

Abstract
Objective:
Mutations in the ATXN2 gene (CAG expansions ≥32 repeats) can be a rare cause of Parkinson’s disease and amyotrophic lateral sclerosis (ALS). We recently reported that the stress granule (SG) protein Staufen1 (STAU1) was overabundant in neurodegenerative disorder spinocerebellar ataxia type 2 (SCA2) patient cells, animal models, and ALS-TDP-43 fibroblasts, and provided a link between SG formation and autophagy. We aimed to test if STAU1 overabundance has a role in the pathogenesis of other neurodegenerative diseases.
Methods:
With multiple neurodegenerative patient-derived cell models, animal models, and human postmortem ALS tissue, we evaluate STAU1 function using biochemical and immunohistological analyses.
Results:
We demonstrate STAU1 overabundance and increased total and phosphorylated mammalian target of rapamycin (mTOR) in fibroblast cells from patients with ALS with mutations in TDP-43, patients with dementia with PSEN1 mutations, a patient with parkinsonism with MAPT mutation, Huntington’s disease (HD) mutations, and SCA2 mutations. Increased STAU1 levels and mTOR activity were seen in human ALS spinal cord tissues as well as in animal models. Changes in STAU1 and mTOR protein levels were post-transcriptional. Exogenous expression of STAU1 in wildtype cells was sufficient to activate mTOR and downstream targets and form SGs. Targeting STAU1 by RNAi normalized mTOR, suggesting a potential role for therapy in diseases associated with STAU1 overabundance.
Interpretation:
STAU1 overabundance in neurodegeneration is a common phenomenon associated with hyperactive mTOR. Targeting STAU1 with ASOs or miRNA viral vectors may represent a novel, efficacious therapy for neurodegenerative diseases characterized by overabundant STAU1.
CAG repeat expansion mutations in the ATXN2 cause spinocerebellar ataxia type 2 (SCA2), a multisystem neurodegeneration that primarily affects the cerebellum and its connections.1 Full-length mutant expansions ≥32 repeats can be a rare cause of Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS).2 Long normal repeats in ATXN2 are also associated with the development of ALS, and CAG32-repeat alleles increase ALS risk close to 10-fold.3,4 Supporting ATXN2 as a therapeutic target for SCA2 and ALS, we treated SCA2 mice with antisense oligonucleotides (ASOs) that lower ATXN2 expression, resulting in delayed neurodegeneration at the cellular, biochemical, and slice physiology level.5 Similarly, treatment of a TDP-43 ALS mouse model with ASOs against mouse Atxn2 improved neurodegeneration.6 These studies led to the development of BIIB105, an ATXN2 ASO therapeutic now in a phase I clinical trial for ALS (ClinicalTrials.gov Identifier: NCT04494256; https://clinicaltrials.gov/ct2/show/NCT04494256).
The potential for targeting ATXN2 not only in the mendelian disease SCA2, but also as a disease modifier for ALS prompted us to identify other potential therapeutic targets by searching for proteins interacting with ATXN2. Using immunoprecipitation and mass spectrometry, we identified STAU1 as an ATXN2 interactor.7 STAU1 is a double-stranded RNA binding protein (dsRBP) and a component of stress granules (SGs) that can influence RNA stability and translation.8,9 It can bind to a large number of mRNAs via the 3’UTR and initiate STAU-mediated decay (SMD), a process similar to nonsense-mediated decay.8,10
To our surprise, the degree of interaction between wildtype and mutant ATXN2 and STAU1 was not different, but STAU1 levels were greatly elevated in cells from patients with SCA2, a patient with ALS patient with the TDP-43G298S mutation, and in SCA2 mouse models.7 Reduction of Stau1 in vivo via genetic interaction improved biochemical phenotypes and motor performance in an SCA2 mouse model.7 STAU1 overabundance was also observed in cells after exposure to a number of stressors and amplified the proapoptotic activation of the unfolded protein response (UPR).11 STAU1 was necessary and sufficient to mediate caspase-3 cleavage in wildtype cells.11
In the present work, we studied STAU1 levels in the spinal cords from patients with ALS, cells derived from 12 patients with multiple neurodegenerative diseases, and 2 animal models of neurodegeneration and found that they all presented significantly elevated STAU1 levels. This was associated with hyperactive mammalian target of rapamycin (mTOR) signaling that could be normalized by STAU1 silencing. We show STAU1 colocalized with G3BP1, an SG-marker protein in C9orf72-ALS/frontotemporal degeneration (FTD) cells. Our results describe STAU1 overabundance as a common feature across multiple neurodegenerative diseases of varied origin and suggest a functional link between STAU1 and mTOR signaling that could be exploited in therapeutic development.
Materials and Methods
DNA Constructs
The plasmid constructs used in this study were 3XFlag-tagged STAU17 or wildtype TDP-43,7 GFP, DsRED, and STAU1-DsRED. The pRK5-EGFP-Tau was a gift from Karen Ashe (Addgene, Plasmid #46904). The pBacMam2-DiEx-LIC-N-flag_huntingtin_full-length_Q23 (Addgene, Plasmid #111723) and pBacMam2-DiEx-LIC-C-flag_huntingtin_full-length_Q66 (Addgene, Plasmid #111750) were gifts from Cheryl Arrowsmith. Mutant TDP-43 cDNAs (TDP-43G298S and TDP-43A382T) were polymerase chain reaction (PCR)-amplified from cDNA of patients with ALS fibroblasts (FBs) with TDP-43 mutations (G298S #ND32947 and A382T #ND41003; Coriell Cell Repositories, Camden, NJ, USA). TDP-43G348C, and C-terminal fragment of TDP-43 (TDP-43CTF) constructs were generated using TDP-43 cDNA as template.7 TDP-43 with mutated nuclear localization signals (TDP-43ΔNLS) was PCR amplified from genomic DNA from hTDP-43ΔNLS mouse.12 The PCR products were cloned into pCMV-3XFlag plasmid (Agilent Technologies, Santa Clara, CA, USA). To generate a STAU1-tagged DsRED construct, the STAU1 coding sequence along with in frame GFP sequence was cloned into the pcDNA3 plasmid (Thermo Fisher Scientific). All constructs were verified by sequencing. 3XFlag is referred to as Flag in the text and figures.
siRNAs and Reagents
The siRNAs used in this study are as follows: All Star Negative Control siRNA (Qiagen, Cat #1027280), human siSTAU1: 5′-CCUAUAACUACAACAUGAGdTdT-3′,7 human siC9orf72 #1: 5′-CAUUAAUCUUUGAUGGAAAdTdT-3′,13 human siC9orf72 #2: 5′-GGAAGAAUAUGGAUGCAUAdTdT-3′,13 and human siHTT/REP: 5′-GCUGCUGCUGCUGCUGCUGdTdT-3′.14 All siRNA oligonucleotides were synthesized by Invitrogen, USA. The oligonucleotides were deprotected and the complementary strands were annealed.
Cell Line Authentication
In order to adhere with the National Institutes of Health (NIH) guideline on scientific rigor in conducting biomedical research (NOT-OD-15-103) on the use of biological and/or chemical resources, we authenticated our cell lines utilizing STR analysis on 24 loci, including amelogenin for sex identification. The kit used for this was the GenePrint 24 system (Promega).
Cell Culture, Transfections, and Human Tissue Specimens
All human subjects gave written consent, and all procedures were approved by the Institutional Review Board (IRB) at the University of Utah. Five SCA2 patient-derived skin FBs were established from local patients. The following primary human FBs were obtained from the Coriell Cell Repositories (Camden, NJ, USA): healthy subjects (#ND29510, #ND34769, and #ND38530), patients with ALS with TDP-43 mutations (G298S #ND32947 and A382T #ND41003), 2 from patients at risk of FTD with C9orf72 expansions (#ND42504 and #ND42506), 1 from a patient with parkinsonism with a C9orf72 expansion (#ND40069), 3 from patients with Huntington’s disease (HD; Q44 #ND31038, Q57 #ND33392, and Q66 #ND40536), 3 from patients with Alzheimer’s disease (AD; PSEN1 mutations: M146I #ND34730, E184D #ND34732, and PSEN1 Intron4 Var #ND41001), and 1 from a patient with parkinsonism with an MAPT mutation (N279K #ND40074). Human bone osteosarcoma epithelial cells (U2OS) were obtained from ATCC Cat #HTB-96. U2OS and FBs were cultured and maintained as previously described.7 Spinal cord tissues were received from the Target ALS Postmortem Tissue Core (Johns Hopkins School of Medicine, Department of Neurology, Baltimore, MD, USA). These included 3 non-ALS/FTD control specimens (JHU 96, JHU 101, and JHU 123), 4 ALS/FTD specimens with C9orf72 expansions (JHU 88, JHU 92, JHU 119, and JHU 120), and 4 specimens from patients with sporadic ALS (JHU 102, JHU 111, JHU 113, and JHU 115). Cells were transfected with siRNAs as described.7 For overexpression of recombinant proteins, HEK-293 cells were plated on 6-well dishes and incubated overnight. The cells were then transfected with plasmid DNAs and harvested 48 hours post-transfection, and processed as 2 aliquots for protein and RNA analyses as needed. For siRNA experiments, cells were transfected with siRNAs using lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Prior standardization experiments showed that maximum silencing was achieved 4 to 5 days post-transfection.
Mice
ATXN2Q127 (Pcp2-ATXN2[Q127]),15 and B6;SJL-Tg(Thy1-TARDBP)4Singh/J mice16 (Jackson Laboratories; Stock 012836) were maintained as described.7
Antibodies
Antibodies used for Western blotting and their dilutions were as follows: mouse anti-Ataxin-2 antibody [Clone 22/Ataxin-2; 1:4000; BD Biosciences, Cat #611378], rabbit anti-Staufen [1:5000; Novus biologicals, NBP1-33202], LC3B antibody [1:7000; Novus biologicals, NB100-2220], TDP-43 antibody [1:7000; Proteintech, Cat #10782-2-AP], SQSTM1/p62 antibody [1:4000; Cell Signaling, Cat #5114], mTOR antibody [1:4000; Cell Signaling, Cat #2972], Phospho-mTOR antibody [Ser2448; 1:3000; Cell Signaling, Cat #2971], monoclonal anti-FLAG M2 antibody [1:10,000; Sigma-Aldrich, F3165], Huntingtin [D7F7] XP rabbit monoclonal antibody [1:3000; Cell Signaling, Cat #5656], C9orf72 rabbit polyclonal antibody [1:5000; ProteinTech, Cat #22637-1-AP], GFP antibody [B-2; 1:2,000; Santa Cruz, sc-9996], GAPDH (14C10) rabbit monoclonal antibody [1:8000; Cell Signaling, Cat #2118], and monoclonal anti-β-Actin–peroxidase [clone AC-15; 1:30,000; Sigma-Aldrich, A3854]. Secondary antibodies: peroxidase-conjugated AffiniPure goat anti-rabbit IgG [H + L; 1:5000; Jackson ImmunoResearch Laboratories, Cat #111-035-144], and goat anti-mouse IgG [Fab specific] Peroxidase [1: 5000; Sigma-Aldrich/Millipore, Cat #A2304-1ML].
Immunofluorescent Microscopy
U2OS cells expressing STAU1-DsRED and FBs were plated on coverslips overnight. Cells were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS), permeabilized with 0.3% Triton X-100, and processed for immunostaining using corresponding primary and fluorescent secondary antibodies. Paraffin-embedded spinal cord tissue slices were received from the Target ALS Postmortem Tissue Core (Johns Hopkins School of Medicine, Department of Neurology, Baltimore, MD, USA). Human tissues were maintained and processed under standard conditions consistent with NIH guidelines and conformed to an approved University of Utah IRB protocol. Sections were deparaffinized using the standard method, and blocked/permeabilized with 5% goat serum, 0.3% Triton X-100 in PBS, and processed for immunostaining. The nuclei were stained with DAPI followed by mounting with Fluoromount-G (Southern Biotech, Cat #0100-01). Images were acquired using confocal microscopy (Nikon A1 Confocal microscope) in the University of Utah cell imaging core laboratory and analyzed by Nikon EZ-C1 or NIS-Elements AR version 4.5 software. Antibody dilutions for cells and tissue immunostainings were STAU1 antibody (C-4) [1:200; Santa Cruz, sc-390820], rabbit anti-Staufen antibody [1:200; Novus biologicals, NBP1-33202], G3BP1 monoclonal antibody [M01J], clone 2F3 [1:1000; Abnova, Cat #H00010146-M01J], and G3BP1 antibody [1:500; Novus biologicals, NBP1-18922]. Fluorescent secondary antibodies were: Goat anti-Mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 594 (1:1000; Thermo Fisher Scientific, Cat #A32742), goat anti-mouse IgG (H + L) antibody, DyLight-488 (1:1,000; Thermo Fisher Scientific, Cat #35502), goat anti-Rabbit IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488 (1:1,000; Thermo Fisher Scientific, Cat #A32731), and goat anti-Rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 594 (1:1000; Thermo Fisher Scientific, Cat# A11037). The binary threshold function of Nikon’s NIS-Elements software was used to quantify the number of granules per nucleus for human spinal cord tissues. Objects were selected by using thresholds of area and intensity of light. Thresholds were set for DAPI, TRITC, and FITC, and NIS-Elements intersection feature counted the number of objects where FITC and TRITC were both present. To assess STAU1 stress granules in C9orf72 patient FBs, normal (#ND38530) and C9orf72 (#ND42504) cells were treated with vehicle or 0.5 mM sodium arsenite for 30 minutes then labeled with anti-G3BP1 and ant-STAU1 antibodies (primary and secondary antibodies as above) and mounted. The number of colocalized granules was determined by automated microscopy using a Nikon Ti-E inverted microscope with a 20× objective. Using NIS-Elements, images were subjected to 2D deconvolution by the Richardson–Lucy method. Per condition, 24 to 29 frames were imaged. The average ± SD cells evaluated per condition was 697 ± 108 with total STAU1-G3BP1 colocalized granule numbers ranging from 566 (untreated normal cells) to 1,966 (arsenite treated C9orf72 cells). The binary threshold function of NIS-Elements was used to quantify the numbers of granules per nucleus.
Preparation of Protein Lysates and Western Blotting
Cellular extracts were prepared by a single-step lysis method.7 The harvested cells were suspended in SDS-PAGE sample buffer (Laemmli sample buffer [Bio-Rad, Cat #161-0737]) and then boiled for 5 minutes. Equal amounts of the extracts were used for Western blot analyses. Human spinal cord tissue and mouse cerebellar or spinal cord protein extracts were prepared by homogenization of tissues in extraction buffer (25 mM Tris–HCl pH 7.6, 250 mM NaCl, 0.5% Nonidet P-40, 2 mM EDTA, 2 mM MgCl2, 0.5 M urea, and protease inhibitors [Sigma-Aldrich, P-8340]) followed by centrifugation at 4°C for 20 minutes at 14,000 revolutions per minute (RPM). Only supernatants were used for Western blotting. Protein extracts were resolved by SDS-PAGE and transferred to Hybond P membranes (Amersham Bioscience, USA), and then processed for Western blotting according to our published protocol.7 Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Cat #WBKLSO500) was used to visualize the signals, which were detected on the ChemiDoc MP imager (Bio-Rad). For some blots, we used a film developing system, and band intensities were quantified by ImageJ software analyses after inversion of the images. Relative protein abundances were expressed as ratios to ACTB or GAPDH, or Actb.
RNA Expression Analyses by Quantitative Real Time PCR
Quantitative real time (qRT)-PCR was performed in QuantStudio 12K (Life Technologies, USA) with the Power SYBR Green PCR Master Mix (Applied Biosystems, USA) or with commercial TaqMan assay (Cat #4331182: Hs00244999_m1 for STAU1 probe and Hs01060665_g1 for ACTB probe, Thermo Fisher Scientific) as described.7 The forward (F) and reverse (R) primer sequences for qRT-PCR are; ACTB-F: 5′-GAAAATCTGGCACCACACCT-3′ and ACTB-R: 5′-TAGCACAGCCTGGATAGCAA-3′, mTOR-F: 5′-CAGAAGGTGGAGGTGTTTGAG-3′ and mTOR-R: 5′-TGACATGACCGCTAAAGAACG-3′, Stau1-F: 5′-AGTACATGCTCCTTACAGAACG-3′ and Stau1-R: 5′-TGATGCCCAACCTTTACCTG-3′, mTor-F: 5′-ATTCAATCCATAGCCCCGTC-3′ and mTor-R: 5′-TGCATCACTCGTTCATCCTG-3′, Actb-F: 5′-CGTCGACAACGGCTCCGGCATG-3′ and Actb-R: 5′-GGGCCTCGTCACCCACATAGGAG-3′.
Statistical Analysis
One-way analysis of variance (ANOVA) was used to determine significant differences among groups with Bonferroni post hoc tests for multiple comparisons. Significance levels are as follows: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ns = p > 0.05. Means ± SD are presented throughout unless otherwise specified. Statistical tests were carried out using GraphPad Prism version 8.
Results
STAU1 Levels Are Increased in Neurodegenerative Diseases
Our prior work identified STAU1 overabundance in the cells of patients with SCA2, ATXN2-transgenic animal models, and ALS-TDP-43 FBs. These studies also revealed that reduction of STAU1 improved neurodegenerative phenotypes in vitro and in vivo in SCA2 models.7,11 We have now expanded our study to evaluate STAU1’s role in neurodegenerative diseases using multiple cell and animal models, with emphasis on mTOR and autophagy. We used FB cell lines from patients with ALS with mutations in TDP-43 (G298S and A382T), patients with dementia with 3 different PSEN1 mutations (M146I, E184D, and Intron4 Var), and an MAPT mutation (N279K; Fig 1A). We also obtained cell lines from 3 patients with different CAG repeat expansions in the HTT gene with 44, 57, and 66 CAG repeats, and 5 additional FBs from SCA2 patients with 35, 42, and 45 CAG repeats in ATXN2 (Fig 1B). All FB cell lines showed a significant overabundance of STAU1, ranging from 3-fold to 6-fold depending on the disease and the respective mutation (Fig 1C). We had previously found that STAU1 was primarily degraded through the autophagosome, presumably owing to its association with RNA granules. We, therefore, tested if autophagy was impaired in cells with endogenously elevated STAU1. A master regulator of autophagy is mTOR, and its activation inhibits autophagy in cells from yeast to human.17 We, therefore, determined the level of mTOR by quantitative Western blot analysis. In all cell lines, total and active phospho-mTOR levels were increased, as were levels of p62 and LC3-II, consistent with significantly impaired autophagy (Fig 1A, B). Quantified data show that total mTOR elevations ranged from 3 to 4-fold (Fig 1D).
FIGURE 1:

Increased STAU1 levels and abnormal mTOR signaling in neurodegenerative disease patient cell lines. (A–D) Western blot analysis of FB protein extracts from patients with ALS with mutations in TDP-43, AD mutations ([PSEN1 mutations: M146I, E184D and PSEN1 Intron4 Var]), a parkinsonism patient with MAPT mutation (N279K) (A), HD mutations (Q44, Q57 and Q66 repeats), and SCA2 mutations (3 Q35, Q42 and Q45 repeats) (B). All show increased levels of STAU1, mTOR, P-mTOR, p62, and LC3-II compared to controls. Each lane represents an individual patient cell line. STAU1 and mTOR protein levels were normalized to ACTB and quantification of average fold changes for STAU1 and mTOR are shown in C, D. The representative blots of 3 independent experiments are shown. (E, F) STAU1 and mTOR mRNA levels are unaltered in patient’s cell lines. qRT-PCR analyses of RNA extracts from the FB cells in (A, B). Ordinary one-way ANOVA followed by Bonferroni tests for multiple comparisons. Data are mean ± SD, ns = p > 0.05, ***p < 0.001. AD = Alzheimer’s disease; ALS = amyotrophic lateral sclerosis; ANOVA = analysis of variance; FBs = fibroblasts; HD = Huntington’s disease; mTOR = mammalian target of rapamycin; qRT-PCR = quantitative real time polymerase chain reaction; SCA2 = spinocerebellar ataxia type 2.
We had previously shown that STAU1 overabundance in cells expressing mutant ATXN2[Q58] was not transcriptionally mediated but was due to slowed degradation by autophagy.7 We, therefore, determined the mRNA levels of STAU1 and mTOR by quantitative RT-PCR. As we had observed in prior analyses, STAU1 and mTOR mRNA levels were not altered in the presence of mutant disease proteins (Fig 1E, F).
Repeat expansion mutations in the C9orf72 gene are a major cause of familial as well as sporadic ALS.18,19 We obtained 3 FBs from the Coriell Cell Repositories (Camden, NJ, USA) to analyze STAU1 and mTOR. FBs from patients with an expansion in the C9orf72 gene showed a 4.5 to 6-fold increase in STAU1 protein levels (Fig 2A, B). Levels of mTOR protein were also significantly elevated (see Fig 2A, B). Although the proteins were elevated, STAU1 and mTOR mRNAs remained unchanged in C9orf72 FBs (Fig 2C). Of note, the lowest increase of STAU1 protein observed in C9orf72 FBs was still greater than that in any of the other cell lines that we examined. To determine whether STAU1 elevations were also present in ALS tissues, we analyzed spinal cords from 4 patients with C9orf72 and patients with sporadic ALS (sALS; Fig 2D, E). All tissues had significantly elevated STAU1 levels ranging from 3 to 5-fold, and mTOR protein levels were also significantly elevated. There were no obvious differences between tissues with a known C9orf72 mutation versus those classified as sporadic, with regard to abnormal STAU1 and mTOR levels.
FIGURE 2:
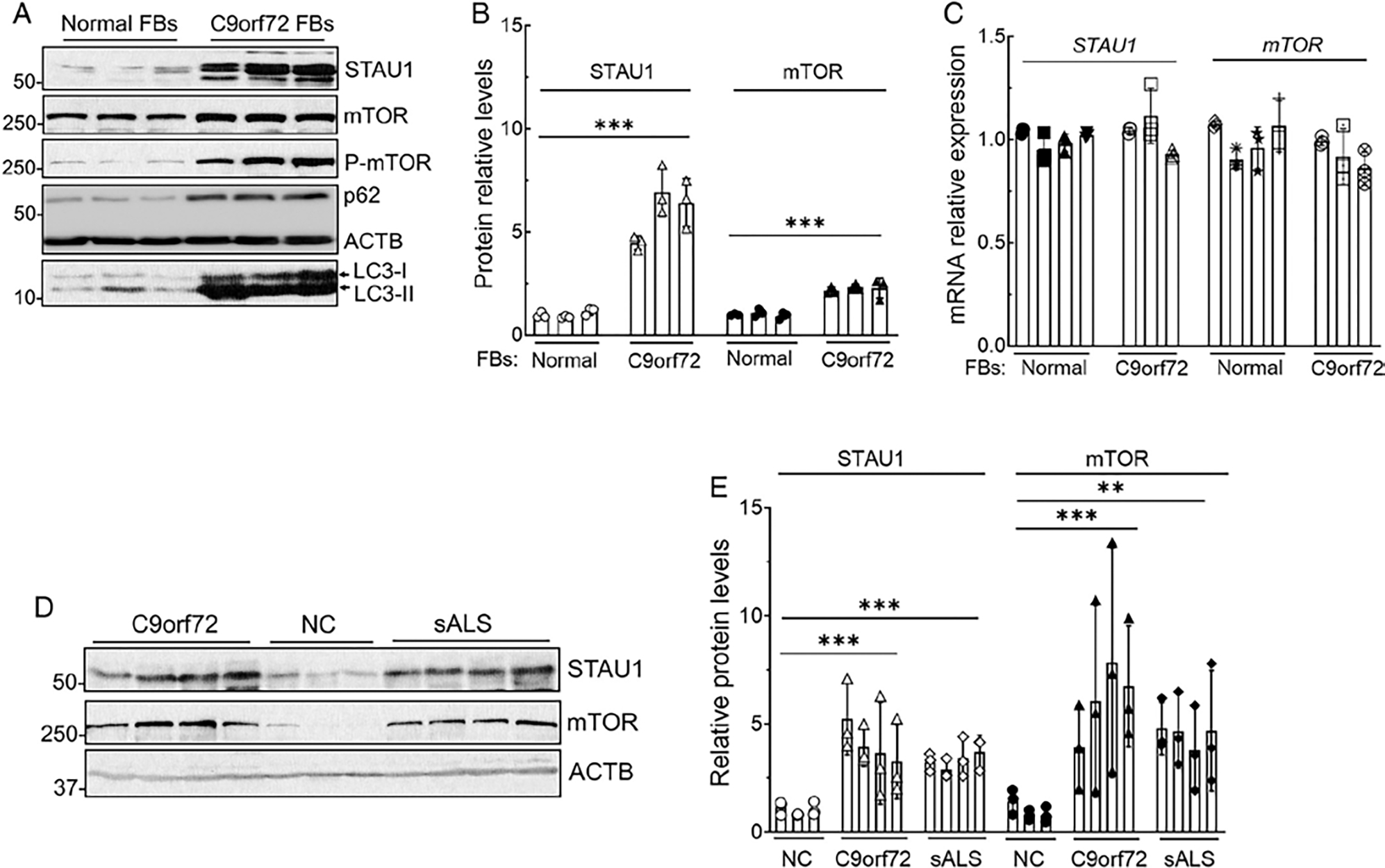
STAU1 and mTOR overabundance in patient cell lines with C9orf72 expansion and patients with sporadic forms of ALS (sALS). Western blot analysis of protein extracts from FBs with C9orf72 expansions showing increased STAU1, mTOR, P-mTOR, p62, and LC3-II levels compared to control FBs (A). (B) Quantification of STAU1 and mTOR average fold changes. (C) STAU1 and mTOR mRNA levels are unaltered in C9orf72 FBs. qRT-PCR analyses of the C9orf72 FBs revealed unaltered mRNA levels for STAU1 and mTOR compared with control cells. (D, E) Spinal cord tissues of patients with C9orf72 expansion and sALS showing increased STAU1 and mTOR levels compared to non-ALS controls (NC) on Western blots (D). Each lane represents individual patient lines or tissues. Protein levels were normalized to ACTB, and quantified STAU1 and mTOR average fold changes are shown in (E). Ordinary one-way ANOVA followed by Bonferroni tests for multiple comparisons. Data are mean ± SD, ns = p > 0.05, **p < 0.01 ***p < 0.001. ALS = amyotrophic lateral sclerosis; ANOVA = analysis of variance; FBs = fibroblasts; mTOR = mammalian target of rapamycin; qRT-PCR = quantitative real time polymerase chain reaction.
STAU1 is Recruited to SG-like Structures in ALS/FTD with C9orf72 Expansions
The precise role of STAU1 in stress granules is controversial. STAU1 is not necessary for the formation of SGs, but a role for STAU1 in SG assembly and disassembly has also been suggested.20,21 As C9orf72 mutation appeared to result in the greatest increase in STAU1 (see Fig 2B vs Fig 1C), we analyzed the number of SGs as defined by positivity for G3BP1 in culture and in the fixed spinal cord tissue. Under physiological conditions (without added stress), C9orf72 FBs demonstrated constitutive SG-like aggregates positive for both STAU1 and G3BP1 that were rarely observed in normal FBs (Fig 3A). Following quantitative analysis of the number of granules positive for both STAU1 and G3BP1, we found that the numbers of aggregates were significantly higher for C9orf72 FBs over normal FBs (Fig 3B). When cells were stressed with arsenite (0.5 mM for 30 minutes), the numbers of STAU1-G3BP1 granules were further increased for both normal and C9orf72 FBs, but granule numbers were significantly greater in C9orf72 FBs versus normal cells (see Fig 3A, B). We then investigated STAU1 granules in vivo. Double- immunostaining for STAU1 and G3BP1 demonstrated that both colocalized to aggregates in spinal cord tissues from individuals with C9orf72-ALS/FTD but were rarely observed in non-neurological controls (see Fig 3C, D). Our observation supports the presence of STAU1 pathological inclusions in ALS, similar to our previous finding of STAU1 in Purkinje cell inclusion bodies in patients’ with SCA2 brains.7
FIGURE 3:
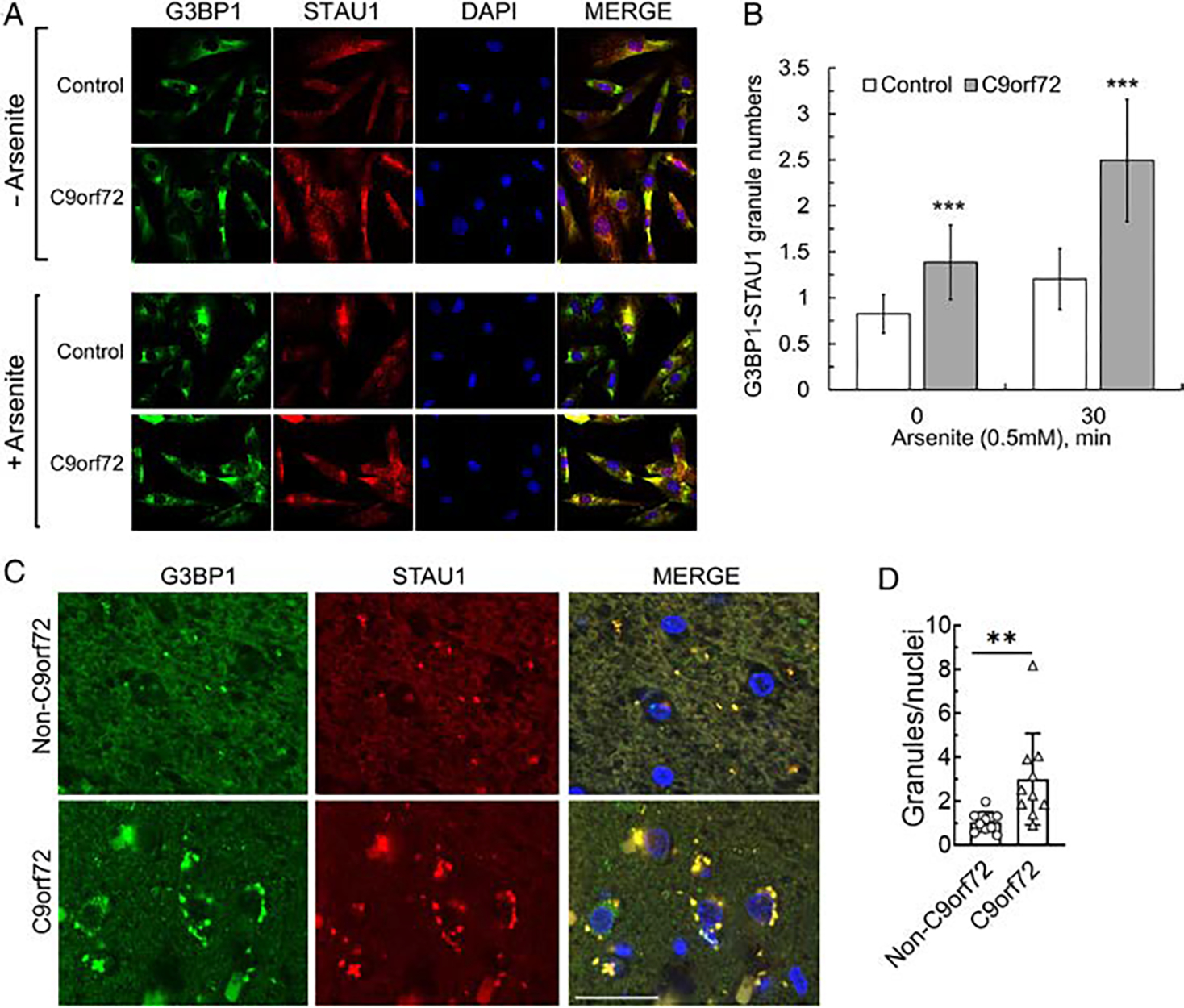
STAU1 forms SG-like structures. (A, B) Co-localization of STAU1 with G3BP1 in SG-like structures in FTD-C9orf72 FBs. Immunostaining of C9orf72 FBs with antibodies against STAU1 and G3BP1 show constitutive colocalization of STAU1 with G3BP1 under non-stress condition. Following exposure of the cells to sodium arsenite (0.5 mM for 30 minutes), both normal and C9orf72 FBs are positive for STAU1-G3BP1 granules, and the number of granules in C9orf72 FBs are significantly greater than in arsenite-induced stressed normal FBs. A total of 24–29 frames per condition were captured by an automated Nikon Ti-E inverted microscope with a 20 times objective. Data are mean ± SD, ***p < 0.001, Student t test. Scale bar, 30 μM. (C–D) Colocalized STAU1 (red) and G3BP1 (green) to aggregates in spinal cord tissue specimens of patients with C9orf72 expansion, that are absent in an unaffected control. Quantification of STAU1-G3BP1 aggregates in motor neurons of 353 number of C9orf72 and 361 number of controls are shown. Data are mean ± SD, **p < 0.01, Student t test. Scale bar, 30 μM. FBs = fibroblasts.
Stau1 Overabundance and Abnormal mTor Activity in Mouse Models of Neurodegeneration Models
Although STAU1 overabundance and mTOR activity were seen in a number of cell lines from patients with various neurodegenerative conditions, those analyses used cells that were actively dividing. In addition to patient spinal cord tissues, we wanted to validate our observations further in mouse models with neuronal targeting of transgenes. We examined Stau1 and mTor signaling in cerebellar extracts from Pcp2-ATXN2 [Q127] (ATXN2Q127) mice15 and cerebellar and spinal cord extracts from mice overexpressing wildtype Thy1-TDP-43 (TDP-43Tg/+),16 2 mouse models utilized in preclinical studies.5,6 In all 3 cases, Stau1 abundance was significantly increased by 2.5 to 3-times of the amount seen in wildtype littermate animals (Fig 4A–F). Levels of activated mTor were similarly increased (see Fig 4A–F), along with phosphorylation of mTor kinase and downstream substrates p62 and LC3-II. Similar to patient FBs, whereas the proteins were elevated, the Stau1 and mTor mRNA levels were not changed (Fig 4G, H).
FIGURE 4:
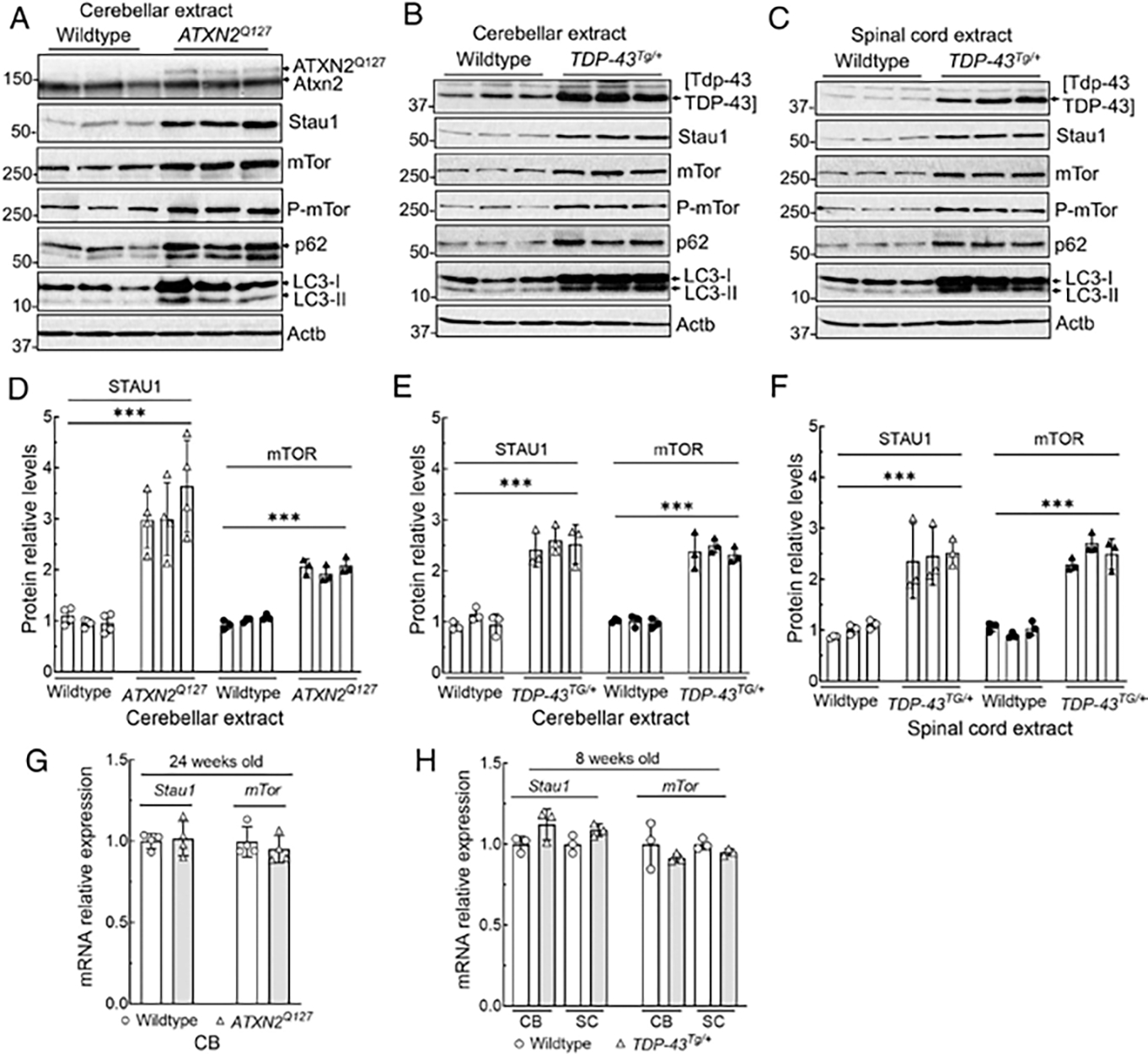
CNS tissues from mice transgenic for ATXN2Q127 or TDP-43 have increased Stau1 levels and abnormal mTor activity. (A–F) Western blot analyses show increased Stau1, mTor, P-mTor, p62, and LC3-II levels compared with wildtype controls in cerebella of ATXN2Q127 mice (24 weeks of age; n = 3) (A), and in cerebella (B) and spinal cords (C) of TDP-43Tg/+ hemizygous mice (8 weeks of age; n = 3). Each lane represents an individual mouse. Stau1 and mTor protein levels were normalized to Actb, and quantified average fold changes for Stau1 and mTor are shown in (D–F). Blots are from 3 replicate experiments. (G, H) Stau1 and mTor RNA levels are unaltered in neurodegenerative disease tissues. qRT-PCR analyses of Stau1, and mTor mRNAs from cerebella and spinal cords from ATXN2Q127 mice (24 weeks of age; n = 4) (G) and TDP-43Tg/+ hemizygous mice (8 weeks of age; n = 3) (H) compared to wildtype littermates. Gene expression levels were normalized to Actb. Ordinary one-way ANOVA followed by Bonferroni tests for multiple comparisons. Data are mean ± SD, ns = p > 0.05, ***p < 0.001. ANOVA = analysis of variance; CNS = central nervous system; CB = cerebellum; mTor = mammalian target of rapamycin; qRT-PCR = quantitative real time polymerase chain reaction; SC = spinal cord.
Disease-Linked Genes Modulate STAU1 Level
Although STAU1 overabundance appeared to be present in a number of neurodegenerative disease FBs, mouse models, and human tissues, we wanted to determine whether STAU1 overabundance was sufficient and necessary to cause abnormal autophagy and granule formation. We used several independent approaches to establish a causal relationship from disease proteins to STAU1 overabundance leading to hyperactive mTOR. First, we replicated the effects of mutant disease proteins by CRISPR/Cas9 modification of the ATXN2 CAG repeat, or by exogenous expression of wildtype or mutant TDP-43 or HTT in HEK-293 cells. Second, we silenced disease gene expression in patient FBs using the respective siRNA. Third, to show that STAU1 overabundance was sufficient to change mTOR activity, we exogenously expressed STAU1 in wildtype HEK-293 cells. In addition, we expressed DsRED-tagged STAU1 in U2OS cells to evaluate the effect of STAU1 on granule formation phenotypes by immunofluorescent microscopy. Finally, we treated patient cells with STAU1 siRNA and determined the effects on mTOR signaling.
Using CRISPR/Cas9 technology, we introduced a mutant ATXN2 allele into HEK-293 cells, ATXN2-Q22/587 or ATXN2-Q22/37 (Fig 5A). Compared with wildtype HEK-293 cells, the presence of endogenous mutant ATXN2 resulted in STAU1 overabundance and hyperactive mTOR signaling. For other disease proteins, we used exogenous expression of the respective protein tagged with either 3XFlag or GFP. These included wildtype and mutant TDP-43 proteins, a C-terminal TDP-43 fragment (CTF), TDP-43 with mutated nuclear localization signal (TDP-43ΔNLS; Fig 5B–D), as well as mutant MAPT and huntingtin [Q66]7,12,22–24 (Fig 5E, F). In all cases, STAU1 steady-state levels increased as did mTOR, p62, and LC3-II.
FIGURE 5:
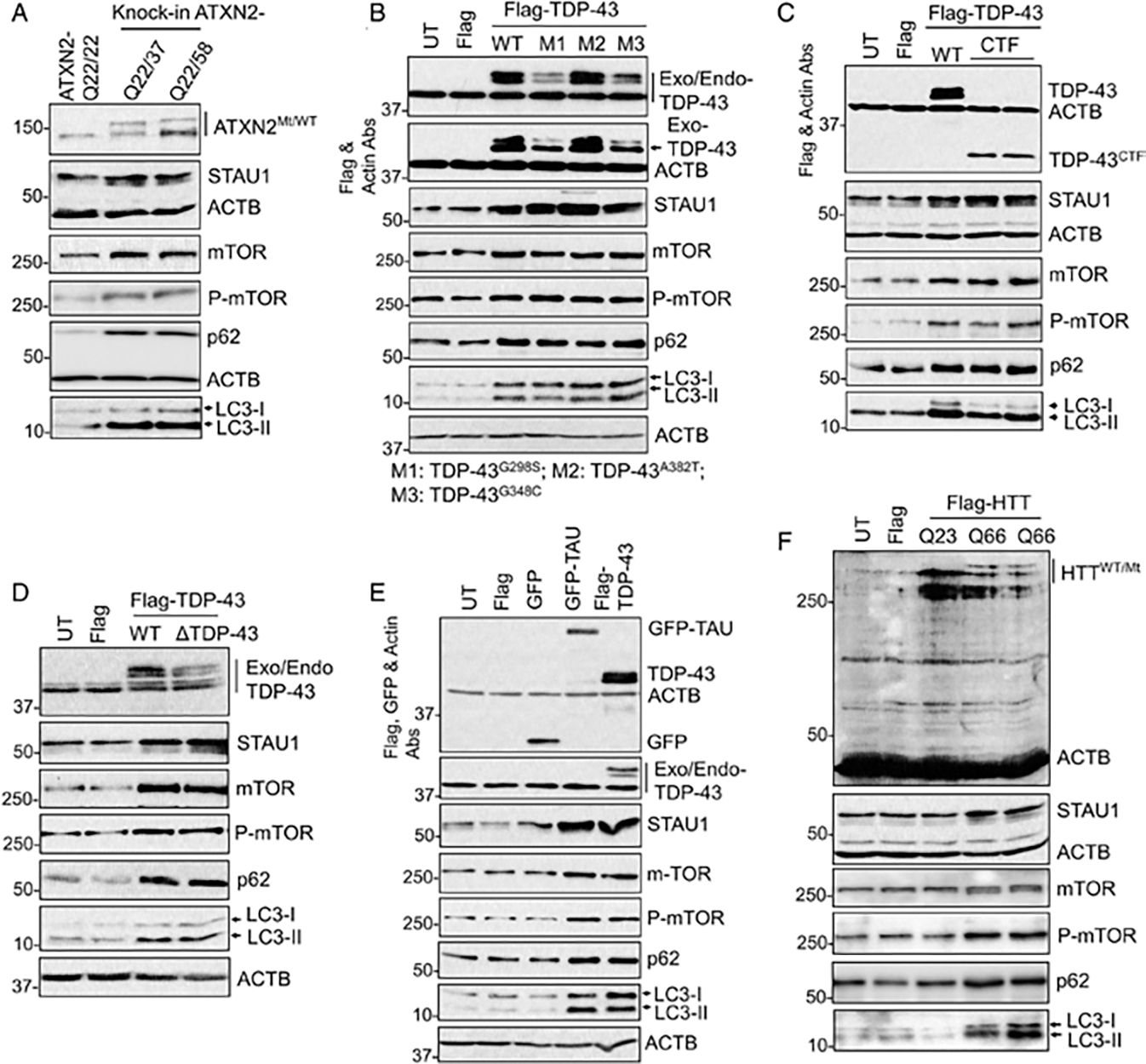
Expression of disease-linked genes recapitulates STAU1 abundances and mTOR activation. (A) CRISPR/Cas9 edited ATXN2-Q37 or ATXN2-Q58 KI cells showing increased STAU1, mTOR levels, and mTOR activity (increased P-mTOR, p62, and LC3-II levels on western blot. (B–F) Exogenous expression of disease-linked genes recapitulates STAU1 overabundance and mTOR activation. HEK-293 cells were transfected with the indicated plasmid constructs for 48 hours followed by Western blotting. Expression of exogenous wildtype TDP-43 or mutant TDP-43 (G298S, A382T, G348C and CTF) and TDP-43ΔNLS (B–D) or TAU (E) or HTT-Q66 repeats (F) resulted in increased STAU1 levels and mTOR activation. ACTB was used as loading control, and representative blots of 3 independent experiments are shown. Abs = antibodies; KI = knock in; mTOR = mammalian target of rapamycin.
To test whether STAU1 overabundance was associated with mTOR activation, we used RNAi to knockdown the levels of the respective disease proteins. We used 2 C9orf72 FB lines and 2 HD lines with 57 and 66 CAG repeats (Fig 6A, B). Control and disease FBs were each treated with a scrambled control siRNA or an siRNA to C9ORF72 or HTT mRNA. The siRNA treatments reduced the target protein levels to those of the control FBs. As quantification of 3 independent experiments in Figure 6C and 6D shows, STAU1 and mTOR protein levels dropped to the levels seen in control FBs.
FIGURE 6:
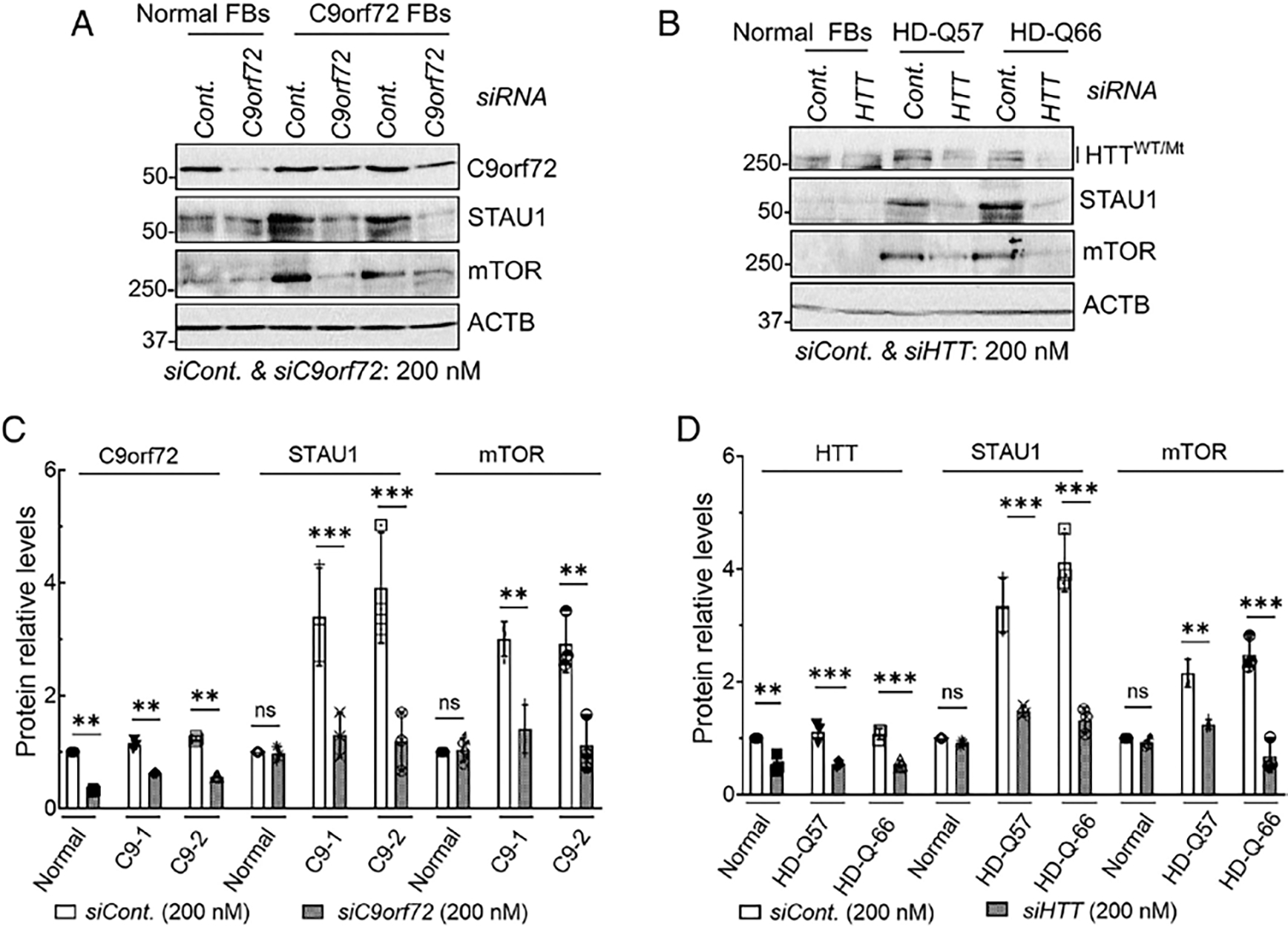
Silencing of disease-linked genes in patient fibroblasts lowers STAU1 levels. (A–D) Normal and FTD-C9orf72 FBs were treated with siRNA to C9orf72 for 4 days (A). Wildtype and HD (HTT with Q57 and Q66 repeats) were transfected with HTT RNAi for 4 days and analyzed by Western blotting (B). Reducing expression of disease-linked genes results in lowered STAU1 and mTOR levels. ACTB is used as a loading control, and the blot is a representative example from 3 replicate experiments. Quantified average fold changes for STAU1 and mTOR are shown in (C, D). Ordinary one-way ANOVA followed by Bonferroni tests for multiple comparisons. Data are mean ± SD, ns = p > 0.05, **p < 0.01 ***p < 0.001. ANOVA = analysis of variance; FBs = fibroblasts; mTOR = mammalian target of rapamycin.
STAU1 Recapitulates mTOR Elevation and Forms Cytoplasmic SG-Like Aggregates
We next asked whether overexpression of STAU1 by itself would change mTOR levels. We exogenously expressed Flag-tagged STAU1 in HEK-293 cells (Fig 7A). Moderate overexpression of STAU1 by itself in normal cells replicated the profile that we had observed with overexpression of disease proteins (see Fig 5). Thus, we saw an increase of mTOR and P-mTOR, and downstream targets. Overabundance of mTOR was post-transcriptional as mTOR mRNA levels did not change (Fig 7B). Consistent with this, mTOR protein and mRNA levels were also found to be invariant in RNAi-mediated STAU1 reduction in HEK-293 cells (Fig 7C, D), indicating that STAU1-mediated mTOR abundance is associated with post-transcriptional modification.
FIGURE 7:
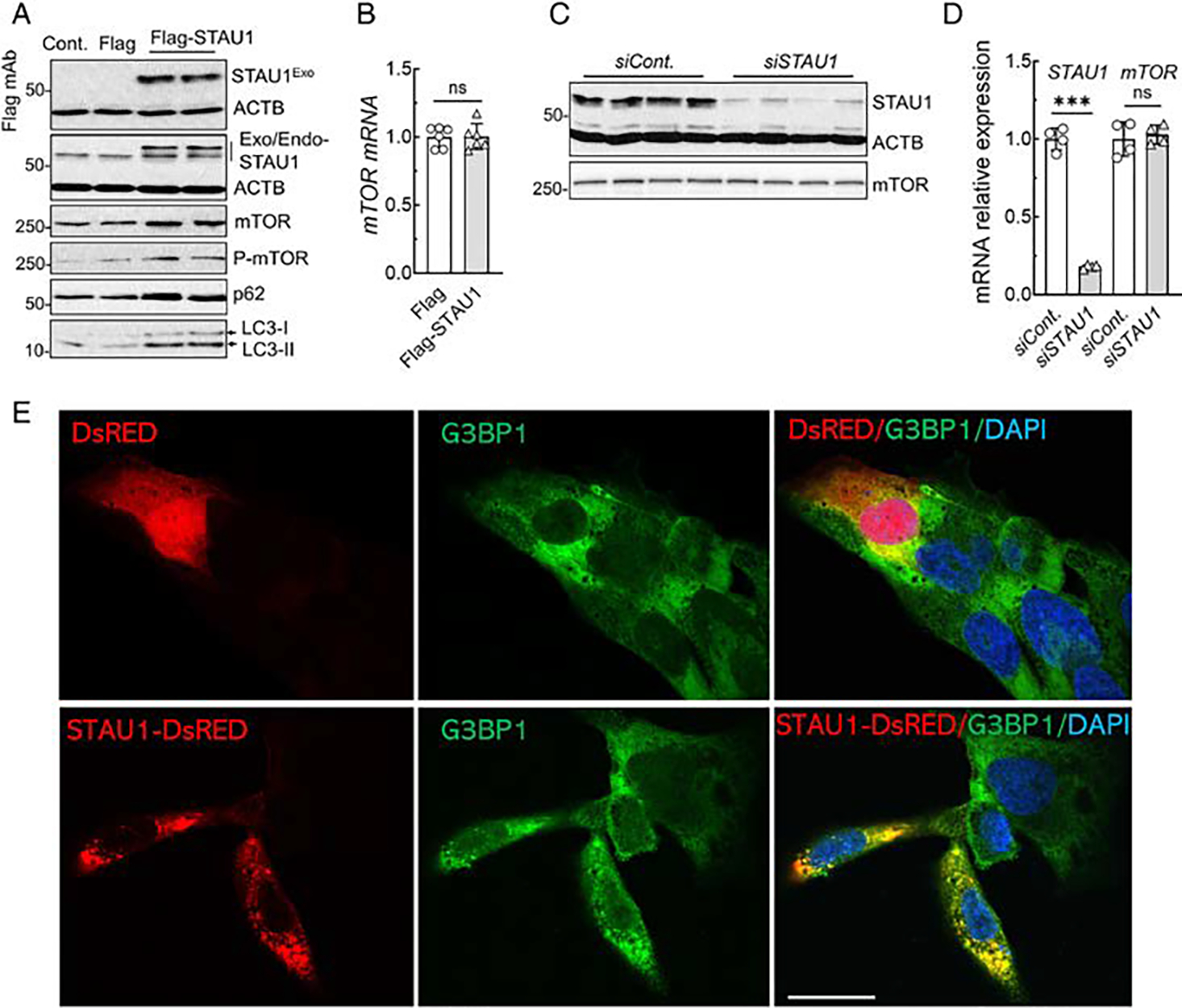
STAU1 is sufficient to recapitulate mTOR activation and form cytoplasmic SG-like aggregates. (A, B) STAU1 overexpression elevates mTOR. (A) HEK-293 cells exogenously expressing Flag-tagged STAU1 were analyzed 48 hours post-transfection by Western blotting and showed increased levels of mTOR, P-mTOR, p62, and LC3-II. (B) mTOR mRNA levels determined by qRT-PCR were unchanged. RNA expression levels were normalized to ACTB. (C, D) STAU1 RNAi-mediated reduction of STAU1 levels does not change mTOR protein and mRNA levels. HEK-293 cells were transfected with STAU1 RNAi and analyzed 4 days post-transfection by Western blotting (C) and qRT-PCR (D). Ordinary one-way ANOVA followed by Bonferroni’s multiple comparisons test. Data are mean ± SD, ns = p > 0.05, ***p < 0.001. (E) STAU1 forms cytoplasmic SG-like aggregates. U2OS cells were transfected with STAU1-DsRED or DsRED plasmids for 48 hours followed by immunostaining with G3BP1, SG marker. Cells expressing STAU1-DsRED, but not DsRED, form spontaneous cytoplasmic SG-like aggregates to recruit G3BP1 (green). Scale bar, 30 μM. mTOR = mammalian target of rapamycin; qRT-PCR = quantitative real time polymerase chain reaction; SG = stress granule.
We previously showed that STAU1 formed constitutive SG-like structures in SCA2 models, and that lowering Staufen1 levels reduced Staufen1 aggregates in an SCA2 animal model.7 We now show that constitutive SG-like aggregates are positive for both STAU1 and G3BP1 in C9orf72 patient cells and tissues (see Fig 3). To investigate if STAU1 is sufficient to induce SG-like structures, we exogenously expressed STAU1-DsRED in U2OS (osteosarcoma epithelial) cells, a cell line frequently used for SG analysis. At 48 hours after transfection, STAU1-DsRED expressing cells, but not DsRED, demonstrated constitutive cytoplasmic aggregates positive for G3BP1, a marker for SGs (Fig 7E), indicating that STAU1 by itself was sufficient to drive SG formation in U2OS cells.
Reduced STAU1 Level Lowers mTOR Activity
If STAU1 is directly involved in altered autophagy in the presence of mutant proteins, then knockdown of STAU1 should normalize mTOR and downstream targets. In order to test this hypothesis, we used siRNA to STAU1 in our ATXN2-Q22/Q58 knock-in cells as well as in cells overexpressing wildtype and mutant TDP-43. Treatment of cells with 100 nM or 200 nM STAU1 siRNA normalized mTOR, P-mTOR, p62, and LC3-II to levels equal to or lower than those seen in wildtype cells (Fig 8A, B). As C9orf72 FBs showed the greatest increase of STAU1, we also treated 2 C9orf72 FB lines with 200 nM STAU1 siRNA. This resulted in reduction of STAU1 to levels seen in normal FBs as well as normalization of mTOR, P-mTOR, p62, and LC3-II (Fig 8C). Quantification of STAU1 and mTOR protein levels of 3 independent experiments are shown in Figure 8D to F. Thus, reducing STAU1 levels rescued the effect of mutant genes on mTOR activity observed in neurodegenerative states.
FIGURE 8:
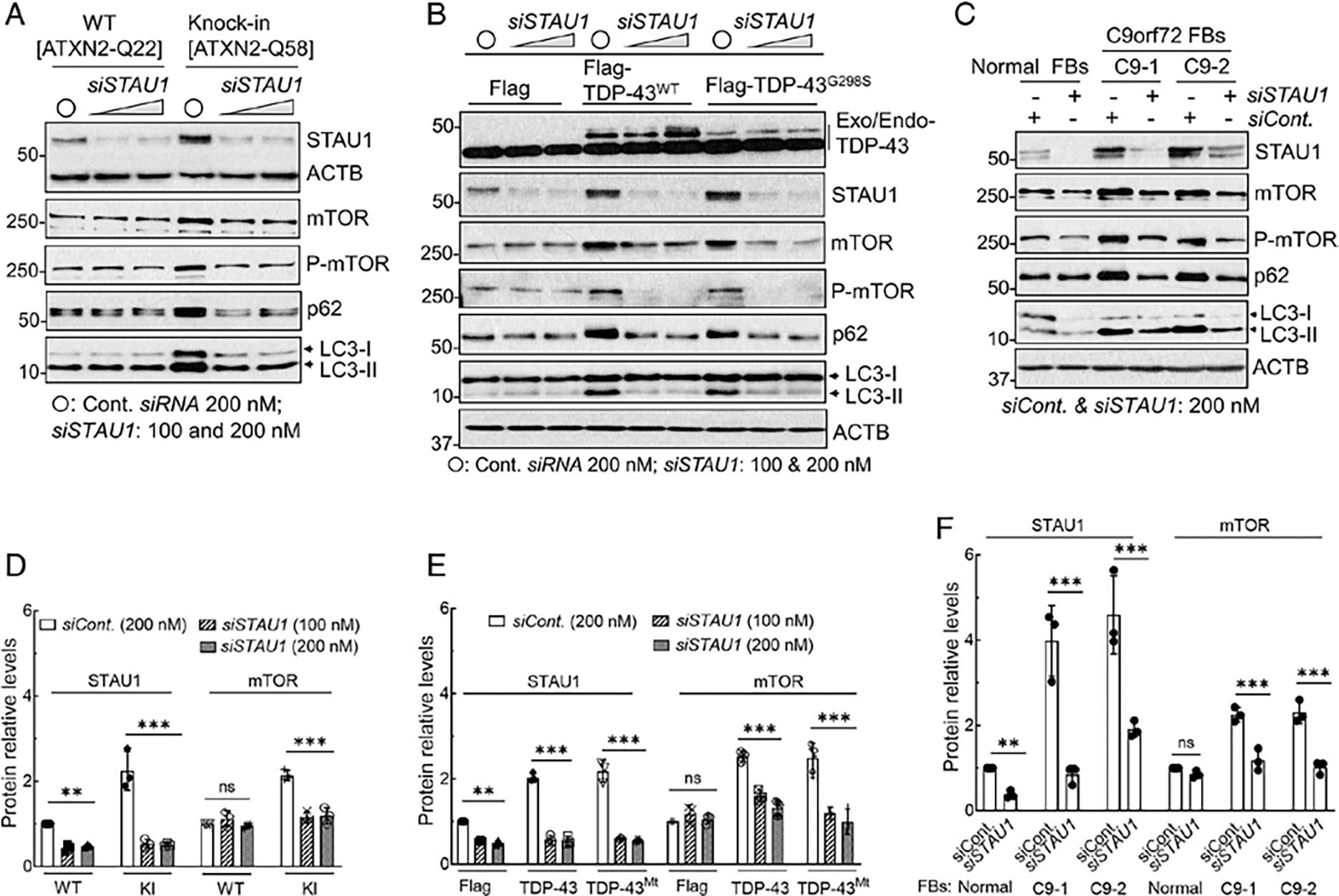
Silencing of STAU1 reduces mTOR activation in SCA2 and ALS/FTD cellular models. (A) STAU1 depletion lowers mTOR activity concurrently with mTOR targets and restores autophagic pathway proteins in ATXN2-Q22/58 KI cells. Cells were transfected with STAU1 RNAi for 4 days and analyzed by Western blotting. Lowering STAU1 in ATXN2-Q22/58 KI cells result in decreased mTOR and P-mTOR levels, as well as decreased p62 and LC3-II levels, indicating restored autophagy activity. (B, C) STAU1 depletion lowers mTOR levels and restores autophagic pathway proteins in cells with TDP-43 mutation (B) or FTD-C9orf72 FBs (C). (B) Western blots of HEK-293 cells expressing Flag-tagged wildtype or mutant TDP-43 or empty vectors followed by transfection with STAU1 RNAi for 4 days. (C) C9orf72 FBs (C9-1 and C9-2) were transfected with STAU1 RNAi for 4 days and analyzed by Western blotting. Lowering STAU1 levels results in decreased mTOR, p62, and LC3-II levels compared to cells treated with control RNAi. ACTB is used as a loading control, and quantification of STAU1 and mTOR levels are shown in (D–F). The blots are from 3 replicate experiments. Ordinary one-way ANOVA followed by Bonferroni tests for multiple comparisons. Data are mean ± SD, ns = p > 0.05, **p < 0.01, ***p < 0.001. ALS = amyotrophic lateral sclerosis; ANOVA = analysis of variance; FB = fibroblast; FTD = frontotemporal degeneration; KI = knock in; mTOR = mammalian target of rapamycin; WT = wildtype.
Discussion
Identifying protein interactors has been an important approach to understand pathways altered by the presence of mutant disease proteins. We identified A2BP1/RBFOX1, an RNA binding protein (RBP) that functions in mRNA splicing, shortly after cloning of the ATXN2 gene.1 More recently, a direct link to ALS was found by showing that ATXN2 interacted with another RBP, TDP-43, which is involved in multiple neurodegenerative diseases.3 We have since used immunoprecipitation and mass spectroscopy to identify other ATXN2 interactors.7 One of the proteins precipitated by both wildtype and mutant ATXN2 was STAU1, another RBP. Although there was no difference in the strength of interaction between wildtype and mutant ATXN2, STAU1 was overabundant in cells in the presence of mutant ATXN2. We showed that STAU1 overabundance was not due to transcriptional changes but was due to decreased autophagic degradation.7
Staufen is a well-studied protein in fly development.25 Owing to its ability to recognize double-stranded RNA, it helps to establish RNA gradients in the oocyte and early embryo. Through gene duplication, 2 Staufen genes exist in mammal cells, whereby Staufen2 (STAU2) is more similar to fly Staufen (Stau) than STAU1. Staufen proteins have a number of functions as RBPs. These include RNA transport,26 STAU1-mediated RNA decay via binding to the 3’UTR of target RNAs,8 or enhanced translation, when STAU1 binds to the 5’UTR of specific RNAs.27 STAU1 is also known to localize to stress granules.7
Mutations in heterogeneous nuclear ribonucleoproteins and TIA1, an SG protein, can cause ALS and frontotemporal lobar degeneration (FTLD).28,29 The role of STAU proteins, however, in neurodegeneration has received relatively little attention. Direct mutations in STAU1 have not been described, but the several-fold increase in steady-state levels in cells and animals with ATXN2 mutations suggested a potentially important role in disease.
We now show that STAU1 overabundance is not limited to the relatively rare disease of SCA2. We used FB cell lines from patients with a diverse group of mendelian mutations in genes leading to AD, ALS, FTLD, and HD. All of these cell lines showed a remarkable increase in steady state levels of STAU1 protein, but not STAU1 mRNA. The increase was particularly marked in cells with C9orf72 or PSEN1 mutations. STAU1 overabundance was also evident in extracts of cerebellum and spinal cord of animals expressing mutant ATXN2 or TDP-43 and in human spinal cord tissue obtained from patients with sporadic and C9orf72-mutated ALS.
Concomitant with STAU1 overabundance, we observed that the mTOR protein, but not its mRNA, was also increased in abundance, as was its phosphorylated form. The hyperactive mTOR kinase was physiologically active and led to increased p62. These changes predict inhibition of autophagy, which is also consistent with increased LC3-II levels as a sign of reduced autophagosome-lysosome fusion. Impaired autophagy has been observed in a number of neurodegenerative models.30–33 More studies will be needed to directly assess autophagic flux in these cell and animal models.
Several experimental observations support that STAU1 is necessary and sufficient to drive neurodegeneration. Previously, we observed that overexpression of STAU1 in HEK-293 cells resulted in reduced Purkinje cell protein 2 (Pcp2) and Calbindin 1 (Calb1) mRNA abundance, replicating observations in ATXN2Q127 mouse cerebellar extracts with elevated Stau1.7 We also saw that STAU1 overexpression in HEK-293 cells activated the PERK pathway of the UPR, leading to increased cleaved caspase 3.11 We now show that STAU1 overexpression in HEK-293 cells increased mTOR and p62 abundance associated with increased LC3-II, and that STAU1 overexpression led to production of STAU1-positive SGs in U2OS cells (see Fig 7).
Complementary to STAU1-induced SGs, we have also demonstrated increased production of STAU1-G3BP1 positive SGs in patients with arsenite treated ALS/FTD-C9orf72 FB cells. Autophagy dysfunction affects clearance of toxic aggregate-prone proteins in multiple neurodegenerative diseases, and induction of autophagy increases the autophagic degradation of these toxic proteins and reduces their toxicity.34 Our observations of STAU1 overabundance across neurodegenerative models, and STAU1 mediated mTOR activation, and SG production support the hypothesis that STAU1 has a role in neurodegeneration. It is possible that STAU1 is more directly involved in autophagic feedback regulation on aggregate-prone disease proteins.
This parallels our observations for STAU1 in modifying UPR signaling. We studied HEK-293 cells expressing STAU1 in the absence of disease-related mutations. Exogenous STAU1 expression caused a substantial increase in levels of the eIF2α kinase PERK, p-PERK, and p-eIF2α. STAU1 overexpression by itself was able to activate caspase 3.11
Overabundance of STAU1 in disease models predicts that abnormal RNA granules would be a prominent feature of the associated diseases. Staufen has more classically been considered a marker of neuronal granules, that functions in trafficking of RNAs in dendrites along microtubules.35 More recently, STAU1 was shown to participate functionally in neuronal trafficking in concert with TDP-43 and Fragile X mental retardation protein (FMRP). Anterograde trafficking of RNA granules in dendrites is dependent on TDP-43 and FMRP that interacts with kinesin1, and is impaired by TDP-43 mutations,36,37 whereas STAU1 is critical for retrograde trafficking via its interaction with the dynein/dynactin complex.37 STAU1/FMRP-positive neuronal granules appear to be distinct from stress granules as the latter appear prominently with disease-related stress, whereas neuronal granules are critical for the normal trafficking of RNAs to the base of dendritic spines, where mRNA cargo is off-loaded and then loaded into ribosomes.37 Indeed, actively translocating STAU1/FMRP granules are structurally and functionally more related to p-bodies than stress granules. STAU1 granules are diverse, representing RNP subclasses, evidenced by 55% of dendritic STAU1 granules that lack FMRP labeling.38 These observations predict that neuronal granule trafficking would be disabled by STAU1 overexpression leading to aggregate formation.
Observations that we made on granule numbers depending on STAU1 abundance are similar to those seen in a previous study by Barbee et al.38 They observed increased colocalization of STAU1 and FMRP, and increased particle size, and reduced particle numbers with the overexpression of either protein.38 In our study, greater STAU1/G3BP1 colocalization to granules occurred in patients with C9orf72 FBs versus control in response to arsenite treatment as well as different time courses of G3BP1-positive and STAU1-positive granule numbers following stress. This latter observation might reflect the diversity of STAU1 in RNP granules as characterized by Barbee et al.38
The importance of STAU1 in causing the observed cellular changes was emphasized by the fact that knockdown of STAU1 with siRNA in patient-derived C9orf72 cell lines normalized mTOR and downstream targets. These observations are in agreement with prior studies that showed a reduction of caspase activation in Stau1 deficient mouse neurons in response to stressors and in cells expressing mutant ATXN2-Q22/58.11 Finally, following our observation that STAU1 localized with ATXN2 in inclusion bodies of SCA2 patient Purkinje cells (PCs),7 we now show that STAU1 localized in G3BP1 positive granules in patients with ALS spinal cords. Taken together, our data indicate that STAU1 accumulations in neurodegenerative disease tissues are accompanied by defective autophagy mediated by hyperactive mTOR.
STAU1 may not function as a sensor of cellular stress, but as an amplifier of stress that is sensed by a number of different pathways. STAU1 can modify its own degradation via it signaling through the mTOR-kinase pathway. Thus, any stressors that inhibit autophagy will engage the feedforward amplification through STAU1. It is also likely that other pathways directly converge on STAU1 and cause changes in STAU1 abundance or cellular localization by post-translational modifications.39 Although a positive feedforward loop may be useful in embryonic development, such as generation of borders during pattern formation; this process appears to be maladaptive for adult neurons.40 In the case of PCs, a second deleterious positive feedback loop exists that involves elevated intracellular calcium and enhanced mGluR1 function.41,42 Although a direct link has not been established, raising of intracellular calcium with ionomycin in HEK-293 cells resulted in a doubling of STAU1 abundance.11
In vivo genetic interaction experiments in SCA2 mice showed that Stau1 haploinsufficiency reduced ATXN2 aggregates in PCs and improved motor function.7 The presence of overabundant STAU1 and hyperactive mTOR in mouse models of SCA2 and TDP-43-neurodegeneration will allow the investigation of STAU1 ASO- and RNAi-mediated therapies on biochemical, morphologic, and functional neurodegenerative phenotypes.
In summary, STAU1 overabundance in neurodegeneration appears to be a more common phenomenon than previously appreciated and is associated with hyperactive mTOR. In the presence of disease mutations, STAU1 overabundance drives mTOR hyperactivation. Future preclinical studies will need to evaluate whether targeting STAU1 with ASOs or miRNA viral vectors can not only improve in vitro but also in vivo neurodegenerative phenotypes and whether STAU1 can serve as another biomarker in neurodegenerative diseases.
Acknowledgments
The authors thank Christopher Thomas Nelson, Kaylee Esparza Dayton, and Brian S. Marshall for technical assistance and mouse handlings. We thank T. Alexander Price and Erika Aoyama for part of cellular and tissue immunostaining, and immunohistological image analyses.
Funding:
This work was supported by National Institutes of Neurological Disorders and Stroke (NINDS) grants R37NS033123 and R56NS33123 to S.M.P., R01NS097903 to D.R.S., and U01NS103883 and R21NS103009 to D.R.S. and S.M.P. S.M.P. also received grant support from the Target ALS Foundation, and D.R.S. received grant support from the Harrington Discovery Institute. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Potential Conflicts of Interest
The authors declare no competing interests.
References
- 1.Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14:269–276. [DOI] [PubMed] [Google Scholar]
- 2.Scoles DR, Pulst SM. Spinocerebellar ataxia type 2. Adv Exp Med Biol 2018;1049:175–195. 10.1007/978-3-319-71779-1_8. [DOI] [PubMed] [Google Scholar]
- 3.Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neuenschwander AG, Thai KK, Figueroa KF, Pulst SM. Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol 2014;71:1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scoles DR, Meera P, Schneider MD, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017;544:362–366. 10.1038/nature22044 Epub 2017 Apr 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017;544:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paul S, Dansithong W, Figueroa KP, et al. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat Commun 2018;9:3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park E, Maquat LE. Staufen-mediated mRNA decay. Wiley Interdiscip Rev RNA 2013;4:423–435. 10.1002/wrna.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heraud-Farlow JE, Kiebler MA. The multifunctional Staufen proteins: conserved roles from neurogenesis to synaptic plasticity. Trends Neurosci 2014;37:470–479. 10.1016/j.tins.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugimoto Y, Vigilante A, Darbo E, et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen1. Nature 2015;519:491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandelman M, Dansithong W, Figueroa KP, et al. Staufen1 amplifies proapoptotic activation of the unfolded protein response. Cell Death Differ 2020;27:2942–2951. 10.1038/s41418-020-0553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker AK, Spiller KJ, Ge G, et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 2015;130:643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chitiprolu M, Jagow C, Tremblay V, et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun 2018;9:2794. 10.1038/s41467-018-05273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J, Matsui M, Gagnon KT, et al. Allele-specific silencing of mutant huntingtin and ataxin 3 genes by targeting expanded CAG repeats in mRNAs. Nat Biotechnol 2009;27:478–484. 10.1038/nbt.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 2013;22:271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 2010;107:3858–3863. 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274–293. 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 2016;17:383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gitler AD, Tsuiji H. There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res 2016;1647:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas MG, Tosar LJM, Desbats MA, et al. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J Cell Sci 2009;122:563–573. 10.1242/jcs.038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler JR, Matheny T, Jain S, et al. Distinct stages in stress granule assembly and disassembly. Elife 2016;5:e18413. 10.7554/eLife.18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igaz LM, Kwong LK, Chen-Plotkin A, et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem 2009;28:8516–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoover BR, Kwong LK, Chen-Plotkin A, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010;68:1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harding RJ, Loppnau P, Ackloo S, et al. Design and characterization of mutant and wildtype huntingtin proteins produced from a toolkit of scalable eukaryotic expression systems. J Biol Chem 2019;294:6986–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St Johnston D, Beuchle D, Nὔsslein-Volhard C. Staufen, a gene required to localize maternal RNAs in the Drosophila egg. Cell 1991; 66:51–63. [DOI] [PubMed] [Google Scholar]
- 26.Kiebler MA, Hemraj I, Verkade P, et al. The mammalian staufen protein localizes to the somatodendritic domain of cultured hippocampal neurons: implications for its involvement in mRNA transport. J Neurosci 1999;19:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dugré-Brisson S, Elvira G, Boulay K, et al. Interaction of Staufen1 with the 5′ end of mRNA facilitates translation of these RNAs. Nucleic Acids Res 2005;33:4797–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467–473. 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackenzie IR, Nicholson AM, Mohona Sarkar M, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017;95:808–816.e9. 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 2012;109:15024–15029. 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 2014;10:677–685. 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in flyand mouse models of Huntington disease. Nat Genet 2004;36:585–595. [DOI] [PubMed] [Google Scholar]
- 33.Tsai PT, Hull C, Chu Y, et al. Autistic like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012;488:647–651. 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain Res 1649;2016:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferrandon D, Elphick L, Nüsslein-Volhard C, Johnston DS. Staufen protein associates with the 3’UTR of bicoid mRNA to form particles that move in a microtubule-dependent manner. Cell 1994;79:1221–1232. 10.1016/0092-8674(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 36.Alami NH, Smith RB, Carrasco MA, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014;81:536–543. 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu JF, Majumder P, Chatterjee B, et al. TDP-43 regulates coupled dendritic mRNA transport-translation processes in co-operation with FMRP and Staufen1. Cell Rep 2019;29:3118–3133.e6. 10.1016/j.celrep.2019.10.061. [DOI] [PubMed] [Google Scholar]
- 38.Barbee SA, Estes PS, Cziko AM, et al. Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 2006;52:997–1009. 10.1016/j.neuron.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruzzone L, Argüelles C, Sanial M, et al. Regulation of the RNA-binding protein Smaug by the GPCR smoothened via the kinase fused. EMBO Rep 2020;21:e48425. 10.15252/embr.201948425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller-McNicoll M, Rossbach O, Hui J, Medenbach J. Auto-regulatory feedback by RNA-binding proteins. J Mol Cell Biol 2019;11:930–939. 10.1093/jmcb/mjz043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meera P, Pulst SM, Otis T. A positive feedback loop linking enhanced mGluR function and basal calcium in spinocerebellar ataxia type 2. Elife 2017;6:e26377. 10.7554/eLife.26377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meera P, Pulst SM, Otis TS. Cellular and circuit mechanisms underlying spinocerebellar ataxias. J Physiol 2016;594:4653–4660. 10.1113/JP271897. [DOI] [PMC free article] [PubMed] [Google Scholar]