

Abstract
Bacillus subtilis is the leading model Gram-positive bacterium, and a widely used chassis for industrial protein production. However, B. subtilis research is limited by a lack of inducible promoter systems with low leakiness and high dynamic range. Here, we engineer an inducible promoter system based on the T7 RNA Polymerase, the lactose repressor LacI, and a chimeric T7lac promoter PT7lac, integrated as a single copy in the B. subtilis genome. In the absence of IPTG, LacI strongly represses T7RNAP and PT7lac and minimizes leakiness. Addition of IPTG de-represses PT7lac and simultaneously induces expression of T7RNAP, resulting in very high output expression. Using green fluorescent and β-galactosidase reporter proteins, we estimate that LacI-T7 can regulate expression with a dynamic range of over 10,000, by far the largest reported for an inducible B. subtilis promoter system. Furthermore, LacI-T7 responds to similar IPTG concentrations and with similar kinetics as the widely used Phy-spank IPTG-inducible system, which we show has a dynamic range of at most 300 in a similar genetic context. Due to its superior performance, our LacI-T7 system should have broad applications in fundamental B. subtilis biology studies and biotechnological applications.
Keywords: Bacillus subtilis, protein expression, inducible promoter system, IPTG, T7 RNA Polymerase
Graphical Abstract
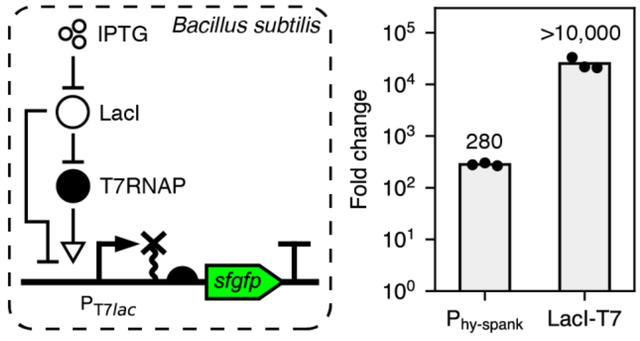
Bacillus subtilis is a model organism for studying Gram-positive bacterial biology and systems biology of cellular differentiation1, stress responses2, and multicellular organization3. Additionally, B. subtilis is among the most widely used hosts for protein production in the biotechnology industry due to its ability to secrete proteins into the cell medium, its non-pathogenic GRAS (generally recognized as safe) designation, and its high genetic tractability. For example, B. subtilis is used for large-scale production of lipases, proteases, and amylases, among other industrially-relevant proteins4,5.
Although many libraries of synthetic biological parts, such as constitutive promoters, RBSs, and degradation tags, have been reported for B. subtilis6–8, high-quality inducible expression systems with low leakiness and high expression are still needed. For example, biotechnology applications require inducible promoters capable of switching between a low production state for early-stage culturing and a high production state that maximizes protein yield during fermentation. Such parts are particularly important when the recombinant protein or metabolic pathway of interest are toxic to the host cells when overproduced9. Typical inducible promoters in B. subtilis have dynamic ranges (ratio of output protein expression in the presence versus absence of inducer) of at most a few hundred10–15. While a B. subtilis bacitracin-inducible promoter with a 1,000-fold range has been reported16, it requires antibiotic selection to maintain a multicopy plasmid, and its activity is transient and shuts down less than two hours after induction, likely due to an endogenous bacitracin stress response. In contrast, for the Gram-negative bacterium Escherichia coli, inducible promoters have been engineered with dynamic ranges greater than 1,00017–19 or even 10,00020. Thus, improved B. subtilis inducible promoter systems are needed.
To address this issue, we engineered a stringent (i.e. non-leaky) and highly-inducible LacI-T7 promoter system for B. subtilis. Our system utilizes the hybrid PT7lac promoter17 to express a gene of interest, and the IPTG-inducible promoter Phy-spank21 to express the T7 RNA Polymerase (T7 RNAP) (Figure 1A). In the absence of inducer, LacI, which is constitutively expressed from the PpenP promoter10, represses both expression of T7 RNAP and transcription from PT7lac. This dual repression is expected to minimize leaky expression of the gene of interest. Upon addition of IPTG, LacI activity is inhibited, and the newly produced T7 RNAP strongly transcribes the gene of interest from de-repressed PT7lac (Figure 1A). Our LacI-T7 design is conceptually similar to some variants of the commercial E. coli pET expression system, where IPTG also induces both expression of T7 RNAP and de-repression of PT7lac17. However, our system uses B. subtilis-specific promoters (other than PT7lac) and ribosome-binding sites (RBSs). Additionally, while a few B. subtilis gene expression systems based on T7 RNAP have been previously reported12,15, they use the LacI-independent PT7 instead of PT7lac, which result in dynamic ranges of less than 50.
Figure 1. LacI-T7 design and performance.
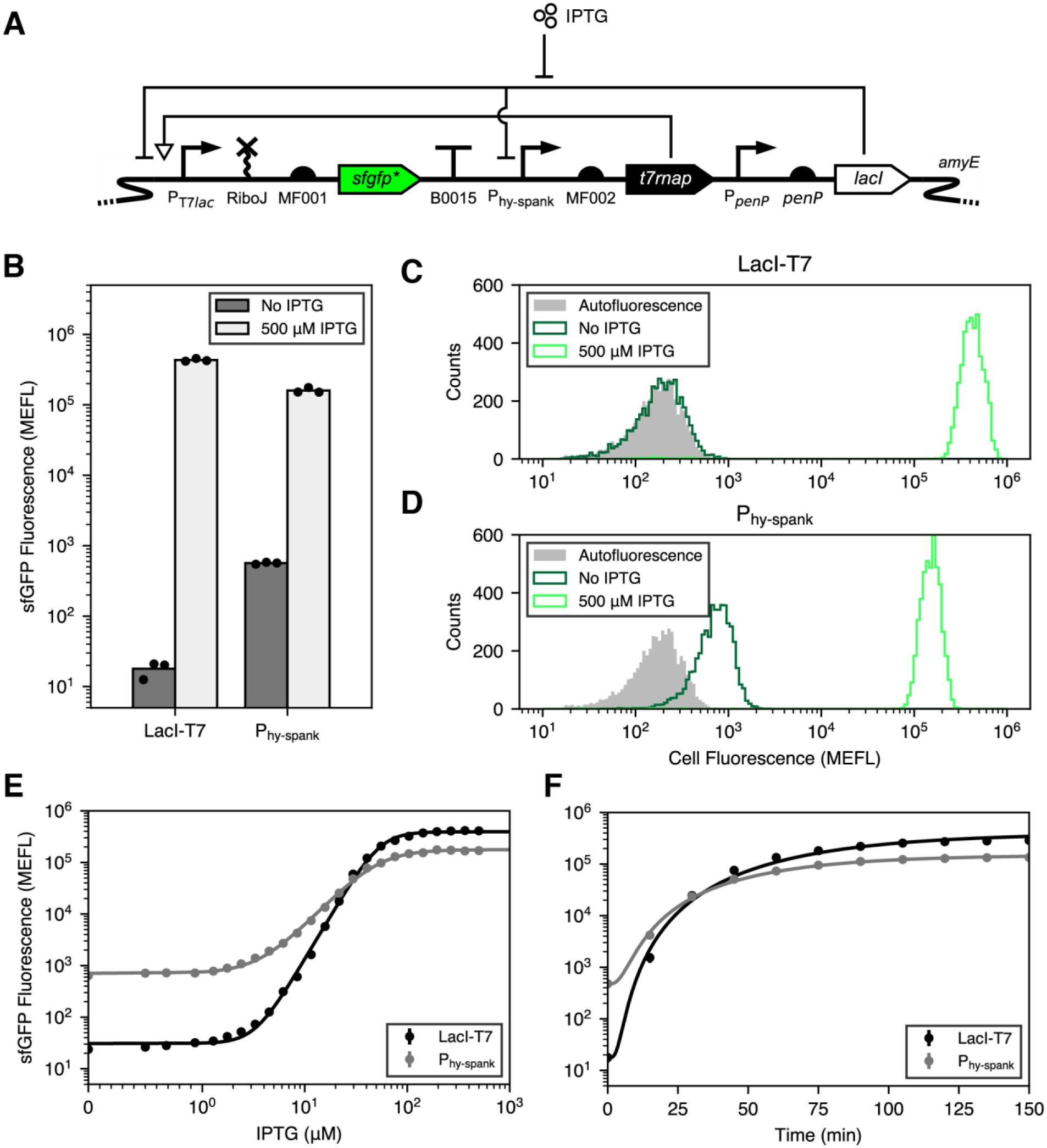
(A) Genetic device schematic of LacI-T7 with regulatory interactions shown. (B) sfGFP fluorescence from LacI-T7 and Phy-spank in the absence or presence of IPTG. Fluorescence is shown in calibrated Molecules of Equivalent Fluorescein (MEFL) units. Bars represent the mean of three experimental replicates run on separate days. Dots show the values of the individual replicates. (Methods). (C and D) Representative flow cytometry histograms of autofluorescence control (B. subtilis lacking any sfgfp gene), and those with sfgfp under either LacI-T7 (C) or Phy-spank (D), in the presence or absence of IPTG. (E) sfGFP Fluorescence from LacI-T7 and Phy-spank as a function of IPTG concentration. (F) sfGFP fluourescence from LacI-T7 and Phy-spank after addition of saturating IPTG. Dots and error bars show the mean and standard deviation, respectively, of three experiments run on separate days. Error bars are, in most cases, smaller than the size of the dots, and thus not visible. Black lines represent model fits (Methods).
To validate our design, we placed a sfgfp reporter gene22 with a codon-optimized N-terminal fragment (sfgfp*, Methods) under control of LacI-T7, the self-cleaving ribozyme RiboJ23, and RBS MF001 (Methods), and integrated it into the non-essential amyE locus of the B. subtilis genome as a single copy (Figure 1A, Methods). Then, we utilized flow cytometry to characterize sfGFP fluorescence levels in the absence and presence of IPTG. We found that, in the absence of inducer, sfGFP fluorescence equals 17.9 ± 4.7 molecules of equivalent fluorescein (MEFL)24 (Figure 1B and C, Methods). In contrast, sfGFP levels in the presence of IPTG are 432,000 ± 20,000 MEFL (Figure 1B and C), resulting in a dynamic range of 25,300 ± 6,900. Remarkably, only a small growth slowdown was observed under inducing conditions (cell division time: 25.15 ± 0.46 minutes without IPTG, 30.2 ± 1.1 minutes with IPTG) (Supplementary Figure 1A). Polyacrylamide gel electrophoresis (PAGE) analysis of total cellular protein indicated that sfGFP is the mostly highly expressed protein in the cell in the presence of IPTG (Supplementary Figure 2). To validate that this high dynamic range is preserved when expressing a different gene of interest, we placed the lacZ reporter gene under LacI-T7 and measured β-galactosidase activity in the absence and presence of IPTG. This resulted in a dynamic range of 11,000 ± 1,200 (Supplementary Figure 3). The difference between this value and the one obtained with sfGFP might be explained by uncertainty in reporter measurements under non-inducing conditions, which result in signals that are close to background. Again, we observed only a mild growth slowdown (cell division time: 23.84 ± 0.22 minutes without IPTG, 28.49 ± 0.98 minutes with IPTG) (Supplementary Figure 1B). We conclude that LacI-T7 can regulate expression of several genes with a dynamic range greater than 10,000-fold with little toxicity.
Phy-spank is a variant of the IPTG-inducible promoter Pspac optimized for higher expression and dynamic range10,21,25, and is perhaps the most widely used B. subtilis inducible promoter system1,2,26. We constructed a second B. subtilis strain wherein sfgfp* was expressed under the control of Phy-spank, RiboJ, and RBS MF001 to compare its performance to that of LacI-T7 (Methods). We found that Phy-spank exhibits much greater leakiness (565 ± 18 MEFL) and a lower maximal output (160,000 ± 14,000 MEFL) than LacI-T7, resulting in a dynamic range of only 282 ± 18 (Figure 1B, D). Next, we measured the steady state transfer function of both systems by growing the corresponding strains under different concentrations of IPTG. In both cases, sfGFP fluorescence increases as a function of IPTG concentration in a manner well-approximated by a Hill function (Figure 1E). Remarkably, both systems exhibit similar IPTG detection thresholds (50% activation concentrations: LacI-T7: 54.1 ± 1.6 μM, Phy-spank: 50.2 ± 1.5 μM). On the other hand, the Hill coefficient of the LacI-T7 system is larger (3.377 ± 0.051, compared to 2.120 ± 0.033 for Phy-spank), indicating that protein expression is more sensitive to changes in IPTG levels in the responsive range of the system. Finally, we characterized the response dynamics of both systems after an instantaneous addition of saturating IPTG (Figure 1F). As expected, both responses show an exponential-like increase until saturation. However, LacI-T7 responds slightly slower (t1/2 = 82.1 ± 6.5 min for LacI-T7, 62.1 ± 1.1 min for Phy-spank), consistent with the need to produce an intermediate protein (T7RNAP) before expression of the reporter gene. In conclusion, LacI-T7 exhibits lower leakiness, higher maximal expression output, similar sensitivity to inducer, and much higher dynamic range than the widely used Phy-spank system, albeit with a slightly slower response time.
The superior performance of LacI-T7 appears to arise from its unique design features. First, high maximal expression results from the use of T7 RNAP, a strong viral RNA polymerase which is capable of re-directing all bacterial resources towards expression of a single output gene27. In contrast, previous expression systems based on endogenous promoters are limited by the native transcriptional machinery and are subject to competition with other endogenous promoters. Second, leaky expression in the absence of inducer is reduced via the dual repression activity of LacI. In E. coli, a similar design has been shown to reduce basal expression by more than an order of magnitude compared to an unmodified PT7 output promoter17.
LacI-T7 should be useful in quantitative studies of B. subtilis biology. In particular, its stable single-copy chromosomal location and its low leakiness are desirable for analyses of ultrasensitive or excitable networks where low amounts of excess protein can cause cells to undergo dramatically different differentiation programs28.
We also expect LacI-T7 to be useful for heterologous protein expression applications. High expression from PT7lac should enable high yields of both cytoplasmic and secreted proteins. Additionally, low leakiness in the absence of inducer should allow for fast initial cell growth, even with potentially toxic proteins. Furthermore, LacI-T7 is integrated into the B. subtilis genome, and thus will not suffer from plasmid instability issues29 or require strong selective pressure to be maintained. Because it relies on the orthogonal T7 polymerase, LacI-T7 could also be ported to other industrially-relevant Bacillus species or strains with little additional work.
In conclusion, we have engineered a B. subtilis inducible promoter system with superior performance to current alternatives. Our system addresses important needs in fundamental and applied B. subtilis research.
Methods
DNA and strain construction
All cloning and experiments were performed in B. subtilis strain PY7930. Primers were ordered from Integrated DNA Technologies, Inc. Phy-spank was amplified from integration plasmid pDR11121. PT7lac was constructed via oligo annealing and extension. Synthetic RBS MF001 was obtained from integration plasmid pMF3531. Genomic homology fragments required for chromosomal integration were amplified from the purified genome of B. subtilis PY79. A list of genetic parts, along with their sequences, can be found in Supplementary Table S1.
All systems were built as linear double-stranded integration module (IM), as we have previously described32. IMs contain the DNA of interest and a selection marker flanked by 1.5kb-long sequences homologous to the amyE locus of the B. subtilis genome where chromosomal integration via double crossover occurs. IMs were assembled from PCR-amplified parts using Golden Gate33. The resulting Golden Gate product was amplified using NEB Phusion DNA Polymerase and gel purified to obtain the IM. 500 ng was transformed into competent B. subtilis using standard transformation methods. The transformants were plated on selective media. Colonies were picked the next day and grown in LB media at 37 °C and 250 RPM for a few hours. Finally, freezer stocks were prepared with 700 μL culture and 300 μL 60% glycerol, and stored at −80 °C. This method avoids sub-cloning of integration plasmids in E. coli, as long as enough PCR-amplified DNA can be obtained. The complete sequences of all IMs constructed in this study can be found in Genbank via the following accession numbers: Phy-spank-sfgfp: MN005205, LacI-T7-sfgfp: MN005204, LacI-T7-lacZ: MN005206.
For DNA sequence verification, an overnight LB culture was grown from a freezer stock, and 2 μL saturated culture was used as template for a 50 μL PCR reaction, either with Taq or Phusion DNA Polymerase. PCR products obtained in this fashion were gel-purified and sent for sequence verification to Genewiz, Inc.
Codon optimization of sfgfp
Codon optimization of the N-terminal sequence of the sfgfp ORF was performed to decrease secondary structure with the RBS and increase translation efficiency. To do so, for each of the first 15 codons of the original sfgfp sequence22, a synonymous codon was chosen to reduce GC and increase AU content, with A preferred over U, with no regard for codon frequency. These changes were confirmed to increase the mRNA secondary structure free energy (and thus decrease secondary structure stability) via Nupack34, by using the sequence from the transcription start site up to the 90th nucleotide residue of the ORF. The complete optimized sfgfp* sequence can be found in Supplementary Table S1.
Media and Experimental Protocols
We used a modified M9 medium for all experiments. 1L 5xM9 salts at pH ~ 6.8 were prepared with 64 g Na2HPO4.7H2O, 15 g KH2PO4, 2.5 g NaCl, 5 g NH4Cl, 9.2 mL 6M HCl, and up to 1 L dH2O. For 1 L M9, we used 200 mL 5x M9 salts, 20 mL 10% casamino acids, 6.67 mL 60% glycerol, 1 mL 50 mM FeCl3/100 mM C6H8O7 solution, 2 mL 50 mM MnSO4, 2 mL 1M MgSO4, 100 μL 1M CaCl2, and dH2O up to 1 L.
For experiments in Figure 1B–D, an overnight LB culture was started from the freezer stock of each relevant strain. The next day, saturated cultures (OD600 ~ 3) were diluted 105-fold in M9. Media was distributed in culture tubes (3 mL per tube), inoculated with the appropriate inducers (0 μM or 500 μM IPTG), and incubated in a shaker operating at 250 rpm and 37 °C, until the OD600 reached between 0.08 and 0.15 (around 6 hours). Culture tubes were then transferred to an ice-water bath. 100 μL of each sample was transferred to a flow cytometry tube containing 1 mL PBS for measurement.
For the experiment in Supplementary Figure 1, saturated overnight cultures of the two LacI-T7 strains were diluted 105-fold in M9 media and distributed in two containers with 19 mL media each. IPTG was added to 500 μM for only one container per strain. Media from each container was then distributed into six culture tubes (3 mL per tube) and incubated at 250 rpm and 37 °C. At the indicated timepoints, one tube per strain and IPTG condition was removed from the shaker, its OD600 was measured using a spectrophotometer (Varian Cary 50 Bio), and the tube was discarded.
For the experiment in Supplementary Figure 2, saturated overnight cultures of each relevant strain were diluted 105-fold in 50 mL M9 media, distributed in two 250 mL flasks each, and incubated in a shaker until an OD600 of around 0.3. Then, IPTG was added to 1 mM for only one flask of each strain, and incubation continued for three additional hours. Whole cell lysates of each culture were prepared by sonication. Total proteins were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE, 18%, 30:0.15 acrylamide:bis ratio) and detected by Coomassie brilliant blue staining. Immunodetection was carried out using polyclonal antibodies against GFP35 and σA 36. Primary antibodies were detected using an alkaline phosphatase coupled secondary antibody (Anti-rabbit IgG, Promega). A colorimetric detection system using nitro blue tetrazolium-5bromo-4-chloro-3-indoyl phosphate (NBT/BCIP, Promega) as a substrate was utilized to detect the proteins of interest.
For experiments in Supplementary Figure 3, saturated overnight cultures of each relevant strain were diluted 105-fold in M9 media, distributed in culture tubes, inoculated with inducers, and incubated in a shaker until an appropriate OD600, as described above. β-galactosidase assays were performed on each sample as previously described37. The reported β-galactosidase activity values were obtained by subtracting the measured activity of a wild-type PY79 negative control sample measured the same day from the measured activity of each sample.
For experiments in Figure 1E, saturated overnight cultures of each relevant strain were diluted 105-fold in M9 media as above. Next, media was distributed across wells of a flat-bottom 24-well plate (Fisher 07-200-84), IPTG was added, and plates were sealed with adhesive foil (VWR 60941–126). Incubation and flow cytometry measurements were conducted as above.
For experiments in Figure 1F, saturated overnight cultures of each relevant strain were diluted 105-fold in 25 mL M9 media contained in a 250 mL Erlenmeyer glass flask. Flasks were incubated as described above for 5:30h, after which IPTG was added to 500 μM. Samples of 100 μL were removed from each flask every 15 minutes and placed in flow cytometry tubes with 1mL PBS located in an ice-water bath. Flow cytometry measurements were conducted at the end of sample acquisition, as described above.
Flow Cytometry Analysis
The sfGFP fluorescence distribution of each culture was measured using a BD FACScan flow cytometer with an excitation source of 488 nm and an emission window of 510/21 nm. 10,000 events were collected per sample. A suspension of calibration beads (Spherotech RCP-30–5A) in PBS was measured with each experiment. After data acquisition, raw .fcs flow cytometry files were processed using FlowCal24. Cell populations were gated by forward scatter/side scatter density (Supplementary Figure 4) retaining 50% of the total number of events. Next, fluorescence of each gated event in arbitrary units was converted into standardized MEFL values using the calibration bead data. The total cellular fluorescence of each culture sample was then obtained by calculating the median MEFL fluorescence of all gated events in that sample. Finally, the reported sfGFP fluorescence values were obtained by subtracting the total cellular fluorescence of a wild-type PY79 sample measured the same day from each sample’s total cellular fluorescence. Numerical sfGFP fluorescence values of every sample and replicate can be found in Supplementary Data 1.
Statistical analysis
Each experiment was replicated three times over different days. Fluorescence of each sample is reported as the mean ± standard deviation of the sfGFP fluorescence from three experiments. A one-sample Student’s t-test was conducted for every sample to evaluate whether sfGFP fluorescence was significantly different from zero (one-sided, p < 0.05). Fluorescence of samples that failed this test are reported as “Not Detected” or “N.D.”.
Growth model fitting
OD600 data in Supplementary Figure 1 was fitted to an exponential growth model of the form:
Where x(t) is the OD600 of the culture at time t, x0 is the initial OD600, and td is the doubling time. Fitting was performed using the LmFit python package38 with the Levenberg-Marquardt algorithm. To adequately fit low and high OD600 values, the error to minimize was defined as the difference between the logarithm of an OD600 datapoint and the logarithm of the model prediction.
Transfer function fitting
Steady state transfer functions in Figure 1E were fitted to a Hill Function of the form:
Here, y is the observed sfGFP fluorescence in MEFL, which has a minimum value of y0 in the absence of inducer and a maximum of y0 + Δy under saturating conditions, x is IPTG concentration in μM, K1/2 is the inducer concentration for half-maximum activation, and n is the Hill coefficient. Fitting was performed using the LmFit as described above. Experimental data from three replicates were combined and fitted simultaneously. Fitted parameter values and their uncertainties can be found in Supplementary Table S2 (Phy-spank-sfgfp) and Supplementary Table S3 (LacI-T7-sfgfp).
Kinetic model fitting
The response of the Phy-spank system to a step inducer addition was modeled as a differential equation system of the form:
Here, p(t) represents the sfGFP production rate, g(t) represents immature sfGFP, and G(t) is the fully mature, observed sfGFP. Their dynamics are determined by rate constants kp, kg, and kd, the last of which corresponds to the cell growth rate. Finally, c is the system input. Units for c, p(t), and G(t) have been chosen such that, in steady state, c = pss = gss = Gss, and thus c determines the steady state output fluorescence.
Similarly, the response of the LacI-T7 system to a step inducer addition was modeled as a differential equation system of the form:
Where the additional term r(t) represents the T7 RNAP, whose dynamics are determined by kd (cell division) just as with G(t). Fitting was performed using LmFit as described above.
Supplementary Material
Acknowledgements
This work was supported by a Michel Systems Biology Innovation Award from Rice University and the National Institutes of Health (1R21AI115014-01A1). O.A.I. also acknowledges support from the National Science Foundation (MCB-1616755) and the Welch Foundation (Grant C-1995).
Footnotes
Conflict of Interest Statement
A provisional patent on the B. subtilis LacI-T7 system is being filed by Rice University with J.J.T. and S.M.C. as inventors.
References
- 1.Narula J et al. Chromosomal Arrangement of Phosphorelay Genes Couples Sporulation and DNA Replication. Cell 162, 328–337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Locke JCW, Young JW, Fontes M, Jiménez MJH & Elowitz MB Stochastic Pulse Regulation in Bacterial Stress Response. Science 334, 366–369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prindle A et al. Ion channels enable electrical communication in bacterial communities. Nature 527, 59–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westers L, Westers H & Quax WJ Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim. Biophys. Acta BBA - Mol. Cell Res 1694, 299–310 (2004). [DOI] [PubMed] [Google Scholar]
- 5.van Dijl J & Hecker M Bacillus subtilis: from soil bacterium to super-secreting cell factory. Microb. Cell Factories 12, 3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radeck J et al. The Bacillus BioBrick Box: generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng 7, 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guiziou S et al. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res 44, 7495–7508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popp PF, Dotzler M, Radeck J, Bartels J & Mascher T The Bacillus BioBrick Box 2.0: expanding the genetic toolbox for the standardized work with Bacillus subtilis. Sci. Rep 7, 15058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu S-C & Wong S-L Engineering of a Bacillus subtilis Strain with Adjustable Levels of Intracellular Biotin for Secretory Production of Functional Streptavidin. Appl Env. Microbiol 68, 1102–1108 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yansura DG & Henner DJ Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis. Proc. Natl. Acad. Sci 81, 439–443 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim L, Mogk A & Schumann W A xylose-inducible Bacillus subtilis integration vector and its application. Gene 181, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Conrad B, Savchenko RS, Breves R & Hofemeister J A T7 promoter-specific, inducible protein expression system forBacillus subtilis. Mol. Gen. Genet. MGG 250, 230–236 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Bhavsar AP, Zhao X & Brown ED Development and Characterization of a Xylose-Dependent System for Expression of Cloned Genes inBacillus subtilis: Conditional Complementation of a Teichoic Acid Mutant. Appl Env. Microbiol 67, 403–410 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bongers RS, Veening J-W, Wieringen MV, Kuipers OP & Kleerebezem M Development and Characterization of a Subtilin-Regulated Expression System in Bacillus subtilis: Strict Control of Gene Expression by Addition of Subtilin. Appl Env. Microbiol 71, 8818–8824 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen PT, Shaw J-F, Chao Y-P, David Ho T-H & Yu S-M Construction of Chromosomally Located T7 Expression System for Production of Heterologous Secreted Proteins in Bacillus subtilis. J. Agric. Food Chem 58, 5392–5399 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Toymentseva AA, Schrecke K, Sharipova MR & Mascher T The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter. Microb. Cell Factories 11, 143 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubendorf JW & Studier FW Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol 219, 45–59 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Guzman LM, Belin D, Carson MJ & Beckwith J Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol 177, 4121–4130 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutz R & Bujard H Independent and Tight Regulation of Transcriptional Units in Escherichia Coli Via the LacR/O, the TetR/O and AraC/I1-I2 Regulatory Elements. Nucleic Acids Res 25, 1203–1210 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X et al. An extraordinary stringent and sensitive light-switchable gene expression system for bacterial cells. Cell Res 26, 854–857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Britton RA et al. Genome-Wide Analysis of the Stationary-Phase Sigma Factor (Sigma-H) Regulon of Bacillus subtilis. J. Bacteriol 184, 4881–4890 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC & Waldo GS Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol 24, 79–88 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Lou C, Stanton B, Chen Y-J, Munsky B & Voigt CA Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol 30, 1137–1142 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castillo-Hair SM et al. FlowCal: A User-Friendly, Open Source Software Tool for Automatically Converting Flow Cytometry Data from Arbitrary to Calibrated Units. ACS Synth. Biol 5, 774–780 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quisel JD, Burkholder WF & Grossman AD In Vivo Effects of Sporulation Kinases on Mutant Spo0A Proteins in Bacillus subtilis. J. Bacteriol 183, 6573–6578 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Gestel J, Weissing FJ, Kuipers OP & Kovács ÁT Density of founder cells affects spatial pattern formation and cooperation in Bacillus subtilis biofilms. ISME J 8, 2069–2079 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Studier FW & Moffatt BA Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol 189, 113–130 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Süel GM, Garcia-Ojalvo J, Liberman LM & Elowitz MB An excitable gene regulatory circuit induces transient cellular differentiation. Nature 440, 545–550 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Haima P, Bron S & Venema G The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol. Gen. Genet. MGG 209, 335–342 (1987). [DOI] [PubMed] [Google Scholar]
- 30.Zeigler DR et al. The Origins of 168, W23, and Other Bacillus subtilis Legacy Strains. J. Bacteriol 190, 6983–6995 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujita M & Losick R An investigation into the compartmentalization of the sporulation transcription factor σE in Bacillus subtilis. Mol. Microbiol 43, 27–38 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Landry BP, Palanki R, Dyulgyarov N, Hartsough LA & Tabor JJ Phosphatase activity tunes two-component system sensor detection threshold. Nat. Commun 9, 1433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engler C, Gruetzner R, Kandzia R & Marillonnet S Golden Gate Shuffling: A One-Pot DNA Shuffling Method Based on Type IIs Restriction Enzymes. PLoS ONE 4, e5553 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zadeh JN et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem 32, 170–173 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Eswaramoorthy P, Guo T & Fujita M In Vivo Domain-Based Functional Analysis of the Major Sporulation Sensor Kinase, KinA, in Bacillus subtilis. J. Bacteriol 191, 5358–5368 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujita M Temporal and selective association of multiple sigma factors with RNA polymerase during sporulation in Bacillus subtilis. Genes Cells Devoted Mol. Cell. Mech 5, 79–88 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Harwood CR & Cutting SM Molecular biological methods for Bacillus (Wiley, 1990). [Google Scholar]
- 38.Newville M et al. Lmfit: Non-Linear Least-Square Minimization and Curve-Fitting for Python. Astrophys. Source Code Libr (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.