Data Analysis (CRC Press, 2018).
- 104.Foster ZSL, et al. Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLOS Comput. Biol. 2017;13:e1005404. doi: 10.1371/journal.pcbi.1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data and the MAGs generated during the current study are available from NCBI under bioproject accession number PRJNA595749. Supplementary Data 1 lists the taxonomic classifications of the carbapenem biosynthesis KEGG pathway identified within the MAGs. A reporting summary for this article is available as a Supplementary Information file (Supplementary Data 2). Supplementary Data 3 provides the clinical characteristics of all babies including details on diet, antibiotic and growth tracking. A description of the HGT events between reconstructed genomes and the manual validation of phage-conferred AMR is available in Supplementary Data 4, while the accession numbers for the Gasparrini et al.45 sequence data are listed in Supplementary Data 5. Legends for the supplementary figures are provided in the Supplementary material file. A description of the AMR and HGT analyses including pre-processing steps along with the scripts and config files can be found at GitLab: https://git-r3lab.uni.lu/susheel.busi/cosmic2.