

Abstract
Background
Patients with cancers that exhibit extraordinarily high somatic mutation numbers are ideal candidates for immunotherapy and enable identifying tumor-specific peptides through stimulation of tumor-reactive T cells (Tc).
Methods
Colorectal cancers (CRC) HROC113 and HROC285 were selected based on high TMB, microsatellite instability and HLA class I expression. Their HLA ligandome was characterized using mass spectrometry, compared with the HLA ligand atlas and HLA class I-binding affinity was predicted. Cryptic peptides were identified using Peptide-PRISM. Patients’ Tc were isolated from either peripheral blood (pTc) or tumor material (tumor-infiltrating Tc, TiTc) and expanded. In addition, B-lymphoblastoid cells (B-LCL) were generated and used as antigen-presenting cells. pTc and TiTc were stimulated twice for 7 days using peptide pool-loaded B-LCL. Subsequently, interferon gamma (IFNγ) release was quantified by ELISpot. Finally, cytotoxicity against autologous tumor cells was assessed in a degranulation assay.
Results
100 tumor-specific candidate peptides—97 cryptic peptides and 3 classically mutated neoantigens—were selected. The neoantigens originated from single nucleotide substitutions in the genes IQGAP1, CTNNB1, and TRIT1. Cryptic and neoantigenic peptides inducing IFNγ secretion of Tc were further investigated. Stimulation of pTc and TiTc with neoantigens and selected cryptic peptides resulted in increased release of cytotoxic granules in the presence of autologous tumor cells, substantiating their improved tumor cell recognition. Tetramer staining showed an enhanced number of pTc and TiTc specific for the IQGAP1 neoantigen. Subpopulation analysis prior to peptide stimulation revealed that pTc mainly consisted of memory Tc, whereas TiTc constituted primarily of effector and effector memory Tc. This allows to infer that TiTc reacting to neoantigens and cryptic peptides must be present within the tumor microenvironment.
Conclusion
These results prove that the analyzed CRC present both mutated neoantigenic and cryptic peptides on their HLA class I molecules. Moreover, stimulation with these peptides significantly strengthened tumor cell recognition by Tc. Since the overall number of neoantigenic peptides identifiable by HLA ligandome analysis hitherto is small, our data emphasize the relevance of increasing the target scope for cancer vaccines by the cryptic peptide category.
Keywords: Antigens, Neoplasm; T-Lymphocytes; Lymphocytes, Tumor-Infiltrating; Gastrointestinal Neoplasms; Antigen Presentation
WHAT IS ALREADY KNOWN ON THIS TOPIC.
Tumor-specific peptides presented via HLA class I molecules are obvious candidates for cancer vaccine designs. HLA ligandome analyses are the only way to identify cryptic peptides translated from genomic regions, presumed to not be translated—in addition to the classical mutation-derived neoantigens.
WHAT THIS STUDY ADDS
We newly identified peptides, three of which are derived from immunogenic neoantigens and a substantial number of cryptic peptides with the potential for T cell activation and tumor cell recognition. Mutated neoantigens as well as cryptic peptides are presented on the tumor cells and recognized by autologous T cells.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Cryptic peptides add to the overall number of tumor-specific epitopes and thus constitute promising candidates for future cancer vaccination developments in addition to mutated or non-mutated neoantigens and other tumor specific peptides.
Background
Immunotherapeutic agents and strategies are most promising, adding to the armamentarium for effectively treating cancers. Even though much effort was put into broadening the applicability of immunotherapies over the last decade, immunotherapeutic treatments are not yet available for all patients with cancer. Especially solid tumors remain difficult to target via immunotherapy. This is dismal, since they account for more than 90% of cancer-related deaths.1 Optimal tumor-specific immune responses are thought to be based on the recognition of mutated neoantigens, resulting in a very effective distinction between malignant tumor and healthy tissue cells.2 This distinction is more successful in tumors with high mutational burden, compared with neoplasms with few mutations.3 A high number of mutations is found first and foremost in melanoma and lung cancer.4 But also colorectal cancers (CRC) with microsatellite instability (MSI) and DNA polymerase epsilon mutations possess a high TMB.4 However, not only mutated neoantigens are excellent targets for immunotherapeutic treatment strategies. There are also other sources for tumor-specific antigens, including cryptic peptides, which can be used as targets for cancer vaccinations.5 Cryptic peptides derive from non-canonical protein translation—especially from non-coding RNAs, intronic and intergenic regions, physiologically untranslated regions and shifted reading frames in coding sequences—and represent up to 15% of the tumor cell ligandome.6–11 These anomalous translational processes might produce short intracellular peptides, which are almost exclusively presented on HLA class I molecules.7 Their aberrant occurrence likely prevents self-tolerance and thus makes them promising targets for cancer vaccine development.12
Considering both somatic mutations and aberrant expression events, we here aimed at the unbiased identification of patient-individual antigenic peptides capable of inducing anti-tumoral T cell (Tc) responses. We chose two CRC cell models of the MSI subtype established from the Lynch syndrome patients HROC113 and HROC285. Genetic and HLA ligandome analyses resulted in the identification of 100 candidate peptides, consisting of mutated neoantigens and cryptic peptides. These peptides were screened for pre-existing autologous Tc reactivity. Furthermore, we tested the ability of stimulated peripheral Tc (pTc) and tumor-infiltrating Tc (TiTc) to release cytotoxic granules in response to autologous tumor cells. This approach allowed to conclude that the identified neoantigenic and cryptic peptides are presented by tumor cells and can therefore serve as valid Tc targets.
Methods
Cell culture
Patient-derived CRC cell lines HROC113 and HROC285 T0 M2 from the HROC collection13 were cultured in DMEM/Hams F12 medium supplemented with 10% FCS and 2 mM L-glutamine. To increase the expression of HLA molecules on the surface, tumor cells were treated for 48 hours with 200 IU/mL interferon gamma (IFNγ) (Imukin, Boehringer Ingelheim, Ingelheim am Rhein, Germany) for selected experiments. B-lymphoblastoid cell lines (B-LCL) were cultivated in DMEM/Hams F12 medium, 10% human AB serum (Institute for Transfusion Medicine, University of Rostock, Rostock, Germany), penicillin, streptomycin, amphotericin B, and 2 mM L-glutamine. Cell culture was performed at 37°C and 5% CO2 in vessels selected according to experimental needs. All cell culture reagents were obtained from PAN Biotech, Aidenbach, Germany, unless stated otherwise.
Whole-exome sequencing
Whole exome sequencing was performed as described previously.14 In short, genomic DNA was extracted using QIAmp DNA Mini Kit (Qiagen, Hilden, Germany). Enrichment was carried out with the SureSelect Human All Exon kit (Agilent, Santa Clara, California, USA). The generated libraries were sequenced on the HiSeq4000 platform (Illumina, San Diego, California, USA) using the 150 bp paired-end protocol and targeting for an average coverage of at least 100 x.
Variant calling, annotation and prioritization was based on a set of publicly available and Centogene in-house tools. By parallel analysis of normal tissue, germline polymorphisms were excluded.
Isolation of naturally presented HLA ligands and characterization by mass spectrometry (HLA Ligandomics)
HLA ligands were isolated and subsequently characterized through tandem-mass spectrometry, as described previously.15–18 In brief, HLA class I molecules were isolated from HROC113 and HROC285 T0 M2 cell lysates by immunoaffinity purification using the pan–HLA-class I monoclonal antibody W6/3219 (produced in house) with cyanogen bromide-activated sepharose columns (Sigma-Aldrich, Burlington, Massachusetts, USA) and subsequently eluted with trifluoroacetic acid.15 Eluted HLA ligands were purified by ultrafiltration using centrifugal filter units (Amicon; Millipore, Burlington, Massachusetts, USA) and desalted using ZipTip C18 pipette tips (Millipore, Billerica, Massachusetts, USA). Subsequently peptides were separated by reversed-phase nanoflow uHPLC (UltiMate 3000 RSLCnano System, ThermoFisher Scientific, Waltham, Massachusetts, USA) using a 75 µm×2 cm trapping column (Acclaim PepMap RSLC; Thermo Fisher Scientific) and a gradient ranging from 2.4% to 32.0% acetonitrile over the course of 90 min. Eluting peptides were analyzed once or in multiple technical replicates in an online coupled Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific) using a top-speed collision-induced dissociation method.
Ligandomics data analysis
MHC I peptides were identified using Peptide-PRISM,6 reanalysis was done using a recently published database of translated open reading frames (ORF) obtained from ribosome profiling data (nuORFdb).11 De novo peptide sequencing was performed with PEAKS X (Bioinformatics Solutions, Waterloo, Canada).20 Raw data were refined with the following settings: (1) Merge Options: no merge; (2) Precursor Options: corrected; (3) Charge Options: 1–6; (4) Filter Options: no filter; (5) Process: true; (6) Default: true; (7) Associate Chimera: yes; Parent Mass Error Tolerance was set to 10ppm, Fragment Mass Error Tolerance to 0.02 Da, and Enzyme to none. The following post-translational modifications were used: Oxidation (M), pyro-Glu from Q (N-term Q), and carbamidomethylation (C) with a maximum of three modifications allowed per peptide. Up to 10 de novo sequencing candidates were reported for each identified fragment ion mass spectrum, with their corresponding average local confidence score. As chimeric spectra option of PEAKS X was used, two or more TOP10 candidate lists could possibly be assigned to a single fragment ion spectrum. All de novo sequence candidates were matched against the six-frame translated human genome (GRCh37) and the three-frame translated transcriptome (ENSEMBL release 75) using Peptide-PRISM. All detected single nucleotide variants (SNV) and indels obtained from mutation calling were considered for the Peptide-PRISM search. Results were filtered to category-specific <10% false discovery rate (FDR). NetMHCpan 4.0 was used to predict binding affinities for all identified MHC I peptides.21 A cut-off of 0.5% rank for strong and 2% rank for weak binders was used. Sequence-specific hydrophobicity indices were calculated using SSRCalc.22
The raw data generated and analyzed during the current study as well as results have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRoteomics IDEntifications (PRIDE) database partner repository23 with the dataset identifier PXD037587. The data sets reanalyzed with Peptide-PRISM using ENSEMBL release 90 with the same settings as described above are also available online through the PRIDE database (dataset identifiers: PXD014017,24 PXD016582,25 PXD02175526 and PXD00489427).
Characterization of Tc subsets
The subpopulations of CD4+ and CD8+ Tc were identified via flow cytometry using the definitions shown in table 1. Antibodies were purchased from Immunotools (Frisoythe, Germany) and Biolegend (San Diego, USA) and the measurements were performed using a BD FACSVerse (BD Biosciences, Franklin Lakes, USA). Data were analyzed by FCSalyzer 0.9.21-alpha (https://sourceforge.net/projects/fcsalyzer).
Table 1.
Definitions of T cell subpopulations
| Marker combination | T cell subpopulation |
| CD3+/CD62L+/CD45RO-/CD95– | Naïve Tc |
| CD3+/CD62L+/CD45RO-/CD95+ | Stem cell-like memory Tc |
| CD3+/CD62L+/CD45RO+/CD95+ | Central memory Tc |
| CD3+/CD62L–/CD45RO+/CD95+ | Effector memory Tc |
| CD3+/CD62L–/CD45RO-/CD95+ | Effector Tc |
Tc expansion
Peripheral blood mononuclear cells (PBMC) were isolated from patients’ heparinized blood by ficoll density gradient centrifugation. pTc were isolated with the Pan Tc isolation Kit (Miltenyi Biotec, Bergisch-Gladbach, Germany). For the expansion of pTc, a rapid expansion protocol (REP)28 was adapted. Briefly, pTc were cocultured with irradiated (40 Gy) healthy donors’ PBMC in a ratio of 1:200 in 50 mL REP medium (TexMACS medium (Miltenyi Biotec), 3% human AB serum, 3000 IU/mL interleukine 2 (Proleukin, Novartis, Basel, Switzerland), penicillin, streptomycin and amphotericin B) supplemented with 0,1% OKT3 cell culture supernatant in a standing T75 cell culture flask per 1×106 pTc. At day 5 of coculture, ¾ of the medium was replaced with fresh REP medium. Starting at day 7, cells were counted daily and pTc were adjusted to a concentration of 1.0×106 cells/mL REP medium. The cumulative growth factor was calculated using the following formula: .
Expansion was ended at day 14 and pTc were frozen or directly used for further experiments.
For the expansion of TiTc, vitally frozen tumor pieces were minced with crossed scalpels. Assuming a concentration of approximately 2–4 x 106 TiTc per cm³ tumor tissue, irradiated PBMC were added at a ratio of 1:200. Subsequent cultivation steps followed the same protocol as used for pTc (see above).
Tc stimulation
B-LCL used as antigen presenting cells were generated from patients’ PBMC using Epstein-Barr-Virus containing cell culture supernatant from B95/8 cells.29 For peptide-loading, 3×106 B-LCL were incubated at 37°C with 10 µg peptide pools in 1 mL serum free medium for 1 hour. Thereafter, B-LCL were irradiated with 30 Gy and seeded together with expanded Tc in a ratio of 1:4. Medium supplemented with 1xITS solution IV (PAN Biotech) and 300 IU/mL interleukine 2 was used for stimulation. Lymphocytes were seeded into a 24-well plate with 1×106 Tc per well in 2 mL of stimulation medium. Tc were counted every 7 days and freshly prepared peptide-loaded B-LCL were added in the 1:4 ratio. Tc stimulated with B-LCL without any peptide served as controls.
IFNγ-ELISpot
For detection of IFNγ secretion, the human IFNγ-ELISpotBASIC (Mabtech, Nacka Strand, Sweden) was performed. Per well, 20,000 Tc and 10,000 B-LCL were seeded in a total of 200 µL Tc medium on the antibody-coated membrane plate; 4 µg peptide was added. Tc incubated with B-LCL but without peptide served as negative, Tc incubated with 50 ng/mL PMA and 1 µg/mL ionomycin (both Merck, Darmstadt, Germany) as positive control. After 16–20 hours at 37°C and 5% CO2, medium was discarded and the plate was handled according to the manufacturer’s instructions. In the end, occurring spots indicating IFNγ-secreting Tc were counted using an ImmunoSpot Analyzer (CTL, Cleveland, USA).
Degranulation assay
The ability of Tc to degranulate was tested with the degranulation assay.30 A total of 100,000 IFNγ-treated tumor cells and 200,000 pTc or TiTc were seeded in Tc medium containing FITC anti-human CD107a (Biolegend) per well of a 96-well plate precoated with anti-human CD28 (Immunotools). After 1 hour of incubation, 1 µg/mL Brefeldin A (MedChemExpress, Monmouth Junction, USA) was added, followed by another 4 hours incubation. Flow cytometry staining was performed using the Inside Stain Kit (Miltenyi Biotec), APC anti-human CD8 (Immunotools) and PE anti-human IFNγ (Biolegend). Measurements were performed at a BD FACSCalibur and gates were adjusted to detect CD8+/CD107a+/IFNγ+ cells. Percentage of triple-positive Tc was normalized to the triple-positive population of Tc not incubated with tumor cells.
Tetramer staining
The PE-IQGAP1neo tetramer was from Miltenyi Biotec. The tetramers of TRIT1neo, Pep 8, Pep 18 and Pep 32 conjugated with PE and APC were prepared with the FLEX-T Kit HLA-B*08:01 (Biolegend). Tc were washed with wash buffer containing PBS, 0.5% BSA, and 2 mM EDTA, centrifuged at 400 g for 5 min and stained with tetramer for 30 min at room temperature. After two further washing steps, the cells were analyzed with a BD FACSCalibur.
Results
Mass spectrometric identification of tumor-specific HLA-I peptides
Adherent cultures of HROC113 and HROC285 T0 M2 cells either treated with IFNγ or untreated were collected without enzymatic detachment methods and immediately snap frozen for subsequent HLA-ligand analysis. Using Peptide-PRISM,6 a computational tool that enables sensitive and reliable identification of conventional as well as neoantigenic and cryptic HLA-I peptides, 8564 conventional and 98 cryptic HLA-I peptides were identified for HROC113. For HROC285, 2374 conventional and 31 cryptic HLA-I peptides were identified (figure 1A; online supplemental table S1).
Figure 1.
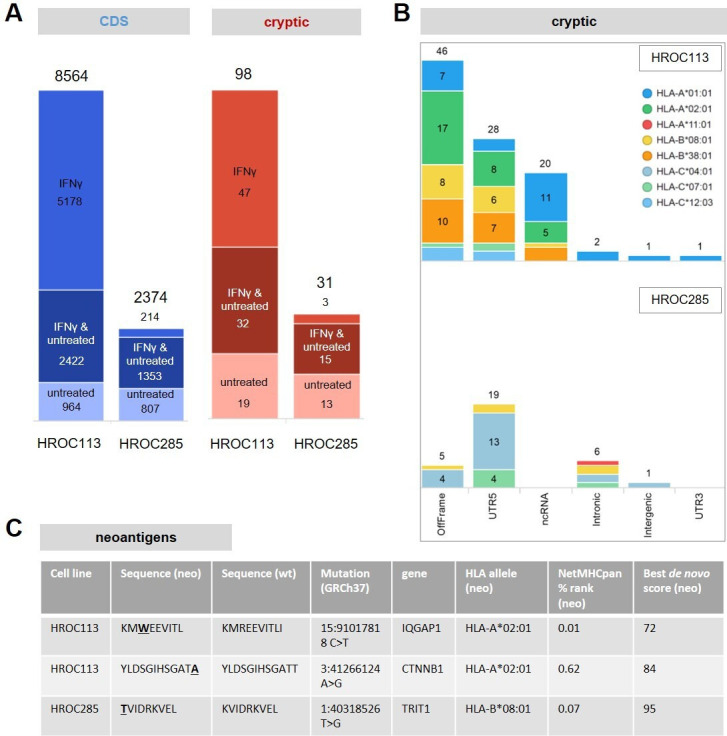
HLA-I immunopeptidomes of HROC113 and HROC285. (A) Total number of identified conventional (CDS) and cryptic HLA-I peptides with and without IFNγ treatment (false discovery rate: <10%, NetMHCpan rank: <2%). (B) Identified cryptic peptides identified from HROC113 and HROC285 by category. HLA peptides were assigned to the HLA allele with the lowest rank value. (C) Identified neoantigens derived from non-synonymous SNVs. SNVs, single-nucleotide variants.
jitc-2022-005651supp002.xlsx (3.4MB, xlsx)
HROC113 showed a typical distribution of conventional HLA peptides among HLA alleles, with the highest number of peptides presented on HLA-A and HLA-B, significantly less on HLA-C and 9% NetMHCpan-predicted non-binders. HROC285 presented by far the highest number of peptides on HLA-C*04:01 (55%) and had a higher percentage (28%) of NetMHCpan-predicted non-binders (online supplemental figure S1A). Although the two cell lines have three HLA alleles in common, only a single cryptic peptide (Pep 28, FHDPLTLKF) was shared among the two cell lines. Gibbs clustering analysis with GibbsCluster 2.031 of the identified HLA peptides of HROC285 T0 M2 revealed two peptide binding motifs (online supplemental figure 2). The first motif resembled the binding motif of HLA-C*04:01, the second motif did not resemble any of the patients HLA alleles. In addition, HLA-B-presented peptides were strongly induced by IFNγ for HROC113, as has been described recently,32 but not for HROC285 T0 M2 (online supplemental figure S1A). Cryptic peptides showed similar HLA allele distribution and IFNγ responsiveness as conventional HLA peptides (online supplemental figure S1B). The overall percentage of cryptic peptides was low for both cell lines (~1%), possibly because both cell lines predominantly express HLA alleles presenting a low percentage of cryptic peptides, such as HLA-A*01:01 and HLA-A*02:01.5 Most cryptic peptides derived either from 5’-UTR or from a non-canonical reading frame (OffFrame), in accordance with previous findings5 (figure 1B).
jitc-2022-005651supp001.pdf (1.8MB, pdf)
To verify that the FDR for cryptic is comparable to that for conventional peptides, we performed a variety of quality controls. We compared the de novo score distribution, the percentage of NetMHCpan-predicted non-binders, the peptide length distribution, the correlation of the calculated hydrophobicity with the experimental retention time, and the median intensity of cryptic with that of conventional peptides. All comparisons clearly indicated that cryptic peptides were presented as efficiently and identified with the same reliability as conventional peptides (online supplemental figure S3).
To identify neoantigenic peptides derived from SNV or indels, we included all somatic mutations obtained from mutation calling for the analysis. Although more than 1400 frameshift mutations for each of the two cell lines were considered, no frameshift-derived neoantigenic peptide was detected. However, two neoantigens derived from non-synonymous SNV were identified for HROC113 and one for HROC285 (figure 1C). These neoantigens were detected in tumor cells with and without IFNγ treatment. The p.Arg321Trp mutation of IQGAP1 in HROC113 resulted in the neoantigen KMWEEVITL (wildtype KMREEVITL). A second neoantigen was detected in this CRC cell line translated from p.Thr41Ala in CTNNB1 leading to the neoantigen YLDSGIHSGATA (wildtype YLDSGIHSGATT). For HROC285, a single neoantigen from TRIT1 (p.Lys66Thr), translating into TVIDRKVEL (wildtype KVIDRKVEL) was detected together with the wild type peptide. Contrarily, only the mutated neoantigens but not the wild type peptides were identified for HROC113.
Whereas mutated neoantigens derived from somatic mutations are tumor-specific per se, cryptic peptides are also detectable in healthy tissue,33 and only a small proportion of cryptic peptides can be expected to be tumor-specific. Therefore, we filtered out peptides previously observed in benign tissues included in the HLA ligand atlas.33 Fifty-four of the initial 128 cryptic peptides found in HROC113 and HROC285 were thus filtered out. Another 23 cryptic peptides identified in HROC113 or HROC285 with a de novo score below the applied FDR score cut-off were included into the functional analysis, because they (1) were identified in ligandomics data of other tumor samples with a higher de novo score (data not shown), (2) were not in the benign HLA ligand atlas data set, and (3) were predicted as binders to one of the corresponding patients’ HLA alleles. Thus, our final candidate list comprised 97 cryptic HLA peptides and 3 mutated neoantigens (online supplemental table S2). More than 60% (59) of the 97 selected cryptic candidate peptides were derived from translated ORF as classified by ribosome profiling using nuORFdb (online supplemental table S2).
jitc-2022-005651supp003.xlsx (33.9KB, xlsx)
All 100 candidate peptides showed excellent correlation of the calculated hydrophobicity with the experimental retention time (online supplemental figure S4).
Expansion of Tc populations
Polyclonal expansion of pTc and TiTc resulted in cumulative growth factors of 100 and 160 for pTc and 210 and 500 for TiTc for patients HROC113 and HROC285, respectively. Flow cytometric analysis of the Tc subpopulations revealed, that distribution of CD4+ and CD8+ Tc generally shifted during expansion in favor of CD8+ Tc (figure 2). This effect is attributable to the rapid expansion protocol, as it was not only observed in pTc from the two patients HROC113 and HROC285, but also in more than 20 other colorectal and pancreatic patients with cancer as well as in healthy individuals (data not shown). The expanded pTc are mainly composed of memory Tc. The largest populations among pTc in HROC113 were CD8+ central memory Tc (49.6%) and CD8+ stem cell-like memory Tc (25.8%), followed by CD4+ central memory Tc (13.5%) and CD4+ stem cell-like memory Tc (4.5%). A minority among pTc of HROC113 was identified as effector memory Tc (CD8+: 2.5%; CD4+: 1.9%) or effector Tc (CD8+: 0.5%; CD4+: 0.1%). A similar distribution pattern was observed in the pTc of HROC285. More than 90% of pTc were assigned to the stem cell-like memory Tc category (CD8+: 68.4%; CD4+: 24.7%). The central memory compartment made up 4.2% and 1.6% among the CD8+ and CD4+ Tc in HROC285, respectively. Effector memory as well as effector cells represent only 1% of Tc in HROC285.
Figure 2.

T cell populations. Expanded pTc and TiTc were classified by the detection of CD3, CD4, CD8, CD45RO, CD62L and CD95 via flow cytometry. The following subpopulations could be discriminated: naïve Tc (TN, CD3+/CD62L+/CD45RO-/CD95-), stem cell-like memory Tc (TSCM, CD3+/CD62L+/CD45RO-/CD95+), central memory Tc (TCM, CD3+/CD62L+/CD45RO+/CD95+), effector memory Tc (TEM, CD3+/CD62L-/CD45RO+/CD95+) and effector Tc (TEFF, CD3+/CD62L-/CD45RO-/CD95+). pTc, peripheral blood Tc; TiTc, tumor-infiltrating Tc.
Overall, the TiTc were composed of more effector (memory) Tc, but central memory as well as stem cell-like memory constituted only a minority among TiTc compared with the pTc (figure 2). Moreover, up to 10% of TiTc were identified as CD4+/CD8+ double positive cells. TiTc in HROC113 consisted of more CD4+ (50.9%) than CD8+ (30.8%) effector Tc; and this pattern was even stronger in TiTc HROC285. Here, 85.9% of TiTc were positive for CD4 with a larger amount of stem cell-like memory (27.4%) and central memory Tc (29.0%) than effector memory (16.3%) and effector Tc (13.2%).
Neoantigenic and cryptic peptides can induce IFNγ secretion in pTc and TiTc
TiTc and pTc were stimulated with different pools of peptides and IFNγ secretion was analyzed after 14–28 days. The goal was to investigate the potential of these identified peptides to induce an immune response. The peptide pools contained 8–12 mers and the composition was based on the predicted binding affinity to the respective HLA molecules: ‘weak’ and ‘strong’ peptide pools for HROC285 and ‘weak’, ‘strong I’ and ‘strong II’ peptide pools for HROC113 due to the high number of identified peptides (online supplemental table S2). An additional peptide stimulation was performed using the identified mutated neoantigen(s). Overall, the response of TiTc, compared with pTc, was stronger as indicated by higher numbers of spots in the IFNγ ELISpot (online supplemental figure S5). Furthermore, distinct response patterns were clearly discernible already after 14 days in the stimulated TiTc. In comparison, pTc required 28 days of stimulation. Samples with significant more spots than the negative control (t-test, p<0.05) hinted at peptides able to induce an IFNγ response in Tc.
Even though the ‘weak’ peptide pool also resulted in the faintest Tc response, three peptides were detected to induce a Tc response in the TiTc HROC113 (figure 3). Stimulations using the ‘strong’ peptide pools in TiTc from HROC113 led to IFNγ secretion ascribable to 12 of 56 tumor-specific cryptic peptides, but only one peptide of the ‘strong’ peptide pool induced an IFNγ release in pTc HROC113. The two analyzed neoantigens both reached significant numbers of reactive pTc (pIQGAP1neo=0.04; pCTNNB1neo=0.02) as well as TiTc (pIQGAP1neo=0.01; pCTNNB1neo=0.04) in HROC113.
Figure 3.

Screening for IFNγ secretion after stimulation with cryptic peptides and neoantigens. pTc and TiTc of HROC113 (A, B) and HROC285 (C, D) were stimulated with peptide pools for 28 and 14 days, respectively. Number of spots was normalized to controls. N=2–3, Significance was assessed by t-test. Striped columns: peptides with weak binding prediction; color-filled columns: peptides with strong binding prediction; orange columns: significantly increased IFNγ release (p<0.05, determined by t-test). pTc, peripheral blood Tc; TiTc, tumor-infiltrating Tc.
In pTc and TiTc of HROC285, fewer peptides triggered IFNγ release. None of the peptides of the ‘weak’ peptide pool induced a specific Tc response (figure 3). However, 2 of the 15 peptides of the ‘strong’ peptide pool elicited an IFNγ response in pTc and TiTc of HROC285. One of these peptides induced IFNγ release in pTc and in TiTc HROC285. The reactivity against the mutated neoantigen was higher in the pTc than in the TiTc stimulation approach of HROC285.
Overall, the cryptic peptides inducing a strong Tc response were bioinformatically mainly predicted to strongly bind to the respective HLA molecules, irrespective of the HLA allele per se. In addition, no correlation between translational origin (off frame, untranslated 5’ or 3’, non-coding RNA, intergenic or intronic regions) and stimulation potential became obvious.
All peptides inducing a strong Tc response validated by IFNγ secretion in this screening (table 2) were combined in one pool of immunogenic cryptic peptides per cell line and included into subsequent experiments.
Table 2.
Immunogenic cryptic peptides
| Tumor | Sequence | Gene | Symbol | Category | HLA binder | Filtered HLA allele |
| HROC113 | ALPEVQKQV | ENSG00000138085 | ATRAID | UTR5 | Strong binder | HLA-A*02:01 |
| HROC113 | ETDIEMETRY | ENSG00000253738 | GS1-251I9.4 | ncRNA | Strong binder | HLA-A*01:01 |
| HROC113 | HHSDWGNIM | ENSG00000204616 | TRIM31 | OffFrame | Strong binder | HLA-B*38:01 |
| HROC113 | HHSDWGNIMW | ENSG00000204616 | TRIM31 | OffFrame | Strong binder | HLA-B*38:01 |
| HROC285 | LFDYEVRL | ENSG00000034063 | UHRF1 | ncRNA | Strong binder | HLA-C*04:01 |
| HROC113 | LLDIDLDKY | ENSG00000259345 | RP11-624L4.1 | ncRNA | Strong binder | HLA-A*01:01 |
| HROC113 | MAALRALL | ENSG00000072121 | ZFYVE26 | UTR5 | Strong binder | HLA-B*08:01 |
| HROC113 | MARARAVAA | ENSG00000197006 | METTL9 | UTR5 | Strong binder | HLA-B*08:01 |
| HROC113 | NMKQRTERL | ENSG00000165322 | ARHGAP12 | OffFrame | Strong binder | HLA-B*08:01 |
| HROC113 | NSKKRLNTL | ENSG00000119335 | SET | OffFrame | Strong binder | HLA-B*08:01 |
| HROC113 | QTELSQLLK | ENSG00000145741 | BTF3 | UTR3 | Weak binder | HLA-A*01:01 |
| HROC113 | RLATLKSTV | ENSG00000120756 | PLS1 | UTR5 | Weak binder | HLA-A*02:01 |
| HROC285 | SLNIRTPIL | ENSG00000167635 | ZNF146 | OffFrame | Strong binder | HLA-B*08:01 |
| HROC113 | TLKDRNFQI | ENSG00000198561 | CTNND1 | UTR5 | Strong binder | HLA-B*08:01 |
| HROC113 | VPLTRILTL | ENSG00000140105 | WARS | OffFrame | Strong binder | HLA-B*08:01 |
| HROC113 | WAAPFPKLL | ENSG00000110851 | PRDM4 | UTR5 | Weak binder | HLA-C*12:03 |
| HROC113 | YHSSTDSLI | ENSG00000168036 | CTNNB1 | UTR5 | Strong binder | HLA-B*38:01 |
| HROC113 | YSSEIWDLY | ENSG00000100852 | ARHGAP5 | UTR5 | Strong binder | HLA-A*01:01 |
ncRNA, non-coding RNA; UTR3, 3’ untranslated region; UTR5, 5’ untranslated region.
Stimulation with cryptic peptides can induce degranulation in Tc and TiTc
After 14 days of stimulation with immunogenic cryptic peptides, the reactivity of pTc and TiTc against autologous tumor cells was assessed in a degranulation assay (online supplemental figure S6). Stimulated pTc HROC113 showed an increased degranulation, represented by significantly (p=0.0008) higher numbers of CD8+/CD107a+/IFNγ+ cells compared with unstimulated Tc from HROC113 (figure 4). This effect was validated in TiTc from HROC113 with 2-fold increase in degranulation observed in peptide-stimulated TiTc compared with the unstimulated control (p=0.0025).
Figure 4.
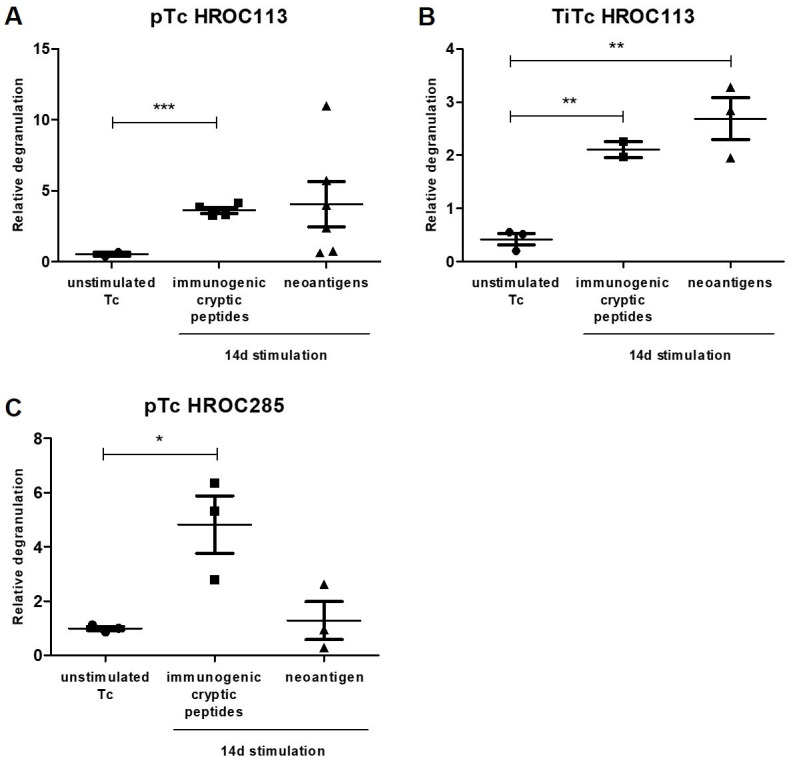
Recognition of autologous tumor cells. After 14 days of stimulation with neoantigenic or immunogenic cryptic peptides, degranulation of pTc and TiTc HROC113 (A, B) and Tc HROC285 (C) was investigated. pTc and TiTc were cocultured for 5 hours with autologous tumor cells and degranulating cells, defined as CD8+/CD107a+/IFNγ+, were detected by subsequent flow cytometric measurement. Statistical results are based on t-test indicating *p<0.05, **p<0.01, ***p<0.001. pTc, peripheral blood Tc; TiTc, tumor-infiltrating Tc.
An activating effect induced by the stimulation with immunogenic cryptic peptides was also observed in pTc from HROC285 (figure 4). Here, a 5-fold increase in degranulating cytotoxic T lymphocytes was detected (p=0.0226). The amount of cytotoxic TiTc from HROC285 decreased with further stimulation and no stimulation-induced degranulation was detected in the remaining CD8+ Tc (mean 2%). Because of the unusual high proportion of CD8- Tc in this stimulation, we also looked for degranulating CD8- Tc, but did not detect any.
Recurrency of tumor-specific cryptic peptides
To determine the recurrency of the cryptic peptides, we re-analyzed two immunopeptidome data sets of CRC organoids,24 25 one of the CRC cell line HCT11626 and one melanoma data set of tumor samples27 with Peptide-PRISM. In contrast to neoantigens, which derive from patient-specific somatic mutations and are therefore rarely recurrent, tumor-specific cryptic peptides might be shared among HLA-matched patients. Indeed, 49 of the 97 cryptic candidate peptides were identified in at least two samples from different patients (online supplemental figures S7; table S3). The HLA-A*02:01-restricted peptide ALPEVQKQV derived from all-trans retinoic acid-induced differentiation factor (ATRAID, peptide #1) had the highest recurrence frequency. This antigen was identified in eleven samples of three different patients with CRC and in all samples of four melanoma patients. ALPEVQKQV was identified in three of four HLA-A*02:01 positive patients with CRC. Ribosome profiling data from nuORFdb provided evidence that the underlying ORF is indeed translated (online supplemental figure S8). The peptide ALPEVQKQV might either be derived from the 5’-UTR of isoform 2 of ATRAID (UniProt ID Q6UW56-2; ATRAID-202, ENST00000405489.7) or from a potential transcript with retained intron (online supplemental figure S8). The C-terminus of the peptide corresponds to the last amino acid of the corresponding ORF before the stop codon, which means that no proteasomal processing is required to generate the peptides’ C-terminus. Tumor specificity of this peptide might rely on tumor-specific transcription, tumor-specific splicing, or tumor-specific translation from the corresponding cryptic ORF.
Stimulation with neoantigens can induce degranulation in Tc and TiTc
pTc and TiTc of HROC113 were stimulated for 14 days with the neoantigenic peptides derived from mutations in the genes IQGAP1 and CTNNB1. The Tc response was again analyzed in a degranulation assay. Here a 4-fold increase of cytotoxic Tc releasing their granules was observed for stimulation approaches using the mutated neoantigen-stimulated pTc compared with unstimulated pTc (figure 4). This effect was validated in TiTc from HROC113 and a highly significant difference between neoantigen-stimulated and unstimulated TiTc HROC113 was detected (p=0.0051).
Induction of CD8+/CD107a+/IFNγ+ cells by neoantigen stimulation in pTc from HROC285 was less extensive than in the HROC113 approach (figure 4). Again, no CD8+/CD107a+/IFNγ+ TiTc could be detected in HROC285.
Tetramer staining
Stimulation efficiency of tumor-specific mutated neoantigens was determined by staining of pTc and TiTc with tetramers containing the two neoantigenic peptides with high binding prediction (IQGAP1neo and TRIT1neo) prior to and after stimulation. A remarkable increase of Tc specific for IQGAP1neo was detected in pTc and TiTc from HROC113 after 14 days of stimulation (figure 5A, B). Here, a clear distinction between Tc receptive for neoantigen stimulation and Tc resistant to stimulation in the pTc from HROC113 was observed. In pTc from HROC285, the neoantigen stimulation led to a slight increase of TRIT1neo-specific Tc (p=0.1) (figure 5C). In contrast, TiTc from HROC285 showed inconsistent results concerning numbers of TRIT1neo-specifc T lymphocytes after the stimulation period, at times surpassing 5% but with a high variance of the replicates (figure 5D).
Figure 5.

T cells specific for neoantigens and cryptic peptides. At days 0 and 14 of stimulation pTc and TiTc of HROC113 were stained with tetramers specific for the neoantigen IQGAP1neo (A, B) and pTc and TiTc of HROC285 with tetramers specific for the neoantigen TRIT1neo (C, D). The amount of Tc specific the cryptic peptides #8, #18 and #32 was determined in pTc and TiTc HROC113 stimulated with B-LCL loaded with a pool of immunogenic cryptic peptides (stim) or without any peptide (ctrl) (E, F). B-LCL, B-lymphoblastoid cells; pTc, peripheral blood Tc; TiTc, tumor-infiltrating Tc.
The proliferation of Tc specific for cryptic peptides was also determined by tetramer staining. The three immunogenic cryptic peptides #8, #18 and #32 identified from HROC113 were selected based on their results in the ELISpot and recurrency analysis. Peptide-stimulated Tc and TiTc HROC113 partially showed an increased population of Tc specific for cryptic peptides, especially #32 (figure 5E, F).
Discussion
Patients with cancer with extraordinarily high numbers of somatic tumor mutations, resulting in many neoantigens, are ideal candidates for immunotherapy.34 35 Therefore, we selected two patients with MSI CRC with thousands of genetic aberrations. Identification of candidate peptides was based on expressed protein sequence modification, presence in HLA ligandome analysis and predicted binding to HLA molecules. Here, we could identify only two mutated neoantigens for HROC113—one with strong and one with weak binding prediction—and merely one for HROC285. Further studies confirmed a similarly low number of mutated neoantigens per patient with current methodology.36–39
All three mutated neoantigens identified in this study arise from single nucleotide substitutions resulting in modified amino acid sequences. IQGAP1 and TRIT1, the origins of the two neoantigens with strong HLA binding prediction, are rather uncommon mutational sites, whereas CTNNB1 is mutated in 25% of MSI CRC.40 Unfortunately, this neoantigen had the lowest binding prediction. Nonetheless, we included all three neoantigens in all sets of experiments.
In addition to mutated neoantigens, we also aimed at detecting tumor-specific cryptic peptides by choosing an immunopeptidomic approach, which has been proven successful before.7 41–43 Ligandomic analyses revealed 10,938 conventional and 128 cryptic peptides in HROC113 and HROC285 T0 M2; 97 of the latter were tumor-specific. Quality parameters like de novo score distribution, percentage of NetMHCpan-predicted non-binders, peptide length distribution and correlation of the calculated hydrophobicity with the experimental retention time confirmed that the identification of cryptic peptides is not inferior to conventional peptides. They may be ideal new targets for immunotherapy, as Tc responses, represented by IFNγ secretion, have been demonstrated before.7 42–44
Patient-derived Tc are classically the bottle-neck of immunogenic epitope identification; thus we polyclonally expanded pTc and TiTc. Subpopulation determination in the expanded pTc revealed first and foremost an increase in the CD8+ Tc population. High amounts of cytotoxic Tc form a perfect basis for peptide stimulation and raised expectations of tumor recognition by such stimulated Tc. Furthermore, the thorough subdivision of pTc revealed that most were stem cell-like memory and central memory Tc. This again was a positive portent for high tumor reactivity since especially central memory and stem cell-like memory Tc have proven superiority in cancer cell eradication to effector (memory) Tc.45
A completely different distribution was observed for the expanded TiTc from HROC113 and HROC285. After 14 days of expansion, effector and effector memory cells prevailed, strengthening the assumption of tumor-reactive Tc present in the original tumor microenvironment. Due to limited availability of biologic material, a comparison of subpopulation distribution to TiTc before the polyclonal expansion was not possible. The high quantity of CD4+ TiTc, especially in the TiTc from HROC285, underlines the importance of helper Tc activity but also hints at the possibility of cytotoxic CD4+ being present within the tumor. Moreover, the presence of CD4+/CD8+ double positive cells was striking in both TiTc populations. However, the role of these double positive Tc is still not conclusively defined, assuming potential cytotoxic as well as immunosuppressive capabilities.46
Both mutated neoantigens and 16 of 70 cryptic peptides of HROC113 triggering a significant pTc or TiTc reaction in ELISpot were mainly predicted to be strong HLA binders. Tc responses to these peptides could be replicated in more functional degranulation experiments. These results thus confirm also for cryptic, not only for neoantigenic peptides, presentation on CRC cells, induction of Tc proliferation and specific recognition by peptide-stimulated autologous Tc. Successful stimulation was detected in pTc and TiTc of HROC113, leading to the assumption that both memory as well as effector Tc are receptive for stimulation with tumor-specific peptides. Remarkably, these results indicate that Tc recognizing cryptic peptides are present in the tumor microenvironment, thereby emphasizing their future potential for cancer vaccine development. Of note, 49 of the 97 cryptic peptides of the present analysis were shared between one of the HROC cases and at least one additional tumor sample. Even more importantly, this was also true for the immunogenic cryptic peptides, where seven of the 16 peptides induced Tc activation and were found in further tumor samples. Such cryptic peptides shared by different patients’ tumors would make them ideal candidates for off-the-shelf cancer vaccine approaches.7 The efficiency of cryptic peptide containing cancer vaccines was already demonstrated in a mouse model.43 Tokita et al observed reduced CRC growth after vaccination with a mixture of three cryptic peptides, but not with individual peptides.43
The distinct degranulation of Tc and TiTc from HROC113 in response to the neoantigenic peptides lead to the classification as indeed immunogenic patient-individual neoantigens. This is further confirmed by the increasing amount of Tc with receptors for IQGAP1neo in HROC113 Tc and TiTc with stimulation time; as identified by tetramer staining. In addition, the positive results observed in the degranulation assay validate the presentation of IQGAP1neo on the HLA molecules expressed by HROC113 cells. Furthermore, as the composition of TiTc HROC113 is dominated by effector Tc which probably proliferated under mutated neoantigen stimulation, we presume a pre-existing neoantigen-specific immunity within the tumor. Still, it is important to consider the heterogeneic reactivity especially in the Tc from HROC113 and the very weak effect for both pTc and TiTc from HROC285. The human Tc receptor repertoire consists of approximately 1×108 Tc clones,47 but our experiments were performed with only up to 1×107 Tc, definitely not representing entirety of Tc clones. This might, partially, explain the negative results, as it is well possible, that Tc with receptors specific for tested peptides were simply not present among the cells used for some experiments.
In conclusion, we identified tumor-specific neoantigenic and cryptic peptides by sequencing combined with ligandomics analysis. Stimulations of pTc and TiTc confirmed multiple predicted peptides as truly immunogenic. The fact that functional assays were performed using patient’s tumor cells and autologous Tc sets this study apart from others and is the first study demonstrating the immunogenic potential of cryptic peptides in a human ex vivo setting. Furthermore, the results from these degranulation assays prove that peptide-primed pTc and TiTc recognized target cells which were not pulsed with the respective peptides but processed and presented them naturally. Beside validating the immunogenicity of cryptic peptides, we also identified three patient-individual neoantigens with the potential to induce significant Tc responses with increased release of cytotoxic granules in the presence of matching tumor cells. These results should, therefore, be a strong impetus for considering cryptic peptides besides mutation derived neoantigens for the incorporation of in future cancer vaccine development approaches.
jitc-2022-005651supp004.xlsx (53.6KB, xlsx)
Acknowledgments
The authors express their gratitude to the patients for participating in the HROC collection. The authors want to thank Wendy Bergmann and the whole team of the Core Facility for Cell Sorting and Cell Analysis of the University Medical Center Rostock for expertise in flow cytometry.
Footnotes
Contributors: Design and conceptualization of the study was directed by ML. Experimental design, data interpretation and formal analysis of WES data was performed by SK, BA, KDK, AW and PB with supervision and conceptualization by PB. Experimental design, data interpretation and formal analysis of ligandome data was conducted by MG, MWL and H-GR with supervision and conceptualization by MWL and H-GR. Experimental support and protocol expertise was provided by EO, MM, NM-T and AD. Identification, validation and reanalysis of cryptic peptides was performed by AS. Experimental design, data interpretation and formal analysis of T cell experiments were executed by SS and JS. The initial draft and figures were prepared by SS and AS; CSM, ML, SS, AS, MG, MWL and H-GR reviewed and edited the manuscript. ML accepts full responsibility for the finished work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding: This research was in part funded by grant number TBI-V-1-241-VBW-084 and TBI-V-240- VBW-084 from the state Mecklenburg-Vorpommern.
Competing interests: MWL is an inventor of patents owned by Immatics Biotechnologies and has acted as a speaker and paid consultant in cancer immunology for Boehringer Ingelheim.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available in a public, open access repository. Data were uploaded to PRIDE (PXD037587).
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by Ethics Committee of the University of Rostock University Medical Centre (II HV 43/2004 and A 45/2007). Participants gave informed consent to participate in the study before taking part.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2.Wagner S, Mullins CS, Linnebacher M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J Gastroenterol 2018;24:5418–32. 10.3748/wjg.v24.i48.5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rooney MS, Shukla SA, Wu CJ, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. 10.1016/j.cell.2014.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 5.Marijt KA, Blijleven L, Verdegaal EME, et al. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J Exp Med 2018;215:2325–37. 10.1084/jem.20180577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erhard F, Dölken L, Schilling B, et al. Identification of the cryptic HLA-I immunopeptidome. Cancer Immunol Res 2020;8:1018–26. 10.1158/2326-6066.CIR-19-0886 [DOI] [PubMed] [Google Scholar]
- 7.Marcu A, Schlosser A, Keupp A, et al. Natural and cryptic peptides dominate the immunopeptidome of atypical teratoid rhabdoid tumors. J Immunother Cancer 2021;9:e003404 https://jitc.bmj.com/content/9/10/e003404.long 10.1136/jitc-2021-003404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laumont CM, Daouda T, Laverdure J-P, et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun 2016;7:10238. 10.1038/ncomms10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haen SP, Löffler MW, Rammensee H-G, et al. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol 2020;17:595–610. 10.1038/s41571-020-0387-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong C, Müller M, Pak H, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun 2020;11:1293. 10.1038/s41467-020-14968-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouspenskaia T, Law T, Clauser KR, et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol 2022;40:209–17. 10.1038/s41587-021-01021-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laumont CM, Perreault C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell Mol Life Sci 2018;75:607–21. 10.1007/s00018-017-2628-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullins CS, Micheel B, Matschos S, et al. Integrated biobanking and tumor model establishment of human colorectal carcinoma provides excellent tools for preclinical research. Cancers 2019;11. 10.3390/cancers11101520. [Epub ahead of print: 09 10 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 2017;25:176–82. 10.1038/ejhg.2016.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowalewski DJ, Stevanović S. Biochemical large-scale identification of MHC class I ligands. Methods Mol Biol 2013;960:145–57. 10.1007/978-1-62703-218-6_12 [DOI] [PubMed] [Google Scholar]
- 16.Nelde A, Kowalewski DJ, Stevanović S. Purification and identification of naturally presented MHC class I and II ligands. Methods Mol Biol 2019;1988:123–36. 10.1007/978-1-4939-9450-2_10 [DOI] [PubMed] [Google Scholar]
- 17.Löffler MW, Kowalewski DJ, Backert L, et al. Mapping the HLA Ligandome of colorectal cancer reveals an imprint of malignant cell transformation. Cancer Res 2018;78:4627–41. 10.1158/0008-5472.CAN-17-1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Löffler MW, Mohr C, Bichmann L, et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med 2019;11:28. 10.1186/s13073-019-0636-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnstable CJ, Bodmer WF, Brown G, et al. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 1978;14:9–20. 10.1016/0092-8674(78)90296-9 [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Xin L, Shan B, et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics 2012;11:M111.010587. 10.1074/mcp.M111.010587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurtz V, Paul S, Andreatta M, et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 2017;199:3360–8. 10.4049/jimmunol.1700893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krokhin OV, Spicer V. Peptide retention standards and hydrophobicity indexes in reversed-phase high-performance liquid chromatography of peptides. Anal Chem 2009;81:9522–30. 10.1021/ac9016693 [DOI] [PubMed] [Google Scholar]
- 23.Perez-Riverol Y, Bai J, Bandla C, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022;50:D543–52. 10.1093/nar/gkab1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newey A, Griffiths B, Michaux J, et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J Immunother Cancer 2019;7:309. 10.1186/s40425-019-0769-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demmers LC, Kretzschmar K, Van Hoeck A, et al. Single-Cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat Commun 2020;11:5338. 10.1038/s41467-020-19142-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker JP, Helm D, Rettel M, et al. Nmd inhibition by 5-azacytidine augments presentation of immunogenic frameshift-derived neoepitopes. iScience 2021;24:102389. 10.1016/j.isci.2021.102389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassani-Sternberg M, Bräunlein E, Klar R, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 2016;7:13. 10.1038/ncomms13404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dudley ME, Wunderlich JR, Shelton TE, et al. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 2003;26:332–42. 10.1097/00002371-200307000-00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tosato G, Cohen JI. Generation of Epstein-Barr Virus (EBV)-immortalized B cell lines. Curr Protoc Immunol 2007;Chapter 7:Unit 7.22. 10.1002/0471142735.im0722s76 [DOI] [PubMed] [Google Scholar]
- 30.Cattaneo CM, Dijkstra KK, Fanchi LF, et al. Tumor organoid-T-cell coculture systems. Nat Protoc 2020;15:15–39. 10.1038/s41596-019-0232-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreatta M, Lund O, Nielsen M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics 2013;29:8–14. 10.1093/bioinformatics/bts621 [DOI] [PubMed] [Google Scholar]
- 32.Javitt A, Barnea E, Kramer MP, et al. Pro-Inflammatory cytokines alter the immunopeptidome landscape by modulation of HLA-B expression. Front Immunol 2019;10:141. 10.3389/fimmu.2019.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcu A, Bichmann L, Kuchenbecker L, et al. HLA ligand atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J Immunother Cancer 2021;9:e002071 https://jitc.bmj.com/content/9/4/e002071 10.1136/jitc-2020-002071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cristescu R, Aurora-Garg D, Albright A, et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer 2022;10:e003091 https://jitc.bmj.com/content/10/1/e003091 10.1136/jitc-2021-003091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang T, Chen X, Zhang H, et al. Prognostic role of tumor mutational burden in cancer patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Front Oncol 2021;11:706652. 10.3389/fonc.2021.706652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24:743–50. 10.1101/gr.165985.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalaora S, Barnea E, Merhavi-Shoham E, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016;7:5110–7. 10.18632/oncotarget.6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran E, Ahmadzadeh M, Lu Y-C, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350:1387–90. 10.1126/science.aad1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirama T, Tokita S, Nakatsugawa M, et al. Proteogenomic identification of an immunogenic HLA class I neoantigen in mismatch repair-deficient colorectal cancer tissue. JCI Insight 2021;6. 10.1172/jci.insight.146356. [Epub ahead of print: 22 07 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirabelli-Primdahl L, Gryfe R, Kim H, et al. Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res 1999;59:3346–51. [PubMed] [Google Scholar]
- 41.Cleyle J, Hardy M-P, Minati R, et al. Immunopeptidomic analyses of colorectal cancers with and without microsatellite instability. Mol Cell Proteomics 2022;21:100228. 10.1016/j.mcpro.2022.100228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kikuchi Y, Tokita S, Hirama T, et al. CD8+ T-cell Immune Surveillance against a Tumor Antigen Encoded by the Oncogenic Long Noncoding RNA PVT1. Cancer Immunol Res 2021;9:1342–53. 10.1158/2326-6066.CIR-20-0964 [DOI] [PubMed] [Google Scholar]
- 43.Tokita S, Kanaseki T, Torigoe T. Therapeutic potential of cancer vaccine based on MHC class I cryptic peptides derived from non-coding regions. Immuno 2021;1:424–31. 10.3390/immuno1040030 [DOI] [Google Scholar]
- 44.Bredenbeck A, Losch FO, Sharav T, et al. Identification of noncanonical melanoma-associated T cell epitopes for cancer immunotherapy. J Immunol 2005;174:6716–24. 10.4049/jimmunol.174.11.6716 [DOI] [PubMed] [Google Scholar]
- 45.Liu Q, Sun Z, Chen L. Memory T cells: strategies for optimizing tumor immunotherapy. Protein Cell 2020;11:549–64. 10.1007/s13238-020-00707-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overgaard NH, Jung J-W, Steptoe RJ, et al. CD4+/CD8+ double-positive T cells: more than just a developmental stage? J Leukoc Biol 2015;97:31–8. 10.1189/jlb.1RU0814-382 [DOI] [PubMed] [Google Scholar]
- 47.Qi Q, Liu Y, Cheng Y, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A 2014;111:13139–44. 10.1073/pnas.1409155111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-005651supp002.xlsx (3.4MB, xlsx)
jitc-2022-005651supp001.pdf (1.8MB, pdf)
jitc-2022-005651supp003.xlsx (33.9KB, xlsx)
jitc-2022-005651supp004.xlsx (53.6KB, xlsx)
Data Availability Statement
Data are available in a public, open access repository. Data were uploaded to PRIDE (PXD037587).