

Abstract
Autophagy has emerged as the prime machinery for implementing organelle quality control. In the context of mitophagy, the ubiquitin E3 ligase Parkin tags impaired mitochondria with ubiquitin to activate autophagic degradation. Although ubiquitination is essential for mitophagy, it is unclear how ubiquitinated mitochondria activate autophagosome assembly locally to ensure efficient destruction. Here, we report that Parkin activates lipid remodeling on mitochondria targeted for autophagic destruction. Mitochondrial Parkin induces the production of phosphatidic acid (PA) and its subsequent conversion to diacylglycerol (DAG) by recruiting phospholipase D2 and activating the PA phosphatase, Lipin‐1. The production of DAG requires mitochondrial ubiquitination and ubiquitin‐binding autophagy receptors, NDP52 and optineurin (OPTN). Autophagic receptors, via Golgi‐derived vesicles, deliver an autophagic activator, EndoB1, to ubiquitinated mitochondria. Inhibition of Lipin‐1, NDP52/OPTN, or EndoB1 results in a failure to produce mitochondrial DAG, autophagosomes, and mitochondrial clearance, while exogenous cell‐permeable DAG can induce autophagosome production. Thus, mitochondrial DAG production acts downstream of Parkin to enable the local assembly of autophagosomes for the efficient disposal of ubiquitinated mitochondria.
Keywords: diacylglycerol, Lipin‐1, mitophagy, Parkin, PLD2
Subject Categories: Autophagy & Cell Death, Membranes & Trafficking, Metabolism
Parkin orchestrates mitophagy by coordinating mitochondrial lipid remodeling and sequential production of mitochondrial phosphatic acid and diacylglycerol.
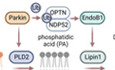
Introduction
Deterioration of organelle integrity, most notably mitochondria and lysosomes, is a prominent feature of aging and aging‐associated neurodegenerative diseases (Zhang et al, 2015; Nixon, 2020). The discovery that the ubiquitin E3 ligase Parkin, the causative gene of early‐onset Parkinson's disease (AR‐JP), ubiquitinates and thereby tags impaired mitochondria for autophagic destruction has established a paradigm for organelle quality control (QC; Geisler et al, 2010; Lee et al, 2010; Matsuda et al, 2010; Narendra et al, 2010). Similar to mitophagy, damaged lysosomes are also tagged by ubiquitin, followed by autophagic degradation (Hung et al, 2013; Maejima et al, 2013; Papadopoulos et al, 2017). Thus, organelle ubiquitination is a primary mechanism that enables the selective removal of damaged organelles by QC autophagy.
In mitophagy, ubiquitination serves at least two functions: marking mitochondrial outer membrane proteins, most notably mitofusins (MFN1 and MFN2), for proteasome‐mediated degradation (Tanaka et al, 2010; Chan et al, 2011). MFN degradation enables the production of smaller mitochondrial units for packaging into autophagosomes. Mitochondrial ubiquitination also recruits multiple ubiquitin‐binding autophagic receptors, including Optineurin (OPTN), NDP52, and p62/SQSTM1 (Lee et al, 2010; Wong & Holzbaur, 2014; Lazarou et al, 2015). Through their bivalent binding activity for ubiquitin and LC3‐ a key component of the autophagosome, these adaptor proteins are thought to bring together ubiquitinated cargos and autophagosomes (Pankiv et al, 2007). The simple autophagic receptor hypothesis, however, does not account for the findings that autophagosomes are synthesized “de novo” at mitochondria tagged for destruction (Itakura et al, 2012; Yang & Yang, 2013). Indeed, OPTN and NDP52 can also affect kinases, including TBK1 and Ulk1, important for autophagy (Richter et al, 2016; Vargas et al, 2019; Goodall et al, 2022). The critical question of how Parkin, mitochondrial ubiquitination, and autophagic receptors coordinate local autophagosome biogenesis to sequester condemned mitochondria remains to be fully elucidated.
In this report, we present evidence that mitochondrial lipids are integral components and effectors of Parkin‐mediated mitophagy. We showed that the recruitment of Parkin to depolarized mitochondria initiates a series of lipid remodeling events characterized by the sequential production of mitochondrial phosphatidic acid (PA) and diacylglycerol (DAG). Parkin binds and recruits phospholipase D2 (PLD2) to generate mitochondrial PA, while ubiquitin‐dependent recruitment of OPTN and NDP52 activates the PA phosphatase Lipin‐1, resulting in mitochondrial DAG production. We present evidence that OPTN and NDP52 deliver a lipid remodeling factor, EndoB1, to ubiquitinated mitochondria and activate DAG synthesis. Mitochondrial DAG, in turn, stimulates the local production of autophagosomes, providing an efficient mechanism for eliminating impaired mitochondria. Our findings uncover mitochondrial lipid remodeling and DAG production as key effectors of mitochondrial ubiquitination that activate local autophagosome assembly and imply a critical role of the Golgi complex in enforcing mitochondrial QC.
Results and Discussion
Parkin stimulates mitochondrial PA and DAG accumulation
Evidence suggests that mitochondria tagged by Parkin lose mitofusin and undergo fission to reduce their dimension in order to be sequestered by autophagosomes (Poole et al, 2008; Tanaka et al, 2010; Chan et al, 2011). Mitochondrial dynamics is also affected by membrane lipid composition, where mitochondrial phosphatidic acid (PA) and diacylglycerol (DAG) stimulate fusion and fission, respectively (Huang et al, 2011). We set out to investigate whether mitochondrial PA or DAG is involved in mitochondrial fission/fusion associated with Parkin‐mediated mitophagy. To this end, we employed specific fluorescent reporters that bind PA (EGFP‐Raf1‐PABD, referred to as PA reporter; Rizzo et al, 2000) or DAG (C1bδ‐CFP/YFP, referred to as YFP‐DAGR), which have been used extensively to monitor PA and DAG production in cells (Giorgione et al, 2006; Huang et al, 2011). The PA reporter showed diffused cytosolic distribution in Parkin‐expressing cells under basal conditions (Fig 1A, DMSO). When these cells were challenged by high‐dose carbonyl cyanide 5‐chloro‐phenylhydrazone (CCCP, 10 μM) to activate global mitophagy, prominent accumulation of the PA reporter on Parkin‐tagged mitochondria was observed as early as 1 h after CCCP treatment (Fig 1A). A different PA‐binding reporter (Spo20p‐GFP) (Kassas et al, 2012) showed a similar response to CCCP treatment (Fig EV1A). These findings indicate that PA accumulates on depolarized mitochondria tagged by Parkin.
Figure 1. Parkin stimulates PA and DAG accumulation on depolarized mitochondria.
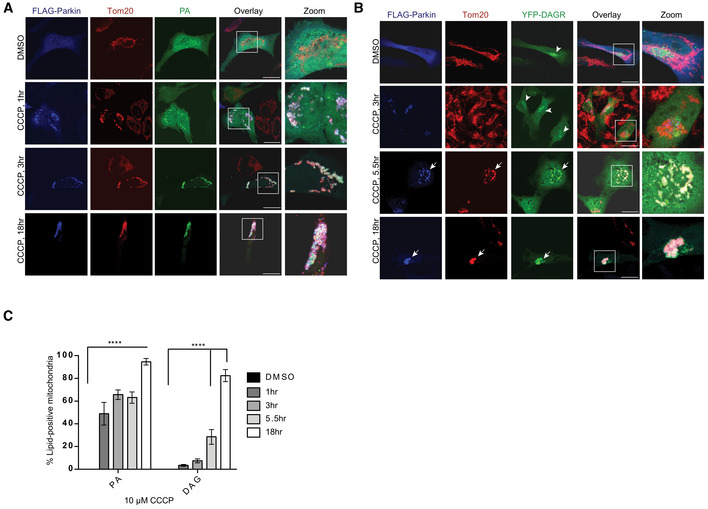
- PA accumulated on Parkin‐positive mitochondria after 1 h of 10 μM CCCP treatment, with a more dramatic increase by 3 h and 18 h. Scale bar = 25 μM, and zoom is 9×.
- Quantification of imaging experiments shown in (A) and (B). Bars represent mean with SEM from three biological independent experiments; two‐way ANOVA analysis was performed for statistical analysis (***P < 0.0001).
- DAG (diacylglycerol) accumulated on Parkin‐positive mitochondria after 5.5 h of 10 μM CCCP treatment. Arrowheads and arrows point to the DAGR concentration at the Golgi and mitochondria, respectively. Scale bar = 25 μM, and zoom is 9×.
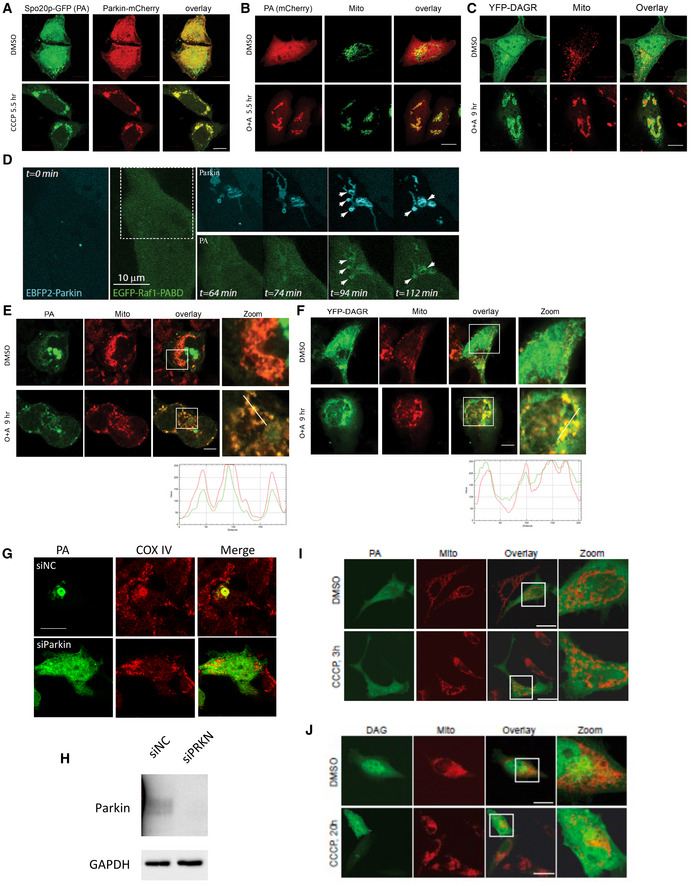
Figure EV1. Mitophagy activation stimulates mitochondrial PA and DAG production.
-
AHela cells were transfected with the Spo20p‐GFP PA reporter and mCherry‐Parkin followed by CCCP treatment for 5.5 h. Note that CCCP treatment led to the recruitment of Spo20p‐GFP to Parkin‐positive mitochondria. Scale bar = 10 μM.
-
B, CHeLa cells were transfected with Raf1‐PABD‐mCherry PA reporter or YFP‐DAGR with Parkin‐FLAG followed by antimycin (4 μM) and oligomycin (10 μM; O + A) treatment to activate mitophagy. Note that Raf1‐PABD‐mCherry PA reporter (B) and YFP‐DAGR (C) both translocated to mitochondria (TOM20) after the treatment. Scale bar = 10 μM.
-
DHela cells expressing KR‐dMito and EBFP2‐Parkin (cyan) were illuminated at 559 nm to induce mitophagy. Images were acquired at indicated time points after illumination. Note that PA accumulation on Parkin‐positive mitochondria is induced by photodamage (t = 94 min and 112 min). Arrowheads point to the co‐accumulation of Parkin and the PA reporter on damaged mitochondria. Scale bar = 10 μM.
-
E, FSH‐SY5Y cells were transfected with (E) the Raf1‐PABD‐GFP PA reporter or (F) YFP‐DAGR followed by antimycin (4 μM) and oligomycin (10 μM) or DMSO treatment, as indicated. Both reporters became enriched in the mitochondria (TOM20). Line scan analysis (Image J software) under the images indicates colocalization between the lipid reporters (green) and mitochondria (red) corresponding to the lines drawn in the images. Scale bar = 10 μm.
-
GSh‐Sy5Y cells were transfected with control (siNC) or Parkin siRNA (siParkin) followed by the PA‐GFP reporter and treated with CCCP (5.5 h). Note that Parkin KD prevented mitochondrial (COX IV) aggregation and PA concentration. Scale bar = 10 μM.
-
HImmunoblotting confirmed Parkin KD's efficiency.
-
I, JHela cells were transfected with PA reporter and DAGR‐YFP without Parkin, by CCCP treatment, as indicated. Note that there is no accumulation of PA and DAGR reporters on mitochondria after CCCP treatment (by a Tom20 antibody). Scale bar = 10 μM.
To assess whether PA on Parkin‐tagged mitochondria is further processed to DAG, we employed a high‐affinity DAG reporter (YFP‐DAGR). Under basal conditions, YFP‐DAGR mainly labeled perinuclear structures corresponding to the Golgi apparatus, which contains abundant DAG (Baron & Malhotra, 2002) (Fig 1B, arrowheads). Upon mitophagy activation, dramatic accumulation of YFP‐DAGR on Parkin‐tagged mitochondria was observed ~ 5.5 h after CCCP treatment (Fig 1B, arrows), a kinetic slower than that of the PA receptor (Quantified in Fig 1C). Mitophagy activated by different methods, such as antimycin/oligomycin treatment (Fig EV1B and C) or focal photodamage (Fig EV1D), similarly induced concentration of the PA or DAG reporter on mitochondria. Mitophagy‐induced mitochondrial translocation of the PA and DAG reporter was also detected in SH‐SY5Y cells expressing endogenous Parkin, and this accumulation was suppressed in Parkin knockdown cells (Fig EV1E–H). Indeed, neither reporter accumulated on mitochondria in the absence of Parkin (Fig EV1I and J). The lipid‐reporter assays indicate that PA and DAG are produced and accumulate on impaired mitochondria tagged by Parkin.
PLD2 and Lipin‐1 catalyze sequential production of mitochondrial PA and DAG
Phosphatidic acid is primarily produced by phospholipase D (PLD) from membrane phospholipids. We investigated whether PLD1 and PLD2, the two principal PLD members, are involved in mitochondrial PA production. Immuno‐localization analysis showed that PLD2, but not PLD1, translocated from the cytosol to Parkin‐tagged mitochondria induced by CCCP (Fig 2A) or oligomycin/antimycin treatment (Fig EV2A and B). Immunoblotting confirmed that endogenous PLD2, along with Parkin, became enriched in the mitochondrial fractions after CCCP treatment (Fig 2B). Co‐immunoprecipitation indicates that Parkin interacts with PLD2, suggesting a mechanism by which PLD2 is recruited to Parkin‐tagged mitochondria (Fig EV2C). To investigate whether PLD2 is responsible for mitochondrial PA production, we inhibited PLD2 by a chemical inhibitor, VU 0364739 (Lavieri et al, 2010), or an siRNA that effectively suppressed PLD2 (Fig EV2G). Both treatments significantly suppressed mitochondrial accumulation of the PA reporter (> 3‐fold; Figs 2C and D, and EV2D and E). These findings indicate that Parkin recruits PLD2 to catalyze mitochondrial PA production.
Figure 2. PLD2 and lipin1 work in tandem to stimulate mitochondrial PA and DAG production.
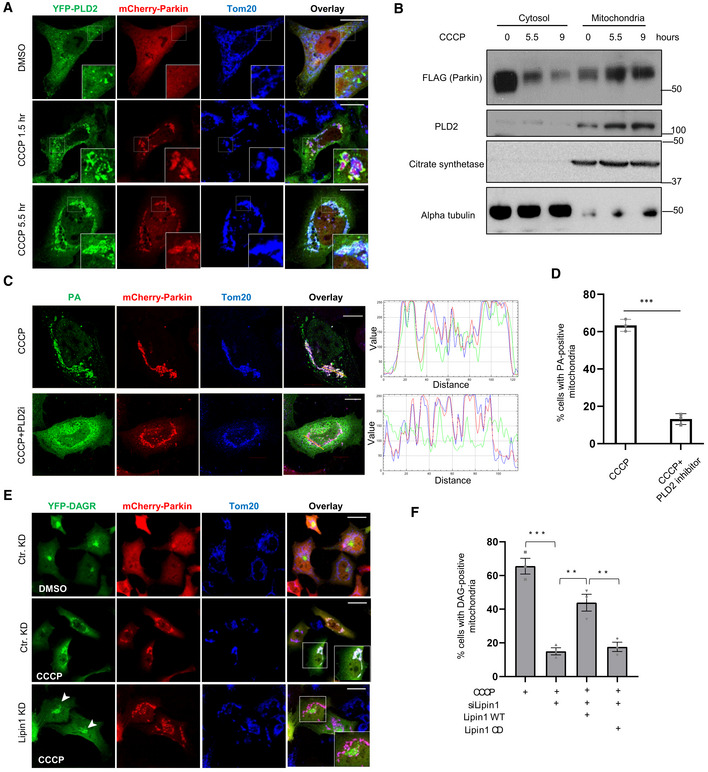
- HeLa cells were transfected with expression plasmids for Parkin (mCherry‐Parkin, Red) and YFP‐PLD2 (Green) and treated with DMSO or 10 μM CCCP for the indicated time. Mitochondria were visualized via anti‐Tom20 staining (blue). Note that YFP‐PLD2 accumulated on Parkin‐positive mitochondria after CCCP treatment. Scale bar = 25 μM, and zoom is 9×.
- HEK‐293T cells were transfected with Parkin‐FLAG and DAGR‐YFP followed by CCCP treatment for indicated times. Mitochondrial and cytosolic fractions were purified and analyzed by Western blots by indicated antibodies. Citrate synthetase and α‐tubulin were used as a mitochondrial and cytosolic marker, respectively. Note that PLD2 and Parkin levels were elevated in the mitochondrial fraction in response to CCCP treatment.
- Hela cells were co‐transfected with mCherry‐Parkin and the PA reporter, followed by CCCP treatment alone or with a PLD2 inhibitor VU 0364739 (3 μM) for 5.5 h. Line scan analysis (Image J software) corresponding to the line drawn in the images indicates colocalization between the PA reporter (green) and mitochondria (red). Note that VU 0364739 suppressed mitochondrial PA accumulation. Scale bar = 10 μM.
- Quantification of the numbers of cells with the PA reporter‐positive mitochondria shown in (C). Asterisks indicate statistical significance (***P < 0.001, Student's t‐test) from three biological independent experiments. The bars indicate mean ± SEM.
- Hela cells were transfected with a lipin1 siRNA, followed by the DAG reporter and mCherry‐Parkin, treated with CCCP (10 μM) for 9 h, and then subject to image analysis. Scale bar = 25 μm, and zoom is 4×. Lipin‐1 KD reduced mitochondrial DAGR, but not Golgi‐DAGR, accumulation (arrowheads).
- The numbers of cells with DAG reporter‐positive mitochondria shown in (E) and in Lipin‐1 knockdown cells transfected with siRNA‐resistant wild‐type (WT) and catalytically dead (CD; D712E; D714E) mutant cDNA. Note that Lipin1 KD reduced mitochondrial DAG accumulation, which can be significantly restored by the re‐expression of WT, but not CD mutant, Lipin‐1. The graph shows the means with SEM (error bars) from three biological independent experiments. Asterisks indicate statistical significance by one‐way ANOVA (**P < 0.01, ***P < 0.001).
Figure EV2. PLD1 is not translocated to mitochondria, and Lipin‐1 is dispensable for PA production.
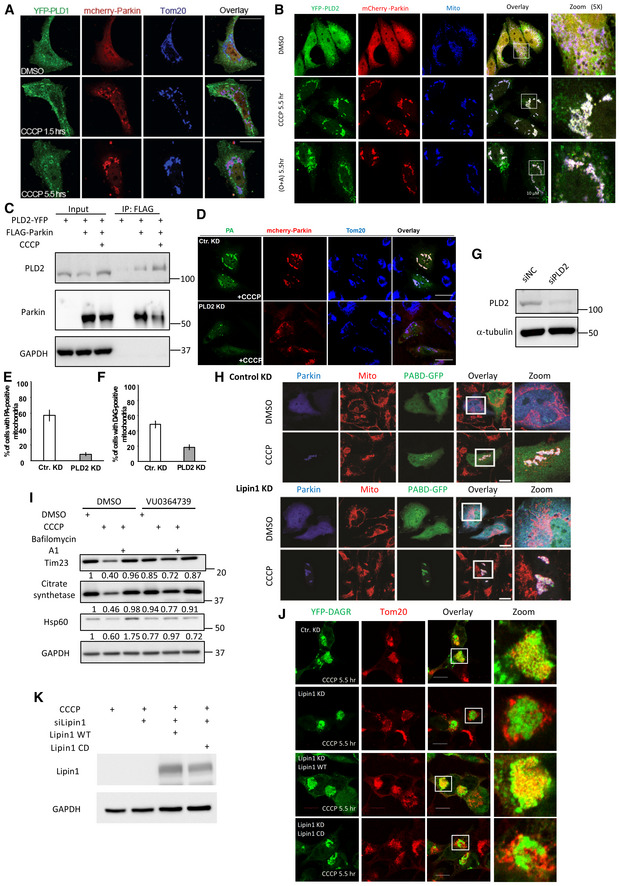
-
AHela cells were transfected with YFP‐PLD1 and mCherry‐Parkin followed by CCCP (10 μM) treatment at indicated time points. Note that PLD1 is not translocated to mitochondria upon CCCP treatment. Scale bar = 10 μM.
-
BHela cells were transfected with YFP‐PLD2 and mCherry‐Parkin, followed by CCCP or oligomycin/antimycin treatment for the indicated time. Note that PLD2 became colocalized with Parkin and mitochondria (TOM20) under both treatment conditions. Scale bar = 10 μM.
-
CHEK‐293T cells expressing PLD2‐YFP and FLAG‐Parkin cDNA were treated with CCCP for 5.5 h. Parkin was then pulled down by a FLAG antibody from cell lysates and blotted with a PLD2 antibody. Note that PLD2 interaction with Parkin was enhanced by CCCP.
-
D–GHela cells were transfected with siRNA for PLD2 followed by expression plasmids for mCherry‐Parkin and the PA or GFP reporter, and subject to DMSO or CCCP treatment, as indicated. PLD2 knockdown inhibited both mitochondrial PA reporter (D‐E) and DAG reporter accumulation (F). n = 3 biological replicated. The bars indicate mean ± SEM. PLD2 knockdown efficiency was confirmed by immunoblotting (G).
-
HHela cells were transfected with Lipin‐1 siRNA followed by plasmids for FLAG‐Parkin and the PA reporter and treated with DMSO or CCCP, as indicated. Note that PA accumulated on mitochondria (TOM20) in Lipin‐1 knockdown cells. Scale bar is 25 μM, and zoom is 3×.
-
IHela cells stably expressing mCherry‐Parkin were treated with PLD2 inhibitor (VU0364739, 3 μM) and subjected to CCCP (10 μM) and Bafilomycin A1 (0.1 μM, lysosomal inhibitor) for 18 h. Note that PLD2 inhibition rescued mitochondrial protein degradation. The band intensity of mitochondrial proteins relative to control untreated conditions was determined by the Image J. software.
-
JHela cells were transfected with Lipin‐1 siRNA followed by plasmids for FLAG‐Parkin, DAGR‐YFP, and Lipin‐1‐WT or Lipin‐1‐catalytic dead (CD) mutant and treated with CCCP (5.5 h), as indicated. Note that Lipin‐1‐WT, but not the CD mutant, restored mitochondrial DAGR‐YFP accumulation induced by CCCP. Scale bar: 10 mm.
-
KLipin1 knockdown and overexpression efficiency was confirmed by immunoblotting (G).
Phospholipase D2 inhibition also markedly inhibited mitochondrial YFP‐DAGR accumulation (Fig EV2F), indicating that mitochondrial PA is converted to DAG, a reaction mainly catalyzed by a lipid phosphatase, Lipin‐1 (Han et al, 2006; Huang et al, 2011). Indeed, Lipin‐1 knockdown led to a dramatic loss of DAGR‐YFP from CCCP‐induced Parkin‐tagged mitochondria (Fig 2E, Lipin‐1 KD) without affecting Golgi‐DAG (Fig 2E, arrowheads). Lipin‐1 KD did not inhibit the mitochondrial accumulation of the PA reporter (Fig EV2H). Importantly, the re‐expression of a siRNA‐resistant wild‐type (WT) Lipin‐1, but not an inactive catalytic mutant (Lipin‐1 CD), significantly restored mitochondrial DAGR‐YFP accumulation (Figs 2F and EV2J and K). Together, these findings support the proposal that PLD2 and Lipin‐1 work in tandem to generate PA and DAG on impaired mitochondria tagged by Parkin.
Lipin1‐dependent DAG is required for efficient mitophagy and mitophagosome production
To determine the role of mitochondria DAG, we knocked down Lipin‐1 and assessed mitophagy. Compared with control cells (Ctr KD), Lipin‐1 knockdown cells were less efficient in mitochondrial clearance, as shown by the retention of mitochondria following CCCP treatment (Fig 3A, quantified in B). Immunoblotting analysis confirmed that autophagy‐dependent degradation of multiple mitochondrial markers was inhibited in Lipin‐1 KD cells (Fig 3C). PLD2 inhibition, which suppressed the production of mitochondrial PA and DAG, also reduced the efficiency of mitophagy (Fig EV2I). Collectively, these results indicate that mitochondrial DAG production is required for efficient mitophagy.
Figure 3. Lipin‐1‐mediated mitochondrial DAG production is required for mitophagosome production.
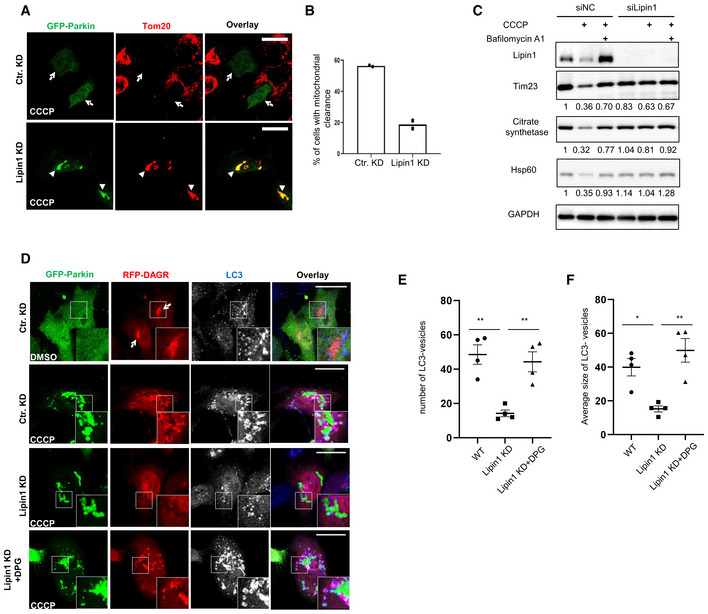
-
A, BHela cells were transfected with a Lipin1 or control (cKD) siRNA followed by a GFP‐Parkin expression plasmid. Transfected cells were treated with CCCP at 10 μM for 18 h, and subject to immunostaining. (B) Quantification of Parkin‐positive cells in (A) that have lost a majority of mitochondria (marked by TOM20, arrows). Note that mitochondrial clearance is reduced in Lipin‐1 knockdown cells (arrowheads in (A)). Scale bar = 25 μM.
-
CControl or lipin1 knockdown Hela cells stably expressing parkin‐mCherry were treated with DMSO or CCCP (10 μM for 18 h) and Bafilomycin A1 (1 μM, lysosomal inhibitor) treatment, followed by immunoblotting with indicated antibodies and quantified by the Image J. software. Note that lipin1 silencing suppressed CCCP‐induced mitochondrial protein degradation. Lipin‐1‐mediated mitochondrial DAG production is required for mitophagosome production.
-
D–FHela cells were transfected with control or a Lipin1 siRNA followed by the expression plasmids of GFP‐Parkin and RFP‐DAGR. Transfected cells were treated with DMSO or CCCP at 10 μM for 9 h, with or without further incubation with 1,2‐Dipalmitoyl‐sn‐glycerol (DPG) at 100 μM, as indicated. Autophagosome formation was assessed by an LC3 antibody (pseudocolored in white in single‐channel and blue in the overlay images). Arrows point to DAGR concentration at the Golgi under basal conditions. The number (E) and size (F, arbitrary unit) of LC3 vesicles were quantified. n = 4 biological replicates. The bars indicate mean ± SEM. Asterisks indicate statistical significance by one‐way ANOVA (*P < 0.05, **P < 0.01). Scale bar = 25 μM and zoom is 5×. Note that both the number and size of LC3‐vesicles in Lipin‐1 knockdown cells were much smaller than those in WT cells, and these defects were corrected by DPG treatment.
We noticed that YFP‐DAGR on mitochondria appeared to retard mitochondrial clearance, as indicated by the continuous accumulation of YFP‐DAGR‐positive mitochondria after prolonged CCCP treatment (Fig 1C, 18 h). This finding is consistent with the notion that high‐affinity DAGR can mask mitochondrial DAG from the downstream effectors. To follow the fate of DAG at a later stage of mitophagy, we employed another DAG reporter with a lower binding affinity, C1(2)‐mRFP (referred to as RFP‐DAGR; Giorgione et al, 2006; Adachi et al, 2009). Like YFP‐DAGR, RFP‐DAGR mainly labeled the Golgi complex under basal conditions (Fig 3D, top panels, arrowhead). However, unlike the predominant mitochondrial concentration of YFP‐DAGR upon CCCP treatment, RFP‐DAGR labeled many vesicle‐like structures that encompassed dispersed Parkin‐tagged mitochondria (Fig 3D, second Panels). We noted that the YFP‐DAGR reporter also labeled similar structures when expressed at a lower concentration (Fig EV3). The DAGR vesicles with internal mitochondria are reminiscent of autophagosomes. We, therefore, immunostained for the autophagosome marker LC3. This analysis demonstrated extensive co‐localization of LC3 with RFP‐DAGR (Fig 3D, second Panels) and YFP‐DAGR (Fig EV3), indicating that DAG‐positive vesicles are autophagosomes with sequestered mitochondria (i.e., mitophagosomes). Consistent with this proposal, live cell imaging showed that DAGR‐positive vesicles formed around and sequestered Parkin‐tagged mitochondria (Movie EV1). Significantly, CCCP‐induced RFP‐DAGR vesicles and LC3 vesicles were suppressed in Lipin‐1 KD cells (Fig 3D, third panels; quantified in Fig 3E). These results indicate that Parkin‐induced and Lipin‐1‐dependent DAG activates mitophagosome biogenesis. To test this hypothesis, we treated Lipin‐1 KD cells with a cell‐permeable DAG, 1,2‐dipalmitoyl‐sn‐glycerol (DPG) (Zhang et al, 2014). As shown in Fig 3D, DPG supplement (+DPG panels) markedly stimulated the production of LC3‐positive vesicles, many of which contained mitochondria. DPG treatment increased both the number and size of autophagosomes in Lipin‐1 KD cells (Quantified in Fig 3E and F). Collectively, these findings indicate that Lipin‐1‐dependent DAG production stimulates the formation of autophagosomes that sequester mitochondria.
Figure EV3. RFP‐DAGR and YFP‐DAGR can label similar Golgi and mitophagosome structures.
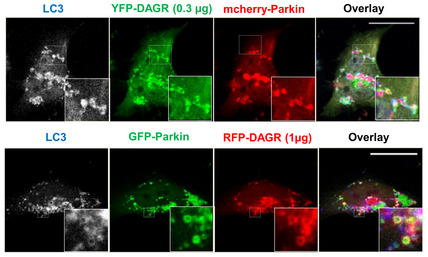
Hela cells were transfected with mCherry‐Parkin and 0.3 μg of YFP‐DAGR (top panels) or GFP‐Parkin and 1 μg of RFP‐DAGR (bottom panels), followed by CCCP treatment. The formation of autophagosomes was assessed by immunostaining with an LC3 antibody. Note that, similar to RFP‐DAGR, under lower expression levels, YFP‐DAGR also surrounded LC3‐positive and Parkin‐tagged mitochondria. Scale bar is 25 μM, and zoom is 5× (top panel), 10× (bottom panel).
Mitochondrial DAG production requires ubiquitin‐binding autophagy receptors
Parkin activates mitophagy by catalyzing mitochondrial ubiquitination (Lee et al, 2010; Vives‐Bauza et al, 2010). To determine whether mitochondrial ubiquitination regulates mitochondrial lipid remodeling, we assessed Parkin‐T240R and T415N mutants‐ two PD mutations deficient in catalyzing mitochondrial ubiquitination and mitophagy (Lee et al, 2010). As shown in Fig 4A, although Parkin‐T240R and Parkin‐T415N were active in promoting mitochondrial PA reporter translocation, both mutants were deficient in mobilizing the YFP‐DAGR reporter to mitochondria in response to CCCP (Fig 4B, quantified in C). These results indicate that DAG production requires Parkin‐mediated mitochondrial ubiquitination.
Figure 4. Requirement of OPTN, NDP52 for DAG production and exogenous DAG restores mitophagy.
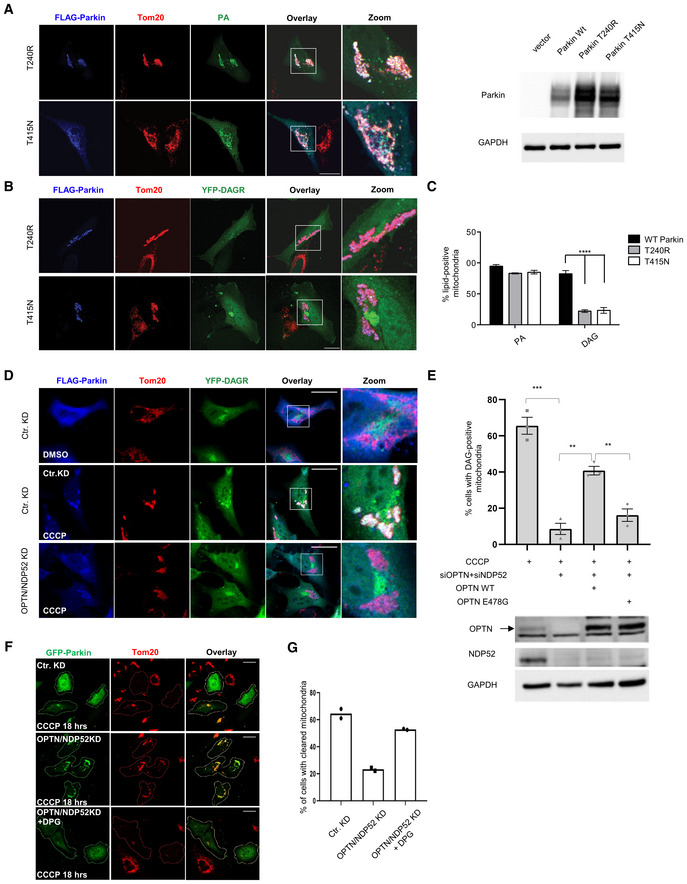
-
AE3 ligase‐deficient Parkin mutants (T240R and T415N) have no effect on PA accumulation (green) on depolarized, Parkin‐positive mitochondria. The expression of WT and Parkin mutants was determined by Immunoblotting. Scale bar = 25 μM, and zoom is 3×.
-
BDAG accumulation depends upon intact Parkin E3 ligase activity. Scale bar = 25 μm, and zoom is 3×.
-
CQuantification of imaging experiments shown in (A) and (B). Bars represent the mean with SEM; two‐way ANOVA analysis was performed for statistical analysis (***P < 0.001). n = 3 biological replicates.
-
DHela cells were transfected with control or optineurin‐ and NDP52‐ siRNAs and expression plasmids of FLAG‐Parkin and YFP‐DAGR. These cells were treated with DMSO or 10 μM CCCP for 5.5 h, as indicated. Note that OPTN/NDP52 double knockdown prevented mitochondrial YFP‐DAGR accumulation. (Scale bar = 25 μM and zoom is 3×).
-
EHela OPTN/NDP52 knockdown cells were transfected with a siRNA‐resistant wild‐type or ubiquitin‐binding deficient E478G OPTN mutant, as indicated. The percentage of cells with mitochondrial YFP‐DAGR following CCCP treatment was quantified. Asterisks indicate statistical significance by one‐way ANOVA (**P < 0.01, ***P < 0.001) from three biological independent experiments. The bars indicate mean ± SEM. Bottom panel: Expression of OPTN, OPTN E478, and NDP52 was determined by immunoblotting.
-
F, GHela OPTN/NDP52 knockdown cells were incubated with 1,2‐Dipalmitoyl‐sn‐glycerol (DPG) followed by CCCP treatment for 18 h. Mitophagy efficiency was assessed by TOM20 staining and quantified in (G) (n = 2 biological replicates). Note that DPG significantly restored mitophagy in OPTN/NDP52 knockdown cells. Scale bar = 10 μM.
Mitochondrial ubiquitin recruits ubiquitin‐binding autophagic receptors, including OPTN and NDP52 (Wong & Holzbaur, 2014; Lazarou et al, 2015). We next determined whether OPTN and NDP52 are involved in mitochondrial DAG production. We found that although efficient knockdown of OPTN and NDP52 individually had no effect (Fig EV4A), double knockdown abrogated mitochondrial YFP‐DAGR (Figs 4D and EV4B and C)‐ a finding that is consistent with the reported redundant functions of OPTN and NDP52 in mitophagy (Lazarou et al, 2015). Importantly, the re‐expression of a siRNA‐resistant WT OPTN, but not a ubiquitin‐binding deficient (E478G) mutant OPTN (Wong & Holzbaur, 2014), significantly restored mitochondrial DAGR recruitment in OPTN/NDP52 KD cells (Figs 4E and EV4D). Importantly, exogenous DPG treatment markedly increased mitophagy in OPTN/NDP52 KD cells (Fig 4F and G). These findings show that a critical function of OPTN and NDP52 in mitophagy is to promote mitochondrial DAG production.
Figure EV4. The effect of optineurin (OPTN) or NDP52 single knockdown on mitochondrial DAG production and double knockdown on mito‐autophagosome (mitophagosome) formation.
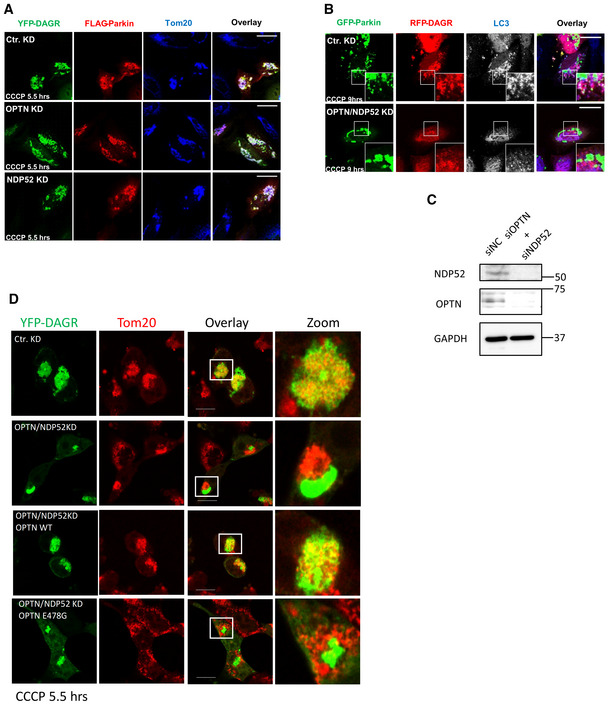
- Hela cells were transfected with control siRNA, OPTN‐siRNA, or NDP52‐siRNA. Knockdown cells were subsequently transfected with the DAGR‐YFP reporter and FLAG‐Parkin, followed by CCCP treatment. Note that DAGR accumulated on mitochondria (Tom20) in OPTN and NDP52 single knockdown cells. Scale bar = 25 μM.
- OPTN and NDP52 double knockdown cells were transfected with GFP‐Parkin and RFP‐DAGR, followed by CCCP treatment (10 μM) for 9 h. Autophagosomes were assessed by immunostaining with an LC3 antibody. Note that DAG‐positive LC3 vesicle production was reduced in OPTN and NDP52 double knockdown cells. Scale bar = 25 μM, and zoom is 5×.
- Knockdown efficiency for OPTN and NDP52 was confirmed by immunoblotting.
- Hela cells were transfected with OPTN‐siRNA and NDP52‐siRNA followed by plasmids for FLAG‐Parkin, DAGR‐YFP, and OPTN‐WT or OPTN‐E478G mutant and treated with CCCP (5.5 h), as indicated. Note that OPTN‐WT, but not the ubiquitin‐binding deficient E478G mutant, restored mitochondrial DAGR‐YFP accumulation induced by CCCP. Scale bar: 10 mm.
Mitochondrial DAG production requires Endophilin B1 (EndoB1)
Although OPTN and NDP52 are widely known as autophagic receptors, they were initially identified as perinuclear Golgi‐associated factors that regulate Golgi vesicle transport (Sahlender et al, 2005; Morriswood et al, 2007). These findings suggest that OPTN and NDP52 might deliver Golgi factor(s) to affect mitochondrial DAG production. We searched for potential Golgi factor(s) that are known to modify membrane lipid composition and regulate mitophagy and identified Endophilin B1 (EndoB1, also known as Bif‐1), a Golgi‐associated protein that possesses an intrinsic PA‐binding activity (Zhang et al, 2011) and can affect mitophagy (Takahashi et al, 2013). We found that EndoB1 was concentrated on the perinuclear OPTN‐positive vesicles under basal conditions (Fig 5A, top panels). In response to CCCP treatment, endogenous EndoB1 became co‐localized with Parkin‐ and OPTN‐tagged mitochondria (Fig 5A, bottom panels) and enriched in the mitochondrial fractions (Fig 5B). Further supporting EndoB1 as a potential cargo delivered to mitochondria by OPTN/NDP52, mitochondrial recruitment of EndoB1 was markedly reduced in OPTN/NDP52 KD cells (Fig 5C). Importantly, efficient EndoB1 knockdown by a siRNA (Fig EV5C) significantly suppressed the mitochondrial accumulation of YFP‐DAGR (Fig 5D and E) and the production of DAGR‐positive LC3 vesicles (Fig EV5B) but did not affect the PA reporter (Fig EV5A). These findings suggest that EndoB1 is a cargo and effector of OPTN and NDP52 that promotes mitochondrial DAG production and mitophagy.
Figure 5. OPTN and NDP52‐dependent EndoB1 mitochondrial recruitment is required for mitochondrial DAG production.
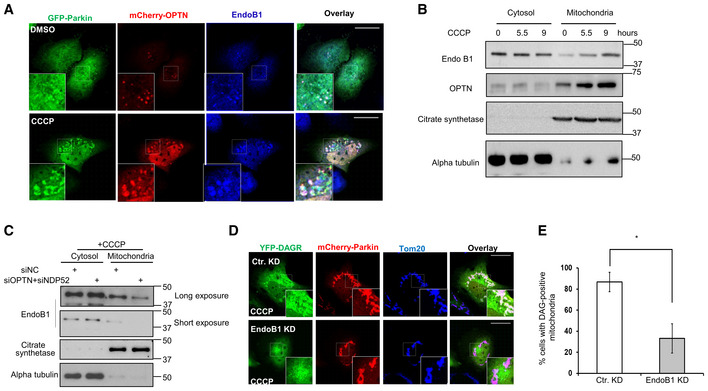
-
AHela cells were co‐transfected with expression plasmids for mCherry‐OPTN and GFP‐Parkin and treated with DMSO or CCCP for 5.5 h. Mitochondria were visualized by immunostaining with a Tom20 antibody and EndoB1 localization was assessed by an EndoB1 antibody. Note that EndoB1 (Blue) translocates to Parkin‐ and OPTN‐tagged mitochondria in response to CCCP treatment. Scale bar = 25 μM, and zoom is 9×.
-
BMitochondrial and cytosolic fractions obtained from control and CCCP treated cells, as described and analyzed in Fig 2B, were immunoblotted for EndoB1 and OPTN, as indicated.
-
CCytosolic and mitochondrial fractions purified from control and OPTN/NDP52 knockdown cells treated with CCCP were immunoblotted with indicated antibodies. Note that mitochondrial EndoB1 levels were reduced in OPTN/NDP52 knockdown cells.
-
D, EHela cells were transfected with an EndoB1 siRNA, followed by the expression plasmids for mCherry‐Parkin and YFP‐DAGR, and CCCP treatment (10 μM) for 9 h. Cells with mitochondrial YFP‐DAGR were quantified in (E) (*P < 0.05, Student's t‐test, n = 3 biological replicates). Scale bar = 25 μM and zoom is 5×. Note that knockdown of EndoB1 suppressed mitochondrial DAG production.
Figure EV5. Effect of EndoB1 knockdown on mitochondrial PA and mito‐autophagosome (mitophagosome) production.
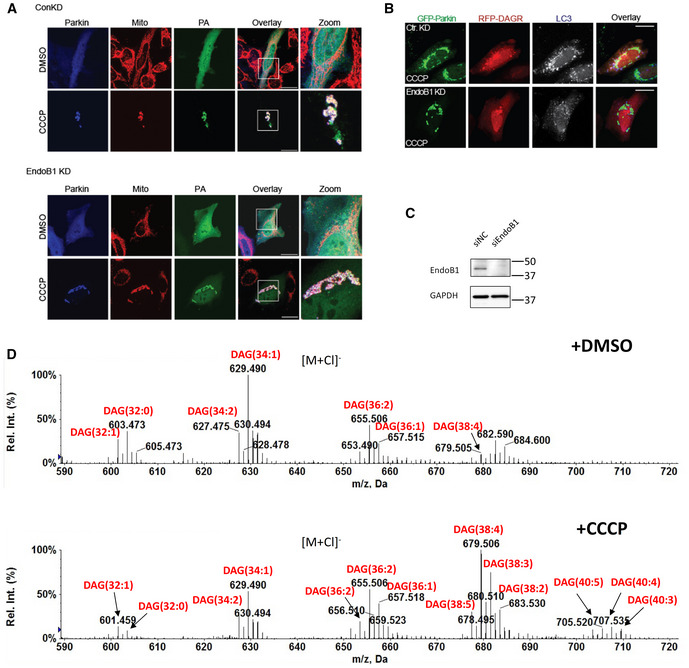
- EndoB1 knockdown cells were transfected with FLAG‐Parkin and the PA reporter, followed by CCCP treatment. Mitochondria was assessed by immunostaining with a Tom20 antibody. Note the prominent accumulation of the PA reporter (PA) on mitochondria in EndoB1 knockdown cells. Scale bar = 25 μM and zoom is 3×.
- EndoB1 knockdown cells were transfected with GFP‐Parkin and a DAG reporter RFP‐DAGR, followed by CCCP treatment. Autophagosomes were assessed by immunostaining with an LC3 antibody. Note that the abundance of DAG‐positive LC3 vesicles was reduced in EndoB1 knockdown cells. Scale bar = 25 μM.
- Control and EndoB1 KD cells were subjected to immunoblotting by EndoB1 and GAPDH antibodies to confirm knockdown efficiency.
- Mitochondria purified from control, and CCCP treated (5 h) Parkin‐expressing HeLa cells were subjected to the lipidomic analysis by LC/MS. CCCP treatment resulted in the increase and alteration of fatty acyl compositions of mitochondrial DAGs. Most notably, the levels of DAGs containing longer, polyunsaturated fatty acids (e.g., C38:4 and C38:3 as well as 36:2 and 36:1) are drastically increased after CCCP treatment. Negative ion mass spectra showing the [M + Cl]− ions of DAGs in mitochondria isolated from cells treated with either DMSO or CCCP. The numbers of total acyl chain carbon atoms and double bonds of DAGs are denoted in parentheses.
In organelle QC autophagy, protein ubiquitination plays a central role in targeting damaged organelles for autophagic destruction. However, the mechanism by which ubiquitination activates autophagy is not known. In this report, we have identified mitochondrial lipid remodeling as a novel effector that activates autophagy locally on mitochondria tagged for destruction. Our evidence indicates that mitochondrial Parkin orchestrates sequential production of PA and DAG to enable local autophagosome production. Autophagy‐activating lipid remodeling involves at least two enzymes, whereby mitochondrial recruitment of PLD2 by Parkin catalyzes mitochondrial PA production. At the same time, ubiquitin‐binding autophagic receptors OPTN/NDP52 and Golgi‐derived EndoB1 activate Lipin‐1‐dependent PA to DAG conversion. Our analysis revealed that one essential function of Parkin‐dependent DAG is to promote mitophagosome production. Supporting the instructive role of DAG in autophagosome biogenesis, a cell‐permeable DAG (DPG) can stimulate LC3‐positive vesicle production in Lipin‐1 KD cells (Fig 3). Our study indicates that local production of DAG on Parkin‐ and ubiquitin‐tagged mitochondria activates de novo autophagosome production, thereby ensuring “condemned” mitochondria are efficiently “restricted” by autophagosomes.
Our analysis indicates that mitochondrial ubiquitination activates local DAG production by recruiting ubiquitin‐binding OPTN and NDP52 (Fig 4). Importantly, autophagosome assembly and mitophagy defects in OPTN/NDP52 double knockdown cells can be significantly reversed by exogenous DAG (Fig 4F and G). Thus, a key function of OPTN/NDP52 in mitophagy is stimulating DAG production. Interestingly, OPTN/NDP52, known as LC3‐binding autophagic receptors, can recruit LC3 to damaged mitochondria without directly binding LC3 (Padman et al, 2019). Based on our findings, we speculate that OPTN/NDP52 could indirectly recruit LC3 by activating DAG‐dependent mitophagosome assembly on ubiquitinated mitochondria‐ an activity that a cell‐permeable DAG can mimic.
We identified Golgi‐associated EndoB1 as an effector of OPTN/NDP52 required for mitochondrial DAG and LC3 vesicle production (Figs 5 and EV5B). As we have not been able to detect Lipin‐1 on Parkin‐tagged mitochondria, we speculate that Lipin‐1 affects mitochondrial DAG production via mitochondrial EndoB1, whose recruitment to mitochondria depends on OPTN and NDP52. Supporting these findings, aberrant accumulation of mitochondria was observed in Lipin‐1 and EndoB1 knockout mice (Takahashi et al, 2013; Zhang et al, 2014). Importantly, EndoB1 was previously shown to activate the autophagy initiating PI3 kinase, Vps34, by an unknown mechanism (Takahashi et al, 2007). Our study suggests that Golgi‐derived EndoB1 activates Vps34 and autophagy via DAG production. Consistent with this proposal, Vps34‐dependent recruitment of PI3P‐binding WIPI1 and DFPC1 to Parkin‐tagged mitochondria is significantly diminished in OPTN/NDP52 knockout cells (Lazarou et al, 2015). Whether OPTN/NDP52 or EndoB1 also utilize DAG to regulate additional factors critical for mitophagy, for example, Ulk1 (Vargas et al, 2019) and TBK1 (Richter et al, 2016), is a crucial question that requires further investigation. It is notable that OPTN, NDP52, and EndoB1 are all associated with perinuclear Golgi complex/vesicles (Sahlender et al, 2005; Morriswood et al, 2007). EndoB1 is required for the fission of Golgi membrane induced by starvation (Takahashi et al, 2011). As mitochondria tagged by Parkin frequently form perinuclear aggregates before sequestration by autophagosomes (Lee et al, 2010; Vives‐Bauza et al, 2010; Chan et al, 2011), perinuclear concentration might grant damaged mitochondria better access to the Golgi OPTN/NDP52/EndoB1 vesicles for efficient autophagic processing. Indeed, growing evidence supports the critical role of the Golgi complex in autophagy and neurodegeneration (Martinez‐Menarguez et al, 2019; De Tito et al, 2020). However, the involvement of non‐Golgi pools of EndoB1, OPTN, and NDP52 in mitophagy cannot be excluded.
How mitochondrial DAG activates mitophagy remains to be determined. By mass spectrometry‐based lipid analysis, we have detected a marked accumulation of selected DAG species on purified mitochondria tagged for autophagy (Fig EV5D). This finding not only confirms mitochondrial DAG production during mitophagy but also reveals a preferential accumulation of DAG species with longer acyl chains (C38:4 and C38:3 as well as 36:2 and 36:1) compared with shorter acyl chains (C32 and C34; Fig EV5D). C38:4 DAG (1‐stearoyl‐2‐arachidonoyl‐sn‐glycerol; SAG) and C36:2 (1‐stearoyl‐2‐linoleoyl‐sn‐glycerol; SLG) are both potent PKC activators (Madani et al, 2001). Intriguingly, PKC regulates autophagy in yeast (Shahnazari et al, 2010). DAG also activates PKD and PKD activity was reduced in the skeletal muscle of Lipin‐1 knockout mice (Zhang et al, 2014). Whether PKC or PKD are effectors of mitochondrial DAG requires future studies. We noted that mitophagosomes are labeled by the DAGR (Figs 3D and EV3), indicating that autophagosomes are also enriched for DAG. The function of autophagosomal DAG and its relationship to mitochondrial DAG remains to be determined. However, because both types of DAG depend on Lipin‐1 for production (Figs 2E and F, and 3D–F), we speculate that mitochondrial DAG is transferred onto autophagosomes, where DAG might have additional roles in autophagosome maturation.
In conclusion, our study has uncovered mitochondrial lipid remodeling and DAG production as new effectors in the autophagic clearance of impaired mitochondria. We found that photo‐damaged lysosomes targeted for autophagic clearance (Hung et al, 2013) also accumulated the PA reporter (W.Y.Y, Unpublished observation). Furthermore, DAG accumulation was reported on salmonella tagged by LC3 (Shahnazari et al, 2010). Thus, local lipid remodeling might play a broad and critical role in autophagy‐dependent organelle quality control (QC) and antibacterial defense.
Materials and Methods
Antibodies and reagents
The following primary antibodies were used: anti‐Tom20 (Santa Cruz sc‐11415), anti‐cytochrome c (BD Bioscience 556432), anti‐GAPDH (Cell Signaling 14C10, 2118), anti‐actin (Sigma AC‐15, A1978), anti‐EndoB1 (R&D, AF7456), anti‐Tim23 (BD Bioscience), anti‐Mfn2 (Santa Cruz sc‐50331), anti‐Mfn1 (Santa Cruz sc‐166644), anti‐LC3 (MBL International, M152‐3), anti‐FLAG (M2 Sigma F7425), anti‐lipin‐1 (Cell Signaling 5195), anti‐PLD2 (Scbt sc‐515744), anti‐Citrate synthetase (GTX110624, GeneTex), Hsp60 (Cell Signaling, 4870), and anti‐Parkin (77924, Abcam) Secondary antibodies were from Jackson Immunochem or Invitrogen.
The following reagents were used: DMSO (Sigma D8418), carbonyl cyanide 5‐chloro‐phenylhydrazone (CCCP) (Sigma), PLD2 inhibitor VU 0364739 hydrochloride (Tocris 4171), Cytochalasin B (Sigma), and 1,2‐dipalmitoyl‐sn‐glycerol (Sigma D9135).
Cell culture, plasmids, and transfection
Hela cells were obtained from Duke Cell Culture Facility (Durham, NC, USA). Hela cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO‐11995) supplemented with 10% fetal bovine serum and 1 × antibiotics (penicillin, 10,000 UI/ml and streptomycin, 10,000 UI/ml). These cell lines have been authenticated by STR DNA profiling and validated to be mycoplasma‐free and before being frozen by the Duke Cell Culture Facility (Durham, NC, USA). All cells were maintained at 37°C and 5% CO2.
The following plasmids/siRNAs/shRNAs were used: GFP‐Parkin and mutants as previously described (Lee et al, 2010); mcherry‐Parkin (Yang & Yang, 2013); GFP‐Raf1‐PABD and CFP/YFP‐DAGR (Huang et al, 2011); mcherry‐optineurin (Wong & Holzbaur, 2014); YFP‐PLD1 and YFP‐PLD2 (Gifts from W.G. Zhang). mCherry‐Raf1‐PABD was generated by sub‐cloning the PABD domain from GFP‐Raf1‐PABD to mCherry2(C1). Expression plasmids for WT and catalytic mutant mouse Lipin‐1 were obtained from Addgene. Lipin‐1 siRNA 5′‐GAAUGGAAUGCCAGCUGAA‐3′ and 3′‐UUCAGCUGGCAUUCCAUUC‐5′ (Invitrogen); HSS118307 (Sigma); optineurin siRNA (Invitrogen 4392420), NDP52‐siRNA 5′‐UUCAGUUGAAGCAGCUCUGUCUCCC‐3′ (Thurston et al, 2009), EndoB1 siRNA 5′‐UGUUUAUACGACUUGGAGCUU‐3′ and 3′‐AAGCUCCAAGUCGUAUAAACA‐5′ (Invitrogen), control siRNA (Ambion). Expression plasmids were transfected in HeLa and YFP‐Parkin HeLa using Xtreme Gene 9 (Roche) according to the manufacturer's directions. Cells were transfected and treated 24–48 h later. shRNA plasmids were transfected as stated above, but cells were not treated until 3–5 days later. siRNAs were transfected using RNAi MAX (Invitrogen) according to the manufacturer's directions and cells treated 48–72 h later.
Immunofluorescence, microscopy, and quantification
Cells were seeded on coverslips, transfected, treated as indicated, and fixed in 4% PFA for 15 min. Coverslips were rinsed in PBS, permeabilized with 0.2% Triton‐X 100 in PBS for 5 min, blocked in 10% BSA for 20 min in a humidified chamber, and incubated in primary antibodies diluted in 10% BSA overnight at 4°C, followed by secondary antibodies in 10% BSA for 30 min. Coverslips were mounted on slides using Fluormount G (Southern Biotech).
Fixed sample and live cell imaging were performed on a Leica SP5 confocal microscope using 100×/1.4–0.70NA or 40× oil objective (Leica Plan Apochromat). Z‐stack images were acquired using the Leica LAS AF program software, and maximum projections were used in analyses and figures. Changes to brightness and contrast were performed in ImageJ. Number and size of vesicles were measured and quantified using the particle analysis module in Image J. For all immunofluorescent quantifications, at least 35 to 50 cells were counted for 2–3 independent experiments. Graphs represent means +/− SEM. For comparison of two conditions, a two‐tailed, unequal student's t‐test was performed with P < 0.05 considered significant. For all other comparisons, a one‐way or two‐way ANOVA was conducted.
Preparation of mitochondrial fractions
The isolation of mitochondria from cultured cells was performed by following the manufacturer's protocol (#89874, ThermoFisher). In brief, after CCCP treatment, the cells harvested from a 10 cm plate (~ 3 × 104 cells) were first resuspended in reagent A. Cell membrane was then lysed using a reagent‐based method by adding reagent B. After removing cell debris by low‐speed centrifugation, the mitochondrial fractions were collected in pellets with high‐speed centrifugation. The remaining supernatant is cytosol fraction. Mitochondrial fractions were further subject to Western blots or lipidomic analysis.
Western blots
HeLa or YFP‐Parkin HeLa were seeded onto 10 cm or 6 cm dishes, transfected as outlined above, and treated as described. For western blot analysis, whole cell lysates were collected using 170 mM NETN buffer, incubated at 4°C for 30 min, spun, supernatant collected, and protein concentration measured using the BCA assay (Thermo Scientific). Samples were normalized, diluted in XT buffer with a reducing agent (BioRad), and boiled for 5 min at 100°C. 4–20% TGX gels (BioRad) were used, followed by transfer to nitrocellulose membranes. Primary antibodies were added to 2% BSA, incubated overnight at 4°C, followed by rinsing, secondary antibody incubation for 1 h at room temperature, rinsing, and developing using either ECL PicoWest (Thermo Scientific) or ECL Pro Lightning (Perkin‐Elmer). Films were scanned, cropped, and adjusted for brightness and contrast in Photoshop. Density of bands was measured using Image J.
Lipid extraction and LC/MS analysis
Lipid extraction of purified mitochondria (form HeLa‐Parkin cells transfected with DAGR‐YFP, which preserved DAG from being metabolized) was performed using a modified Bligh‐Dyer method as previously decried (Tan et al, 2012). For LC/MS analysis, the dried lipid extracts were dissolved in chloroform/methanol (2:1, v/v) and injected for normal phase LC/MS analysis on an Agilent 1200 Quaternary LC system equipped with an Ascentis Silica HPLC column (5 μm, 25 cm × 2.1 mm, Sigma‐Aldrich, St. Louis, MO). Mobile phase A consisted of chloroform/methanol/aqueous ammonium hydroxide (800:195:5, v/v/v); mobile phase B consisted of; mobile phase C consisted of. The elution program consisted of the following: 100% mobile phase A (chloroform/methanol/aqueous ammonium hydroxide; 800:195:5, v/v/v) was held isocratically for 2 min and then linearly increased to 100% mobile phase B (chloroform/methanol/water/aqueous ammonium hydroxide; 600:340:50:5, v/v/v/v) over 14 min and held at 100% B for 11 min. The LC gradient was then changed to 100% mobile phase C (chloroform/methanol/water/aqueous ammonium hydroxide; 450:450:95:5, v/v/v/v) over 3 min and held at 100% C for 3 min, and finally returned to 100% A over 0.5 min and held at 100% A for 5 min. The LC eluent (with a total flow rate of 300 μl/min) was introduced into the ESI source of a high‐resolution TripleTOF5600 mass spectrometer (Sciex, Framingham, MA). Instrumental settings for negative ion ESI/MS analysis of lipid species were as follows: IS = −4,500 V; CUR = 20 psi; GSI = 20 psi; DP = −55 V; and FP = −150 V. The MS/MS analysis used nitrogen as the collision gas. Data analysis was performed using Analyst TF1.5 software (Sciex, Framingham, MA).
Author contributions
Chao‐Chieh Lin: Conceptualization; data curation; formal analysis; funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing. Jin Yan: Conceptualization; data curation; formal analysis; funding acquisition; validation; investigation; methodology; writing – original draft. Meghan Kapur: Conceptualization; data curation; formal analysis; funding acquisition; investigation; writing – original draft. Kristi Norris: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft. Cheng‐Wei Hsieh: Methodology. De Huang: Methodology. Nicolas Vitale: Methodology. Kah Leong Lim: Conceptualization. Ziqiang Guan: Methodology. Xiao‐fan Wang: Conceptualization; funding acquisition. Jen‐Tsan Chi: Conceptualization; funding acquisition. Wei Yuan Yang: Conceptualization; methodology. Tso‐Pang Yao: Conceptualization; resources; supervision; funding acquisition; investigation; writing – original draft; writing – review and editing.
Disclosure and competing interest statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
PDF+
Acknowledgements
We thank Drs. M. Frohman and I. Kojima for PA and DAG reporters, W.G. Zhang for PLD1/2‐YFP, E.F. Holzbaur for OPTN‐Cherry, V. Bennett for TGN38‐GFP expression plasmids, and Dr. M. Frohman for PLD6 KO MEFs. We thank Dr. T. Slotkin for the advice on the statistical analysis and YS Gao on the image analysis. This work was supported, in part, by 2R01‐NS054022 and 1R01GM144497 (NIH), to T.‐P.Y.
EMBO reports (2022) 23: e55191
Data availability
All data supporting the findings of this study are available from the authors upon request. No data were generated, which require deposition in public databases.
References
- Adachi E, Kazoe Y, Sato Y, Suzuki Y, Urano T, Ueyama T, Saito N, Nikolaev VO, Lohse MJ, Tominaga M et al (2009) A technique for monitoring multiple signals with a combination of prism‐based total internal reflection fluorescence microscopy and epifluorescence microscopy. Pflugers Arch 459: 227–234 [DOI] [PubMed] [Google Scholar]
- Baron CL, Malhotra V (2002) Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295: 325–328 [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Tito S, Hervas JH, van Vliet AR, Tooze SA (2020) The Golgi as an assembly line to the autophagosome. Trends Biochem Sci 45: 484–496 [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- Giorgione JR, Lin JH, McCammon JA, Newton AC (2006) Increased membrane affinity of the C1 domain of protein kinase Cdelta compensates for the lack of involvement of its C2 domain in membrane recruitment. J Biol Chem 281: 1660–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall EA, Kraus F, Harper JW (2022) Mechanisms underlying ubiquitin‐driven selective mitochondrial and bacterial autophagy. Mol Cell 82: 1501–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GS, Wu WI, Carman GM (2006) The Saccharomyces cerevisiae Lipin homolog is a Mg2+−dependent phosphatidate phosphatase enzyme. J Biol Chem 281: 9210–9218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, Morris AJ, Frohman MA (2011) piRNA‐associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial‐surface lipid signaling. Dev Cell 20: 376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Koyama‐Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin‐mediated mitophagy. J Cell Sci 125: 1488–1499 [DOI] [PubMed] [Google Scholar]
- Kassas N, Tryoen‐Toth P, Corrotte M, Thahouly T, Bader MF, Grant NJ, Vitale N (2012) Genetically encoded probes for phosphatidic acid. Methods Cell Biol 108: 445–459 [DOI] [PubMed] [Google Scholar]
- Lavieri RR, Scott SA, Selvy PE, Kim K, Jadhav S, Morrison RD, Daniels JS, Brown HA, Lindsley CW (2010) Design, synthesis, and biological evaluation of halogenated N‐(2‐(4‐oxo‐1‐phenyl‐1,3,8‐triazaspiro[4.5]decan‐8‐yl)ethyl)benzamides: discovery of an isoform‐selective small molecule phospholipase D2 inhibitor. J Med Chem 53: 6706–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP (2010) Disease‐causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6‐dependent mitophagy. J Cell Biol 189: 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani S, Hichami A, Legrand A, Belleville J, Khan NA (2001) Implication of acyl chain of diacylglycerols in activation of different isoforms of protein kinase C. FASEB J 15: 2595–2601 [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y et al (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Menarguez JA, Tomas M, Martinez‐Martinez N, Martinez‐Alonso E (2019) Golgi fragmentation in neurodegenerative diseases: is there a common cause? Cell 8: 748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, Ryzhakov G, Puri C, Arden SD, Roberts R, Dendrou C, Kendrick‐Jones J, Buss F (2007) T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J Cell Sci 120: 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2020) The aging lysosome: an essential catalyst for late‐onset neurodegenerative diseases. Biochim Biophys Acta Proteins Proteomics 1868: 140443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M (2019) LC3/GABARAPs drive ubiquitin‐independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun 10: 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K et al (2017) VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36: 135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105: 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA et al (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113: 4039–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, Shome K, Watkins SC, Romero G (2000) The recruitment of Raf‐1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem 275: 23911–23918 [DOI] [PubMed] [Google Scholar]
- Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, Kendrick‐Jones J, Buss F (2005) Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol 169: 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, Nakayama K, Klionsky DJ, Brumell JH (2010) A diacylglycerol‐dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 8: 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ et al (2007) Bif‐1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9: 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Hori T, Cooper TK, Liao J, Desai N, Serfass JM, Young MM, Park S, Izu Y, Wang HG (2013) Bif‐1 haploinsufficiency promotes chromosomal instability and accelerates Myc‐driven lymphomagenesis via suppression of mitophagy. Blood 121: 1622–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Meyerkord CL, Hori T, Runkle K, Fox TE, Kester M, Loughran TP, Wang HG (2011) Bif‐1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy 7: 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z (2012) Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc Natl Acad Sci U S A 109: 16504–16509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F (2009) The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin‐coated bacteria. Nat Immunol 10: 1215–1221 [DOI] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74: 347–362.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives‐Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS et al (2010) PINK1‐dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci U S A 111: E4439–E4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Yang WY (2013) Bit‐by‐bit autophagic removal of parkin‐labelled mitochondria. Nat Commun 4: 2428 [DOI] [PubMed] [Google Scholar]
- Zhang C, Li A, Gao S, Zhang X, Xiao H (2011) The TIP30 protein complex, arachidonic acid and coenzyme a are required for vesicle membrane fusion. PLoS One 6: e21233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CW, Hang L, Yao TP, Lim KL (2015) Parkin regulation and neurodegenerative disorders. Front Aging Neurosci 7: 248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Verity MA, Reue K (2014) Lipin‐1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab 20: 267–279 [DOI] [PMC free article] [PubMed] [Google Scholar]