

Abstract
Germinal centers (GC) are microstructures where B cells that have been activated by antigen can improve the affinity of their B cell receptors and differentiate into memory B cells (MBCs) or antibody‐secreting plasma cells. Here, we have addressed the role of activation‐induced deaminase (AID), which initiates somatic hypermutation and class switch recombination, in the terminal differentiation of GC B cells. By combining single cell transcriptome and immunoglobulin clonal analysis in a mouse model that traces AID‐experienced cells, we have identified a novel subset of late‐prePB cells (L‐prePB), which shares the strongest clonal relationships with plasmablasts (PBs). Mice lacking AID have various alterations in the size and expression profiles of transcriptional clusters. We find that AID deficiency leads to a reduced proportion of L‐prePB cells and severely impairs transitions between the L‐prePB and the PB subsets. Thus, AID shapes the differentiation fate of GC B cells by enabling PB generation from a prePB state.
Keywords: activation‐induced deaminase, germinal center, plasma cell, single cell sequencing
Subject Categories: Development; DNA Replication, Recombination & Repair; Immunology
A combination of single cell transcriptome and immunoglobulin gene sequencing identifies a novel pre‐plasmablast/plasma cell (L‐prePB) subset. Activation‐induced deaminase (AID) deficiency reduces this L‐prePB subset and the transition toward PB/PC cells.
Introduction
During the immune response, B cells that have been stimulated by antigen with T cell help can engage in the germinal center (GC) reaction, where they can differentiate into either memory B cells (MBC) or high‐affinity plasma cells (PC). GCs are key to the efficiency of the immune response and underlie the mechanism of action of most vaccination strategies. In GCs, B cells proliferate, modify their immunoglobulin genes by somatic hypermutation (SHM), are selected by affinity maturation, and terminally differentiate into alternate fates (Allen et al, 2007; Victora & Nussenzweig, 2012; Mesin et al, 2016; Shlomchik et al, 2019; Laidlaw & Cyster, 2020).
Activation‐induced deaminase (AID) initiates SHM and CSR (Muramatsu et al, 2000; Revy et al, 2000) with the deamination of cytosines on the DNA of immunoglobulin genes, which can be subsequently processed by various molecular pathways (Methot & Di Noia, 2017). In the case of SHM, AID deaminates the antigen recognizing, variable region of immunoglobulin genes, generating mutations that can give rise to variants with altered affinity for antigen (Methot & Di Noia, 2017). In CSR, AID‐induced deaminations promote a recombination reaction between switch regions—highly repetitive sequences that precede constant regions—thus promoting the exchange of IgM/IgD isotypes with IgG, IgE, or IgA isotypes encoded by downstream constant genes at the immunoglobulin heavy (IgH) locus (Methot & Di Noia, 2017). Therefore, immunoglobulin diversification by AID is central to the GC reaction.
Germinal centers comprise two different compartments, the dark zone (DZ), where B cells proliferate and undergo SHM in their variable genes, and the light zone (LZ), where B cells are selected in the context of T follicular helper cells and follicular dendritic cells (Allen et al, 2007; Victora & Nussenzweig, 2012). CSR can take place in the GC but frequently occurs prior to its entry into the GC (Roco et al, 2019). B cells perfection affinity maturation with iterative cycles of mutation and proliferation in the DZ and positive selection in the LZ (Victora et al, 2010; Victora & Nussenzweig, 2012, 2022).
Notably, differentiation into the MBC and the PC fates from the GC is not stochastic; instead, higher affinity B cells preferentially differentiate into PCs, while MBCs generally show lower affinity for antigen (Phan et al, 2006; Taylor et al, 2015; Mesin et al, 2016; Shinnakasu et al, 2016; Kräutler et al, 2017; Suan et al, 2017; Viant et al, 2020, 2021). Accordingly, the average frequency of SHM is higher in PCs than in MBCs (Shinnakasu et al, 2016; Weisel et al, 2016; Laidlaw et al, 2020). Likewise, BCR isotype influences the outcome of GC differentiation toward the PC or MBC fates (Kometani et al, 2013; Gitlin et al, 2016; King et al, 2021). This skewed selection into the MBC versus PC fate ensures high‐affinity protection by the PC effector compartment while preserving an MBC reservoir with a broader range of affinities, which could be critical to provide a rapid defense against closely‐related pathogens, as previously proposed (Kaji et al, 2012; Viant et al, 2020; Victora & Nussenzweig, 2022).
Activation‐induced deaminase deficiency does not only promote a complete block in CSR and SHM, but also it causes lymphoid hyperplasia, both in mouse and man (Muramatsu et al, 2000; Revy et al, 2000), indicating a role of AID in B cell homeostasis. Indeed, AID−/− mice have an increased number of GC B cells upon immunization (Muramatsu et al, 2000). Interestingly, AID−/− GC B cells show a reduced apoptosis rate (Zaheen et al, 2009), accumulate in the LZ, and do not efficiently form PCs in mixed bone marrow chimeras (Boulianne et al, 2013). However, the contribution of AID to shaping B cell fate in GCs is not well understood.
Here, we have approached this question by combining single cell transcriptome analysis with single cell V(D)J analysis of B cells from wild‐type and AID‐deficient mice. To that end, we have used a genetic mouse model that irreversibly labels cells that have expressed AID. We found that AID‐experienced B cells clustered into 8 distinct transcriptional clusters, including a novel L‐prePB cluster, which shares a strong clonal relationship with PB. The GC response in AID deficient mice showed alterations in cluster proportions and transcriptome differences in some of these clusters. Further, clonal relationships were profoundly altered in AID deficient mice, where the connection between L‐prePB and PB clusters was severely impaired. Thus, our data reveal a critical role of AID in shaping the ultimate fate of B cell differentiation in GCs.
Results
Single cell analysis of the GC response identifies 8 transcriptional clusters
To map GC differentiation at the single cell level, we made use of the AicdaCre +/ki ; R26tdTom +/ki (hereafter AicdaCre/+) mouse model, which allows genetic tracing of cells that have expressed AID. In this model, the cDNA encoding the Tomato (Tom) fluorescent protein was inserted in the Rosa26 endogenous locus preceded by a transcriptional stop sequence flanked by loxP sites (R26tdTom allele). The Cre recombinase was inserted in the endogenous Aicda locus (AicdaCre allele; Robbiani et al, 2008; Rommel et al, 2013). In AicdaCre/+ mice, activation of the Aicda locus promotes Cre expression and excision of the transcriptional stop at the R26tdTom allele, unleashing the expression of the Tom protein. Thus, B cells that have been activated for AID expression, become irreversibly Tom+ (Fig EV1A). To trigger a GC immune response, we first adoptively transferred CD4+ T cells from OT‐II mice, which harbor a TCR recognizing a peptide from the ovalbumin (OVA) protein, into AicdaCre/+ mice. Mice were immunized with OVA 1 and 15 days after the transfer of OT‐II cells. Mice were sacrificed for analysis 15 days after the second immunization (Fig 1A). Flow cytometry analysis showed that OVA immunization expectedly resulted in the generation of Tom+ cells, which comprised GC B cells (Tom+ CD138− GL7+), plasmablasts (PB) and PCs (Tom+ CD138+) and putative memory B (pMem) cells (Tom+ CD138− GL7− CD38+), with various proportions of switched cells (complete gating strategy in Figs 1B and C, and EV1B and C). ELISA analysis showed an accumulation of anti‐OVA IgG antibody titers (Fig EV1D).
Figure EV1. Mouse model and experimental design.
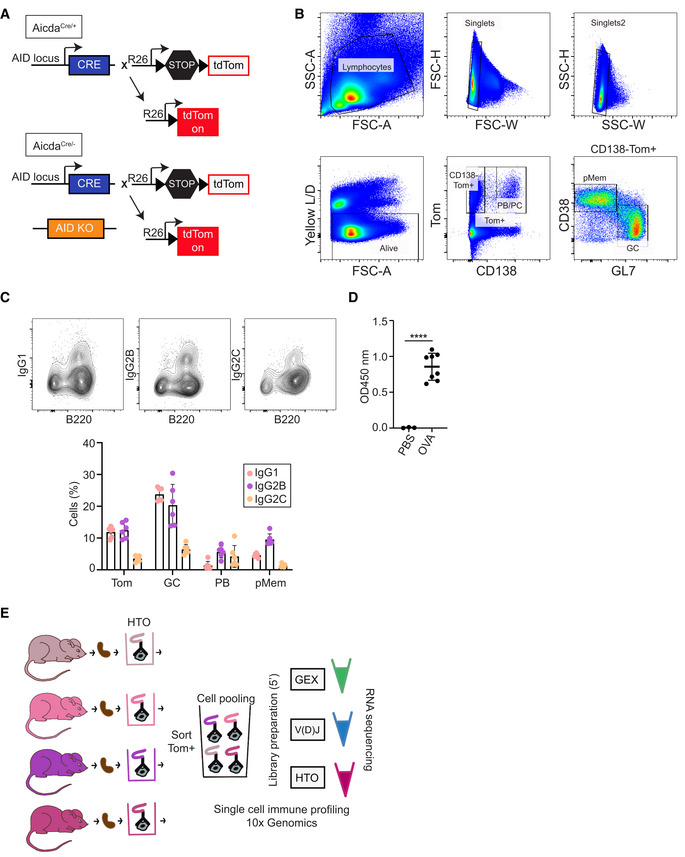
- Genetic model to study the GC reaction with a fluorescent tracer. AicdaCre+/ki; R26tdTom+/ki (AicdaCre/+), and AicdaCre−/ki; R26tdTom+/ki (AicdaCre/−) mice are shown.
- Complete gating strategy for B cell subsets analyzed in Fig 1B and C.
- FACS representative plots and quantification of IgG1, IgG2B, and IgG2C within B cell populations in Fig 1C (n = 6 AicdaCre/+ mice).
- Antibody titers specific for OVA were measured in the plasma of control AicdaCre/+ mice (PBS; n = 3) and immunized AicdaCre/+ mice (OVA; n = 8) by ELISA. Statistics were calculated with the paired t‐test. ****P < 0.0001. n indicates biological replicates.
- Experimental approach followed for single cell RNA sequencing.
Data information: Bars and error bars indicate mean ± standard deviation.
Figure 1. Single cell RNA sequencing of AID‐labeled Tomato+ (Tom+) cells identifies eight cell clusters.
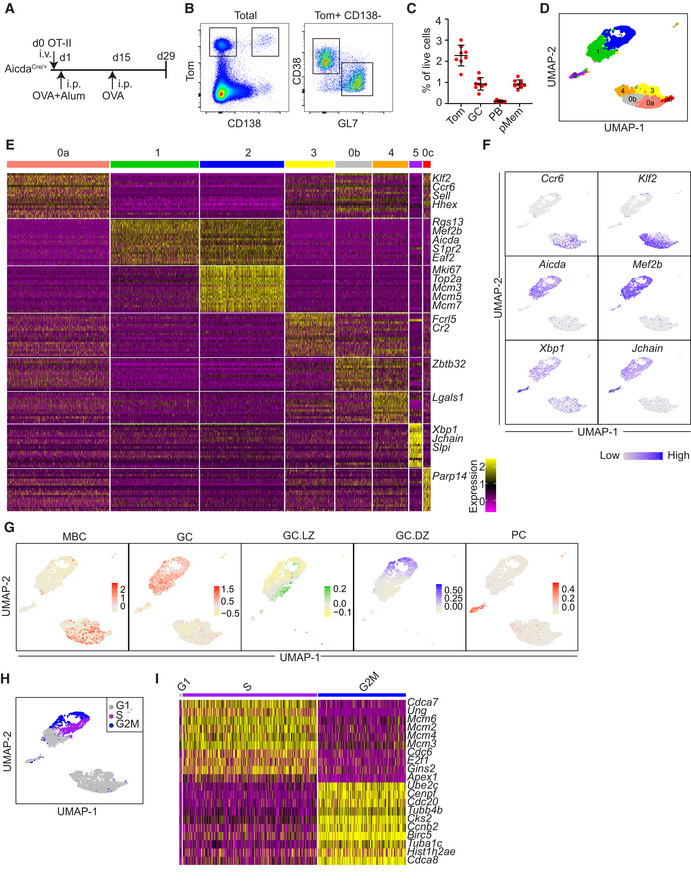
- Immunization protocol. AicdaCre/+ mice were immunized intraperitoneally (i.p.) with OVA in alum (n = 8) 1 day after OT‐II CD4+ T cell transfer. Two weeks later, mice were boosted with OVA i.p.
- Representative flow cytometry plots of spleen Tom+ cells, germinal center B cells (GC; Tom+ CD138− GL7+), plasma cells/plasmablasts (PB; Tom+ CD138+) and putative memory B cells (pMem; Tom+ CD138− GL7− CD38+).
- Quantification of flow cytometry analysis of immunized mice as shown in A and B (n = 8 mice per group). Percentages of the different subsets within total live cells are shown. Data information: Bars and error bars indicate mean ± standard deviation.
- Splenic Tom+ cells from two immunized AicdaCre/+ mice were analyzed by single cell RNA sequencing (scRNA‐seq) using the 10x Genomics platform. Cells were clustered based on transcriptomic data and mapped to a UMAP plot. Clusters are labeled from 0 to 5 according to decreasing cell numbers. Cluster 0 was subclusterized in 0a, 0b and 0c (Fig EV2A).
- Heatmap showing expression of the top 20 upregulated genes within the clusters identified in D. Yellow indicates higher gene expression. Representative gene names are indicated on the right.
- UMAP plots showing expression of representative genes of the different B cells clusters, as shown in D. Blue color indicates higher gene expression.
- UMAP plot showing the cell cycle phase of individual cells in the different clusters as shown in D.
- Heatmap of G1, S, and G2M subclusters of GC.DZ cells showing expression of the top 10 upregulated genes in S and G2M phases. Gene expression information was obtained for 4,061 AicdaCre/+ Tom+ cells. Cluster 0: 1425 [0a: 998, 0b:354, 0c:73], 1: 867, 2: 820, 3: 480, 4:345, 5:124. See Materials and Methods for details.
To analyze the B cell immune response at the single cell level we performed 10× Genomics analysis in Tom+ spleen cells isolated from OT‐II transferred AicdaCre/+ mice 15 days after the boost OVA immunization (Fig 1A). Two individual immunized mice were multiplexed by hashtag labeling (HTO, see Materials and Methods) and gene expression and V(D)J sequencing of individual cells was performed (Fig EV1E). Seurat clustering of gene expression sequencing of individual Tom+ cells initially identified 6 independent clusters, labeled from 0 to 5 according to cluster size (Fig EV2A). Further subclustering of cluster 0 resulted in 8 distinct transcriptional clusters, as explained below (Fig 1D).
Figure EV2. Cluster analysis.
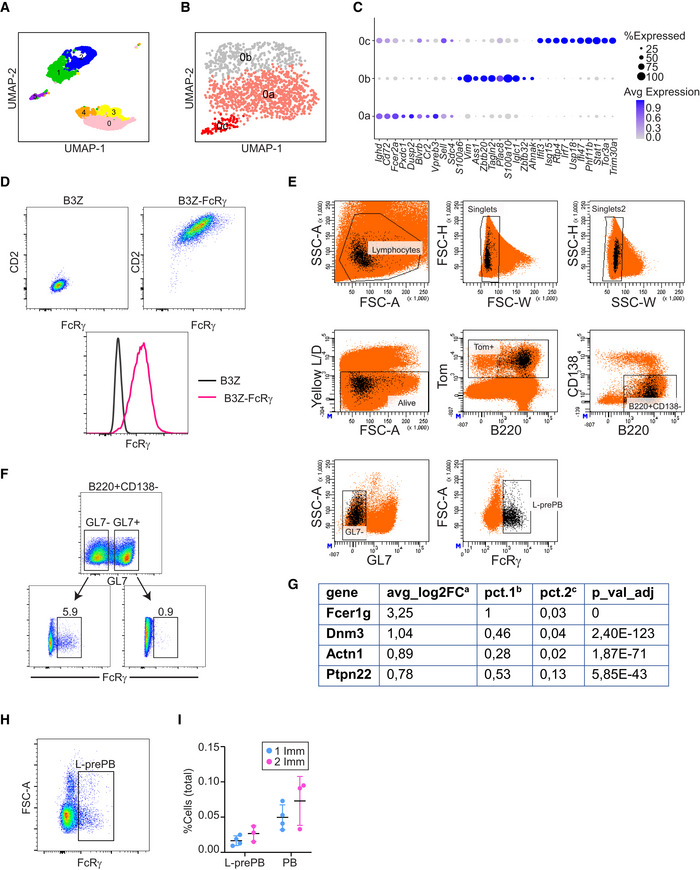
- UMAP plot showing transcriptional clusters obtained before cluster 0 subclustering.
- UMAP plot showing three transcriptionally distinct subclusters (0a, 0b, and 0c).
- Dot plot depicting the expression levels of the top 10 upregulated genes in clusters 0a, 0b, and 0c.
- Anti‐FcRγ antibody test. Representative flow cytometry plots of B3Z (NIH) parental cells (left) and FcRγ‐CD2‐transfected cell lines stained with anti‐FcRγ and anti‐CD2 antibodies.
- Gating strategy for L‐prePB identification by flow cytometry and cell sorting. L‐prePB backgating shown in black.
- Expression analysis of the indicated genes obtained by scRNA‐seq shown in Fig 3A. aAverage log2 fold change between the two groups being compared. bProportion of cells expressing the indicated gene within FcRγ+ cells. cProportion of cells expressing the indicated gene within the non‐FcRγ+ cells.
- FACS representative plot for L‐prePB staining in the spleen of AicdaCre/+ mice 2 weeks after a single OVA immunization.
- Anti‐FcRγ staining in GL7− and GL7+ cells within live, singlets, Tom+, CD138−, B220+ gated cells.
- AicdaCre/+ mice (n = 7) were immunized with OVA following the protocol in Fig 1A. Four mice were sacrificed 2 weeks after the first immunization. The proportion of prePB (Tom+ B220+ CD138− GL7− FcRγ+) and PB (Tom+ CD138+) cells was determined by flow cytometry within total live cells.
Data information: Bars and error bars indicate mean ± standard deviation.
Clusters 1 and 2 showed high levels of Aicda, S1pr2, or Mef2b and were both enriched in GC B cell signature (Fig 1E–G). Cluster 1 and cluster 2 were distinctly enriched for LZ and DZ signatures, as defined before (Victora et al, 2010, 2012; Fig 1G; Dataset EV1). We found that the vast majority of proliferating Tom+ cells were contained in cluster 2, and conversely, virtually all the cells (99%, 811/820 cells) in cluster 2 were in the S+G2M phases of the cell cycle (Fig 1H and I). UMAP projection precisely distinguished between cells in the S phase expressing high levels of replication genes (Mcm2, Mcm3, Mcm4, Mcm6, Cdc6, etc) and cells in the G2 and M phases, with high expression of mitotic genes (Cdc20, Ccnb2, Cdca8, etc; Fig 1H and I). Thus, cluster 1 was designated as LZ GC B cells (GC.LZ) and cluster 2 was designated as DZ GC B cells (GC.DZ).
Cluster 5 displayed high levels of Xbp1, Jchain, and immunoglobulin genes (Fig 1E and F; Dataset EV1) and was enriched in cells expressing the PB/PC signature as defined in the Immunological Genome Project gene set (Heng et al, 2008; Fig 1G) and was thus designated as PB.
Cluster 0 showed high expression of Klf2 and Ccr6 (Fig 1E and F; Dataset EV1), previously associated with the MBC transcriptional program (Suan et al, 2017; Laidlaw et al, 2020). Signature enrichment analysis of MBC TFs (Glaros et al, 2021) further supported the MBC identity of cluster 0 (Fig 1G). Subclusterization of cluster 0 identified three transcriptionally distinct populations: one major subset (0a) with highest expression levels of Klf2, Ccr6, and Hhex, one subset (0b) with highest levels of Zbtb32 and Vim, and a minor subset (0c) with highest levels of Irf7 and Isg15 (Figs 1D and E, and EV2B and C; Dataset EV1). We found that subcluster 0a was enriched in the gene signature of a recently identified subset of MBCs that derive from activated B cells (Viant et al, 2021); conversely, subcluster 0b was enriched in a distinct MBC signature of cells that originate from highly proliferative GC precursors (Viant et al, 2021; Fig 2A). Thus, we have identified two major subsets of MBCs: cluster 0a, hereafter, Mem.Act, and cluster 0b, hereafter, Mem.GC. Finally, 0c is a minor subcluster showing more association with activated B cell‐derived MBC signature and will be labeled as Mem.Act2.
Figure 2. Identification of two prePB clusters.
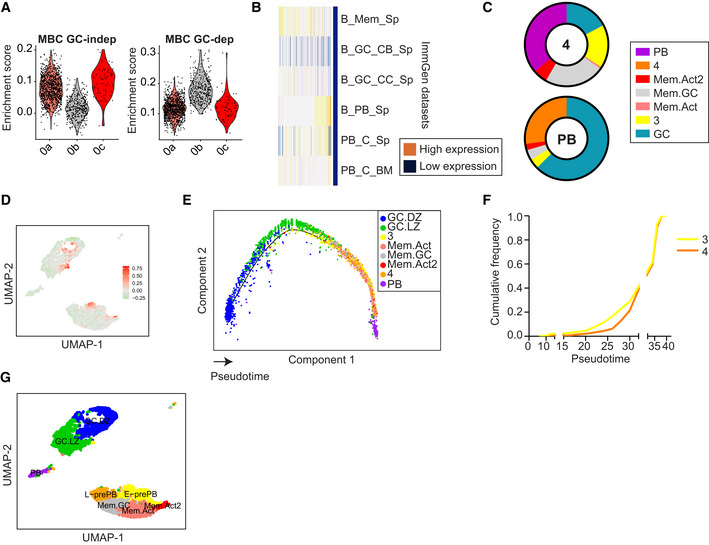
- Enrichment scores of gene signatures derived from GC‐independent and GC‐dependent memory B cell populations described in (Viant et al, 2021).
- Heatmap showing expression levels of cluster 4 markers in different Immunological Genome Project (ImmGen) datasets (Heng et al, 2008).
- Pie charts depicting the proportion of shared markers between cluster 4 and PB cluster with the rest of the clusters, as analyzed by Quickmarkers.
- UMAP plot showing enrichment score for the top 100 upregulated genes in Fraction 1 prePB cells (Ise et al, 2018).
- Monocle Pseudotime analysis of the cells displayed in Fig 1D. The projection is colored by cluster identity and the cells are ordered by Pseudotime.
- Cumulative frequency of cluster 3 and cluster 4 cells across Pseudotime.
- UMAP plot of cells in Fig 1D with assigned cell identities.
Identification of two prePB clusters
Clusters 3 and 4 were identified as distinct clusters that could not obviously be assigned as MBC, GC, or PC cells (Fig 1G; Dataset EV1). However, the transcriptional profile of cluster 4 partially resembled PB and MBC Immunological Genome Project gene sets (Heng et al, 2008; Fig 2B). To get insights into the transcriptional inter‐relationships of cluster 4 with other clusters, we performed an analysis of transcriptional transitions using QuickMarkers (Dataset EV2). This approach quantifies the proportion of markers in a given cluster shared with the highest frequency by the rest of the clusters. We found that cluster 4 showed the highest transition probability with the PB cluster, and conversely, the PB cluster showed a high transcriptional transition probability with cluster 4, only second to GC cells (Fig 2C). These analyses suggested that cluster 4 could represent a subset related with PB differentiation. This prompted us to ask whether this subset was similar to a previously described subset of prePB cells (Fraction 1; Ise et al, 2018). Interestingly, gene signature analysis using the 100 most highly expressed genes by Fraction 1 prePB cells (Ise et al, 2018) did not bear a high enrichment in cluster 4 (Fig 2D). Instead, we found that the highest enrichment of the Fraction 1 prePB signature was split into two transcriptional clusters. Expectedly, one of them was a subset of the LZ cluster, in agreement with the initial definition of Fraction 1 prePB cells (Fig 2D). Notably, the other high enrichment hit of Fraction 1 prePB cells was with our cluster 3. Together, these analyses suggested that clusters 3 and 4 could represent 2 distinct subsets of prePB cells.
To further assess the differentiation trajectories of the identified transcriptional clusters, we performed Monocle pseudotime analysis (Fig 2E). We expectedly found that GC.DZ and GC.LZ clusters represented the earliest differentiation states in the pseudotime analysis, while the PB cluster was the end differentiation state of the analysis. MBC clusters were interspread between GC and PB clusters. Interestingly, cluster 3 showed as an earlier state than cluster 4 (Fig 2E and F). Thus, cluster 3 was tentatively labeled as early‐prePB (E‐prePB) while cluster 4 was tentatively labeled as late‐prePB (L‐prePB).
We conclude that single cell transcriptome analysis identified 8 distinct GC‐related populations, including GC.DZ, GC.LZ, PB, 3 clusters of MBC, and 2 clusters of putative prePB cells, namely, E‐prePB, which are closely related to previously described fraction 1 prePB cells (Ise et al, 2018), and a novel prePB cluster that we have named L‐prePB (Fig 2G).
Characterization of the L‐prePB cluster
To gain insights into the identity of cells in the L‐prePB cluster, we first identified genes in our transcriptome data that could be potentially used as markers for L‐prePB cells. We found that FcRγ, Actn1, Dnm3, and Ptpn22 are expressed by cells in the L‐prePB cluster but very rarely expressed in other clusters (Fig 3A). Interestingly, all FcRγ, Actn1, Dnm3, and Ptpn22 are more highly expressed in PC/PB than in GC or MBC subsets, as defined in the Immunological Genome Project (Heng et al, 2008; Fig 3B), supporting the transcriptional link between L‐prePB and PB cells. Further analysis of our transcriptome data with the Combinatorial Marker Detection from Single Cell Transcriptomic Data (COMET tool; Delaney et al, 2019) identified FcRγ as the best putative marker for the L‐prePB cluster. FcRγ (high‐affinity immunoglobulin epsilon receptor subunit gamma) is a component of various membrane receptors, including high‐affinity IgE receptor, endowed with a signal transducing ITAM motif and a short extracellular segment. To assess the identity of L‐prePB cells by flow cytometry, we first performed intracellular staining of spleen cells from mice immunized as shown in Fig 1A using an anti‐FcRγ antibody. This antibody was tested in B3Z cells transfected with an FcRγ‐CD2 fusion protein following the same fixation/permeabilization protocol (Fig EV2D). We found that a fraction of non‐PC, non‐GC, Tom+ B cells (Tom+ B220+ CD138− GL7− CD19+) expressed FcRγ (Fig 3C), consistent with the presence of L‐prePB cells (complete gating strategy in Fig EV2E). We also confirmed the absence of FcRγ+ cells within the GC compartment (Fig EV2F). We next isolated FcRγ+ cells by cell sorting and performed quantitative RT–PCR of the L‐prePB markers identified in our transcriptome data. We found that all FcRγ, Actn1, Dnm3, and Ptpn22 genes were more highly expressed in FcRγ+ cells than in total Tom+ cells, confirming that FcRγ+ cells are part of the L‐prePB cluster (Figs 3D and EV2G). Finally, we verified that FcRγ+ cells were also detectable in mice who received a single immunization challenge (Fig EV2H and I). Thus, we conclude that the L‐prePB cluster represents a common B cell subset associated with T cell‐dependent responses that can be identified by the expression of FcRγ.
Figure 3. L‐prePB cells can be identified by the expression of FcRγ.
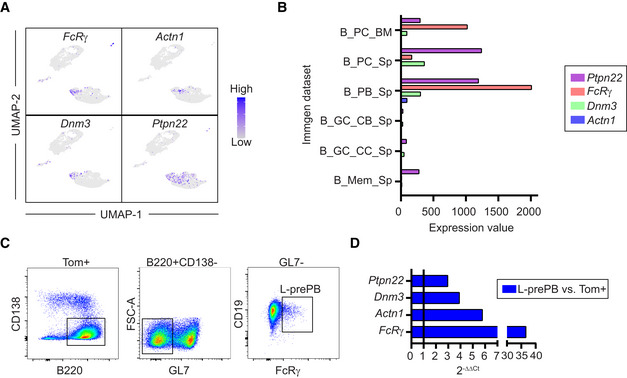
- UMAP plots showing expression of representative genes of the L‐prePB cluster. Blue color indicates higher gene expression.
- Barplot showing expression levels of representative L‐prePB markers in different Immgen datasets (Heng et al, 2008).
- Gating strategy for L‐prePB identification by flow cytometry.
- Validation of the markers shown in A using RT–qPCR. Relative quantification comparing expression levels in prePB and total Tom+ cells by 2−ΔΔCt method.
Single cell clonal analysis of the GC response
To further characterize the identified transcriptional clusters, we first analyzed SHM in V(D)J transcripts of individual cells. We found that the mutation load at the IgH variable region widely varied across cells from different populations, with the highest mutation frequency found in both GC.DZ and GC.LZ B cells (Figs 4A–C and EV3A). Within MBC clusters, Mem.Act and Mem.Act2 harbored lower mutation frequencies than Mem.GC, in agreement with their labeling as MBCs from activated B cell and GC origin, respectively (Viant et al, 2021; Figs 4A–C and EV3A). Finally, we found that cells in the L‐prePB and PB clusters had similar mutation frequencies (Figs 4A–C and EV3A), and that mutation load was significantly higher in L‐prePB than in the E‐prePB compartment. CSR analysis on V(D)J transcripts generally mirrored the SHM results, with the highest proportion of isotype switched cells within the GC.DZ and GC.LZ clusters (Figs 4D and E, and EV3B–D).
Figure 4. Single cell analysis of SHM, CSR, and clonal relationships during the GC reaction.
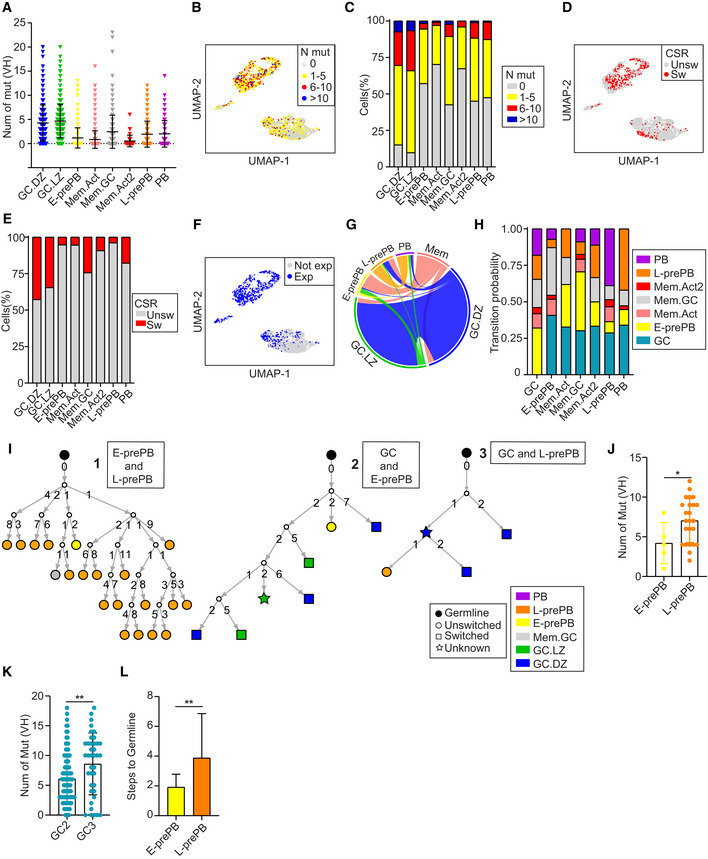
- Total number of somatic mutations at the IgH variable region (VH) in individual cells from the different B cell clusters. Symbols represent individual cells. Statistics were calculated with the Kruskal–Wallis test using the Dunn's multiple comparison test (P‐values shown in Fig EV3A).
- UMAP plot showing mutational load in single cells of the clusters defined in Fig 1D.
- Quantification of data shown in B.
- UMAP plot showing CSR of individual cells (Unsw, unswitched; Sw, switched).
- Proportion of CSR in individual clusters (Unsw, unswitched; Sw, switched).
- UMAP plot showing the distribution of clonally expanded cells among clusters (Exp, expanded; Not exp, not expanded).
- Circos plot displaying pairwise clonal overlap between B cell clusters. Only clones shared between 2 clusters are shown. For the sake of clarity, Mem.Act, Mem.GC and Mem.Act2 clusters are shown together as Mem.
- Transition probabilities among the different clusters, based on the frequency of clonal sharing between 2 or more clusters.
- Representative phylogenetic trees of the three categories analyzed in panels J‐L. Type 1 trees represent clonal families containing both E‐ and L‐prePB cells. Types 2 and 3 contain GC B cells and either E‐ or L‐prePB cells, respectively. Numbers in branches depict the mutation load acquired in each diversification event. Each branch represents a diversification step, as quantified in panel L.
- Quantification of the number of mutations in E‐ (n = 5 cells) and L‐prePB (n = 23 cells) belonging to clones of type 1 trees (n = 3). n refers in all cases to biological replicates.
- Quantification of the number of mutations in GC B cells in type 2 trees (n = 12 trees; GC2, n = 98 cells) and type 3 trees (n = 9 trees; GC3, n = 39 cells). n refers in all cases to biological replicates.
- Quantification of the diversification steps of E‐prePB cells (n = 13) and L‐prePB cells (n = 17) belonging to clones from type 2 trees (n = 12) and type 3 trees (n = 9). n refers in all cases to biological replicates.
Data information: Bars and error bars indicate mean ± standard deviation. Statistics were calculated with an unpaired t‐test. *P ≤ 0.05, **P < 0.01. V(D)J information was obtained for 2,677 AicdaCre/+ Tom+ cells (see Materials and Methods for details).
Figure EV3. SHM and CSR analysis.
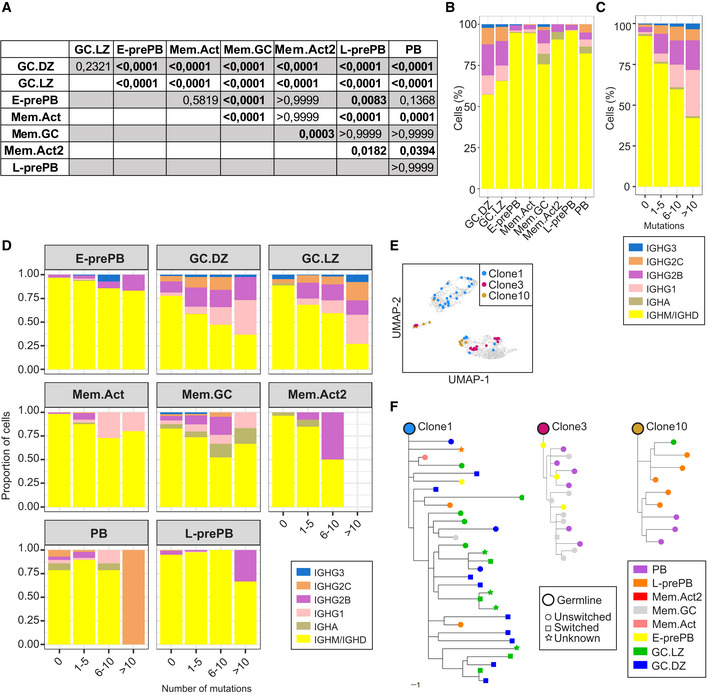
-
AP‐values for the SHM data shown in Fig 4A. Statistics were calculated with the Kruskal–Wallis test.
-
BQuantification of the different isotypes within B cell clusters.
-
C, DIsotype quantification in total B cells (C) and individual clusters (D) according to their different mutational load.
-
EUMAP plot showing 3 representative expanded clones (clone1, clone3, and clone10).
-
FTrees showing phylogenetic relationships between IgH sequences of clone1, clone3, and clone10 of panel E. Scale bar: 1 mutation.
To identify clonal relationships among the different clusters we performed the analysis of BCR sequences using the Immcantation pipeline. Cells were assigned to the same clone when they shared identical V(D)J segments and identical CDR3 lengths in both their IgH and IgL chains. We found that GC.DZ and GC.LZ clusters were highly enriched in clonally expanded cells (Fig 4F). Likewise, clonal sharing was most frequently observed between the GC.DZ and the GC.LZ clusters (Fig 4G) (note that the circos plot represents all Mem clusters together for the sake of clarity) and that only clonal sharing between two clusters is depicted. These findings reflect the high proliferation rate at the DZ and the iterative transitions of B cells between the DZ and the LZ in GC. Our data also revealed clonal sharing among other clusters. For instance, clone 1 (containing 31 cells) was found several times in the GC.DZ and GC.LZ clusters, both in unswitched and in switched versions, but it was also found in the E‐prePB and L‐prePB clusters (Fig EV3E and F). By contrast, we found that clone 3 was shared between the E‐prePB and the PB clusters (Fig EV3E and F). Thus, clone 3 probably represents the end differentiation state of a clonal expansion that is already extinguished at the GC; by contrast, clone 1 must have been at full expansion in the GC at the time of analysis, but we can also identify cells that have already transited to the E‐prePB or the L‐prePB clusters.
V(D)J clonal sharing (Fig 4G) allows us to calculate the transition probabilities across differentiation states (Fig 4H; Dataset EV3). We observed that the GC cluster interconnects with all other clusters, suggesting that GC cells can feed other subsets, and possibly also receive cells from different MBC subsets (Fig 4G and H). Interestingly, the PB cluster shows the most prominent relationships with the GC and the L‐prePB cluster. This finding indicates that, while differentiation into the PB cluster can directly occur from the GC and Mem clusters, L‐prePB is a very common intermediate state preceding the generation of PC/PB.
To deepen into the clonal relationships of E‐prePB and L‐prePB cells, we analyzed the phylogenetic trees of all the clones that contained E‐prePB or L‐prePB cells (representative trees are shown in Fig 4I). Branches of the trees represent diversification steps and numbers on branches indicate the number of mutations acquired at that step. Trees were classified as type 1 when they contained both E‐prePB and L‐prePB cells; type 2, when they contained GC B cells and E‐prePB cells; and type 3, when they contained GC B cells and L‐prePB cells. We found that, within the same clone, E‐prePB cells harbored fewer mutations than L‐prePB cells (Fig 4J), in agreement with our analysis on total mutation frequency (Fig 4A–C). Interestingly, this mutation difference was also found in their GC counterparts (trees type 2 and 3), i.e., GC cells in trees shared with E‐prePB (GC2, type 2 trees) have fewer mutations than GC cells in trees shared with L‐prePB (GC3, type 3 trees; Fig 4K). In turn, lower mutation load in E‐prePB‐containing clones correlates with fewer diversification steps from the inferred germline cell (Fig 4L). These results indicate that E‐prePB cells represent earlier, less diversified cells than L‐prePB cells.
Transcriptional clusters are shifted in AID deficient mice
To address the role of AID in GC differentiation at the single cell level, we generated AicdaCre −/ki; R26tdTom +/ki mice (hereafter AicdaCre/− mice), where one Aicda allele is disrupted by the Cre recombinase and the other one is a knock‐out allele (Fig EV1A; Muramatsu et al, 2000). Thus, AID deficient, GC‐associated B cells can be genetically traced by the expression of the Tom protein. Control AicdaCre/+ mice and AID deficient AicdaCre/− mice were immunized with OVA as above (Fig 1A) and the GC response in the spleen was assessed by flow cytometry. We found that immunized AicdaCre/− spleens had a higher proportion of Tom+ cells, in agreement with the GC expansion found in AID deficient mice (Fig 5A; Muramatsu et al, 2000; Zaheen et al, 2009). Within Tom+ cells, the proportion of GC was not altered. Likewise, the proportion of Mem cells (Tom+ CD138− GL7− CD38+ FcRγ−) was not significantly different in AID proficient and deficient mice, while the proportion of PB was reduced (from 5 to 2.9%), consistently with previous reports (Boulianne et al, 2013; Fig 5B). Interestingly, Tom+ CD138− GL7− FcRγ+ L‐prePB cells were also reduced in AID deficient mice (Fig 5B, from 2.2 to 1.4%). In addition, the DZ to LZ ratio was reduced in AicdaCre/− mice, as previously reported (Boulianne et al, 2013; Fig 5C and D). Immunization of AicdaCre/+ and AicdaCre/− mice with NP‐CGG also revealed a reduction in the fraction of L‐prePB cells in AID deficient mice (from 1.8% in AicdaCre/+ mice to 0.9% in AicdaCre/− mice), confirming that this effect is not specific for our immunization protocol with OVA (Fig EV4A–D).
Figure 5. AID deficiency alters GC differentiation.
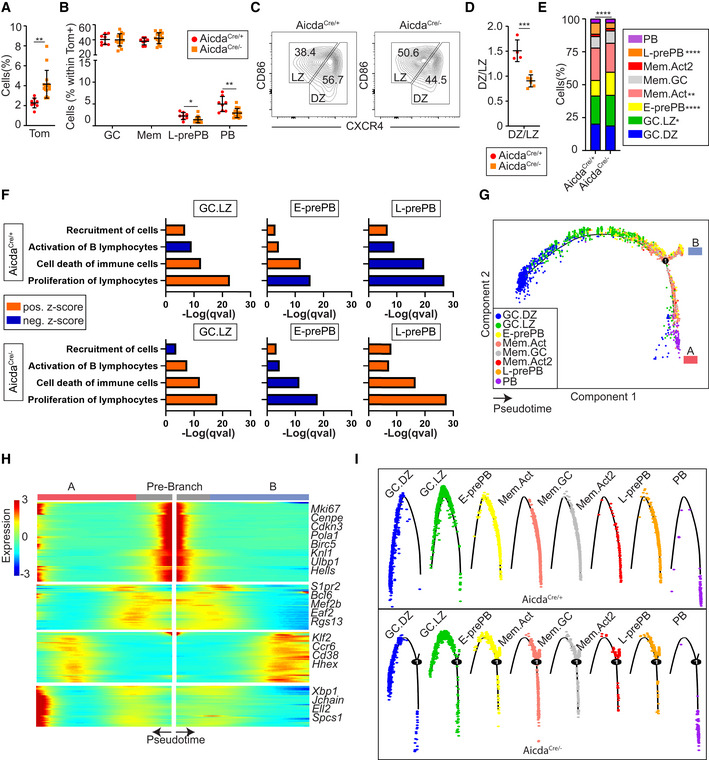
- AicdaCre/− mice were immunized with OVA following the protocol in Fig 1A (n = 15). Spleen Tom+ cells were quantified and compared with OVA‐immunized AicdaCre/+ mice.
- The proportion of GC B cells (Tom+ B220+ CD138− GL7+), PB (Tom+ CD138+), Mem (Tom+ B220+ CD138− GL7− CD38+ FcRγ−), and L‐prePB (Tom+ B220+ CD138− GL7− FcRγ+) was determined by flow cytometry within total Tom+ cells in AicdaCre/+ (n = 8) and AicdaCre/− (n = 15) mice.
- Representative flow cytometry plots of dark zone (DZ; B220+ Tom+ GL7+ CXCR4hiCD86lo) and light zone (LZ; B220+ Tom+ GL7+ CXCR4loCD86hi) GC B cells.
- Quantification of DZ/LZ ratio for AicdaCre/+ (n = 4) and AicdaCre/− (n = 6) mice immunized with OVA.
- Spleen Tom+ cells from two immunized AicdaCre/− mice were analyzed by scRNA‐seq. Cluster labels from AicdaCre/+ mice (Fig 2G) were transferred to AicdaCre/− cells. Bar plot showing the proportions of the different B cell clusters in AicdaCre/+ and AicdaCre/−.
- Comparative pathway enrichment analysis of differentially expressed genes (DEGs) in GC.LZ, E‐prePB, and L‐prePB clusters in AicdaCre/+ and AicdaCre/− mice. Analysis was performed with Ingenuity Pathway Analysis (IPA). Terms are colored by z‐score (activation status). Positive values of z‐score indicate the activation of the pathway, while negative values indicate pathway inhibition. Benjamini‐Hochberg adjusted P‐values (qval) are represented.
- Monocle Pseudotime analysis of AicdaCre/− cells. The projection is colored by cluster identity. Number in the circle identifies the branching point for branches A and B.
- Heatmap of Pseudotime gene expression changes, showing representative DEGs between branches A and B. Genes are clustered hierarchically in modules with similar branch‐dependent expression patterns.
- Mapping of cluster identities in the Pseudotime plots of AicdaCre/+ (upper) and AicdaCre/− (bottom) cells.
Data information: Bars and error bars indicate mean ± standard deviation. Statistics were calculated with an unpaired t‐test (A, B, and D), chi‐square (E, all clusters), and Fisher test (E, individual clusters). *P ≤ 0.05, **P < 0.01 and ***P < 0.001. Gene expression information was obtained for 3,923 AicdaCre/− Tom+ cells. Cluster 0: 1301 [0a: 862, 0b: 368, 0c: 71], 1: 913, 2: 738, 3: 689, 4: 169, 5: 114. (See Materials and Methods for details).
Figure EV4. L‐prePB identification in NP‐CGG immunized mice.
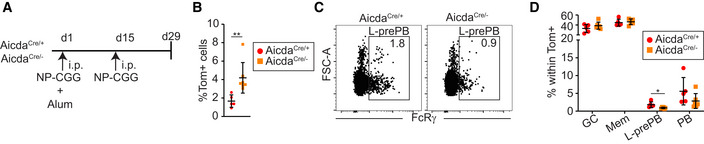
- Immunization protocol. AicdaCre/+ (n = 5) and AicdaCre/− (n = 7) mice were immunized intraperitoneally (i.p.) with NP‐CGG in alum. Two weeks later, mice were boosted with NP‐CGG i.p.
- Quantification of spleen Tom+ cells in NP‐CGG immunized AicdaCre/+ (n = 5) and AicdaCre/− (n = 7) mice.
- Representative flow cytometry plots of L‐prePB staining (Tom+ B220+ CD138− GL7− FcRγ+) in AicdaCre/+ and AicdaCre/− mice.
- The proportion of GC B cells (B220+ Tom+ CD138− GL7+), PB (Tom+ CD138+), Mem (Tom+ B220+ CD138− GL7− CD38+ FcRγ−), and L‐prePB (Tom+ B220+ CD138− GL7− FcRγ+) was determined by flow cytometry within total Tom+ cells in AicdaCre/+ (n = 5) and AicdaCre/− (n = 7) mice.
Data information: Bars and error bars indicate mean ± standard deviation. Statistics were calculated with an unpaired t‐test. *P ≤ 0.05, **P < 0.01.
To further characterize the GC disturbances in AID deficient mice, we performed a single cell analysis of Tom+ cells using 10x Genomics. Transfer of the identified cell type labels from AicdaCre/+ mice onto AicdaCre/− transcriptome data showed that AicdaCre/− mice had altered proportions of several subsets (Fig 5E). Specifically, in addition to confirming the increase in GC.LZ cells, we found that the proportion L‐prePB cells was severely diminished, in agreement with the flow cytometry data shown in Fig 5B, and that the proportion of E‐prePB cells was increased in AicdaCre/− mice (Fig 5E).
Changes in subset proportions were accompanied by transcriptional alterations in several biological pathways (Fig 5F). Notably, in AicdaCre/− mice the apoptotic signaling pathway was reduced in E‐prePB cells but increased in L‐prePB cells (Fig 5F), suggesting that AID deficiency plays a critical role in prePB cell survival and thus in the transition to PB differentiation. In line with this idea, pseudotime analysis of AID deficient cells revealed a branch point in a late GC state (Fig 5G) that corresponds with distinct cell fates (Fig 5H). Thus, the mainstream branch (A) leads to the generation of PB cells, while branch B fails to give rise to the PB state and is instead biased toward the MBC fate (Fig 5I). Together, these results indicate that AID deficiency has profound consequences in B cell fate and differentiation during the GC response, by compromising the generation of L‐prePB cells and instead promoting the accumulation of E‐prePBs.
AID deficiency alters B cell differentiation during the GC response
To approach the consequences of AID deficiency in the clonal relationships and in the differentiation fate in GCs, we combined transcriptomics with V(D)J analysis in single cells. Clonal analysis of Tom+ cells showed that AicdaCre/− mice had more and larger expanded clones (Fig 6A and B). Increased clonal expansion in AicdaCre/− mice was not common to all cell clusters; rather, it was specifically observed in GC.DZ, E‐prePB, Mem.Act and Mem.GC clusters (Fig 6C), indicating that AID can impact on proliferation, survival, or differentiation at distinct stages of the GC reaction, in agreement with our transcriptome data. Analysis of clonal sharing between different clusters in AicdaCre/− mice expectedly showed the greatest interconnection between GC.DZ and GC.LZ clusters, similarly to our findings in AicdaCre/+ mice (Figs 6D and EV5A and B). Pairwise clonal relationships between clusters were represented in circos plots where GC.LZ and GC.LZ cells are pooled into a single GC subset, so that their pairwise relationships do not dominate the representation. We found major alterations in AicdaCre/− mice compared with AicdaCre/+ mice (Fig 6E); most notably, the clonal sharing of L‐prePB and PB cells was severely reduced and the E‐prePB to PB was increased in AicdaCre/− mice (Fig 6E). To specifically quantify these alterations, we used clonal sharing frequencies (Dataset EV3) to calculate the transition probabilities between any given cluster and all the rest (Figs 6F and G, and EV5C). We found that, in AicdaCre/− mice, the L‐prePB to PB transition is very much reduced, and instead, the transition probability between E‐prePB and PB is enhanced. These results indicate that, in the absence of AID, differentiation into PB is inefficient due to a block at the L‐prePB state, and this deficiency is partially compensated by an increased differentiation of PB from the E‐prePB state.
Figure 6. Altered B cell fate decisions in AID deficient mice.
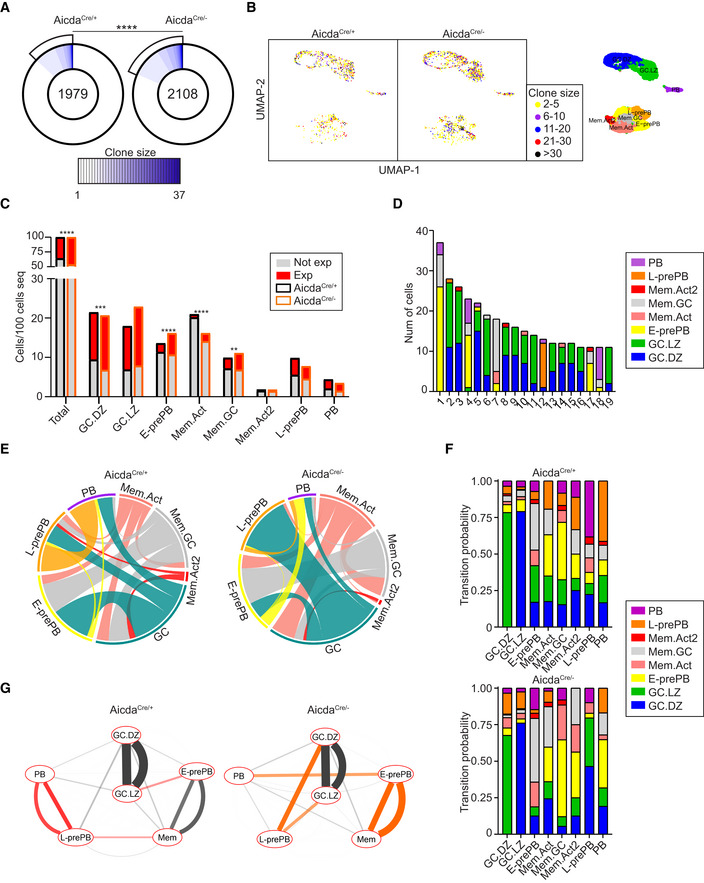
- Pie charts depicting the clonal size of Tom+ cells from immunized AicdaCre/+ and AicdaCre/− mice. Clones are defined as cells sharing identical IGH and IGL V and J sequences and CDR3 lengths. Number in the inner circle indicates the total number of unique sequences. White represents sequences found only once, and blue gradient represents increasing clonal sizes. Pie sector sizes are proportional to clone sizes. The two‐sided Fisher's exact test was used to compare the proportions of expanded and non‐expanded clones. ****P < 0.0001.
- UMAP plot showing clone sizes in AicdaCre/+ and AicdaCre/− mice. Cells are colored according to the size of the clone they belong to. Unique events are not shown. For reference, UMAP plot of transcriptional clusters is shown on the right.
- Clonal expansion in transcriptional clusters. Quantification of expanded and non‐expanded cells is shown for individual clusters, normalized to the total number of cells sequenced per genotype. The two‐sided Fisher's exact test was used to compare the proportions of expanded versus non‐expanded clones in each cluster. **P < 0.01, ***P < 0.001 and ****P < 0.0001.
- Circos plots showing pairwise clonal sharing between B cell clusters in AicdaCre/+ and AicdaCre/− mice. GC.LZ and GC.DZ populations were grouped and shown as GC for the sake of simplicity. Complete clonal sharing relationships are shown in Fig EV5B.
- Transition probabilities among the different clusters in AicdaCre/+ and AicdaCre/−, calculated as the frequency of clonal sharing between 2 or more clusters.
- Cytoscape representation of cluster interactions in AicdaCre/+ and AicdaCre/− immune response, based on their clonal sharing probabilities. For each pairwise transition, two lines are shown, which depict the probabilities with respect to each cluster of the pair. Red and orange connecting lines show the most increased cluster relationships in AicdaCre/+ and AicdaCre/− mice, respectively. Mem.Act, Mem.GC, and Mem.Act2 clusters are shown together for simplicity. See Fig EV5C for complete interactions. V(D)J information was obtained for 3,178 AicdaCre/− Tom+ cells (see Materials and Methods for details).
Figure EV5. Comparative clonal analysis of AicdaCre/+ and AicdaCre/− mice.
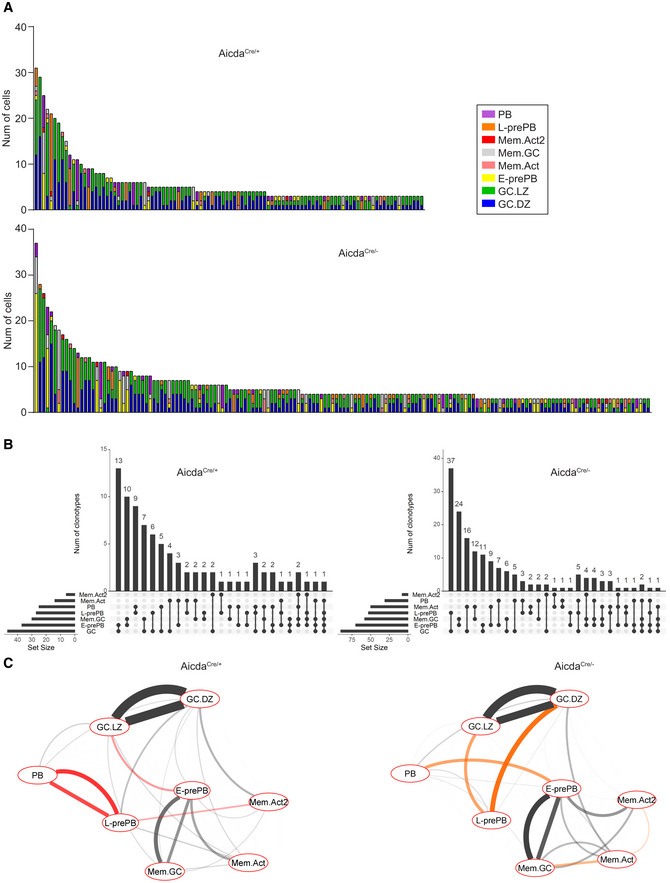
- Bar plot depicting the contribution of the different transcriptional clusters in expanded clones with more than 2 cells in AicdaCre/+ (top) and AicdaCre/− (bottom) mice.
- UpSet plots showing quantification of clonal overlap between clusters identified in Fig 1D in AicdaCre/+ (left) and AicdaCre/− (right). GC.LZ and GC.DZ populations were grouped and shown as GC for the sake of clarity.
- Cytoscape representation of all cluster interactions in AicdaCre/+ and AicdaCre/− immune response, based on their clonal sharing probabilities. Red and orange connecting lines show the most increased cluster relationships in AicdaCre/+ and AicdaCre/− mice, respectively.
Discussion
In this study, we have examined the role of AID in GC differentiation by combining single cell transcriptome and V(D)J sequencing. Several recent papers have performed a single cell analysis of GCs both in mouse (Laidlaw et al, 2020; Riedel et al, 2020; Wong et al, 2020; Nakagawa et al, 2021) and human (Holmes et al, 2020; King et al, 2021), employing distinct isolation strategies. Here we have made use of an AID‐based genetic tracer to isolate B cells involved in the immune response. This tracing approach labeled both GC‐derived cells and extrafollicular activated B cells and provided the best strategy to precisely address the loss of AID in those cells that have been programmed for AID expression. In addition, the combination of transcriptome and V(D)J analysis in single cells has allowed us to establish detailed clonal and cell‐fate relationships among different cell subsets.
Our study has identified 8 distinct transcriptional clusters within AID‐labeled cells, with very accurate identification of the LZ and DZ compartments as previously described (Victora et al, 2010; Victora & Nussenzweig, 2012), and the plasmablast/PC subset. Interestingly, we found that GC B cells harbored the highest mutational load of all clusters, indicating that many GC B cells that do not differentiate to either the PC or the MBC fates instead proceed with iterative cycles of SHM at the GC. In addition, we identified 3 Mem subclusters, which indicates a heterogeneity within the MBC compartment that has been previously observed by others (Shlomchik et al, 2019), also in single cell sequencing analyses (Laidlaw et al, 2020; Riedel et al, 2020; King et al, 2021). Indeed, making use of a lineage‐tracking approach, a recent paper distinguished between two types of MBCs, i.e., activated B cell‐derived and GC‐derived, which differ in isotype, overall gene expression, somatic hypermutation, and antigen affinity (Viant et al, 2021). Very interestingly, we have identified a cluster of MBCs (Mem.GC) that shares a high transcriptional similarity with GC‐derived MBCs and two clusters (Mem.Act and the minor cluster Mem.Act2), which resemble activated B cell‐derived MBCs. In agreement with this, GC cells share more clones with Mem.GC than with Mem.Act. Moreover, Mem.GC harbors a higher mutational load and CSR than Mem.Act cells, further confirming their alikeness with the MBC subsets described before (Viant et al, 2021). Although additional work is needed to understand precisely the differentiation and functional properties of these MBC subsets, our study shows that our AID‐tracing approach has been able to capture distinct MBC populations and their distinct molecular features.
Interestingly, we identified an L‐prePB transcriptional cluster that shared the largest clonal similarities with the PB cluster, thus indicating that L‐prePB cells can be PB immediate precursors. L‐prePB cells can be identified by flow cytometry based on their expression of FcRγ and show the highest transcriptional proximity with PB, but they do not show particularly high levels of Prdm1 or Irf4. Given the distal transcriptional similarities with our Mem clusters, we cannot rule out that L‐prePB are recent MBC emigrants skewed to PB differentiation. However, we speculate that L‐prePB represents a transitional stage between GC cells and PB, such that neither the GC nor the PB transcriptional programs are accurately captured. In addition, we have identified our E‐prePB cluster as a non‐GC counterpart of the Fraction 1 prePB subset reported before (Ise et al, 2018), which, like L‐prePB cells, seem distantly related to MBCs. Interestingly, E‐prePB cells harbor less diversified immunoglobulin genes than L‐prePB, suggesting an earlier emergence from the GC. Therefore, our results have identified two distinct prePB subsets.
Here we have confirmed the finding that AID deficiency impairs PB/PC differentiation and decreases the DZ/LZ ratio at GC (Zaheen et al, 2009; Boulianne et al, 2013). In addition, our single cell analysis has shown that AID deficiency severely reduces the proportion of L‐prePB cells while favoring the accumulation of E‐prePB. These alterations underlie a complex differentiation shift, which includes changes in transcriptional programs governing B cell survival and proliferation. Of note, since AID itself is not a transcriptional regulator, it is safe to argue that the observed transcriptional changes in AicdaCre/− mice are a consequence of secondary immunoglobulin diversification mediated by AID. We propose that the absence of SHM and CSR in AID deficient mice impinges on antibody affinity and immunoglobulin isotype‐dependent checkpoints, both of which are important in favoring the B cell choice toward the PB/PC fate versus the MBC fate.
Our study has established for the first time a direct link between these transcriptional shifts and the clonal expansion and relationships among different clusters. We show here that a high proportion of PB cells share clonal relationships with L‐prePB cells, indicating that PB cells frequently differentiate from L‐prePB cells. This is further reinforced by the finding that in AID deficient mice, the proportion of clonal relationships between L‐prePB and PB is decreased, and the L‐prePB subset is very much reduced. The results thus suggest that L‐prePB are close to the transitional stage where the selective events leading to the PB/PC fate take place. In addition, we find that in the absence of AID, the E‐prePB cluster is very much expanded and shows increased clonal relationships with the PB cluster, suggesting that the differentiation into PB in AID deficient mice is partially compensated by an alternative differentiation track.
While further research is needed to pinpoint the exact molecular mechanisms underlying our observations, this study provides the first single cell map of the function of AID in determining GC differentiation fates.
Materials & Methods
Mice
AicdaCre +/ki mice (Rommel et al, 2013) and Rosa26tdTomato +/ki mice are from Jackson laboratories (007770 and 007909). AicdaCre +/ki ;Rosa26tdTomato +/ki mice were generated by breeding AicdaCre +/ki to Rosa26tdTomato +/ki mice. C57BL/6‐Tg (TcraTcrb) 425Cbn/J mice (OT‐II) express a TCR specific for the OVA peptide (amino acid residues 323–339; Jackson Laboratories 004194). Aicda −/− mice were a generous gift of Dr Tasuku Honjo (Muramatsu et al, 2000). AicdaCre/− mice were generated by crossing AicdaCre ki/ki ;Rosa26tdTomato ki/ki mice with Aicda +/− mice. Male and female mice were used throughout the study. Mice analyzed in this study were at least 6 weeks old and kept under specific pathogen‐free conditions. Animals were housed in the CNIC animal facility under specific pathogen‐free conditions. All animal procedures conformed to EU Directive 2010/63EU and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under RD 53/2013. The procedures have been reviewed by the Institutional Animal Care and Use Committee (IACUC) of CNIC and approved by Consejeria de Medio Ambiente, Administración Local y Ordenación del Territorio of Comunidad de Madrid.
Cell transfer and immunization
CD4+ OT‐II spleen T cells were enriched by negative selection with the EasySep™ Mouse CD4+ T Cell Isolation Kit (STEMCELL) and 5 × 104 cells were transferred intravenously to groups of 10‐week‐old AicdaCre/+ and AicdaCre/− mice. 16 h after the transference, mice were immunized intraperitoneally with 50 μg of OVA (Sigma‐Aldrich) in PBS, precipitated in alum (Imject Alum, Thermo Scientific) in a ratio of 2:1. Booster immunization was performed 2 weeks later, by intraperitoneal injection of 50 μg of OVA in PBS. Mice were euthanized 2 weeks after the second immunization. For NP‐CGG immunization, mice were immunized intraperitoneally with 30 μg of NP‐CGG (Biosearch‐Technologies) precipitated in alum in a ratio of 2:1. Booster immunization was performed 2 weeks later, by intraperitoneal injection of 30 μg of NP‐CGG in PBS. Mice were euthanized 2 weeks after the second immunization.
Flow cytometry
Single cell suspensions were obtained from spleens and erythrocytes were lysed (ACK Lysing Buffer). Fc receptors were blocked with anti‐mouse CD16/CD32 antibodies (Cat#553142 BD Pharmingen™) and cells were stained with fluorophore or biotin‐conjugated anti‐mouse antibodies to detect B220 (Cat#562922 RA3‐6B2), GL7 (Cat#144610 GL7), CD138 (Cat#563193 281‐2), CD38 (Cat#25–0381‐82 HIT2), FcRγ (Cat#FCABS400F polyclonal), IgG1 (Cat#553441 A85‐1), IgG2B (Cat#406706 RMG2b‐1), IgG2C (Cat#STAR135F polyclonal), CXCR4 (Cat#13‐9991‐81 2B11) and CD86 (Cat#558703 GL1). Intracellular staining of FcRγ was performed with FOXP3 staining kit (Cat#00‐5521‐00 eBioscience). Streptavidin was used for biotin‐conjugated antibodies. Live cells were detected by 7AAD (Cat#559925 BD Pharmingen) or LIVE/DEAD Fixable Yellow Dead Cell Stain (Cat#L34959 Thermo Fisher) staining. B3Z cells transfected with an FcRγ‐CD2 fusion protein were kindly provided by Salvador Iborra (Iborra et al, 2016). The parental B3Z cell line was used as a negative control for FcRγ expression. Intracellular staining of FcRγ (Cat#FCABS400F, polyclonal) was performed with FOXP3 staining kit (Cat#00‐5521‐00, eBioscience), after surface staining with anti‐CD2 antibody (Cat# 201305, OX‐34) in both parental and transfected B3Z cells. Samples were acquired on LSRFortessa and analyzed with FlowJo V10.4.2 software.
RT–qPCR
RNA was extracted from FACS sorted fixed and permeabilized LprePB (Tom+ B220+ CD138− GL7− FcRγ+) and total Tom+ cells from OVA‐immunized AicdaCre/+ mice using the Qiagen rNeasy FFPE kit. cDNA was synthesized using High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA was quantified by SYBR green assay (Applied Biosystems) and normalized to GAPDH expression. The following primers were used: GAPDH (forward) 5′‐TGA AGC AGG CAT CTG AGG G‐3′, (reverse) 5′‐CGA AGG TGG AAG AGT GGG AG‐3′; FcRγ (forward) 5′‐GCT GTC TAC ACG GGC CTG AA‐3′, (reverse) 5′‐ACT GGG GTG GTT TCT CAT GC‐3′, Actn1 (forward) 5′‐GTT GGG TCC CGA AGA GTT CA‐3′, (reverse) 5′‐CAA ACT CTG CCT CTC CCT GG‐3′; Dnm3 (forward) 5′‐GCC GGG GTC TAC CCA GAT A‐3′, (reverse) 5′‐ TGC GTA TGG TCT CCA CTT GC‐3′; Ptpn22 (forward) 5′‐ GTG TGG CAG CAA AAG ACC AA‐3′, (reverse) 5′‐TCT GTC ATC CGT TGG CCA GT‐3′.
Cell sorting and scRNA‐seq
Four different TotalSeq™‐C antibodies were used for Hash Tag Oligonucleotide (HTO) labeling before cell sorting (Biolegend, M1/42M30‐F11) following the manufacturer's standard protocol. HTO‐stained Tom+ spleen cells were purified from immunized mice using Sy3200 Cell Sorter (SonyBiotechnology). After sorting, Tom+ cells from the four mice were pooled together and diluted at a concentration of 1,500 cells/μl. 17,500 cells were used for the Chromium 10× Genomics System following the manufacturer's protocol (Chromium Single Cell 5′ Reagent Kit, v1.1 Chemistry). Single cell cDNA was separated into three aliquots for HTO, scRNA‐seq and V(D)J‐enriched library generation from the same input sample. The three libraries were sequenced with the HiSeq4000 System (Illumina).
scRNA‐seq data analysis
Cell Ranger mouse mm10 genome reference (v3.0.0 November 18, 2018) was used for the alignment of FASTQ files using 10× Genomics Cell Ranger software v3.1.0 (Zheng et al, 2017). A unique count table of gene expression for the four samples was generated. HTO antibodies were used for sample demultiplexing using Seurat R package (Butler et al, 2018; v3.1.5). An average of 57,343 reads per cell and a median value of 1,748 expressed genes per cell were obtained. The estimated number of total cells recovered was 10,454. Mice were grouped by genotype for further analyses. Single cell gene expression analysis was performed with Seurat. UMI counts measuring gene expression were log‐normalized and cells with mitochondrial reads > 8% of all transcripts were removed. A small number of non‐B cells were identified with SingleR (Aran et al, 2019) using Immgen (Heng et al, 2008) and filtered out. After quality filtering, there were a total of 8,116 B cells from the 4 samples (AicdaCre/+: 2126 and 1935; AicdaCre/−: 2090 and 1833).
The top 2,000 variable genes were identified with the “FindVariableFeatures” function and scaled. UMAP projection plots were generated using the top 30 principal components. Clusters were identified with “FindClusters” function with a resolution of 0.25. Upregulated marker genes for each cluster were inferred with “FindAllMarkers” function (Wilcoxon test), using normalized expression values. Top 20 markers per cluster were visualized using “DoHeatmap” function. Cluster identities were assigned based on the expression of markers for different B cell types and using “MyGeneSet” tool from the Immunological Genome Project Databrowser. Markers used for the assignment of cluster identities included: Aicda, Fas, Mef2b, Bcl6, S1pr2, and Rgs13 for GC B cells; Klf2, Ccr6, Sell, Zbtb32, and Hhex for MBCs, Xbp1, Scd1, Jchain, and Slpi for PBs; DZ and LZ signatures (Victora et al, 2012). Signature enrichment was assessed using the “AddModuleScore” function in Seurat package. Further subclustering of the “Mem” cluster was performed and applied to the main UMAP projection. Cell cycle scores were calculated using the “CellCycleScoring” function in the Seurat package using previously defined gene signatures for each cell cycle stage. Second best‐hit marker analysis was performed using the function “QuickMarkers” in the SoupX package 1.5.0 (Young & Behjati, 2020), with a gene frequency threshold of 10%.
Ingenuity Pathway Analysis (IPA) was used the identify the distinct biological functions altered between AicdaCre/+ and AicdaCre/− within the specified transcriptomic clusters.
Pseudotime analysis
Pseudotime analysis was performed using the R package Monocle 2.16.0 (Trapnell et al, 2014). A single cell trajectory was constructed by Discriminative Dimensionality Reduction with Trees (DDRTree) algorithm using all genes differentially expressed among Seurat clusters. Cells were ordered across the trajectory and pseudotime was calculated. DEGs over pseudotime were identified and clustered by their pseudotime expression patterns. For differential expression between branches A and B in AicdaCre/− mice, BEAM function in Monocle package was used. The top 500 DEGs were used for the branched heatmap.
BCR assignment
FASTQ files were preprocessed using the “cellranger vdj” command from 10× Genomics Cell Ranger v3.1.0 for alignment against the Cell Ranger mouse mm10 vdj reference (v3.1.0 July 24, 2019). The resulting FASTA file was split by sample using HTO data and output files were converted to Change‐O format. Downstream processing and analyses were performed following the Immcantation pipeline (Gupta et al, 2015). V and J gene usage was determined using IgBlast on the IMGT database and sequences annotated as “nonfunctional” by IgBLAST were removed from further analysis. A total of 5,856 cells passed the Immcantation quality filter for both V(D)J and GEX (AicdaCre/+: 1436 and 1241; AicdaCre/−: 1698 and 1480).
Clonal group assignment of sequences was made according to the following requirements: identical V and J gene usage and identical CDR3 length for the IgH and the IgL genes.
Clonal analysis
Clonal groups were split when differing IgL V and J genes were identified. Germline sequences were reconstructed for each clonal lineage and silent and nonsilent mutations were quantified as deviations from the inferred germline. Clone sizes were determined with “countClones” function of Alakazam R package (v1.0.2). Clonal analysis was performed for each mouse separately; mice of the same genotype were grouped together after clonal assignment.
Clonal overlap among B cells clusters was quantified and plotted with the UpSetR package (Conway et al, 2017; v1.4.0). Circos plots were generated using pairwise sharing data obtained from UpSetR, with circlize package (Gu et al, 2014; v0.4.11). Transition probabilities were manually calculated within each transcriptional cluster for each shared clone present in that cluster. The probability of transition from/to the cluster of interest to the rest of the clusters was calculated based on the presence/absence of the same clone in different clusters. Average transition probabilities were depicted. Interaction networks were generated with Cytoscape (Shannon et al, 2003). Only transitions with probabilities greater than 0.1 are shown. Transition probabilities with differences greater than 0.15 between AicdaCre/+ and AicdaCre/− mice are colored.
Lineage trees were built with IgPhyML, which builds maximum likelihood trees with B cell‐specific models (HLP19 model). Only clones with more than 2 cells were considered and identical sequences were collapsed. Trees were visualized with Alakazam and ape (v5.4‐1) R packages.
Mutational load was calculated using the “ObservedMutations” function of sHazaM R package (v1.0.2) by counting the number of nucleotide mismatches from the germline sequence in the heavy chain variable segment leading up to the CDR3.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 8. Error bars represent standard deviation of the mean. Normality of the data was assessed with the Anderson‐Darling test. For data following a normal distribution, a two‐tailed unpaired Student's t‐test was used for comparing two experimental groups. The Fisher and chi‐square tests were used for categorical variables. The Kruskal–Wallis test was used for comparing not normally distributed data. P‐values < 0.05 were considered statistically significant.
Author contributions
Carmen Gómez‐Escolar: Data curation; formal analysis; investigation; visualization; methodology; writing – review and editing. Alvaro Serrano‐Navarro: Data curation; methodology. Alberto Benguria: Methodology. Ana Dopazo: Resources. Fatima Sanchez‐Cabo: Resources; visualization; methodology. Almudena R Ramiro: Conceptualization; resources; formal analysis; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
PDF+
Acknowledgements
We thank all the members of the B lymphocyte Biology lab for helpful suggestions, Ana Rodriguez‐Ronchel for the elaboration of the graphical abstract, Sonia Mur for technical assistance, Virginia G de Yebenes for critical reading of our manuscript, Julia Merkenschlager, Carlos Torroja, and Enrique Vazquez for their advice on single cell sequencing and analysis, the CNIC Flow Cytometry for assistance on cell analysis and separation and the CNIC Genomics Unit for single cell sequencing. We also thank Sergio Roa, Alicia G Arroyo, Salvador Iborra, and David Sancho for kindly sharing mouse lines and reagents with us. CG‐E is supported by a fellowship awarded by La Caixa España in 2017 and AS‐N is an FPI Severo Ochoa fellow (PRE2018‐083475). AB, FS‐C, and ARR are supported by CNIC. This project was funded by grants from the Spanish Ministerio de Economía, Industria y Competitividad (SAF2016‐75511‐R), the Spanish Ministerio de Ciencia e Innovación (PID2019‐106773RB‐I00/AEI/10.13039/501100011033) and the “la Caixa” Banking Foundation under the project code HR17‐00247 to ARR. FS‐C is supported by the project RT2018‐102084‐B‐I00 financed by MCIN/AEI/10.13039/5011000110033/ and by FEDER Una Manera de hacer Europa and by Ayuda EQC2021‐007294‐P financed by MCIN/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the Ministerio de Ciencia e Innovación (MCIN), and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence, CEX2020‐001041‐S funded by MICIN/AEI/10.13039/501100011033.
EMBO reports (2022) 23: e55000
Data availability
The datasets produced in this study are available in the following database: scRNA‐seq data: Gene Expression Omnibus GSE189775 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE189775).
References
- Allen CD, Okada T, Cyster JG (2007) Germinal‐center organization and cellular dynamics. Immunity 27: 190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR et al (2019) Reference‐based analysis of lung single‐cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulianne B, Rojas OL, Haddad D, Zaheen A, Kapelnikov A, Nguyen T, Li C, Hakem R, Gommerman JL, Martin A (2013) AID and caspase 8 shape the germinal center response through apoptosis. J Immunol 191: 5840–5847 [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33: 2938–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney C, Schnell A, Cammarata LV, Yao‐Smith A, Regev A, Kuchroo VK, Singer M (2019) Combinatorial prediction of marker panels from single‐cell transcriptomic data. Mol Syst Biol 15: e9005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, Nussenzweig MC (2016) Independent roles of switching and hypermutation in the development and persistence of B lymphocyte memory. Immunity 44: 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaros V, Rauschmeier R, Artemov AV, Reinhardt A, Ols S, Emmanouilidi A, Gustafsson C, You Y, Mirabello C, Björklund ÅK et al (2021) Limited access to antigen drives generation of early B cell memory while restraining the plasmablast response. Immunity 54: 2005–2023.e2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, Brors B (2014) Circlize implements and enhances circular visualization in R. Bioinformatics 30: 2811–2812 [DOI] [PubMed] [Google Scholar]
- Gupta NT, Vander Heiden JA, Uduman M, Gadala‐Maria D, Yaari G, Kleinstein SH (2015) Change‐O: a toolkit for analyzing large‐scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31: 3356–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, Painter MW, Immunological Genome Project Consortium (2008) The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9: 1091–1094 [DOI] [PubMed] [Google Scholar]
- Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, Nitzan M, Grunstein E, Pasqualucci L, Dalla‐Favera R et al (2020) Single‐cell analysis of germinal‐center B cells informs on lymphoma cell of origin and outcome. J Exp Med 217: e20200483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra S, Martínez‐López M, Cueto FJ, Conde‐Garrosa R, Del Fresno C, Izquierdo HM, Abram CL, Mori D, Campos‐Martín Y, Reguera RM et al (2016) Leishmania uses Mincle to target an inhibitory ITAM signaling pathway in dendritic cells that dampens adaptive immunity to infection. Immunity 45: 788–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T et al (2018) T follicular helper cell‐germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity 48: 702–715.e4 [DOI] [PubMed] [Google Scholar]
- Kaji T, Ishige A, Hikida M, Taka J, Hijikata A, Kubo M, Nagashima T, Takahashi Y, Kurosaki T, Okada M et al (2012) Distinct cellular pathways select germline‐encoded and somatically mutated antibodies into immunological memory. J Exp Med 209: 2079–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, James LK (2021) Single‐cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol 6: eabe6291 [DOI] [PubMed] [Google Scholar]
- Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, Furukawa K, Koseki H, Takemori T, Kurosaki T (2013) Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39: 136–147 [DOI] [PubMed] [Google Scholar]
- Kräutler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, Sundling C, Kaplan W, Schofield P, Jackson J et al (2017) Differentiation of germinal center B cells into plasma cells is initiated by high‐affinity antigen and completed by Tfh cells. J Exp Med 214: 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Cyster JG (2020) Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol 21: 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG (2020) The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol 21: 1082–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, Victora GD (2016) Germinal center B cell dynamics. Immunity 45: 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, Di Noia JM (2017) Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol 133: 37–87 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T (2000) Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563 [DOI] [PubMed] [Google Scholar]
- Nakagawa R, Toboso‐Navasa A, Schips M, Young G, Bhaw‐Rosun L, Llorian‐Sopena M, Chakravarty P, Sesay AK, Kassiotis G, Meyer‐Hermann M et al (2021) Permissive selection followed by affinity‐based proliferation of GC light zone B cells dictates cell fate and ensures clonal breadth. Proc Natl Acad Sci U S A 118: e2016425118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R (2006) High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med 203: 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq‐Labelouse R, Gennery A et al (2000) Activation‐induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper‐IgM syndrome (HIGM2). Cell 102: 565–575 [DOI] [PubMed] [Google Scholar]
- Riedel R, Addo R, Ferreira‐Gomes M, Heinz GA, Heinrich F, Kummer J, Greiff V, Schulz D, Klaeden C, Cornelis R et al (2020) Discrete populations of isotype‐switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat Commun 11: 2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Bothmer A, Callen E, Reina‐San‐Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A et al (2008) AID is required for the chromosomal breaks in c‐myc that lead to c‐myc/IgH translocations. Cell 135: 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez‐Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J et al (2019) Class‐switch recombination occurs infrequently in germinal centers. Immunity 51: 337–350.e337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel PC, Bosque D, Gitlin AD, Croft GF, Heintz N, Casellas R, Nussenzweig MC, Kriaucionis S, Robbiani DF (2013) Fate mapping for activation‐induced cytidine deaminase (AID) marks non‐lymphoid cells during mouse development. PLoS One 8: e69208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, Kurosaki T (2016) Regulated selection of germinal‐center cells into the memory B cell compartment. Nat Immunol 17: 861–869 [DOI] [PubMed] [Google Scholar]
- Shlomchik MJ, Luo W, Weisel F (2019) Linking signaling and selection in the germinal center. Immunol Rev 288: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suan D, Kräutler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M et al (2017) CCR6 defines memory B cell precursors in mouse and human germinal centers, revealing light‐zone location and predominant low antigen affinity. Immunity 47: 1142–1153.e1144 [DOI] [PubMed] [Google Scholar]
- Taylor JJ, Pape KA, Steach HR, Jenkins MK (2015) Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naïve B cell. Science 347: 784–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant C, Weymar GHJ, Escolano A, Chen S, Hartweger H, Cipolla M, Gazumyan A, Nussenzweig MC (2020) Antibody affinity shapes the choice between memory and germinal center B cell fates. Cell 183: 1298–1311.e1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant C, Wirthmiller T, ElTanbouly MA, Chen ST, Cipolla M, Ramos V, Oliveira TY, Stamatatos L, Nussenzweig MC (2021) Germinal center‐dependent and ‐independent memory B cells produced throughout the immune response. J Exp Med 218: e20202489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC (2012) Germinal centers. Annu Rev Immunol 30: 429–457 [DOI] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC (2022) Germinal centers. Annu Rev Immunol 40: 413–442 [DOI] [PubMed] [Google Scholar]
- Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer‐Hermann M, Dustin ML, Nussenzweig MC (2010) Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 143: 592–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Dominguez‐Sola D, Holmes AB, Deroubaix S, Dalla‐Favera R, Nussenzweig MC (2012) Identification of human germinal center light and dark zone cells and their relationship to human B‐cell lymphomas. Blood 120: 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel FJ, Zuccarino‐Catania GV, Chikina M, Shlomchik MJ (2016) A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity 44: 116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R, Belk JA, Govero J, Uhrlaub JL, Reinartz D, Zhao H, Errico JM, D'Souza L, Ripperger TJ, Nikolich‐Zugich J et al (2020) Affinity‐restricted memory B cells dominate recall responses to heterologous flaviviruses. Immunity 53: 1078–1094.e1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Behjati S (2020) SoupX removes ambient RNA contamination from droplet‐based single‐cell RNA sequencing data. Gigascience 9: giaa151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A (2009) AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood 114: 547–554 [DOI] [PubMed] [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049 [DOI] [PMC free article] [PubMed] [Google Scholar]