

Abstract
Maintenance of desmin intermediate filaments (IF) is vital for muscle plasticity and function, and their perturbed integrity due to accelerated loss or aggregation causes atrophy and myopathies. Calpain‐1‐mediated disassembly of ubiquitinated desmin IF is a prerequisite for desmin loss, myofibril breakdown, and atrophy. Because calpain‐1 does not harbor a bona fide ubiquitin‐binding domain, the precise mechanism for desmin IF disassembly remains unknown. Here, we demonstrate that the AAA‐ATPase, ATAD1, is required to facilitate disassembly and turnover of ubiquitinated desmin IF. We identified PLAA and UBXN4 as ATAD1's interacting partners, and their downregulation attenuated desmin loss upon denervation. The ATAD1‐PLAA‐UBXN4 complex binds desmin filaments and promotes a release of phosphorylated and ubiquitinated species into the cytosol, presenting ATAD1 as the only known AAA‐ATPase that preferentially acts on phosphorylated substrates. Desmin filaments disassembly was accelerated by the coordinated functions of Atad1 and calpain‐1, which interact in muscle. Thus, by extracting ubiquitinated desmin from the insoluble filament, ATAD1 may expose calpain‐1 cleavage sites on desmin, consequently enhancing desmin solubilization and degradation in the cytosol.
Keywords: ATAD1, desmin, intermediate filaments, muscle atrophy, protein degradation
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Musculoskeletal System; Post-translational Modifications & Proteolysis
The AAA‐ATPase ATAD1 promotes the disassembly and turnover of desmin filaments in a complex with the ubiquitin‐binding proteins UBXN4 and PLAA. Downregulation of ATAD1 prevents desmin loss and attenuates muscle atrophy upon denervation.
Introduction
Desmin intermediate filaments (IF) are critical for muscle architecture and function (Agnetti et al, 2021). By linking the contractile myofibrils to the sarcolemma and cellular organelles (i.e., mitochondria, nucleus, T‐tubules, sarcoplasmic reticulum, and motor endplate; Bär et al, 2004), this cytoskeletal network contributes to muscle structural and cellular integrity, force transmission, and mitochondrial homeostasis (Milner et al, 2000). Mutations in desmin cause myopathies in humans (Milner et al, 1996; Bär et al, 2004; Mavroidis et al, 2008) and its loss promotes muscle wasting (Helliwell et al, 1989; Boudriau et al, 1996; Cohen et al, 2012; Thottakara et al, 2015; Volodin et al, 2017; Aweida et al, 2018). Our previous studies in animal models suggested that the loss of desmin filaments in muscles atrophying due to neuronal damage (i.e., muscle denervation; Volodin et al, 2017) or systemic catabolic states (e.g., fasting, type‐2 diabetes; Cohen et al, 2012; Aweida et al, 2018; Eid‐Mutlak et al, 2020), is an initial key event triggering myofibril destruction by the ubiquitin proteasome system (UPS) (Solomon & Goldberg, 1996). The resulting excessive myofibril breakdown is a hallmark of muscle atrophy, and accounts for the associated frailty, disability, and morbidity in aging or disease (Jackman & Kandarian, 2004; Cohen et al, 2014b).
We recently discovered that phosphorylation of serine residues within desmin's head domain is required for desmin IF ubiquitination by the ubiquitin ligase TRIM32, and subsequent depolymerization (Aweida et al, 2018). Phosphorylation of desmin IF is catalyzed by glycogen synthase kinase 3‐β (GSK3‐β) (Agnetti et al, 2014; Aweida et al, 2018), which is activated primarily by the fall in PI3K‐AKT signaling (e.g. in fasting or type‐2 diabetes). Although calpain‐1 binds phosphorylated and ubiquitinated desmin filaments, cleavage by this enzyme probably does not require desmin ubiquitination because calpain‐1 does not harbor a bona fide ubiquitin‐binding domain. In addition, filaments per se are not accessible to the catalytic core of the proteasome (Solomon & Goldberg, 1996), and therefore must disassemble before degradation in the cytosol (Aweida & Cohen, 2021). For example, the AAA‐ATPase, p97/VCP disassembles ubiquitinated filamentous myofibrils and promotes their loss in muscles atrophying due to denervation or fasting (Piccirillo & Goldberg, 2012; Volodin et al, 2017). However, desmin IF are lost by a mechanism not requiring p97/VCP (Volodin et al, 2017). We show here that their degradation requires a distinct AAA‐ATPase, ATAD1.
ATAD1 (also called THORASE) belongs to the ATPases Associated with diverse cellular Activities (AAA) superfamily of proteins that form homohexameric ring‐structured complexes (Vale, 2000; Zhang & Mao, 2020). In humans, a homozygous frameshift mutation in Atad1 causes encephalopathy, stiffness, and arthrogryposis (Piard et al, 2018), but the effects of this mutation on ATAD1 function are unclear. Prior investigations have suggested a role for ATAD1 in regulation of synaptic plasticity, learning and memory (Zhang et al, 2011; Li et al, 2016), and other studies proposed that the ATAD1 yeast homolog, Msp1, is involved in the turnover of mislocalized mitochondrial membrane proteins (Wohlever et al, 2017; Li et al, 2019). Although ATAD1 appears to have important roles, its precise cellular functions remain largely obscure, and its interacting partners are not yet known. We demonstrate here that upon muscle denervation, ATAD1 catalyzes the disassembly and loss of desmin IF, which ultimately lead to myofibril breakdown and atrophy (Cohen et al, 2012; Volodin et al, 2017; Aweida et al, 2018). Phosphorylation of desmin IF is required for ATAD1 binding, presenting ATAD1 as the only known AAA‐ATPase that preferentially acts on phosphorylated substrates. We identified PLAA and UBXN4 as ATAD1's interacting partners, and propose that ATAD1‐PLAA‐UBXN4 complex cooperates with calpain‐1 to facilitate desmin IF solubilization, and subsequent degradation in the cytosol.
Results
ATAD1 promotes muscle wasting on denervation
The Ca2+‐dependent protease calpain‐1 promotes degradation of phosphorylated and ubiquitinated desmin IF during atrophy (Aweida et al, 2018). Because calpain‐1 does not harbor a bona fide ubiquitin‐binding domain, we sought to identify the factors that cooperate with calpain‐1 in promoting desmin IF disassembly and loss. The AAA‐ATPase, p97/VCP, which extracts ubiquitinated proteins from the myofibril, does not seem to be required for loss of ubiquitinated desmin filaments (Volodin et al, 2017). By incubating muscle pellets (containing insoluble desmin filaments and myofibrils) with a nonhydrolyzable ATP analogue, AMP‐PNP, we recently identified enzymes that utilize ATP for their activity and act on desmin IF (Aweida et al, 2018). The addition of AMP‐PNP facilitated the identification of such enzymes by mass spectrometry because they were sequestered to desmin but could not act on it (Aweida et al, 2018). The only identified AAA‐ATPase enzymes on our dataset were ATAD1 and ATAD3. ATAD1 was twofold more abundant in the insoluble fraction of 3 days denervated muscles than in innervated controls, while ATAD3 showed similar abundance in denervated and control muscles. To determine whether these AAA‐ATPase enzymes also bind calpain‐1, we immunoprecipitated calpain‐1 from 7 days denervated muscle homogenates, when desmin depolymerization by calpain‐1 is accelerated (Volodin et al, 2017; Aweida et al, 2018), and analyzed protein precipitates by mass spectrometry. Interestingly, ATAD1 was the only AAA‐ATPase that bound calpain‐1, and was also one of the most abundant proteins in the sample (1,000 times more abundant in the calpain‐1 immunoprecipitation sample than in IgG control). These findings were unexpected because ATAD1 has been considerably understudied, and there had been no prior reports of ATAD1 in muscle. In denervated skeletal muscle, however, ATAD1 binds both the insoluble filaments and calpain‐1, and is critical for desmin filament loss and atrophy (see below).
To clarify the role of ATAD1, we first determined whether it is induced during atrophy. The levels of mRNA for Atad1 increased significantly at 7 and 10 days after denervation (Fig 1A), just when desmin depolymerization by calpain‐1 is rapid (Aweida et al, 2018), but returned to basal levels at 14 days, when the content of phosphorylated and ubiquitinated desmin filaments is substantially low and myofibril disassembly by p97/VCP is accelerated (Volodin et al, 2017). To test whether ATAD1 is important for atrophy, we suppressed its expression by the electroporation of a specific shRNA into mouse Tibialis Anterior (TA) muscle. Two shRNAs had been generated, shAtad1‐1 and shAtad1‐2, which reduced Atad1 mRNA levels in mouse muscles below the levels in shLacz expressing controls (Fig 1B), and here we used the potent shAtad1‐1. The downregulation of Atad1 was sufficient to attenuate muscle fiber atrophy because at 14 days after denervation the cross‐sectional area of 975 fibers expressing shAtad1 was significantly larger than that of 975 nontransfected denervated fibers in the same muscle (n = 8 mice) (Fig 1C, Table 1; Appendix Fig S1). Therefore, ATAD1 is important for muscle atrophy induced by denervation.
Figure 1. ATAD1 promotes muscle atrophy on denervation.
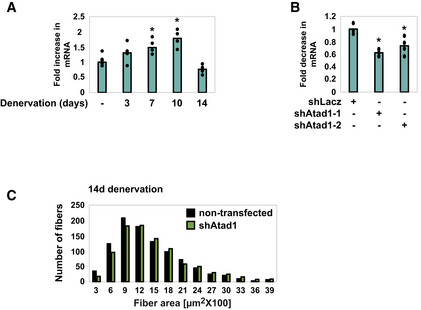
- Atad1 is induced upon muscle denervation. RT‐PCR of mRNA preparations from innervated muscles and ones denervated for 3, 7, 10, and 14 days using specific primers for Atad1. Data are plotted as the mean fold change relative to innervated control ± SEM. n = 4 mice, biological replicates. *P (7 days) = 0.03, P (10 days) = 0.002 vs. innervated. Data are representative of two independent experiments.
- shRNA‐mediated knockdown of Atad1 in 14 days denervated TA muscles. RT‐PCR of mRNA preparations from 14 days denervated muscles expressing shLacz control or two different shRNAs against Atad1, shAtad1‐1 and shAtad1‐2, using specific primers for Atad1. Data are plotted as the mean fold change relative to shLacz control ± SEM. n = 4 mice, biological replicates. *P (shAtad‐1) = 0.015, P (shAtad‐2) = 0.046 vs. shLacz. Data are representative of one experiment.
- Atad1 downregulation attenuates fiber atrophy on denervation. Measurements of cross‐sectional areas of 973 fibers expressing shAtad1 (green bars) vs. 973 nontransfected fibers (black bars) in the same muscle. n = 8 mice, biological replicates. Bar: 50 μm.
Table 1.
Statistical analysis of fiber size measurements.
| Nontransfected | Transfected | % change | |
|---|---|---|---|
| 14 days shAtad1 | |||
| Median | 1,369 | 1,503 | 9.79 |
| Skewness | 2.41 | 2.81 | 16.59 |
| Brunner‐Manzel test | P‐value = 0.000547 | ||
| A‐statistic | 0.45 | ||
| Innervated shAtad1 | |||
| Median | 2,926 | 3,330 | 13.80 |
| Skewness | 0.82 | 1.04 | 26.82 |
| Brunner‐Manzel test | P‐value = 0.0023 | ||
| A‐statistic | 0.44 | ||
Summary statistics of fiber size analyses presented in Figs 1C and 4F based on our recent methodology paper (Gilda et al, 2021). With regard to A‐statistics, if 0 ≤ A < 0.5 then dataset1 (nontransfected) is stochastically less than dataset2 (transfected). The A‐statistics is a direct measure of the fiber size effect (Gilda et al, 2021), and it shows beneficial effect on cell size by shAtad1. Such an effect can be simply missed by traditional measurements of median, average, and Student's t‐test.
ATAD1 binds phosphorylated desmin filaments and promotes their disassembly
To investigate if this reduction in fiber atrophy occurred through the attenuation of desmin IF loss, which should reduce overall proteolysis (Cohen, 2020; Agnetti et al, 2021; Aweida & Cohen, 2021), we initially confirmed our mass spectrometry data (Aweida et al, 2018) and determined whether ATAD1 in fact binds desmin filaments in vivo. For this purpose, we isolated desmin filaments from muscles at different times after denervation, and analyzed by SDS–PAGE and immunoblotting. Ubiquitination of desmin IF increased at 3 days after denervation (Fig 2A), as we had previously shown (Volodin et al, 2017), but was markedly reduced at 14 days after denervation, when ubiquitinated desmin is degraded (Volodin et al, 2017). It is noteworthy that the amount of ubiquitinated desmin IF at 7 days after denervation did not differ significantly from that in innervated controls (Fig 2A) because at this time after denervation, depolymerization of ubiquitinated desmin IF is accelerated (Volodin et al, 2017). Interestingly, at 3 days after denervation, when desmin filaments are rapidly ubiquitinated, ATAD1 accumulated on desmin IF (Fig 2B). However, at 7 days after denervation, when desmin IF depolymerization by calpain‐1 is accelerated (Volodin et al, 2017; Aweida et al, 2018), ATAD1 association with desmin filament was markedly reduced; instead, ATAD1 mainly accumulated in the cytosol (Fig 2B), most likely bound to ubiquitinated species of desmin (see below).
Figure 2. ATAD1 binds phosphorylated desmin filaments and promotes their disassembly.
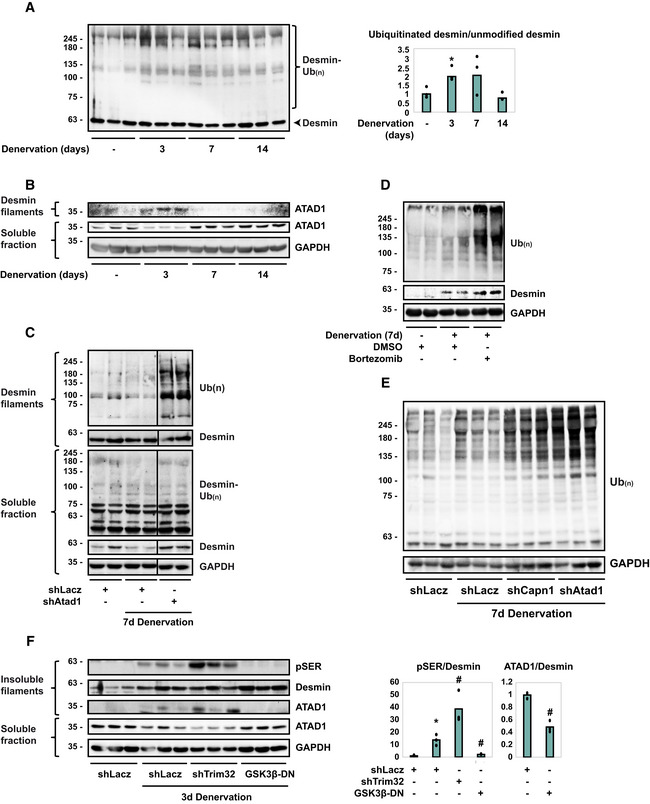
- Desmin ubiquitination increases at 3 days after denervation. Left: desmin filaments isolated from muscles at 0, 3, 7, or 14 days after denervation were analyzed by immunoblotting. Right: densitometric measurement of presented blots. Mean ratios of ubiquitinated desmin to unmodified desmin ± SEM are depicted in a graph. n = 3 mice, biological replicates. *P = 0.002 vs. innervated. Data are representative of two independent experiments.
- ATAD1 binds desmin filaments at 3 days after denervation, and at 7 days after denervation it is released from the pellet and accumulates in the soluble fraction. Desmin filaments and soluble fractions isolated from innervated and denervated muscles were analyzed by immunoblotting. n = 3 mice, biological replicates. Data are representative of two independent experiments.
- Downregulation of Atad1 prevents depolymerization of ubiquitinated desmin filaments. Desmin filaments and soluble fractions from innervated and 7 days denervated TA muscles expressing shLacz or shAtad1 were analyzed by immunoblotting. Black line indicates the removal of intervening lanes for presentation purposes. n = 2 mice, biological replicates. Data are representative of two independent experiments.
- Desmin is degraded by the proteasome in atrophying muscles. Soluble fractions from innervated and 7 days denervated TA muscles of mice injected with Bortezomib or DMSO were analyzed by SDS–PAGE and immunoblotting. n = 2 mice, biological replicates. Data are representative of one experiment.
- Atad1 downregulation results in accumulation of ubiquitinated soluble proteins. Soluble fractions from innervated and 7 days denervated TA muscles expressing shLacz, shCapn1 or shAtad1 were analyzed by SDS–PAGE and immunoblotting. n = 3 mice, biological replicates. Data are representative of two independent experiments.
- Desmin filament phosphorylation is a prerequisite for ATAD1 binding. Left: insoluble and soluble fractions from innervated or 3 days denervated muscles expressing shLacz, shTrim32, or GSK3‐β‐DN were analyzed by immunoblotting. ATAD1 was detected on isolated desmin filaments. Right: densitometric measurement of presented blots. Mean ratios of pSER to desmin and ATAD1 to desmin ± SEM are depicted in a graph. n = 3 mice, biological replicates. pSER to desmin ratio: *P = 0.002 vs. shLacz in innervated; #, P (shTrim32) = 0.02, P (GSK3‐β‐DN) = 0.03 vs. shLacz in denervated. ATAD1 to desmin ratio: # P = 0.005 vs. shLacz in denervated. Data are representative of two independent experiments.
Source data are available online for this figure.
To test more directly whether ATAD1 is required for desmin filament depolymerization and loss, we employed a similar approach as we used to analyze disassembly of filamentous myofibrils by p97/VCP (Volodin et al, 2017). We determined the effects of ATAD1 downregulation on the total amount of ubiquitinated desmin in the soluble and insoluble fractions of 7 days denervated muscles, just when disassembly of desmin IF is accelerated (Volodin et al, 2017). Downregulation of ATAD1 with shAtad1 prevented disassembly of desmin IF because ubiquitinated desmin accumulated as insoluble filaments (Fig 2C). In fact, the amount of ubiquitinated filaments in these shAtad1‐expressing muscles exceeded the amounts observed in shLacz expressing denervated controls. Thus, the ubiquitination of desmin IF increases on denervation (Fig 2A and Volodin et al, 2017), and ATAD1 catalyzes the solubilization and degradation of these ubiquitinated proteins.
In addition, desmin was released from the cytoskeleton into the cytosol as ubiquitinated species, and it was then degraded (Fig 2C), but not when endogenous ATAD1 was downregulated with shAtad1 (Fig 2C). Degradation of soluble desmin was validated in vivo by the injection of mice with the proteasome inhibitor, Bortezomib (3 mg/kg body weight), which prevented desmin degradation in denervated (7 days) muscles and led to accumulation of desmin in its ubiquitinated form (compare to DMSO injected controls, Fig 2D). Interestingly, downregulation of either Atad1 or calpain‐1 (shCapn1 is described in Aweida et al, 2018) led to a dramatic increase in the content of overall ubiquitin conjugates in the cytosol (Fig 2E), indicating that on denervation, degradation of soluble proteins also depends on ATAD1 and calpain‐1 functions. Thus, ubiquitinated desmin seems to be released from the filament into the cytosol for degradation, and this step requires ATAD1.
ATAD1 binds desmin IF early after denervation (3 days), when desmin filaments are phosphorylated by GSK3‐β and then ubiquitinated by the ubiquitin ligase, TRIM32 (Aweida et al, 2018). To determine if these posttranslational modifications of desmin filaments are required for association with ATAD1, we purified desmin filaments from innervated and 3 days denervated muscles expressing GSK3‐β dominant negative (GSK3‐β‐DN), Trim32 shRNA (shTrim32), or shLacz control and analyzed by immunoblotting (GSK3‐β‐DN and shTrim32 were described in Cohen et al, 2014a; Volodin et al, 2017; Aweida et al, 2018). At 3 days after denervation desmin phosphorylation increased and inhibition of GSK3‐β with GSK3‐β‐DN prevented this phosphorylation (Fig 2F), as reported (Aweida et al, 2018). Loss of desmin IF by TRIM32‐mediated ubiquitination requires desmin phosphorylation (Cohen et al, 2012; Volodin et al, 2017; Aweida et al, 2018). Consistently, downregulation of Trim32 prevented this loss, and desmin accumulated as phosphorylated filaments (Fig 2F). Desmin IF phosphorylation seems to be important for ATAD1 binding because the association of ATAD1 with desmin increased correlatively as phosphorylated desmin filaments accumulated, if by the induction of atrophy or by the downregulation of Trim32 (Fig 2F). Moreover, ATAD1 was less bound to desmin in muscles where GSK3‐β was inactive (Fig 2F). Therefore, desmin IF phosphorylation is a prerequisite for ATAD1 binding.
Isolation of ATAD1 binding partners from muscle homogenates
To investigate if ATAD1 cooperates with adaptor proteins to promote desmin IF disassembly, we searched for its interacting partners in vivo using three independent mass spectrometry‐based proteomic approaches. The first approach we employed was size‐exclusion chromatography (SEC) of 7 days denervated muscles because at this time after denervation ubiquitinated desmin IF are solubilized by calpain‐1 (Volodin et al, 2017; Aweida et al, 2018), and ATAD1 accumulates in the soluble phase (Fig 2B) bound to calpain‐1 (see also below Fig 3D). To facilitate the identification of low abundant proteins, mouse muscles (1 g) were cross‐linked in 1% PFA, homogenized, and after 10 min of incubation the cross‐linking reaction was quenched with glycine (0.125 M). Ammonium sulfate precipitates were then solubilized and fractionated by a gel filtration column (Superdex 200 10/300 GL), and protein eluates were analyzed by SDS–PAGE, immunoblotting, and silver staining (Fig 3A and B). ATAD1 was predominantly recovered in fraction #8, together with desmin and additional low and high molecular weight proteins (Fig 3B). Mass spectrometry analysis of this fraction in two biological replicates of denervated muscles identified ATAD1 and several UPS components, including E1 and ubiquitin, E2s, both RING and HECT domain E3s, proteasome subunits, deubiquitinating enzymes, calpain‐1 and ‐2, and SUMO activating enzyme (Table 2). Only UPS components that were identified with ≥2 unique peptides were categorized based on function using DAVID annotation tool.
Figure 3. PLAA and UBXN4 are binding partners of ATAD1 in denervated muscle.
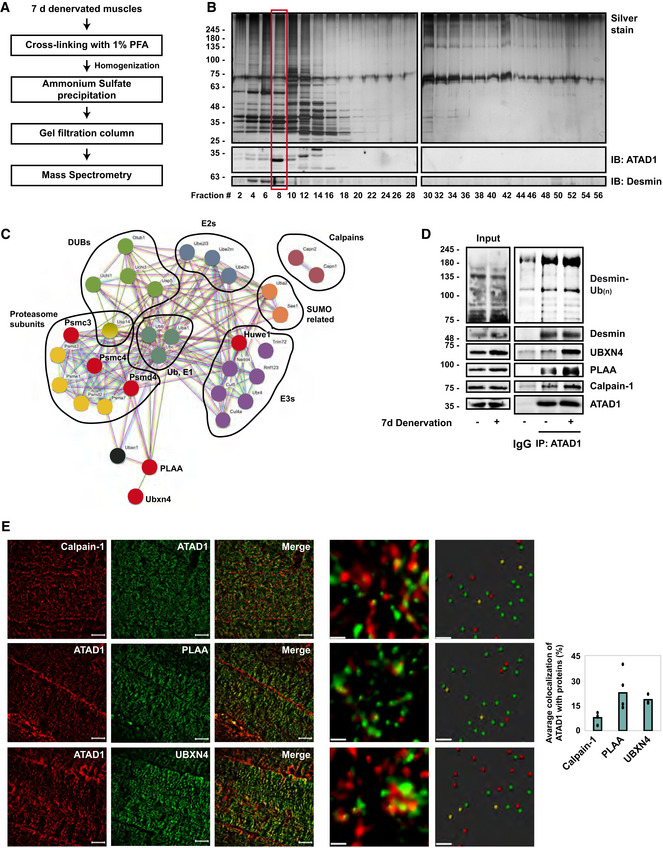
- Scheme of ATAD1's purification from muscle and isolation of its binding partners.
- Analysis of size‐exclusion chromatography fractions by SDS–PAGE, silver staining (top panel) or immunoblotting (lower panel). Red box: The ATAD1‐containing fraction was subjected to mass spectrometry analysis. n = 2 mice, biological replicates. Data are representative of two independent experiments. Data are representative of two independent experiments.
- Interaction networks for UPS components and related proteins identified by three independent mass spectrometry‐based proteomics approaches using the STRING database. UPS enzymes are grouped based on function. Labeled in red are proteins that were at least 3 times more abundant on desmin filaments at 3 days after denervation.
- ATAD1 and its partners bind ubiquitinated desmin, and these associations increase after denervation. ATAD1 was immunoprecipitated from innervated and 7 days denervated muscle homogenates, and protein precipitates were analyzed by immunoblotting. Data are representative of two independent experiments. Data are representative of one experiment.
- ATAD1 colocalizes with calpain‐1, PLAA and UBXN4 in muscle. Left: Structured illumination microscopy (SIM) images (bar, 10 μm) of 7 days denervated muscle longitudinal sections stained with the indicated antibodies. Middle: representative analysis of SIM images using the spots module of the Imaris software (bar, 0.5 μm). Only spots that were within a distance threshold of < 100 nm were considered colocalized. Right: average colocalization of ATAD1 with the indicated proteins. Data are represented as mean ± SEM. n = 3 mice, biological replicates. Data are representative of one experiment.
Source data are available online for this figure.
Table 2.
ATAD1 binds a variety of UPS components in muscle.
| Protein | Gene | #Unique peptides | Fold change (3 days denervated/innervated) | ||
|---|---|---|---|---|---|
| SEC | ATAD1 IP | Bound to desmin filaments at 3 days after denervation | |||
| Ubiquitin‐like modifier‐activating enzyme 1 | UBA1 | 37 | 3 | ||
| Ubiquitin‐conjugating enzyme E2 L3 | UBE2L3 | 3 | 2 | 5 | |
| Ubiquitin‐conjugating enzyme E2 N | UBE2N | 2 | 5 | ||
| NEDD8‐conjugating enzyme Ubc12 | UBE2M | 2 | 7 | ||
| E3 ubiquitin‐protein ligase UBR4 | UBR4 | 4 | 86 | ||
| E3 ubiquitin‐protein ligase NEDD4 | NEDD4 | 4 | 5 | ||
| Tripartite motif‐containing protein 72 | TRIM72 | 2 | 10 | 40 | |
| E3 ubiquitin‐protein ligase RNF123 | RNF123 | 19 | 3 | 6 | |
| E3 ubiquitin‐protein ligase HUWE1 | HUWE1 | 5 | 50 | 3.7 | |
| Cullin‐5 | CUL5 | 6 | 6 | ||
| Cullin‐4A | CUL4A | 2 | |||
| Polyubiquitin‐B | UBB | 6 | 7 | ||
| Polyubiquitin‐C | UBC | 5 | 6 | ||
| 26 S proteasome non‐ATPase regulatory subunit 4 | PSMD4 | 4 | 5 | 4.5 | |
| Proteasome subunit alpha type‐7 | PSMA7 | 3 | 5 | ||
| Proteasome activator complex subunit 1 | PSME1 | 3 | |||
| 26 S protease regulatory subunit 6A | PSMC3 | 2 | 24 | 8.6 | |
| 26 S proteasome regulatory subunit 6B | PSMC4 | 4 | 24 | 6.7 | |
| 26 S proteasome non‐ATPase regulatory subunit 2 | PSMD2 | 4 | 3 | ||
| 26 S proteasome non‐ATPase regulatory subunit 3 | PSMD3 | 4 | 37 | ||
| Ubiquitin carboxyl‐terminal hydrolase 14 | USP14 | 4 | 4 | ||
| Ubiquitin carboxyl‐terminal hydrolase 5 | USP5 | 3 | 2 | ||
| Ubiquitin carboxyl‐terminal hydrolase isozyme L1 | UCHL1 | 2 | |||
| Ubiquitin carboxyl‐terminal hydrolase isozyme L3 | UCHL3 | 2 | |||
| Ubiquitin thioesterase OTUB1 | OTUB1 | 2 | 7 | ||
| Calpain‐1 catalytic subunit | CAPN1 | 12 | 12 | 24 | |
| Calpain‐2 catalytic subunit | CAPN2 | 2 | 2 | 18 | |
| SUMO‐activating enzyme subunit 1 | SAE1 | 2 | |||
| SUMO‐activating enzyme subunit 2 | UBA2 | 3 | |||
| Phospholipase A‐2‐activating protein | PLAA | 4 | 13 | 4.9 | |
| UBX domain‐containing protein 4 | UBXN4 | 4 | 4.7 | ||
| UBX domain‐containing protein 1 | UBXN1 | 2 | |||
ATAD1's interacting partners were identified by three independent mass spectrometry‐based proteomics approaches. Only UPS components that were identified with ≥2 unique peptides were categorized based on function using DAVID annotation tool. Two proteomics approaches were oriented specifically towards identifying ATAD1‐binding partners: SEC (427 total proteins were identified), and ATAD1 immunoprecipitation (592 total proteins were identified). These lists of UPS components were compared to our previous kinase‐trap assay dataset (Aweida et al, 2018, 1,552 total proteins were identified) and the number of unique peptides only for proteins that overlapped is listed here. The kinase trap assay was used to identify proteins that utilize ATP for their function and act on desmin, and ATAD1 was one of the most abundant proteins in the sample.
To confirm these findings by an independent approach, we immunoprecipitated ATAD1 from 3 days denervated muscle homogenates (6,000 g supernatant), and washed the resulting precipitates extensively with buffer containing 500 mM of NaCl to remove nonspecific or weakly associated proteins. The ATAD1‐bound proteins were identified by mass spectrometry, which revealed several of the aforementioned components, and additional UPS components as well as Phospholipase A‐2‐activating protein (PLAA) (Doa1/Ufd3 in yeast) (Table 2), which contains WD40 repeats that can bind ubiquitinated proteins (Pashkova et al, 2010).
To corroborate these findings further, we employed an additional strategy and determined if any of these proteins accumulate on desmin filaments together with ATAD1 at 3 days after denervation (Fig 2B). As mentioned above, using AMP‐PNP and mass spectrometry, we previously identified enzymes that utilize ATP for their activity and bind with high affinity to desmin filaments (Aweida et al, 2018). Searching this list of enzymes, we found an overlap with almost all proteins identified by SEC and ATAD1 immunoprecipitation analyses (Fig 3A–C and Table 2). Using the STRING database, we generated a network based on established annotation for protein interactors and found a significant interconnectivity among the vast majority of the identified enzymes. The exception was calpain‐1 and ‐2, which have no known interactions with the other components on our dataset (Fig 3C). Here, we present evidence that calpain‐1 interacts with several components on this network to promote disassembly of desmin filaments (see below Fig 3D).
Out of the 32 identified proteins in our dataset, six were at least threefold more abundant on desmin filaments at 3 days after denervation (Table 2, and labeled in red in Fig 3C), the time when ATAD1 accumulates on desmin (Fig 2B), including three proteasome subunits PSMD4 (Rpn10), PSMC4 (Rpt3), and PSMC3 (Rpt5), the ubiquitin ligase HUWE1, PLAA, and UBXN4 (UBXD2/erasin), which contains a Ubiquitin regulatory X (UBX) domain and is known to promote recruitment of ubiquitinated substrates to AAA‐ATPase complexes (e.g., p97/VCP (Lim et al, 2009)).
Downregulation of Plaa or Ubxn4 prevents desmin solubilization and degradation, and loss of soluble ubiquitinated proteins
To clarify the mechanism for ATAD1‐mediated disassembly of phosphorylated and ubiquitinated desmin filaments, we focused our attention on UBXN4 and PLAA because these components mediate degradation of ubiquitinated proteins (Ren et al, 2008; Lim et al, 2009), and harbor ubiquitin‐binding domains (WD40 repeats in PLAA and UBX domain in UBXN4). We initially validated our mass spectrometry data and investigated if UBXN4 and PLAA in fact form an intact complex with ATAD1 in muscle. UBXN4 and PLAA could be coprecipitated with ATAD1 from innervated muscle, and these associations increased after denervation (Fig 3D). This protein assembly, which also contained calpain‐1, bound ubiquitinated desmin filaments, and muscle denervation enhanced these associations (Fig 3D), suggesting that UBXN4 and PLAA may be adaptor proteins that link ubiquitinated desmin to ATAD1 complex to promote desmin IF disassembly. These new associations were further validated by an immunofluorescence staining of longitudinal sections from 7 days denervated muscles and super‐resolution structured illumination microscopy (SIM), which demonstrated colocalization of ATAD1 with calpain‐1, PLAA, and UBXN4 (Fig 3E). To confirm that these proteins in fact colocalize, we measured the average colocalization of ATAD1 with calpain‐1, PLAA, and UBXN4 using the spots detection and colocalization analysis of the Imaris software (Fig 3E). Only spots that were within a distance threshold of <100 nm were considered colocalized (Fig 3E, graph).
To determine if PLAA and UBXN4 are required for desmin IF loss, we analyzed the reduction in the amount of ubiquitinated desmin filaments upon denervation after electroporation of PLAA and UBXN4 shRNAs (shPlaa and shUbxn4, respectively), which efficiently reduced Plaa and Ubxn4 expression (Fig 4A). In accord with prior findings (Volodin et al, 2017), the amount of ubiquitinated desmin filaments decreased in muscle at 14 days after denervation due to their degradation (Fig 4B). However, downregulation of either Plaa or Ubxn4 blocked this decrease, and instead desmin accumulated as insoluble phosphorylated and ubiquitinated filaments, which exceeded the amounts observed in innervated muscles (Fig 4B). Thus, phosphorylation and ubiquitination of desmin IF increase on denervation, as reported (Volodin et al, 2017), and PLAA and UBXN4 promote the solubilization and degradation of these filaments.
Figure 4. PLAA and UBXN4 promote disassembly of ubiquitinated desmin filaments.
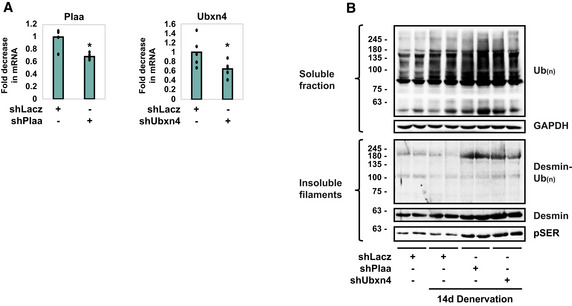
- shRNA‐mediated knockdown of Plaa and Ubxn4 in HEK293 cells. RT‐PCR of mRNA preparations from HEK293 cells expressing shLacz, shPlaa, or shUbxn4 using specific primers for PLAA or UBXN4. Data are plotted as the mean fold change relative to control ± SEM. n = 5 technical replicates (cells were plated in 5 wells per DNA plasmid). *P (shPlaa) z = 0.006, P (shUbxn4) = 0.015 vs. shLacz. Data are representative of one experiment.
- Downregulation of Plaa or Ubxn4 prevents the loss of desmin and soluble proteins in denervated muscle. Insoluble and soluble fractions from 14 days denervated muscles expressing shLacz, shPlaa, or shUbxn4 were analyzed by immunoblotting. n = 2 mice, biological replicates. Data are representative of one experiment.
Source data are available online for this figure.
Our prior findings imply that stabilization of desmin filaments attenuates overall proteolysis (Volodin et al, 2017; Aweida et al, 2018). Accordingly, the muscle's content of soluble ubiquitin conjugates increased at 14 days after denervation compared with innervated controls, as we had shown (Volodin et al, 2017). However, the downregulation of Plaa or Ubxn4 caused a more dramatic increase in the total content of ubiquitin conjugates to levels that exceeded those in denervated muscles (Fig 4B), indicating that, similar to ATAD1 (Fig 2E), PLAA and UBXN4 are also required for degradation of proteins in the cytosol.
ATAD1 and calpain‐1 cooperatively promote desmin filament disassembly and loss
Because ATAD1 in denervated muscles was bound to calpain‐1, and since both enzymes promote dissociation of phosphorylated desmin filaments, we investigated whether they cooperate in promoting desmin IF loss. Muscles were electroporated with either shCapn1 or shAtad1, or with both constructs (Fig 5A and B), and the insoluble fraction was analyzed by SDS–PAGE and immunoblotting (Fig 5C). Expressing either shCapn1 or shAtad1 attenuated disassembly and loss of phosphorylated desmin IF on denervation, and desmin accumulated as insoluble phosphorylated filaments (Fig 5C). Interestingly, this beneficial effect on desmin was much larger by simultaneously downregulating both enzymes, which caused a more dramatic accumulation of phosphorylated desmin IF (Fig 5C). Simultaneous downregulation of both Atad1 and calpain‐1 also affected soluble ubiquitin conjugates, which accumulated in the cytosol to a greater extent than when each enzyme was downregulated alone (Fig 5D). Thus, ATAD1 and calpain‐1 cooperate in an additive fashion in promoting desmin IF loss and protein degradation during atrophy.
Figure 5. ATAD1 and calpain‐1 cooperatively promote desmin filament disassembly and loss.
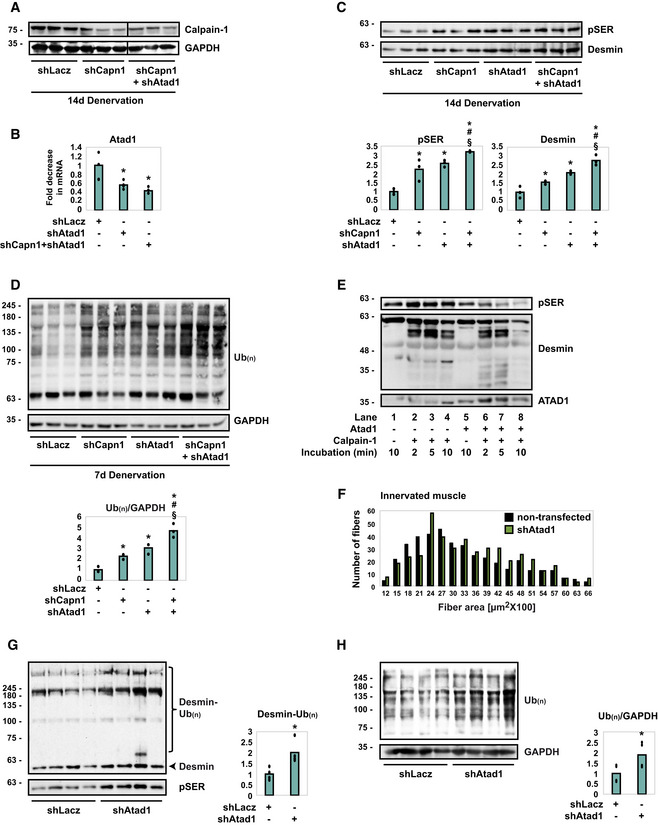
- shRNA‐mediated knockdown of calpain‐1 in 14 days denervated TA muscles. Homogenates from denervated muscles expressing shLacz, shCapn1 or co‐expressing shCapn1 and shAtad1 were analyzed by immunoblotting. Black line indicates the removal of intervening lanes for presentation purposes. n = 3 mice, biological replicates. Data are representative of two independent experiments.
- shRNA‐mediated knockdown of Atad1 in 14 days denervated TA muscles. RT‐PCR of mRNA preparations from denervated muscles expressing shLacz, shCapn1 or co‐expressing shCapn1 and shAtad1 using specific primers for Atad1. Data are plotted as the mean fold change relative to control ± SEM. n = 4 mice, biological replicates. *P (shAtad1) = 0.03, P (shCapn1 + shAtad1) = 0.03 vs. denervated shLacz. Data are representative of one experiment.
- On denervation, ATAD1 and calpain‐1 cooperate in an additive fashion in promoting desmin IF disassembly and loss. Top: desmin filaments isolated from denervated muscles expressing shLacz, shCapn1, shAtad1 or co‐expressing shCapn1 and shAtad1 were analyzed by immunoblotting. Bottom: densitometric measurement of presented blots. Mean ± SEM. n = 3 mice, biological replicates. pSER: *P (shCapn1) = 0.048, P (shAtad1) = 0.005, P (shCapn1 + shAtad1) = 0.001 vs. shLacz; # P = 0.049 vs. shCapn1 in atrophy; § P = 0.02 vs. shAtad1 in atrophy. Desmin: *P (shCapn1) = 0.049, P (shAtad1) = 0.02, P (shCapn1 + shAtad1) = 0.0002 vs. shLacz; # P = 0.009 vs. shCapn1 in atrophy; § P = 0.04 vs. shAtad1 in atrophy. Data are representative of one experiment.
- ATAD1 and calpain‐1 cooperate in an additive fashion in promoting loss of soluble proteins in denervated muscle. Left: homogenates from denervated muscles expressing shLacz, shCapn1, shAtad1 or co‐expressing shCapn1 and shAtad1 were analyzed by immunoblotting. Bottom: densitometric measurement of presented blots. The mean ratios of total ubiquitin conjugates to GAPDH ± SEM is presented. n = 3 mice, biological replicates. *P (shCapn1) = 0.008, P (shAtad1) = 0.007, P (shCapn1 + shAtad1) = 0.009 vs. shLacz; # P = 0.02 vs. shCapn1 in atrophy; § P = 0.035 vs. shAtad1 in atrophy. Data are representative of one experiment.
- Cleavage of desmin filaments by calpain‐1 is facilitated in the presence of ATAD1. The insoluble fraction from 3 days denervated muscle was subjected to cleavage by recombinant calpain‐1 in the presence (lanes 5–8) or absence (lanes 1–4) of ATAD1 (ATAD1 was immunoprecipitated from mouse muscle). Data are representative of two independent experiments. Data are representative of two independent experiments.
- Atad1 downregulation promotes normal muscle growth. Measurements of cross‐sectional areas of 423 fibers expressing shAtad1 (green bars) vs. 423 nontransfected fibers (black bars) in the same muscle. n = 3 mice, biological replicates.
- Downregulation of Atad1 in normal muscle causes accumulation of ubiquitinated desmin filaments. Left: insoluble fractions from normal muscles expressing shLacz or shAtad1 were analyzed by immunoblotting. Right: densitometric measurement of presented blots. Mean ± SEM. n = 4 mice, biological replicates. *P = 0.03 vs. shLacz. Data are representative of one experiment.
- Downregulation of Atad1 in normal muscle causes accumulation of ubiquitinated soluble proteins. Left: homogenates from normal muscles expressing shLacz or shAtad1 were analyzed by immunoblotting. Right: densitometric measurement of presented blots. The mean ratios of total ubiquitin conjugates to GAPDH ± SEM is presented. n = 4 mice, biological replicates. *P = 0.048 vs. shLacz. Data are representative of one experiment.
Source data are available online for this figure.
To test this idea more directly, we assayed in vitro the susceptibility of phosphorylated desmin filaments to cleavage by calpain‐1 in the presence of ATAD1. The insoluble fraction from 3 days denervated muscles, in which GSK3‐β was catalyzing desmin IF phosphorylation, was isolated and subjected to cleavage by recombinant calpain‐1, with or without the addition of ATAD1. ATAD1 was immunoprecipitated from 7 days denervated muscles, when it is found bound to calpain‐1 and other critical interacting partners (Fig 3C–E). As we had shown before (Aweida et al, 2018), desmin filaments were efficiently processed by calpain‐1 in vitro as indicated by the appearance of short desmin fragments over time (Fig 5E, lanes 2–4). However, in the presence of ATAD1, cleavage of phosphorylated desmin filaments by calpain‐1 was substantially more efficient, as even smaller fragments of desmin appeared in shorter incubation times, which correlated with a rapid reduction in the amount of phosphorylated desmin (Fig 5E, compare lanes 6–7 and 2–3). In addition, by 10 min of incubation, phosphorylated monomeric desmin and its cleavage products were virtually absent in the reaction containing ATAD1, most likely due to their processive cleavage by calpain‐1 (Fig 5E, compare lanes 8 and 4), suggesting that desmin is more sensitive to cleavage by calpain‐1 in the presence of ATAD1. Thus, on denervation, desmin filament depolymerization by calpain‐1 is facilitated by ATAD1.
Atad1 downregulation attenuates basal turnover of desmin filaments and of soluble proteins
Because downregulation of Atad1 reduced atrophy on denervation, we determined whether it also affects the basal turnover of muscle proteins. Downregulation of ATAD1 by in vivo electroporation of shAtad1 into normal TA muscles induced muscle fiber growth because the cross‐sectional area of 423 transfected fibers (also express GFP) was significantly larger than that of the surrounding 423 nontransfected ones (Fig 5F, Table 1; Appendix Fig S2). ATAD1 attenuated normal muscle growth most likely by promoting the loss of desmin filaments and of soluble proteins because in normal muscles expressing shAtad1 phosphorylated desmin filaments accumulated as ubiquitinated species (Fig 5G) and ubiquitinated proteins also accumulated in the cytosol (Fig 5H). Thus, ATAD1 seems to function in normal postnatal muscle to limit fiber growth, and suppression of its activity alone can induce muscle hypertrophy.
Discussion
In this study, we uncovered a new critical cellular role for ATAD1, a AAA‐ATPase with previously unknown cofactors and very few identified roles. We identified and validated in vivo two interacting partners for ATAD1, UBXN4, and PLAA, and revealed that the ATAD1‐PLAA‐UBXN4 complex regulates muscle mass by promoting desmin filaments depolymerization and loss. On denervation, ATAD1 binds and cooperates with calpain‐1 to facilitate desmin IF depolymerization, and both enzymes are essential for desmin loss. These enzymes are also necessary for the degradation of ubiquitinated proteins in the cytosol of denervated muscles, or their actions allow other proteolytic enzymes to act. In either case, ATAD1 and calpain‐1 are clearly general enzymes affecting insoluble and soluble proteins in an additive manner. Moreover, our studies implicate ATAD1 in the slower turnover of desmin IF and soluble proteins in normal muscle. Consequently, suppression of ATAD1 activity alone not only attenuates atrophy but can also induce muscle growth.
Reducing ATAD1 or calpain‐1 (Aweida et al, 2018) functions by shRNA attenuated the decrease in fiber diameter on denervation. The stabilization of desmin IF in these muscles coincided with accumulation of ubiquitinated proteins in the cytosol, consistent with our prior findings demonstrating that desmin IF loss in atrophy promotes overall proteolysis (Cohen et al, 2012; Volodin et al, 2017; Aweida et al, 2018). The reduced structural integrity of desmin filaments on denervation is likely the key step in the destabilization of insoluble proteins (e.g. myofibrils) during atrophy, leading to the enhanced solubilization and degradation in the cytosol. Degradation of soluble desmin in the cytosol also requires ATAD1 (Fig 2C). Such soluble pools of desmin and its homolog vimentin are small as these proteins mostly exist in the cell assembled within filaments (Soellner et al, 1985; Cohen et al, 2012; Aweida et al, 2018). Possibly, this soluble pool of desmin functions either as precursors to the mature filament or as components released during filament turnover. Because desmin IF disassembly is prevented in atrophying muscles expressing shAtad1, the soluble desmin that accumulates in the cytosol of these muscles likely represents new precursors to the mature filament. In addition to the essential roles of ATAD1 and calpain‐1 in atrophy, the accelerated proteolysis during atrophy seems to require both enzymes and additional cofactors (e.g., UBXN4, PLAA) to function with them, as well as posttranslational modifications to enhance the susceptibility of desmin filaments to these enzymes. For example, desmin filaments must be modified by phosphorylation before their disassembly by ATAD1 and calpain‐1 (Fig 2F; Aweida et al, 2018).
Because ATAD1 is important for muscle atrophy and for normal muscle growth, identification of its cofactors was essential to understand the mechanisms of protein homeostasis and loss. We used three independent mass spectrometry‐based proteomics approaches and identified a number of UPS components as novel interacting partners for ATAD1 in vivo, all of which are important for protein degradation by the proteasome (Table 2). Using these analyses, we identified and validated UBXN4 and PLAA as ATAD1 partners, and the ATAD1‐PLAA‐UBXN4 complex as responsible for depolymerization and loss of ubiquitinated desmin IF in skeletal muscle. Precisely how ubiquitination enables ATAD1‐driven desmin IF disassembly is an important question for further research. Although ATAD1 can bind ubiquitin (Fig 3C and Table 2), we found that this AAA‐ATPase associates with phosphorylated desmin, whether they are polyubiquitinated (e.g., in atrophying muscles) or not (e.g., in atrophying muscles expressing shTrim32), suggesting that ATAD1 binds substrates either before or during their polyubiquitination. In fact, ATAD1 accumulates on desmin filaments in muscles expressing shTrim32 suggesting that ubiquitination by TRIM32 is not required for ATAD1 binding but is rather important for its release from desmin IF. In other words, ubiquitination by TRIM32 is likely important for ATAD1‐mediated extraction of ubiquitinated desmin from the insoluble filaments into the cytosol, where desmin is degraded by the proteasome (Fig 2D). TRIM32 was not identified as ATAD1 partner by our mass spectrometry analyses, suggesting that the association of this E3 with desmin filaments is not dependent on ATAD1. It will be important to determine if ubiquitination by TRIM32 is sufficient for ATAD1‐mediated disassembly of desmin filaments, or whether the ubiquitin chains generated by TRIM32 are extended further by ubiquitin ligases that are recruited by ATAD1 to desmin (e.g., HUWE1, see Table 2). Desmin was recently reported to be ubiquitinated by the ubiquitin ligase MURF1 in normal mouse muscles overexpressing Murf1, though this ubiquitination of desmin did not lead to degradation (Baehr et al, 2021). We could not precipitate MURF1 with ATAD1 from denervated muscle extracts, and it remains to be determined if MURF1 acts on desmin in vivo during atrophy.
It is noteworthy that co‐fractionation with ATAD1 using three approaches did not identify the exact same subset of putative adaptors (Table 2), suggesting a dynamic association of ATAD1 with its cofactors. It is also possible that distinct ATAD1's interacting partners may have been missed by our analyses, especially if specific stimuli are required to facilitate such interaction. Future studies will determine how these adaptor proteins function with ATAD1 to promote substrate recruitment and processing, and whether their function is regulated by substrates or other cofactors. These various binding partners could be precipitated with ATAD1 from the cytosolic fraction, and could also bind desmin in the insoluble pellet of normal and atrophying muscles (Table 2). ATAD1 appears to be recruited to desmin filaments early during atrophy to promote desmin release during filament turnover. This AAA‐ATPase complex clearly binds desmin at 3 days after denervation when desmin filaments are intact, and 4 days later, when desmin IF solubilization is rapid (Volodin et al, 2017; Aweida et al, 2018), ATAD1 and its binding partners accumulate in the cytosol, bound to soluble ubiquitinated desmin (Fig 3D). In addition, ATAD1 catalyzes desmin IF disassembly only when these filaments are phosphorylated, presenting ATAD1 as the only known AAA‐ATPase that acts preferentially on phosphorylated substrates. It is likely that ATAD1's interacting partners, which contain a phospho‐Serine/Threonine‐binding domain, mediate ATAD1 association with phosphorylated desmin IF. Although calpain‐1 binds preferentially to phosphorylated desmin IF (Aweida et al, 2018) and is present in ATAD1 complex, it is not required for ATAD1 recruitment to desmin IF because in denervated muscles expressing shCapn1, ATAD1 accumulated on desmin filaments (Appendix Fig S3). Thus, calpain‐1 facilitates the release of ATAD1 from desmin IF, which is consistent with the cooperative roles of these enzymes in promoting desmin filament disassembly.
The previously known cellular roles for ATAD1 are regulation of synaptic plasticity in the brain (Zhang et al, 2011; Li et al, 2016), and extraction of mislocalized proteins from mitochondrial membrane (Wohlever et al, 2017; Li et al, 2019). Our findings that ATAD1 disassembles ubiquitinated intermediate filaments in skeletal muscle present a novel function for this AAA‐ATPase complex, which is similar to p97/VCP complex that also extracts ubiquitinated proteins from larger structures (Piccirillo & Goldberg, 2012; Stach & Freemont, 2017; Volodin et al, 2017). ATAD1 is expressed in various tissues (Appendix Fig S4) and may thus have many cellular roles that are probably dictated by the specific cofactors that it binds. This study is the first to identify ATAD1's binding partners, and to demonstrate that ATAD1‐PLAA‐UBXN4 complex binds the insoluble phosphorylated desmin IF and promote the release of ubiquitinated species into the cytosol. Accordingly, downregulation of either ATAD1, PLAA, or UBXN4 was sufficient to prevent solubilization and loss of ubiquitinated desmin filaments. PLAA and UBXN4 are also known cofactors for p97/VCP (Liang et al, 2006; Papadopoulos et al, 2017), a AAA‐ATPase that was not in our datasets, indicating that p97/VCP adaptors can bind and function with other AAA‐ATPases. It is plausible that ATAD1 binds additional factors, as our mass spectrometry data strongly suggest, whose specific cellular roles merit further study. It is also likely that such factors will be required for ATAD1‐mediated removal of mislocalized proteins from the mitochondrial membrane (Wohlever et al, 2017). For example, UBXN4 contains a putative transmembrane segment (Sasagawa et al, 2010), and via association with ATAD1 may anchor and recruit ATAD1 complex to mitochondrial membrane proteins. Mitochondria are located in close proximity to the myofibrils in muscle, and are linked to each other and to other cellular organelles via desmin IF (Milner et al, 2000). Thus, in skeletal muscle, the ATAD1‐PLAA‐UBXN4 complex may serve a dual role in the extraction of proteins from desmin filaments and the mitochondria.
The strongest evidence for the importance of ATAD1 and calpain‐1 cooperation for the accelerated loss of desmin was our finding that simultaneous downregulation of both enzymes inhibited desmin loss to a greater extent than when each enzyme was downregulated alone. In addition, our data indicate that desmin filaments are more sensitive to cleavage by calpain‐1 in vitro in the presence of ATAD1 than in samples containing calpain‐1 alone. Thus, the present findings uncover a novel mechanism involving a cooperation between an AAA‐ATPase and a protease to facilitate filament loss. Because ATAD1 and calpain‐1 also affect ubiquitinated soluble proteins in an additive fashion (Fig 5D), and since these enzymes are ubiquitously expressed, this new mechanism probably contributes to proteolysis in other cells, and may also apply to other AAA‐ATPases and proteases.
Desmin loss is a critical event leading to myofibril destruction and muscle atrophy, therefore, identifying key regulators of this process should be of major therapeutic promise to treat muscle wasting. ATAD1, along with the previously identified key enzymes the protein kinase GSK3‐β, the ubiquitin ligase TRIM32, and the Ca2+‐specific protease calpain‐1, appear to act on the desmin cytoskeleton in a specific order to promote its loss (Aweida et al, 2018). We propose that ATAD1 binds desmin filaments early after denervation (at 3 days), and together with its interacting partners promote a slow dissociation of desmin filaments (Fig 6). Then, at 7 days after denervation, when Ca2+ levels rise and calpain‐1 is activated, desmin depolymerization is accelerated by the cooperative functions of calpain‐1 and ATAD1. We propose that these two enzymes function in a “Pull and Chop” mechanism, in which the ATAD1 complex extracts ubiquitinated desmin filaments, exposing on desmin cleavage sites for calpain‐1, consequently facilitating desmin cleavage, solubilization, and degradation by the proteasome. Cleavage by calpain‐1 may expose degrons on desmin to facilitate its degradation, as has been proposed for other proteases (Varshavsky, 2019). Such cooperation may be important to facilitate protein degradation during rapid types of atrophy, for example, during fasting or denervation, where muscle loss occurs over days, and may be less relevant in prolonged types of atrophy such as during aging where muscle loss is slow and gradual.
Figure 6. Proposed mechanism for desmin filament loss during denervation‐induced atrophy.
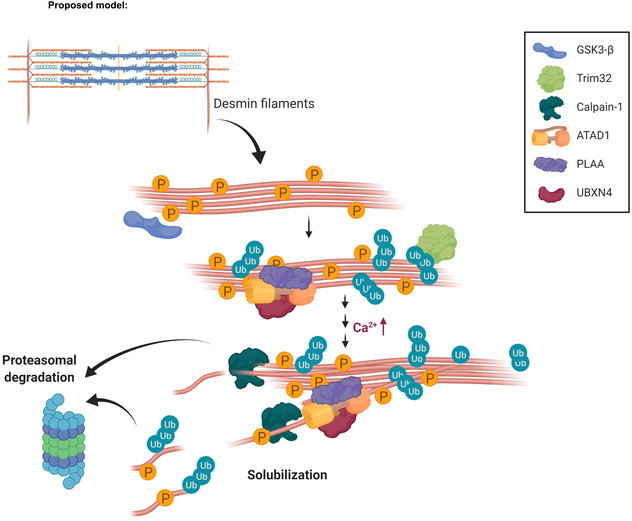
Early after denervation (at 3 days), ATAD1 binds phosphorylated and ubiquitinated desmin filaments and together with its interacting partners promote a slow dissociation of desmin IF. Then, at a more delayed phase, when calcium levels rise and calpain‐1 is activated, desmin filament depolymerization is accelerated by the cooperative functions of calpain‐1 and ATAD1. We propose that these two enzymes function in a “Pull and Chop” mechanism, in which the ATAD1 complex extracts ubiquitinated desmin filaments, exposing on desmin cleavage sites for calpain‐1, consequently facilitating desmin cleavage, solubilization, and subsequent degradation by the proteasome. The illustration was created with BioRender.com.
Materials and Methods
Animal work
All animal experiments were consistent with Israel Council on Animal Experiments guidelines and approved by the Institutional regulations of Animal Care and Use. Specialized personnel provided mice care in the institutional animal facility. We used adult wild‐type Hsd:ICR male mice (~ 30 g) (Envigo), which are 6 weeks old in all experiments. The mice were housed in an SPF animal facility consisting of surgery suites, generous animal housing rooms, fully automated cage washing and waste disposal systems, and trained technicians and on‐site veterinarians available to help researchers in all aspects of animal care, handling and pathology.
Muscle denervation was performed by sectioning the sciatic nerve on one limb, while the contralateral leg served a control. Muscles were excised at 3, 7, 10, or 14 days after denervation. To inhibit proteasome activity, mice were injected with Bortezomib (3 mg/kg body weight, i.p.) or DMSO, and an hour later were sacrificed.
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells (previously described in Ziv et al, 2006) were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Transient transfection was performed using PolyJet™ In Vitro DNA Transfection Reagent (SL100688, SignaGen Laboratories) according to manufacturer's instructions. Cells were plated in a 6‐well plate in five technical replicates, and on the following day were transfected with 2 μg of DNA plasmids encoding either shPlaa or shUbxn4. Cells were collected and RNA extracted 48 h after transfection.
Antibodies, constructs, and materials
Oligos encoding shRNA against ATAD1, PLAA, and UBXN4 were cloned into pcDNA 6.2GW/EmGFP‐miR vector using Invitrogen's BLOCK‐iT RNAi expression kit as described (Volodin et al, 2017; Goldbraikh et al, 2020). shCapn1, shTrim32, shLacz, and the plasmid encoding HA‐tagged GSK3‐β dominant negative (K85A catalytically dead mutant) were described and validated before (Aweida et al, 2018). A plasmid encoding 6His‐Calpain‐1 was a kind gift from Dr. Shoji Hata, Tokyo Metropolitan Institute of Medical Science, Tokyo, JAPAN. ATAD1 (#ab94583) (for immunofluorescence and immunoblotting) and desmin (#ab8592) antibodies were from Abcam. Anti PLAA (#sc‐390454) was from Santa Cruz. A second Desmin antibody (#AB_528195) from Developmental Studies Hybridoma bank was developed by D.A. Fischman, Cornell University Medical College. β‐Dystroglycan antibody (#AB_2618140) was from Developmental Studies Hybridoma bank (developed by G.E. Morris, Wolfson Centre for Inherited Neuromuscular Disease, RJAH Orthopedic Hospital). GAPDH (#G8795), UBXN4 (#HPA036325), ATAD1 (#SAB2107561) (for immunofluorescence), and calpain‐1 (#C5736) (for immunofluorescence) antibodies were from Sigma. Anti calpain‐1 (#2556) (for immunoblotting) was from Cell Signaling, anti‐phospho‐Threonine/Serine (#PP2551) from ECM Biosciences, and Wheat Germ Agglutinin (WGA)(W7024) from Invitrogen. Anti‐mono and polyubiquitin conjugates (#BML‐PW1210) was from Enzo, and TRIM32 antibody was kindly provided by Dr. Knoblich (Institute of Molecular Biotechnology, Vienna, Austria).
In vivo transfection
In vivo electroporation experiments were performed in adult CD‐1 male mice (~ 30 g) as reported (Goldbraikh et al, 2020; Gilda et al, 2021). Briefly, 20 μg of plasmid DNA were injected into adult mouse TA muscles, and a mild electric pulse was applied using two electrodes (12 V, 5 pulses, 200 ms intervals). For fiber size analysis, muscle cross‐sections were fixed in 4% paraformaldehyde (PFA) and fiber membrane was stained with WGA or β‐Dystroglycan antibody (1:30, see details under immunofluorescence). Cross‐sectional areas of muscle sections (20 μm) were analyzed using Imaris software (Bitplane). Images were collected using a Nikon Ni‐U upright fluorescence microscope with Plan Fluor 20 × 0.5 NA objective lens and a Hamamatsu C8484‐03 cooled CCD camera, at room temperature. For biochemical analyses, muscles that are at least 60–70% transfected were used.
Fractionation of muscle tissue
To obtain whole cell extracts, muscles were homogenized in lysis buffer (20 mM of Tris pH 7.6, 5 mM of EGTA, 100 mM of KCl, 1% Triton X‐100, 1 mM of PMSF, 10 mM of Sodium Pyrophosphate, 3 mM of Benzamidine, 10 μg/ml of Leupeptin, 10 μg/ml Aprotinin, 50 mM of NaF, and 2 mM of Sodium OrthoVanadate), and incubated for 1 h at 4°C. After centrifugation at 6,000 g for 20 min at 4°C, the supernatant (i.e., soluble fraction) was stored at −80°C. The insoluble pellet was washed once with homogenization buffer and twice with suspension buffer (20 mM of Tris pH 7.6, 100 mM of KCl, 1 mM of DTT, and 1 mM of PMSF), and after a final centrifugation at 6,000 g for 10 min at 4°C, the insoluble pellet (i.e., purified myofibrils and desmin filaments) was re‐suspended in storage buffer (20 mM of Tris pH 7.6, 100 mM of KCl, 1 mM of DTT, and 20% glycerol) and kept at −80°C. To isolate desmin filaments, 30 μg of the insoluble muscle pellet were resuspended in ice‐cold extraction buffer (0.6 M of KCl, 1% Triton X‐100, 2 mM of EDTA, 2 mM of PMSF, 1× PBS, 10 μg/ml of leupeptin, 3 mM of benzamidine, 10 μg/ml of Aprotinin, 50 mM of NaF, 2 mM of Sodium OrthoVanadate, and 10 mM of Sodium Pyrophosphate) for 10 min on ice. After centrifugation at 6,000 g for 10 min at 4°C, the pellet was resuspended in 20 mM of Tris pH 7.6 and analyzed by SDS–PAGE and immunoblotting.
Protein analysis
For immunoblotting, soluble (25 μg) or insoluble (3 μg) fractions were resolved by SDS–PAGE, transferred onto PVDF membranes and immunoblotted with specific primary antibodies, and secondary antibodies conjugated to HRP.
For immunoprecipitation, 300 μg of muscle soluble fraction were incubated with a specific primary antibody (the control sample contained nonspecific IgG) overnight at 4°C, and protein A/G agarose was then added for 4 h. To remove nonspecific or weakly associated proteins, the precipitates were washed extensively with 10 bed volumes of each of the following buffers: buffer A (50 mM of Tris–HCl, pH 8, 500 mM of NaCl, 0.1% SDS, 0.1% Triton, 5 mM of EDTA), buffer B (50 mM of Tris–HCl, pH 8, 150 mM of NaCl, 0.1% SDS, 0.1% Triton, 5 mM of EDTA), and buffer C (50 mM of Tris–HCl, pH 8, 0.1% Triton, 5 mM of EDTA). Protein precipitates were eluted with protein loading buffer containing 50 mM of DTT, and were analyzed by immunoblotting.
Immunofluorescence labeling of frozen muscle sections
Frozen longitudinal sections of mouse TA were cut at 10 μm, fixed in 4% PFA for 10 min at room temperature (RT), and washed in PBST (PBS, 0.1% Triton X‐100) three times. For antigen retrieval, slides were incubated in 10 mM of Citrate buffer (9 ml of 0.1 M Citric acid, 41 ml of 0.1 M Sodium citrate, 450 ml water, pH 6.0) on low heat (lowest power in microwave) for 20 min, and then left to cool down at RT (~ 2 h). Following one wash in PBST, the slides were incubated in blocking solution (0.2% BSA, 5% goat serum, and 5% donkey serum in PBST) for 1 h at RT. Immunofluorescence was performed using ATAD1 (1:50), Calpain‐1 (1:50), PLAA (1:20), or UBXN4 (1:50) antibodies overnight at 4°C, followed by 2 h incubation at RT with secondary antibodies conjugated to Alexa Fluor 568 or 647 (1:400). Primary antibodies were diluted in blocking solution, and secondary antibodies were diluted in 0.2% BSA/PBST. Sections were imaged using the Elyra 7 eLS lattice SIM super resolution microscope by Zeiss with a pco.edge sCMOS camera. A X63 1.46 NA oil immersion objective with 561 and 642 nm lasers. 16‐bit 2D image data sets were collected with 13 phases. The SIM^2 image processing tool by Zeiss was used.
Colocalization analysis by Imaris
Colocalization measurements were performed using the Imaris software (Bitplane, ver 9.31), after setting an appropriate threshold that was kept constant during the entire analysis. At least three images per protein staining were analyzed. Imaris segmented the stained proteins with the “Spots” module. The spots were set to 100 nm in diameter that is below the light diffraction limit but above the SIM^2 super resolution imaging limit (60 nm). Finally, colocalized spots were identified by using imaris extension “coloc spots” and a distance threshold between close spots was set to 100 nm from each other.
Purification of recombinant calpain‐1
6His‐tagged calpain‐1 was purified from BL21 bacteria (OD 0.6) grown with 0.2 mM IPTG at 17°C overnight. After centrifugation (4,000 g at 4°C for 20 min), the pellet was lysed with lysis buffer (50 mM of NaH2PO4, 300 mM of NaCl, and 10 mM of imidazole) and a microfluidizer, and centrifuged at 10,000 g at 4°C for 20 min. The obtained supernatant was incubated with a nickel column for 1 h at 4°C, the column was washed twice with wash buffer (50 mM of NaH2PO4, 300 mM of NaCl, and 20 mM of imidazole), and 6His‐calpain‐1 was eluted with elution buffer (50 mM of NaH2PO4, 300 mM of NaCl, and 250 mM of imidazole). After dialysis against 50 mM of Tris, pH 7.6, overnight at 4°C, 1 mM of DTT and 10% glycerol were added, and purified calpain‐1 was stored at −80°C.
In vitro cleavage assay by calpain‐1
ATAD1 was immunoprecipitated from 300 μg of soluble fraction from 7 days denervated TA muscle (see Protein analysis). Two sets of tubes containing 30 μg of the myofibrillar fraction (see Fractionation of muscle tissue) from denervated muscles (3 days), were incubated with 6.5 μg of purified 6His‐Calpain‐1 in reaction buffer (100 mM of Hepes, 10 mM of DTT, and 5 mM of CaCl2) for 2, 5 or 10 min at 30°C, in the presence or absence of ATAD1. Negative control samples also contained 50 mM EGTA. Desmin cleavage by calpain‐1 was assessed by SDS–PAGE and immunoblotting.
Identification of ATAD1's interacting partners using SEC
Seven days denervated lower limb muscles (1 g) were homogenized in 10 volumes of 1% PFA/PBS (v/w), and incubated for 10 min at RT. The reaction was quenched by the addition of 0.125 M glycine for 5 min at RT, centrifuged at 3,000 g for 5 min at 4°C, and the supernatant was adjusted to 1× homogenization buffer (see fractionation of muscle tissue. Adjustment was performed using 10× homogenization buffer). Then, proteins in the supernatant were precipitated with 40% ammonium sulfate and centrifuged at 20,000 g for 30 min at 4°C. The pellet was resuspended in 0.5 ml of buffer A (50 mM of Tris pH 8, 0.5 M of NaCl) 5 mM of EGTA, 1% Triton X‐100, 1 mM of PMSF, 10 mM of Sodium Pyrophosphate, 3 mM of Benzamidine, 10 μg/ml of Leupeptin, 10 μg/ml of Aprotinin, 50 mM of NaF, and 2 mM of Sodium OrthoVanadate, and loaded onto a Superdex 200 10/300 Gel Filtration column (GE Healthcare Life Sciences, UK) equilibrated with 50 mM of Tris pH 8, 0.5 M of NaCl. Proteins were eluted at a flow rate of 0.5 ml/min and 500 μl of fractions were collected. Even fractions were analyzed by silver staining and immunoblotting.
Protein identification by mass spectrometry
For mass spectrometry analysis, protein bands were excised from gel, reduced with 3 mM of DTT in 100 mM of ammonium bicarbonate [ABC] (60°C, 30 min), modified with 10 mM of Iodoacetamide in 100 mM of ABC (at the dark, RT, 30 min), and digested in 10% Acetonitrile and 10 mM of ABC with modified trypsin (Promega) at a 1:10 enzyme‐to‐substrate ratio overnight at 37°C. The resulted peptides were desalted using C18 tips (Homemade stage tips, Empore), dried, and re‐suspended in 0.1% Formic acid. The peptides were then resolved by reverse‐phase chromatography on 0.075 × 180‐mm fused silica capillaries (J&W) packed with Reprosil reversed phase material (Dr. Maisch GmbH, Germany), and were eluted with linear 60 min gradient of 5–28%, 15 min gradient of 28–95%, and 15 min at 95% acetonitrile with 0.1% formic acid in water at flow rates of 0.15 μl/min. Mass spectrometry was performed by Q Exactive plus mass spectrometer (Thermo) in a positive mode using repetitively full MS scan followed by collision induces dissociation (HCD) of the 10 most dominant ions selected from the first MS scan.
The mass spectrometry data was analyzed using Proteome Discoverer 1.4 software with Sequest (Thermo) and Mascot (Matrix Science) algorithms against mouse uniport database with mass tolerance of 10 ppm for the precursor masses and 0.05 amu for the fragment ions. Minimal peptide length was set to six amino acids and a maximum of two mis‐cleavages was allowed. Peptide‐ and protein‐level false discovery rates (FDRs) were filtered to 1% using the target‐decoy strategy. Protein tables were filtered to eliminate the identifications from the reverse database and from common contaminants. Semi quantitation was done by calculating the peak area of each peptide based its extracted ion currents (XICs) and the area of the protein is the average of the three most intense peptides from each protein.
Real‐Time qPCR
Total RNA was isolated from muscle using TRI reagent (T9424; Sigma‐Aldrich) and served as a template for the synthesis of cDNA by reverse transcription. Real‐time qPCR was performed on mouse target genes using specific primers (Appendix Table S1) and the Perfecta SYBR Green qPCR Kit (95073–012; Quanta Biosciences) according to the manufacturer's protocol.
Statistical analysis and image acquisition
Data are presented as mean ± SEM. The statistical significance was accessed with one‐tailed paired Student's t test. No blinding was done. Muscle sections for fiber size analysis were imaged at room temperature with a Nikon Ni‐U upright fluorescence microscope with Plan Fluor 20x 0.5NA objective lens and a Hamamatsu C8484‐03 cooled CCD camera. Image acquisition and processing was performed using the Metamorph or the Imaris software (Bitplane). Statistics on fiber size distributions was performed using A‐statistics and Brunner‐Manzel test as described in our recent methodology paper (Gilda et al, 2021). Muscle sections in Fig 3E were imaged using the Elyra 7 eLS lattice SIM super resolution microscope by Zeiss with a pco.edge sCMOS camera. A X63 1.46 NA oil immersion objective with 561 nm and 642 nm lasers. 16‐bit 2D image data sets were collected with 13 phases. The SIM^2 image processing tool by Zeiss was used. Black and white images were processed with Adobe Photoshop CS5, version 12.1x64 software. Quantity One algorithm (Bio‐Rad Laboratories, version 29.0) was used for densitometric measurements of intensity of protein bands.
Author contributions
Dina Aweida: Data curation; formal analysis; validation; investigation; writing—original draft; project administration. Shenhav Cohen: Conceptualization; supervision; funding acquisition; validation; project administration; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
This project was supported by grants from the Israel Science Foundation (grant no. 1068/19), Israel Ministry of Health (grant no. 3‐16061), and the Niedersachsen‐Deutsche (grant no. ZN3008) to S. Cohen. Additional funds were received from the Russell Berrie Nanotechnology Institute, Technion to S. Cohen. We thank the Smoler Proteomics Center at Technion for the mass spectrometry analysis. The illustration in Fig 6 was created with BioRender.com. We thank the technicians and veterinarians at the Technion Gutwirth animal facility for their assistance with animal care, handling, and pathology.
EMBO reports (2022) 23: e55175
Data availability
There are no public datasets associated to the results in the paper.
References
- Agnetti G, Halperin VL, Kirk JA, Chakir K, Guo Y, Lund L, Nicolini F, Gherli T, Guarnieri C, Caldarera CM et al (2014) Desmin modifications associate with amyloid‐like oligomers deposition in heart failure. Cardiovasc Res 102: 24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnetti G, Herrmann H, Cohen S (2021) New roles for desmin in maintenance of muscle homeostasis. FEBS J 289: 2755–2770 [DOI] [PubMed] [Google Scholar]
- Aweida D, Cohen S (2021) Breakdown of filamentous myofibrils by the UPS‐step by step. Biomolecules 11: 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aweida D, Rudesky I, Volodin A, Shimko E, Cohen S (2018) GSK3‐β promotes calpain‐1‐mediated desmin filament depolymerization and myofibril loss in atrophy. J Cell Biol 217: 3698–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, Hughes DC, Lynch SA, Van Haver D, Maia TM, Marshall AG, Radoshevich L, Impens F, Waddell DS, Bodine SC (2021) Identification of the MuRF1 skeletal muscle Ubiquitylome through quantitative proteomics. Function (Oxf) 2: zqab029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär H, Strelkov SV, Sjöberg G, Aebi U, Herrmann H (2004) The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol 148: 137–152 [DOI] [PubMed] [Google Scholar]
- Boudriau S, Côté CH, Vincent M, Houle P, Tremblay RR, Rogers PA (1996) Remodeling of the cytoskeletal lattice in denervated skeletal muscle. Muscle Nerve 19: 1383–1390 [DOI] [PubMed] [Google Scholar]
- Cohen S (2020) Role of calpains in promoting desmin filaments depolymerization and muscle atrophy. Biochim Biophys Acta Mol Cell Res 1867: 118788 [DOI] [PubMed] [Google Scholar]
- Cohen S, Zhai B, Gygi SP, Goldberg AL (2012) Ubiquitylation by Trim32 causes coupled loss of desmin, Z‐bands, and thin filaments in muscle atrophy. J Cell Biol 198: 575–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Lee D, Zhai B, Gygi SP, Goldberg AL (2014a) Trim32 reduces PI3K‐Akt‐FoxO signaling in muscle atrophy by promoting plakoglobin‐PI3K dissociation. J Cell Biol 204: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Nathan JA, Goldberg AL (2014b) Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14: 58–74 [DOI] [PubMed] [Google Scholar]
- Eid‐Mutlak Y, Aweida D, Volodin A, Ayalon B, Dahan N, Parnis A, Cohen S (2020) A signaling hub of insulin receptor, dystrophin glycoprotein complex and plakoglobin regulates muscle size. Nat Commun 11: 1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilda JE, Ko JH, Elfassy AY, Tropp N, Parnis A, Ayalon B, Jhe W, Cohen S (2021) A semiautomated measurement of muscle fiber size using the Imaris software. Am J Physiol Cell Physiol 321: C615–C631 [DOI] [PubMed] [Google Scholar]
- Goldbraikh D, Neufeld D, Eid‐Mutlak Y, Lasry I, Gilda J, Parnis A, Cohen S (2020) USP1 deubiquitinates Akt to inhibit PI3K‐Akt‐FoxO signaling in muscle during prolonged starvation. EMBO Rep 21: e48791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell TR, Gunhan O, Edwards RH (1989) Lectin binding and desmin expression during necrosis, regeneration, and neurogenic atrophy of human skeletal muscle. J Pathol 159: 43–51 [DOI] [PubMed] [Google Scholar]
- Jackman RW, Kandarian SC (2004) The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 287: C834–C843 [DOI] [PubMed] [Google Scholar]
- Li H, Liu K, Lei R, Li Q, Wang XX, Wu Q, An P, Zhang J, Zhu M, Xu Z et al (2016) Transferrin receptor controls AMPA receptor trafficking efficiency and synaptic plasticity. Sci Rep 6: 21019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zheng J, Wu X, Jiang H (2019) Mitochondrial AAA‐ATPase Msp1 detects mislocalized tail‐anchored proteins through a dual‐recognition mechanism. EMBO Rep 20: e46989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Yin C, Doong H, Fang S, Peterhoff C, Nixon RA, Monteiro MJ (2006) Characterization of erasin (UBXD2): a new ER protein that promotes ER‐associated protein degradation. J Cell Sci 119: 4011–4024 [DOI] [PubMed] [Google Scholar]
- Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S et al (2009) Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol 187: 201–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavroidis M, Panagopoulou P, Kostavasili I, Weisleder N, Capetanaki Y (2008) A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z‐disc localization. FASEB J 22: 3318–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y (1996) Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 134: 1255–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y (2000) Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 150: 1283–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K et al (2017) VCP/p97 cooperates with YOD 1, UBXD 1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36: 135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S, Piper RC (2010) WD40 repeat propellers define a ubiquitin‐binding domain that regulates turnover of F box proteins. Mol Cell 40: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piard J, Umanah GKE, Harms FL, Abalde‐Atristain L, Amram D, Chang M, Chen R, Alawi M, Salpietro V, Rees MI et al (2018) A homozygous ATAD1 mutation impairs postsynaptic AMPA receptor trafficking and causes a lethal encephalopathy. Brain 141: 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo R, Goldberg AL (2012) The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J 31: 3334–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Pashkova N, Winistorfer S, Piper RC (2008) DOA1/UFD3 plays a role in sorting ubiquitinated membrane proteins into multivesicular bodies. J Biol Chem 283: 21599–21611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa Y, Yamanaka K, Saito‐Sasagawa Y, Ogura T (2010) Caenorhabditis elegans UBX cofactors for CDC‐48/p97 control spermatogenesis. Genes Cells 15: 1201–1215 [DOI] [PubMed] [Google Scholar]
- Soellner P, Quinlan RA, Franke WW (1985) Identification of a distinct soluble subunit of an intermediate filament protein: tetrameric vimentin from living cells. Proc Natl Acad Sci USA 82: 7929–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon V, Goldberg AL (1996) Importance of the ATP‐ubiquitin‐proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem 271: 26690–26697 [DOI] [PubMed] [Google Scholar]
- Stach L, Freemont PS (2017) The AAA+ ATPase p97, a cellular multitool. Biochem J 474: 2953–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thottakara T, Friedrich FW, Reischmann S, Braumann S, Schlossarek S, Krämer E, Juhr D, Schlüter H, van der Velden J, Münch J et al (2015) The E3 ubiquitin ligase Asb2β is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J Mol Cell Cardiol 87: 214–224 [DOI] [PubMed] [Google Scholar]
- Vale RD (2000) AAA proteins: lords of the ring. J Cell Biol 150: 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2019) N‐degron and C‐degron pathways of protein degradation. Proc Natl Acad Sci USA 116: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volodin A, Kosti I, Goldberg AL, Cohen S (2017) Myofibril breakdown during atrophy is a delayed response requiring the transcription factor PAX4 and desmin depolymerization. Proc Natl Acad Sci USA 114: E1375–E1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlever ML, Mateja A, McGilvray PT, Day KJ, Keenan RJ (2017) Msp1 is a membrane protein dislocase for tail‐anchored proteins. Mol Cell 67: 194–202.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mao Y (2020) AAA+ ATPases in protein degradation: structures, functions and mechanisms. Biomolecules 10: 629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Chi Z, Keuss MJ, Pai YME, Kang HC, Shin JH, Bugayenko A, Wang H, Xiong Y et al (2011) The AAA+ ATPase thorase regulates AMPA receptor‐dependent synaptic plasticity and behavior. Cell 145: 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv I, Fuchs Y, Preger E, Shabtay A, Harduf H, Zilpa T, Dym N, Ron D (2006) The human sef-a isoform utilizes different mechanisms to regulate receptor tyrosine kinase signaling pathways and subsequent cell fate. J Biol Chem 281: 39225–39235 [DOI] [PubMed] [Google Scholar]