

Abstract
FBXW7, which encodes a substrate‐specific receptor of an SCF E3 ligase complex, is a frequently mutated human tumor suppressor gene known to regulate the post‐translational stability of various proteins involved in cellular proliferation. Here, using genome‐wide CRISPR screens, we report a novel synthetic lethal genetic interaction between FBXW7 and CCNL1 and describe CCNL1 as a new substrate of the SCF‐FBXW7 E3 ligase. Further analysis showed that the CCNL1–CDK11 complex is critical at the G2‐M phase of the cell cycle since defective CCNL1 accumulation, resulting from FBXW7 mutation, leads to shorter mitotic time. Cells harboring FBXW7 loss‐of‐function mutations are hypersensitive to treatment with a CDK11 inhibitor, highlighting a genetic vulnerability that could be leveraged for cancer treatment.
Keywords: CDK11, cell cycle, cyclin L1, FBXW7, mitosis
Subject Categories: Cancer, Cell Cycle, Post-translational Modifications & Proteolysis
The candidate oncogene CCNL1 is regulated by FBXW7 through ubiquitination and degradation in a Cul1‐dependent manner. FBXW7‐mutant cells depend on CCNL1 for growth, sensitizing them to OTS964, an inhibitor of CCNL1 kinase partner CDK11.
Introduction
Fbox and WD‐repeat domain‐containing protein 7 (FBXW7) is a substrate‐specific recognition module of a Skp1‐Cul1‐Fbox (SCF) E3 ligase complex named SCFFBXW7 (Welcker & Clurman, 2008). SCF E3 ligases target several proto‐oncogenes for ubiquitin‐dependent proteasomal degradation to regulate cell cycle progression and proliferation (Koepp, 2001; Reed et al, 2004; Welcker et al, 2004b; Wei et al, 2005; Yeh et al, 2018). One of the better‐characterized substrates of SCFFBXW7 is cyclin E, a master regulator of the G1‐S cell cycle phase transition (Ohtsubo et al, 1995; Koepp, 2001). Loss of FBXW7 and concomitant accumulation of cyclin E deregulates the cell cycle (Reed et al, 2004), enhances DNA replication (Minella et al, 2008), and causes genomic instability across many different cell types (Spruck et al, 1999; Rajagopalan et al, 2004; Loeb et al, 2005; Minella et al, 2007). c‐Myc is another well‐studied substrate of SCFFBXW7 (Welcker et al, 2004a, 2004b). Although not directly implicated in the regulation of cell cycle phase transition, c‐Myc inhibits many inhibitors of the cell cycle including p21 and p27, which negatively regulate the G1‐S transition checkpoint (Mateyak et al, 1999; Perez‐Roger et al, 1999).
Given its fundamental roles in cell cycle control, FBXW7 inactivating mutations that lead to substrate stabilization are common across a wide range of cancers, with the highest mutational frequency in uterine, cervical, and intestinal cancers (Yeh et al, 2018). The most common mutations are within FBXW7 exons encoding WD repeats, which function as the substrate‐recognition domain. These mutations lead to defective post‐translational control of proto‐oncogene abundance and hence promote cancer progression. Biochemical studies have confirmed that the WD‐repeat hotspot mutations (R465C/H/L, R479P/Q/*, and R505C/G/H) are loss of function mutations that disrupt substrate‐binding (Orlicky et al, 2003; Tang et al, 2007). In addition to cyclin E and c‐Myc, SCFFBXW7 has also been implicated in the control of other potentially oncogenic substrates, including c‐Jun (Nateri et al, 2004; Wei et al, 2005), and Notch1 (Gupta‐Rossi et al, 2001; Oberg et al, 2001; Tetzlaff et al, 2004; O'Neil et al, 2007).
Currently categorized as a transcriptional or non‐canonical cyclin, CCNL1 (cyclin L1) was demonstrated to functionally regulate the spliceosome, along with its serine/threonine kinase partner cyclin‐dependent kinase 11 (CDK11) (Chen et al, 2006, 2007; Loyer & Trembley, 2020). Consistent with the understanding of cyclin‐CDK biology, the primary role of CCNL1 is to promote CDK11 activity (Loyer & Trembley, 2020). CCNL1 has been proposed as a candidate oncogene in head and neck cancer due to high levels of chromosomal amplification that correlates with poor overall survival in patients (Redon et al, 2002; Sticht et al, 2005; Muller et al, 2006). Amplification of CCNL1 is also associated with poor prognosis in uterine cancer (Mitra et al, 2010). CDK11 has several isoforms, with the p58 version generated via the usage of an internal ribosomal entry site within CDK11 mRNA transcripts at the G2/M cell cycle transition (Cornelis et al, 2000). Recently, the CCNL1‐CDK11 complex has been implicated in cytokinesis, with CDK11‐p58 kinase activity required for abscission, the final stage of mitosis (Renshaw et al, 2019). Despite a poor understanding of the role of CCNL1 in cancer initiation and progression, several studies have established CDK11 as an important regulator of cancer cell proliferation and that its loss of function is lethal in many cancer types (Liu et al, 2016; Ahmed et al, 2019). A novel selective CDK11 inhibitor, OTS964, was recently serendipitously identified (Lin et al, 2019) and offers a potentially tractable therapeutic opportunity for cancers, perhaps in contexts where tumor cells are reliant on CDK11 activity.
In the present study, we combine drug selection with gene‐editing of pancreatic cancer cells to rewire their growth dependency in such a way as to be exquisitely reliant on the loss of FBXW7 activity. This engineered model was required to ensure that cell growth was dependent on this single FBXW7 mutation in order to hone in on the molecular mechanisms underlying this frequent cancer‐causing alteration. Using genome‐wide CRISPR fitness screens performed in isogenic cell lines, we uncovered a novel synthetic lethal interaction between FBXW7 and CCNL1. Given that SCFFBXW7 is known to control the levels of other cyclins, we showed that CCNL1 is a novel substrate for this E3 ligase. Our findings suggest that the deregulation of this axis is frequent in human cancers and it culminates in the hyperactivation of CCNL1's kinase partner CDK11, thereby uncovering a novel therapeutic opportunity.
Results
Loss of FBXW7 induces resistance to Wnt‐inhibition in a Wnt‐addicted cell line
A subset of pancreatic adenocarcinoma harbor inactivating mutations within RNF43, a negative regulator of the Wnt‐ßcatenin pathway (Jiang et al, 2013). As a result of RNF43 mutation, cells express high levels of Frizzled receptors and are exquisitely dependent on autocrine Wnt‐ßcatenin signaling for growth as highlighted by their hypersensitivity to LGK974, a small molecule inhibitor of porcupine (PORCN) that blocks secretion and function of Wnt proteins (Fig 1A and B), as well as to anti‐Frizzled blocking antibodies (Steinhart et al, 2017). To pre‐emptively study mechanisms of resistance to Wnt inhibitors, we conducted a genome‐wide CRISPR suppressor screen, in the RNF43 mutant cell line HPAF‐II. This experiment identified gene knockouts that overcome the growth arrest phenotype induced by LGK974 (Fig 1C and D). Predictably, the screen identified the well‐known negative regulators of ßcatenin signaling, AXIN1, APC, and CSNK1A1 (Fig 1D) as these mutations all lead to ligand‐independent ßcatenin stabilization and regulation of gene expression and hence bypass the requirement for autocrine Wnt ligands (Amit et al, 2002; Liu et al, 2002; Su et al, 2008; Steinhart et al, 2017). The genes above represented nearly 20% of the total amount of next‐generation sequencing reads in the suppressor screen highlighting their strong negative regulatory functions during Wnt‐ ßcatenin signaling. In addition to these genes, the screen also uncovered tumor suppressor Fbox protein FBXW7, which functions as a substrate‐recognition subunit within an Skp1‐Cullin‐Fbox (SCF) E3 ligase complex and is a well‐studied tumor suppressor gene (Mao et al, 2004; Yeh et al, 2018). APC and FBXW7 HPAF‐II knockout cells were generated using CRISPR and selected using selective pressure with LGK974 (Figs 1E and EV1A and B). Strikingly, whereas FBXW7 knockout cells were confirmed to be resistant to LGK974‐mediated cell cycle arrest (Fig 1F), with relative confluence reduced to 9.1% in the wild‐type, 28% in FBXW7 −/− and unchanged in APC −/− in the presence of LGK974 (Fig 1F), only APC −/− cells maintained high ßcatenin levels in the presence of LGK974 (Fig 1G). These data confirm that in HPAF‐II cells, FBXW7 itself is not regulating ßcatenin levels as was reported in a different context (Jiang et al, 2016). We conclude that FBXW7 −/− cells are partially resistant to PORCN inhibitor‐mediated growth arrest by a mechanism that does not involve reactivation of downstream ßcatenin signaling.
Figure 1. FBXW7 loss of function bypasses the requirement for autocrine Wnt signaling in Wnt‐addicted PDAC cells.
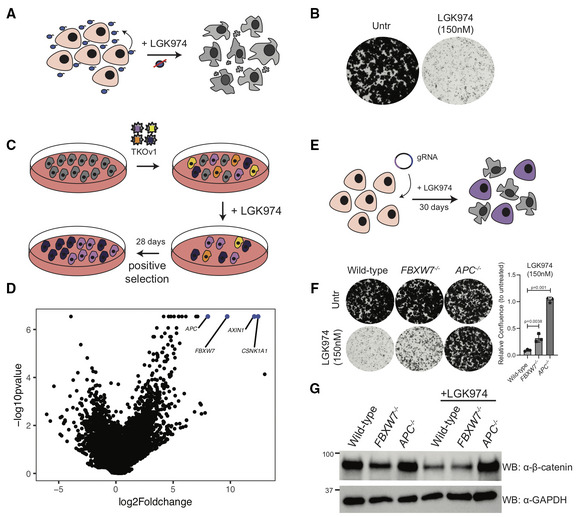
- HPAF‐II cells contain an autocrine Wnt‐signaling loop, where Wnt secreted by cells is required for the cells to grow. Treatment with the Porcupine (PORCN) inhibitor LGK974 (which inhibits Wnt secretion) leads to cell cycle arrest.
- Clonogenic growth assay in control HPAF‐II cells or in cells treated with 150 nM LGK974 for 10 days, representative of three independent replicates.
- Schematic representation of the positive selection screen conducted to identify genes involved in LGK974 sensitivity.
- Volcano plot with results of the positive selection screen identifying FBXW7 along with AXIN1, CSNK1A1, and APC as genes overcoming LGK974‐induced cell cycle arrest when knocked out.
- Schematic representation of FBXW7 −/− and APC −/− cell line generation in HPAF‐II using LGK974 treatment to enrich for edited cells.
- Clonogenic growth assay of indicated HPAF‐II cells left untreated or in the presence of 150 nM LGK974 for 10 days; representative of three independent replicates. Quantification of n = 3 biological replicates, one‐way ANOVA, mean ± SEM.
- Immunoblot of cytoplasmic βcatenin expression from lysates of HPAF‐II cells from indicated genotype, treated with vehicle or 100 nM LGK974 for 48 h, representative of three biological replicates.
Figure EV1. Verification of FBXW7 and APC knockout & genome‐wide screen quality control.
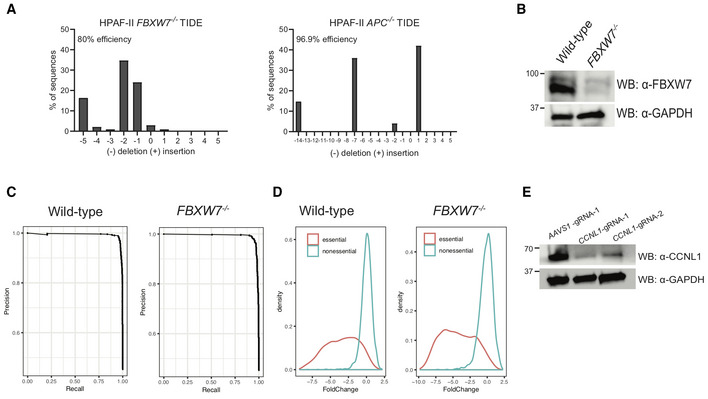
- TIDE analysis of HPAF‐II FBXW7 −/− cell line and HPAF‐II APC −/− cell line.
- Immunoblot of lysates extracted from HPAF‐II FBXW7 −/− cell lines demonstrating knockout of FBXW7 protein expression.
- Fold‐change plots of HPAF‐II wild‐type and FBXW7 −/− genome‐wide screens demonstrating a change in essential genes at T24 of the screen.
- Precision‐recall curves of HPAF‐II wild‐type and FBXW7 −/− genome‐wide screens demonstrating training sets of essential and nonessential genes performed appropriately in the BAGEL algorithm.
- Immunoblot of lysates extracted from FBXW7 −/− cells following treatment with gRNAs targeting CCNL1.
CCNL1 loss of function is synthetic lethal with FBXW7 mutation
To reveal the growth mechanisms dysregulated in FBXW7 −/− cells that underlie the resistance to LGK974 treatment, we performed isogenic CRISPR fitness screens in wild‐type and FBXW7 −/− HPAF‐II cells (Fig 2A). We then used the BAGEL algorithm to calculate a Bayes factor (BF) for each gene (Hart & Moffat, 2016). BF is a confidence score that the knockout of a specific gene causes a decrease in fitness where high BF indicates increased confidence that the knockout of the gene results in a decrease in fitness. We then derived a differential fitness score for each gene by subtracting the BF scores obtained in FBXW7 −/− and wild‐type cells and plotted the differential Z‐score (Figs 2B and EV1C and D). Confirming that FBXW7 −/− cells have evaded a requirement for Wnt‐ßcatenin signaling, several of the genes we previously identified as fitness genes in HPAF‐II cells (FZD5, PORCN, TCF7L2, WLS, CTNNB1; Steinhart et al, 2017) have negative Z‐scores, indicating a greater fitness defect observed in wild‐type cells. Interestingly, the gene with the highest positive differential Z‐score was the poorly characterized cyclin family member cyclin L1 (CCNL1) (Fig 2B and individual CCNL1 gRNA dropouts comparison in Fig 2C). To validate these results, we performed multicolor cell competition assays with HPAF‐II wild‐type or HPAF‐II FBXW7 −/− mutant cells expressing a control gRNA targeting AAVS1 (labeled with mCherry) or two independent gRNAs targeting CCNL1 (labeled with GFP) and showed that mCherry cells outcompeted GFP cells at a much faster rate in the absence of FBXW7, with a 50% reduction in GFP‐expressing cells by day 4, with wild‐type cells largely unaffected until day 16 (Figs 2D and EV1E). Supporting these results, when infected with lentivirus encoding for Cas9 and gRNAs targeting CCNL1, HPAF‐II FBXW7 −/− mutant cells proliferated at a much slower rate when compared to wild‐type HPAF‐II cells (Figs 2E and EV1E). We conclude that loss of CCNL1 is synthetic lethal with FBXW7 mutation.
Figure 2. Genome‐wide CRISPR screen performed in isogenic wild‐type and FBXW7 −/− HPAF‐II cells identifies a synthetic lethal genetic interaction.
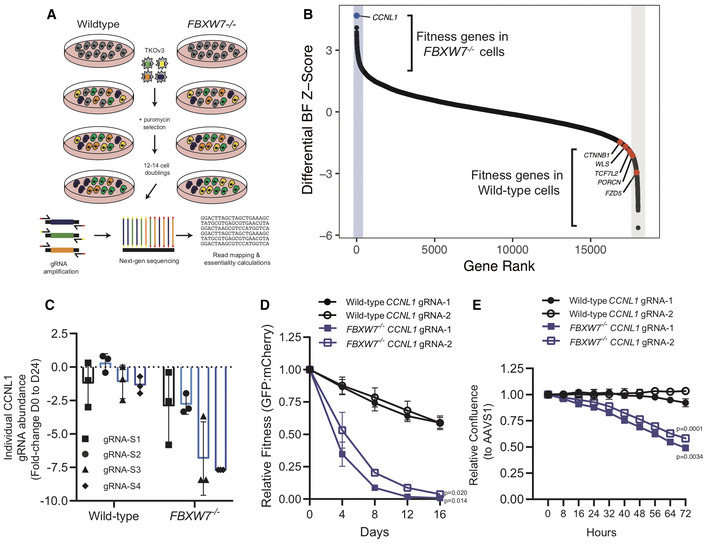
- Schematic representation of genome‐wide CRISPR‐Cas9 dropout screens performed in isogenic wild‐type and FBXW7 −/− HPAF‐II cell lines.
- Differential Bayes Factor Z‐score plot comparing wild‐type and FBXW7 −/− genome‐wide dropout screens.
- Fold‐change abundance of individual gRNA targeting CCNL1 during the genome‐wide dropout screens from day 0 to day 24, n = 3 technical replicates per gRNA, mean ± SEM.
- Multicolour competition assay in both wild‐type and FBXW7 −/− cell lines, using mCherry‐AAVS1 and GFP‐GOI, normalized to AAVS1 control cells at each time‐point (n = 3 independent replicates), mean ± SEM, one‐way ANOVA.
- Proliferation assays in wild‐type and FBXW7 −/− cell lines show that knockout of CCNL1 preferentially affects FBXW7 −/− cells, normalized to AAVS1 control (n = 3 independent replicates), mean ± SEM, one‐way ANOVA.
CCNL1 is a substrate of the SCFFBXW7 E3 ligase
The well‐established role of SCFFBXW7 in regulating cyclin E stability hinted that one potential mechanism underlying the observed synthetic lethality phenotype is that CCNL1 is a novel substrate of the SCFFBXW7 E3 ligase. In support, we noted increased steady‐state CCNL1 levels in FBXW7 −/− cells when compared to wild‐type cells (Figs 3A and EV2A). Cycloheximide chase further revealed that the half‐life of CCNL1 was extended in FBXW7 −/− vs wild‐type HPAF‐II cells (Fig 3B and C). Expression of a dominant‐negative Cul1 mutant (Van Rechem et al, 2011) induced stabilization of CCNL1 at steady state, further indicating that an SCF complex is involved in the regulation of CCNL1 (Figs 3D and EV2B). We next scanned the amino acid sequence of CCNL1 for the presence of a canonical Cdc4 phosphodegron (CPD) motif present in the majority of FBXW7 substrates (Nash et al, 2001; Orlicky et al, 2003) and identified a TPXXS sequence at position 325–329 (Fig 3E). Analysis of CPD peptide binding to purified FBXW7 using fluorescence polarization assays revealed a requirement for dual phosphorylation at both T325 and S329 within the CCNL1 CPD motif (Fig 3F), similar to what is seen for the FBXW7‐Jun interaction (Wei et al, 2005), but in contrast to the FBXW7‐cyclin E interaction where phosphorylation at the threonine residue is sufficient for binding, with maximal binding requiring a diphoshorylated degron (Hao et al, 2007, and Fig 3F). Given this requirement for dual phosphorylation, binding of CCNL1 to FBXW7 may require a second degron or contact site that could boost the affinity, as described with many well‐studied substrates including cyclin E and Myc. Mutation of the TPXXS motif to VPXXA in the context of full‐length CCNL1 extended the half‐life of CCNL1 compared with wild‐type proteins following cycloheximide chase (Fig 3G and H). Co‐expression of HA‐CCNL1 with FLAG‐FBXW7 further reduced the half‐life of CCNL1, while co‐expression with the substrate‐binding mutant FBXW7R465C fully stabilized CCNL1 expression (Fig EV2G and H), demonstrating a direct role of FBXW7 in regulating CCNL1 expression.
Figure 3. CCNL1 protein stability is mediated by SCFFBXW7 .
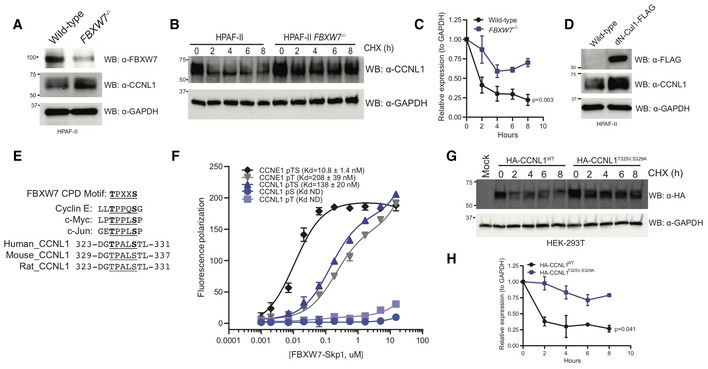
- Immunoblot of CCNL1 and FBXW7 expression in HPAF‐II wild‐type and FBXW7 −/− cells, representative blot of three independent replicates.
- Immunoblot of CCNL1 expression following a cycloheximide chase in HPAF‐II wild‐type and FBXW7 −/− cells, representative blot of three independent replicates.
- Quantification of cycloheximide chase in (B), mean ± SEM of three independent replicates. Two‐way ANOVA at T8.
- Immunoblot of CCNL1 expression in HPAF‐II wild‐type and dN‐Cul1 expressing cells, representative blot of three independent replicates.
- Sequence alignment of well‐characterized CPD degron motifs, and identification of a potential FBXW7 phosphodegron motif in a conserved region of CCNL1.
- Fluorescence polarization assay of CCNL1 and cyclin E peptides binding FBXW7‐Skp1 complex, three independent replicates, mean ± SEM.
- Immunoblot of lysates following cycloheximide treatment of HEK293T cells expressing wild‐type or degron‐mutated CCNL1, representative blot of three independent replicates.
- Quantification of cycloheximide chase in (G), mean ± SEM of three independent replicates. Two‐way ANOVA at T8.
Figure EV2. CCNL1 degradation requires Cul1 and is blocked when substrate‐binding deficient FBXW7R465C is co‐expressed.
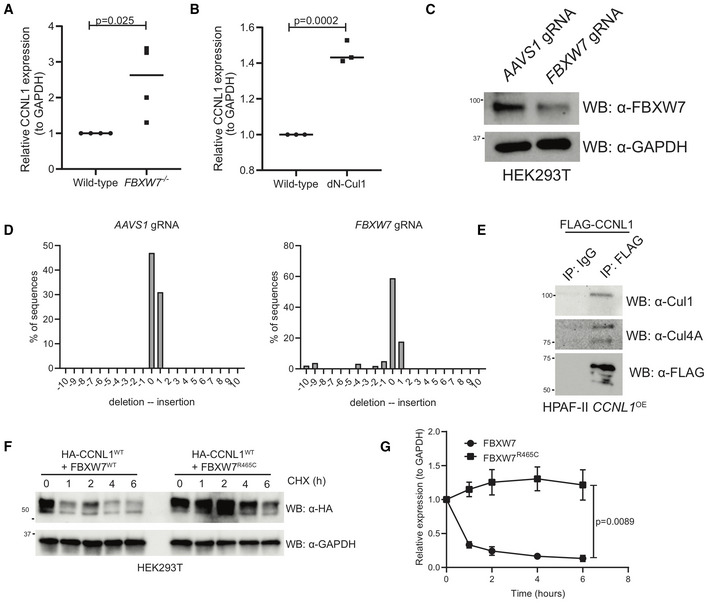
- Quantification of immunoblots in Fig 3A, mean ± SEM, students t‐test, n = 3 independent biological replicates.
- Quantification of immunoblots in Fig 3D, mean ± SEM, students t‐test, n = 3 independent biological replicates.
- Immunoblot analysis of HEK293T cells expressing gRNAs against AAVS1 or FBXW7.
- TIDE analysis of HEK293T cells expressing gRNAs against AAVS1 or FBXW7.
- Immunoblot of immunoprecipitation of FLAG‐CCNL1 overexpressed in HPAF‐II cells, detecting endogenous Cul1 and Cul4A. Representative image of three independent replicates.
- Immunoblot of lysates following cycloheximide treatment of HEK293T cells expressing HA‐CCNL1 and FLAG‐FBXW7 or FLAG‐FBXW7R465C. Representative blot of three independent replicates.
- Quantification of cycloheximide chase in (F), mean ± SEM of three independent replicates, t‐test at T6.
To verify that CCNL1 and FBXW7 are indeed interacting in cells, we performed an immunoprecipitation assay using overexpression of either FLAG‐CCNL1 or FLAG‐FBXW7, and detected interactions with endogenous FBXW7 and CCNL1, respectively (Fig 4A and B). We next wished to perform in vitro ubiquitination assays but were faced with a roadblock in our multiple attempts to purify CCNL1 from either Sf9 or E. coli cultures. We therefore employed a cellular ubiquitination assay in HEK293T cells to confirm CCNL1 ubiquitination and the role of T325 and S329 in the process (Fig 4C). A second cellular ubiquitination assay in HEK293T cells expressing a control gRNA targeting AAVS1, or a gRNA targeting FBXW7 demonstrated that reducing FBXW7 expression reduced ubiquitination of CCNL1 (Figs 4D and EV2C and D). Using HEK293T cells expressing an FBXW7‐targeting gRNA, we added a gRNA‐resistant FLAG‐FBXW7 cDNA and assessed CCNL1 ubiquitination. The results indicated that re‐expression of FBXW7 rescued CCNL1 ubiquitination (Fig 4E). We detected an interaction between CCNL1 and endogenous CUL1 in HPAF‐II cells, further supporting a role of the SCF complex in CCNL1 degradation (Fig EV2E). Interestingly, CUL4A was also co‐immunoprecipitated with CCNL1 perhaps suggesting a role for this cullin in the regulation of CCNL1 stability, similar to another FBXW7 substrate—Jun—which is degraded by both CUL1‐ and CUL4A‐based E3 ligases (Cang et al, 2007; Fig EV2E). We conclude that CCNL1 is a bona fide substrate of the SCFFBXW7 complex.
Figure 4. FBXW7 is involved in the ubiquitination of CCNL1.
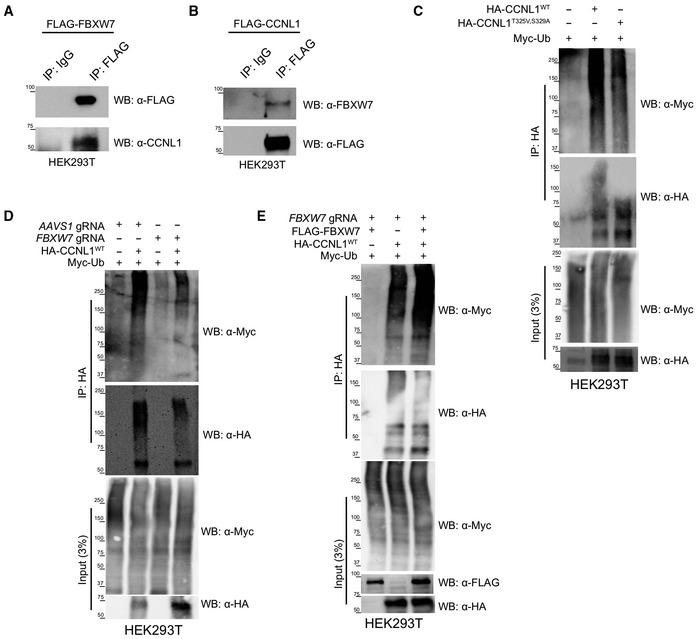
- Immunoblot of immunoprecipitation of FLAG‐FBXW7 overexpressed in HEK293T cells, detecting endogenous CCNL1. Representative of three independent replicates.
- Immunoblot of immunoprecipitation of FLAG‐CCNL1 overexpressed in HEK293T cells, detecting endogenous FBXW7. Representative of three independent replicates.
- Immunoblot of cellular ubiquitination assay demonstrating the requirement of the 325‐TPALS‐329 degron for ubiquitination of CCNL1. Representative of three independent replicates.
- Immunoblot of cellular ubiquitination assay demonstrating the requirement of FBXW7 for ubiquitination of CCNL1. Representative of three independent replicates.
- Immunoblot of cellular ubiquitination assay showing re‐expression of FBXW7 in knockout cells leads to increased CCNL1 ubiquitination. Representative of three independent replicates.
FBXW7 regulates G2‐M progression through regulation of CCNL1 stability
To identify whether CCNL1 has a role in cell cycle progression like other SCF substrates, a cell cycle profile experiment was performed following the release of cells that were first arrested in mitosis using nocodazole. In wild‐type cells, CCNL1 expression oscillates in a pattern similar to cyclin B, supporting a potential role of CCNL1 in the G2‐M phase of the cell cycle (Fig 5A and B). By contrast, in FBXW7 −/− cells the cycling of CCNL1 levels is lost, supporting the role of FBXW7 in targeting CCNL1 for degradation in a cell cycle‐dependent manner (Fig 5A and B). Considering the previously described role of CCNL1 and CDK11 in cytokinesis (Renshaw et al, 2019), we assessed whether upregulation of CCNL1 through either FBXW7 loss of function or CCNL1 overexpression affected normal progression through mitosis. First, to assess the cell cycle dynamics during logarithmic growth, cell cycle profiles for HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells were generated by flow cytometry. This assay identified a decreased proportion of cells in the G2‐M phase in both FBXW7 −/− and CCNL1 OE cell lines, suggestive of a shortened G2‐M phase (Figs 5C and EV3A). Next, using the Eg5 kinesin inhibitor monastrol (Mayer et al, 1999) GFP‐tubulin labeled cells were arrested in prometaphase overnight and released before live‐cell imaging to measure the timing of mitosis progression. Consistent with the hyperactivity of CCNL1‐CDK11 complexes, FBXW7 −/− and CCNL1 OE cell lines completed cell division with a mean of 295 and 255 min, respectively, while wild‐type cells took an average of 350 min following monastrol washout (Fig 5D and E; Movies [Link], [Link]; Renshaw et al, 2019). Importantly, the expression level of CCNL1 correlated with mitosis duration (Fig 5E and F). To further validate this shortened mitosis, the PIP‐FUCCI reporter (Grant et al, 2018; Fig EV3B) was employed to assess mitotic timing following nocodazole treatment. Indeed, FBXW7 −/− and CCNL1 OE cells exited mitosis faster following nocodazole treatment as detected by quantification of mCherry fluorescence (labeling cells in S and G2/M phases) at the bulk population level (Fig EV3C), as well as in individual cells exiting mitosis (Fig EV3D).
Figure 5. CCNL1 regulates mitotic timing.
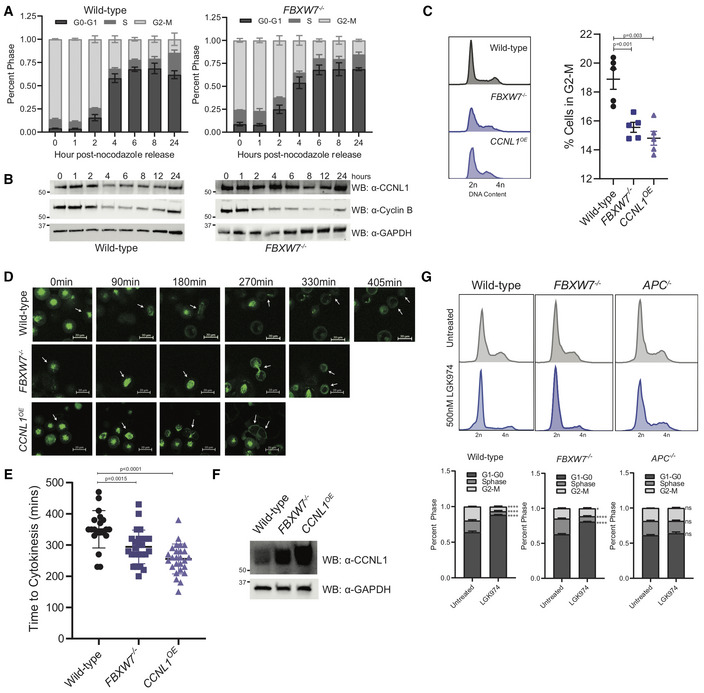
- Cell cycle profile of nocodazole treated and released HPAF‐II wild‐type and FBXW7 −/− cells measured by flow cytometry, n = 3 independent replicates, mean ± SEM.
- Immunoblot of HPAF‐II wild‐type and FBXW7 −/− cells released from overnight nocodazole treatment at indicated time points, representative of three independent replicates.
- Representative cell cycle profile of HPAF‐II wild‐type, FBXW7 −/−, and CCNL1 OE cells stained with propidium iodide, with quantification of G2‐M phase. Three independent replicates, mean ± SEM, one‐way ANOVA.
- Representative images of GFP‐tubulin labeled wild‐type, FBXW7 −/−, and CCNL1 OE HPAF‐II cells monitoring cellular progression through cytokinesis following arrest in prometaphase using monastrol (150 μM for 18 h). Arrows indicate tracked cell, 20 μm scale bars.
- Quantification of cytokinetic timing, n = 20, 25, 26 (wild‐type, FBXW7 −/−, CCNL1 OE ) pooled from three independent experiments, one‐way ANOVA, mean ± SEM.
- Immunoblot of wild‐type, FBXW7 −/− and CCNL1 OE HPAF‐II cell lines demonstrating varying levels of CCNL1.
- Cell cycle profiles of wild‐type, FBXW7 −/− and APC −/− HPAF‐II cells following treatment with 500 nM LGK974 for 24 h. Representative images of three independent biological replicates, mean ± SEM, 2‐way ANOVA, *P = 0.044, ****P < 0.001.
Figure EV3. HPAF‐II FBXW7−/− and CCNL1OE cells exit mitosis faster than wild‐type.
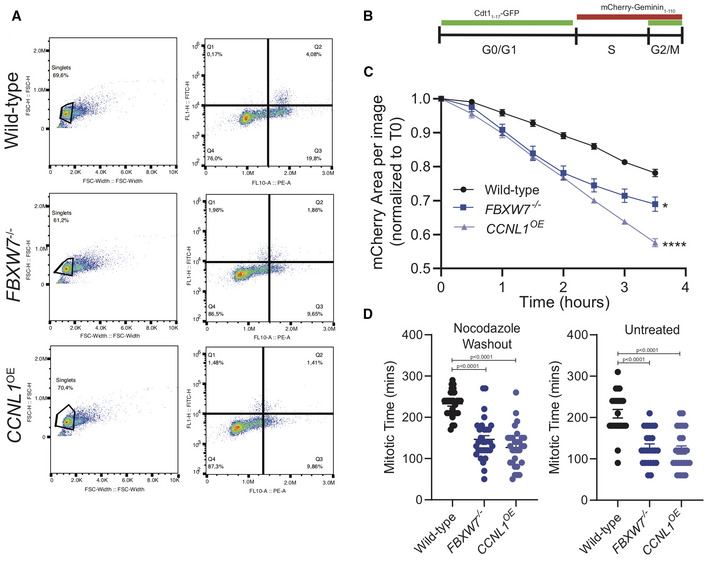
- Representative images of gating strategy for HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells to determine cell cycle distribution.
- Schematic of PIP‐FUCCI reporter marker expression through three major cell cycle phases.
- Live‐cell imaging of HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells expressing PIP‐FUCCI reporter treated with nocodazole overnight and released. Images collected over 3.5 h. Reduction in total population mCherry expression imaged over time, quantified in the Incucyte. Three independent replicates, mean ± SEM, two‐way ANOVA, *P = 0.0258, ****P < 0.0001.
- Live‐cell imaging of HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells expressing PIP‐FUCCI reporter either untreated or treated with nocodazole overnight and released. Measurement of individual cells as they lose mCherry expression. n = 15 cells per replicate, three independent replicates, one‐way ANOVA, mean ± SEM.
Considering a role for CCNL1 in mediating the final stages of mitosis, we wanted to assess the cell cycle profiles of HPAF‐II wild‐type, FBXW7 −/− and APC −/− cells following treatment with LGK974, previously reported to arrest cells in G0 (Steinhart et al, 2017). Indeed, HPAF‐II FBXW7 −/− cells showed a higher proportion of actively dividing G2‐M cells, with an average of 6.5% of G2‐M cells in the wild‐type and 11% G2‐M cells in the FBXW7 −/− cell line following LGK974 treatment, suggesting that cells harboring an FBXW7‐knockout or loss of function (LOF) mutation may bypass LGK974‐induced cell cycle arrest by maintaining a pool of actively dividing cells (Figs 5G and EV4A).
Figure EV4. Genome‐wide chemogenomic screen identifies CCNL1 and CDK11 are targets of OTS964.
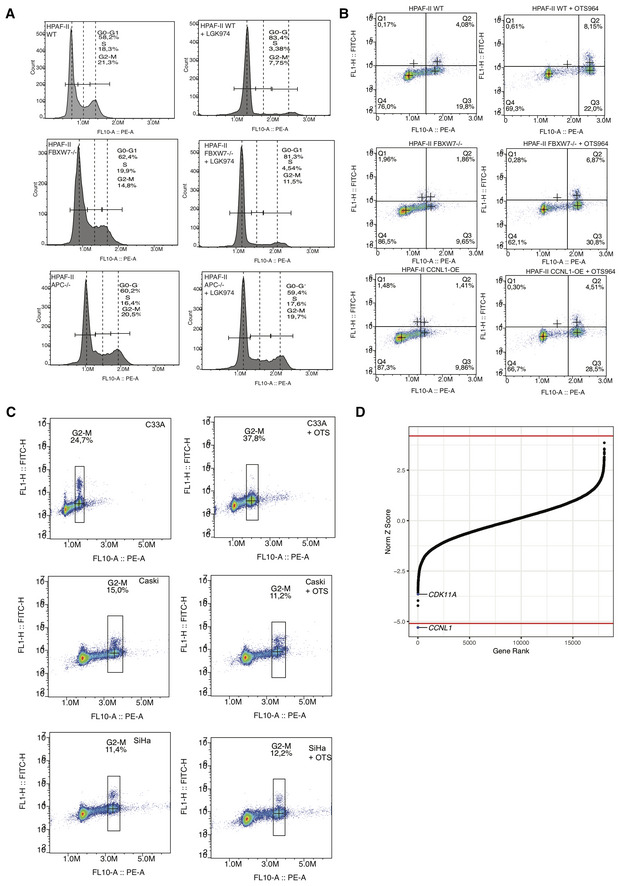
- Representative gating strategy for HPAF‐II wild‐type, FBXW7 −/− and APC −/− cells, with and without LGK974 treatment, to determine cell cycle distribution.
- Representative gating strategy for HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells, with and without OTS964 treatment, to determine cell cycle distribution.
- Representative images of gating strategy for C33A, Caski, and SiHa cells, with and without OTS964 treatment, to determine cell cycle distribution.
- Normalized Z‐score calculated in DrugZ plotted against gene rank for C33A chemogenomic screen with OTS964. Negative score indicates gene knockout synergistic with OTS964; positive score indicates gene knockouts resistant to OTS964. Red line marks the cutoff of FDR < 0.05.
FBXW7 loss of function and CCNL1 overexpression sensitize cells to CDK11 inhibitor OTS964
A kinase inhibitor currently in preclinical development, OTS964, was recently identified to target CDK11, the cyclin‐dependent kinase (CDK) partner of CCNL1 (Lin et al, 2019). Considering the requirement for CCNL1 in FBXW7 −/− HPAF‐II cells, we aimed to assess whether inhibiting CDK11 using OTS964 could target this synthetic lethal interaction. FBXW7 −/− HPAF‐II cells were isolated and treated with OTS964 in a clonogenic growth assay, and were shown to be hypersensitive to CDK11 inhibition when compared to wild‐type cells (Fig 6A and C). Similarly, two independent CCNL1 OE clones demonstrated enhanced sensitivity to OTS964 when compared to wild‐type cells, suggesting that CCNL1 expression through FBXW7 LOF or genetic amplification sensitizes cells to CDK11 inhibition (Fig 6B and C).
Figure 6. Cells harboring FBXW7‐LOF mutations or CCNL1 overexpression are highly sensitive to CDK11 inhibitor, OTS964.
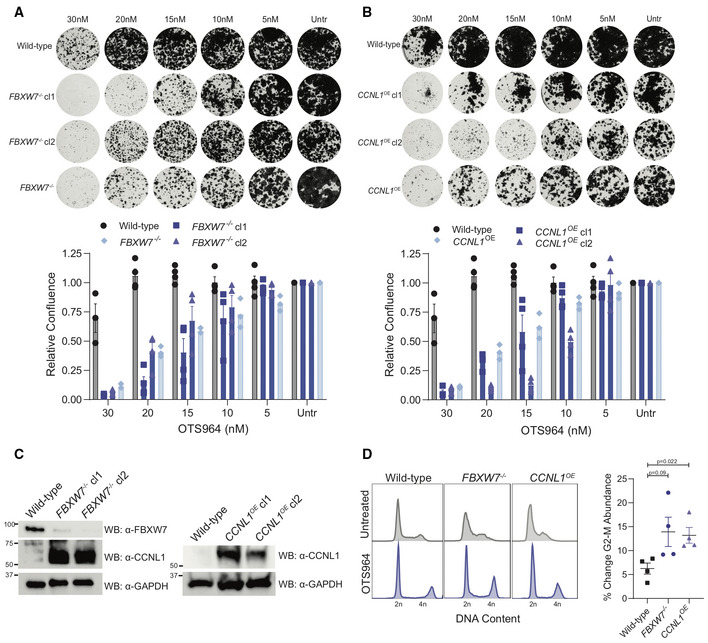
- Clonogenic growth assays for HPAF‐II wild‐type, FBXW7 −/− polyclonal, and FBXW7 −/− clones in the presence of various OTS964 doses for 14 days. Representative images of four independent replicates, quantified by crystal violet absorbance at A595 and plotted mean ± SEM.
- Clonogenic growth assays for HPAF‐II wild‐type, CCNL1 OE polyclonal, and CCNL1 OE clones in the presence of various doses of OTS964 for 14 days. Representative images of four independent replicates, quantified by crystal violet absorbance at A595 and plotted mean ± SEM.
- Immunoblot for the indicated proteins from lysates extracted from the individual FBXW7 −/− and CCNL1 OE clones.
- Cell cycle distribution plots with and without 24 h OTS964 treatment, representative of four independent replicates. Normalized to untreated, mean ± SEM, one‐way ANOVA.
To understand the mechanism behind OTS964 sensitivity, cell cycle profiles of wild‐type, FBXW7 −/−, and CCNL1 OE cells were obtained by flow cytometry following 24 h of OTS964 treatment. We identified that OTS964 leads to the accumulation of cells in the G2‐M phase of the cell cycle, consistent with previous findings (Lin et al, 2019). Interestingly, G2‐M accumulation following OTS964 treatment was higher in FBXW7 −/− and CCNL1 OE cells when compared to wild‐type cells suggesting that hyperactivity of CCNL1:CDK11 complexes in G2‐M in these genotypes represents a tractable therapeutic vulnerability (Figs 6D and EV4B). We conclude that loss of post‐translational control of CCNL1 levels is an oncogenic event that can be preferentially targeted by CDK11 inhibitor in cancer cells and that CCNL1 levels could represent a biomarker to stratify patients or predict patient response.
FBXW7 mutation and CCNL1 amplification are mutually exclusive
Considering the high mutation burden of FBXW7 across a wide range of cancers, and the observation that CCNL1 amplification occurs in many cancer types, we performed an analysis using cBioPortal to determine the probability of a tumor harboring both FBXW7 alteration or CCNL1 amplification. This analysis identified that very few tumors harbor alterations in both FBXW7 and CCNL1, indicating mutual exclusivity and suggesting that these genes function in the same pathway, and that alteration in both would be functionally redundant (Fig 7A).
Figure 7. Cervical cancer cell lines exhibit differential sensitivity to OTS964 depending on FBXW7 mutational status.
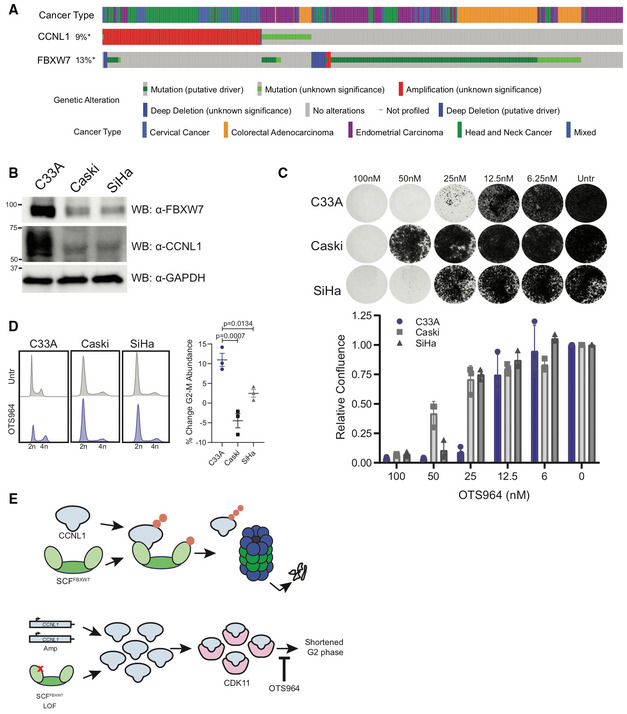
- Oncoprint from cBioPortal demonstrates mutual exclusivity between FBXW7 and CCNL1 alterations.
- Immunoblot of lysates from cervical cell lines demonstrating C33A cells (FBXW7 R465H) express high levels of CCNL1, representative of three independent replicates.
- Clonogenic growth assay of C33A, Caski, and SiHa cells in presence of various doses of OTS964 for 14 days. Representative images of three independent replicates, quantified by crystal violet absorbance at A595 and plotted mean ± SEM.
- Cell cycle distribution plots with and without 24 h OTS964 treatment, representative of three independent replicates. Normalized to untreated, mean ± SEM, one‐way ANOVA.
- Model of proposed mechanism. SCF‐FBXW7 mediated ubiquitination of CCNL1 (indicated by orange circles) promotes proteasomal degradation. LOF mutations in FBXW7 or CCNL1 amplification result in an accumulation of CCNL1/CDK11 complex which shortens the G2 phase of the cell cycle and causes increased sensitivity to OTS964.
FBXW7‐CCNL1‐CDK11 axis is therapeutically relevant in cervical cancer cell lines
Given the high prevalence of FBXW7 mutations and CCNL1 amplification in cervical cancers (Fig 7A), we next screened a genotypically diverse panel of cervical cancer cell lines for their susceptibility to OTS964. The C33A cell line carries a heterozygous FBXW7 R465H LOF mutation and exhibited high levels of CCNL1 expression when compared to Caski and SiHa, which are wild‐type at this locus (Fig 7B). Supporting a deregulation of the CCNL1‐CDK11 axis, the C33A cell line exhibited increased sensitivity to the CDK11 inhibitor OTS964 than the other FBXW7 WT lines Caski and SiHa (Fig 7C). C33A cells showed an accumulation of cells in the G2‐M phase of the cell cycle upon the addition of OTS964, whereas Caski and SiHa cell lines show minimal effect (Figs 7D and EV4C)—validating the sensitivity of this cell line to OTS964 and perturbation of the G2‐M transition. Further, to confirm the on‐target toxicity of OTS964 in C33A cells, we performed a genome‐wide chemogenomic CRISPR screen using an IC50 dose of OTS964; this screen identified CDK11A and CCNL1 as important in mediating response to OTS964 (Fig EV4D). We conclude that CCNL1 levels could represent a biomarker to predict response to CDK11 inhibitors for the treatment of cancers that harbor LOF mutations in FBXW7 or CCNL1 amplification (Fig 7E).
Discussion
The study of the tumor suppressor gene FBXW7 as substrate‐specific receptor of an SCF complex has garnered significant interest, and various oncogenic substrates of FBXW7 have been identified since its discovery (Koepp, 2001; Welcker et al, 2004a, 2004b; Wei et al, 2005; Takada et al, 2017). Mechanistically, the substrate phosphodegron CPD motif has also been studied in depth, which describes the I/L‐I/L/P‐TPXXS motif as the canonical binding moiety required for substrate recognition, in which phosphorylation at the threonine and/or serine residues were described to be important for FBXW7 binding (Nash et al, 2001). The degron motif identified and validated in this work for CCNL1 matches the consensus CPD motif, with the exception that CCNL1 does not contain the I/L‐I/L/P sequence directly upstream of the TPXXS sequence. Our work highlighted a requirement for dual phosphorylation at both the threonine and serine residues within the CCNL1 CPD for recognition by FBXW7, similar to what was observed for c‐Jun (Wei et al, 2005). This contrasts with other FBXW7 substrates such as cyclin E (Fig 3E) and c‐Myc (Welcker et al, 2004b) where phosphorylation at only one site is sufficient for peptide binding. The functional implications of this differential requirement (single vs dual phosphorylation) are currently unknown but suggest a possible additional layer of regulation through the activity of a priming kinase, other signaling events, or additional contact sites. Challenges with expressing and purifying CCNL1 are a core limitation of our study, preventing our ability to further dissect the direct binding interaction between FBXW7 and CCNL1. Additionally, these data support the role of FBXW7 in regulating the G2‐M transition through control of CCNL1 protein levels—a novel role for this ubiquitous E3 ligase.
Despite early evidence that CCNL1 functions as an oncogene in a subset of cancers (Redon et al, 2002; Sticht et al, 2005; Muller et al, 2006), relatively little progress has been made toward understanding the molecular mechanisms underlying its role in tumor development or cell cycle progression. In this study, using an engineered cell model of FBXW7 LOF and cervical cancer cell lines, harboring wild‐type and mutated genotypes for FBXW7, we confirmed the mitotic role of CCNL1 (Renshaw et al, 2019) and revealed a molecular basis for deregulation of the FBXW7‐CCNL1 axis in cancer. Indeed, both FBXW7 LOF mutations and CCNL1 amplification would be predicted to result in the upregulation of CCNL1 levels and hyperactivation of CDK11. Interestingly, FBXW7 mutations and CCNL1 amplification events are mutually exclusive and are highly prevalent in many cancer types. Cervical squamous cell carcinoma appears to be a cancer with frequent alteration in this axis (38% of FBXW7 mutations and 10% of CCNL1 amplification), which could benefit from CDK11 inhibitor treatment or from other therapies directed at this pathway. Uterine carcinosarcoma (UCS), an aggressive gynecological cancer with poor prognosis and treatment options, also presents with high frequency of FBXW7 mutations (38%). This, combined with recent studies in engineered mouse models that suggest concurrent loss of function in FBXW7 and PTEN is a specific driver of UCS tumorigenesis and aggressive tumor behavior (Cuevas et al, 2019) provide further rationale for targeting this molecular perturbation in this disease. CCNL1 amplification, on the other hand, most frequently occurs in squamous cell cancers such as primary lung squamous cell carcinoma (19%), cervical squamous cell carcinoma (10%), as well as head and neck squamous cell carcinoma (9%) (Fig 6A). The CCNL1 gene resides on chromosome 3q25, a region commonly amplified along with PIK3CA (Redon et al, 2001) a known human oncogene. Determining whether CCNL1 amplification is merely a collateral “passenger” event or is required along with PIK3CA for cancer initiation or tumor progression remains to be determined. Whether these tumors are more sensitive to CDK11 inhibitors, such as OTS964, in patients that harbor amplification of the 3q25 genomic region also remains to be tested.
Precision oncology is a burgeoning field currently limited by the scarcity of genetic vulnerabilities identified across multiple cancer types. In addition, the identification of biomarkers that may predict response to targeted treatments is especially important for stratifying patients for clinical trials to assess the true benefit of a new therapeutic modality. This work has identified a novel synthetic lethal genetic interaction that has the potential to impact a broad range of cancer types. In this study, we have demonstrated that CCNL1 expression in cells can predict sensitivity to a novel CDK11 inhibitor, OTS964. Originally identified as a novel inhibitor of the kinase TOPK (T‐LAK cell originating protein kinase; Matsuo et al, 2014), it is unlikely that the activity of OTS964 in our HPAF‐II model is based on TOPK targeting, considering this gene is nonessential in both HPAF‐II wild‐type and FBXW7 −/− cells and that a chemogenomic CRISPR screen confirmed the on‐target activity of OTS964 (Fig EV4D). The importance of CDK11 in cancer progression has been predicted for many years, and our study outlines a novel targeting strategy to guide the use of CDK11 inhibitors.
This work has identified CCNL1 as a novel substrate of tumor suppressor FBXW7, with implications on the growth requirement of cells and tumors harboring FBXW7 mutations. Through uncontrolled CCNL1 expression, the mitotic phase of the cell cycle is shortened as a result of increased CDK11 activity, which sensitizes cells to its inhibition. CCNL1 is therefore another FBXW7 substrate along with cyclin E, c‐Myc, and c‐Jun, which have all been linked to cancer. Understanding the individual roles of these substrates and the cellular and cancer contexts where their deregulation contributes to cancer initiation and progression will be important future work needed to realize the potential of various targeted therapies inhibiting these signaling axes.
Materials and Methods
Cell culture and lentivirus production
HPAF‐II, HEK293T, SiHa, and C33A cells (ATCC) were cultured in DMEM (Gibco) + 10% FBS (Gibco) and 1% antibiotic and antimycotic (Gibco), Caski (ATCC) cells were cultured in RPMI (Gibco) + 10% FBS and 1% antibiotic and antimycotic, all at 37°C and 5% humidity. Cells were routinely tested for mycoplasma (Lonza), and authenticated by STR profiling at The Center for Applied Genomics at Sickkids Hospital, Toronto. HEK293T cells were seeded to 60% confluence, and the following day transfected with 6 μg target plasmid, 6 μg pSPAX (Addgene #12260), and 1 μg pMD2.G (Addgene #12259) in 60 μg polyethylenimine (Sigma‐Aldrich) and Opti‐MEM (Gibco). Twenty‐four hours post‐transfection, media was replaced. Lentivirus was harvested 48 h post‐transfection, filtered through a 0.45 μm filter, and aliquoted and stored at −80°C prior to use.
Cell treatments
Cycloheximide chase: Following overnight serum starvation, cells were released into 50 μg/ml (HEK293T) or 100 μg/ml (HPAF‐II) cycloheximide (Sigma‐Aldrich) for the indicated time points. Clonogenic assays: OTS964 (Selleck Chemicals) was used to treat cells at indicated concentrations for 14 days, with media refreshed every 3–4 days. MG132 (Sigma‐Aldrich) was used at 10 μM in HEK293T cells for 10 h, and 1 μM in HPAF‐II cells for 18 h. Nocodazole (Cell Signaling Technologies) was used at 150 μg/ml for 18 h. Monastrol was used at 150 μM for 18 h. Cells were treated with 500 nM LGK974 for 24 h to assess cell cycle dynamics.
Genome‐wide CRISPR screens
Positive selection
HPAF‐II cells expressing Cas9 were infected with the Toronto knockout library version 1 (TKOv1)—a pooled sgRNA lentiviral library (Hart et al, 2015) at a multiplicity of infection of 0.3, in the presence of 8 μg/ml polybrene (Sigma‐Aldrich) for 24 h. Cells were treated with 2 μg/ml puromycin (Life Technologies) for 48 h. Seven days postselection, cells were split into treatment groups—one using an LD90 dose of LGK974 at 20 nM, and the second a DMSO (Sigma‐Aldrich) control; duplicates were included for both treatment arms. LGK974 treatment was harvested on day 28, and DMSO treatment on day 31. Genomic DNA was extracted using the QIAmp DNA Blood Maxi Kit (Qiagen). Genomic DNA samples were amplified, and barcoded using i5 and i7 adaptor primers for Illumina next‐generation sequencing. Barcoded PCRs were sequenced with the Illumina HiSeq2500. Sequenced gRNAs were mapped to the TKOv1 library using MaGECK 0.5.3, and read counts were normalized by total reads per sample before averaging biological replicates and determining gRNA enrichment.
Dropout
HPAF‐II WT and FBXW7 −/− cells were infected with the Toronto knockout library version 3 (TKOv3)—a pooled sgRNA lentiviral library (Hart et al, 2017) at a multiplicity of infection of 0.3, in the presence of 8 μg/ml polybrene (Sigma) for 24 h. Cells were treated with 2 μg/ml puromycin for 48 h. Following selection, pooled cells were split into three replicates, and passed every 4 days for 24 days, maintaining 18 million cells per replicate. Cell pellets at T = 0, 12, and 24 days were collected, and genomic DNA was extracted using the QIAmp DNA Blood Maxi Kit (Qiagen). Genomic DNA samples were amplified, and barcoded using i5 and i7 adaptor primers for Illumina next‐generation sequencing. Barcoded PCRs were sequenced with the Illumina HiSeq2500 with read depths of 200‐fold coverage. Sequenced gRNAs were mapped to the TKOv3 library using MaGECK 0.5.3 (Li et al, 2014). Read counts were normalized, and the fold change of gRNA distribution compared with T = 0 was calculated using the BAGEL package (Hart & Moffat, 2016). BAGEL analysis was performed, and Bayes factors were compared between HPAF‐II wild‐type and FBXW7 −/− cells. Z‐scores of differential Bayes factors between wild‐type and FBXW7 −/− were calculated.
Chemogenomic
C33A cells were infected with the Toronto knockout library version 3 (TKOv3) at a multiplicity of infection of 0.3, in the presence of 8 μg/ml polybrene (Sigma) for 24 h. Cells were treated with 2 μg/ml puromycin for 48 h. Following selection, pooled cells were split into two arms with two replicates per arm. The first arm was treated with DMSO for 16 days, the second arm was treated with 95 nM of OTS964 for 16 days. Cell pellets at T = 0, 12 and 24 days were collected, and genomic DNA was extracted using the QIAmp DNA Blood Maxi Kit (Qiagen). Genomic DNA samples were amplified, and barcoded using i5 and i7 adaptor primers for Illumina next‐generation sequencing. Barcoded PCRs were sequenced with the Illumina HiSeq2500 with read depths of 200‐fold coverage. Sequenced gRNAs were mapped to the TKOv3 library using MaGECK 0.5.3 (Li et al, 2014). gRNAs inducing resistance or synthetic lethal with OTS964 treatment were assessed using the DrugZ algorithm (Colic et al, 2019).
Generation of FBXW7 and APC mutant cell line
HPAF‐II were transfected via electroporation using the Neon system (ThermoFisher Scientific) under the following conditions; 2 μg of DNA (pX330 [Addgene #42230]—sgFBXW7 or sgAPC, see Oligo table) 1,150 V, 30 ms and 2 pulses. Cells recovered for 2 days in full media before the addition of 100 nM LGK974 (Cayman Chemicals). The polyclonal cell lines were validated for editing using TIDE (tracking of insertions and deletions; Brinkman et al, 2014).
Generation of CCNL1 overexpressing cell line
HPAF‐II wild‐type cells were infected with lentivirus carrying a FLAG‐CCNL1 cDNA, in the pLenti‐puro vector (Addgene #39481). Cells were infected with an ~ 0.3 MOI of lentivirus overnight in the presence of 8 μg/ml polybrene. The following day, virus‐containing media was removed, and cells were selected in 2 μg/ml puromycin for 48 h.
Generation of GFP‐tubulin cell lines
HPAF‐II wild‐type, FBXW7 −/− and CCNL1 OE cells were infected with lentivirus‐carrying tubulin‐GFP cDNA, in the pLKO.1 vector (a kind gift from Dr Jason Moffat). Cells were infected with an ~ 0.3 MOI of lentivirus overnight in the presence of 8 μg/ml polybrene. The following day, virus‐containing media was removed, and cells were selected in 2 μg/ml puromycin for 48 h.
Clone isolation for HPAF‐II FBXW7 −/− and CCNL1 OE
HPAF‐II FBXW7 −/− and CCNL1 OE cells were seeded at 0.5 cells/well in multiple 96‐well plates. Single clones were expanded and tested for FBXW7 knockout and CCNL1 expression by western blot. Two clones were chosen and moved forward to clonogenic growth assays.
Cell competition assay
HPAF‐II wild‐type and FBXW7 −/− cells expressing Cas9 were infected with pLentiguide‐2A‐GFP or pLentiGuide‐2A‐mCherry‐AAVS1 (kind gifts from Dr Daniel Durocher, Lunenfeld‐Tanenbaum Research Institute) lentivirus at an MOI of ~ 0.3 in the presence of 8 μg/ml polybrene. Cells were infected overnight and treated with 2 μg/ml puromycin for 48 h. Following selection cells were left to recover for 24 h. Cells transduced with pLentiguide‐2A‐GFP targeting AAVS1 or CCNL1 were mixed 1:1 with pLentiGuide‐2A‐mCherry‐AAVS1 expressing cells, and GFP:mCherry ratios were measured by flow cytometry (Beckman Coulter CytoFLEX) every 4 days for 16 days. Relative fitness was normalized to AAVS1‐infected cells.
Proliferation assay
HPAF‐II wild‐type and FBXW7 −/− cells expressing Cas9 were infected with pLentiguide‐2A‐GFP virus stocks at an MOI of ~ 0.3 in the presence of 8 μg/ml polybrene. Cells were infected overnight and treated with 2 μg/ml puromycin for 48 h. Following selection, fresh media was added, and cells were left for 24 h to grow. Cells were seeded to ~ 2,500 cells/well in triplicate in a 96‐well plate and left overnight to attach. Plates were moved to the Incucyte (Sartorius) and confluence was tracked over time. Cell confluence in each line was normalized to AAVS1‐infected cells.
Western blotting
All samples were lysed in 4× Laemmli Sample Buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 1% β‐mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue). Lysates were sonicated, boiled, and centrifuged to pellet insoluble material. Approximately 10 μg of protein was loaded per sample on a 4–15% SDS–PAGE Stain‐Free TGX precast gel (BioRad). Gels were run at 150 V for approximately 60 min. Gels were transferred to methanol‐activated PVDF (BioRad) at 90 V for 120 min. Membranes were blocked in 5% milk in Tris‐buffered saline (pH 7.4) + 1% Tween‐20 (TBS‐T) for 1 h, and incubated with corresponding primary antibodies overnight (see Table 1). The following day, membranes were washed 4 times in TBS‐T, and incubated with corresponding secondary antibodies for 1 h, in 5% milk in TBS‐T, at room temperature with agitation. Membranes were washed, and detected using SuperSignal West Pico PLUS chemiluminescent substrate (ThermoFisher) and imaged on the Chemidoc‐MP (BioRad).
Table 1.
Antibody and oligo list.
| Antibody list | ||
|---|---|---|
| Target | Vendor | Cat# |
| Cyclin L1 (Rabbit) | Bethyl Laboratories | A302‐058A |
| FBXW7 (Rabbit) | Bethyl Laboratories | A301‐720A |
| HA‐tag (Rb) | Cell Signaling Technologies | 3724S |
| HA‐tag (Ms) | Cell Signaling Technologies | 2367S |
| Myc‐tag | Cell Signaling Technologies | 2276S |
| GAPDH | Thermo Fisher | AM4300 |
| FLAG‐tag | Thermo Fisher | MA1‐91878 |
| βCatenin | Cell Signaling Technologies | 8480S |
| phospho‐H3 (Ser10) | Cell Signaling Technologies | 9701S |
| Cul1 | Cell Signaling Technologies | 4995S |
| Cul4A | Cell Signaling Technologies | 2699S |
| Oligo list | |
|---|---|
| Oligo | Sequence |
| sgCCNL1‐1 | AAGTTATCAAAGCAGAGAGG |
| sgCCNL1‐2 | TTGAAATCGAACAAACACAT |
| sgPSMD1 | GACCAGAGCCACAATAAGCCA |
| sgAAVS1 | GTCCCCTCCACCCCACAGTG |
| sgFBXW7 | TGGTTCTGAGGTCCGCTCTT |
| FBXW7_TIDE_F | TCACCTTTCCATTCCATTCAGAGT |
| FBXW7_TIDE_R | GAGAAAGGAAGAAATGTCATAACCA |
| sgAPC | ATTTTTAGGTACTTCTCGCT |
| APC_TIDE_F | GGCTGCCACTTGCAAAGTTTC |
| APC_TIDE_R | GATGACTTTGTTGGCATGGCAG |
| sgFBXW7‐Br | ACAGAATTGATACTAACTGG |
| FBXW7_br_TIDE_F | GGGATTGATGAACCATTGCACA |
| FBXW7_br_TIDE_R | GCATTATTTTTCCTGGCTGACGAA |
Cytoplasmic fractionation
800,000 cells from each condition were lysed in ice‐cold cytoplasmic extraction buffer (10 mM HEPES pH8, 1.5 mM MgCl2, 10 mM NaCl, 0.5 mM DTT, 1 mM EDTA) and incubated on ice for 15 min. NP‐40 was added to a final concentration of 0.05%, lysates were mixed thoroughly, and the insoluble fraction was collected by centrifugation. Cytoplasmic fraction was quantified using the Qubit (ThermoFisher) Protein quantification kit, and stored at −80°C until western blotting.
Quantification
All western blot quantification was performed by densitometry in ImageJ (FIJI).
Live‐cell imaging
GFP‐tubulin‐expressing cells were plated into 8‐well chamber slides and left overnight to adhere. Cells were then incubated with 150 μM monastrol (Selleck Chemicals) overnight. The following day, monastrol was washed out, and cells were imaged every 10 min for 8 h on the Evos FL Auto2 (ThermoFisher) at 20× magnification, at 37°C with 5% CO2. Representative movies were imaged at 37°C and 5% CO2 on a laser scanning confocal microscope (LSM700, Carl Zeiss) at 8‐bit with Plan‐Apochromat 63×/1.4NA oil immersion objective using Zen software. Z‐stacks were captured every 15 min for 8 h. Images were compiled in ImageJ (FIJI).
PIP‐FUCCI imaging
PIP‐FUCCI expressing cells were seeded into 6‐well plates. Cells were then treated with 150 nM nocodazole overnight. The following day, nocodazole was removed, and cells were imaged every 30 min in the Incucyte (Sartorius). mCherry expression was quantified within the incucyte software and plotted over time.
Two‐dimensional cell cycle flow cytometry
Cells were grown in logarithmic proliferation and harvested using 0.025% Trypsin–EDTA. Cells were washed in ice‐cold PBS, fixed in 70% ethanol under vortex, and stored at −20°C overnight. The following day, cells were washed 2× in ice‐cold PBS and solubilized in PBS + 1% BSA + 0.15% Triton‐X for 15 min on ice. Cells were washed and incubated with anti‐phosphoH3 (Ser10) (CST) antibody for 1.5 h on ice. Cells were washed 2× in PBS + 1% BSA and incubated with anti‐Rb‐Alexa488 (ThermoFisher) for 1 h on ice. Cells were washed and incubated with 20 μg/ml RNAse A (Invitrogen) and 50 μg/ml propidium iodide (BioShop) for 30 min prior to acquisition on a Beckman Coulter CytoFLEX flow cytometer. Cells were gated for singlets, and the cell cycle phase was determined using the intensity in the PE channel. G2‐M cells were quantified by gating on all 4N within the pH3+ region.
Nocodazole release cell cycle flow cytometry
Cells were treated with 150 μg/ml nocodazole (Cell Signaling Technology) for 18 h. Following synchronization, cells were released into the full medium. Samples were collected for western blotting and flow cytometry at indicated time points. For flow cytometry, samples were trypsinized, collected, and washed twice in PBS. Cells were fixed with ice‐cold 70% ethanol under vortex, and stored at −20°C. Cells were washed twice in PBS and stained in 50 μg/ml propidium iodide (BioShop) in 25 nM RNAse A (Invitrogen) in PBS. Samples were run on a Beckman Coulter CytoFLEX flow cytometer. Cells were gated for singlets, and the cell cycle phase was determined using the intensity of propidium iodide in the PE channel.
Immunoprecipitations
Following treatments, 15 cm plates were scraped on ice in 1 ml PBS and cells collected. Pellets were stored at −80°C until processing. For nondenaturing lysis, pellets were resuspended in RIPA buffer (0.1% SDS, 0.1% NP‐40, 2 mM EDTA,150 mM NaCl, 50 mM Tris–HCl pH7.6, 0.5% sodium deoxycholate, 1× protease inhibitor, 10 mM NaF, 0.25 mM NaOVO3), for denaturing lysis, SDS was increased to 1%. Lysates were sonicated and cleared at 20,000 g for 20 min. Antibodies or FLAG‐beads (Sigma‐Aldrich) were added to lysates (denaturing lysates first diluted to 0.1% SDS) and incubated at 4°C with end‐over‐end rotation for 3 h. Pre‐equilibrated Protein‐G‐conjugated agarose beads (Roche) were added for 1 h. Beads were collected, washed several times in lysis buffer, and boiled at 95°C for 5 min in 4× Laemmli buffer. Samples were stored at −20°C until western blotting.
Cellular ubiquitination assay
HEK293T cells were transfected with plasmids carrying indicated cDNAs, using PEI. The medium was changed the following day. On day 2, cells were starved overnight through the removal of FBS from media. Following overnight starvation, cells were treated with 10 μM MG132 (in full media) for 8 h. Cells were scraped in ice‐cold PBS, and stored at −80°C prior to processing. Lysates were processed as per Immunoprecipitation protocol (denaturing lysis), with the following adjustments: Following lysis, samples were boiled at 90°C for 10 min, prior to sonication. Elution from Protein‐G beads was performed by boiling at 55°C for 5 min.
Fluorescence polarization assay
FITC‐CCNL1321–332 peptides (numbering according to Uniprot Q9UK58‐1) were purchased from GenScript and FITC‐cyclin E377–384 peptides (numbering according to Uniprot P24864‐3) were purchased from BioBasic. Experiments were performed by combining 25 nM FITC‐conjugated peptides and the indicated amount of Skp1‐FBXW7263–707 complex in buffer containing 25 mM HEPES pH 7.5, 100 mM NaCl, 5 mM DTT, 0.01% Brij‐35 and 0.1 mg/ml BSA. Mixed samples (25 μl total volume) were incubated for 30 min in 384‐well, black, flat‐bottom, low‐flange plates (Corning, 3573). Fluorescence intensities were measured using a BioTek Synergy Neo plate reader with excitation and absorbance at 485/528 nm, respectively. Fluorescence polarization was calculated with the Gen5 Data Analysis Software. Binding constants for three independent experiments were calculated using GraphPad Prism v8.2.1 (GraphPad) with mean and standard deviation being reported in the figure.
Statistical tests
All statistical analyses were performed in GraphPad Prism. Data are represented as a mean ± SEM of at least three independent biological replicates.
Author contributions
Siobhan O'Brien: Conceptualization; data curation; formal analysis; investigation; visualization; writing—original draft; writing—review and editing. Susan Kelso: Formal analysis; investigation; writing—review and editing. Zachary Steinhart: Conceptualization; investigation. Stephen Orlicky: Investigation; methodology. Monika Mis: Conceptualization; investigation; methodology; writing—review and editing. Yunhye Kim: Investigation. Sichun Lin: Investigation. Frank Sicheri: Supervision; funding acquisition; writing—original draft; writing—review and editing. Stephane Angers: Conceptualization; supervision; funding acquisition; writing—original draft; writing—review and editing.
Disclosure and competing interests statement
SO, ZS, and SA are inventors on a patent involving the use of OTS964 in FBXW7‐mutated cancers (WO2021108927A1). FS is a founder and consultant at Repare Therapeutics.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Dataset EV1
PDF+
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research (PJT‐148691 to SA; FDN 143277 to FS), the Canadian Cancer Society (CCSRI)‐Impact grant to FS (704116) and SA (705045), and the Terry Fox Research Institute (to FS). FS is a Canada Research Chair. SO was supported by the Ontario Graduate Scholarship and the Centre for Pharmaceutical Oncology scholarships. The authors would like to thank Andrew Wilde, Michael Ohh, and Brian Raught for reagents, Kin Chan at the LTRI sequencing facility, Azza Al‐Mahrouki at the CPO facility, and all members of the Angers lab for support and helpful discussions.
EMBO reports (2022) 23: e55044
Data availability
This study includes no data deposited in external repositories. All CRISPR screening data can be found in Dataset EV1.
References
- Ahmed RL, Shaughnessy DP, Knutson TP, Vogel RI, Ahmed K, Kren BT, Trembley JH (2019) CDK11 loss induces cell cycle dysfunction and death of BRAF and NRAS melanoma cells. Pharmaceuticals 12: 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben‐Shushan E, Mann M, Ben‐Neriah Y, Alkalay I (2002) Axin‐mediated CKI phosphorylation of beta‐catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16: 1066–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Y, Zhang J, Nicholas SA, Kim AL, Zhou P, Goff SP (2007) DDB1 is essential for genomic stability in developing epidermis. Proc Natl Acad Sci USA 104: 2733–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐H, Wang Y‐C, Fann M‐J (2006) Identification and characterization of the CDK12/Cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol 26: 2736–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐H, Wong Y‐H, Geneviere A‐M, Fann M‐J (2007) CDK13/CDC2L5 interacts with L‐type cyclins and regulates alternative splicing. Biochem Biophys Res Commun 354: 735–740 [DOI] [PubMed] [Google Scholar]
- Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D et al (2019) Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med 11: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R (2000) Identification and characterization of a novel cell cycle–regulated internal ribosome entry site. Mol Cell 5: 597–605 [DOI] [PubMed] [Google Scholar]
- Cuevas IC, Sahoo SS, Kumar A, Zhang H, Westcott J, Aguilar M, Cortez JD, Sullivan SA, Xing C, Hayes DN et al (2019) Fbxw7 is a driver of uterine carcinosarcoma by promoting epithelial‐mesenchymal transition. Proc Natl Acad Sci USA 116: 25880–25890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GD, Kedziora KM, Limas JC, Cook JG, Purvis JE (2018) Accurate delineation of cell cycle phase transitions in living cells with PIP‐FUCCI. Cell Cycle 17: 2496–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta‐Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israël A (2001) Functional interaction between SEL‐10, an F‐box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem 276: 34371–34378 [DOI] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP (2007) Structure of a Fbw7‐Skp1‐cyclin E complex: multisite‐phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell 26: 131–143 [DOI] [PubMed] [Google Scholar]
- Hart T, Moffat J (2016) BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics 17: 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet‐Turcotte A, Sun S et al (2015) High‐resolution CRISPR screens reveal fitness genes and genotype‐specific cancer liabilities. Cell 163: 1515–1526 [DOI] [PubMed] [Google Scholar]
- Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A et al (2017) Evaluation and design of genome‐wide CRISPR/SpCas9 knockout screens. G3 (Bethesda) 7: 2719–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Hao H‐X, Growney JD, Woolfenden S, Bottiglio C, Ng N, Lu B, Hsieh MH, Bagdasarian L, Meyer R et al (2013) Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 110: 12649–12654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J‐X, Sun C‐Y, Tian S, Yu C, Chen M‐Y, Zhang H (2016) Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting β‐catenin for degradation in pancreatic cancer. Tumour Biol 37: 13893–13902 [DOI] [PubMed] [Google Scholar]
- Koepp DM (2001) Phosphorylation‐dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294: 173–177 [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS (2014) MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, Sausville EL, Lukow DA, Liu L, Chait AR et al (2019) Off‐target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 11: eaaw8412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 108: 837–847 [DOI] [PubMed] [Google Scholar]
- Liu X, Gao Y, Shen J, Yang W, Choy E, Mankin H, Hornicek FJ, Duan Z (2016) Cyclin‐dependent kinase 11 (CDK11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol Cancer Ther 15: 1691–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb KR, Kostner H, Firpo E, Norwood T, D Tsuchiya K, Clurman BE, Roberts JM (2005) A mouse model for cyclin E‐dependent genetic instability and tumorigenesis. Cancer Cell 8: 35–47 [DOI] [PubMed] [Google Scholar]
- Loyer P, Trembley JH (2020) Roles of CDK/Cyclin complexes in transcription and pre‐mRNA splicing: cyclins L and CDK11 at the cross‐roads of cell cycle and regulation of gene expression. Semin Cell Dev Biol 107: 36–45 [DOI] [PubMed] [Google Scholar]
- Mao J‐H, Perez‐Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A (2004) Fbxw7/Cdc4 is a p53‐dependent, haploinsufficient tumour suppressor gene. Nature 432: 775–779 [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Sedivy JM (1999) c‐Myc regulates cyclin D‐Cdk4 and ‐Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol 19: 4672–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Park J‐H, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, Nakamura Y (2014) TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci Transl Med 6: 259ra145 [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ (1999) Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype‐based screen. Science 286: 971–974 [DOI] [PubMed] [Google Scholar]
- Minella AC, Grim JE, Welcker M, Clurman BE (2007) p53 and SCFFbw7 cooperatively restrain cyclin E‐associated genome instability. Oncogene 26: 6948–6953 [DOI] [PubMed] [Google Scholar]
- Minella AC, Loeb KR, Knecht A, Welcker M, Varnum‐Finney BJ, Bernstein ID, Roberts JM, Clurman BE (2008) Cyclin E phosphorylation regulates cell proliferation in hematopoietic and epithelial lineages in vivo. Genes Dev 22: 1677–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Mazumder Indra D, Basu PS, Mondal RK, Roy A, Roychoudhury S, Panda CK (2010) Amplification of CyclinL1 in uterine cervical carcinoma has prognostic implications. Mol Carcinog 49: 935–943 [DOI] [PubMed] [Google Scholar]
- Muller D, Millon R, Théobald S, Hussenet T, Wasylyk B, du Manoir S, Abecassis J (2006) Cyclin L1 (CCNL1) gene alterations in human head and neck squamous cell carcinoma. Br J Cancer 94: 1041–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M (2001) Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414: 514–521 [DOI] [PubMed] [Google Scholar]
- Nateri AS, Riera‐Sans L, Da Costa C, Behrens A (2004) The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 303: 1374–1378 [DOI] [PubMed] [Google Scholar]
- Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U (2001) The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel‐10 homolog. J Biol Chem 276: 35847–35853 [DOI] [PubMed] [Google Scholar]
- Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M (1995) Human cyclin E, a nuclear protein essential for the G1‐to‐S phase transition. Mol Cell Biol 15: 2612–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R et al (2007) FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma‐secretase inhibitors. J Exp Med 204: 1813–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- Perez‐Roger I, Kim SH, Griffiths B, Sewing A, Land H (1999) Cyclins D1 and D2 mediate myc‐induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 18: 5310–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C (2004) Inactivation of hCDC4 can cause chromosomal instability. Nature 428: 77–81 [DOI] [PubMed] [Google Scholar]
- Redon R, Muller D, Caulee K, Wanherdrick K, Abecassis J, du Manoir S (2001) A simple specific pattern of chromosomal aberrations at early stages of head and neck squamous cell carcinomas: PIK3CA but not p63 gene as a likely target of 3q26‐qter gains. Cancer Res 61: 4122–4129 [PubMed] [Google Scholar]
- Redon R, Hussenet T, Bour G, Caulee K, Jost B, Muller D, Abecassis J, du Manoir S (2002) Amplicon mapping and transcriptional analysis pinpoint cyclin L as a candidate oncogene in head and neck cancer. Cancer Res 62: 6211–6217 [PubMed] [Google Scholar]
- Reed SE, Spruck CH, Sangfelt O, van Drogen F, Mueller‐Holzner E, Widschwendter M, Zetterberg A, Reed SI (2004) Mutation of hCDC4 leads to cell cycle deregulation of cyclin E in cancer. Cancer Res 64: 795–800 [DOI] [PubMed] [Google Scholar]
- Renshaw MJ, Panagiotou TC, Lavoie BD, Wilde A (2019) CDK11p58–cyclin L1β regulates abscission site assembly. J Biol Chem 294: 18639–18649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck CH, Won K‐A, Reed SI (1999) Deregulated cyclin E induces chromosome instability. Nature 401: 297–300 [DOI] [PubMed] [Google Scholar]
- Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, Robitaille M, Brown KR, Jaksani S, Overmeer R et al (2017) Genome‐wide CRISPR screens reveal a Wnt‐FZD5 signaling circuit as a druggable vulnerability of RNF43‐mutant pancreatic tumors. Nat Med 23: 60–68 [DOI] [PubMed] [Google Scholar]
- Sticht C, Hofele C, Flechtenmacher C, Bosch FX, Freier K, Lichter P, Joos S (2005) Amplification of Cyclin L1 is associated with lymph node metastases in head and neck squamous cell carcinoma (HNSCC). Br J Cancer 92: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, Schreiber EM, Day BW, Liu B (2008) APC is essential for targeting phosphorylated beta‐catenin to the SCFbeta‐TrCP ubiquitin ligase. Mol Cell 32: 652–661 [DOI] [PubMed] [Google Scholar]
- Takada M, Zhang W, Suzuki A, Kuroda TS, Yu Z, Inuzuka H, Gao D, Wan L, Zhuang M, Hu L et al (2017) FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2‐mediated phosphorylation of CENP‐A. Cancer Res 77: 4881–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Orlicky S, Lin Z, Willems A, Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F, Tyers M (2007) Suprafacial orientation of the SCFCdc4 dimer accommodates multiple geometries for substrate ubiquitination. Cell 129: 1165–1176 [DOI] [PubMed] [Google Scholar]
- Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ (2004) Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F‐box protein. Proc Natl Acad Sci USA 101: 3338–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rechem C, Black JC, Abbas T, Allen A, Rinehart CA, Yuan G‐C, Dutta A, Whetstine JR (2011) The SKP1‐Cul1‐F‐box and leucine‐rich repeat protein 4 (SCF‐FbxL4) ubiquitin ligase regulates lysine demethylase 4A (KDM4A)/Jumonji domain‐containing 2A (JMJD2A) protein. J Biol Chem 286: 30462–30470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG (2005) The v‐Jun point mutation allows c‐Jun to escape GSK3‐dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8: 25–33 [DOI] [PubMed] [Google Scholar]
- Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83–93 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Grim JA, Eisenman RN, Clurman BE (2004a) A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c‐Myc and cell size. Curr Biol 14: 1852–1857 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Grim JA, Harper JW, Eisenman RN, Clurman BE (2004b) The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation‐dependent c‐Myc protein degradation. Proc Natl Acad Sci USA 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh C‐H, Bellon M, Nicot C (2018) FBXW7: a critical tumor suppressor of human cancers. Mol Cancer 17: 115 [DOI] [PMC free article] [PubMed] [Google Scholar]