

Abstract
Methylation of the mRNA 5′ cap by cellular methyltransferases enables efficient translation and avoids recognition by innate immune factors. Coronaviruses encode viral 2′‐O‐methyltransferases to shield their RNA from host factors. Here, we generate recombinant SARS–CoV‐2 harboring a catalytically inactive 2′‐O‐methyltransferase Nsp16, Nsp16mut, and analyze viral replication in human lung epithelial cells. Although replication is only slightly attenuated, we find SARS–CoV‐2 Nsp16mut to be highly immunogenic, resulting in a strongly enhanced release of type I interferon upon infection. The elevated immunogenicity of Nsp16mut is absent in cells lacking the RNA sensor MDA5. In addition, we report that Nsp16mut is highly sensitive to type I IFN treatment and demonstrate that this strong antiviral effect of type I IFN is mediated by the restriction factor IFIT1. Together, we describe a dual role for the 2′‐O‐methyltransferase Nsp16 during SARS–CoV‐2 replication in avoiding efficient recognition by MDA5 and in shielding its RNA from interferon‐induced antiviral responses, thereby identifying Nsp16 as a promising target for generating attenuated and highly immunogenic SARS–CoV‐2 strains and as a potential candidate for therapeutic intervention.
Keywords: 2′‐O‐methyltransferase, IFIT1, MDA5, Nsp16, SARS–CoV‐2
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Replication of SARS‐CoV‐2 harboring inactive Nsp16 is attenuated and very sensitive to type I IFN treatment. Nsp16 counteracts intrinsic restriction and sensing mechanisms, rendering Nsp16‐deficient viruses good candidates for live attenuated vaccine development.

Introduction
To replicate successfully within a cell, viruses have to cope with various intrinsic antiviral host factors and immune sensors. Severe acute respiratory syndrome–Coronavirus‐2 (SARS–CoV‐2) is an enveloped virus harboring a positive‐stranded (+) RNA genome of approximately 30 kb. Upon entry, the SARS–CoV‐2 RNA genome is translated to generate nonstructural proteins (Nsp) from two open reading frames, ORF1a and ORF1b. A ribosomal frameshift (−1) upstream of ORF1a allows for continuous translation of ORF1b and results in a large polypeptide (pp1ab), which is subsequently cleaved into 16 proteins (Nsp) by viral proteases. In addition, genomic RNA serves as template for transcription of minus‐strand (−) RNA intermediates by the viral RNA‐dependent RNA polymerase (RdRp; Nsp12). The (−) strand RNA is then used by the polymerase to generate (+) strand genomic RNA, subgenomic RNAs encoding the structural proteins M, E, S, and N, as well as several accessory proteins (ORF 3–10). Upon infection, viral RNAs make up the majority of transcripts within the cells, with nucleocapsid (N) RNA being the most abundant (Kim et al, 2020).
Mammalian cells detect invading viruses by so‐called pattern recognition receptors (PRRs), which target nucleic acids of unusual location, form, or quantity (Roers et al, 2016). Key to intracellular sensing of RNA viruses is the family of retinoic acid‐inducible gene I (RIG‐I)–like receptors (RLRs) with its three known members: RIG‐I, melanoma differentiation‐associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2; Rehwinkel & Gack, 2020). While LGP2 is considered to be of mainly regulatory function, RIG‐I and MDA5 recognize immunostimulatory RNA. Upon multimerization, both initiate downstream signaling events via the mitochondrial antiviral‐signaling protein (MAVS), which culminate in type I interferon (IFN) expression and the induction of antiviral effector proteins (Rehwinkel & Gack, 2020). Eukaryotic mRNAs are modified by adding N7‐methylated guanosine on the first nucleotide through reverse triphosphate linkage resulting in the 5′ m7G cap structure (cap0). The m7G cap is crucial for the initiation of protein synthesis, protection from exonucleases, RNA splicing, and many more processes (Ramanathan et al, 2016). Cellular RLRs have been shown to detect uncapped, double‐stranded 5′ RNA ends with accessible triphosphate (PPP) or biphosphate (PP) residues (Hornung et al, 2006; Kato et al, 2006; Pichlmair et al, 2006). In addition, RNA transcripts in higher eukaryotes are methylated by cellular methyltransferases (MTases) at the 2′‐O position of the first (cap1) and sometimes the second (cap2) ribose (Ghosh & Lima, 2010). The lack of 2′‐O‐methylation has been shown to facilitate discrimination between self and nonself transcripts by triggering RIG‐I‐ or MDA5–dependent immune responses (Daffis et al, 2010; Zust et al, 2011; Schuberth‐Wagner et al, 2015). The difference in substrate specificity between RIG‐I and MDA5 is not completely understood, however, RIG‐I seems to preferentially detect shorter molecules, while MDA5 targets longer RNA transcripts (Kato et al, 2008). RNA viruses employ different strategies to avoid detection by host RLRs. Besides encoding for proteins that directly antagonize innate immune sensors, 5′ capping of viral RNA is used by many viruses to mask their transcripts from RLR detection. Some viruses, such as the influenza A virus, “steal” the 5′ end of capped host RNA and ligate it to their own viral RNA, a process called cap snatching. Others, such as coronaviruses, encode their own enzymes to modify the 5′ ends of their RNAs accordingly.
The SARS–CoV‐2 replication has been shown to trigger an MDA5‐dependent immune response in lung epithelial cells (Rebendenne et al, 2021; Sampaio et al, 2021; Thorne et al, 2021; Yin et al, 2021). In addition, RIG‐I has also been suggested to play a role in SARS–CoV‐2 sensing (Thorne et al, 2021). Independent of the RLR involved, the SARS–CoV‐2‐triggered immune response seemed to be delayed and the subsequently initiated type I IFN response was not sufficient to substantially block viral replication. This suggests that SARS–CoV‐2 encodes viral antagonists that dampen RLR‐sensing as well as type I IFN responses. Indeed, several in vitro screening assays by different groups identified multiple viral proteins that could possibly counteract these pathways (Setaro & Gaglia, 2021). SARS–CoV‐2 encodes the methyltransferase Nsp16 within ORF1ab. Together with its noncatalytic binding partner Nsp10, Nsp16 methylates nascent, cap0‐transcripts at the 2′‐OH position of the first ribose using S‐adenosyl‐L‐methionine (SAM) as methyl donor. The crystal structure of Nsp16/Nsp10 together with nucleic acid analogs and SAM donor has been solved, suggesting that Nsp16 of SARS–CoV‐2 acts as methyltransferase (Krafcikova et al, 2020; Rosas‐Lemus et al, 2020a; preprint: Rosas‐Lemus et al, 2020b; Viswanathan et al, 2020; Viswanathan et al, 2021). Interestingly, overexpression of Nsp16 did not interfere with innate immune signaling or sensing in the aforementioned screening assays (Setaro & Gaglia, 2021). Nsp16 of human Coronavirus 229E, however, has been shown to protect viral replication from an MDA5‐mediated immune response and the cellular restriction factor IFIT1 (Zust et al, 2011). Similarly, the replication of SARS–CoV‐1 lacking functional Nsp16 has been found to be attenuated in vitro and in a mouse model (Menachery et al, 2014). Menachery and colleagues also identified IFIT1 and MDA5 as important host factors restricting the replication of SARS–CoV‐1 in the absence of Nsp16 (Menachery et al, 2014).
Within this manuscript, we describe the important role of the 2′‐O‐methyltransferase Nsp16 during SARS–CoV‐2 replication and identify its enzymatic activity to be crucial for viral immune evasion. We generated recombinant SARS–CoV‐2 harboring enzymatically inactive Nsp16, Nsp16 D130A/K170A (Nsp16mut), and analyzed viral replication as well as the role of Nsp16 in innate sensing in human lung epithelial cells. Although the introduced mutations rendered Nsp16 catalytically inactive, we found the replication of SARS–CoV‐2 Nsp16mut to be only slightly attenuated in the human lung epithelial cells. This is particularly interesting since replication of the Nsp16mut virus triggered a strongly enhanced MDA5‐mediated innate immune response compared to the wt virus. In addition, we discovered that the replication of SARS–CoV‐2 lacking an active methyltransferase was highly sensitive to type I IFN pretreatment in control cells but not in IFIT1 KO cells. Together, our article demonstrates that the methyltransferase Nsp16 of SARS–CoV2 shields viral transcripts from detection by MDA5 and counteracts viral inhibition by IFIT1.
Results
Attenuated replication of recombinant SARS–CoV‐2 lacking functional Nsp16 in the human lung epithelial cell lines
Previously, we generated recombinant SARS–CoV‐2 by cloning a passage‐free SARS–CoV‐2 genome into a bacterial artificial chromosome (BAC), resulting in pBelo‐SARS–CoV‐2 (Herrmann et al, 2021). To generate recombinant virus containing nonfunctional Nsp16 (SARS–CoV‐2 Nsp16mut), we designed genome fragments encoding the mutations D130A and K170A within the active center of Nsp16 (Fig 1A; Habjan et al, 2013; Menachery et al, 2017). We used Lambda‐based red recombination and Gibson assembly to introduce the modified Nsp16 fragment into pBSCoV2, resulting in the bacmid pBSCoV2–Nsp16mut (Fig 1B). We then produced recombinant, replication‐competent SARS–CoV‐2 Nsp16mut by transient transfection of the bacmid into 293T cells expressing the SARS–CoV‐2 receptor ACE2 (Herrmann et al, 2021), followed by transfer of the supernatant onto susceptible Caco‐2 cells lacking the RNA sensor MDA5 (Caco‐2 MDA5‐KO) (Rebendenne et al, 2021; Sampaio et al, 2021; Yin et al, 2021). Three days postinfection (dpi), we quantified the virus titer in the supernatant by RT–qPCR or by focus forming assay (FFA). Of note, the number of infectious units (IUs)/ml quantified by FFA was on average 5‐ to 10‐fold lower than the number of RNA copies/ml supernatant, depending on virus strain and stock sample (Fig EV1A). We only detected minor differences in virus production between recombinant SARS–CoV‐2 wt and Nsp16mut, independent of the read out, and used the recombinant viruses throughout the manuscript to characterize the role of Nsp16 during viral replication. First, we compared viral replication kinetics of SARS–CoV‐2 wt and Nsp16mut on various human epithelial cell lines (Fig 2). We tested replication in Caco‐2 cells, a colorectal cancer cell line, as well as the two lung epithelial cell lines Calu‐3 and A549 transiently expressing the receptor ACE2 upon lentiviral transduction. Importantly, Calu‐3 cells have been shown to mirror the innate immune response of primary human airway epithelial (HAE) cells upon infection and are therefore considered a suitable model for analyzing innate host responses to SARS–CoV‐2 replication (Rebendenne et al, 2021; Sampaio et al, 2021; Yin et al, 2021). First, we infected the different cell lines with low doses of wt and Nsp16mut virus and compared replication by quantifying viral genome copies in the supernatant by RT–qPCR or FFA at various time points postinfection. We found that replication of both viruses was detectable already at 1 dpi and peaked at 3 dpi. In Calu‐3 lung epithelial cells, SARS–CoV‐2 Nsp16mut replication was slightly attenuated suggesting an important role of the methyltransferase Nsp16 during viral replication (Fig 2A). Quantification of viral replication by determining RNA copies or IUs in the supernatant of infected cells was very similar for both viruses (Fig 2A). Thus, we continued to quantify RNA copies/ml supernatant as a measure of viral replication for the remaining manuscript. In A549‐Ace2 cells, the difference between wt and Nsp16mut replication was less pronounced (Fig 2B) and in colorectal Caco‐2 cells both viruses replicated with similar efficiency (Figs 2C and EV1B). Therefore, we continued to use Calu‐3 and A549 lung epithelial cells for the following experiments. To test whether the diminished replication of Nsp16mut virus can be saturated by high viral loads, we infected Calu‐3 cells with increasing amounts of wt and Nsp16mut virus and quantified SARS–CoV‐2 RNA and infectious virus particles in the supernatant over the course of 3 days (Figs 2D and EV1C). We found that wt virus replicated to higher titers than Nsp16mut virus independently of viral input titers, indicating that the diminished replication because of the lack of Nsp16 cannot be overcome by high‐viral loads.
Figure 1. Generation of recombinant SARS–CoV‐2 harboring inactivating mutations in Nsp16.
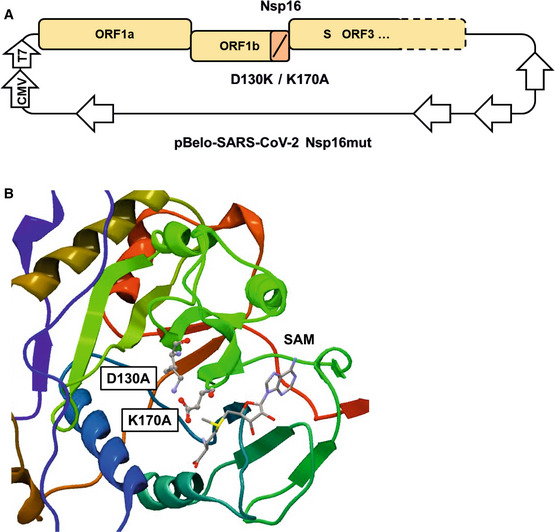
- Schematic depiction of Nsp16 modifications within the bacterial artificial chromosome pBelo‐SARS–CoV‐2 Nsp16mut. Not drawn to scale.
- Inactivating mutations of aspartic acid residue 130 to Alanine (D130A) and lysine residue 170 to Alanine (K170A) within the active center of Nsp16, based on the publicly available crystal structure of Nsp16 (PDB 6W4H). SAM, S‐Adenosyl methionine.
Figure EV1. Quantification of SARS–CoV‐2 wt and Nsp16mut infectivity using a focus forming assay.
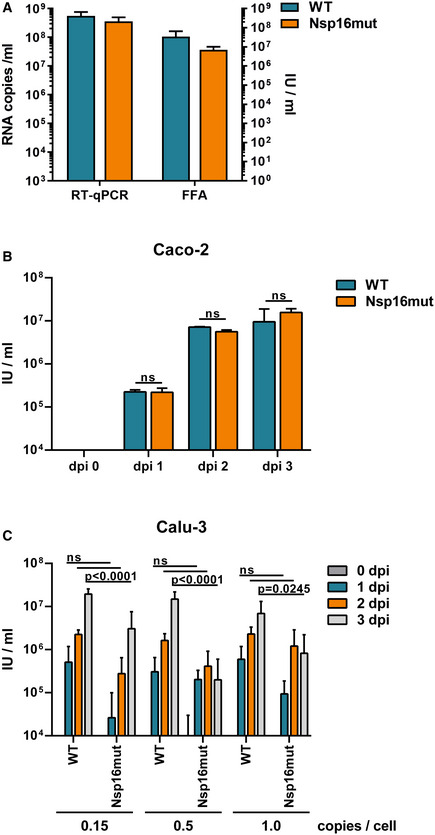
- Quantification of SARS–CoV‐2 WT and Nsp16mut virus stocks using RT–qPCR or focus forming assay. Supernatant from transfected Caco2‐MDA5 KO cells was collected at 3 dpi and quantified by RT–qPCR or focus forming assay (FFA). RT–qPCR: SARS–CoV‐2 WT: 5.59 × 108 copies/ml; Nsp16mut 3.56 × 108 copies/ml. FFA: SARS‐CoV‐2 WT: 1.0 × 108 IU/ml; Nsp16mut 3.7 × 107 IU/ml. Results are plotted as mean of triplicate reactions (technical replicates) with error bars representing the SD. One out of two independent experiments is shown (n = 2).
- Quantification of SARS–CoV‐2 WT and Nsp16mut infecrtion of Caco‐2 cells using focus forming assay. Results are plotted as mean of triplicate infections (biological replicates) with error bars representing the SD. One out of three independent experiments is shown (n = 3).
- Quantification of Calu‐3 cell infection with increasing amounts of SARS–CoV‐2 WT and Nsp16mut using focus forming assay. Results are plotted as mean of triplicate infections (biological replicates) with error bars representing the SD. One out of three independent experiments is shown (n = 3).
Source data are available online for this figure.
Figure 2. Attenuated replication of recombinant SARS–CoV‐2 Nsp16mut on human lung epithelial cell lines.
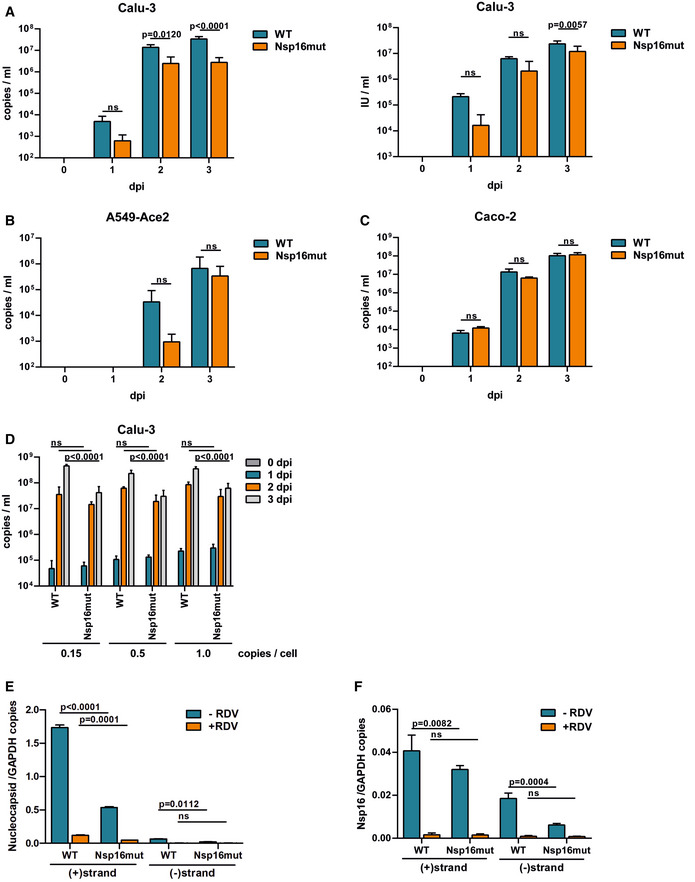
-
ACalu‐3 cells were infected in triplicates with normalized amounts of recombinant SARS–CoV‐2 wt (WT) or Nsp16mut at 0.015 RNA copies/cell (left) or 0.003 IU/cell (right).
-
BA549 cells were transduced with lentiviral particles encoding the SARS–CoV‐2 receptor ACE2 and infected in triplicates with SARS–CoV‐2 wt (WT) or Nsp16mut at 0.025 RNA copies/cell the next day.
-
CCaco‐2 cells were infected with SARS–CoV‐2 wt (WT) or Nsp16mut at 0.015 RNA copies/cell.
-
DCalu‐3 cells were infected with the indicated increasing amounts of SARS–CoV‐2 wt (WT) or Nsp16mut (copies/well).
-
E, FCalu‐3 cells were inoculated with SARS–CoV‐2 wt or Nsp16mut at 0.02 RNA copies/cell in the presence or absence of 200 nM Remdesivir (RDV). At 24 h postinfection, cells were washed and intracellular levels of Nucleocapsid (E) or Nsp16 (F) transcripts were quantified by droplet digital PCR using minus (−) or plus (+) strand‐specific oligos. SARS‐CoV‐2 transcripts were normalized to GAPDH transcript copy number. Results are plotted as mean of triplicate PCR reactions (technical repeats) with error bars representing the SD. One out of three independent biological repeats is shown (n = 3).
Data information: Statistical analysis was performed using two‐way ANOVA followed by Bonferroni's multiple comparisons test. ns, not significant (P > 0.05); dpi, days postinfection. (A–D) Virus release into the supernatant was quantified at the indicated time points by RT–qPCR targeting viral polymerase RdRp or by focus forming assay. Results are plotted as mean of triplicate infections (biological replicates) with error bars representing the SD. One out of three independent experiments is shown (n = 3).
Source data are available online for this figure.
The 5′ capping of mammalian mRNA enhances its stability and, together with 2′‐O‐methylation of the first ribose, shields RNA from innate host factors such as the antiviral effector protein IFIT1, which has been shown to sequester 2′‐O‐unmethylated mRNA upon detection (Habjan et al, 2013). To determine the effect of enzymatically inactive Nsp16 on SARS–CoV‐2 RNA levels, we analyzed viral transcripts upon infection with SARS–CoV‐2 wt and Nsp16mut in Calu‐3 cells (Fig 2E and F). At 24 hpi, we quantified (+) and (−) stranded viral RNAs by strand‐specific digital droplet PCR with specific probes targeting either Nucleocapsid (N) (Fig 2E) or Nsp16 (Fig 2F) transcripts. In line with previous publications, we found a high number of N transcripts at 24 hpi for both viruses, whereas levels of Nsp16‐containing ORF1ab transcripts were 30‐ to 50‐fold less abundant. For both the targets, we found a 3‐ to 5‐fold reduced accumulation of (+) and (−) strand transcripts in Nsp16mut virus infected cells compared to wt infected cells. Of note, we did not observe differences in the number of GAPDH housekeeping transcripts between wt and Nsp16mut infections. However, transcripts were slightly elevated in the presence of RDV, which is in line with the reported inhibitory influence of SARS–CoV‐2 on cellular transcripts (Fig EV2). Treatment with the viral polymerase inhibitor Remdesivir (RDV) drastically reduced SARS–CoV‐2 RNA level, demonstrating that the difference between Nsp16mut and wt RNA level is due to newly transcribed molecules rather than viral input (Fig 2E and F). Thus, the slightly reduced viral RNA level found upon Nsp16mut infection resemble the attenuated replication of Nsp16mut determined by RT–qPCR (Fig 2B).
Figure EV2. Quantification of GAPDH and SARS–CoV‐2 Nsp16 (+) strand transcripts upon infection of Calu‐3 cells with SARS–CoV‐2 WT and Nsp16mut by ddPCR.
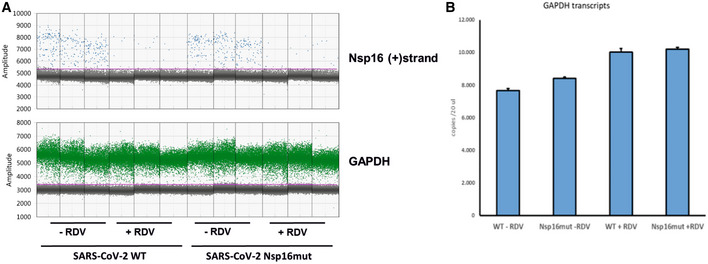
- As in Fig 2, Calu‐3 cells were inoculated with SARS–CoV‐2 wt (WT) or Nsp16mut at 0.02 RNA copies/cell in the presence or absence of 200 nM Remdesivir (RDV). At 24 h postinfection, cells were washed and intracellular levels of Nsp16 transcripts were quantified by droplet digital PCR using plus (+) strand‐specific or GAPDH‐specific oligos.
- Average GAPDH transcript copy numbers for each condition are plotted (biological replicates, n = 3).
Source data are available online for this figure.
SARS–CoV‐2 Nsp16 acts as 2′‐O‐methyltransferase in vitro and upon SARS–CoV‐2 infection
To test whether Nsp16 of SARS–CoV‐2 acts as methyltransferase and whether the amino acid exchanges D130A and K170A introduced in Nsp16 abrogate its enzymatic function, we purified recombinant Nsp16 wt and mutant protein from HEK293F cells (Fig EV3) and analyzed the activity of both proteins in in vitro methyltransferase (MTase) assays. Therefore, we incubated in vitro transcribed Cap0 RNA with wt or mutant Nsp16 together with recombinant SARS–CoV‐2 Nsp10, and measured the transfer of tritium‐labeled methyl groups from SAM[3H] donor molecules to Cap0 RNA (Fig 3A). We found that recombinant Nsp16/Nsp10 complexes efficiently methylated Cap0 RNA, whereas Nsp16 bearing the mutations D130A and K170A was enzymatically inactive (Fig 3A). Next, we asked whether Nsp16 is enzymatically active within recombinant SARS–CoV‐2 and whether the mutations introduced in Nsp16mut abrogate its MTase activity. Thus, we harvested total cellular mRNA of SARS–CoV‐2 wt and Nsp16mut‐infected cells at one dpi and analyzed the methylation status of the isolated RNA upon incubation with recombinant Vaccinia virus (VACV) methyltransferase VP39 (Fig 3B). We found that VACV VP39 methylated recipient mRNA isolated from SARS–CoV‐2 Nsp16mut‐infected cells with significantly higher efficiency than mRNA isolated from wt virus infected cells (Fig 3B), suggesting a higher abundancy of 2′‐O‐unmethylated RNA molecules in Nsp16mut infected cells. Thus, we conclude that Nsp16 of SARS–CoV‐2 acts as 2′‐O‐MTase in vitro as well as during viral replication and that the introduced mutations in SARS–CoV‐2 Nsp16mut render it enzymatically inactive.
Figure EV3. Purification of recombinant Nsp10, Nsp16, and Nsp16mut.
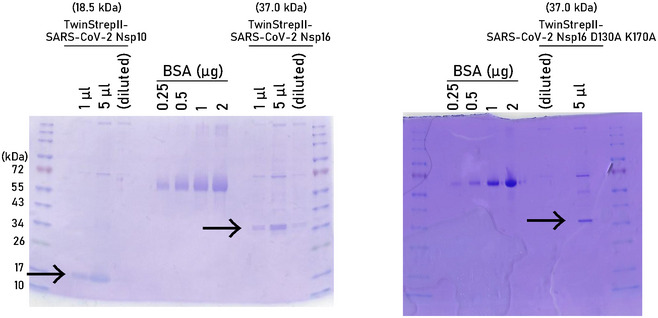
Coomassie staining of SDS–PAGE. Arrows indicate size of purified recombinant proteins used in invtro methyltransferase assays shown in Fig 3. Weak additional bands could not be detected by anti‐Strep‐antibody by immunoblot.
Source data are available online for this figure.
Figure 3. Mutations in the active center render Nsp16 catalytically inactive.
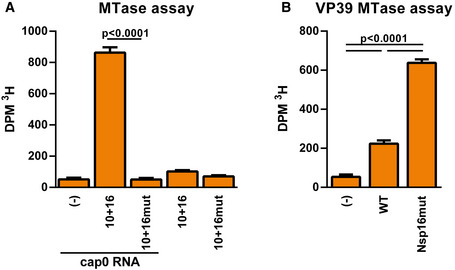
- In vitro Methyltransferase activity. Recombinant proteins Nsp10 and Nsp16 wt or Nsp16mut were combined with cap0 RNA and Adenosyl‐L‐methionine ([methyl‐3H] SAM) for 90 min at 37°C. Methylation of cap0 RNA was quantified using a scintillation counter (Beckmann). (−) Control did not contain any recombinant protein. Results are plotted as mean of triplicate reactions with error bars representing SD. One out of three independent biological repeats is shown.
- MTase activity of Nsp16 upon viral infection. Cellular mRNA was purified 24 h postinfection with SARS–CoV‐2 wt or Nsp16mut (triplicate infections, biological replicates). Purified mRNA was incubated with 100 U of the recombinant MTase VACV VP39 (NEB) and [methyl‐3H] SAM for 90 min at 37°C. Methylation of mRNA from SARS–CoV‐2 infected cells was quantified using a scintillation counter (DPM3H; Beckmann). (−) Control did not contain cellular RNA. Results are plotted as mean of triplicate infections with error bars representing SD. One out of three independent experiments is shown (n = 3).
Data information: Statistical analysis was done using a one‐way ANOVA followed by Tukey's multiple comparisons test.
Source data are available online for this figure.
Replication of SARS–CoV‐2 Nsp16mut is highly interferon sensitive
The SARS–CoV‐2 replication induces type I and type III IFN release in cell culture and animal models. Nevertheless, SARS–CoV‐2 replicates to relative high titers despite the presence of type I IFN (Rebendenne et al, 2021; Sampaio et al, 2021; Yin et al, 2021), suggesting that SARS–CoV‐2 infection inhibits antiviral responses mediated by type I IFN and might block innate immune signaling pathways. However, pretreatment of cells with type I IFN has been shown to limit SARS–CoV‐2 replication in vitro (Rebendenne et al, 2021; Sampaio et al, 2021; Yin et al, 2021). To test the sensitivity of Nsp16mut to type I IFN, we infected Calu‐3 cells with SARS–CoV‐2 wt and Nsp16mut after pretreating the cells with increasing doses of type I IFN (IFNα2a) 18 h prior to infection (Fig 4A). Previously, wt SARS–CoV‐2 replication was found to be limited by pretreatment with high concentrations of type I IFN (1,000 U/ml) (Rebendenne et al, 2021). Interestingly, we found that on cells pretreated with low amounts of type I IFN (10–100 U/ml) SARS–CoV‐2 wt replication was only slightly affected, whereas the replication of Nsp16mut was drastically reduced already at low amounts of type I IFN (Fig 4A). Pretreatment with increasing amounts of type I IFN was monitored by analyzing the expression of the interferon‐stimulated gene (ISG) IFIT1 (Fig 4A). These findings confirm the inhibitory effect of type I IFN pretreatment on SARS–CoV‐2 replication and reveal an increased sensitivity of attenuated SARS–CoV‐2 Nsp16mut toward type I IFN.
Figure 4. Replication of Nsp16mut is highly sensitive to type I interferon.
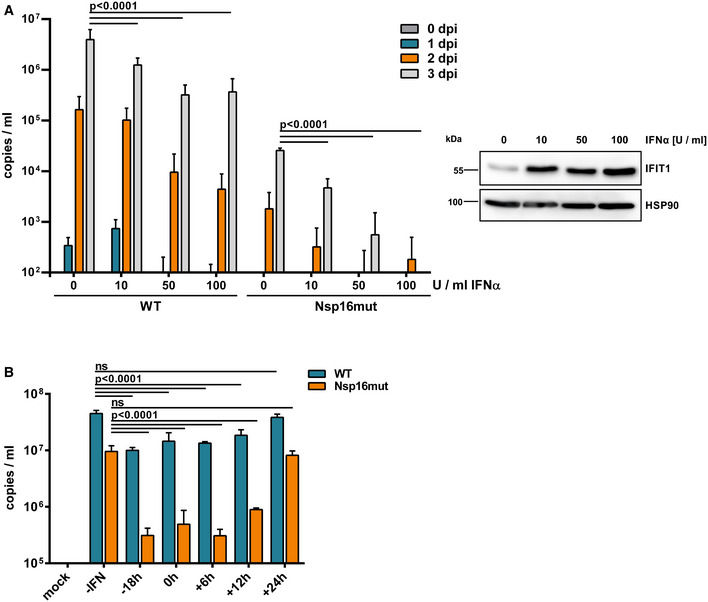
- Calu‐3 cells were incubated with the indicated amounts of type I IFN 24 h prior to infection with normalized amounts of SARS–CoV‐2 wt (WT) or Nsp16mut at 0.015 RNA copies/cell. Viral load in the supernatant was quantified at the indicated time points by RT–qPCR targeting viral polymerase RdRp. Results are plotted as mean of triplicate infections with error bars representing the SD (biological replicates). One out of three independent experiments is shown (n = 3). Three days postinfection, protein expression of IFIT1 was analyzed by immunoblot to control for type I IFN treatment. Membranes were probed with antibodies targeting IFIT1 and the housekeeping gene HSP90.
- Calu‐3 cells were incubated with medium (‐IFN) or 100 U/well type I IFN at the indicated time points prior‐ or postinfection. Cells were infected with normalized amounts of SARS–CoV‐2 wt (WT) or Nsp16mut at 0.015 RNA copies/cell. Viral load in the supernatant was quantified at 3 dpi by RT–qPCR targeting viral polymerase RdRp. Results are plotted as mean of triplicate infections with error bars representing the SD (biological replicates). One out of three independent experiments is shown (n = 3).
Data information: Statistical analysis was done using two‐way ANOVA followed by Bonferroni's multiple comparisons test. ns, not significant (P > 0.05).
Source data are available online for this figure.
Previously, it has been shown that exposure to type I IFN at time points later than 24 hpi did not inhibit SARS–CoV‐2 replication anymore (Rebendenne et al, 2021), indicating that SARS–CoV‐2 efficiently interferes with IFN‐mediated antiviral responses once infection is established. For example, it has been shown that SARS–CoV‐2 Orf6 interferes with interferon signaling by blocking nuclear transfer of STAT complexes (Miorin et al, 2020). We next asked whether the enhanced sensitivity of Nsp16mut toward type I IFN also depends on the time point of exposure. Therefore, we quantified viral replication on Calu‐3 cells, which were treated with 100 U/ml type I IFN at different time points pre or postinfection, over the course of 3 days by RT–qPCR (Fig 4B). We found that type I IFN treatment prior to infection or at early time points postinfection drastically reduced Nsp16mut replication. However, similar to wt virus infection, exposure to type I IFN as late as 24 h postinfection did not affect SARS–CoV‐2 Nsp16mut replication efficiency anymore (Fig 4B). Our findings confirm earlier studies on SARS–CoV‐1 lacking Nsp16 (Menachery et al, 2014) and suggest that the enhanced IFN‐sensitivity of Nsp16mut virus is due to the induction of an antiviral effector molecule, which would otherwise be blocked by viral proteins expressed upon successful infection of the cell.
SARS–CoV‐2 Nsp16mut replication is not affected by MDA5 or RIG‐I expression
Upon infection, SARS–CoV‐2 has been reported to be sensed by the intracellular RIG‐I like receptors (RLRs) MDA5 (Rebendenne et al, 2021; Sampaio et al, 2021; Thorne et al, 2021; Yin et al, 2021) and RIG‐I (Thorne et al, 2021; Yamada et al, 2021), which then culminates in type I IFN release. Since, we found Nsp16mut replication to be attenuated compared to wt virus, we asked whether Nsp16mut might be more sensitive to MDA5‐ or RIG‐I‐mediated antiviral responses. Thus, we compared wt virus and Nsp16mut replication on Calu‐3 cells lacking the receptors MDA5 (MDA5 KO) or RIG‐I (RIG‐I KO) with control luciferase KO cells (Figs 5A and B, and EV4). Similar to previous reports, we found that the replication of SARS–CoV‐2 wt was not enhanced in the absence of either intrinsic RNA sensor (Fig 5A); we even observed a small reduction in infectivity. For Nsp16mut replication, we found a 10‐ to 100‐fold decrease in viral load compared with wt virus infection on control cells (Fig 5B). However, the difference between Nsp16mut and wt virus replication was still detectable in the absence of the RNA‐sensors MDA5 or RIG‐I (Fig 5B). These findings suggest that, similar to wt virus replication, the presence or absence of the intrinsic RNA sensors MDA5 or RIG‐I does not directly affect Nsp16mut replication. This might be explained by the activity of other viral proteins, such as Orf6 (Miorin et al, 2020), that block type I FN signaling once infection is established (Fig 4B).
Figure 5. SARS–CoV‐2 infection triggers an enhanced MDA5‐dependent immune response in the absence of active Nsp16.

-
ACalu‐3 cells lacking MDA5 (MDA5 KO), RIG‐I (RIG‐I KO), or luciferase KO control cells (Ctrl) were infected with normalized amounts of SARS–CoV‐2 wt (WT) with 0.015 RNA copies/cell. Viral load in the supernatant was quantified at the indicated time points by RT–qPCR targeting viral polymerase RdRp. Results are plotted as mean of triplicate infections with error bars representing the SD (biological replicates).
-
BCalu‐3 cells lacking MDA5 (MDA5 KO), RIG‐I (RIG‐I KO), cGAS (cGAS KO), or control KO cells (Ctrl) were inoculated with normalized amounts of Nsp16mut with 0.015 RNA copies/cell. Viral load in the supernatant was quantified at the indicated time points by RT–qPCR targeting viral polymerase RdRp. Results are plotted as mean of triplicate infections with error bars representing the SD (biological replicates).
-
CCalu‐3 cells lacking MDA5 (MDA5 KO), RIG‐I (RIG‐I KO), or control KO cells (Ctrl) were seeded into 96well plates and infected with increasing amounts of SARS–CoV‐2 wt (WT) or Nsp16mut in triplicates (biological replicates). Type I IFN release was quantified after 48 h by incubating supernatants with HEK‐Blue IFN‐α/β reporter cells. SEAP activity in the supernatant of the reporter cells is shown as mean of quadruplicates with error bars representing SD.
-
DCalu‐3 cells (Ctrl) were seeded into 96 well plates and infected with 10 RNA copies/cell of SARS–CoV‐2 wt (WT) or Nsp16mut in the presence of increasing amounts of Remdesivir (RDV). Type I IFN release was quantified after 48 h by IFN bioassay on HEK‐Blue IFN‐α/β and is depicted as mean of quadruplicates with error bars representing SD. In parallel, infectivity of SARS–CoV‐2 wt and Nsp16 in the presence of increasing amounts of RDV was determined at the indicated time points by RT–qPCR targeting viral polymerase RdRp transcripts.
-
E–GCalu‐3 cells were mock infected or infected with SARS–CoV‐2 wt or Nsp16mut at 0.15 copies/cell. At 24 hpi, total RNA was extracted from lysed cells and expression of the indicated ISGs was quantified using RT–qPCR. ISG levels were normalized on GAPDH transcript expression. Results are plotted as mean of triplicate infections with error bars representing SD (biological replicates). One out of three independent experiments is shown (n = 3).
Data information: Statistical analysis was performed using two‐way ANOVA followed by Bonferroni's multiple comparisons test. ns, not significant (P > 0.05).
Source data are available online for this figure.
Figure EV4. Confirmation of RIG‐I and MDA5 KO in Calu‐3 cells by immunoblot.
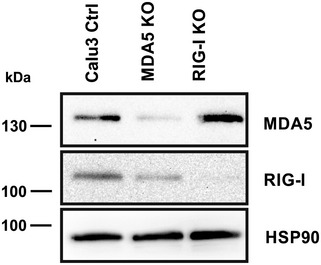
Expression of IFIT1 in the different Calu‐3 cell lines used in this study was analyzed by immunoblot. Membranes were probed with antibodies targeting IFIT1 and the housekeeping gene HSP90.
Source data are available online for this figure.
Enhanced sensing of SARS–CoV‐2 Nsp16mut is mediated by MDA5
Since SARS–CoV‐2 replication is known to trigger the release of type I IFN, we next asked whether Nsp16mut virus induces a similar response. Thus, we infected Calu‐3 control cells, MDA5 KO cells, as well as RIG‐I KO cells with either wt or Nsp16mut virus and measured type I IFN release into the supernatant using HEK–Blue IFNα/β reporter cells (Fig 5C). We observed a strongly enhanced type I IFN release upon Nsp16mut infection compared to wt infection (Fig 5C). Similar to wt virus infection, the enhanced IFN release upon Nsp16mut infection was strongly reduced in MDA5 KO cells, with only marginal levels of type I IFN present. In contrast, similar type I IFN concentrations were found in the supernatant of Calu‐3 wt and RIG‐I KO cells infected with Nsp16mut, suggesting that MDA5 rather than RIG‐I is the major receptor sensing 2′‐O‐unmethylated SARS–CoV‐2 RNA in the absence of Nsp16 (Fig 5C), which confirms earlier findings on SARS–CoV‐1 lacking Nsp16 (Menachery et al, 2014). Next, we asked whether newly generated RNA or incoming viral genomic RNA triggers the enhanced response in the absence of Nsp16 (Fig 5D). Thus, we infected Calu‐3 cells with wt and Nsp16mut viruses in the presence of increasing amounts of the viral polymerase inhibitor RDV. As expected, RDV blocked wt and Nsp16mut replication in a dose‐dependent manner over the course of 3 days (Fig 5D). Interestingly, we found that the enhanced release of type I IFN upon SARS–CoV‐2 Nsp16mut infection negatively correlated with the amount of RDV used, with 3.0 μM of the inhibitor completely abolishing the response (Fig 5D). This suggests that newly generated viral RNAs rather than incoming viral RNA genomes are triggering the enhanced type I IFN release in the absence of Nsp16.
Next, we compared the innate immune response towards wt SARS–CoV‐2 and virus lacking functional Nsp16 on the transcriptional level by quantifying IFN‐stimulated transcripts upon infection by RT–qPCR (Fig 5E–G). We infected Calu‐3 Ctrl or MDA5 KO cells with wt virus or Nsp16mut and analyzed transcript levels of IFNβ, ISG15, and MxA as surrogate for immune activation. In line with the results of the IFN release assays we found that ISG transcript levels were slightly upregulated 24 h post SARS–CoV‐2 wt infection in Calu‐3 Ctrl but not in MDA5 KO cells. Upon infection with SARS–CoV‐2 lacking Nsp16, we found ISG transcripts to be strongly enhanced with MxA levels being 10‐fold, ISG15 levels being 3‐fold, and IFN β levels being 20‐fold higher than after wt virus infection (Fig 5E–G). Interestingly, the increase in ISG transcripts upon Nsp16mut infection was absent in MDA5 KO cells, strongly indicating that the enhanced response towards viral RNA lacking 2′‐O‐methylation mainly depends on the RNA sensor MDA5. In addition, we found that the infection with SARS–CoV‐2 wt as well as Nsp16mut in the absence of MDA5 resulted in a slight upregulation of ISG expression, which is more pronounced upon mutant virus infection (Fig 5E and G). This suggests that an additional receptor, such as RIG‐I, might sense SARS–CoV‐2 replication in the absence of MDA5, albeit to a rather low level.
The enhanced type I IFN sensitivity of SARS–CoV‐2 Nsp16mut is IFIT1‐dependent
Members of the interferon‐induced protein with tetratricopeptide repeats (IFIT) protein family are highly expressed in response to type I IFN signaling and have been shown to interfere with viral protein synthesis and to modulate innate immune signaling (Diamond, 2014). In particular, IFIT1 has been reported by various independent groups to interfere with viral replication in the absence of a viral 2‐O′‐MTase activity by interacting with unmethylated viral mRNA, thereby distinguishing viral from host mRNA (Daffis et al, 2010; Zust et al, 2011; Habjan et al, 2013; Menachery et al, 2014). To test whether the high IFN sensitivity of SARS–CoV‐2 lacking functional Nsp16 is due to the activity of IFIT1, we preincubated ACE2‐expressing A549 control cells and IFIT1 KO cells with increasing amounts of type I IFN 18 h prior to infection with wt or Nsp16mut virus (Figs 6A and EV5). We found SARS–CoV‐2 wt replication to be reduced by increasing amounts of type I IFN in A549 control cells as well as in IFIT1 KO cells at 3 dpi by RT–qPCR. In case of Nsp16mut infection, however, we found a very strong block to viral replication upon treatment with increasing doses of type I IFN (Fig 6A), which negatively correlated with increasing IFIT1 expression levels (Fig 6B). Strikingly, the dose‐dependent inhibitory effect of type I IFN on Nsp16mut was absent in cells lacking the restriction factor IFIT1. In the absence of IFIT1, SARS–CoV‐2 Nsp16mut replicated with similar kinetics and to a similar yield as wt virus (Fig 6A and B) and preincubation with increasing amounts of type I IFN had only a weak effect on SARS–CoV‐2 Nsp16mut replication. Similarly, we generated Calu‐3 IFIT1 KO cells as well as control KO cells by lentiviral transduction followed by antibiotic selection (Fig 6C and D). Although the polyclonal cell line showed residual IFIT1 expression, the type I IFN‐mediated block to Nsp16mut replication was almost completely abrogated in both IFIT1 KO cell lines (Fig 6C). The findings suggest that IFIT1 is the main restriction factor that blocks SARS–CoV‐2 replication in response to type I IFN but is counteracted by the viral 2′‐O‐methyltransferase Nsp16.
Figure 6. The high sensitivity of SARS–CoV‐2 Nsp16mut to type I IFN is mediated by IFIT1.
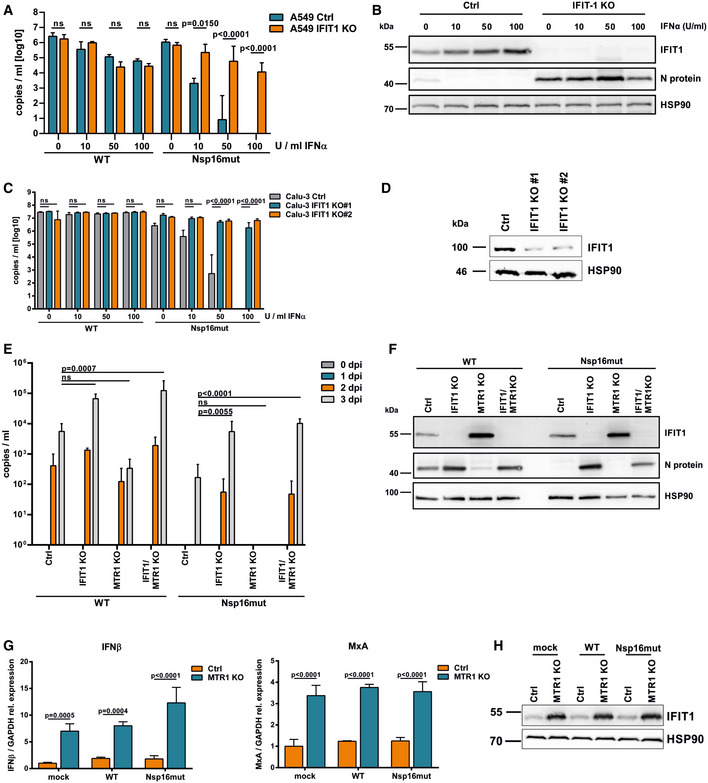
- A549 luciferase KO (Ctrl) or IFIT1 KO cells were transduced with lentiviral particles encoding ACE2 followed by pretreatment with increasing amounts of type I IFN (IFNα2a) as indicated (10, 50, or 100 U/ml). At 24 h postinterferon treatment, cells were infected with SARS‐CoV‐2 wt or Nsp16mut at 0.025 RNA copies/cell. Virus release was quantified at 3 days postinfection by RT–qPCR targeting RdRp. Results are plotted as mean of triplicate infections with error bars representing the SD (biological repliactes). One out of three independent experiments is shown.
- Three days after SARS–CoV‐2 Nsp16mut infection of A549 cells described in (A), expression of IFIT1 and viral nucleocapsid protein N was analyzed by immunoblot. Membranes were probed with antibodies targeting IFIT1, SARS–CoV‐2 N protein, and the housekeeping gene HSP90.
- Calu‐3 control cells (Ctrl) or two different IFIT1 KO cell lines (IFIT1 KO #1, #2) were incubated with increasing amounts of type I IFN (10, 50, or 100 U/ml) 18 h prior to infection with SARS–CoV‐2 wt and Nsp16mut at 0.015 RNA copies/ml. Virus release was quantified at 3 dpi by RT–qPCR targeting RdRp. Results are plotted as mean of triplicate infections with error bars representing SD (biological replicates). One out of three independent experiments is shown.
- IFIT1 expression levels in Calu‐3 Ctrl, IFIT1 KO #1 and #2 lines treated with type I IFN (24 h; 100 U/ml) were analyzed by immunoblot. Membranes were probed with antibodies targeting IFIT1 or the housekeeping gene HSP90.
- A549 cells lacking IFIT1 (IFIT1 KO), the cellular methyltransferase MTR1 (MTR1 KO), both proteins (IFIT1/MTR1 KO), or control cells (Ctrl) were transduced with lentiviral particles encoding ACE2 and infected with SARS–CoV‐2 wt (WT) or Nsp16mut at 0.025 RNA copies/cell the next day. Virus release into the supernatant was quantified at the indicated time points by RT–qPCR. Results are plotted as mean of triplicate infections with error bars representing the SD. One out of three independent experiments is shown.
- Three days postinfection, A549 cells described in (E) were analyzed by immunoblot. Membranes were probed with antibodies targeting IFIT1, SARS–CoV‐2 N protein, or the housekeeping gene HSP90.
- A549 Ctrl and MTR1 KO cells were infected with SARS–CoV‐2 wt or Nsp16 mut virus at 0.015 RNA copies/cell or mock treated (triplicates, biological repeats). At 24 hpi, total RNA was extracted from lysed cells and expression of MxA and IFNβ transcripts was quantified using RT–qPCR. ISG levels were normalized on GAPDH transcripts. Data are plotted as mean of triplicate infections with error bars representing the SD.
- IFIT1 expression was analyzed in mock treated, SARS–CoV‐2 wt (WT) or Nsp16mut infected cells at 48 hpi by immunoblot. Membranes were probed with antibodies targeting IFIT1 and the housekeeping gene HSP90.
Data information: Statistical analysis was performed using two‐way ANOVA followed by Bonferroni's multiple comparisons test. ns, not significant (P > 0.05).
Source data are available online for this figure.
Figure EV5. Confirmation of MTR1 KO in A549 cells by immunoblot.
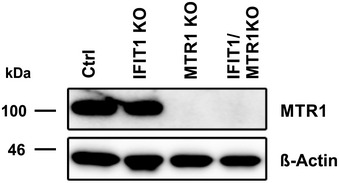
Expression of MTR1 in the different A549 cell lines used in Fig 6 was analyzed by immunoblot. Membranes were probed with antibodies targeting IFIT1 and the housekeeping gene HSP90.
Source data are available online for this figure.
In addition to Nsp16, we analyzed the impact of the cellular methyltransferase MTR1 on SARS–CoV‐2 replication (Bergant et al, 2022; Figs 6E and F, and EV5). Upon infection of A549 cells, we found that SARS–CoV‐2 wt infectivity was reduced by ~10‐fold in the absence of MTR1. In stark contrast, Nsp16mut replication in cells lacking MTR1 was almost completely abrogated. These results highlight the crucial role of RNA 2′‐O‐methylation for viral replication and demonstrate not only the importance of Nsp16 for efficient replication but also suggest that the activity of the viral 2′‐O‐methyltransferase Nsp16 can, at least partly, compensate for the lack of the cellular enzyme, and vice versa. The very similar phenotypes mediated by the lack of one of the enzymes further underline the importance of 2′‐O‐methylation for viral replication and provides further evidence that Nsp16 acts as 2′‐O‐methyltransferase during viral replication.
To confirm the importance of 2′‐O‐methylation for evading IFIT1 restriction, we generated A549 double KO cells lacking MTR1 and the restriction factor IFIT1 using CRISPR/Cas9 (Fig 6E and F). We asked whether IFIT1 plays a role in the diminished replication of SARS–CoV‐2 in the absence of the cellular enzyme. We found that the reduction of SARS–CoV‐2 wt replication in MTR1 KO cells was relieved and, even more impressively, that the complete block to SARS–CoV‐2 Nsp16mut replication in MTR1 KO cells could also be reverted by the additional KO of IFIT1.
Interestingly, for both viruses we found stronger IFIT1 expression levels upon infection of MTR1 KO cells compared to control cell infection (Fig 6F), suggesting that the absence of the cellular methyltransferase contributes to immune activation (Fig 6F). To test whether the restricted replication of wt and especially Nsp16mut virus in MTR1 KO cells might be due to immune activation prior to infection, we determined ISG RNA expression and IFIT1 effector protein expression in uninfected and infected MTR1 KO and control cells (Fig 6G and H). Interestingly, we found upregulated ISG transcripts (MxA and IFNβ) (Fig 6G) as well as enhanced IFIT1 protein level (Fig 6H) already in uninfected MTR1 KO cells. Upon infection, both levels did not increase any further. This suggests that the absence of the cellular methyltransferase MTR1 triggers an immune activation in A549 cells resulting in IFIT1 effector protein upregulation. While SARS–CoV‐2 wt is slightly affected, replication of Nsp16mut is highly sensitive to IFIT1 and its replication in MTR1 KO cells is therefore blocked with high efficacy.
Discussion
In COVID‐19, a timely and robust type I IFN response is associated with a mild course of the disease, which coincides with rapid clearance of the virus (Lowery et al, 2021). In severe cases, however, the response seems delayed, leading to prolonged viral replication and hinting towards efficient viral strategies to avoid and counteract type I IFN responses. Here, we generated recombinant SARS–CoV‐2 lacking the 2′‐O‐methyltransferase Nsp16 (Nsp16mut) and report a dual role for Nsp16 in counteracting innate immune mechanisms during viral replication. Analyzing SARS–CoV‐2 Nsp16mut, harboring an inactive viral methyltransferase, revealed a slightly attenuated replication in human lung epithelial cell lines. Although viral replication in the absence of Nsp16 may also be restricted by other factors, such as decreased RNA stability or reduced translation efficacy of RNAs lacking methylated 5′ cap structures, it has previously been shown for other viruses that viral RNA lacking 2′‐O‐methylation is targeted by the antiviral restriction factor IFIT1 (Daffis et al, 2010; Zust et al, 2011; Habjan et al, 2013). IFIT1 is one of the most strongly induced ISGs and known to recognize and sequester 2′‐O‐unmethylated RNA (Diamond, 2014). Fittingly, we found that the restriction to SARS–CoV‐2 lacking Nsp16 was strongly enhanced upon type I IFN treatment. In addition, SARS–CoV‐2 Nsp16mut proofed to be highly IFN‐sensitive and its replication was already efficiently blocked by low amounts of type I IFN, which only minimally affected wt replication. The IFN‐mediated block to Nsp16mut, and to a lesser degree to wt virus, was almost completely absent in IFIT1 KO cells, suggesting that in SARS–CoV‐2 replication IFIT1 is the main effector protein mediating the IFN‐dependent block to viral replication and is counteracted by Nsp16 (Fig 6A and C). Of note, we also observed small inhibitory effects of type I IFN on the replication of wt virus, which seemed to be independent of IFIT1 expression, and on the replication of Nsp16mut virus in IFIT1 KO cells. Since both the effects were similar in magnitude, it is tempting to speculate that the effect of type I IFN on wt virus is mediated by upregulation of additional, unknown ISGs targeting SARS–CoV‐2 replication. In addition, we found that the absence of the cellular 2′‐O‐methyltransferase MTR1 resulted in reduced replication of wt SARS–CoV‐2 and in a complete block to Nsp16mut replication (Fig 6E; Bergant et al, 2022). This further underlines the importance of RNA 2′‐O‐methylation for viral replication and suggests that the cellular methyltransferase MTR1 can partly compensate for the loss of the viral enzyme in SARS–CoV‐2 Nsp16mut. Importantly, the partial restriction to Nsp16mut replication in wt cells as well as the complete block in MTR1 KO cells could both be lifted by the additional KO of IFIT1 (Fig 6E). This finding indicates that both Nsp16 and MTR1 are protecting viral RNA from IFIT1 restriction and therefore confirms Nsp16 as active 2′‐O‐methyltransferase during viral replication.
In agreement with previous studies for wt virus (Rebendenne et al, 2021), we found that low levels of type I IFN only blocked SARS–CoV‐2 Nsp16mut replication when added prior to or shortly after initial infection but not at later time points, indicating that the viral mechanisms to counteract IFN‐mediated responses, such as blocking IFN signaling, are still intact in SARS–CoV‐Nsp16mut (Fig 4B). The intracellular RNA sensor MDA5 is known to be activated upon wt SARS–CoV‐2 infection, resulting in the induction of a type I IFN‐mediated innate immune response (Rebendenne et al, 2021). Surprisingly, we found that Nsp16mut replication triggers an even stronger MDA5‐dependent immune response than wt virus, despite lower replication capacity, including a higher upregulation of ISG transcripts and an increased release of type I IFN from infected cells (Fig 5). We found that blocking the viral polymerase RdRp abolished IFN release upon infection, suggesting that newly transcribed RNA lacking 5′ cap methylation rather than incoming genomic RNA triggers MDA5 sensing. The substrate specificity of MDA5 is less clear than of RIG‐I, which is known to sense short 5′ uncapped dsRNA molecules. In the absence of MDA5, we found only residual immune induction upon Nsp16 infection leaving room for the idea that RIG‐I might have a minor role in Nsp16mut sensing, but also demonstrating the important role of MDA5 as the main receptor triggered by Nsp16mut virus infection. So far, MDA5 was known to sense long dsRNA molecules, however, no involvement of 5′ cap methylation status in sensing has been reported. Future research will elucidate whether potential cellular cofactors or regulators of MDA5 are necessary to sense SARS–CoV‐2 RNA and to compare the role of the cap methylation in the interaction with possible regulators and RNA substrates. Our finding that MDA5 is activated by 2′‐O‐unmethylated RNA might provide a new handle to decipher MDA5‐mediating RNA sensing and help to clarify the differences between the closely related RLRs RIG‐I and MDA5 in substrate specificity. Similar to wt virus replication, we found that the attenuated replication of Nsp16 could not be rescued by the absence of MDA5, indicating that the enhanced type I IFN response triggered by MDA5 is not sufficient to block Nsp16 replication (Fig 5B). This is in line with the idea that, similar to wt virus, SARS–CoV‐2 lacking Nsp16 still efficiently blocks type I IFN signaling by expression of various viral immune antagonists such as Nsp1 (Thoms et al, 2020; Hayn et al, 2021). However, the role of MDA5‐sensing during replication might be more pronounced in vivo and it will be interesting to analyze the impact of MDA5‐mediated immune responses on SARS–CoV‐2 wt and Nsp16mut replication in animal models such as MDA5 KO mice. An attenuated replication of SARS–CoV‐1 lacking Nsp16 in a mouse model has already been observed (Menachery et al, 2014). Interestingly, in this infection model the difference in replication between wt and Nsp16‐mutated virus was absent in mice lacking MDA5 (Menachery et al, 2014), indicating the important role for MDA5 in coronavirus sensing, especially in the absence of Nsp16. Our findings on the role of Nsp16 in shielding viral RNA from the restriction factor IFIT1 in combination with its enhanced immunogenicity identifies Nsp16 as a potential candidate gene for the development of an attenuated live vaccine. Since the replication of SARS–CoV‐2 Nsp16mut in cell culture is only weakly affected, large quantities of the highly IFN‐sensitive virus could be generated. Replication of virus lacking Nsp16 might be more easily controlled by type I IFN responses in vivo. In addition, since Nsp16mut virus proofed to be more immunogenic in cell culture, it might also generate enhanced innate immune responses compared in vivo. These properties make SARS–CoV‐2 lacking Nsp16 an interesting first candidate for further in vivo experiments, especially in combination with additional mutations affecting the viral counteraction of IFN‐mediated responses and antiviral pathways. In addition, the important role of Nsp16 as well as the cellular 2′‐O‐methyltransferase MTR1 for viral replication identifies both MTases as potential targets for pharmaceutical intervention (Fig 6E–H). Small compound inhibitors targeting the active center of both Nsp16 and MTR1 resulted in reduced replication capacity of wt virus but completely blocked Nsp16mut virus replication. These results confirm the finding that the cellular MTase MTR1 can partly complement the lack of Nsp16 during viral replication. The findings document the high sensitivity of Nsp16mut viruses to MTase inhibitors, making these inhibitors an important safety feature upon potential live vaccine approaches using SARS–CoV‐2 lacking Nsp16.
Materials and Methods
Cell culture and reagents
Human embryonic kidney (HEK)‐293T, human alveolar adenocarcinoma A549, and Vero E6 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, 41965), 10% fetal bovine serum (Gibco, 10270), 100 U/ml penicillin, and 100 mg/ml streptomycin (Gibco, 15140) at 37°C and 5% CO2. Caco‐2 cells were maintained in DMEM, 1% non‐essential amino acids (NEAA, Gibco, 11140), 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. Calu‐3 cells were kept in Eagle's Minimum Essential Medium (MEM), containing 1% NEAA, 10% fetal bovine serum, 1 mM Na‐Pyruvate (Gibco, 11360), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. HEK‐Blue IFN‐α/β cells were maintained in complete Dulbecco's modified Eagle medium (DMEM) cultured with 100 μg/ml zeocin and 30 μg/ml blasticidin. A549 IFIT1 KO cells were generated by CRISPR‐Cas9 genome editing. Lentiviral particles encoding a sgRNA targeting IFIT1 (5′‐AGGCATTTCATCGTCATCAA‐3′) or firefly luciferase (Ctrl), the Cas9 protein, and a puromycin resistance gene were used to infect A549 wt cells. After 3 days, transduced cells were selected by adding puromycin followed by single clone isolation. The KO was confirmed by immunoblot and sequence analysis. A549 MTR1 KO, IFIT1 KO, and MTR1KO/IFIT1 KO have been described previously (Bergant et al, 2022). Calu‐3 and Caco‐2 KO cells (RIG I KO (5′‐CACCGCAGGATGTAGGTAGGGTCCA‐3′), MDA5 KO (5′‐CACCGAACTGCCT GCATGTTCCCGG‐3′), IFIT1 KO (5′‐AGGCATTTCATCGTCATCAA‐3′), or luciferase Ctrl KO) were generated by CRISPR‐Cas9 genome editing, as described above. Cells were transduced with lentiviral particles harboring the respective guide RNAs followed by puromycin selection.
Viruses
Recombinant SARS–CoV‐2 was generated by transfecting the bacterial artificial chromosome (BAC) pB‐SCoV2, which encodes a passage‐free SARS–CoV‐2 genome (Pangolin B1; GISAID EPI_ISL_2732373), into HEK 293T cells overexpressing viral N protein, ACE2 receptor, and T7 RNA polymerase, as described previously (Herrmann et al, 2021). The BAC pBeloCoV2‐Nsp16mut harboring the inactivating mutations D130A, K170A within the Nsp16 2′‐O‐methyltransferase gene was generated by serial recombination steps introducing a gene block (nt 20.296–21.192; synthesized by IDT), carrying the mutations corresponding to the inactivating mutations D130A, K170A in the 2′‐O‐methyltransferase Nsp16. (NEBuilder HiFi DNA Assembly Cloning Kit, E5520). DNA was transformed into E. coli GS1783, and the resulting colonies were screened by restriction digestion. Clones with restriction patterns matching the full‐length SARS–CoV‐2 genome were selected and confirmed by next‐generation sequencing. Three days posttransfection, viral supernatant was transferred to Caco‐2 MDA5 KO cells. Viral load within the purified supernatant was quantified either by RT–qPCR or by focus forming assay (FFA) 3 days postinfection of Caco‐2 cells. For the production of ACE2‐containing lentiviral particles, HEK 293T cells were cotransfected with vesicular stomatitis virus glycoprotein expression plasmid (pMD‐G), HIV gag/pol lentiviral vector (p8.9QV) and pSACE2 encoding for the ACE‐2 receptor at a mass ratio of 1:1:1.5 using calcium phosphate transfection. After 48 h, supernatant was harvested, passed through 0.4 μm pore size filters, aliquoted, and stored at −80°C. Transduction of A549 cells was performed 24 h prior to SARS–CoV‐2 infection. Remaining ACE2‐containing viral particles were removed from the supernatant by medium exchange prior to infection. To generate lentiviral particles encoding the CRISPR/Cas9 system, HEK 293T cells were transfected with pMD‐G, p8.9QV, and pLentiCRISPRv2 encoding guide RNAs targeting the indicated host factors using the calcium phosphate method. After 48 h, supernatant was collected, passed through 0.4 μm pore size filters, aliquoted, and stored at −80°C.
SARS–CoV‐2 infection
A549, Caco‐2, or Calu‐3 cells were infected in triplicates or quadruplicates with either recombinant SARS–CoV‐2 wt or Nsp16mut using the indicated amounts of viral RNA copies or IUs per cell. After 6 h at 37°C, cells were washed with medium twice, followed by sampling the supernatant (0 dpi time point). Supernatant from infected cells was collected at the indicated time points (0–3 dpi) and inactivated for 20 min at 95°C. Viral load in the supernatant was quantified by RT–qPCR on an ABI Prism 7500 cycler (Applied Biosystems) using the Luna Universal Probe One‐Step RT–qPCR Kit (NEB), 5 μl of supernatant, forward primer (GTGAAATGGTCATGTGTGGCGG), reverse primer (CAAATGTTAAAAACACTATTAGCATA), as well as a probe (VIC‐caggtggaacctcatcaggagatgc‐BMN‐Q535) targeting viral polymerase. A standard curve was generated by serial dilution of SARS–CoV‐2 RNA. Alternatively, infectivity of cellular supernatants (IU/ml) was determined using a focus forming assay (FFA). Briefly, Caco‐2 target cells were infected with increasing amounts of viral supernatant in a 96 well format. After 1 h, medium was replaced with low‐viscosity overlay medium (MEM) containing 2.4% Avicel (microcrystalline cellulose), 1% Hepes, 1% Glutamax, 0.2% Gentamycin, 2% NaHCO3, and 10% FCS. After 24 h, cells were washed, permeabilized (0.5% Triton), and probed with anti SARS–CoV‐2 Spike antibodies TRES618 and TRES219 (250 ng/ml) and anti‐mouse IgG Alexa488 (Cell Signaling 4408S) (Peter et al, 2022). Infectivity was quantified using the ImageXpress Pico Automated Cell Imaging System (Molecular Devices). For IFN treatment, 10–100 U/ml type I IFN (IFNα2a, PBL‐11100‐1) were applied 18 h prior to infection or as indicated.
Immunoblot analysis
Cells were lysed in NP‐40 lysis buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5% NP‐40, Halt Protease Inhibitor). Protein content of the lysates was determined by Bradford assay (Carl Roth). Samples were separated by SDS–PAGE, transferred onto Immobilon‐P PVDF membranes (Merck), and probed with primary antibodies targeting endogenous IFIT‐1 (Cell Signaling, 14769), SARS–CoV2 Nucleocapsid protein (novubio, NB100‐56576), and the house keeping genes HSP90 α/β (Santa Cruz, sc‐13119) or GAPDH (Cell Signaling, 2118). Membranes were probed with anti‐mouse or anti‐rabbit HRP‐labeled secondary antibodies (Cell Signaling).
Digital droplet PCR
Calu‐3 cells were infected with recombinant SARS–CoV‐2 wt or Nsp16mut (0.02 copies/cell) in presence or absence of 200 nM Remdesivir (RDV, Gilead). After 24 h, total cellular RNA was extracted using the NucleoSpin RNA Mini Kit (Machery & Nagel). To quantify strand‐specific viral transcripts, isolated RNA was reverse transcribed (Superscript II RT, Life Technologies) using strand‐specific oligos for Nsp16 (plus strand: 5′‐AACAACTCTGTTGTTTTCTC‐3′; minus strand: 5′‐GGGTGTTGCTATGCCTAATC‐3′) or Nucleocapsid protein (plus strand: 5′‐CACTGCTCATGGATTGTTGC‐3′; minus strand: 5′‐AATCAGCGAAATGCACCCCG‐3′) and GAPDH (5′‐CTGTAGCCAAATTCGTTGTC‐3′). Upon cDNA synthesis, remaining RNA in RNA/DNA duplexes was removed by addition of RNaseH (NEB). The following ddPCR reactions included 100 pg of strand‐specific cDNA, 2x ddPCR Supermix (BioRad), 900 nM oligos targeting transcripts of Nsp16 (Forward: 5′‐AATTATGGTGATAGTGCAAC‐3′; Reverse: 5′‐CCACTGTCTTAAAACAGCTG‐3′) or Nucleocapsid (Forward: 5′‐CAGTAACCAGAATGGAGAAC‐3′; Reverse: 5′‐GGTCATCTGGACTGCTATTG‐3′) and 250 nM of FAM‐labeled fluorescent probes (Nsp16: 5′‐FAM‐CATTAACATTAGCTGTACCC‐BHQ1‐3′; Nucleocapsid: 5′‐FAM.ATACTGCGTCTTGGTTCACC‐BHQ1‐3′). Nsp16 and Nucleocapsid copy numbers where normalized on GAPDH copy numbers using 900 nM of forward (5′‐AAGGACTCATGACCACAGTC‐3′) and reverse (5′‐AAGGCCATGCCAGTGAGCTTC‐3′) oligos as well as a HEX‐labeled fluorescent probe (5′‐HEX‐CAGAACATCATCCCTGCCTC‐BHQ‐1‐3′). Droplets were generated using the QX200 Droplet Generator (BioRad). ddPCR reactions were run on a T100 Thermal cycler (BioRad) and analyzed with a QX200 Plate Reader using the Quanta Soft Analysis Software (BioRad).
Quantitative PCR of ISG transcripts
Total RNA of Calu‐3 cells infected with SARS–CoV‐2 wt or Nsp16mut (0.15 copies/cell) or mock infected cells was isolated 24 hpi using the NucleoSpin RNA Mini Kit (Machery & Nagel) according to manufacturer's instructions. To quantify transcript level of the interferon‐stimulated genes (ISGs) MxA, ISG15, and IFN β by RT–qPCR, 2.5 μg of total RNA was reverse transcribed using oligo‐dT primer and Superscript II RT (Life Technologies). Quantitative PCR was performed in triplicates using the Maxima SYBR Green qPCR Mastermix (Life Technologies) on an ABI Prism 7500 cycler (Applied Biosystems). Expression levels of ISGs were normalized to GAPDH using the 2−ΔΔCt method. Oligonucleotides for qPCR: MxA 5′‐TCCAGCCACCATTCCAAG‐3′ (forward) and 5′‐CAACAAGTTAAATGGTATCACAGAGC‐3′ (reverse); ISG15 5′‐GTCTGGCTGTCCACCCGAGC‐3′ (forward) and 5′‐CGTGCTGCCGGGGCCCAGGC‐3′ (reverse); IFN‐beta 5′‐CTTT GCTATTTTCAGACAAGATTCA‐3′ (forward) and 5′‐GCCAGGAGGTTCTCAACAAT‐3′ (reverse), GAPDH 5′‐GGAGTCCCTGCCACACTCAG‐3′ (forward) and 5′‐GGTCTACATGGCAACTGTGAGG‐3′ (reverse).
IFN bioassays
HEK‐Blue IFN‐α/β (InvivoGen) were seeded at 50,000 cells per 96‐well. The following day, supernatant from infected or control cells was added. A reference curve was generated in parallel by serial dilutions of type I IFN in complete DMEM. After 24 h, 40 μl of HEK‐Blue IFN‐α/β supernatant was added to 40 μl PNPP (p‐nitrophenyl phosphate) substrate and incubated at RT for 30–90 min. Absorbance was measured at 405 nm using a plate reader. The reference curve was used to quantitate type I IFN concentrations in the supernatant of infected cells.
Recombinant protein production and purification
Recombinant SARS–CoV‐2 Nsp10, Nsp16, and Nsp16 D130A K170A (NSP16mut) proteins were expressed in Expi293F cells using the ExpiFectamine Transfection Kit (Gibco, A14524) and purified using an affinity tag TwinStrep‐tag II. Cell pellets were lysed with 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% NP‐40, 1 mM DTT, 2 μg/ml leupeptin, 1 mM PMSF, and 1 mM vanadate, and disrupted by three freeze and thaw cycles. After removal of cell debris by centrifugation, the supernatant was mixed with Strep‐Tactin Superflow Agarose (Merck, 71592‐4) in an open column (Pierce, 29920), washed, and eluted with biotin‐containing buffer (Merck, 71613‐3 (100 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 2.5 mM desthiobiotin, pH 8.0)). Protein concentration was determined using BSA standard protein (Pierce, 23210) with Coomassie blue staining.
In vitro RNA transcription
Cap0 RNA was synthesized using the HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB, E2050S) with the cap analog m7G(5′)ppp(5′)A (NEB, S1405) according to manufacturer's instructions. Forward (5′‐TAATACGACTCACTATA‐3′) and reverse (5′‐CACTTTCACTTCTCCCTTTCAGTTTCCCTATAGTGAGTCGTATTA‐3′) oligos were annealed to serve as DNA template. The synthesized RNA, m7GpppApG(pN27), was used in the methyltransferase activity assay.
Methyltransferase activity assay
Methyltransferases (2.5 U/μl VACV VP39 (NEB, M0366S) or 1.7 μM Nsp16 and 0.3 μM Nsp10 from SARS–CoV‐2) were incubated together with RNA (70 ng/μl mRNA from SARS–CoV‐2 infected cells or 10 μM m7GpppApG(pN27)), and 0.3 or 1.2 μM (0.005 or 0.02 μCi/μl) adenosyl‐L‐methionine (SAM[3H]) (PerkinElmer, NET155V250UC) were incubated at 37°C overnight in reaction buffer (50 mM Tris–HCl (pH 8.0), 5 mM KCl, 1 mM MgCl2, 1 mM DTT). The reaction sample was purified using a mini Quick Oligo column (Roche, 11814397001) to remove free SAM[3H]. The purified sample was diluted in ULTIMA GOLD (PerkinElmer, 6013329) and quantified using a scintillation counter LS6500 (Beckman Coulter).
Biosafety
All experiments involving infectious recombinant SARS–CoV‐2 wt or Nsp16mut virus were carried out under biosafety level 3 conditions at the Institute of Clinical and Molecular Virology, Universitätsklinikum Erlangen. Experiments with recombinant SARS–CoV‐2 were approved by federal (ZKBS AZ 45110.2084) and local authorities (Regierung von Unterfranken, 55.1‐8791.27.‐28‐20 und 27‐29‐20).
Author contributions
Alina Russ: Data curation; formal analysis; investigation; visualization; methodology. Sabine Wittmann: Data curation; investigation; methodology. Yuta Tsukamoto: Investigation; methodology. Alexandra Herrmann: Investigation. Janina Deutschmann: Validation; investigation; visualization; methodology. Justine Lagisquet: Investigation; methodology. Armin Ensser: Investigation; methodology. Hiroki Kato: Conceptualization; funding acquisition; investigation; methodology. Thomas Gramberg: Conceptualization; formal analysis; supervision; visualization; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
PDF+
Acknowledgements
We thank Stephan Pöhlmann (Deutsches Primatenzentrum, Münster) und Stephan Ludwig (University of Münster) for kindly providing Calu‐3 cells. Caco‐2 cells were a kind gift from Michael Schindler (University of Tübingen) and Konstantin M Sparrer (University of Ulm). We thank Florian Simon (University Hospital Erlangen) for support. TG, AR, SW, JD, and JL were funded by BMBF SenseCoV2 01KI20172A, DFG 401821119/GRK 2504, and the coronavirus research fund of the Bavarian State Ministry of Science and the Arts. HK and YT were supported by grants of the DFG under Germany's Excellence Strategy‐EXC ‐ 390873048, and the DFG ‐ Grant No. 369799452 ‐ Project number 404459591. AE and AH were supported by BMBF SenseCoV2 01KI20172A; DFG Fokus COVID‐19, EN 423/7‐1; IZKF Erlangen project A81; and Coronavirus research grants by the Bavarian State Ministry of Science and the Arts and Bavarian State Ministry of Health Bay‐VOC. Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2022) 23: e55648
Data availability
This study includes no data deposited in external repositories.
References
- Bergant V, Yamada S, Grass V, Tsukamoto Y, Lavacca T, Krey K, Muhlhofer MT, Wittmann S, Ensser A, Herrmann A et al (2022) Attenuation of SARS‐CoV‐2 replication and associated inflammation by concomitant targeting of viral and host cap 2′‐O‐ribose methyltransferases. EMBO J 41: e111608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H et al (2010) 2′‐O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468: 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS (2014) IFIT1: a dual sensor and effector molecule that detects non‐2′‐O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev 25: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Lima CD (2010) Enzymology of RNA cap synthesis. Wiley Interdiscip Rev RNA 1: 152–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, Mann A, Kindler E, Gil‐Cruz C, Ziebuhr J et al (2013) Sequestration by IFIT1 impairs translation of 2′O‐unmethylated capped RNA. PLoS Pathog 9: e1003663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, Hunszinger V, Zech F, Prelli Bozzo C, Aftab W et al (2021) Systematic functional analysis of SARS‐CoV‐2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep 35: 109126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann A, Jungnickl D, Cordsmeier A, Peter AS, Uberla K, Ensser A (2021) Cloning of a passage‐free SARS‐CoV‐2 genome and mutagenesis using red recombination. Int J Mol Sci 22: 10188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M et al (2006) 5′‐triphosphate RNA is the ligand for RIG‐I. Science 314: 994–997 [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al (2006) Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 441: 101–105 [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo‐Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S (2008) Length‐dependent recognition of double‐stranded ribonucleic acids by retinoic acid‐inducible gene‐I and melanoma differentiation‐associated gene 5. J Exp Med 205: 1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H (2020) The architecture of SARS‐CoV‐2 transcriptome. Cell 181: 914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafcikova P, Silhan J, Nencka R, Boura E (2020) Structural analysis of the SARS‐CoV‐2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat Commun 11: 3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery SA, Sariol A, Perlman S (2021) Innate immune and inflammatory responses to SARS‐CoV‐2: implications for COVID‐19. Cell Host Microbe 29: 1052–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery VD, Yount BL Jr, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS (2014) Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′‐o‐methyltransferase activity. J Virol 88: 4251–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery VD, Gralinski LE, Mitchell HD, Dinnon KH 3rd, Leist SR, Yount BL Jr, Graham RL, McAnarney ET, Stratton KG, Cockrell AS et al (2017) Middle East respiratory syndrome coronavirus nonstructural protein 16 is necessary for interferon resistance and viral pathogenesis. mSphere 2: e00346‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miorin L, Kehrer T, Sanchez‐Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E et al (2020) SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA 117: 28344–28354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter AS, Roth E, Schulz SR, Fraedrich K, Steinmetz T, Damm D, Hauke M, Richel E, Mueller‐Schmucker S, Habenicht K et al (2022) A pair of noncompeting neutralizing human monoclonal antibodies protecting from disease in a SARS‐CoV‐2 infection model. Eur J Immunol 52: 770–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis ESC (2006) RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5′‐phosphates. Science 314: 997–1001 [DOI] [PubMed] [Google Scholar]
- Ramanathan A, Robb GB, Chan SH (2016) mRNA capping: biological functions and applications. Nucleic Acids Res 44: 7511–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebendenne A, Valadao ALC, Tauziet M, Maarifi G, Bonaventure B, McKellar J, Planes R, Nisole S, Arnaud‐Arnould M, Moncorge O et al (2021) SARS‐CoV‐2 triggers an MDA‐5‐dependent interferon response which is unable to control replication in lung epithelial cells. J Virol 95: e02415‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Gack MU (2020) RIG‐I‐like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20: 537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roers A, Hiller B, Hornung V (2016) Recognition of endogenous nucleic acids by the innate immune system. Immunity 44: 739–754 [DOI] [PubMed] [Google Scholar]
- Rosas‐Lemus M, Minasov G, Shuvalova L, Inniss NL, Kiryukhina O, Brunzelle J, Satchell KJF (2020a) High‐resolution structures of the SARS‐CoV‐2 2′‐O‐methyltransferase reveal strategies for structure‐based inhibitor design. Sci Signal 13: eabe1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas‐Lemus M, Minasov G, Shuvalova L, Inniss NL, Kiryukhina O, Wiersum G, Kim Y, Jedrzejczak R, Maltseva NI, Endres M et al (2020b) The crystal structure of nsp10‐nsp16 heterodimer from SARS‐CoV‐2 in complex with S‐adenosylmethionine. bioRxiv 10.1101/2020.04.17.047498 [PREPRINT] [DOI]
- Sampaio NG, Chauveau L, Hertzog J, Bridgeman A, Fowler G, Moonen JP, Dupont M, Russell RA, Noerenberg M, Rehwinkel J (2021) The RNA sensor MDA5 detects SARS‐CoV‐2 infection. Sci Rep 11: 13638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth‐Wagner C, Ludwig J, Bruder AK, Herzner AM, Zillinger T, Goldeck M, Schmidt T, Schmid‐Burgk JL, Kerber R, Wolter S et al (2015) A conserved histidine in the RNA sensor RIG‐I controls immune tolerance to N1‐2′O‐methylated self RNA. Immunity 43: 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setaro AC, Gaglia MM (2021) All hands on deck: SARS‐CoV‐2 proteins that block early anti‐viral interferon responses. Curr Res Virol Sci 2: 100015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens‐Kiani T, Cheng J et al (2020) Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science 369: 1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne LG, Reuschl AK, Zuliani‐Alvarez L, Whelan MVX, Turner J, Noursadeghi M, Jolly C, Towers GJ (2021) SARS‐CoV‐2 sensing by RIG‐I and MDA5 links epithelial infection to macrophage inflammation. EMBO J 40: e107826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan T, Arya S, Chan SH, Qi S, Dai N, Misra A, Park JG, Oladunni F, Kovalskyy D, Hromas RA et al (2020) Structural basis of RNA cap modification by SARS‐CoV‐2. Nat Commun 11: 3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan T, Misra A, Chan SH, Qi S, Dai N, Arya S, Martinez‐Sobrido L, Gupta YK (2021) A metal ion orients SARS‐CoV‐2 mRNA to ensure accurate 2′‐O methylation of its first nucleotide. Nat Commun 12: 3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Sato S, Sotoyama Y, Orba Y, Sawa H, Yamauchi H, Sasaki M, Takaoka A (2021) RIG‐I triggers a signaling‐abortive anti‐SARS‐CoV‐2 defense in human lung cells. Nat Immunol 22: 820–828 [DOI] [PubMed] [Google Scholar]
- Yin X, Riva L, Pu Y, Martin‐Sancho L, Kanamune J, Yamamoto Y, Sakai K, Gotoh S, Miorin L, De Jesus PD et al (2021) MDA5 governs the innate immune response to SARS‐CoV‐2 in lung epithelial cells. Cell Rep 34: 108628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zust R, Cervantes‐Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS et al (2011) Ribose 2′‐O‐methylation provides a molecular signature for the distinction of self and non‐self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12: 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]