

Abstract
Enzymes with hydroxymethylpyrimidine/phosphomethylpyrimidine kinase activity (HMPPK) are essential in the vitamin B1 (thiamine pyrophosphate) biosynthesis and recycling pathways. In contrast, enzymes with pyridoxal kinase activity (PLK) produce pyridoxal phosphate (vitamin B6), an essential cofactor for various biochemical reactions. In the ATP‐dependent vitamin kinases family, the members of PLK/HMPPK‐like subfamily have both enzymatic activities. It has been proposed that the promiscuous PLK activity of ancestral HMPPK enzymes could have been the starting point for this activity. In earlier work, we reconstructed the ancestral sequences of this family and characterized the substrate specificity of the common ancestor between PLK/HMPPK‐like and HMPPK enzymes (AncC). From these studies, the Gln45Met mutation was proposed as a critical event for the PLK activity emergence. Here, we crystallize and determine the AncC structure by X‐ray crystallography and assess the role of the Gln45Met mutation by site‐directed mutagenesis. Kinetic characterization of this mutant shows a significant increase in the PL affinity. Through molecular dynamics simulation and MM/PBSA calculations some residues, important for substrate interactions and catalysis, were identified in the wild type and in the mutated ancestor. Interestingly, a strong epistatic interaction responsible for the evolutionary pathway of the PLK activity in PLK/HMPPK‐like enzymes was revealed. Also, other putative mutations relevant to PLK activity in modern PLK/HMPPK‐like enzymes were identified.
Keywords: ancestral enzymes, enzyme promiscuity, epistatic interaction, hydroxymethyl pyrimidine kinases, pyridoxal kinases
Short abstract
PDB Code(s): 7L07;
Abbreviations
- AncB
PLK/HMPPK‐like ancestral protein
- AncS
HMPPK ancestral protein
- ATP
adenosine triphosphate
- HMP
4‐amino‐5‐hydroxymethyl‐2‐methyl pyrimidine
- HMP‐P
4‐amino‐5‐phosphohydroxymethyl‐2‐methyl pyrimidine
- HMPPK
vitamin kinase family members able to catalyze HMPK and HMPPK reactions
- HMP Kinase
4‐amino‐5‐hydroxymethyl‐2‐methyl pyrimidine kinase activity
- HMPP Kinase
4‐amino‐5‐phosphohydroxymethyl‐2‐methyl pyrimidine kinase activity
- MM/PBSA
Molecular Mechanics Poisson‐Boltzmann Surface Area
- NPT
constant number of atom, pressure and temperature
- PL
pyridoxal
- PLP
pyridoxal phosphate
- PLK
pyridoxal kinaseR
- PLK/HMPPK‐like
vitamin kinase family members able to catalyze PLK and HMPK reactions
- THZ
5‐(2‐hydroxyethyl)‐4‐methylthiazole
- THZ Kinase
hydroxyethylthiazole kinase activity
1. INTRODUCTION
Enzyme promiscuity has received considerable attention in recent years because these catalytic activities, irrelevant in a given physiological context, may be the starting point for new enzyme activities. 1 , 2 Promiscuous activities can become relevant if the physiological context changes are accompanied by mutations that enhance these secondary activities. This situation can result in the appearance of a bifunctional enzyme with two physiologically relevant catalytic activities. In some cases, the new activity is incompatible with the native function, and then the native activity is lost. 3 However, bifunctionality has been proposed as the most probable mechanism in the appearance of new enzyme functions, since after a gene duplication event, the ancestral function will be preserved while the duplicate gene can freely evolve to a new function. 1 Then, the existence of bifunctional enzymes represents a valuable tool for exploring the evolutive pathway and the structural determinants involved in the appearance of new enzyme functions. The ribokinase superfamily constitutes an interesting model for the study of the evolution of enzyme functions due to the highly conserved catalytic machinery and the great diversity of substrates that their members display. 4 , 5 , 6 The ATP‐dependent vitamin kinase family belongs to the ribokinase superfamily and, unlike the members of the other two families (ribokinase and ADP‐dependent family), its members lack a small domain that functions as the active site lid in the other families. Moreover, members of the ATP‐dependent vitamin kinase show diverse activities, like pyridoxal kinase (PLK), hydroxyethylthiazole kinase (THZ Kinase), hydroxymethylpyrimidine kinase (HMP Kinase) and phosphomethylpyrimidine kinase activity (HMPP Kinase).
Enzymes with PLK activity catalyze the phosphorylation of pyridoxal (PL), producing pyridoxal phosphate (PLP) (Figure 1), the active form of vitamin B6, 7 which is an essential cofactor for a variety of biochemical reactions including synthesis, catabolism, and amino acid interconversion. 8 Enzymes with hydroxymethylpyrimidine kinase activity (Figure 1) are crucial to the vitamin B1 (thiamine pyrophosphate) biosynthesis and recycling pathways. Based on its evolutionary history, a new classification was recently proposed for these enzymes, which recognizes two groups. 9 One of them, encoded by the thiD gene and denoted “HMPPK,” catalyzes two ATP‐dependent phosphorylation reactions, first the phosphorylation of 4‐amino‐5‐hydroxymethyl‐2‐methylpyrimidine (HMP) to produce HMP phosphate (HMP‐P) (EC 2.7.1.49), and then this product is again phosphorylated to produce HMP pyrophosphate (HMP‐PP) 9 , 10 (EC 2.7.4.7). Members of the other group, named PLK/HMPPK‐like, can catalyze either the phosphorylation of pyridoxal (EC 2.7.1.35) or the phosphorylation of HMP 9 , 11 , 12 (EC 2.7.1.49), but are not able to catalyze HMP‐P phosphorylation.
FIGURE 1.
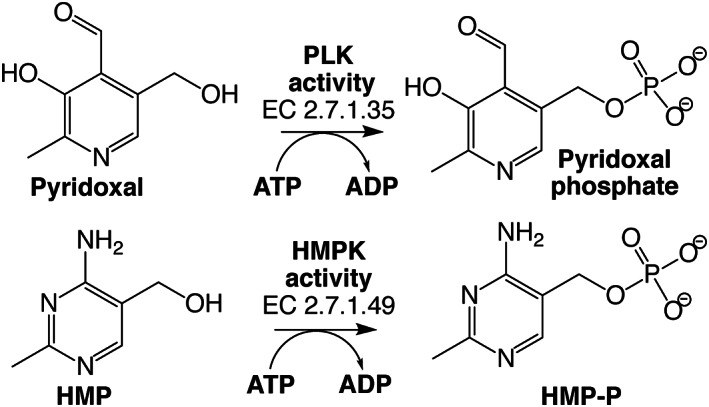
Reactions catalyzed by PLK/HMPPK‐like enzymes. Pyridoxal kinase (PLK) activity phosphorylates pyridoxal using ATP as the phosphoryl donor to form pyridoxal phosphate, the active form of vitamin B6. While hydroxymethyl pyrimidine kinase (HMPK) activity also uses ATP to phosphorylate HMP, producing HMP‐P, an intermediate of vitamin B1 synthesis
A structural comparison of the PLK/HMPPK‐like enzyme's active site from Bacillus subtilis (PDB 2I5B) with those from the HMPPKs from Salmonella Typhimurium (PDB 1JXI) and Thermus thermophilus 11 (PDB 1UB0) reveals a very similar active‐site pocket with near 80% identical residues. In contrast, its comparison to canonical PLKs from Escherichia coli, encoded by the pdxK (PDB 2DDW) or pdxY gene (PDB 1TD2), shows only 20% of conservation of the active site residues, despite sharing the fold. This suggests that the PLK activity of the PLK/HMPPK‐like enzymes could arise by a convergent evolution event, not related to this family's canonical PLK activity.
By employing the ancestral reconstruction methodology, we inferred the sequence of three ancestors of this family: the ancestor of the HMPPK enzymes (AncS), the ancestor of the PLK/HMPPK‐like enzymes (AncB), and the last common ancestor of both groups (AncC) (Figure 2). The resurrection and kinetic characterization of AncC, showed that the ancestral enzyme was specific for HMP, although it presents promiscuity for PL, having a low affinity for this substrate. 13
FIGURE 2.
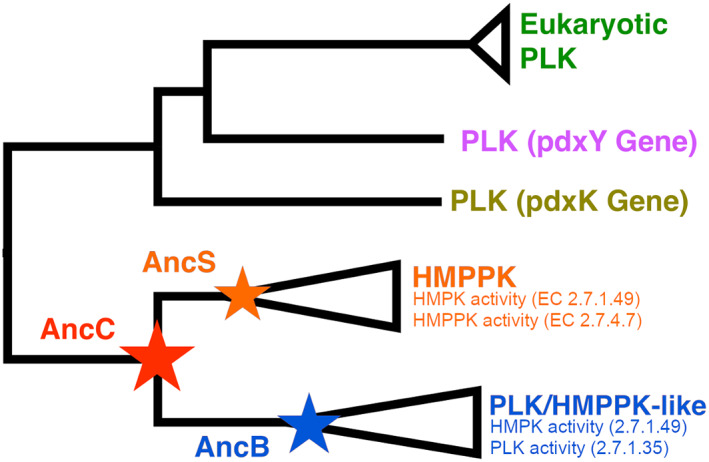
Phylogenetic relationship of canonical PLK (pdxK and pdxY genes), HMPPK and PLK/HMPPK‐like enzymes. Reconstructed ancestral sequences for the family are shown with stars; red for AncC, orange for AncS, and blue for AncB
Ancestral enzyme reconstruction has been successfully used to obtain information about the evolutionary history of protein families like ATP‐dependent vitamin kinases. It has also been proposed as a tool for protein engineering due to the high stability and soluble expression of ancestral proteins. 14 In this work, taking advantage of these characteristics, the ancestral enzyme AncC was crystallized, and its structure was determined by X‐ray crystallography. Also site‐directed mutation in AncC of a key residue for the appearance of PLK activity and kinetic characterization employing HMP and PL as substrates were performed. Additionally, using molecular dynamics simulations and MM/PBSA calculations, the enzyme‐substrate interactions in the wild type and in the mutated ancestor were evaluated. In addition to key residues for binding and catalysis with HMP/PL, a strong epistatic interaction responsible for the evolutionary pathway of the PLK activity in PLK/HMPPK‐like enzymes, and a putative mutation relevant for PLK activity in modern PLK/HMPPK‐like enzymes, were identified.
2. MATERIALS AND METHODS
2.1. Protein purification
Recombinant ancestral protein AncC was expressed in E. coli BL21(DE3) and purified by Ni‐NTA affinity chromatography. Cultures were grown in LB broth at 37°C until OD 0.6 was reached. Protein expression was induced with 1 mM IPTG at 37°C overnight. Afterward, the cells were pelleted, sonicated and centrifuged to isolate the soluble fraction. The supernatant was loaded onto a Ni‐NTA affinity chromatography purification and elluted with a linear gradient between 20 and mM imidazole. Fractions with enzyme activity were collected, dialyzed and concentrated in storage buffer (50 mM Tris‐HCl pH 7.5, 5% glycerol and 10 mM de β‐mercaptoethanol).
2.2. Crystallization and diffraction
Crystallization conditions for AncC were screened through high‐throughput assays performed by the sitting drop vapor‐diffusion method by mixing 0.8 μl protein (9 mg/ml, 20 mM tris 7.8, 10 mM MgCl, 10 mM ADP, 1 mM AlCl3 and 30 mM NaF) with 0.8 μl of the crystallization condition. The screens were implemented in the ARI Crystal Gryphon (Arts Robbins Instruments LLC). Crystals suitable for diffraction were obtained in the JCSG+ screen (condition B8), composed of 0.2 M Magnesium chloride hexahydrate, 0.1 M Tris pH 7.0 and 10% wt/vol PEG 8000. The crystals were flash‐frozen and diffracted at 100 K at the MX2 beamline of the Brazilian Synchrotron Light Laboratory (LNLS Campinas‐ SP) using a PILATUS 2M detector (Dectris Ltd.) and an X‐ray wavelength of 1.459 Å.
2.3. Data reduction and structure refinement
The data were integrated with the program XDS 15 and scaled and merged using AIMLESS 16 from the CCP4 suite. 17 The preliminary phases were estimated by molecular replacement using MOLREP 18 and the apo structure 4C5K from Staphylococcus aureus 12 as a search model. A solution was found in the space group P6522 with only one molecule in the asymmetric unit. Iterative cycles of manual refinement in COOT 19 and automated refinement in PHENIX 20 were conducted, until a final model containing 214 of 281 residues and an R factor of 0.20 (0.21 R‐free) was obtained. Validation was performed with Molprobity. 21 Details of data collection and data processing statistics are presented in Table 2.
TABLE 2.
Summary of data collection and refinement statistics
| Data collection | |
|---|---|
| Diffraction source | MX2, LNLS Brazil. |
| Wavelength (Å) | 1.45867 |
| Resolution range (Å) | 46.58–2.00 (2.07–2.00) |
| Space group | P 65 2 2 |
| Cell dimensions (Å) a b c‐ (°) α β γ | 65.6 65.6243.7–90.0 90.0120.0 |
| Total reflections | 152,893 (9871) |
| Unique reflections | 22,065 (2142) |
| Multiplicity | 10.9 (6.2) |
| Completeness (%) | 99.91 (99.86) |
| Mean I/sigma(I) | 13.5 (2.06) |
| R‐merge | 0.145 (1.07) |
| R‐pim | 0.046 (0.352) |
| CC1/2 | 0.998 (0.793) |
| Refinement | |
| R‐work | 0.2075 |
| R‐free | 0.2152 |
| Number of atoms | 1732 |
| Protein | 1,598 |
| B‐factors | |
| Average | 27.22 |
| Macromolecule | 26.08 |
| Solvent | 39.76 |
| Bonds lengths (Å) | 0.007 |
| Bond angles (°) | 1.02 |
| Ramachandran plot | |
| Favored (%) | 98.54 |
| Allowed (%) | 1.46 |
| Outliers (%) | 0.00 |
Note: Highest‐resolution shell are shown in parentheses.
2.4. Site‐directed mutagenesis
The Gln45Met mutation of AncC was generated by the Gene Taylor method. Briefly, primers were designed to overlap and introduce a codon change leading to the Gln45Met mutation. Primers were synthesized by IDT (Integrated Device Technology, inc) and the PCR reaction mix contained PFU polymerase, 25 ng template DNA, 0.2 mM dNTPS and 0.1 μM primers. The vector was transformed into DH5α cells and plasmid were purified by E.Z.N.A. Plasmid Mini Kit II (Omega Bio‐tek) and further sequenced by IDT to verify the mutation.
2.5. Enzyme activity assays
Enzyme activities were measured in spectrophotometric continuous assays as previously described. 13 Briefly, HMP kinase reaction was measured by coupling the ADP production to NAD+ reduction at 340 nm, in the presence of PK (pyruvate kinase) and LDH (lactate dehydrogenase). The reaction mix contained 25 mM Tris‐HCl pH 7.5, 10 mM HMP, 15 mM MgCl2, 125 mM KCl, 10 mM ATP, 0.3 mM PEP, 0.24 mM NADH, 1.7 U/ml of LDH and 0.8 U/ml of PK.
PLK activity was measured directly by following PLP formation at 388 nm. The reaction mixture contained 5 mM PL, ATP 10 mM, MgCl2 15 mM, PIPES 25 mM pH 6.5. Variable PL concentrations were used to construct the saturation curves shown in Figure S2.
The initial rates were determined at 37°C for both reactions, enzyme unit (U) was defined as μmol of product per minute (μmol/min) and a molar extinction coefficient of 6,220 M‐1s‐1 and 2,886 M‐1s‐1 were used for NADH and PLP, respectively. Michaelis Menten or Substrate inhibition models were adjusted to the data with the program Graphad Prism to calculate the kinetic parameters K M , k cat and K i .
2.6. Comparative homology Modelling of missing loops and β2‐β3‐hairpin
Missing loops in AncC (109 to 116, 178 to 188, 201 to 216 and 244 to 281) and substrates were modeled with software Modeller v9.24, 22 remaining residues were not modelled to preserve the crystallographic structure of AncC. Loops models were built using the crystallographic structures of HMPPK bound to HMP in a closed conformation (PDB‐ID: 1JXI 10 ), and PLK/HMPPK‐like bound to PL (PDB‐ID: 4C5L 12 ) as templates. The Residues corresponding to positions 32 to 57 that are located in the β2‐β3‐hairpin were modelled to match with each ligand‐bound state. Additionally, the ligand was modeled in a bound state to AncC. The cofactors ATP and Mg2+ ion, and the water molecules coordinating this ion, were modeled using the structure of pyridoxal kinase from Lactobacillus plantarum (PDB‐ID: 3IBQ) as a template. Twenty models were generated with Modeller and the best models was chosen according to the DOPE Score and validation performed with Molprobity, 21 Ramachandran plots are shown in Figure S5.
2.7. Molecular dynamics
Molecular dynamics simulations were performed with AMBER18, 23 using the force field ff14SB for proteins, 24 and force field GAFF2 25 for HMP and PL as substrates. The partial charges on substrates were obtained with the AM1‐BCC method. Protonation states of PL were calculated at pH 6.5 according to the experimental assays. The force field parameters for ATP and Mg2+ were taken from the Bryce Amber Parameter Database. 26 , 27
Two molecular enzyme‐substrate systems were built with tleap from AmberTools19, one for AncC‐MgATP‐HMP and other for AncC‐MgATP‐PL. Both molecular systems were solvated in a cubic box with TIP3P 28 water model with a buffer distance of 12 Å from solute. NaCl counterions were added in order to neutralize the system and achieve a final concentration of 0.15 M. The energy of both molecular systems were minimized in one stage using 5,000 steps of steepest descent, followed by 10,000 steps of conjugate gradient method without any restraints performed with pmemd.cuda. After achieving convergence of the system energy, a heating step was performed increasing the temperature gradually from 0 to 300 K by 2 ns with a time step of 2 fs. The Shake Algorithm 29 was used to restrain the hydrogen bond, and long‐range electrostatic interactions were treated with Particle Mesh Ewald (PME) method, 30 with a nonbonded cutoff of 8.0 Å. During the heating step, a restraint of 0.5 kcal/mol over the protein backbone was applied. After heating protocol, four independent molecular dynamic simulations, 100ns each, were performed for enzyme‐substrate complexes using the ensemble NPT. The pressure and temperature of molecular systems were maintained at 1 atm and 300 K, respectively, using the Langevin thermostat and barostat with a step of 2.0 ps‐1. Two simulations were performed without restrictions, while in the remaining two, the restraint was maintained on the backbone, and a distance restraint of 4.0 Å was added between atoms OE1(Gln45) and N4A(HMP substrate) with a force of 20 kcal/mol*A2 (Figure S3).
2.8. MM/PBSA calculation
In order to calculate the binding free energy (ΔGbind) for each substrate (HMP and PL) to AncC, and the per‐residue contribution, the g_mmpbsa tools 31 was used. The g_mmpbsa module is based on molecular mechanics Poisson−Boltzmann surface area (MM/PBSA). 32 The average of two independent restricted MD simulations of 100 ns each were used to estimate the ΔGbind, and the residue contribution. This was performed on enzyme‐substrate structures taken every 1000 ps from each MD simulation trajectory, and taking the average and standard deviation of ΔGbind, and its residue contribution values. The MM/PBSA approach uses three energetic terms to calculate the binding free energy: the molecular mechanics' potential energy in vacuum, the polar‐solvation energy (PB), and non‐polar solvation energy (SA) terms.
3. RESULTS AND DISCUSSION
3.1. Sequence comparison between ancestral and modern enzymes of the ATP‐dependent vitamin kinases family
Sequence comparison of the AncC with other ancestral sequences (AncS and AncB) and modern enzymes from the ATP‐dependent vitamin kinases family, including PLK/HMPPK‐like enzymes and HMPPK enzymes (Figure 3), shows that the metal‐binding motifs NXXE, DPV, and the catalytic GXGC motif, are strictly conserved in all of them. However, an inspection of active site residues of specific HMPPK and PLK/HMPPK‐like enzymes pointed out two residues as responsible for substrate specificity: Gln45 and Val50 (according to AncC numbering). These residues are conserved in specific HMPPK, while in the PKL/HMPPK‐like lineage are replaced by methionine and histidine, respectively. 13 Interestingly, the AncC has Gln45 and His50 in these positions, which could represent an intermediate situation towards the evolution of HMPPK and PKL/HMPPK‐like enzymes (Figure 3), since the Gln45Met mutation has been proposed as a key event for the emergence of the PLK activity in PLK/HMPPK‐like enzymes. 13
FIGURE 3.
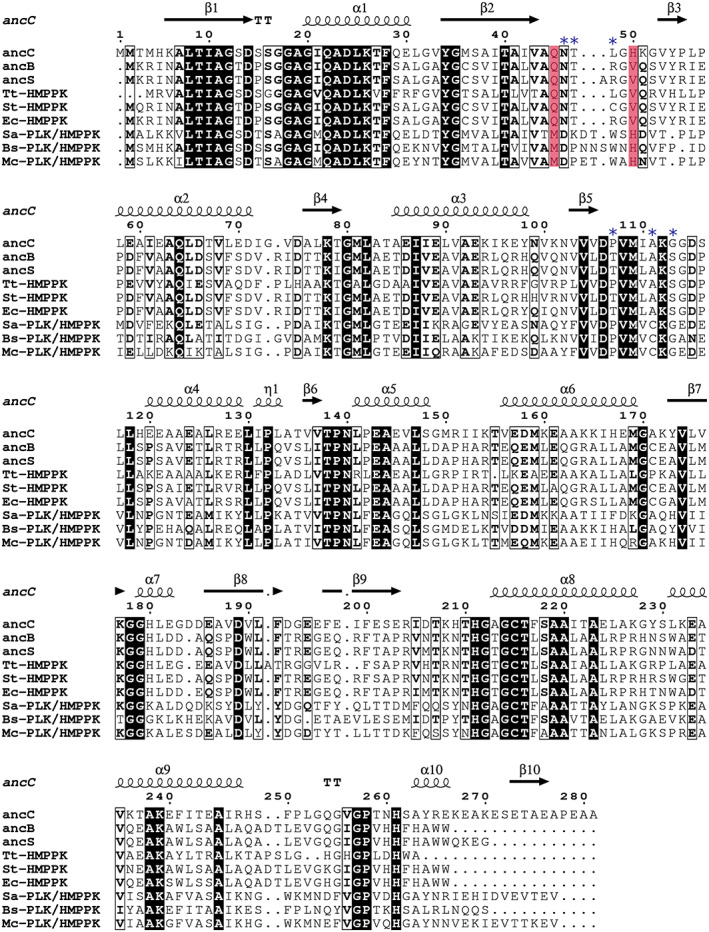
Sequence alignment of ancestral enzymes and representative sequences of PLK/HMPPK‐like and HMPPK enzymes. Tt‐HMPPK (Thermus thermophilus), St‐HMPPK (Salmonella typhimurium), Ec‐HMPPK (Escherichia coli), Sa‐PLK/HMPPK (Staphylococcus aureus), Bs‐PLK/HMPPK (Bacillus subtilis) and Mc‐PLK/HMPPK (Macrococcus caseolyticus). Secondary structure annotation corresponds to the AncC structure. The proposed residues that determine substrate specificity are highlighted in red. Blue asterisks indicate a new set of active site residues involved in ligand recognition
However, despite the presence of His50 in the AncC ancestor, this enzyme is specific for HMP, which suggests that the presence of this residue only is not enough to account for the PLK activity display by PLK/HMPPK‐like enzymes, at least in the AncC ancestor context.
3.2. Kinetic characterization of the AncC Gln45Met mutant
To assess the role of Gln45 in the evolutionary emergence of the PLK activity in PLK/HMPPK‐like enzymes, this residue was mutated by a methionine in the AncC. The mutant enzyme was purified (Figure S1) and kinetically characterized. Saturation curves showed substrate‐inhibition behavior with HMP and hyperbolic behavior with PL and Mg‐ATP (Figure S2). The mutation results in a significant improvement in the PL affinity, whereas the K m value for HMP remains the same. However, the mutation diminishes the k cat with PL, approximately 25‐fold, with only a slight effect on the k cat with HMP kinase activity. K i values show that the mutation causes a decrease in substrate inhibition in the HMP kinase activity and an absence of an inhibitory behavior for PL and ATP‐Mg. Although the mutated residue does not contact the ATP molecule, the mutant enzyme shows an increase in the k cat value from 0.080 to 0.22 s‐1, suggesting the attainment of a more favorable conformation for catalysis (Table 1). These results highlight the relevance of Met45 in the affinity for PL and in the transition from HMP specific to bifunctional PLK/HMPPK‐like enzymes. Nonetheless, considering the low k cat with PL as substrate, it is reasonable to assume that other mutations will be required to achieve the full PLK activity in PLK/HMPPK‐like enzymes.
TABLE 1.
Kinetic parameters for the AncC wild type and Gln45Met mutant
| AncC13 | AncC Gln45Met | |||||
|---|---|---|---|---|---|---|
| K M (mM) | k cat (s −1) | K i (mM) | K M (mM) | k cat (s −1) | K i (mM) | |
| HMP | 7 ± 2 | 0.40 ± 0.06 | 31 ± 17 | 6 ± 1 | 0.28 ± 0.03 | 68 ± 21 |
| PL | 28 ± 9 | 0.20 ± 0.04 | 85 ± 54 | 1.3 ± 0.4 | 0.008 ± 0.001 | N/A |
| ATP‐Mg a | 1 ± 0.2 | 0.080 ± 0.005 | 44 ± 19 | 1.3 ± 0.2 | 0.22 ± 0.01 | N/A |
Note: Mean ± SEM are shown.
Abbreviation: N/A, not applicable.
a10 mM of HMP was used as co‐substrate.
3.3. Structure of the last common ancestor between PLK/HMPPK‐like and HMPPK enzymes
To obtain structural information about substrate evolution specificity in enzymes from the ATP‐dependent vitamin kinases, crystallization trials of the AncC in the presence of substrates were carried out. However, only the apo conformation was solved since no substrate electron density could be associated with the active site (Table 2). As expected, the overall structure of AncC revealed the distinctive Rossman fold of the ribokinase superfamily (Figure 4). The structure is formed by a single large domain, composed of a central β‐sheet of eight parallel strands surrounded by eight helices, arranged three and five on each side of the sheet. Also, regions corresponding to the residues 109‐115, 178‐188, 201‐216 and 244 to 281 could not be solved (loops β5‐α4, α7‐β8, β9 ‐ α8 and C‐terminal). These regions would be highly dynamic loops that could adopt ordered conformations upon substrate binding events, as reported for other members of the family 10 Comparison of AncC structure against modern homologous proteins with ligands (PDB‐ID: 1JXI and 4C5N) revealed a general conserved substrate‐binding site for HMP/PL and ATP. According to this comparison, the active site of AncC is located in a cavity between the central β‐sheet and the α‐helices. HMP would be bound between the β1‐β5 sheet, where the β2‐β3 hairpin would act as a lid, while Mg‐ATP would be bound between the β6‐β9 sheets. However, among ribokinase superfamily members exist important differences regarding the small domain. In the ADP‐dependent kinases, the small domain is formed by β‐sheets and α‐helices insertions, while in the ribokinase family it only contains β‐sheets. On the other hand, this domain is almost absent in the vitamin kinase family, where only one β‐hairpin is found. Protection of the transition state in this later family (and AncC) could be achieved by the β2‐β3 hairpin and the β5‐α4 flexible loop, as previously proposed. 11 , 12
FIGURE 4.
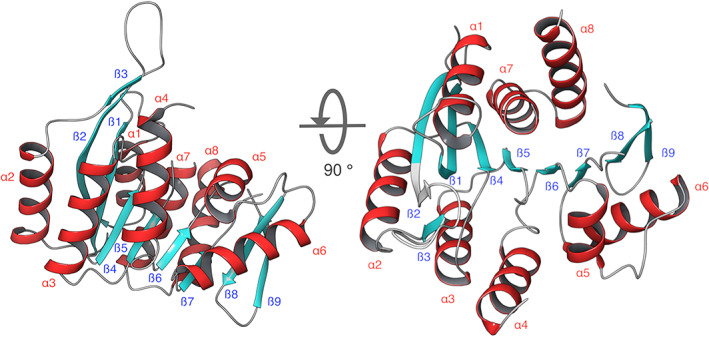
X‐Ray Structure determination of AncC. Two different orientations are shown, where the alpha helices are shown in red and beta sheets in cyan. The figures were prepared using the software Maestro v12.6 (academic version from Schrödinger Inc.). The secondary structure elements are numbered from N to C terminal
3.4. Molecular dynamics simulations of AncC‐ATP‐HMP/PL complexes
A structural comparison of modern vitamin kinases in the presence of substrates show that binding of HMP and PL produces a conformational change that mainly involves β2‐β3‐hairpin and the loop β5‐α4. In the HMPPK structure from S. typhimurium bound to HMP, the β2‐β3‐hairpin is in a close conformation whereas, in the PLK/HMPPK‐like structure from S. aureus (PDB‐ID: 4C5L) bound to PL, the β2‐β3‐hairpin is in a semi‐closed conformation. In the apo crystallographic structure of AncC, the β2‐β3‐hairpin is in an open conformation. Considering the different conformation of these loops in either PL or HMP binding, two models were built. Also, to correctly model the cofactor ATP‐Mg complex within the AncC binding site in a pre‐catalytic conformation, the crystallographic structure of PLK from Lactobacillus plantarum (PDB‐ID: 3IBQ) was used as a template. Then, enzyme structural models were minimized with their respective ligands, and MD simulations of ancestor AncC‐ATP in complex with HMP and PL were performed.
MD trajectory analysis revealed a stable Mg‐ATP configuration along all the simulation time, with the Mg2+ located between the β and γ‐phosphate of ATP (Figure 5a). ATP's adenine ring was found in a hydrophobic environment, where residues Ala187, Arg204, Ile205, Ile205, Thr207, Thr210 and Ile246 were located. Additionally, the hydroxyl group of Thr210 makes a hydrogen bond with N7, and the carbonyl of the main chain of Ile205 forms a polar interaction with N6 of the adenine ring. The ribose moiety contacts residues Asp189, Phe217, Lys239, and Ile242, where Asp189 and Lys 239 form a hydrogen bond network with hydroxyls 3′ and 2′. Phosphates α, β and γ make polar contact with Lys112, Asn140, Lys177, His180, Gly212 and Cys215 (main chain nitrogen), where Cys215 is located in the anion hole motif (211‐214 GAGC). This residue has been proposed as the catalytic base that deprotonates the hydroxyl group of the phosphate acceptor, making a direct nucleophilic attack on the γ‐phosphate of ATP in an in‐line displacement mechanism. 33 Besides this anion hole formed by amides of GXGC, negative charges of the transition state could be stabilized by a positive charge of conserved lysines or arginines present in the small domain. 33 , 34 Since AncC does not have a small domain, Lys112 present in loop β5‐α4 could perform this stabilization of the transition state.
FIGURE 5.
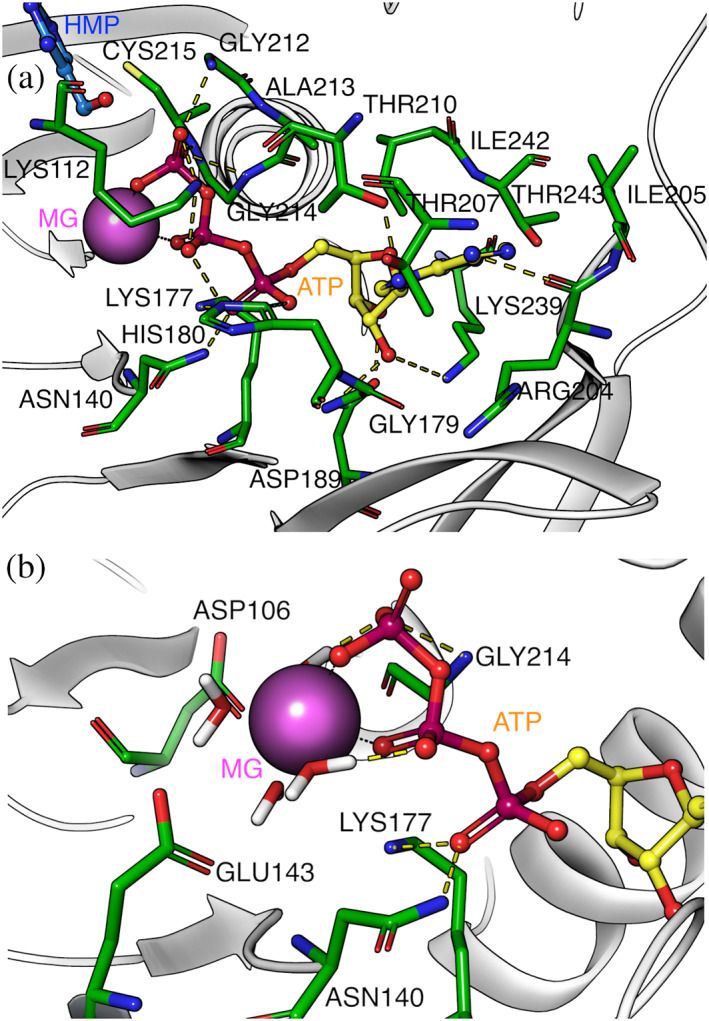
Mg‐ATP binding site of the AncC structure. (a) ATP binding site and residues within 3 Å from the ligand. (b) residues within 3 Å from the Mg2+ ion and first water coordination sphere. Magnesium ion is represented as a purple sphere and ATP is depicted in sticks
During the MD simulation, it was possible to observe stable coordination of Mg2+ with water molecules (Figure 5b), where the oxygen atoms stabilizing the ion were located at ~2.1Å. Residues that contact the water molecules are Asp106, Asn140, Glu143, and Lys177 (Figure 5b), where Asp106 and Asn140‐Glu143 are part of the metal‐binding DPV and NXXE motifs, respectively. 35 However, in MD simulations of AncC‐HMP and AncC‐PL complex, the substrates lost their pre‐catalytic conformation, evidenced by a >6 Å distance between gamma phosphate and substrate hydroxyl. The simulations showed high RMSF, mainly for the β2‐β3 hairpin and loops of the phosphoryl acceptor binding site (HMP and PL) (Figure S3). To analyze the pre‐catalytic conformations of these substrates, MD simulations with weak restrictions (0.5 kcal/mol*Å2) on the protein backbone were performed, achieving stable simulations of the pre‐catalytic complexes.
The averaged ΔGbind values of AncC in complex with PL or HMP were calculated with MM/PBSA method to assess the stability of the complexes and as well as the residues that contribute differentially to substrates binding. Moreover, the averaged ΔGbind obtained for both substrates were compared and correlated with the K m values experimentally determined.
Results from MM/PBSA calculations show that HMP binding is more favorable than PL (Table 3), with a ΔGbind value of −32.7 kJ/mol, while for PL this value is −16.0 kJ/mol. Interestingly, for both ligands, the van der Waals contribution is more important than the electrostatic component, with a contribution of −87.9 kJ/Mol for HMP and −99.4 kJ/mol for PL. The cost of polar solvation is more important in PL than in HMP (+150.2 kJ/mol and +76.9 kJ/mol, respectively), although it is mainly compensated by the electrostatic contribution in PL binding, which is −56.0 kJ/mol meanwhile in HMP is only −11.7 kJ/mol.
TABLE 3.
Average MM/PBSA free binding energy (ΔGbind) analysis for HMP and PL in complex with AncC and mutant
| Complex | Van der Waals energy (kJ/Mol) | Electrostatic energy (kJ/Mol) | Polar solvation energy (kJ/Mol) | ΔGbind (kJ/Mol) |
|---|---|---|---|---|
| HMP‐AncC | −87.9 ± 0.9 | −11.7 ± 1.0 | +76.9 ± 2.0 | −32.7 ± 1.8 |
| PL‐AncC | −99.4 ± 0.3 | −56.0 ± 1.8 | +150.2 ± 0.4 | −16.0 ± 2.5 |
| PL‐Gln45Met | −103.6 ± 0.9 | −31.1 ± 6.5 | +118.5 ± 2.1 | −26.9 ± 5.3 |
3.5. Residues involved in HMP and PL binding and catalysis
Residues located at 5 Å from HMP and PL, and that contribute to their binding (attractive and repulsive), are shown in Figure 6. Protein residues in direct contact (3 Å over 50% of simulation time) with both substrates (HMP and PL) are mainly hydrophobic: Val43, Met81, Ala19, Val108, Gly20, His50, Gln45 and Lys112, Ala111.
FIGURE 6.
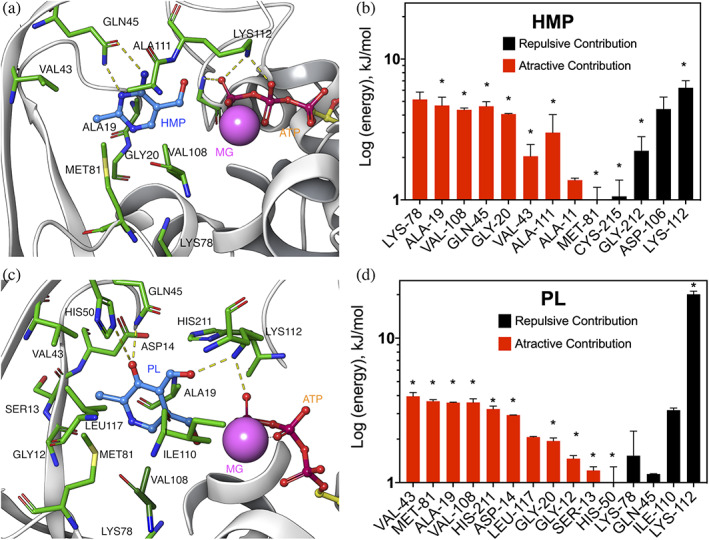
Residues energetic contribution to HMP and PL binding. (a) and (c) HMP and PL binding‐site, respectively. (b) and (d) individual residue contribution to HMP and PL binding, respectively. Residues over 50% of simulation time and over ±1 kJ/mol are shown. Residues at 3 Å, or less, from the ligand are marked with asterisks
Regarding hydrogen bonds for substrate binding, Gln45 is one of the most important residues presenting a favorable interaction with HMP, while for PL presents a non‐favorable interaction, which is in agreement with site‐directed mutagenesis results of this residue. Another predominant amino acid that makes hydrogen bonds with PL and HMP is Lys112, which also interacts with γ‐phosphate of ATP. However, this residue presents the most important repulsive contribution for HMP and PL, mainly due to its high positive polar solvation energy calculated by MM/PBSA. This structural fact highlights its relevance in the stabilization of the negative charges of the transition state, as proposed above. Another interesting residue is His50, which seems to be unimportant for HMP binding (less than 50% of the simulation time is at 5 Å with an interaction energy of −0.3 kJ/mol). However, it established hydrogen bonds with the hydroxyl at C3 of PL, in accordance with the conservation of this residue in PLK/HMPPK‐like enzymes. Nevertheless, its contribution to the stabilization of PL at the active site, seems energetically irrelevant. Noticeably, another residue, Lys78 has one of the most negative interaction energies (attractive) with HMP, although it is in the hydrophobic core of the Rossman domain, rather distant from the substrate. A more detailed analysis shows that this residue remains interacting with the side chain of Cys215 (the catalytic base), suggesting a role in the acid/base deprotonation process of this residue. This is consistent with the estimation of their pKHalf values by PROPKA 36 which point to both as having extremely basic side chains (pKHalf > 12).
3.6. Epistatic interaction between Gln45 and Lys112 on the evolutionary pathway to the PLK/HMPPK‐like lineage
To perform an in‐depth analysis of the Gln45Met mutation effect in pyridoxal binding and its role in the emergence of PLK activity, a MD simulation of this mutant was performed and its ΔGbind value by substrate was estimated using MM/PBSA method. The analysis showed that ΔGbind value for PL (−26.9 kJ/mol) is significantly improved compared to the wild type enzyme (−16.0 kJ/mol), in accordance with its improved K m value for pyridoxal (Table 1 and Table 3).
Estimation of each residue energetic contribution shows that Met45 presents an attractive interaction (−2.4 kJ/mol) compared to Gln45 in the wild type AncC (+1.1 kJ/mol) (Figure 6d). However, since this difference alone cannot explain the improved interaction of the mutant ancestor with PL, the interaction energies of other residues was analyzed (Figure 7). One of the most important energetic contributions came from residue Lys112, which decreases its repulsive contribution from +20.1 kJ/mol in the wild type to +3.8 kJ/mol in the mutant, unveiling a strong epistatic interaction37 between Met45 and Lys112. Besides a favorable interaction with PL, Met45 allows a conformation of PL that avoids a strongly unfavorable interaction with Lys112, a residue that is critical for the transition state stabilization.
FIGURE 7.
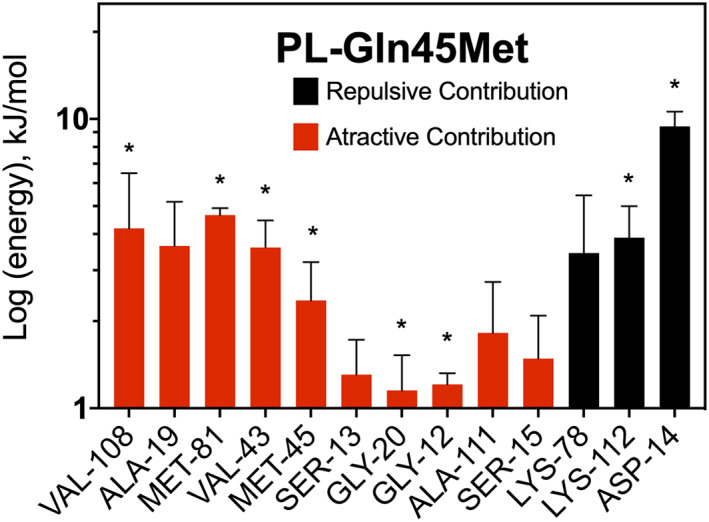
Average MM/PBSA free binding energy (ΔGbind) analysis for the AncC Gln45Met mutant. Selected residues over 50% of simulation time and over ±1 kJ/mol are shown. Residues located within 3 Å from the ligand are indicated with asterisks
Moreover, Lys112 adopts different conformations in MD simulation of AncC‐HMP and AncC‐PL (Figure S4). In AncC‐HMP, this residue interacts mainly with the phosphates's oxygen of the ATP, while in AncC‐PL MD this residue mainly interacts with the aldehyde's oxygens of PL. Interestingly, when we analyze the mutant Gln45Met simulation, the Lys112 adopts a similar conformation that in the AncC‐HMP complex simulation. These results suggest that Gln45 could induce a polar micro‐environment that may favor the interaction of Lys112 with the aldehyde's oxygen of pyridoxal, which counter‐intuitively generated repulsive interaction energy according to the MM/PBSA calculations. On the other side, the Gln45Met mutation may induce a change in this polar micro‐environment due to apolar characteristic, causing Lys112 to interact with the phosphates' oxygen of ATP, avoiding an unfavorable interaction with pyridoxal, how shows the MM/PBSA calculations.
3.7. Other relevant residues for PLK activity in PLK/HMPPK‐like enzymes
Active site residues located at 5 Å from the substrate molecule are similar and highly conserved in specific HMPPK and PLK/HMPPK‐like enzymes, except the Gln45, His50 and Ala111 residues found in AncC. The corresponding residues in HMPPK are Gln, Val, and Ala, while in PLK/HMPPK‐like enzymes are Met, His and Cys. Interestingly, in PLK enzymes, a Cys residue is involved in the hemithioacetal covalent bond with the aldehyde group of pyridoxal. 38 Crystallography and site‐directed mutagenesis studies identified a dual Cys charge relay network, mandatory for PL phosphorylation in the S. aureus enzyme, where the Cys110 forms a hemithioacetal with the aldehyde of PL, which in concert with the catalytic Cys214, increases the nucleophilicity of the hydroxyl group 5′ of the pyridoxal molecule. 12 Nonetheless, in the ancestral sequence of AncC, an alanine(111) residue is found in this position. The absence of this Cys(111) could explain the low catalytic constant (k cat ) for PL in the Gln45Met mutant and pointed out to this residue as a key step for the improvement of this parameter in the evolutionary pathway to modern PLK/HMPPK‐like enzymes.
An expanded analysis of residues conserved in the second interaction shell for PL (over 5 Å and less than 8 Å) was carried out using the multiple sequence alignment described previously (Table S1). 13 This kind of analysis would shed light on residues involved in epistatic interactions or in correlated mutations not directly involved in substrate interactions. In the AncC sequence, the following regions were identified: 43VAQNT47, 48LGHKGV53 and 106DPVMIAKG113, which are associated with Gln45, His50 and Ala111‐Lys112 residues, respectively. The 43VAQNT47 motif is conserved in the HMPPK lineage, whereas in the PLK/HMPPK‐like lineage the consensus motif is 43VAMDP47, where Thr47 is replaced by a Pro residue, which can lead to a more hydrophobic environment and stereochemical restrictions. Regarding the 48LGHKGV53 motif, in the PLK/HMPPK‐like enzymes is 48WXHXVX53, being the main change Leu48 for the voluminous residue Trp, whereas, in HMPPK enzymes, the consensus is 58XGVQSV53, with no significant changes compared to the AncC. Another characteristic of the PLK/HMPPK‐like enzymes, is the insertion of 2 or 3 extra residues between motifs 43VAMDP47 and 48WXHXVX53, making the β2‐β3‐hairpin longer in PLK/HMPPK‐like enzymes. Finally, the 106DPVMIAKG113 motif, located in the β5‐α4 loop, is conserved as 106DPVMVCKG113 in the PLK/HMPPK‐like lineage and as 106DXVMXAKS113 in HMPPK enzymes, changing the serine residue by glycine in this last group. The importance of these motifs for PLK activity could be the subject of future studies.
4. CONCLUSIONS
It has been proposed that the appearance of PLK activity in PLK/HMPPK enzymes involves the Gln45Met mutation in AncC. 13 Here, it is shown that this mutation causes an increase in the affinity for PL and a decrease in k cat , without modifying the kinetic parameters for HMP. Crystallographic structure determination of AncC, and molecular dynamics simulations with PL and HMP, identified key residues for binding and catalysis, such as the β2‐β3‐hairpin, the GXGC anion hole motif and Lys112 located in loop β5‐α4, being the latter critical for transition state stabilization. In silico analysis of the AncC‐Gln45Met mutant showed an epistatic interaction between Gln45 and Lys112, where the increase in PL affinity is mainly due to a decrease in the repulsive interaction between PL and Lys112. Other mutations, such as Ala111Cys are proposed as compensatory mutations for the appearance of PLK activity in the group of the PLK/HMPPK‐like enzymes.
AUTHOR CONTRIBUTIONS
Felipe Gonzalez‐Ordenes: Data curation; formal analysis; investigation; writing‐original draft. Felipe Bravo‐Moraga: Data curation; formal analysis; investigation; writing‐original draft. Evelin Gonzalez: Data curation; formal analysis; investigation. Leslie Hernandez‐Cabello: Data curation; formal analysis; investigation. Jans Alzate‐Morales: Formal analysis; supervision; writing‐review and editing. Guixe Victoria: Conceptualization; supervision; writing‐review and editing. Victor Castro‐Fernandez: Conceptualization; formal analysis; funding acquisition; project administration; supervision; validation; writing‐original draft; writing‐review and editing.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Figure S1 SDS‐polyacrylamide gel electrophoresis for purification of AncC Gln45Met mutant.
Figure S2. Saturation curves of PL, HMP and MgATP.
Figure S3. Conformational dynamics of AncC ligands complexes with and without positional restraints.
Figure S4. Different Lys112 conformations for AncC simulations with HMP and PL.
Figure S5. Ramachandran plots for Anc‐C models.
Table S1. List of residues around HMP and PL at 5 and 8 Angstroms of AncC.
ACKNOWLEDGMENTS
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT Grant 11181133), REDI170497. ANID PhD Fellowship 21191254 to Felipe Gonzalez‐Ordenes. Universidad de Talca PhD Fellowship to Felipe Bravo‐Moraga. The high‐throughput ARI crystallization robot was funded by Fondequip EQM 120208. This research used resources of the Brazilian Synchrotron Light Laboratory (LNLS), an open national facility operated by the Brazilian Centre for Research in Energy and Materials (CNPEM) for the Brazilian Ministry for Science, Technology, Innovations and Communications (MCTIC), proposals 20170963 and 20180298. The MX2 beamline staff is acknowledged for their assistance during the diffraction experiments. Also, we thank the IFSC/CCP4 school 2018, São Carlos, Brazil.
Gonzalez‐Ordenes F, Bravo‐Moraga F, Gonzalez E, et al. Crystal structure and molecular dynamics simulations of a promiscuous ancestor reveal residues and an epistatic interaction involved in substrate binding and catalysis in the ATP‐dependent vitamin kinase family members. Protein Science. 2021;30:842–854. 10.1002/pro.4040
Felipe Gonzalez‐Ordenes and Felipe Bravo‐Moraga contributed equally to this study.
The structure of AncC has been deposited in the Protein Data Bank (PDB) with the following code: 7L07.
Funding information Fondo Nacional de Desarrollo Científico y Tecnológico, Grant/Award Numbers: 11181133, REDI170497; ANID PhD Fellowship, Grant/Award Number: 21191254; Fondequip EQM, Grant/Award Number: 120208
REFERENCES
- 1. Khersonsky O, Tawfik DS. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu Rev Biochem. 2010;79:471–505. [DOI] [PubMed] [Google Scholar]
- 2. Marsh JA, Teichmann SA. Structure, dynamics, assembly, and evolution of protein complexes. Annu Rev Biochem. 2015;84:551–575. [DOI] [PubMed] [Google Scholar]
- 3. Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Dougherty M, Downs DM, Ealick SE. Crystal structure of an aminoimidazole riboside kinase from Salmonella enterica: Implications for the evolution of the ribokinase superfamily. Structure. 2004;12:1809–1821. [DOI] [PubMed] [Google Scholar]
- 5. Guixé V, Merino F. The ADP‐dependent sugar kinase family: Kinetic and evolutionary aspects. IUBMB Life. 2009;61:753–761. [DOI] [PubMed] [Google Scholar]
- 6. Herrera‐Morande A, Castro‐Fernández V, Merino F, et al. Protein topology determines substrate‐binding mechanism in homologous enzymes. Biochim Biophys Acta Gen Subj. 2018;1862:2869–2878. [DOI] [PubMed] [Google Scholar]
- 7. Li M‐H, Kwok F, Chang W‐R, et al. Crystal structure of brain pyridoxal kinase, a novel member of the ribokinase superfamily. J Biol Chem. 2002;277:46385–46390. [DOI] [PubMed] [Google Scholar]
- 8. Li M, Kwok F, Chang W, et al. Conformational changes in the reaction of pyridoxal kinase. J Biol Chem. 2004;279:17459–17465. [DOI] [PubMed] [Google Scholar]
- 9. Cea PA, Araya G, Vallejos G, et al. Characterization of hydroxymethylpyrimidine phosphate kinase from mesophilic and thermophilic bacteria and structural insights into their differential thermal stability. Arch Biochem Biophys. 2020;688:108389. [DOI] [PubMed] [Google Scholar]
- 10. Cheng G, Bennett EM, Begley TP, Ealick SE. Crystal structure of 4‐amino‐5‐hydroxymethyl‐2‐ methylpyrimidine phosphate kinase from salmonella typhimurium at 2.3 Å resolution. Structure. 2002;10:225–235. [DOI] [PubMed] [Google Scholar]
- 11. Newman JA, Das SK, Sedelnikova SE, Rice DW. The crystal structure of an ADP complex of Bacillus subtilis pyridoxal kinase provides evidence for the parallel emergence of enzyme activity during evolution. J Mol Biol. 2006;363:520–530. [DOI] [PubMed] [Google Scholar]
- 12. Nodwell MB, Koch MF, Alte F, Schneider S, Sieber SA. A subfamily of bacterial ribokinases utilizes a hemithioacetal for pyridoxal phosphate salvage. J Am Chem Soc. 2014;136:4992–4999. [DOI] [PubMed] [Google Scholar]
- 13. Castro‐Fernandez V, Bravo‐Moraga F, Ramirez‐Sarmiento CA, Guixe V. Emergence of pyridoxal phosphorylation through a promiscuous ancestor during the evolution of hydroxymethyl pyrimidine kinases. FEBS Lett. 2014;588:3068–3073. [DOI] [PubMed] [Google Scholar]
- 14. Gumulya Y, Gillam EMJ. Exploring the past and the future of protein evolution with ancestral sequence reconstruction: The retro approach to protein engineering. Biochem J. 2017;474:1–19. [DOI] [PubMed] [Google Scholar]
- 15. Kabsch W. XDS. Acta Cryst D. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Cryst D. 2013;69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collaborative Computational Project . Number 4. The CCP4 suite: Programs for protein crystallography. Acta Cryst D. 1994;50:760–763. [DOI] [PubMed] [Google Scholar]
- 18. Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Cryst D. 2010;66:22–25. [DOI] [PubMed] [Google Scholar]
- 19. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 20. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: A comprehensive python‐based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen VB, Arendall WB, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Cryst D. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 23. Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J, Wolf RM, Caldwell JW, Kollman PA. Case5 DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. [DOI] [PubMed] [Google Scholar]
- 26. Meagher KL, Redman LT, Carlson HA. Development of polyphosphate parameters for use with the AMBER force field. J Comput Chem. 2003;24:1016–1025. [DOI] [PubMed] [Google Scholar]
- 27. Allnér O, Nilsson L, Villa A. Magnesium ion–water coordination and exchange in biomolecular simulations. J Chem Theory Comput. 2012;8:1493–1502. [DOI] [PubMed] [Google Scholar]
- 28. Price DJ, Brooks CL. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys. 2004;121:10096–10103. [DOI] [PubMed] [Google Scholar]
- 29. Ryckaert J‐P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n‐alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 30. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 31. Kumari R. Kumar R (2014) g_mmpbsa‐a GROMACS tool for high‐throughput MM‐PBSA calculations. J Chem Inf Model. 2014;54:1951–1962. [DOI] [PubMed] [Google Scholar]
- 32. Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol Inform. 2012;31:114–122. [DOI] [PubMed] [Google Scholar]
- 33. Sigrell JA, Cameron AD, Jones TA, Mowbray SL. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 a resolution: Insights into a new family of kinase structures. Structure. 1998;6:183–193. [DOI] [PubMed] [Google Scholar]
- 34. Cabrera R, Baez M, Pereira HM, Caniuguir A, Garratt RC, Babul J. The crystal complex of phosphofructokinase‐2 of Escherichia coli with fructose‐6‐phosphate: Kinetic and structural analysis of the allosteric ATP inhibition. J Biol Chem. 2011;286:5774–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramírez‐Sarmiento CA, Engelberger F, Guixé V. An evolutionary marker of the ribokinase superfamily is responsible for zinc‐mediated regulation of human pyridoxal kinase. Catalysts. 2020;10:555. [Google Scholar]
- 36. Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. [DOI] [PubMed] [Google Scholar]
- 37. Starr TN, Thornton JW. Epistasis in protein evolution. Protein Sci. 2016;25:1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Safo MK, Musayev FN, Hunt S, Di Salvo ML, Scarsdale N, Schirch V. Crystal structure of the PdxY protein from Escherichia coli. J Bacteriol. 2004;186:8074–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SDS‐polyacrylamide gel electrophoresis for purification of AncC Gln45Met mutant.
Figure S2. Saturation curves of PL, HMP and MgATP.
Figure S3. Conformational dynamics of AncC ligands complexes with and without positional restraints.
Figure S4. Different Lys112 conformations for AncC simulations with HMP and PL.
Figure S5. Ramachandran plots for Anc‐C models.
Table S1. List of residues around HMP and PL at 5 and 8 Angstroms of AncC.