

Abstract
Synthetic molecules capable of DNA binding and mimicking cooperation of transcription factor (TF) pairs have long been considered as a promising tool for manipulating gene expression. Our previous reported Pip-HoGu system, a programmable DNA binder pyrrole–imidazole polyamides (PIPs) conjugated to host−guest moiety, defined a general framework for mimicking cooperative TF pair−DNA interactions. Here, we supplanted the cooperation modules with left-handed (LH) γPNA modules: i.e., PIPs conjugated with nucleic acid-based cooperation system (Pip-NaCo). LH γPNA was chosen due to its bioorthogonality, sequence specific interaction, and high binding affinity toward the partner strand. The cooperativity is highly comparable with natural TF pair-DNA system, with a minimum energetics of cooperation of –3.27 kcal mol–1. Moreover, through changing the linker conjugation site, binding mode, and the length of γPNAs sequence, the cooperative energetics of Pip-NaCo can be tuned independently and reasonably. Current Pip-NaCo platform might also have the potential for precise manipulation of biological processes through the constitution of triple to multiple hetero binding systems.
Keywords: Cooperative binding, Orthogonality, Gap distance, Transcription factor pair, Pyrrole imidazole-polyamide, γ peptide nucleic acid
Graphical Abstract
A powerful cooperative DNA binding system Pip-NaCo (pyrrole-imidazole polyamide conjugated with orthogonal left-handed γPNA) has been developed and showed that the cooperativity is highly comparable with the natural system, with a minimum energetics of cooperation of –3.27 kcal mol–1. The current system, with its properties of orthogonality, tenability, and cooperativity, could be utilized as chemical tool for precise gene regulation.
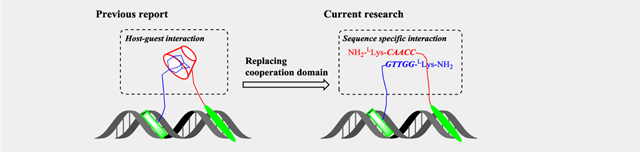
Introduction
Spatial-temporal gene expression are precisely controlled by above 1000 transcription factors (TFs) that recognize around 200 short DNA motifs in mammals.[1] Usually, TFs function as cooperative TF-TF pairs via formation of noncovalently bound homo-/heterodimers which occur in different orientations and/or gap spacings relative to each other.[2] The effect of versatile gap spacings between TF-TF pair on gene activation have been well-characterized,[3] and TF pairs flexibly facilitate mutual binding in diverse binding orientations.[4] For example, the binding site of the C-clamp of T-cell factor (TCF), which is indispensable for specific gene activation via Wnt pathway, can act as a helper by swinging to localize upstream or downstream of the classical high-mobility group (HMG) binding sites.[5] Programmable molecules, e.g., nucleic acid analogues, pyrrole–imidazole polyamides (PIPs), short peptides, and peptide-small molecule covalent conjugates, have been widely applied for disrupting individual TF–DNA interactions.[6] However, they cannot block interactions between collaborative TF pairs and DNA. Therefore, new strategies, especially the incorporation of modules allowing cooperative interactions between DNA binders, are needed to address these challenges in a deliberate and precise manipulation of gene expression patterns.[7]
PIP is currently the best characterized programmable DNA minor-groove binder, and binds according to the rules of Py/Im with C/G, Im/Py with G/C, and Py/Py with A/T and T/A.[8] Recently, we reported a PIP conjugating host-guest cooperation based system, named Pip-HoGu, for targeting cooperative TF pairs (Figure 1).[9] From in vitro and cell-based assays, Pip-HoGu exhibits potent cooperation with spacings of ≤5 nt between two DNA binders. The essence of cooperativity in DNA binding is that, the addition of the partner strand can highly stabilize binding of the overall complexes and the difference in ability to form complexes in the absence/presence of partner strand reflects the magnitude of cooperativity[10]. In addition, the dual binders should prefer to bind the DNA sites containing dual target sites simultaneously in a proper binding orientation, while decreasing the ratio of monomer binding. Moreover, cooperativity should be capable of sequence selectivity to avoid mismatch binding to an extent, and it should also bind degenerate DNA sites with reasonable affinity in some conditions.[4]
Figure 1.

Schematic illustration of the current research design. Based on our previously reported Pip-HoGu system, the host-guest interaction domain was replaced with a nucleic acid-based sequence-specific interaction domain, termed Pip-NaCo.
There are several potential limitations of the previous reported Pip-HoGu system. For example, it is not practical for the case of spacings >5 nt and more significantly, alternative orientations. The cooperation binding energy of host-guest system could not be finely tuned independently.[4] Moreover, the interaction of host-guest moieties is electrostatic and hydrophobic interactions, rather than residue specific interactions.
Here, we expanded the cooperation module from host-guest system to oligonucleotide directed sequence specific recognition moiety.[7b] Peptide nucleic acid (PNA) is an enzymatically stable, tight-binding, synthetically versatile, and informationally interfaced nucleic acid platform.[11] Several groups have made significant headway using γ-backbone PNA modifications, which transform a randomly folded PNA into a preorganized right-handed (RH) or left-handed (LH) helix.[12] More intriguingly, LH γPNA can hybridize to partner strands containing a complementary sequence and matching helical sense; however, they do not cross-hybridize with RH γPNA, DNA, or RNA.[13] Such orthogonal properties and programmability endow LH γPNA with the desired cooperative modules to mimic TF-pair cooperation for molecular assembly and computing while avoiding cross-hybridization with the host’s endogenous genetic materials.
In this context, we envisaged the integration of programmable PIPs with an orthogonal LH γPNA cooperative system, named Pip-NaCo, to mimic the natural versatile binding systems of TF pairs (Figure 1). Distinct from Pip-HoGu, Pip-NaCo cooperation is specific interaction of hydrogen bond with base pairing which could cover as longest spacing as its linker length reach theoretically. Results show a minimum cooperation of –3.27 kcal mol–1, and can flexibly change PIPs-binding orientation and conjugation sites. Furthermore, the tunability of PNA length, orthogonality, and toehold strand displacement performance further empower Pip-NaCo as fascinating tool for mimicking cooperation of transcription factor pairs.
Results and Discussion
The principle of Pip–NaCo system
Two PIPs were designed to target their matching sequences[9] and were individually conjugated with γPNA domains (modified with (L)-diethylene glycol (L-MP) at the γ-site) through a PEG linker (Figure 2).[14] The incorporation of a diethylene glycol unit was confirmed to enhance water solubility and reduce aggregation significantly.[15] The preorganized conformation of single-stranded γPNA and its binding with the respective matching strand could translate into higher affinity and sequence selectivity because of a reduction in the entropic penalty and an increase in backbone rigidity.[12b] The full synthetic procedure and characterization of all conjugates of Pip–NaCo are provided in the Supplementary information. It is noteworthy that PIPs purified by Fmoc–solid phase synthesis were incorporated onto γPNA tails on Boc solid-phase resin.[12d, 16]
Figure 2.

Schematic representation of cooperative interactions of two components of the Pip–NaCo assembly (PP1 and PP2) with dsDNA backbone. Thick solid lines represent the dsDNA backbone of the target site and associated oligonucleotides. The thin module array represents oligonucleotide sequence specific hydrogen bonds. n = gap distance. The dimerization domain of LH L-MPγPNA consisting of 5-nt sequence is showed as colored, bold, and italic letters. (Bottom) Chemical structures of PP1 and PP2.
Pip–NaCo system was designed in a parallel binding orientation, i.e., γPNA duplex is parallel to dsDNA, and γPNA strands meeting each other in the manner of head-to-tail (Figure 2). To our knowledge, Pip–NaCo sets the first example on the application of orthogonal, natural DNA-excluding LH γPNA conjugating with programmable DNA binders.
Conformational study
A circular dichroism (CD) experiment was conducted to determine the effect of PIP conjugation on the conformation of LH γPNA.[13] PP1 and PP2 modified with γ-L-MP have same nucleotide sequence with previous reported LH γPNA that was modified with γ-R-Me but without PIP conjugations.[13] By measuring the CD spectra and comparing with LH γPNA modified with γ-R-Me, we expected that the introduction of PIPs would not disturb the preorganized LH conformation of γPNA. As expected, PP1, PP2, and PP1–PP2 showed similar CD patterns, i.e., positive peak at around 240 nm and negative peak at 265–275 nm, suggesting LH helical conformation (Figures 3, S1). Compared with the respective γPNA sequences (γ-R-Me) without PIP conjugations, PP1 and PP1-PP2 exhibited highly identical CD profiles with unmodified single strand γPNA sequences and their unmodified γPNA duplex sequences, respectively (Figure S1A,C).[17] We conclude that PIP conjugations do not destroy the preorganized LH conformation of γPNA. Moreover, PP2 showed a canonical CD profile of LH conformation, but differed from its resecptive γPNA without PIP conjugation (Figure S1B). Enhancement and stabilization of the preorganization of γPNA by substituting it with γ-L-MP might be one of the mechanisms.[15]
Figure 3.

CD spectra of nonhybridized PP1 and PP2, each at 10.0 μM concentration, and the corresponding PP1–PP2 at 5.0 μM concentration of each strand, recorded at 22 °C. The CD spectrum was recorded from 230–300 nm. CD measurements were prepared in sodium phosphate buffer (10 mM sodium phosphate, 0.1 mM EDTA, 100 mM NaCl, pH 7.2).
PP1 and PP2 showed moderate red-shift of CD signal in comparison with PP1-PP2 duplexes. The CD amplitudes of PP1–PP2 duplexes are relatively higher than the sum of those for the two individual strands, and a third, a subtly positive peak emerges at 285 nm. Those results further support the notion that hybridization is likely to follow Fischer’s “lock and key” hypothesis[18] and the formation of γPNA duplex facilitate and enhance the LH secondary conformations.[19]
Spacing-dependent manner of cooperative binding
Pip–NaCo sequences were applied to the binding affinity assays with DNA sequences of Mode A and B (Figure 4A).[9] The differences between Mode A and B originate from the relative positions of the PP1 and PP2 binding sites. More specifically, in Mode A, the γPNA conjugation sites are close to each other and can form duplexes after covering a short spacing (spacing=gap distance; Figure 4B). However, in negative binding mode B, the two γPNA domains have longer spacings that are equal to the gap distance plus two PIP-binding sites (spacing=gap distance + two PIP-binding sites; Figure 4C, Table S1).
Figure 4.

Spacing-dependent manner of cooperative binding of Pip–NaCo. (A) The DNA oligomers (ODNs) used in the Tm assay, including positive (Mode A, ODN1′P–ODN8P) and negative (Mode B, ODN1′N–ODN8N) binding sequences. The gap distance (green) is the number of base pairs between the binding sites of PP1 (blue) and PP2 (red). Spacing is the distance between two PNA conjugation sites: i.e., spacing equals the gap distance in Mode A, but in Mode B, it equals the gap distance plus two PIP-binding sites. The upper chart shows only the forward DNA strand and omits the complementary DNA strand. (B, C) The gel-shift behavior of all the positive-binding sequences in Mode A (B) and negative-binding sequences in Mode B (C) with PP1–PP2. ODN concentration: 1.0 μM. Compound concentration: 10.0 μM. Black arrow: ODN2P; red: ODN2P/PP1–PP2. Except special illustration, the gel bands were stained with SYBR gold and quantified with a FujiFilm FLA-3000G fluorescent imaging analyzer. Unless otherwise stated, all samples used in the electrophoretic mobility shift assay measurements were prepared in sodium phosphate buffer (10 mM sodium phosphate, 0.1 mM EDTA, 100 mM NaCl, pH 7.2).
An electrophoretic mobility shift assay (EMSA) was conducted to determine the potency of the cooperative binding and how it was influenced by the spacings between the two PIP-binding sites, by direct visualization of the band-shift behavior upon formation of stable complexes.[20] PP1–PP2 was equilibrated with DNA oligomers (ODNs) (Mode A and B) of varying spacings. Because of a PIP-binding steric conflict, no shifted band could be observed for ODNs with a 1 base pair deletion (ODN1′P and ODN1′N) (Figure 4B,C). However, the appearance of a shifted band showed that ODNs in Mode A (0–8 base pair gap distances) display potent cooperative binding. In striking contrast to the Pip–HoGu system (cooperation limited to spacing of 0–5 nt), significant band shifts were also observed for Mode B ODNs with spacing of 12 and 13 base pairs.
Furthermore, the EMSA data showed that, in Mode A, the shifted bands of the middle ODNs (ODN3P, ODN4P, and ODN5P) were weaker than those of the ODNs at both ends. These results can be explained taken together with data from computational studies. Inserting a spacer between two PIP-binding sites will not only shift the linear distance but will also rotate them from the original position. In canonical B-DNA, the addition of 1-nt rotates it 36o alongside the DNA helix and it will have the same orientation again after the insertion of 10 nt. Based on computational studies, PP1 and PP2 are at the greatest angle distance in ODN4P, and further increases in spacings lead to the realignment of two PIPs, which is consistent with the observed results.
Orientation variation of binding sites
DNA-binding proteins can flexibly rearrange their binding orientations when coupled with partner TFs.[2a] We have confirmed that PP1–PP2 possesses strong band-shift ability with ODNs of 0–13 nt spacings, which are long enough to accommodate the diverse binding modes of TF–DNA complexes. Here, we investigated PP1–PP2 complexed with ODNs in two additional binding modes, Modes C and D, to analyze the effects of orientation of PIP-binding sites on cooperative binding (Figure 5A).
Figure 5.

EMSA results illustrating the cooperation of Pip–NaCo in different binding modes. (A) Schematic illustration of PP1–PP2 binding with ODNs in Mode C and D. (B) The gel-shift behavior of PP1–PP2 with ODNs of Mode A-2P (lanes 1–4), Mode A-6P (lanes 5–7), Mode C (lanes 8–10), and Mode D (lanes 11–13). The gap distance (green) is the number of base pairs between the binding sites of PP1 (blue) and PP2 (red). Spacing is the distance between two PNA conjugation sites, i.e., in Mode C, it equals the gap distance plus PP2 binding sites. ODN concentrations: 1.0 μM. Compound concentration: 10.0 μM.
The results shown in Figure 5B suggested that the order of binding affinity of the complexes is Mode A-2P < Mode A-6P < Mode D < Mode C. Because γPNA modules bind head-to-tail, the large size of the dimerization domain imposes unfavorable steric hindrance for Mode A-2P (with a spacing of 2 nt). Such steric hindrance is relieved when the distance increases to six or seven base pairs. Furthermore, Mode C and D both showed higher binding affinity than Mode A-6P, implying that a compact binding mode helps to stabilize the complexes. A slightly higher binding affinity of Mode D (5.0 μM, 29.1%) compared with Mode E (5.0 μM, 15.6%) might be explained by the difference of DNA sequence orientation.[4]
Energetics of cooperative binding
Quantitative EMSAs were performed to analyze the magnitude of cooperativity.[7b, 20] The experimental design involved measuring the equilibrium constants for binding of PP1 to Mode C in the presence and absence of PP2. EMSA results comfirmed that the conjugation of γPNA sequence moderately impairs PIPs binding affinity (Figures S2). Incubation of Mode C with PP1 alone resulted in a very weak band-shift (Figures 6A, S2). The increase in band-shift at low concentrations of PP1 alone and in the presence of 5.0 μM PP2 illustrates the cooperative effect. Compared with weak monomeric binding, γPNA dimerization domain facilitate dimeric binding to their respective biding sites. Fitting a Langmuir binding isotherm yielded the binding isotherms and equilibrium association constants of 1.87 × 104 M–1 (K1) for PP1 binding alone and 4.67 × 106 M–1 (K1,2) for PP1 in the presence of 5.0 μM PP2 (Figure 6B). Based on the free-energy-of-binding equation, we can calculate that the ΔG for PP1 in the presence and absence of PP2 is –9.09 and –5.82 kcal mol–1, respectively. From this, we can estimate that the minimum free energy of interaction (ΔG1,2 – ΔG1) is –3.27 kcal mol–1 (Figure 6C). Therefore, for this system, the presence of partner PP2 enhances the binding affinity of PP1 by a factor of more than 200. Pip-NaCo also showed high sequence selectivity in the assay with 1-bp mismatch DNA sequence (Figure S3).
Figure 6.

Quantitative EMSAs evaluating the cooperation of Pip–NaCo. (A) Quantitative EMSA of Mode C with PP1 at various concentrations (top) and PP1 supplemented with 5.0 μM PP2 (bottom). ODN concentration: 100 nM. Compound concentrations range from 0.1 to 10.0 μM (10-fold concentrations from 100 nM are showed in the figure). FAM labeled forward strand (5’-FAM-AACTAGCCTAATGACGTATAT-3’) used for quantitative assay without SYBR gold staining. (B) Binding isotherms obtained for PP1 alone (■) and in the presence of PP2 (●) using quantitative EMSA. The data points were calculated from the average shift-band intensities of triplicate experiments. (C) Equilibrium association constants and free energies for Mode C with PP1–PP2.
Even though Pip-NaCo show reasonable decrease of binding affinity by mono- or combinatory treatment compared with Pip-HoGu, Pip-NaCo revealed significant improvement on cooperation binding energy (from –2.32 to –3.27 kcal mol–1) and further experiment demonstrated that cooperation strength can be regulated reasonably and flexible on the γPNA modules (see below).
The effect of PNA length on cooperative binding
An important feature of the γPNA-based cooperative system is that the parallel γPNA dimerization domain can be tuned to regulate stabilization through alteration of the length and match/mismatch of PNA sequence. Here, we investigated the influence of PNA length on the cooperation of the Pip–NaCo assembly where the γPNA duplex is parallel to dsDNA. The 5-nt γPNA sequences in PP1 and PP2 were elongated to 7-nt to generate PP4 and PP5, respectively (Figure 7A). After solid-phase synthesis, 5-nt and 7-nt conjugates were evaluated using dimers of either the same γPNA length (5-nt:5-nt or 7-nt:7-nt) or mixed lengths (5-nt:7-nt).
Figure 7.

The effect PNA length on the cooperation of Pip–NaCo. (A) Schematic illustration of Pip–NaCo assembly containing 7-nt γPNA sequences in Mode C. Dashed square frame highlights the inserted nt. (B) The gel-shift behavior of PP1–PP2 (lanes 1, 2), PP1–PP5 (lanes 3, 4), PP2–PP4 (lanes 5, 6), and PP4–PP5 (lanes 7, 8), with Mode C. ODN concentration: 1.0 μM. Compound concentration: 3.0 μM and 10.0 μM.
The data showed the following order of binding affinity to Mode C: PP1–PP2 > PP2–PP4 > PP1–PP5 > PP4–PP5, suggesting that the 7-nt γPNA conjugate destabilizes the binding compared with that of 5-nt γPNA (Figure 7B). These data suggested that γPNA length was an important factor in regulating the binding of the complexes, and that for binding Mode C, a short γPNA might be preferable. Because 5-nt γPNA has shown potent enough duplex binding ability while further increase of γPNA length have weak improvement on cooperation but might significantly deteriorate PIP-DNA binding affinity.[13] One point to emphasize here is that we surmised that the larger size of the parallel form of the γPNA dimerization domain might easily displace PIPs from the DNA minor groove. It might be interesting to explore in the future vertical γPNA binding modes in which γPNA deplex is perpendicular to dsDNA which have the potential to form more stable γPNA-assisted complexes (unpublished work).[7b]
We also studied the influence of the linker conjugation site tethered with PIPs. In comparison with PP2, we designed PP3 in which the linker was conjugated at the tail of PIP2 rather than the γ-turn (Figure S4). The results demonstrated that this minor change in the conjugation site dramatically destabilized the interaction, suggesting that the conjugation site on the γ-turn should be preserved.
Competitive assay
The feature of toehold-mediated strand displacement assay has expanded the application of nucleic acid-based artificial systems.[21] One advantage of the current artificial system derives from the reversibility of γPNA duplex formation depending on the composition of the external environment, e.g., the presence of competitive γPNA strands. Here, we investigated the capabilities of the Pip–NaCo system in a competitive assay. Based on the theory of toehold-mediated strand displacement, a 7-nt PNA5 strand was introduced to displace PP4 binding (Figures 8A, S5). PP2–PP4 complexes with a 5-nt:7-nt γPNA dimerization domain were stabilized with Mode C (lane 1, Figure 8B). Concentration-dependent displacement by γPNA5 was observed during a short incubation, and at a threefold excess of γPNA5, >80% of PP4 was released from PP2-binding complexes (lane 5). This suggested that γPNA-based toehold-mediated strand displacement was of value for future applications in versatile, reversible artificial control systems.
Figure 8.

Toehold-mediated strand displacement assay of Pip–NaCo. (A) Schematic illustration of toehold-mediated strand displacement assay with Pip–NaCo assemblies. γPNA5 is the competitive strand to displace PP2 binding. (B) Toehold-mediated strand displacement assay in EMSA with Mode C. ODN concentration: 1.0 μM. Compound concentrations have been shown in figure.
Conclusions
In summary, the important features of the artificial system Pip-NaCo characterized here are that both recognition domain PIPs and cooperative dimerization domain PNAs are modular, suggesting that they have controllable cooperative energetics. Through changing the linker conjugation site, binding mode, and sequence of PIPs and γPNAs, orientations of binding sites and cooperative-interaction energies can be tuned independently and reasonably. Moreover, the orthogonal properties of LH γPNA have the overwhelming advantage of eliminating the confusion generated by excess endogenous nucleic acids while maintaining its higher dimerization ability with its sequence-specific partner. Most significantly, Pip-NaCo has outstanding cooperative interaction ability compared with natural occurring transcription factor pairs, and it can cover variable orientations of binding sites. Current Pip-NaCo platform also has the potential for precisely manipulating biological processes.
Experimental Section
Full experimental details are provided in the Supporting Information.
Supplementary Material
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant NO. JP16H06356, “Basic Science and Platform Technology Program for Innovative Biological Medicine by Japan Agency for Medical Research and Development (AMED) under Grant Number JP18am0301005”, and “the Platform Project for Supporting Drug Discovery and Life Science Research funded by AMED under Grant Number J18am0101101”. This work was supported in part by the National Institutes of Health (R21NS098102), the National Science Foundation (CHE-1609159), and the DSF Charitable Foundation. MALDI-TOF MS instrumentation at Carnegie Mellon University was partially supported by the National Science Foundation (CHE-9808188, CHE-0130903, and CHE-1039870). We also thank China Scholarship Council for support to Z. Y.
Dedication This paper is dedicated to Professor Isao Saito’s 77th birthday.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.2018xxxxx.
Conflict of interest
The authors have filed a joint patent application on aspects of this work.
References
- [1].Jolma A, Yan J, Whitington T, Toivonen J, Nitta Kazuhiro R., Rastas P, Morgunova E, Enge M, Taipale M, Wei G, Palin K, Vaquerizas Juan M., Vincentelli R, Luscombe Nicholas M., Hughes Timothy R., Lemaire P, Ukkonen E, Kivioja T, Taipale J, Cell 2013, 152, 327–339. [DOI] [PubMed] [Google Scholar]
- [2].a) Morgunova E, Taipale J, Curr. Opin. Struct. Biol 2017, 47, 1–8; [DOI] [PubMed] [Google Scholar]; b) Stampfel G, Kazmar T, Frank O, Wienerroither S, Reiter F, Stark A, Nature 2015, 528, 147–151. [DOI] [PubMed] [Google Scholar]
- [3].Ng CK, Li NX, Chee S, Prabhakar S, Kolatkar PR, Jauch R, Nucleic Acids Res. 2012, 40, 4933–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Enge M, Kivioja T, Morgunova E, Taipale J, Nature 2015, 527, 384–388. [DOI] [PubMed] [Google Scholar]
- [5].a) Hoverter NP, Zeller MD, McQuade MM, Garibaldi A, Busch A, Selwan EM, Hertel KJ, Baldi P, Waterman ML, Nucleic Acids Res. 2014, 42, 13615–13632; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ravindranath AJ, Cadigan KM, Cancers 2016, 8, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB, Nature 1997, 387, 202–205; [DOI] [PubMed] [Google Scholar]; b) Dragulescu-Andrasi A, Rapireddy S, He G, Bhattacharya B, Hyldig-Nielsen JJ, Zon G, Ly DH, J. Am. Chem. Soc 2006, 128, 16104–16112; [DOI] [PubMed] [Google Scholar]; c) Taniguchi J, Pandian GN, Hidaka T, Hashiya K, Bando T, Kim KK, Sugiyama H, Nucleic Acids Res. 2017, 45, 9219–9228; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pazos E, Mosquera J, Vázquez ME, Mascareñas JL, ChemBioChem 2011, 12, 1958–1973; [DOI] [PubMed] [Google Scholar]; e) Wang M, Yu Y, Liang C, Lu A, Zhang G, Int. J. Mol. Sci 2016, 17, 779; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Olalla V, Eugenio VM, J BB, Luis C, J LM, Angew. Chem. Int. Ed. Engl 2007, 46, 6886–6890.17674388 [Google Scholar]
- [7].a) Ueno M, Murakami A, Makino K, Morii T, J. Am. Chem. Soc 1993, 115, 12575–12576; [Google Scholar]; b) Distefano MD, Dervan PB, Proc. Natl. Acad. Sci. U.S.A 1993, 90, 1179–1183; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blanco JB, Dodero VI, Vázquez ME, Mosquera M, Castedo L, Mascareñas JL, Angew. Chem. Int. Ed. Engl 2006, 45, 8210–8214; [DOI] [PubMed] [Google Scholar]; d) Sánchez MI, Mosquera J, Vázquez ME, Mascareñas JL, Angew. Chem. Int. Ed. Engl 2014, 53, 9917–9921; [DOI] [PubMed] [Google Scholar]; e) Chang D, Kim KT, Lindberg E, Winssinger N, Bioconjugate Chem. 2018, 29, 158–163. [DOI] [PubMed] [Google Scholar]
- [8].Trauger JW, Baird EE, Dervan PB, Nature 1996, 382, 559–561. [DOI] [PubMed] [Google Scholar]
- [9].Yu Z, Guo C, Wei Y, Hashiya K, Bando T, Sugiyama H, J. Am. Chem. Soc 2018, 140, 2426–2429. [DOI] [PubMed] [Google Scholar]
- [10].Singleton SF, Dervan PB, Biochemistry 1992, 31, 10995–11003. [DOI] [PubMed] [Google Scholar]
- [11].a) Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE, Nature 1993, 365, 566–568; [DOI] [PubMed] [Google Scholar]; b) Berger O, Gazit E, Pept. Sci 2017, 108, e22930; [Google Scholar]; c) Ellipilli S, Ganesh KN, J. Org. Chem 2015, 80, 9185–9191. [DOI] [PubMed] [Google Scholar]
- [12].a) Sahu B, Chenna V, Lathrop KL, Thomas SM, Zon G, Livak KJ, Ly DH, J. Org. Chem 2009, 74, 1509–1516; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH, J. Am. Chem. Soc 2006, 128, 10258–10267; [DOI] [PubMed] [Google Scholar]; c) Jain DR, V L. Anandi, Lahiri M, Ganesh KN, J. Org. Chem 2014, 79, 9567–9577; [DOI] [PubMed] [Google Scholar]; d) Manna A, Rapireddy S, Sureshkumar G, Ly DH, Tetrahedron 2015, 71, 3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sacui I, Hsieh W-C, Manna A, Sahu B, Ly DH, J. Am. Chem. Soc 2015, 137, 8603–8610. [DOI] [PubMed] [Google Scholar]
- [14].Kameshima W, Ishizuka T, Minoshima M, Yamamoto M, Sugiyama H, Xu Y, Komiyama M, Angew. Chem. Int. Ed. Engl 2013, 52, 13681–13684. [DOI] [PubMed] [Google Scholar]
- [15].Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, Ly DH, J. Org. Chem 2011, 76, 5614–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yu Z, Taniguchi J, Wei Y, Pandian GN, Hashiya K, Bando T, Sugiyama H, Eur. J. Med. Chem 2017, 138, 320–327. [DOI] [PubMed] [Google Scholar]
- [17].Kadhane U, Holm AIS, Hoffmann SV, Nielsen SB, Phys. Rev. E 2008, 77, 021901. [DOI] [PubMed] [Google Scholar]
- [18].Fischer E, Ber. Dtsch. Chem. Ges 1894, 27, 2985–2993. [Google Scholar]
- [19].Wittung P, Eriksson M, Lyng R, Nielsen PE, Norden B, J. Am. Chem. Soc 1995, 117, 10167–10173. [Google Scholar]
- [20].Moretti R, Donato LJ, Brezinski ML, Stafford RL, Hoff H, Thorson JS, Dervan PB, Ansari AZ, ACS. Chem. Biol 2008, 3, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang DY, Seelig G, Nat Chem 2011, 3, 103–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.