

Abstract
Co‐opting host cell protein synthesis is a hallmark of many virus infections. In response, certain host defense proteins limit mRNA translation globally, albeit at the cost of the host cell's own protein synthesis. Here, we describe an interferon‐stimulated helicase, DDX60, that decreases translation from viral internal ribosome entry sites (IRESs). DDX60 acts selectively on type II IRESs of encephalomyocarditis virus (EMCV) and foot and mouth disease virus (FMDV), but not by other IRES types or by 5′ cap. Correspondingly, DDX60 reduces EMCV and FMDV (type II IRES) replication, but not that of poliovirus or bovine enterovirus 1 (BEV‐1; type I IRES). Furthermore, replacing the IRES of poliovirus with a type II IRES is sufficient for DDX60 to inhibit viral replication. Finally, DDX60 selectively modulates the amount of translating ribosomes on viral and in vitro transcribed type II IRES mRNAs, but not 5′ capped mRNA. Our study identifies a novel facet in the repertoire of interferon‐stimulated effector genes, the selective downregulation of translation from viral type II IRES elements.
Keywords: DExDH box helicase, interferon, IRES, protein synthesis
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; RNA Biology; Translation & Protein Quality
Translational shutdown is an effective countermeasure to viral infections, albeit at the cost of the host cell's protein synthesis. DDX60 selectively reduces translation off viral type II internal ribosome entry sites, leaving cap‐mediated translation intact.
Introduction
During viral infection, competition ensues between viruses and their host cells to control the protein synthesis machinery. To initiate mRNA translation in eukaryotes, a covalent m7GpppG 5′ cap structure on host messenger RNAs (mRNAs) enables the recruitment of a translation initiation factor complex that recruits the 40S ribosome subunit (Jackson et al, 2010; Merrick & Pavitt, 2018). The cap structure is recognized by eukaryotic initiation factor (eIF) protein eIF4E, which forms a complex with the scaffold protein eIF4G. Interaction between eIF4G and eIF3 then assembles a 43S preinitiation complex consisting of a 40S ribosomal subunit bound to eIF3, eIF1, eIF1A, and a ternary complex of GTP bound eIF2 and initiator Met‐tRNA i Met, among other factors. This ribosomal complex scans the mRNA in the 5′ to 3′ direction. During scanning, eIF4G‐bound RNA helicase eIF4A and activator protein eIF4B unwind RNA secondary structures in the mRNA until the start codon is identified. Subsequently, eIF1, eIF1A, and eIF5 assist in positioning the 40S ribosomal subunit such that the initiator Met‐tRNA i Met is at the peptidyl (P)‐site of the 40S ribosomal subunit. eIF5 then promotes GTP hydrolysis by eIF2, releasing eIF2 and eIF5 for subsequent cycles of translation initiation. Lastly, the GTPase eIF5B assists in joining the 60S ribosomal subunit to the 40S subunit to form an 80S initiation complex. The poly A‐binding protein (PABP) interacts with the 3′‐poly(A) tail and eIF4G, further promoting mRNA translation initiation.
Viruses evolved diverse mechanisms to compete with and dominate the host protein synthesis machinery, much of it centered on maintaining cap‐dependent mRNA translation or bypassing it completely. Some viruses utilize eukaryotic capping enzymes to add a m7Gppp 5′ cap to their mRNAs, while others encode their own viral capping enzymes to add a 5′ cap that functionally mimics a eukaryotic 5′ cap. A number of viruses naturally have uncapped mRNAs but can “snatch” capped 5′ terminal fragments from host mRNAs (Plotch et al, 1981; Decroly et al, 2012), while others covalently link their uncapped mRNA to a 5′ terminal protein that mechanistically acts like a 5′ cap to recruit translation initiation complex proteins (Goodfellow et al, 2005). Others directly recruit ribosomes to the mRNA and bypass the requirement for 5′ cap recognition using structured RNA elements called IRESs (Jang et al, 1988; Pelletier & Sonenberg, 1988; Stern‐Ginossar et al, 2019).
IRESs assemble the translation initiation apparatus either upstream of or at an initiation codon, independently of a 5′ cap structure (Fraser & Doudna, 2007; Lozano & Martínez‐Salas, 2015; Lee et al, 2017; Yamamoto et al, 2017; Martinez‐Salas et al, 2018). During recruitment of the translation initiation apparatus, often with structural support from host IRES‐transacting factor proteins (ITAFs), IRESs interact with a defined set of eIFs that assist in the recruitment of the 40S ribosomal subunit (Walter et al, 1999; Andreev et al, 2012; Martinez‐Salas et al, 2018). Several subtypes of viral IRESs exist, based on their unique RNA structures, differential requirements for eIFs and ITAFs, and start codon recognition mechanisms (Kaminski et al, 1990; Belsham, 1992; Hunt et al, 1993; Ohlmann & Jackson, 1999; Beales et al, 2003; Lozano & Martínez‐Salas, 2015; Lee et al, 2017; Yamamoto et al, 2017; Martinez‐Salas et al, 2018). Type I IRESs found in picornaviruses such as poliovirus and enterovirus 71 (EV71) employ a ribosomal scanning mechanism for start codon recognition with the assistance of eIFs 1A, 2, 3, 4A, 4B, central domain of 4G, and ITAFs PCBP1/2, PTB, hnRNPA1, and other proteins (Pelletier & Sonenberg, 1988; Thompson & Sarnow, 2003; Sweeney et al, 2014; Martinez‐Salas et al, 2018; Stern‐Ginossar et al, 2019). Type II IRESs, also found in picornaviruses such as EMCV and FMDV, direct ribosome entry at an AUG in the 3′ end of the IRES, or one located a short distance away with the assistance of the eIFs 2, 3, 4A, 4B, central domain of 4G, the ITAF PTB for EMCV and PTB plus Ebp1/ITAF45 for FMDV with additional assistance from eIFs 1 and 1A for translation initiation at a second downstream AUG only in FMDV (Jang et al, 1988; Belsham, 1992; Pestova et al, 1996a, 1996b; Andreev et al, 2007; Martinez‐Salas et al, 2018; Stern‐Ginossar et al, 2019). The type III IRES, found uniquely in the picornavirus hepatitis A virus (HAV), requires an intact heterotrimeric complex of eIF4E, eIF4G, and eIF4A (Avanzino et al, 2017). This is in contrast to all other IRES types, which initiate translation independently of eIF4E (Lozano & Martínez‐Salas, 2015; Martinez‐Salas et al, 2018; Stern‐Ginossar et al, 2019). Type IV IRESs, found in some picornaviruses but originally discovered in flaviviruses such as HCV and bovine viral diarrhea virus (BVDV), recruit the 40S ribosomal subunit close to the start codon without the use of eIFs, and subsequently recruit GTP bound eIF2, initiator Met‐tRNA i Met and eIF3 to facilitate 60S ribosomal subunit joining (Pestova et al, 1998; Fraser & Doudna, 2007). Type V IRESs, found in different genera of picornaviruses, have a three‐dimensional IRES organization resembling a hybrid of type I and type II IRESs and, in some members, have a requirement for the DExH‐box protein DHX29 for efficient translation initiation (Yu et al, 2011; Sweeney et al, 2012; Arhab et al, 2020). Finally, IRESs found in dicistroviruses such as cricket paralysis virus (CrPV), require no eIFs or ITAFs for 40S and 60S ribosomal subunit recruitment, and initiate translation at a noncanonical start codon from the A‐site of the ribosome (Wilson et al, 2000a, 2000b; Jan & Sarnow, 2002).
While viruses must compete for the host's translation machinery, cells respond by enacting different mechanisms to block overall protein synthesis, and in some cases specifically inhibit translation of viral mRNAs. Interferons (IFNs), produced by cells upon viral infection, trigger the expression of a variety of interferon‐stimulated genes (ISGs) that have diverse antiviral functions, some of which target translation (Schneider et al, 2014; Hoffmann et al, 2015; Hopfner & Hornung, 2020; Ficarelli et al, 2021; Li & Wu, 2021). Among them is the double‐stranded RNA activated protein kinase PKR, which phosphorylates the eIF2 α‐subunit to impair GDP to GTP exchange by the eIF2B GTP exchange factor, thus inhibiting global protein synthesis (Stern‐Ginossar et al, 2019). Another mechanism involves the activation of oligoadenylate synthase (OAS), which synthesizes short oligoadenylate polymers to stimulate RNase L to indiscriminately degrade ribosomal RNA, as well as viral and certain host mRNAs (Burke et al, 2019). The interferon‐induced protein with tetratricopeptide repeats (IFIT) family members and interferon‐induced transmembrane protein (IFITM) family members bind specific eIFs to restrict global protein synthesis or recognize structures absent in viral 5′ caps such as 2′O‐methylation (Diamond & Farzan, 2013; Schoggins, 2019). Finally, the zinc finger antiviral protein (ZAP) triggers viral RNA degradation and limits interactions between certain eIFs (Schoggins, 2019). While these mechanisms limit translation of viral mRNAs, and in consequence viral replication, they come at the cost of downregulating host protein synthesis. Here, we describe the ISG DExD/H‐box helicase 60 (DDX60), an RNA helicase that can inhibit viral type II IRES‐driven translation while leaving host 5′ cap‐driven mRNA translation intact.
The antiviral function of DDX60 was initially discovered in a screen for antiviral ISGs, where it was shown to inhibit a reporter HCV (Schoggins et al, 2011). Later studies probing for the antiviral mechanism of DDX60 generated conflicting data. One group found DDX60 to act as a sentinel for the viral RNA recognition receptor retinoic acid‐inducible gene‐I (RIG‐I) (Miyashita et al, 2011), and to promote degradation of viral RNA independently of RIG‐I (Oshiumi et al, 2015). However, another group presented evidence against a role for DDX60 as a sentinel for RIG‐I (Goubau et al, 2015), suggesting instead that DDX60 may enact a specific antiviral mechanism for one or a small group of viruses.
Here, we aimed to clarify the mechanism for DDX60 antiviral activity. We first show that upon IFN‐ß treatment, DDX60 has prolonged and delayed expression dynamics at the mRNA and protein levels, respectively. Through mutagenesis and antiviral assays, we demonstrate that N‐ and C‐terminal regions alongside predicted helicase and ATP binding motifs in DDX60 are important for its antiviral activity. We next use comparative antiviral experiments to show that DDX60 targets type II IRESs found in a group of viruses. We generated in vitro transcribed mRNA reporters to demonstrate that DDX60 specifically inhibits the type II family of IRESs and further show that the type II IRES is sufficient to confer virus inhibition by DDX60. Lastly, we found that DDX60 reduces type II IRES activity by modulating translating ribosome activity both on type II IRES‐driven firefly luciferase (Fluc) mRNA and on viral mRNA during viral infection. Importantly, DDX60 shows neither an effect on the overall translation status of the cell nor an effect on the translation of in vitro synthesized 5′ capped Fluc mRNA. Our work suggests that DDX60 acts as an ISG that inhibits type II IRES‐mediated mRNA translation and can discriminate between 5′ cap‐independent and ‐dependent translation mechanisms. Studying the anti‐IRES mechanism of DDX60 could lead to novel strategies for targeting specific virus translation mechanisms while leaving host translation intact.
Results
DDX60 displays dynamics of a type I ISG at the mRNA and protein level in multiple cell lines
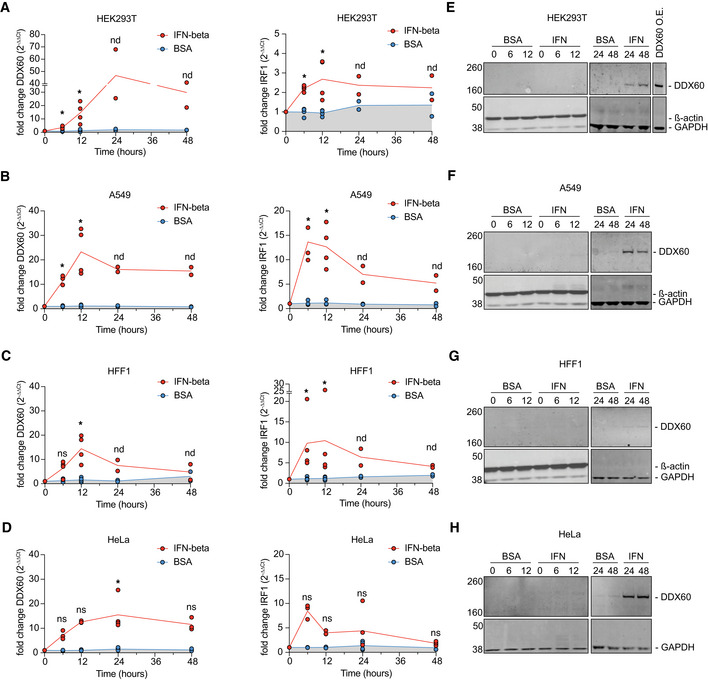
Gene expression of DDX60 at the mRNA level has been shown to be triggered by various stimuli in human cell lines and mouse tissue, including poly(I:C), type I IFN, and virus infections (Miyashita et al, 2011; Goubau et al, 2015). We analyzed the dynamics of DDX60 mRNA in four different human cell lines upon treatment with IFN‐ß and compared it with interferon regulatory factor 1 (IRF1), an IFN‐stimulated transcription factor with broad antiviral function and known expression dynamics (Schoggins et al, 2011; Forero et al, 2019; Feng et al, 2021). We treated three epithelial cell lines (HEK293T, human embryonic kidney; A549, lung adenocarcinoma; and HeLa, cervical adenocarcinoma) as well as primary human foreskin fibroblasts (HFF) with IFN‐ß and analyzed mRNA expression using RT–qPCR. In all cell types tested, both IRF1 and DDX60 expressions increased upon IFN‐ß stimulation (Fig EV1A–D). DDX60 mRNA levels reached higher peaks than those of IRF1, most notably in HEK293T cells (Fig EV1A). While IRF1 mRNA levels returned to baseline (0 h values) at 48‐h poststimulation, DDX60 mRNA levels remained above baseline in all cell types except primary HFF (Fig EV1C). Overall, our IFN stimulation and mRNA analysis demonstrate that DDX60 displays general characteristics of an ISG.
Figure EV1. DDX60 mRNA and protein expression upon interferon treatment.
-
(A–H)(A, E) HEK293T, (B, F) A549, (C, G) HFF1, or (D, H) HeLa cells treated with either 0.1% BSA (carrier control) or 500 U/ml of interferon‐ß for 0, 6, 12, 24, or 48 h. Cells were harvested for mRNA analysis (A–D) using RT‐qPCR or protein analysis (E–H) using Western blot. Panel E includes one HEK293T sample transfected with DDX60 wild type run on the same gel as 24‐ and 48‐h time points to show relative DDX60 protein levels in interferon‐ß treated versus transfected cells. All western blots for 0‐, 6‐, and 12‐h time points were run on the same gel but are separated by cell line for visualization purposes. All western blots for the 24‐ and 48‐h time points were run on the same gel but are separated by cell line for visualization purposes.
Data information: Individual replicates and mean of mRNA fold‐change compared to unstimulated cells. For data points with at least n = 3 replicates *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, non‐significant (ns) using Mann–Whitney ranked test against carrier control values of same time point. For data points with n = 2 replicates statistics were not determined (nd). Representative images of Western blots shown.
Source data are available online for this figure.
We next sought to determine whether protein levels of endogenous DDX60 also change with IFN treatment. We thus analyzed DDX60 protein dynamics in our four IFN‐ß treated cell lines by Western blot. Expectedly, DDX60 protein production increased approximately three‐ to 10‐fold upon IFN‐ß treatment compared with little to no expression at baseline (Fig EV1E–H). However, compared with DDX60 mRNA levels, DDX60 protein levels showed delayed expression dynamics in all cell lines, peaking at 24 or even 48‐h post‐IFN‐ß treatment (Fig EV1E–H).
Together, our findings show that DDX60 is an ISG with very low to undetectable steady‐state levels that then peak at both the RNA and the protein levels after IFN treatment in various human cell culture systems.
DDX60 decreases replication of a bicistronic reporter HCV carrying an EMCV IRES
Previous studies showed that DDX60 inhibits replication of a bicistronic reporter HCV (Schoggins et al, 2011; Oshiumi et al, 2015). To begin determining how DDX60 inhibits HCV, we used InterPro and published literature (Pause & Sonenberg, 1992; Schwer & Meszaros, 2000; Pyle, 2008; Umate et al, 2011; Johnson & Jackson, 2013), to identify putative functional domains and motifs (Fig 1A). We then introduced N‐terminal and C‐terminal truncations and single point mutations in residues predicted to confer ATP binding/hydrolysis and helicase activity to DDX60.
Figure 1. Functional mapping of DDX60 antiviral domains and interrogation of anti‐HCV activity.
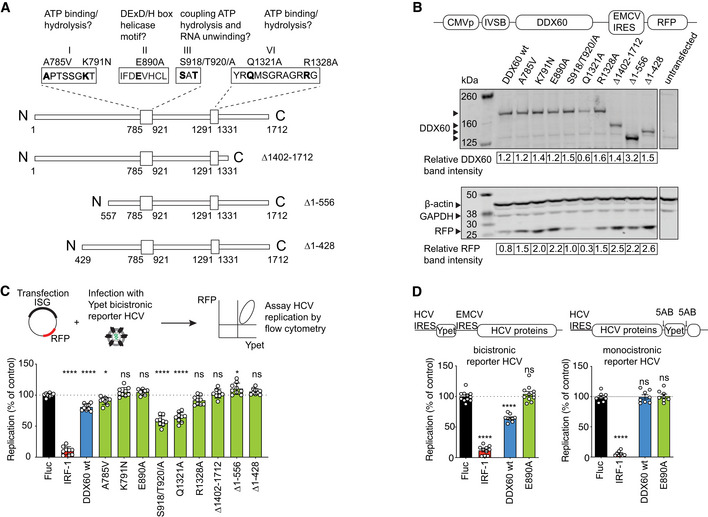
- Schematic of DDX60 protein with putative functional domains. Helicase ATP binding type I domain (amino acids 785–921) and C‐terminal helicase domain (amino acids 1,291–1,331) are shown as larger boxes in linear DDX60 schematic. Amino acids are numbered below. Putative functional motifs (I, II, III, and VI) and mutations made are annotated. The amino acids in bold as well as N‐ and C‐terminal regions were interrogated in antiviral assays.
- Assessment of exogenous DDX60 expression. HEK293T cells transfected with DDX60 wild‐type (wt), or DDX60 mutants and analyzed by Western blot for DDX60, ß‐actin and GAPDH (loading controls), and RFP (reporter). DDX60 and RFP quantification relative to GAPDH from one representative blot are shown below.
- HCV antiviral assays with DDX60 wt or mutant panel. Huh‐7 cells transfected with an RFP containing plasmid backbone encoding either Firefly luciferase (Fluc and negative control), IRF1 (positive antiviral control), DDX60 wt, or DDX60 mutants and infected with HCV‐Ypet, a bicistronic reporter HCV where Ypet reporter protein is driven by HCV IRES and HCV polyprotein consisting of C, E1, E2, p7, NS2, NS3, 4A, 4B, NS5A, and NS5B is driven by EMCV IRES.
- Effect of DDX60 on replication of bicistronic or monocistronic infectious reporter HCVs. Huh‐7 cells transfected as in (C) and infected with either bicistronic HCV‐Ypet (left) or monocistronic HCV J6/JFH‐5AB‐YPet. Ypet reporter in monocistronic HCV is placed in between NS5A and NS5B.
Data information: For (C) and (D), percent of Ypet+ cells in RFP+ cells is scaled to one replicate of Fluc control. Data shows mean ± SD for at least n = 3 biological replicates; ns —not significant, *P < 0.05, ****P < 0.0001, ns, nonsignificant using ANOVA and Dunnett's multiple comparison test against Fluc.
All mutants were ectopically expressed to equal levels as shown by Western blot (Fig 1B). We next used a previously developed virus inhibition assay to assess the antiviral capacity of the different DDX60 constructs (Schoggins et al, 2011). Briefly, we transfected Huh‐7 cells with wild‐type or mutant DDX60 plasmid containing a red fluorescent protein (RFP) marker to monitor transfection efficiency. Firefly luciferease (Fluc) served as negative control, and IRF1 as positive control. We then infected transfected cells with a yellow fluorescent protein (Ypet)‐expressing HCV at a dose yielding approximately 50% infected (Ypet+) cells in Fluc‐expressing cells, as previously determined by flow cytometry‐based infectivity assays (Jones et al, 2010; Schoggins et al, 2011). The percentage of Ypet‐positive (infected) cells within the RFP‐positive (transfected) population at 72‐h postinfection was assayed by flow cytometry. Wild‐type DDX60 reduced the percentage of HCV‐infected cells by approximately 30% relative to Fluc‐negative control. The predicted ATP binding residues and helicase motif are required for full DDX60 antiviral activity (Fig 1C, K791, E890, and R1328). Interestingly, deletions in either N‐ and C‐terminal extensions were also required for efficient antiviral activity (Fig 1C, Δ1–428, Δ1–556, and Δ1,402–1,712). Although DDX60's extensions are void of characterized function, in other RNA helicases, these extensions allow for protein–protein interactions during RNA substrate recognition (Wang et al, 2005; Thoms et al, 2015; Lingaraju et al, 2019).
To characterize the role of essential DDX60 residues and domains in biochemical detail, we next attempted to purify recombinant DDX60 via multiple tagging and protein expression strategies, including yeast and baculovirus systems. However, purification of full‐length DDX60 was unsuccessful due to protein aggregation, resulting in low yields. Attempts to solubilize the protein with different salt and glycerol concentrations were unsuccessful. Work by others had characterized purified truncated versions of DDX60 (Miyashita et al, 2011); however, this was not an option for our study, as both N‐ and C‐terminal regions are required for antiviral function (Fig 1C). We therefore speculate but cannot definitively assign DDX60 residues to have specific enzymatic activities. From here on, we use the minimal DEVH helicase motif mutant, DDX60 E890A, as a loss‐of‐function control in cellular assays.
In these initial experiments, we used the same infectious reporter HCV as a screen for ISGs that initially identified DDX60 to be antiviral (Schoggins et al, 2011). This infectious reporter HCV is bicistronic, as translation of the Ypet reporter is driven by the HCV IRES and translation of the HCV polyprotein is subsequently driven by an inserted EMCV IRES (Fig 1D, left schematic) (Jones et al, 2007, 2010; Schoggins et al, 2011). To validate our findings and rule out artifactual observations due to the use of a reporter virus encoding a foreign viral element, we employed our flow cytometry‐based virus inhibition assay using a infectious monocistronic reporter HCV where translation is initiated by the endogenous HCV IRES and the Ypet is translated as a part of the HCV polyprotein and subsequently excised due to flanking NS5AB cleavage sites (Fig 1D; Jones et al, 2007; Horwitz et al, 2013). While DDX60 successfully downregulated replication of the infectious bicistronic reporter HCV as observed previously (Fig 1C and D, left panel; Schoggins et al, 2011), DDX60 failed to downregulate the infectious monocistronic reporter HCV (Fig 1D, right). Additionally, the EMCV IRES‐driven RFP encoded in our plasmid constructs used in Fig 1B and C showed expression levels that mimicked the expression of our infectious bicistronic Ypet reporter HCV, but 5′ cap‐driven proteins such as ß‐actin, GAPDH, or our DDX60 transgenes of interest did not (Fig 1B). Flow cytometry revealed that reductions in EMCV IRES‐driven RFP was a result of reduced mean fluorescent intensity in RFP‐positive cells and not the percentage of RFP‐positive cells, thereby still enabling gating on cells that express DDX60. As the main distinguishing feature between the two infectious reporter HCVs is the EMCV IRES, we hypothesized that DDX60's antiviral action may be against the EMCV IRES, and not a component of HCV per se.
DDX60 downregulates plasmid‐ and in vitro transcribed RNA‐based reporters translationally driven by type II internal ribosome entry sites
To interrogate DDX60's IRES specificity, we next screened for DDX60's ability to inhibit representatives of type I (poliovirus), type II (EMCV and FMDV), or type IV IRESs (EV71); the IRES of CrPV, HAV, and type V picornavirus IRESs were excluded because of low CrPV IRES activity in mammalian cells (Carter et al, 2008) and lack of the tools discussed below to study the HAV and type V IRESs.
First, we used a plasmid‐based dual luciferase reporter system, where the transfected plasmid is transcribed and the transcript is canonically 5′‐capped in the nucleus by the host cell machinery (Pelletier & Sonenberg, 1988; Honda et al, 2000; Jackson, 2013). In the resulting single bicistronic mRNA, translation of the first cistron, Renilla luciferase (Rluc), is initiated by a canonical 5′ cap mechanism, and translation of the second cistron, Fluc, is initiated by an IRES mechanism. A stop codon separates the Rluc and Fluc genes such that Fluc can only be translated if a cap‐independent IRES allows for translation initiation (see Fig 2A, rightmost panel for schematic of transcript). We chose HEK293T cells for these assays due to their low DDX60 expression in the absence of type I IFN stimulation (Fig EV1A and E) and ease of transfectability. This allowed us to simulate the effects of DDX60 upregulation in the absence of endogenous DDX60 with the caveat that the exogenous DDX60 protein levels are about seven times higher than the amount of endogenous DDX60 detected after 48 h of IFN‐ß treatment (Fig EV1E). We cotransfected HEK293T cells with dual luciferase bicistronic reporter plasmids containing different IRESs along with GFP as a negative control, increasing amounts of IRF1 as a positive control, wild‐type DDX60, or DDX60 E890A, while maintaining equal DNA transfection amounts by supplementing with GFP plasmid. We then analyzed Rluc and Fluc activity from cell lysates. First, we separately analyzed Rluc (cap) and Fluc (IRES) activity relative to our GFP only transfected negative control. DDX60 did not reduce Rluc production driven by 5′ cap or Fluc production driven by EV71 (type I) IRES or HCV (type IV) IRES. By contrast, DDX60 reduced Fluc production driven by EMCV and FMDV (type II) IRES. The positive control IRF1 reduced both 5′ cap‐driven Rluc production and IRES‐driven Fluc production in a dose‐responsive manner regardless of the IRES type (Fig 2A, left). We next normalized the IRES‐driven Fluc activity by the 5′ cap‐driven Rluc activity to account for transfection efficiencies. We hypothesized three scenarios assuming inhibitory effects at the step of translation as depicted on the right in Fig 2A: equal translation of both IRES‐driven Fluc and 5′ cap‐driven Rluc (top), decreased translation of IRES‐driven Fluc compared with 5′ cap‐driven Rluc (center), or greater translation of IRES‐driven Fluc compared with 5′ cap‐driven Rluc (bottom). IRF1‐positive control reduced both IRES‐driven Fluc production and 5′ cap‐driven Rluc production equally, giving a ratio of approximately 1 (Fig 2A). By contrast, DDX60 had a statistically significant dose‐responsive inhibitory effect on the type II IRESs of EMCV and FMDV, but not of the type I IRES of EV71 or type IV IRES of HCV. Importantly, this effect was lost due to DDX60 E890A mutation (Fig 2A), showcasing the specificity of the observed phenotype.
Figure 2. In cell reporter assay for reporters translationally driven by different internal ribosome entry sites.
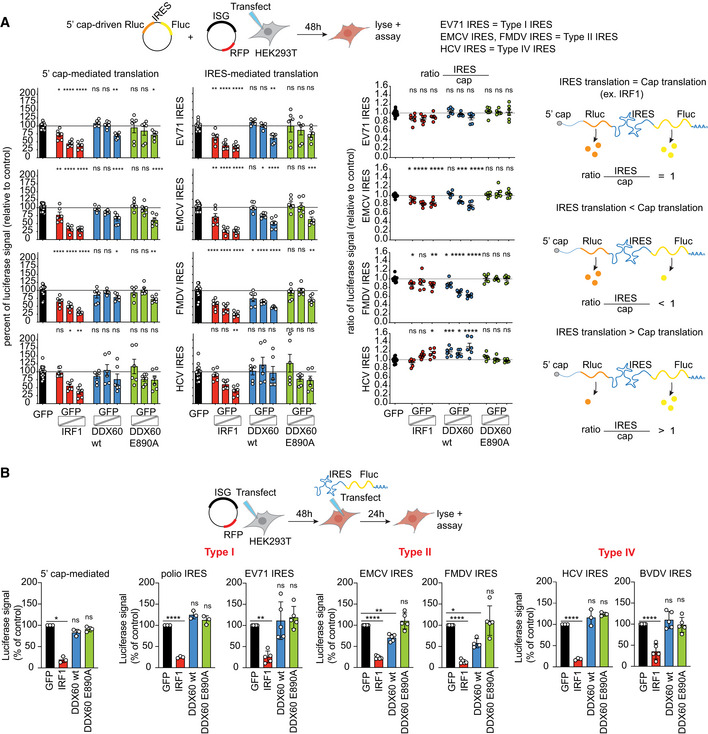
- Plasmid‐based dual luciferase bicistronic reporter assays. HEK293T cells cotransfected with dual luciferase bicistronic reporter plasmid (Renilla luciferase (Rluc) translationally driven by a 5′ cap, and Firefly luciferase (Fluc) translationally driven by different IRESs as indicated) and GFP plasmid (negative control) or increasing amounts of IRF1 (positive control), DDX60 wt, or DDX60 E890A mutant. Total amount of DNA transfected was kept constant by supplementing transfection mixes with GFP plasmid. Luciferase units after cell lysis is plotted as a percentage of GFP transfected cells (left) and ratio of IRES Fluc units over 5′ cap Rluc units (center). Diagram to the right of ratios plot depicts expected ratios of IRES‐driven Fluc units to 5′ cap‐driven Rluc units given either: equal translation of 5′ cap‐driven Rluc and IRES‐driven Fluc (top), greater translation of IRES‐driven Fluc (center), or greater translation of 5′ cap‐driven Rluc (bottom).
- RNA‐based monocistronic luciferase reporter assays. HEK293T cells transfected with GFP (negative control), IRF1 (positive control), DDX60 wt, or DDX60 E890A and subsequently transfected with in vitro transcribed 5′ cap or different IRES‐driven Fluc mRNA constructs as indicated. Luciferase units after cell lysis is plotted as a percentage of GFP transfected cells. Raw data are shown in Fig EV2.
Data information: Data show mean ± SD for at least n = 3 biological replicates; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, nonsignificant using ANOVA and Dunnett's multiple comparison test against GFP.
We noticed that DDX60 may have cis‐acting inhibitory effects on Rluc production when Rluc is linked to EMCV IRES‐driven Fluc (Fig 2A, left bar graphs). Cis‐acting effects may occur if mechanisms of inhibition include either RNA degradation or deterring ribosome accumulation on the entire transcript. To individually assess different translation mechanisms and disentangle results from potential cis‐acting effects, we performed reporter assays with in vitro transcribed monocistronic mRNAs. Translation of these monocistronic mRNAs is driven by either a 5′ cap analog or IRESs from type I, type II, or type IV families. We transfected equimolar amounts of in vitro transcribed Fluc mRNAs to either GFP, IRF1, DDX60 wt, or DDX60 E890A transfected cells and measured Fluc reporter activity. Consistent with our findings from the plasmid‐based system, we found that DDX60 significantly reduced translation of Fluc from mRNAs driven by the type II IRESs of EMCV and FMDV, but not by other IRES types or a 5′ cap (Figs 2B and EV2B). Overall, we conclude that DDX60 downregulates the type II IRES family but not the other IRES types or 5′ cap‐driven translation.
Figure EV2. Monocistronic RNA reporter assays for different internal ribosome entry sites and DDX60 mutant panel.
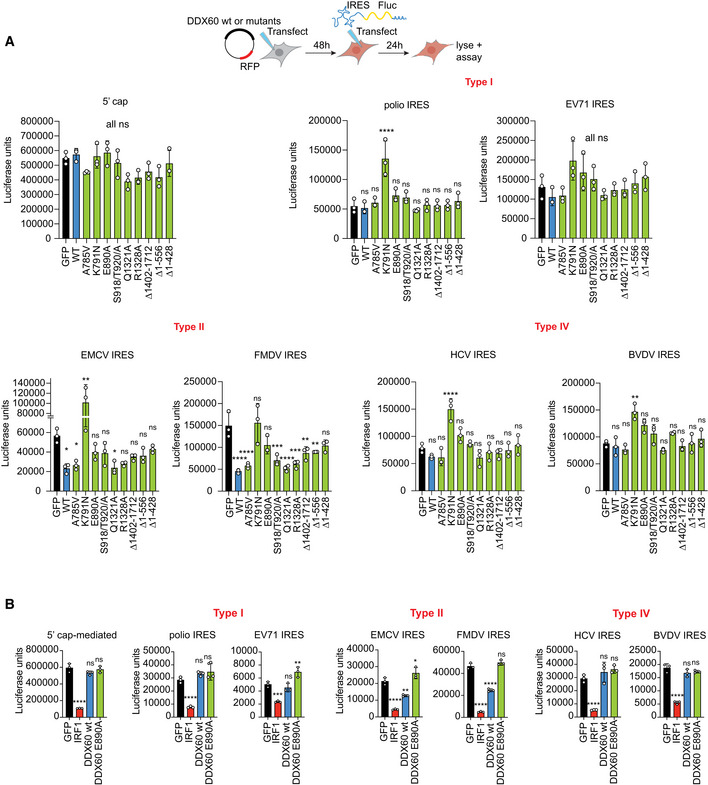
- HEK293T cells transfected as in Fig 2B and luciferase intensity measured from cell lysates. Mean ± SD for n = 3 technical replicates from one representative biological replicate for data shown in Fig 2B; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA and Dunnett's multiple comparison test against GFP.
- HEK293T cells transfected with GFP (negative control), DDX60 wt, or different DDX60 mutants described in Fig 1 and subsequently transfected with in vitro transcribed 5′ cap‐ or different IRES‐driven Fluc mRNA constructs and luciferase intensity measured from cell lysates. Mean ± SD of n = 3 biological replicates; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 using ANOVA and Dunnett's multiple comparison test against GFP.
Next, we asked whether other motifs or regions in DDX60 apart from the helicase motif were important for downregulating different IRESs using our monocistronic RNA reporter system. Probing our existing panel of DDX60 mutants, we found that predicted ATP binding residues, helicase motif, and N‐ and C‐terminal extensions are important for DDX60 antitype II IRES activity (Fig EV2A, type II IRES panels). We additionally found that one DDX60 mutant (domain I K791N, a presumed ATP binding/hydrolysis mutant) increased translation of at least one representative member of all the IRES types tested (Fig EV2A). Future biochemical and structural studies comparing this mutant with wild‐type DDX60 may elucidate the enzymatic activities responsible for the observed effect.
DDX60 specifically decreases infectious titers of viruses that rely on type II IRES‐mediated translation
We next sought to determine whether DDX60 can downregulate type II IRESs in the context of a virus infection. In a first set of experiments, we chose poliovirus as a representative type I IRES‐containing virus and EMCV a representative type II IRES‐containing virus, as we could work with both viruses in our BSL2 environment. HeLa cells were chosen as they are highly permissive to both viruses (Mendelsohn et al, 1989; Jin et al, 1994) and express low levels of endogenous DDX60 (Fig EV1H). We generated HeLa cells stably expressing either wild‐type DDX60, DDX60 E890A, Fluc as a negative control, or IRF1 as a positive control (Fig EV3B). We then performed multicycle growth kinetics with poliovirus or EMCV (Fig 3A and B). Wild‐type DDX60 significantly reduced EMCV titers (type II IRES, Figs 3B and EV3C), but not poliovirus titers relative to DDX60 E890A (type I IRES, Figs 3A and EV3D). The reduction in EMCV titers peaked to a twofold reduction 24‐h postinfection (hpi), and then titers increased to levels observed in our negative control and DDX60 E890A mutant expressing cells at 48 hpi, possibly owing to EMCV titers becoming high enough to overcome any inhibitory effects by DDX60 (Fig EV3D). Poliovirus, on the contrary, was not inhibited by DDX60 at any time point in our assay; its titers remained similar between DDX60, our negative control (Fluc), and DDX60 E890A mutant expressing cells at all time points even while our positive control, IRF1 reduced poliovirus titers starting at 24 hpi (Fig EV3C).
Figure EV3. Growth kinetics of different internal ribosome entry site containing viruses in the presence of DDX60.

-
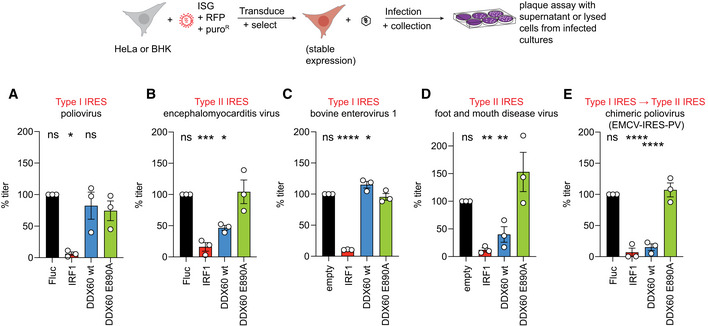
AGrowth kinetics of poliovirus, encephalomyocarditis virus (EMCV), or a chimeric poliovirus with the poliovirus IRES replaced with the IRES of EMCV. HeLa cells were infected with the three viruses at MOI 0.001 and supernatants from infected cultures were collected 0, 1, 8, 24, 32 or 48 hpi. Virus in supernatants was then titrated on HeLa cells. Mean of n = 2 biological replicates.
-
BAssessment of exogenous DDX60 expression in HeLa cells. HeLa cells were transduced with lentivirus carrying either DDX60 wt, DDX60 E890A, IRF1, or Firefly luciferase (Fluc) together with a puromycin resistance gene and selected with puromycin. Cell lysates were analyzed using western blot for DDX60, Fluc, IRF1, and GAPDH (loading control) protein products.
-
C–ETime course of antiviral assays with panel of IRES‐containing viruses. HeLa cells stably expressing Fluc (negative control), IRF1 (positive control), DDX60 wt, or DDX60 E890A were infected with (C) poliovirus, (D) EMCV, or (E) a chimeric poliovirus at a MOI of 0.001. Supernatants from infected cultures were collected at either 0, 1, 8, 24, 32, or 48 hpi and titrated on HeLa cells via plaque assay. Mean of n = 2 biological replicates.
Source data are available online for this figure.
Figure 3. Viral replication assays for a panel of IRES‐containing viruses.
-
A–E(A, B, E) Multicycle infection assays with type I and type II IRES‐containing viruses. HeLa cells stably expressing Firefly luciferase (Fluc) (negative control), IRF1 (positive control), DDX60 wt, or DDX60 E890A and infected with (A) poliovirus, (B) encephalomyocarditis virus, or (E) a chimeric poliovirus with the poliovirus IRES replaced with the IRES of EMCV (EMCV‐IRES‐PV) at MOI 0.001. Supernatants were collected 24‐h postinfection (hpi) (EMCV) or 48 hpi (poliovirus and EMCV‐IRES‐PV) and titers determined via plaque assay on HeLa cells. (C, D) Single‐cycle infection assays with type I and type II IRES‐containing viruses. BHK‐J cells stably expressing empty vector (negative control), IRF1 (positive control), DDX60 wt, or DDX60 E890A and infected with (C) bovine enterovirus‐1, or (D) foot and mouth disease virus at MOI 1. Supernatants were collected 5‐h postinfection (hpi) and titers determined via plaque assay on BHK‐21 clone 13 cells.
Data information: Data show mean ± SEM percent infectious titers relative to Fluc from at least n = 3 biological replicates; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, nonsignificant using ANOVA and Dunnett's multiple comparison test against DDX60 E890A. PFU data are shown in Fig EV3.
Next, we performed infection experiments with another type II IRES‐containing virus, FMDV. In the United States, these experiments are only possible at Plum Island Animal Disease Center's enhanced BSL3 facility. We compared DDX60 action against FMDV to that of action against BEV‐1, which carries a type I IRES. Baby hamster kidney (BHK) cells were chosen, as human cells are not permissive to FMDV or BEV‐1 (Mowat & Chapman, 1962; Ruiz‐Sáenz et al, 2009). BHK‐J cells stably expressing either a negative control empty vector, positive control IRF1, wild‐type DDX60, or DDX60 E890A were infected with either BEV‐1 or FDMV at an MOI of 1 and progeny virus harvested at 5 hpi. Consistent with our findings using EMCV and poliovirus, we detected a statistically significant reduction in titers of FMDV (type II IRES, Fig 3D) but not BEV‐1 (type I IRES, Fig 3C). These results demonstrated that DDX60 decreases infectious titers of viruses that rely on type II IRES‐driven translation.
Next, we tested whether a type II IRES is sufficient to confer sensitivity to DDX60. We generated a chimeric poliovirus replacing its endogenous type I IRES with the type II IRES of EMCV (EMCV‐IRES‐PV). We first characterized EMCV‐IRES‐PV replication in comparison with poliovirus and EMCV. First, we noticed that EMCV‐IRES‐PV generated smaller plaques compared with poliovirus as observed for a similar chimeric virus generated previously (Alexander et al, 1994). In multicycle replication kinetics, EMCV‐IRES‐PV started producing detectable infectious particles in HeLa cells between 8 and 24 hpi. Its titers peaked to approximately 109 PFU/ml at the end point of our assay, 48 hpi (Fig EV3A). This replication dynamic resembled that of poliovirus rather than EMCV. EMCV produced infectious particles of 106 PFU/ml after just 8 hpi with peak titers of approximately 1010 PFU/ml at 24 hpi (Fig EV3A). EMCV‐IRES‐PV and poliovirus both produced titers 10‐ to 100‐fold lower than EMCV for most of the experiment, but eventually reached similar titers as EMCV 48 hpi (Fig EV3A). Next, we analyzed DDX60's ability to inhibit EMCV‐IRES‐PV. In contrast to its parental poliovirus strain, which was resistant to DDX60 (Figs 3A and EV3C), DDX60 reduced EMCV‐IRES‐PV titers beginning at 24 hpi, reaching a 10‐fold titer reduction by the end of our assay at 48 hpi (Figs 3E and EV3E). Remarkably, this inhibitory effect is similar in magnitude to our positive control, IRF1, and is lost due to DDX60 E890A mutation. Together, our data demonstrate that reliance on type II IRES‐driven translation is sufficient to allow DDX60‐mediated inhibition of virus infection.
Abundance of type II IRES‐containing mRNAs is unchanged in the presence of wild‐type or mutant DDX60
Next, we sought to decipher the mechanism by which DDX60 downregulates type II IRESs. As we were unable to purify DDX60 for biochemical assays, we were limited to cell‐based assays for the remainder of the study. DDX60 was previously shown to be most closely related to a family of helicases called superkiller‐2 (Ski‐2)‐like helicases (Miyashita et al, 2011; Goubau et al, 2015). Ski‐2 is a RNA helicase originally discovered in yeast that is important for degrading satellite dsRNA from L‐A double‐stranded RNA virus and for general 3′ to 5′ degradation of yeast mRNAs (Widner & Wickner, 1993; Anderson & Parker, 1998). We first hypothesized that DDX60 decreases the abundance of mRNAs with a type II IRES. To test our hypothesis, we transfected HEK293T cells to express either wild‐type DDX60 or DDX60 E890A, subsequently transfected with equimolar amounts of Fluc mRNAs translationally driven by either a 5′ cap analog, type I IRESs, type II IRESs, or type IV IRESs, and analyzed cell lysates for luciferase signal and Fluc mRNA content in parallel. We found that while wild‐type DDX60 reduced Fluc translation from type II IRES‐driven mRNAs compared with DDX60 E890A, the relative abundance of type II IRES‐driven mRNAs between wild‐type DDX60 and DDX60 E890A in the cells were equivalent (Fig 4A and B). For the other IRES types and 5′ cap‐driven translation, wild‐type DDX60 and DDX60 E890A transfected cells had both equal levels of Fluc translation and mRNA abundance (Fig 4A and B). Our qPCR method has a level of detection of RNA abundance changes by fivefold or more, corresponding to a range of Ct values obtained for our reporter constructs between 16 and 19. Based on this limitation, we conclude that while DDX60 can reduce protein synthesis from type II IRES‐containing mRNAs, it is does not reduce the abundance of such mRNAs by greater than fivefold, with the caveat that DDX60 could be causing RNA abundance changes of less than fivefold.
Figure 4. mRNA abundance for reporters translationally driven by different internal ribosome entry sites, and interferon‐stimulated response element activity in the presence of DDX60.
-
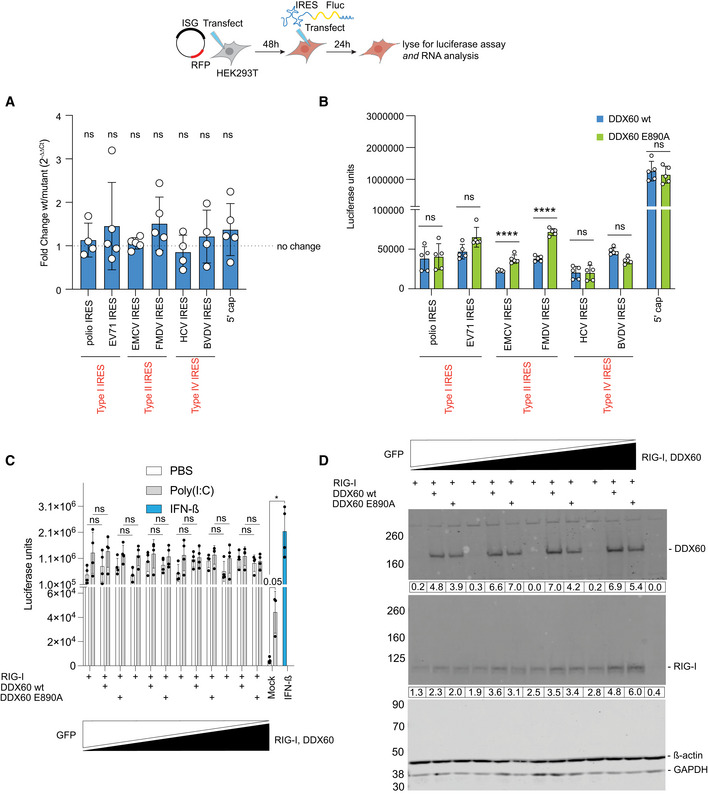
A, BHEK293T cells transfected with DDX60 wt or DDX60 E890A (negative control) and transfected with in vitro transcribed 5′ cap‐ or IRES‐driven Fluc mRNA constructs as indicated and lysed 16 h later. (A) Abundance of luciferase reporter mRNAs assayed using RT–qPCR. Data are representative of at least three biological replicates from experiments performed on separate days. ns, not significant using one sample t‐test comparing mean fold changes to theoretical mean of 1. (B) IRES‐ or cap‐driven translation from the same samples assayed in parallel using luciferase assay.
-
C, DHEK293T cells stably expressing a type I IFN sensitive response element (ISRE) driven Fluc gene were transfected with increasing amounts of RIG‐I, RIG‐I in combination with DDX60 wt, or RIG‐I in combination with DDX60 E890A while supplementing with GFP plasmid to equalize the total amount of DNA transfected. Transfected cells were then treated with either PBS (negative control) or transfected with LMW poly(I:C). In parallel, untransfected cells were treated with either PBS (mock), transfected with LMW poly(I:C), or treated with IFN‐ß (positive control). Cells were subsequently used for a luciferase assay to assess ISRE activity (C) or Western blot for analysis of DDX60, RIG‐I, ß‐actin (loading control), or GAPDH (loading control) protein products (D). Quantification of DDX60 and RIG‐I band intensities relative to ß‐actin are shown below each respective lane in (D).
Data information: (A, B) Mean ± SD from at least n = 3 biological replicates; ****FDR < 0.01% (P < 0.0001) using unpaired t‐test with Welch correction and Benjamini and Yekutieli correction for multiple testing comparing DDX60 wt versus DDX60 E890A transfected cells. ns, nonsignificant. (C) Mean ± SD from at least n = 3 biological replicates; *P < 0.05 using unpaired t‐test with Welch's correction, ns, not significant using repeated measures one‐way ANOVA with Geisser–Greenhouse correction comparing RIG‐I transfected vs RIG‐I + DDX60 wt or RIG‐I + DDX60 E890A transfected cells. (D) representative Western blot from (C).
Source data are available online for this figure.
DDX60 does not enhance IFN signaling to downregulate type II IRESs
Another potential mechanism of DDX60 antiviral action is indirect, through induction of IFN. Previous publications proposed that DDX60 physically interacts with RIG‐I and synergistically enhances IFN production and thus downstream IFN signaling upon recognition of a viral pathogen associated molecular pattern (PAMP) (Miyashita et al, 2011; Oshiumi et al, 2015). However, a second publication did not observe any physical interaction between DDX60 and RIG‐I or enhancement of IFN signaling (Goubau et al, 2015). We tested whether the DDX60‐ and RIG‐I‐mediated enhancement of downstream IFN signaling contributed to its antitype II IRES activity. We used a HEK293 reporter cell line that encodes an interferon‐sensitive response element (ISRE)‐driven Fluc (ISRE:Fluc). Baseline Fluc activity was minimal when cells were treated with PBS but increased 10‐fold upon poly(I:C) transfection and approximately 400‐fold upon 500 U/ml IFN‐ß treatment (Fig 4C, far left three bars). Next, we transfected RIG‐I into our HEK293 reporter cell line and either treated with PBS or transfected with poly(I:C) to trigger signaling through RIG‐I and induce IFN expression. RIG‐I expression alone caused ISRE:Fluc activity to increase approximately 150‐fold compared with untransfected PBS‐treated controls (mock) (Fig 4C). Upon poly(I:C) transfection in addition to RIG‐I expression, ISRE:Fluc activity further increased by approximately twofold compared with RIG‐I expression alone (P = 0.096). We then proceeded to transfect increasing amounts of wild‐type DDX60 or DDX60 E890A in the presence of increasing amounts of transfected RIG‐I. To keep the total amount of DNA transfected per condition equal, we supplemented transfection mixes with GFP plasmid. Both DDX60 and RIG‐I were expressed according to their transfection strategy (Fig 4D). Both wild‐type DDX60 and DDX60 E890A in combination with RIG‐I, but without the presence of poly(I:C), slightly increased ISRE:Fluc activity by about 1.7‐fold compared with RIG‐I transfection alone (Fig 4C, not statistically significant). Adding poly(I:C) to both wild‐type DDX60 or DDX60 E890A and RIG‐I cells did not further increase ISRE:Fluc activity any more than RIG‐I and poly (I:C) only (Fig 4C). This trend remained even when increasing wild‐type DDX60, DDX60 E890A, or RIG‐I levels. If enhanced downstream IFN signaling explained DDX60 antitype II IRES activity, one would expect any enhancement in ISRE:Fluc activity to be diminished due to the DDX60 E890A mutation. Given that we did not observe such a phenomenon suggested that downstream IFN signaling enhancement does not account for DDX60 antitype II IRES activity.
DDX60 binds both 5′ capped and IRES RNA in vitro
Another possible mechanism of selective DDX60 antiviral action is through specific binding to the type II IRES and enacting a steric hinderance effect. Previous studies demonstrated that the closely related helicase, Ski‐2, and the core helicase domains of DDX60 bind both single‐stranded and double‐stranded RNA with equal affinities, suggesting that these helicases can bind diverse RNA substrates (Miyashita et al, 2011; Halbach et al, 2012). Likewise, structural studies of several DEAD‐box RNA helicases suggest that their interaction with RNA is structure‐dependent rather than sequence‐dependent due to their interaction with the RNA sugar‐phosphate backbone (Sengoku et al, 2006; Schütz et al, 2010). We tested whether DDX60 binds type II IRES RNA preferentially as opposed to type I IRES RNA, type IV IRES RNA, or 5′ capped RNA. We devised two complementary pulldown strategies: one pulling down RNA and detecting bound DDX60, and the other pulling down DDX60 and detecting bound RNA.
First, we generated IRES‐containing, biotin‐UTP labeled RNA probes amenable to precipitation using streptavidin. A previous study used this system to identify far upstream element‐binding protein 1 (FBP1) bindings sites in the EV71 IRES (Hung et al, 2016). Our panel of probes included IRESs of poliovirus (type I), EMCV (type II), HCV (type IV), and 5′ capped Fluc RNA, as well as matching unlabeled probes as negative controls. To test whether DDX60 associates with these RNA sequences, we incubated cell lysates from DDX60 expressing HEK293T cells with biotin‐UTP labeled or unlabeled probes, and performed Western blots. We first tested for a protein known to specifically interact with type I and type II IRESs, but not type IV IRESs, the ITAF polypyrimidine tract‐binding protein 1 (PTBP1). We found PTBP1 to be enriched upon precipitation of biotin‐UTP labeled poliovirus IRES and EMCV IRES probes (type I and type II, respectively) compared with matched unlabeled probes but saw no enrichment between labeled and unlabeled probes for HCV IRES and 5′ capped Fluc RNA (Fig EV4A, left blot). Some nonspecific PTBP1 binding observed is attributed to PTBP1 binding to streptavidin beads alone (Fig EV4A, left blot). Additionally, we recapitulated FBP1 binding to EV71 IRES as previously reported (Hung et al, 2016; Fig EV4A, center blot). However, when analyzing DDX60, we found DDX60 to be equally present when precipitating biotin‐UTP labeled or unlabeled probes for all IRES types and 5′ capped Fluc (Fig EV4A, right blot). Unlike PTBP1, this nonspecific precipitation of DDX60 is not due to DDX60 binding to streptavidin beads as DDX60 does not show any binding to streptavidin beads alone (Fig EV4A, left blot). This led us to favor the conclusion that DDX60 is a “sticky,” nonspecific RNA binder with the caveat that in vitro RNA binding does not necessitate binding in cells.
Figure EV4. Probing for a physical interaction between DDX60 and different IRES sequences.

- RNA‐protein binding assays using biotin labeled RNA probes and streptavidin coated beads. DDX60 wt expressing HEK293T cell lysates were combined with either biotin labeled or unlabeled (negative control) IRES RNA sequences (IRESs from poliovirus, EV71, EMCV, or HCV) or 5′ capped Fluc RNA sequence (negative control). After allowing for protein and RNA binding, streptavidin‐coated beads were used to pull down RNA. After washing, RNA bound proteins were visualized using Western blot. Input or DDX60 overexpression (O.E) lanes show DDX60 and other interrogated RNA binding proteins before incubation of cell lysates with interrogated RNAs. Comparisons for enrichment of RNA binding is made by comparing unlabeled and biotin labeled lanes. Visually equivalent band intensities between unlabeled and biotin labeled lanes signify nonspecific binding between an RNA and interrogated protein. Lane labeled beads represents amount of protein that binds to streptavidin coated beads in the absence of any interrogated RNA. RNA binding proteins interrogated include Polypyrimidine Tract Binding Protein 1 (PTBP1) (positive control for binding type I and type II IRESs specifically), Far upstream element binding protein 1 (FBP1) (binds EV71 IRES), and DDX60. Data is representative of n = 2 biological replicates.
- Protein‐RNA binding assays using immunoprecipitated DDX60 and RT‐qPCR for Fluc mRNAs. HEK293T cells transfected with DDX60 wt or left untransfected were transfected with either 5′ cap‐ or EMCV IRES‐driven Fluc mRNA. Cell lysates were then subjected to immunoprecipitation using either DDX60 targeting antibody or isotype control IgG. Bound mRNA was detected using RT–qPCR against Fluc. Efficiency of immunoprecipitation is visualized using western blot against DDX60. Bound mRNA is quantified as a percent of input RNA postimmunoprecipitation with either IgG or anti‐DDX60 antibody in either DDX60 expressing or untransfected cells. Shown are a representative blot and mean of n = 2 biological replicates.
Source data are available online for this figure.
We next validated these findings with the converse strategy. We expressed wild‐type DDX60 in HEK293T cells and subsequently transfected either 5′ cap‐driven Fluc mRNA or EMCV IRES‐driven Fluc mRNA. As a negative control, we transfected 5′ cap‐driven Fluc mRNA or EMCV IRES‐driven Fluc mRNA without expressing DDX60. We then immunoprecipitated using either a DDX60 targeting antibody or isotype control IgG and analyzed Fluc mRNA quantities by RT‐qPCR. We found both 5′ cap‐driven Fluc mRNA and EMCV IRES‐driven Fluc mRNA to be enriched upon DDX60 immunoprecipitation in our DDX60 expressing samples (Fig EV4B, right). By contrast, there was no enrichment of these mRNAs in samples with undetectable DDX60 expression (Fig EV4B, right). This provided additional evidence that DDX60 nonspecifically binds both 5′ capped and EMCV IRES‐containing mRNAs in vitro. Overall, our RNA binding assays led us to conclude that DDX60 does not distinguish type II IRES‐containing mRNAs by differential RNA binding.
DDX60 modulates ribosome occupancy on type II IRES‐containing Fluc mRNAs
Our RNA abundance assay suggested that while DDX60 may not decrease the abundance of type II IRES‐containing mRNAs (Fig 4A), it still diminishes translation from the IRES as seen by the decrease in Fluc protein synthesis (Figs 2 and 4B). This may be a result of DDX60 modulating ribosome binding to the mRNAs—either by reducing initiation, which would lead to fewer ribosomes bound, or by reducing translation elongation, which may lead to an increase in bound, but stalled, ribosomes, or both, which would reduce overall translation activity but without significantly reducing ribosome numbers on mRNA. To test for such changes in ribosome binding, we performed polysome profiling. A greater number of ribosomes bound to mRNA will result in increased abundance of mRNAs in heavier fractions of a sucrose gradient, indicative of increased initiation if elongation rates are unchanged (Panda et al, 2017). Global polysome profiling in untransfected or wild‐type DDX60 expressing HEK293T cells revealed that the overall distribution of mRNA in the different polysome fractions was similar between DDX60‐expressing and control untransfected cells (Fig EV5A), suggesting that DDX60 does not affect cellular translation globally.
Figure EV5. Global translation levels under DDX60 overexpression.

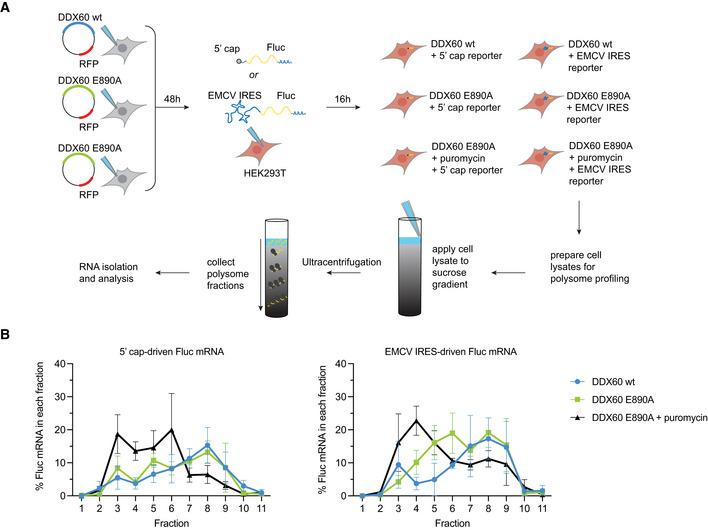
- Polysome profile of total RNA upon DDX60 overexpression. Untransfected or DDX60 wt expressing HEK293T cells were treated with 100 μg/ml of cycloheximide for 15 min to arrest polysomes before being trypsinized and frozen down. All harvested cells per condition were then subjected to polysome profiling by lysing cells in the presence of cycloheximide and protease and phosphatase inhibitors, applying cell lysates to 15–50% sucrose gradients, and subjecting to ultracentrifugation. The centrifuged gradients were run through a fractionator and total RNA in each fraction was measured by UV absorbance (254 nm). Data are an average of two replicate plate of cells per condition.
- Polysome profile of total RNA post transfection of DDX60 and Fluc mRNA reporters. DDX60 wt or DDX60 E890A (negative control) expressing HEK293T cells transfected with in vitro transcribed 5′ cap (left) or EMCV IRES (right) driven Fluc mRNA constructs. Duplicate samples of DDX60 E890A and 5′ cap‐ or EMCV IRES‐driven Fluc mRNA transfected cells were treated with 200 μM puromycin as positive controls for decrease in polysomes. Cells were then treated with 100 μg/ml of cycloheximide for 15 min to arrest polysomes before being trypsinized and frozen down. All harvested cells per condition were then subjected to polysome profiling by lysing cells in the presence of cycloheximide and protease and phosphatase inhibitors, applying cell lysates to 15–50% sucrose gradients, and subjecting to ultracentrifugation. The centrifuged gradients were run through a fractionator and total RNA in each fraction was measured by UV absorbance (254 nm). Mean of n = 3 biological replicates.
- Representative results of electrophoresis analysis of individual fractions. Shown is RNA from one replicate of DDX60 E890A and cap‐fluc transfected cells (control). RNA was analyzed by Bioanalyzer to visualize 28S and 18S ribosome subunit distribution in the different fractions. RIN, RNA integrity number.
We hypothesized that expression of wild‐type DDX60 would selectively modulate the abundance of type II IRES‐containing mRNAs in specific fractions. To test this hypothesis, we performed polysome profiling in HEK293T cells expressing wild‐type DDX60 or our negative control, DDX60 E890A and transfected them with equimolar amounts of either 5′ cap‐driven or EMCV IRES‐driven Fluc mRNA. As positive controls for translation inhibition by disrupting polysomes, we treated duplicate samples of DDX60 E890A plus 5′ cap‐driven Fluc mRNA transfected cells and DDX60 E890A plus EMCV IRES‐driven Fluc mRNA transfected cells with puromycin (Fig 5A; Kudla & Karginov, 2016). As expected, puromycin‐treated samples showed mRNA accumulation in lower fraction numbers (Fig EV5B).
Figure 5. Polysome profile of type II IRES Fluc mRNA in the presence of DDX60.
- Schematic of polysome profiling strategy. HEK293T cells were transfected with DDX60 wt or DDX60 E890A (negative control). 48‐h post‐transfection, cells were transfected with in vitro transcribed 5′ cap‐ or EMCV IRES‐driven Fluc mRNA constructs. 16‐h post‐transfection, duplicate samples were treated with 200 μM puromycin for 20 min as positive controls for decrease in polysomes. Cells were treated with 100 μg/ml of cycloheximide for 15 min to arrest polysomes and subjected to polysome profiling by ultracentrifugation through 15–50% sucrose gradients. Amount of Fluc reporter mRNA from polysome fractions was determined by RT‐qPCR.
- Effect of DDX60 on 5′ cap (left) or EMCV IRES (right) driven Fluc mRNA polysomes.
Data information: Mean percent ± SEM of Fluc mRNA of each fraction relative to Fluc mRNA in all fractions, from n = 3 biological replicates. Full polysome profiles are shown in Fig EV5.
As evident from electrophoresis analysis of RNA isolated from individual polysome fractions, fraction 1 corresponds to free RNAs (tRNAs), fraction 2 to mRNA associated with the 40S subunit of the ribosomes, fraction 3 to mRNA associated with monosomes, and fractions 4–11 to polysomes (Fig EV5C). We next analyzed the distribution of Fluc reporter mRNA in the individual fractions for each condition by RT–qPCR. Puromycin treatment shifted both 5′ cap‐ and EMCV IRES‐driven Fluc mRNAs to lower fraction numbers (3–6 for capped mRNA, 3–5 for EMCV‐IRES mRNA), demonstrating decreased ribosome abundance on the mRNA reporters irrespective of the mode of translation initiation, cap‐mediated or IRES‐mediated. For cap‐driven Fluc mRNA, wild‐type DDX60 did not change Fluc mRNA distribution between fractions—it closely followed the distribution seen with DDX60 E890A (Fig 5B, left graph), consistent with the observation that DDX60 does not arrest the bulk translation (Fig EV5A). For EMCV‐IRES‐driven Fluc mRNA, the profiles for DDX60 and DDX60 E890A diverged in fractions 4–6 (Fig 5B, right graph), albeit not significantly statistically. However, the observed trend of lower EMCV‐IRES‐Fluc mRNA amounts in these specific polysomal fractions together with our data showing significant reduction in Fluc protein production in this condition (Fig 2B) suggests that DDX60 selectively decreases translating ribosomes on type II IRES‐driven mRNAs, thereby causing net reduction in translation.
DDX60 modulates ribosome occupancy on type II IRES viral mRNA but not type I IRES viral mRNA
We next sought to determine whether DDX60 expression changes ribosome abundance on type II IRES mRNA during viral infections. Accordingly, we performed polysome profiling on DDX60 expressing HeLa cells infected with either parental poliovirus (type I IRES) or chimeric EMCV‐IRES‐PV (type II IRES) at MOI of 1 (Fig 6A). Given that poliovirus expresses 2A and 3C proteases that cleave host eIF4G, and eIF5B and PABP, respectively, we expectedly observed that poliovirus infection decreases polysomes, with a concomitant increase in ‐40S, 60 ribosomal subunits and 80S monosomes (Fig 6B, polysome profile above, and total RNA in each fraction shown below for poliovirus versus EMCV‐IRES‐PV infected cells). In EMCV‐IRES‐PV‐infected cells, we observed the presence of polysomes starting at 130 s, corresponding to fraction 4 (Fig 6B, C). We attribute this to the fact that parental type I IRES‐containing poliovirus is resistant to DDX60, but chimeric type II IRES‐containing EMCV‐IRES‐PV is sensitive to DDX60. Therefore, DDX60 expressing EMCV‐IRES‐PV‐infected cells have less viral protein synthesis, which reduces EMCV‐IRES‐PV's ability to install a robust host translation shutoff.
Figure 6. Polysome profile of wild type (type I IRES) poliovirus versus chimeric (type II IRES) poliovirus in the presence of DDX60.
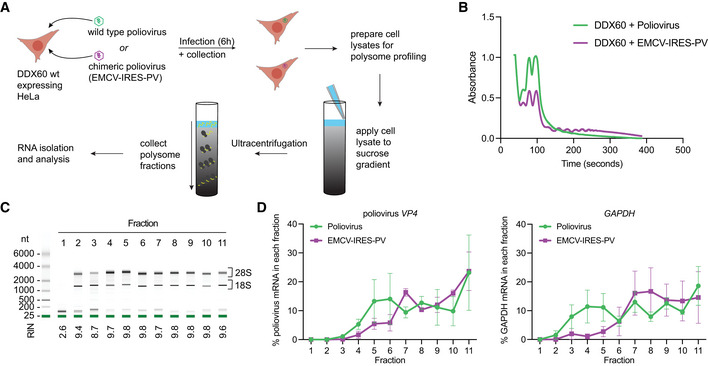
- Schematic of polysome profiling strategy. HeLa cells stably expressing wild type DDX60 were infected with parental poliovirus (type I IRES) or chimeric poliovirus (type II IRES). 6 hpi cells were treated with 100 μg/ml of cycloheximide for 15 min to arrest polysomes and subjected to polysome profiling by ultracentrifugation through 15–50% sucrose gradients. Amount of poliovirus VP4 mRNA from polysome fractions was determined by RT‐qPCR.
- Cells depicted in (A) were subjected to polysome profiling by lysing in the presence of cycloheximide and protease and phosphatase inhibitors, applying cell lysates to 15–50% sucrose gradients, and subjecting to ultracentrifugation. Centrifuged gradients were run through a fractionator and total RNA in each fraction was measured by UV absorbance (254 nm).
- Representative results of RNA electrophoresis analysis of individual fractions. Shown are results from one replicate of DDX60‐transfected, EMCV‐IRES‐PV‐infected cells. RNA was analyzed by Bioanalyzer to visualize 28S, and 18S ribosome subunit distribution. RIN, RNA integrity number.
- Effect of DDX60 expression on parental poliovirus (type I IRES, purple circles) versus chimeric poliovirus (type II IRES, red squares) VP4 mRNA polysomes (left) and cellular GAPDH mRNA polysomes in the same samples.
Data information: Mean percent ± SEM of poliovirus VP4 or GAPDH mRNA in each fraction relative to VP4 or GAPDH mRNA in all fractions from n = 3 replicate plates per condition.
Electrophoresis analysis of RNA isolated from individual polysome fractions revealed that fraction 1 corresponds to free RNAs (tRNAs), fraction 2 to mRNA associated with the 40S subunit of the ribosomes, fraction 3 to mRNA associated with monosomes, and fractions 4–11 to polysomes (Fig 6C) To determine ribosome abundance on type II IRES mRNA vs capped mRNA in detail, we analyzed poliovirus VP4 mRNA levels and host GAPDH mRNA levels in each polysome fraction from DDX60 expressing cells infected with parental poliovirus versus EMCV‐IRES‐PV. In EMCV‐IRES‐PV‐infected cells, we observed a trend of reduced VP4 mRNA in fractions 4–6 (corresponding to light polysomes) as compared with PV‐infected cells (Fig 6D, left graph). This may indicate a reduction in translation initiation on type II IRES mRNA. Interestingly, albeit not statistically significant, we observe an increase of VP4 mRNA in fraction 7 of EMCV‐IRES‐PV‐infected cells. This may represent stalling of ribosomes due to disrupted translation elongation. Together with our data showing significant reduction in EMCV‐IRES‐driven Fluc protein production in the presence of DDX60 (Fig 2B) and reduced replication of EMCV‐IRES‐PV (Fig 3E), this suggests that DDX60 reduces viral type II IRES‐driven protein synthesis by modulating ribosome occupancy on type II IRES‐driven mRNA.
Interestingly, we also observed a trend of reduced ribosome occupancy on cap‐driven GAPDH mRNA in EMCV‐IRES‐PV‐infected cells as compared with parental PV‐infected cells (Fig 6D, right graph, depletion in fractions 3–5 compared with PV‐infected cells). However, we then observe equal ribosome occupancy on cap‐driven GAPDH mRNA in EMCV‐IRES‐PV‐infected cells compared PV‐infected cells (Fig 6D, right graph, fractions 7–11). Therefore, a greater proportion of the total cellular GAPDH mRNA in EMCV‐IRES‐PV‐infected cells compared with PV‐infected cells is concentrated in heavier fractions 7–11. By contrast, in PV‐infected cells, GAPDH mRNA is uniformly distributed between fractions 3 and 11. This indicates to us that GAPDH mRNA is more efficiently translated in EMCV‐IRES‐PV‐infected cells compared with PV‐infected cells, possibly due to DDX60 perturbing EMCV‐IRES‐PV from remodeling cellular translation, which is consistent with our observations in Fig 6B.
Discussion
DDX60 is part of the superfamily‐2 (SF2) DExD/H box RNA helicases that are proposed to use energy from ATP to remodel RNA structures. These helicases contain two tandem helicase core domains with various characteristic sequence motifs, flanked by N‐ and C‐terminal extensions. They impact diverse cellular processes such as transcription, mRNA splicing, translation, and RNA turnover (Pyle, 2008; Ranji & Boris‐Lawrie, 2010; Jankowsky, 2011; Sloan & Bohnsack, 2018). Thus, they are associated with the development of many diseases such as cancer, aging, neurologic and immunologic disorders, and infectious disease (Steimer & Klostermeier, 2012). Indeed, DDX60 expression has been shown to be dysregulated and associated with advanced disease and response to treatment of different cancers, such as oral cancers (Fu et al, 2016; Reyimu et al, 2021), gliomas (Zhang et al, 2021), and breast cancers (Ríos‐Romero et al, 2020; Xin et al, 2020). Additionally, DNA methylation variability in DDX60 is associated with cases of mixed connective tissue disease (Carnero‐Montoro et al, 2019), and DDX60 expression was found to be strongly positive by immunofluorescence in renal biopsy specimens from patients with proliferative lupus nephritis (Karasawa et al, 2022). Lastly, DDX60 has been observed to inhibit viral infections (Miyashita et al, 2011; Schoggins et al, 2011; Goubau et al, 2015; Oshiumi et al, 2015; Ma et al, 2017). Its mechanism of action during virus infection, however, is controversial.
DDX60 was initially shown to be among the strongest inhibitors of a specific reporter HCV (Schoggins et al, 2011). Subsequent studies investigating its antiviral mechanism arrived at differing conclusions. One group suggested that DDX60 enhanced RIG‐I mediated viral RNA sensing and downstream IFN signaling (Miyashita et al, 2011; Oshiumi et al, 2015), while another group suggested that DDX60 did not physically interact with RIG‐I or enhance downstream IFN signaling (Goubau et al, 2015). These discordant observations could be due to differences in knockout mice generation strategies, IFN‐driven reporters used, or other experimental differences. Here, we present an alternate but not mutually exclusive mechanism of action. We show that DDX60 is capable of specifically counteracting viral type II IRES‐mediated translation while leaving host 5′ cap‐mediated translation intact.
Our study is based on the unexpected observation that DDX60 inhibits the replication of a bicistronic reporter HCV containing both HCV and EMCV IRES, but not a monocistronic reporter HCV lacking the EMCV IRES (Fig 1D). The EMCV IRES is a commonly used element that drives dual expression of two independent gene cassettes and was included in early version of reporter HCV (Ghattas et al, 1991; During et al, 1998; Date et al, 2004; Jones et al, 2007, 2010). Utilizing luciferase reporters translationally regulated by different IRESs (Fig 2), comparative virology experiments with different IRES‐containing viruses (Fig 3), and the differential phenotype with two different reporter HCV establish a strong correlation between EMCV (or type II) IRES activity and translational repression by DDX60. Additionally, our functional studies interrogating the effect of DDX60 mutations on type II IRES activity (Fig EV2A) strongly correlate with our bicistronic reporter HCV and EMCV IRES‐driven RFP observations (Fig 1B and C). Altogether, these observations warrant caution in using exogenous translational elements for reporter viruses or protein expression constructs, as it may yield artifactual findings when screening antiviral effectors, particularly with relation to the interferon response.
In the IFN effector function field, ISG function is commonly studied using overexpression of ISGs and corresponding mutants and relative importance of the ISG to the interferon program is studied using an ISG knockdown or knockout approach (Schoggins et al, 2011; Dittmann et al, 2015; Schoggins, 2019). We anticipate that silencing DDX60 would allow the type II IRES to be more active and thus allow greater viral protein synthesis and, subsequently, enhance viral replication. However, performing well‐controlled DDX60 silencing experiments are technically challenging, as, due to low baseline levels of DDX60, they may require prior DDX60 induction by IFN. It is possible that DDX60 induction via picornavirus infection achieves the same result, as picornaviruses induce a strong type I and type III IFN response (Feng et al, 2012; Freundt et al, 2018). Nonetheless, we note that upon infection or interferon treatment, DDX60 would be among hundreds of other ISGs activated. The likelihood of DDX60 depletion alone to inhibit virus infection is low given its modest 0.5‐1‐log reduction in viral titers when DDX60 was overexpressed (Fig 3). We attribute this to the fact that DDX60's mechanism specifically antagonizes the step of translation for few viruses containing a type II IRES. Unlike DDX60, broad transcriptional regulators of antiviral genes such as IRF1 (used as a positive control for many experiments in this study) can activate its own set of antiviral genes augmenting its antiviral effect. Silencing broad regulators of antiviral genes—which are genes that inhibit multiple steps of the virus life cycle, or genes with a potent mechanism or multiple mechanisms to inhibit a single lifecycle step—will likely result in a more profound phenotype than silencing ISGs with a more particular mechanism of action.
One peculiarity we observed was that DDX60 inhibited EMCV at 24 hpi even while EMCV titers could be detected 8–10 hpi (Figs 3B and EV3D, and E). Since DDX60 inhibits type II IRES translation, we anticipated early inhibition of EMCV, as observed for FMDV (Fig 3C). However, another well‐known inhibitor of HCV translation, IFIT1, showed an inhibitory phenotype 96‐h postinfection (Raychoudhuri et al, 2011), even while active HCV replication can be observed as early as 48‐h postinfection (Schoggins et al, 2011). This example suggests that one cannot directly generalize to say that inhibitors of virus translation should always demonstrate an early phenotype. We attribute the observation that the effects of DDX60 become noticeable only after multiple rounds of virus replication to DDX60's overall modest inhibition of virus translation that accumulates over several replication cycles.
In deciphering DDX60's mechanism of type II IRES inhibition, we posited that DDX60 may be triggering the degradation of type II IRES‐containing mRNAs, reducing ribosome binding on said mRNAs, acting indirectly through its previously characterized role as an enhancer of RIG‐I‐mediated downstream IFN activity, or a combination of the three. With the methods used, we were unable to detect a DDX60‐mediated decrease in type II IRES‐driven mRNAs or increase downstream IFN activity in concert with RIG‐I (Fig 4). Instead, DDX60 expression modulated ribosome occupancy on type II IRES‐driven mRNA without impacting overall cellular translation or ribosome accumulation on 5′ capped mRNAs (Figs 5 and EV5). Our polysome profiling did not allow for definite conclusions on whether this is due to inhibition of translation initiation or elongation—both are a possibility. While many DExD/H RNA helicases have a role in regulating mRNA translation, most are described to resolve RNA structures and rearranging 43S complexes to increase translation efficiency (Shen & Pelletier, 2020). Recent studies also implicate these helicases in acting on stalled ribosomes. For example, depletion of the RNA helicase DDX3 decreases ribosome speed along mRNAs, suggesting that it acts to prevent prolonged translation stalls (Padmanabhan et al, 2021). The helicase Ski‐2 in a complex with Ski‐3 and Ski‐8 is able to extract mRNA presumably for 3′ to 5′ degradation from stalled 80S ribosomal complexes (Zinoviev et al, 2020). DDX60 may slow initial ribosome attachment and start codon recognition, stall ribosomes by sterically blocking their progression during translation, or be part of a multisubunit complex that stalls ribosomes.
How DDX60 blocks ribosome accumulation on type II IRES mRNA remains unknown. One possibility is that DDX60 sterically hinders ribosome subunits (i.e., 40S) from either initially binding or fully forming elongation competent ribosomes from progressing along type II IRES mRNA. Our RNA bindings assays (Fig EV4) show that DDX60 binds mRNA indiscriminately, suggesting that RNA binding alone may not be determining IRES specificity. Importantly, the indiscriminate RNA and even DNA binding by DDX60 shown previously was using helicase domains of DDX60 in electrophoretic mobility‐shift assays (EMSAs) (Miyashita et al, 2011). However, N‐ and C‐terminal extensions not included in previous assays may be offering DDX60 some substrate specificity as we show that these extensions are important for type II IRES inhibition (Figs 1 and EV2), and others show that N‐ and C‐terminal extensions contribute to specific substrate recognition for other helicases (Halbach et al, 2012; Thoms et al, 2015; Lingaraju et al, 2019). It is therefore imperative to repeat EMSAs using full‐length purified DDX60. Alternatively, DDX60 may bind several different mRNAs but is displaced by translating or initiating ribosomes more efficiently on noninhibited mRNAs. When DDX60 binds a type II IRES element, it may use its helicase activity to remodel the IRES, thus inhibiting its functions for translation initiation. A combination of steric hinderance and IRES remodeling may also be at play.
Our experiments to test how DDX60 distinguishes type II IRES‐containing RNAs showed that it physically binds to different IRES and 5′ capped RNAs nonspecifically in vitro (Fig EV4). Importantly, we also observed DDX60 binding to GAPDH mRNA. This may be due to a combination of DDX60 expression above physiological levels and cell compartment disruption during cell lysis. This technical limitation may be overcome in future experiments using cross‐linking of proteins to RNA using either chemical agents or UV irradiation prior to cell lysis as done for other RNA binding proteins (Hafner et al, 2010; Huang et al, 2018; Urdaneta et al, 2019; Han et al, 2022). Another possible explanation is that DDX60 has the propensity to bind many IRES types, but only interferes with specific secondary structure(s) found in type II IRESs and not the other IRES types (reviewed in Hellen & Wimmer, 1995; Lozano & Martínez‐Salas, 2015; Martinez‐Salas et al, 2018). Our data demonstrate that poliovirus can be inhibited by DDX60 when its endogenous type I IRES is replaced with the type II IRES of EMCV (Fig 3E), suggesting that DDX60 recognizes and/or interferes with some difference(s) between type I and type II IRESs. One possibility may be that DDX60 binds and interferes with RNA structural domains specific to type II IRESs, such as domains J and K (Hellen & Wimmer, 1995; Martinez‐Salas et al, 2018). UV or chemical cross‐linking‐based RNA binding assays, specific deletions of domains in the EMCV IRES, or specific insertions of domains in the EMCV IRES into the poliovirus IRES can test this possibility. Alternatively, DDX60 may preferentially bind and inhibit type II IRES translation when complexed with other proteins. Indeed, other helicases have been shown to recognize their substrates by interacting with proteins via their helicase domains and N‐ and C‐terminal extensions (Halbach et al, 2012; Thoms et al, 2015; Lingaraju et al, 2019), which we observed to both be important for DDX60 anti‐IRES activity (Figs 1C and EV2A). A compelling possibility is that DDX60 may differentially interact and interfere with eIFs or ITAFs that are specifically involved in promoting type II IRES translation such as eIF4A and central domain of eIF4G, PTB, La protein/SS‐B for EMCV in the context of excessive PTB levels, and Ebp1/ITAF45, Sam68, and C‐terminal cleavage product of hnRNP K (hnRNP K364‐465) for FMDV IRES, to name a few (Hellen & Wimmer, 1995; Kim & Jang, 1999; Lawrence et al, 2012; Lozano & Martínez‐Salas, 2015; Martinez‐Salas et al, 2018; Liu et al, 2020). Alternatively, DDX60 may interact with and enhance activity of ITAFs involved in inhibiting type II IRES translation, such as Gemin5 and full‐length hnRNP K for the case of FMDV (Pacheco et al, 2009; Lee et al, 2017; Liu et al, 2020). Immunoprecipitation and mass‐spectrometry experiments may reveal proteins that interact with DDX60, and subsequent knockdown experiments may reveal the necessity of such factors for DDX60 RNA binding and/or type II IRES inhibition. Alternatively, DDX60 may interact with eIFs shared by type I and type II IRESs, but specifically interfere with the structural rearrangements made on the type II IRES by eIFs such as eIF4A combined with eIF4G (Kolupaeva et al, 2003). Immunoprecipitation of type I versus type II IRES bound eIFs and ITAFs could generate a list of potential proteins that contribute to DDX60 activity against type II IRESs, and in vitro translation, hydroxyl radical, and chemical and enzymatic assays may define possible structural changes in the type II IRES that are blocked by DDX60 but are complicated by the necessity to pull down full‐length DDX60 protein in sufficient amounts.
In our analyses of different DDX60 mutants, we found that predicted ATP binding residues, helicase motif, and N‐ and C‐terminal extensions were all important for DDX60 antiviral and anti‐IRES activity (Figs 1C and EV2A). Intriguingly, the SAT motif of DDX60 was dispensable for antiviral function (Fig 1C). SAT mutants in other DEAD box helicases can bind RNA in an ATP‐dependent manner but lack RNA unwinding activity (Pause & Sonenberg, 1992; Schwer & Meszaros, 2000; Linder, 2006), suggesting that RNA binding or ATPase activity alone may be sufficient for DDX60's antiviral properties. Additionally, we found a putative ATP binding/hydrolysis DDX60 mutant, K791N, to uniformly increase all types of IRES‐driven translation (Fig EV2A). The analogous lysine in the DExD/H box helicase NS3 in HCV is thought to help stabilize ATP binding through interaction with its ß‐phosphate, and in concert with two domain VI arginine residues and a metal ion, stabilize the developing negative charge on the ATP γ‐phosphate during hydrolysis (Gu & Rice, 2009). One can speculate that a K791N mutation alters DDX60 ATP binding and hydrolysis kinetics. What these alterations are and how they contribute to regulating the activity of multiple IRESs, but not cap‐mediated translation remains to be determined. Future work interrogating individual DDX60 mutants through in vitro ATP hydrolysis, RNA, and protein binding assays may reveal the precise enzymatic activities that DDX60 uses to enhance or inhibit different types of IRES‐driven translation. As these experiments must be performed in the context of the full‐length purified DDX60 protein to preserve its differential anti‐IRES function (Figs 1 and 2), they will be technically challenging due to the large size and observed poor solubility of DDX60.
DDX60 is not the only DExD/H box RNA helicase observed to inhibit viral IRES‐mediated translation. The DEAD box helicase DDX21 was recently shown to antagonize FMDV IRES, and the HCV‐like IRESs of classical swine fever virus and Seneca Valley virus (Abdullah et al, 2021). However, unlike DDX60, DDX21 is thought to inhibit these IRESs indirectly through upregulation of IFN‐ß and IL‐8 mRNAs (Abdullah et al, 2021). Likewise, the ability for DDX21 to selectively inhibit IRES‐mediated translation has not been tested. Other antiviral effectors that can restrict IRES‐mediated translation such as PKR and RNase L dampen host mRNA translation as well (Stern‐Ginossar et al, 2019). DDX60 is unique in this regard as it selectively dampens viral type II IRES‐mediated translation.
Often, mechanisms of antiviral genes are antagonized by viral proteins (Beachboard & Horner, 2016; Freundt et al, 2018; Abdullah et al, 2021; Cesaro & Michiels, 2021). An interesting possibility would be if like DDX21, DDX60 is degraded upon infection with a type II IRES‐containing virus. DDX21 is degraded by FMDV 3C proteinase and viral proteins 2B and 2C through lysosomal and caspase‐dependent pathways, respectively (Abdullah et al, 2021). To start addressing if DDX60 is similarly degraded, one can perform an infection time series with a type II IRES virus (i.e., EMCV) and observe for signs of DDX60 proteolysis or degradation via Western blot. If signs of proteolysis or degradation are observed, further assays using different protein pathway degradation inhibitors can uncover the mechanism of degradation.
Recent studies suggest a broader role for DDX60 in translation. One study showed that kinetoplastid‐DDX60 in Trypanosoma and Leishmania species associates with the 43S pre‐initiation complex and possibly alters its conformation upon binding ATP to regulate translation initiation (Bochler et al, 2020). DDX60 homologs exist in every kingdom of eukaryotes (NCBI), and intriguingly, in our hands, human DDX60 can be expressed in animal cells to inhibit animal viruses (Fig 3). The use of human DDX60 in hamster cells is a caveat of our FMDV and BEV‐1 infection experiments, but the mammalian translation apparatus is conserved (Hernández et al, 2010), and observations from this experiment suggest a level of functional conservation for DDX60 in different species, the mammalian translation apparatus, or both. Further work comparing DDX60 sequences and functions from other species may uncover novel and/or conserved DDX60 functions. Another study found that interaction between DDX60 and the m6A reader protein, YTHDF1, is required for YTHDF1 to bind m6A modified Traf6 (tumor necrosis factor receptor‐associated factor 6) mRNA and direct its translation (Zong et al, 2021). Methylation of adenosine nucleotides at the N 6 position (m6A) is the most prevalent posttranscriptional mRNA modification, generally affecting mRNA metabolism and translation (Roundtree et al, 2017; Zhao et al, 2017). Our in vitro transcribed mRNAs lack covalent RNA modifications which may have influenced in vitro DDX60‐RNA interactions we observed (Fig EV4). Thus, future work deciphering the exact role of DDX60 in recognizing m6A modified Traf6 or other m6A modified mRNAs may reveal how DDX60 regulates metabolism or translation of select cellularly transcribed mRNAs.
Lastly, IRESs are not unique to viruses, and many host mRNAs have been found to initiate translation using IRES elements during conditions of stress (Komar & Hatzoglou, 2011; Jackson, 2013). Determining cellular mRNAs bound by DDX60 using an unbiased sequencing‐based approach could decipher common functional or structural themes in DDX60 bound mRNAs. Likewise, a parallel sequencing‐based ribosome profiling approach could reveal the compendium of mRNAs translationally regulated by DDX60.
Overall, our work uncovers DDX60 as host factor that specifically inhibits type II IRES‐mediated translation, but leaves other IRES‐ or host 5′ cap‐mediated translation intact. We show that DDX60 mechanistically acts by reducing ribosomes bound to type II IRES‐driven but not type I IRES or 5′ cap‐driven mRNAs. DDX60 may thus work as an innate immune counter measure to specifically decrease viral protein synthesis while allowing host translation to proceed unencumbered. Future work uncovering the means for DDX60 specificity may unearth novel strategies for selective translational control in the face of a virus infection.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| HEK293T | ATCC | CRL‐3216 |
| A549 | ATCC | CCL‐185 |
| HFF1 | ATCC | SCRC‐1041 |
| HeLa | ATCC | CRL‐1958 |
| HEK293 ISRE Recombinant Reporter Cells | BPS Bioscience | 60510 |
| BHK‐21/J | ATCC | n/a |
| Huh‐7 | Apath | n/a |
| Huh‐7.5 | Apath | n/a |
| Recombinant DNA | ||
| DDX60 WT | GenBank | OM859267 |
| IRF1 | GenBank | OM859269 |
| Fluc | GenBank | OM859268 |
| Bicistronic HCV | Jones et al (2010) | N/A |
| Monocistronic HCV | Horwitz et al (2013) | N/A |
| HCV | Lindenbach et al (2005) | N/A |
| FMDV | Rieder et al (1993) | N/A |
| Bovine Enterovirus (BEV‐1) | Genbank | D00214 |
| Type I Poliovirus | GenBank | OM677908 |
| EMCV | GenBank | OM677909 |
| Chimeric EMCV‐IRES‐PV | GenBank | OM677910 |
| Antibodies | ||
| DDX60 | Abcam | ab139807 |
| IRF1 | Cell Signaling Technology | 8478S |
| GAPDH | Santa Cruz Biotechnology | sc‐47724 |
| ß‐actin | Invitrogen | MA5‐15739 |
| RFP | Invitrogen | MA5‐15257 |
| Fluc | Santa Cruz Biotechnology | sc‐74548 |
| Rabbit IgG | Abcam | Supplied with RIP Kit |
| Oligonucleotides and sequence‐based reagents | ||
| PCR primers | This study | Appendix Table S1 |
| Chemicals, enzymes and other reagents | ||
| DMEM | Corning | 10013CV |
| 10% FBS | Cytiva | SH30071.03 |
| Penicillin + Streptomycin Solution 1× | Corning | 25025CI |
| MEM | Corning | 10‐009‐CV |
| Geneticin Selective Antibiotic (G418 Sulfate) | Thermo Fisher Scientific | 10131027 |
| TransIT®‐LT1 Transfection Reagent | Mirus Bio | MIR 2300 |
| Opti‐MEM | Thermo Fisher Scientific | 51985934 |
| Thermo Scientific Oxoid Skim Milk Powder | Thermo Fisher Scientific | OXLP0031B |
| TBS/0.1% Tween‐20 Solution | Thermo Fisher Scientific | BP337‐500 |
| 0.1% BSA/PBS | Millipore Sigma | IF104 |
| L‐ornithine | Sigma‐Aldrich | P2533 |
| PBS | Fisher Scientific | MT21031CV |
| RLT buffer | Qiagen | 74106 |
| LDS buffer | Invitrogen | B0007 |
| Mercaptoethanol | Sigma‐Aldrich | M3148 |
| cOmplete, Mini, EDTA‐free Protease Inhibitor Cocktail | Roche | 11836170001 |
| Pierce Phophatase Inhibitor Mini Tablets | Thermo Fisher Scientific | A32957 |
| Accumax Cell Aggregate Dissociation Medium | Thermo Fisher Scientific | 00‐4666‐56 |
| Passive Lysis Buffer | Promega | E1941 |
| Promega Luciferase Assay System | Promega | E4550 |
| PBS + Ca and Mg | Corning | 21030CV |
| 2× DMEM | Gibco | 12100046 |
| 4% Avicel, 250 uL DEAE Dextran | Sigma‐Aldrich | 30461 |
| Glutamax | Thermo Fisher Scientific | 35050061 |
| 7.5% Sodium Bicarbonate | Gibco | 25080094 |
| Molecular Grade Biology Water | Fisher Scientific | MT46000CM |
| 2% Formaldehyde | Fisher Scientific | BP531‐500 |
| MagneSphere Paramagnetic Particles | Promega | Z5481 |
| Glycoblue | Thermo Fisher Scientific | AM9515 |
| Complete Growth Media + Cycloheximide | Sigma‐Aldrich | C7698 |
| Cycloheximide Supplemented Protease | Thermo Fisher Scientific | 87786 |
| Phosphatase Inhibitors | Thermo Fisher Scientific | 78420 |
| Riboblock | Thermo Fisher Scientific | EO0382 |
| EDTA | Ambion | AM9260G |
| Invitrogen Anti‐Reverse Cap Analog | Fisher Scientific | AM8045 |
| X‐tremeGENE 9 DNA Transfection Reagent | Roche | 6365809001 |
| NorthernMax‐Gly Sample Loading Dye | Thermo Fisher Scientific | AM8551 |
| Invitrogen Ambion Millennium Markers RNA Markers Formamide | Thermo Fisher Scientific | AM7151 |
| Software | ||
| ImageJ2 | https://imagej.net/software/imagej2/ | |
| Applied Biosystems QuantStudio 3 real‐time PCR | https://www.thermofisher.com | |
| FlowJo Software | https://www.flowjo.com | |
| Other | ||
| Qiagen Rneasy kit | Qiagen | 74106 |
| SuperScript III First‐Strand Synthesis System | Thermo Fisher Scientific | 18080051 |
| Applied Biosystems PowerUp SYBR Green Master Mix | Applied Biosystems | A25918 |
| iBlot 2 Transfer System | Thermo Fisher Scientific | IB24001 |
| Lookout Mycoplasma PCR Kit | Sigma‐Aldrich | MP0035 |
| RNA‐Binding Protein Immunoprecipitation (RIP) Kit | Abcam | ab139807 |
| LSRII‐HTS Flow Cytometer | BD Biosciences | N/A |
| BioTek Synergy Synergy HTX Multi‐Mode Microplate Reader | Agilent | N/A |
| Agilent BioTek Dual Reagent Injector System | Agilent | N/A |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| DynaMag‐2 Magnet | Life Technologies | 12321D |
| Beckman 14 × 89 mm tube | Beckman Coulter | 331372 |
| BR‐188 Density Gradient Franctionation System | Brandel | N/A |
| MEGA script T7 Transcription Kit | Thermo Fisher Scientific | AMB13345 |
| Invitrogen Poly(A) Tailing Kit | Thermo Fisher Scientific | AM1350 |
Methods and Protocols
Cell lines
HEK293T (CRL‐3216), A549 (CCL‐185), HFF1 (SCRC‐1041), and H1 clone of HeLa (CRL‐1958) cells were purchased from ATCC. HEK293 ISRE reporter recombinant cell line was purchased from BPS Bioscience (Catalog #60510). HEK293T and A549 cells were cultured in DMEM (Corning™ 10013CV) supplemented with 10% FBS (Cytiva SH30071.03), penicillin and streptomycin solution (1×, Corning™ 30‐002‐CI), and nonessential amino acid solution (1×, Corning™ 25025CI). HFF1 cells were cultured in DMEM supplemented with 15% FBS, penicillin and streptomycin solution, and nonessential amino acid solution. HeLa and cells were cultured in MEM (Corning™ 10‐009‐CV) supplemented with 10% FBS and penicillin and streptomycin solution. HEK293 ISRE reporter recombinant cells were cultured in MEM supplemented with 10% FBS, penicillin and streptomycin solution, and 400 μg/ml of Geneticin™ Selective Antibiotic (G418 Sulfate) (Thermo Fisher Scientific 10131027). BHK‐21 cells (baby hamster kidney cells strain 21, clone 13, ATCC CL10), obtained from the American Type Culture Collection (ATCC) were used to propagate virus stocks and to measure FMDV and BEV‐1 titers. Cells were cultured in MEM supplemented with 10% FBS. BHK21/J (Leiden) 2nd seed cells were cultured in DMEM (Gibco 11965118) supplemented with 10% FBS and penicillin and streptomycin solution. Huh‐7 (Nakabayashi et al, 1982) and Huh‐7.5 (Blight et al, 2002) were cultured in DMEM (Gibco 11965118) supplemented with 10% FBS and nonessential amino acid solution. All cell lines were tested for mycoplasma using Lookout Mycoplasma PCR kit (Sigma‐Aldrich MP0035).
Viruses
Bicistronic HCV was generated in (Schoggins et al, 2011) from Bi‐YPet‐Jc1FLAG2 described in (Jones et al, 2010). Monocistronic HCV J6/JFH‐5AB‐YPet was derived from Horwitz et al (2013). HCV stocks were generated by electroporation of in vitro transcribed RNA into Huh‐7.5 cells as described in (Lindenbach et al, 2005).
FMDV was generated from the full‐length serotype A12 infectious clone, pRMC35 (Rieder et al, 1993). Viruses were propagated in BHK‐21 cells and were concentrated by polyethylene glycol precipitation and stored at −70°C. Bovine enterovirus 1 (BEV‐1; GenBank accession no. D00214) was obtained from Agricultural Research Service at Plum Island Animal Disease Center.
Type I poliovirus (Mahoney) was rescued from pPVM‐2A‐144‐poliovirus‐GFP (GenBank OM677908) as described in (Burrill et al, 2013). Briefly, pPVM‐2A‐144‐poliovirus‐GFP plasmid DNA was linearized with EcoRI (NEB R3101). Purified linear DNA was then used to in vitro transcribe poliovirus plus stranded RNA using MEGAscript™ T7 Transcription Kit (Thermo Fisher Scientific AM1334) according to the manufacturer's protocol. Purified RNA was then electroporated into HeLa cells. After visible cytopathic effect, media were harvested, and virus purified using ultracentrifugation.
EMCV was rescued from pEC9 (a gift from Ann Palmenberg, GenBank OM677909). pEC9 plasmid was linearized using Sal1 (NEB R3138). Purified linear DNA was then used to in vitro transcribe EMCV plus stranded RNA using MEGAscript™ T7 Transcription Kit according to the manufacturer's protocol. Purified RNA was then electroporated into HeLa cells. After visible cytopathic effect, media were harvested, and virus purified using ultracentrifugation as described in (Burrill et al, 2013).
Chimeric EMCV‐IRES‐PV (GenBank OM677910) was derived from pPVM‐2A‐144‐poliovirus‐GFP. Five PCR segments were amplified from pPVM‐2A‐144‐poliovirus‐GFP using the following primer pairs: ATGGGTGCTCAGGTTTCATCAC and ATATGTGGTCAGATCCTTGGT, ACCAAGGATCTGACCACATATG and AATTGGTGAGGCCTTGTTCCATGGCTTCCGATGACCCAAACACTTC, GGAACAAGGCCTCACCAATT and TTGAAACAAAGCCTCCATACA, TGTATGGAGGCTTTGTTTCAAGG and GTTATTGTCTCATGAGCGGATACA, TGTATCCGCTCATGAGACAATAAC and TCTAAGTTACGGGAAGGGAGTAT. EMCV IRES segment from pEC9 was amplified using primers TACTCCCTTCCCGTAACTTAGATTGAAAGCCGGGGGTGGGA and GTGATGAAACCTGAGCACCCATATTATCATCGTGTTTTTCAAAGGAA (Reagents and Tools Table). All PCR amplified segments were assembled using NEBuilder® HiFi DNA Assembly Master Mix (NEB E2621) according to the manufacturer's protocol. Assembled plasmid was transformed into NEB stable competent E. coli (NEB C3040H). Purified EMCV‐IRES‐PV plasmid DNA was linearized using EcoRI digestion and purified linear DNA was then used to in vitro transcribe EMCV‐IRES‐PV plus stranded RNA using MEGAscript™ T7 Transcription Kit according to the manufacturer's protocol. Purified RNA was then electroporated into HeLa cells. After visible cytopathic effect, media were harvested, and virus purified using ultracentrifugation as described in (Burrill et al, 2013).
Biosafety measures
Work with FMDV and BEV‐1 was performed in high containment biosafety level 3 facility at Plum Island Animal Disease Center in compliance with CDC‐approved select agent protocols. No DURC research was performed as per USDA, ARS policy P&P 621.0 v.3 approved on June 19, 2020. Work with poliovirus, Chimeric EMCV‐IRES‐PV, EMCV, lentiviruses, and human cancer cell lines was performed in a biosafety level 2 facility at NYU Grossman School of Medicine in compliance with protocols approved by the institutional biosafety committee (IBC) and environmental safety and health (EH&S).
Plasmids
DDX60 wild‐type, IRF1, and Fluc plasmid DNA was generated by (Schoggins et al, 2011) in pTRIP.CMV.IVSb.ires.TagRFP‐DEST backbone (GenBank OM859267, OM859269, and OM859268, respectively). Empty vector is pTRIP.CMV.IVSb.ires.TagRFP‐DEST without ccdB suicide gene and Fluc negative control plasmid has Fluc sequence in place of an ISG. DDX60 wild‐type, IRF1, and Fluc DNA was also cloned into a pTRIP.CMV.IVSb.ires.TagRFP‐DEST backbone containing a puromycin selectable marker (pTRIP.CMV.IVSb.ires.TagRFP‐puro‐DEST). GFP plasmid is EGFP cloned into pcDNA DEST40 vector (Thermo Fisher Scientific 12274015). RIG‐I plasmid DNA was generated by (Dittmann et al, 2015) in pSCRPSY lentiviral expression vector. Mutant DDX60 constructs and bicistronic 5′ cap‐driven Rluc and IRES‐driven Fluc constructs (Honda et al, 2000) can be found using GenBank accession numbers: DDX60 A785V: OM859270, DDX60 K791N: OM859271, DDX60 E890A: OM859272, DDX60 S918/T920/A: OM859273, DDX60 Q1321A: OM859274, DDX60 R1328A: OM859275, DDX60 ∆1,402–1,712: OM859276, DDX60 ∆1–556: OM859277, DDX60 ∆1–428: OM859278, Polio IRES: OM859279, EV71 IRES: OM859280, EMCV IRES: OM859281, FMDV IRES: OM859282, HCV IRES: OM859283, BVDV IRES: OM859284.
RNA reporters
5′ cap‐ and IRES‐driven Fluc mRNA reporters were generated from PCR products obtained from bicistronic 5′ cap‐driven Rluc and IRES‐driven Fluc plasmids. First, IRES and Fluc sequences were PCR amplified from each plasmid using primers GGCCTGATATTGAAGAAGATATTGCG and ATAGGGCCCGGATCCTTACAAT. To generate IRES‐driven Fluc PCR products, purified PCR product from this first PCR was used in a second round of PCR using primers CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ATAGGGCCCGGATCCTTACAAT (Appendix Table S1). To generate PCR products for in vitro transcription of 5′ capped Fluc, PCR products from first PCR was amplified using primers CGTGGATAATACGACTCACTATAGGCTAGCCACCACCGGTATGGAAGACGCCAA and ATAGGGCCCGGATCCTTACAAT (Appendix Table S1). These final PCR products were purified and used for in vitro transcription using MEGAscript™ T7 Transcription Kit according to the manufacturer's protocol. The in vitro transcription reaction for the 5′ cap‐driven Fluc reporter included a 1:4 ratio of Invitrogen™ Anti‐Reverse Cap Analog (Fisher Scientific AM8045) and GTP solution (0.4 μl cap analog to 1.6 μl of GTP solution). After in vitro transcription, poly‐A tails were added on using Invitrogen™ Poly(A) Tailing Kit (Thermo Fisher Scientific AM1350) according to the manufacturer's protocol. Final reporter mRNAs were purified using Qiagen RNeasy kit (Qiagen 74106). Quality of mRNA reporters was ensured by sequencing PCR products used for in vitro transcription and running final RNA products on an agarose gel using NorthernMax™‐Gly Sample Loading Dye (Thermo Fisher Scientific AM8551) and Invitrogen™ Ambion Millennium Markers RNA Markers Formamide (Thermo Fisher Scientific AM7151).
To generate biotin labeled and unlabeled Poliovirus IRES, EV71 IRES, EMCV IRES, FMDV IRES, HCV IRES, BVDV IRES, and 5′ capped Fluc probes, PCR products were generated from bicistronic 5′ cap‐driven Rluc and IRES‐driven Fluc plasmids in two rounds. First, IRES and Fluc sequences were PCR amplified from each plasmid using primers GGCCTGATATTGAAGAAGATATTGCG and ATAGGGCCCGGATCCTTACAAT (Appendix Table S1). Purified PCR products from this first PCR was used in a second round of PCR using primers: CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ACCGGTCATTATGATACAATTGTCT (Poliovirus IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ACCGGTCATCGCTTCGTGTT (EV71 IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ACCGGTCATATTATCATCGTGTT (EMCV IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ACCGGTCATGTGTTCAGTGGT (FMDV IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and GCGGTTGGTGTTACGTTTGGT (HCV IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACAATAATTCTAGAGCGGCCGC and ACCGGTTAAAAGTTCATTTGTGATCA (BVDV IRES), CGTGGATAATACGACTCACTATAGGCTAGCCACCACCGGTATGGAAGACGCCAA and ATAGGGCCCGGATCCTTACAAT (5′ capped Fluc). Biotin labeled RNA probes were generated by adding 1.25 μl of 10 mM biotin‐16‐UTP (Millipore Sigma 11388908910) to the in vitro transcription reactions. Unlabeled probes were in vitro transcribed without biotin‐16‐UTP added. Final RNA was purified using Qiagen RNeasy kit. Quality of RNA was ensured by sequencing PCR products used for in vitro transcription and running final RNA products on an agarose gel using NorthernMax™‐Gly Sample Loading Dye and Invitrogen™ Ambion Millennium Markers RNA Markers Formamide.
Interferon‐β treatment time course experiments
HEK293T, A549, HFF1, or HeLa cells were plated at a density of 5E4 (A549 and HFF1) or 1E5 (HEK293T and HeLa) cells/cm2 in a 24‐well plates with two wells for 0.1% BSA/PBS treatment and two wells for 500 U/ml IFN‐ß (dissolved in 0.1% BSA/PBS, Millipore Sigma IF104) treatment. Wells with HEK293T cells were coated with poly‐L‐ornithine (Sigma‐Aldrich P2533) prior to plating. Each cell line was treated with either 0.1% BSA/PBS or 500 U/ml IFN‐ß. 0‐, 6‐, 12‐, 24‐, or 48‐h posttreatment, cells were washed 1× with PBS (Fisher Scientific MT21031CV) and lysed in either 350 μl buffer RLT (supplied with Qiagen 74106) supplemented with 2‐mercaptoethanol at a 1:100 dilution (Sigma‐Aldrich M3148) or 200 μl LDS sample buffer (diluted to 1× with molecular biology grade water, Invitrogen™ B0007) supplemented with cOmplete™, Mini, EDTA‐free Protease Inhibitor Cocktail (Roche 11836170001) and Pierce™ Phosphatase Inhibitor Mini Tablets (Thermo Fisher Scientific A32957). RNA was isolated using Qiagen RNeasy kit (Qiagen 74106) and cDNA synthesized using SuperScript™ III First‐Strand Synthesis System (Thermo Fisher Scientific 18080051). RT–qPCR was performed using Applied Biosystems™ PowerUp™ SYBR™ Green Master Mix (Applied Biosystems™ A25918), Applied Biosystems™ QuantStudio 3 real‐time PCR system, and primers for genes listed in Appendix Table S1 (20 μl reaction volume per well consisting of 3 μl 3 μM primer mix, 10 μl PowerUp™ SYBR™ Green Master Mix, 5 μl cDNA, and 2 μl RNase‐free water). Data were analyzed using delta–delta C t method devised by (Livak & Schmittgen, 2001) after normalizing Ct values to housekeeping gene RPS11. Western blot was performed by boiling samples at 95°C for 5 min and loading samples on a NuPAGE™ 3 to 8%, Tris‐Acetate, 1.5 mm, gel. Proteins were then transferred onto a PVDF membrane using iBlot 2 transfer system (Thermo Fisher Scientific IB24001). Membrane was blocked in 5% Thermo Scientific™ Oxoid™ Skim Milk Powder (Fisher Scientific OXLP0031B) TBS/0.1% Tween‐20 solution (Fisher Scientific BP337‐500). After 3 × 10 min washes with TBS/0.1% Tween‐20, membrane was probed for antibodies of interest using the conditions listed in Appendix Table S1. After primary antibody incubation, membrane was washed with TBS/0.1% Tween‐20 for 3 × 10 min and probed with LI‐COR IRDye® 800CW Donkey anti‐Rabbit IgG and IRDye 680RD Donkey Anti‐Mouse IgG secondary antibodies for 1 h at room temperature. After 3 × 10 min washes with TBS/0.1% Tween‐20, proteins were visualized using Odyssey® DLx Imaging System.
Western blot band intensity quantification
Band intensities were measured using ImageJ2 (2.3.0/1.53f). Region of interests (ROI) were defined for loading control and protein of interest (POI) bands and mean gray values for each band was measured. Background mean gray values using the defined ROIs were also measured from parts of each lane without any protein bands. Inverted pixel densities were calculated by subtracting the mean gray values from 255. Net densities for loading control and POI bands were calculated by subtracting the inverted pixel densities of the background values from the inverted pixel densities of either the loading control or POI bands. Net densities of POI bands were then divided by the net densities of loading control bands to define the final relative quantification values. Relative quantification values were multiplied by an arbitrary scaling factor as necessary to make values range from 0 to 10 (i.e., in Fig 4D all relative quantification values were multiplied by 10).
Western blots to confirm expression of transgenes
HEK293T cells were plated at a density of 1E5 cells/cm2 on a 24‐well poly‐L‐ornithine coated plates. The next day, cells were transfected with 1 μg of DDX60 wt or mutant constructs using TransIT®‐LT1 Transfection Reagent (Mirus Bio MIR 2300) at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 50 μl of Opti‐MEM (Thermo Fisher Scientific 51985034) according to the TransIT®‐LT1 manufacturer's protocol for 24‐well plates. 16–18‐h post‐transfection, media on cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were washed 1× with PBS and lysed in 200 μl LDS sample buffer (diluted to 1× with molecular biology grade water) supplemented with cOmplete™, Mini, EDTA‐free Protease Inhibitor Cocktail and Pierce™ Phosphatase Inhibitor Mini Tablets. Western blot was performed by boiling samples at 95°C for 5 min and loading samples on a NuPAGE™ 3–8%, Tris‐Acetate, 1.5 mm, gel. Proteins were then transferred onto a PVDF membrane using iBlot 2 transfer system. Membrane was blocked in 5% Oxoid™ Skim Milk Powder TBS/0.1% Tween‐20 solution. After 3 × 10 min washes with TBS/0.1% Tween‐20, membrane was probed for antibodies of interest using the conditions listed in Appendix Table S1. After primary antibody incubation, membrane was washed with TBS/0.1% Tween‐20 for 3 × 10 min and probed with LI‐COR IRDye® 800CW Donkey anti‐Rabbit IgG and IRDye 680RD Donkey Anti‐Mouse IgG secondary antibodies for 1 h at room temperature. After 3 × 10 min washes with TBS/0.1% Tween‐20, proteins were visualized using Odyssey® DLx Imaging System. Band intensities were measured using ImageJ2 as described above.
Antiviral assays with HCV reporters
HCV antiviral assays were conducted as described in (Schoggins et al, 2011). Huh‐7 cells were plated on 24‐well plates at a density of 7E4 cells/well. The next day, growth medium was changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 400 ng DNA/well of Fluc, IRF1, DDX60 wt, or DDX60 mutant constructs was transfected using Lipofectamine 2000 (Thermo Fisher Scientific 11668019) in a total volume of 1 ml/well. Plates were centrifuged at 1,000 g for 30 min at 37°C. 5 h later, the medium was changed to DMEM supplemented with 10% FBS, penicillin and streptomycin, and nonessential amino acids. 48 h post‐transfection, cells were washed 1× with PBS and infected with HCV‐Ypet bicistronic or monocistronic HCV J6/JFH‐5AB‐YPet reporter at a dose yielding approximately 50% infected cells as determined by flow‐cytometry‐based infectivity assays on Fluc transfected cells. 72 hpi, adherent cells were collected into 200 μl Accumax Cell Aggregate Dissociation Medium (Thermo Fisher Scientific 00‐4666‐56) and transferred to a 96‐well plates. Cells were pelleted at 1,000 g for 5 min at 4°C and resuspended in 1% paraformaldehyde fixation solution for 1 h. Cells were pelleted once again by centrifugation at 1,000 g for 5 min at 4°C and resuspended in cold PBS supplemented with 3% FBS and stored at 4°C until flow cytometry analysis. Samples were analyzed in a 96‐well‐based high‐throughput manner using an LSRII‐HTS flow cytometer (BD Biosciences). Data were analyzed using FlowJo software with a 0.1% compensation matrix. Percent of Ypet expressing cells in RFP‐positive cells was determined and plotted.
In cell plasmid‐based reporter assays
HEK293T cells were plated on a poly‐L‐ornithine coated 48‐well plates at a density of 6E4 cells/cm2. The next day, cells were cotransfected with 200 ng of bicistronic 5′ cap‐driven Rluc and IRES‐driven Fluc plasmid and 300 ng of GFP alone, or 290 ng GFP + 10 ng IRF1, or 200 ng GFP + 100 ng IRF1, or 300 ng IRF1 alone, or 290 ng GFP + 10 ng DDX60 wt, or 200 ng GFP + 100 ng DDX60 wt, or 300 ng DDX60 wt alone, or 290 ng GFP + 10 ng DDX60 E890A, or 200 ng GFP + 100 ng DDX60 E890A, or 300 ng DDX60 E890A alone. Transfections were performed using X‐tremeGENE 9 DNA transfection reagent (Roche), with 1.2 μl X‐tremeGENE reagent per 500 μg DNA reaction. 16–18‐h post‐transfection, media on cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were lysed using 1× passive lysis buffer (5× passive lysis buffer diluted in PBS) supplied in Promega Dual‐Luciferase™ Reporter assay system (Fisher Scientific PR‐E1980) according to the manufacturer's protocol with the addition of 1 freeze–thaw cycle step, freezing for at least 1 h at −80°C before thaw. Renilla and Firefly luciferase activity was then assayed using Promega Dual‐Luciferase™ Reporter assay system according to the manufacturer's protocol.
In cell RNA‐based reporter assays
HEK293T cells were plated on a poly‐L‐ornithine‐coated 48‐well plates at a density of 3E4 cells/cm2. The next day, cells were transfected with either 500 ng GFP, IRF1, DDX60 wt, or DDX60 mutant constructs using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 30 μl of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol for a 48‐well plates. 16–18‐h post‐transfection, media on cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were transfected with 0.2–0.3 pmol of either 5′ cap‐, Poliovirus IRES‐, EV71 IRES‐, EMCV IRES‐, FMDV IRES‐, HCV IRES‐, or BVDV IRES‐driven Fluc mRNA reporters using TransIT®‐mRNA Transfection Kit (Mirus Bio MIR 2250) at a 1:2 (μg to μl) RNA to lipid reagent ratio in a total volume of 30 μl of Opti‐MEM according to the TransIT®‐mRNA manufacturer's protocol for a 48‐well plates. 16‐h post‐mRNA transfection, cells were lysed using 1× passive lysis buffer (5× passive lysis buffer diluted in PBS) Passive Lysis Buffer (Promega E1941) according to the manufacturer's protocol with the addition of 1 freeze–thaw cycle step, freezing for at least 1 h at −80°C before thaw. Firefly luciferase activity was then assayed using Promega Luciferase Assay System (E4550) according to the manufacturer's protocol. Luminescence activity was measured using BioTek Synergy™ HTX Multi‐Mode Microplate Reader with Agilent BioTek Dual Reagent Injector system.
Generation of stable HeLa cell lines using lentivirus transduction
Lentiviral pseudoparticles generation was carried out as in (Schoggins et al, 2011). 1E6 HEK293T cells in six‐well plates were cotransfected with plasmids expressing the Fluc, IRF1, DDX60 wt, or DDX60 E890A pTRIP.CMV.IVSb.ISG.ires.TagRFP.puro proviral DNA, HIV‐1 gag–pol and VSV‐G in a ratio of 1/0.8/0.2, respectively. For each transfection, 6 μl TransIT®‐LT1 Transfection Reagent was combined with 2 μg total DNA in a total volume of 200 μl Opti‐MEM, allowed to mix for 15 min, and added dropwise to the six‐well plate. Transfections were carried out for 16–18 h, followed by a medium change to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. Supernatants were collected at 48 and 72 h, pooled, cleared by centrifugation and passing through a 0.45 μm filter, and stored at −80°C.
HeLa cells were plated on a six‐well plate at 1E6 cells/well. The next day, cells were transduced with undiluted (DDX60 wt and DDX60 E890A) or 1:50 diluted (Fluc and IRF1) lentiviral pseudoparticles by spinoculation at 1,250 g for 45 min at 37°C in DMEM supplemented with 1.5% FBS, 20 mM HEPES (Gibco™ 15630080), and 4 μg/ml polybrene (Sigma‐Aldrich TR‐1003‐G). Undiluted DDX60 wt and E890A lentiviral pseudoparticles had to be used due to lower viral titers. 5‐h postspinoculation, cell medium was changed to MEM supplemented with 1.5% FBS and penicillin and streptomycin. 48‐h post‐transduction, cells were trypsinized and plated in T175 flasks in MEM supplemented with 1.5% FBS, penicillin and streptomycin, and 2 μg/ml puromycin (Sigma‐Aldrich P8833). Medium was replaced on cells every 48 h until cells were ~ 90% confluent. Afterward, cells were trypsinzed and frozen down in MEM supplemented with 10% FBS, penicillin and streptomycin, and 5% DMSO.
Antiviral assays with IRES‐containing viruses
HeLa cells stably expressing Fluc, IRF1, DDX60 wt, or DDX60 E890A were thawed from frozen vials and passaged once under 2 μg/ml puromycin selection. Cells were then plated on a six‐well plates at a density of 2E4 cells/cm2 in MEM supplemented with 10% FBS, penicillin and streptomycin, and 2 μg/ml puromycin. When plating cells, approximately 5E5 of the remaining cells were lysed for western blot analysis to check for transgene expression (see methods above for collection and western blot details). An additional well was used for seeding untransduced HeLa cells at the same density in MEM supplemented with 10% FBS and penicillin and streptomycin. Two days later, untransduced HeLa cells were trypsinized and counted. Transduced HeLa cells were washed once with PBS + Ca and Mg (Corning™ 21030CV) supplemented with 0.3% BSA (Equitech‐Bio BL62‐0500) and subsequently infected with EMCV, poliovirus, or EMCV‐IRES‐PV at an MOI of 0.001 based on the untransduced HeLa cell count numbers in a volume of 500 μl of PBS + Ca and Mg supplemented with 0.3% BSA. Approximately 50 μl of input virus was kept frozen at −80°C for titration. This is considered the 0 h time point. Infection was carried out for 1 h at 37°C with gentle shaking. After 1 h, virus was aspirated, and cells washed 3× with 1 ml of PBS + Ca and Mg supplemented with 0.3% BSA. After the final wash, medium was changed to 2 ml of MEM supplemented with 1.5% FBS and penicillin and streptomycin. 120 μl of cell medium was taken for each time point and frozen at −80°C. Cell medium was replaced with 120 μl of fresh MEM supplemented with 1.5% FBS and penicillin and streptomycin after each time point to preserve the final volume of cell medium. After the experiment, all equipment and waste were disinfected with 10% bleach.
Virus collected for each time point was titered using plaque assay. For this, HeLa cells were plated on 12‐well plates at 3.5E5 cells/well. The next day, virus time points to be titered were serially diluted from 1E‐1 to 1E‐6 for time points < 24 h and 1E‐3 to 1E‐8 for time points > 24 h in a final volume of 250 μl of PBS + Ca and Mg supplemented with 0.3% BSA. Cells plated on 12‐well plates were washed once with PBS + Ca and Mg supplemented with 0.3% BSA and subsequently infected with 200 μl of serially diluted virus. Infection was carried out for 1 h at 37°C with gentle shaking. After 1 h, virus was aspirated, cells were washed 1× with PBS + Ca and Mg supplemented with 0.3% BSA, and medium was changed two 1 ml of overlay medium. Overlay medium for 2 × 12‐well plates: 12.5 ml 2× DMEM (Gibco™ 12100046), 8.3 ml 4% Avicel, 250 μl DEAE Dextran (Sigma‐Aldrich 30461), 250 μl Glutamax (Thermo Fisher Scientific 35050061), 250 μl nonessential amino acid solution, 250 μl penicillin and streptomycin, 1.23 ml 7.5% sodium bicarbonate (Gibco™ 25080094), and 2 ml molecular biology grade water (Fisher Scientific MT46000CM). Cells were incubated in a 37°C incubator 18–24 h for EMCV time points and 40–48 h for poliovirus and EMCV‐IRES‐PV time points. To stop plaque assays and count viral plaques, cells were fixed by adding 1 ml of 2% formaldehyde (Fisher Scientific BP531‐500) onto the overlay medium for at least 15 min at room temperature. Overlay medium was then aspirated, cells washed 1× with PBS, and stained with 400 μl of crystal violet working solution for at least 15 min at room temperature. Crystal violet working solution consists of 40 ml 1% crystal violet stock solution, 80 ml methanol, and 300 ml water. 1% Crystal violet stock solution consists of 10 g crystal violet, 200 ml ethanol, and 800 ml water. After crystal violet staining, crystal violet working solution was removed (can be reused) and plates plunged into a bucket of water. Plaques were counted for a well with 5–20 plaques and viral titers calculated using the following formula: Titer (PFU/ml) = well plaque count/(0.200 ml × dilution factor).
BHK‐J cells stably expressing empty vector, IRF1, DDX60 wt, or DDX60 E890A were infected with BEV‐1 or FMDV at MOI 1. After 1 h of adsorption, cells were rinsed once with 150 mM NaCl, 20 mM morpholineethanesulfonic acid (MES; pH 6) to inactivate unadsorbed virus and once with MEM to neutralize the MES, followed by addition of MEM and incubation at 37°C. Supernatants and cell lysates were collected at 5 hpi, and titers were determined via plaque assay on BHK‐21 cells.
RNA abundance assay using IRES‐driven Fluc reporter RNAs
HEK293T cells were plated on a poly‐L‐ornithine coated 48‐well plates at a density of 3E4 cells/cm2 (at least two wells per condition). The next day, cells were transfected with either 500 ng of DDX60 wt or DDX60 E890A constructs using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 30 μl of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol for a 48‐well plates. 16–18‐h post‐transfection, media on cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were transfected with 0.2–0.3 pmol of either 5′ cap‐, Poliovirus IRES‐, EV71 IRES‐, EMCV IRES‐, FMDV IRES‐, HCV IRES‐, or BVDV IRES‐driven Fluc mRNA reporters using TransIT®‐mRNA Transfection Kit at a 1:2 (μg to μl) RNA to lipid reagent ratio in a total volume of 30 μl of Opti‐MEM according to the TransIT®‐mRNA manufacturer's protocol for a 48‐well plates. 16‐h post‐mRNA transfection, cells were trypsinzed and half of the cell volume was lysed using 1× passive lysis buffer (5× passive lysis buffer diluted in PBS) Passive Lysis Buffer according to the manufacturer's protocol with the addition of 1 freeze–thaw cycle step, freezing for at least 1 h at −80°C before thaw. Firefly luciferase activity was then assayed using Promega Luciferase Assay System according to the manufacturer's protocol. Luminescence activity was measured using BioTek Synergy™ HTX Multi‐Mode Microplate Reader with Agilent BioTek Dual Reagent Injector system. The other half of the cell volume was lysed using 350 μl buffer RLT (supplied with Qiagen 74106) supplemented with 2‐mercaptoethanol at a 1:100 dilution. RNA was isolated using Qiagen RNeasy kit and cDNA synthesized using SuperScript™ III First‐Strand Synthesis System. RT–qPCR was performed using Applied Biosystems™ PowerUp™ SYBR™ Green Master Mix, Applied Biosystems™ QuantStudio 3 real‐time PCR system, and primers for genes listed in Appendix Table S1 (20 μl reaction volume per well x at least three technical replicates consisting of 3 μl 3 μM primer mix, 10 μl PowerUp™ SYBR™ Green Master Mix, 5 μl cDNA, and 2 μl RNase‐free water). Data were analyzed using delta–delta C t method devised by (Livak & Schmittgen, 2001) after normalizing C t values to housekeeping gene RPS11.
To calculate efficiency of our Fluc qPCR primers (Appendix Table S1), fivefold and 10‐fold serial dilutions were made of Fluc cDNA PCR. Fivefold dilutions ranged from 3E13 to 1E11 calculated copies per ml, and 10‐fold dilutions ranged from 1E10 to 1E4 calculated copies per ml. 5 μl of these dilutions was used for qPCR reaction as described above with at least two technical replicates. C t vs Log2 cDNA copies/ml was plotted (r 2 = 0.98 for fivefold dilutions and 0.99 for 10‐fold dilutions). Slopes of C t vs Log2 cDNA copies/ml were used to calculate PCR efficiency using the formula, Efficiency = 10(−1/slope). Efficiency values were 5.46 for fivefold dilution and 1.68 for 10‐fold dilutions (cutoff value was 0.95, or 95% efficiency).
ISRE reporter assays
HEK293 ISRE reporter recombinant cells were plated on a poly‐L‐ornithine coated 48‐well plates at a density of 6E4 cells/cm2 (at least two wells per condition). The next day, cells were transfected with either: 100 ng RIG‐I + 400 ng of GFP, 125 ng RIG‐I + 375 ng GFP, 150 ng RIG‐I + 350 ng GFP, 175 ng RIG‐I + 325 ng GFP, 100 ng RIG‐I + 100 ng DDX60 wt + 300 ng GFP, 125 ng RIG‐I + 125 ng DDX60 wt + 250 ng GFP, 150 ng RIG‐I + 150 ng DDX60 wt + 200 ng GFP, 175 ng RIG‐I + 175 ng DDX60 wt + 150 ng GFP, 100 ng RIG‐I + 100 ng DDX60 E890A + 300 ng GFP, 125 ng RIG‐I + 125 ng DDX60 E890A + 250 ng GFP, 150 ng RIG‐I + 150 ng DDX60 E890A + 200 ng GFP, or 175 ng RIG‐I + 175 ng DDX60 E890A + 150 ng GFP. Transfections were carried out using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 30 μl of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol for a 48‐well plate. 16–18‐h post‐transfection, media on cells were changed to MEM supplemented with 10% FBS, penicillin and streptomycin, and 400 μg/ml geneticin. Two days post transfection, cells were either treated with PBS or were transfected with 0.641 pmol (~ 250 ng) of low molecular weight poly(I:C) (Invivogen tlrl‐picw) using Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher Scientific 13778075) in a final volume of 30 μl of Opti‐MEM according to RNAiMAX manufacturer's protocol. One well of untransfected cells was also treated with 500 U/ml of IFN‐ß for 16–18 h. 16‐h post‐RNA transfection, one well per condition of cells was lysed using 1× passive lysis buffer (5× passive lysis buffer diluted in PBS) Passive Lysis Buffer according to the manufacturer's protocol with the addition of 1 freeze–thaw cycle step, freezing for at least 1 h at −80°C before thaw. Firefly luciferase activity was then assayed using Promega Luciferase Assay System according to the manufacturer's protocol. Luminescence activity was measured using BioTek Synergy™ HTX Multi‐Mode Microplate Reader with Agilent BioTek Dual Reagent Injector system. Another well per condition of cells was lysed in 100 μl LDS sample buffer (diluted to 1× with molecular biology grade water) supplemented with cOmplete™, Mini, EDTA‐free Protease Inhibitor Cocktail and Pierce™ Phosphatase Inhibitor Mini Tablets. Western blots were run and analyzed for DDX60, RIG‐I, ß‐actin, and GAPDH expression as described in Western blot methods described above.
Biotin labeled RNA pulldown
Biotin labeled RNA pull‐down experiments were adapted from (Hung et al, 2016). HEK293T cells were plated on a poly‐L‐ornithine coated 10‐cm dish at a density of 4E4 cells/cm2. The next day, cells were transfected with 15 μg of DDX60 wt construct using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 1.5 ml of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol for a 10‐cm dish. 16–18‐h post‐transfection, medium on cells was changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were washed with 2 ml of PBS and subsequently lysed at 4°C with 300 μl 3‐[(3‐cholamidopropyl)‐dimethylammonio]‐1‐propanesulfonate (CHAPS) lysis buffer (10 mM Tris–HCl [pH 7.4], 1 mM MgCl2, 1 mM EGTA, 0.5% CHAPS, 10% glycerol, 0.1 mM phenyl‐ methylsulfonyl fluoride [PMSF], 5 mM β‐mercaptoethanol) with gentle shaking for 30 min. Cell lysates were then transferred to a 1.5‐ml tube and centrifuged at 10,000 g for 10 min at 4°C. Supernatants were then transferred to new tubes. Protein collected was quantified using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific 23225) according to the manufacturer's protocol. Samples were then frozen at −80°C.
Before RNA pulldown, cell lysates were precleared to remove proteins that have a background binding to streptavidin MagneSphere paramagnetic particles. First, streptavidin MagneSphere paramagnetic particles (Promega, Z5481) were washed three times with 1 ml of RNA mobility buffer without heparin (5 mM HEPES pH 5.5, 40 mM KCl, 0.1 mM EDTA, 2 mM MgCl2, 2 mM DTT, 1 U RNasin). To wash, place tubes containing streptavidin MagneSphere paramagnetic particles on a magnetic stand (DynaMag™‐2 Magnet, Life Technologies 12321D) for 30s—1 min, remove supernatant, and add 1 ml of RNA mobility buffer without heparin to each tube and invert tubes up and down to resuspend paramagnetic beads. Repeat the mentioned steps for three washes, removing the supernatant on the last wash, but not adding back any RNA mobility buffer. Cell lysates were then precleared by mixing appropriate volume of cell lysate for the number of pulldowns (200 μg protein per pulldown) with streptavidin MagneSphere paramagnetic particles and incubating at room temperature for 10 min on a rocking nutator. The mixture was then placed on a magnetic stand for 30s—1 min and the supernatant was transferred to a new sterile 1.5‐ml tube on ice. The remaining streptavidin MagneSphere paramagnetic particles were resuspended in 25 μl of 2× SDS sample buffer, boiled at 95°C for 10 min, and frozen at −80°C.
For RNA pulldown, 200 μg of DDX60 expressing cell lysate was well mixed with 12.5 pmol of unlabeled or biotin labeled Polio IRES, EMCV IRES, HCV IRES, or 5′ capped Firefly luciferase RNA in RNA mobility buffer with heparin (5 mM HEPES pH 5.5, 40 mM KCl, 0.1 mM EDTA, 2 mM MgCl2, 2 mM DTT, 1 U RNasin, 0.25 mg/ml Heparin). 1% of cell extract was kept for an input sample. The cell lysate and RNA mixture were incubated at 30 degrees for 15 min with gentle shaking. In the meantime, streptavidin MagneSphere paramagnetic particles were washed three times with 1 ml of RNA mobility buffer without heparin (split 1 tube of streptavidin MagneSphere paramagnetic particles for every 2 RNA pulldowns). After the last wash, streptavidin MagneSphere paramagnetic particles were combined and resuspended in 400 μl of RNA mobility buffer without heparin per RNA pulldown. 400 μl of resuspended streptavidin MagneSphere paramagnetic particles was then added to cell lysate and RNA mixture and incubated for 10 min at room temperature rocking on a nutator. Pulled‐down complexes were then washed five times with RNA mobility buffer without heparin. After the last wash, streptavidin MagneSphere paramagnetic particles with bound RNA and protein were resuspended in 25 μl of 2× SDS sample buffer. One volume of 2× SDS sample buffer was also added to one volume of input sample. All samples were boiled at 95°C for 10 min, and frozen at −80°C. To analyze bound proteins, samples were spun down at maximum speed for 1 min and supernatants used for Western blots (see Western blot protocol above for details).
RNA immunoprecipitation assay
RNA immunoprecipitation (RIP) assays were performed using Magna RIP™ RNA‐Binding Protein Immunoprecipitation Kit (EMD Millipore 17‐700). HEK293T cells were plated on 15‐cm dishes coated with poly‐L‐ornithine at a density of 4E4 cells/cm2. The next day, cells were either left untransfected or transfected with 70 μg of DDX60 wt construct using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 3.5 ml of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol. 16–18‐h post‐transfection, cell media on both untransfected and transfected cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. 48‐h post‐transfection, cells were transfected with ~ 30–40 pmol of either 5′ cap or EMCV IRES‐driven Fluc reporter RNA using TransIT®‐mRNA Transfection Kit at a 1:2 (μg to μl) RNA to lipid reagent ratio in a total volume of 3.5 ml of Opti‐MEM according to the TransIT®‐mRNA manufacturer's protocol. 16‐h post‐mRNA transfection, cells were lysed for RIP lysate preparation using the Magna RIP™ RNA‐Binding Protein Immunoprecipitation Kit and protocol. Briefly, 115 μl of RIP lysis buffer with added protease inhibitor (0.5 μl) and RNase inhibitor cocktail (0.25 μl) was prepared for each sample. Cells on the plate were washed twice with 10 ml of ice‐cold PBS. Then, cells were scraped off the plates in 10 ml of ice‐cold PBS per plate and transferred to 15‐ml tubes. Cells were collected by centrifugation for 5 min at 225 g at 4°C. The supernatant was then discarded, and cell pellet was resuspended in an equal volume of RIP lysis buffer and mixed until the mixture appeared homogenous. Lysate was then incubated on ice for 5 min. Cell lysate was dispensed into 1.5‐ml tubes in 200 μl aliquots. 100 μl of cell lysate is used per RIP and a positive and negative antibody is used for each experiment making 200 μl a single use aliquot. All aliquots were frozen at −80°C.
On the day of RIP experiment, the appropriate number of tubes per RIP was prepared and 50 μl of magnetic beads placed into each tube. Magnetic beads were washed twice with 0.5 ml of RIP wash buffer. To wash, magnetic beads were placed on a magnetic stand for 30s—1 min, supernatant discarded, 0.5 ml of RIP wash buffer added, and tubes vortexed briefly (setting 7). After the final wash, magnetic beads in each tube were resuspended in 100 μl of RIP wash buffer. Next, the appropriate about of antibody (5 μg in this case) was added to each tube and antibody‐bead complexes were allowed to form by incubating all tubes on a rotating nutator at room temperature for 30 min (Appendix Table S1). Tubes were then centrifuged briefly and placed on a magnetic stand, supernatant removed, and beads washed twice with RIP wash buffer. After the last wash, 0.5 ml of RIP wash buffer was added to each tube and vortexed briefly (setting 7) before incubating tubes on ice. Next, enough RIP immunoprecipitation buffer was prepared for the appropriate number of RIPs according to Magna RIP™ RNA‐Binding Protein Immunoprecipitation Kit protocol. 900 μl of RIP immunoprecipitation buffer was added to each tube on ice. RIP lysates were then thawed quickly and centrifuged at 21,000 g for 10 min at 4°C. 10 μl of supernatant was used for input samples (one for Western blot and one for RNA preparation) and placed on ice. 100 μl of supernatant was added to the appropriate antibody‐bead complexes. All immunoprecipitation tubes were then left rotating overnight on a nutator at 4°C. An equal volume of 2× SDS was added to Western blot input samples, samples were boiled at 95°C for 5 min, and then stored at −80°C. The RNA input sample was simply frozen at –80°C.
The next day, immunoprecipitation tubes were centrifuged briefly and placed on a magnetic stand. 100 μl of the supernatant was saved as unbound protein fraction for Western blot. 100 μl of 2× SDS was added to the to the supernatant collected for Western blot, boiled at 95°C for 5 min, and stored at −80°C. The rest of the supernatant was discarded. Beads were then washed a total of six times with 0.5 ml of cold RIP wash buffer. 50 μl out of 500 μl of the bead suspension during the last wash was saved from each tube to test the efficiency of immunoprecipitation by Western blot. Proteins were eluted off the 50 μl of beads saved by resuspending the beads in 20 μl of 1× SDS sample buffer following by heating at 95°C for 5 min and storing samples at −80°C. Supernatants from these samples were used during Western blot. The remaining bead suspension was placed on ice until the next step.
To purify RNA from immunoprecipitation samples, proteinase K buffer was prepped for the appropriate number of samples according to Magna RIP™ RNA‐Binding Protein Immunoprecipitation Kit protocol. Beads from each immunoprecipitation samples were then resuspended in 150 μl of proteinase K buffer. RNA input samples were thawed and 107 μl of RIP wash buffer, 15 μl of 10% SDS, and 18 μl of proteinase K was added. All samples were incubated at 55 degrees for 30 min with gentle shaking to digest the protein. After the incubation, tubes were centrifuged briefly, and placed on the magnetic stand. Supernatants containing RNA were transferred into new tubes. To purify RNA, 250 μl of RIP wash buffer and 400 μl of acid phenol:chloroform:isoamyl alcohol (Thermo Scientific AM9720) was added to each tube. Tubes were vortexed at maximum setting for 15 s and centrifuged at 14,000 rpm for 10 min at room temperature to separate the phases. 350 μl of the aqueous phase was removed, placed in a separate tube, and 400 μl of RNase‐free water added to the original tube. Tubes were then once again vortexed for 15 s and centrifuged at 14,000 rpm for 10 min at room temperature to separate the phases. 300 μl of the aqueous phase was combined with the aqueous phase saved earlier. To precipitate RNA, 1/10 volume of 3 M sodium acetate pH 5.1, 1.5 volume of isopropanol, and 1 μL of Glycoblue (Thermo Scientific AM9515) was added to each tube before mixing and incubating the tubes at −80°C overnight. The next day, all tubes were centrifuged at 14,000 rpm for 30 min at 4°C and supernatant discarded. RNA pellet was washed once with 80% ethanol and tubes once again centrifuged at 14,000 rpm for 15 min at 4°C. Supernatants were then discarded carefully and pellets air dried. Pellets were resuspended in 15 μl of Rnase‐free water, and RNA quantified using nanodrop. Equal amounts (in ng) of RNA were used for subsequent cDNA synthesis and qPCR (procedure described in Interferon‐ß treatment time course experiments above). RNA samples were stored at −80°C. To calculate percent input after qPCR analysis, the following formula was used: . Adjusted input C t was calculated using the following formula: Input C t – log2(10) if 10% input was saved.
Polysome profiling experiments with Fluc mRNA
HEK293T cells were plated on 3 × 15‐cm dishes at a density of 1E5 cells/cm2. The next day, one plate was transfected with 70 μg of DDX60 wt construct and two plates were transfected with 70 μg each of DDX60 E890A construct using TransIT®‐LT1 Transfection Reagent at a 1:3 (μg to μl) DNA to lipid reagent ratio in a total volume of 3.5 ml of Opti‐MEM according to the TransIT®‐LT1 manufacturer's protocol. 16–18‐h post‐transfection, DDX60 wt transfected cells were split 1 to 2 from 1 × 15‐cm plate to 2 × 15‐cm plate and DDX60 E890A transfected cells were split 1 to 2 from 2 × 15‐cm plates to 4 × 15‐cm plates. The next day, media on all the cells were changed to DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. Subsequently, one DDX60 wt transfected plate of cells and two DDX60 E890A transfected plate of cells were transfected with ~ 30–40 pmol of 5′ cap‐driven Fluc reporter mRNA and one DDX60 wt transfected plate of cells and two DDX60 E890A transfected plate of cells were transfected with ~ 30–40 pmol of EMCV IRES‐driven Fluc reporter mRNA. mRNA transfections were carried out using TransIT®‐mRNA Transfection Kit at a 1:2 (μg to μl) RNA to lipid reagent ratio in a total volume of 3.5 ml of Opti‐MEM according to the TransIT®‐mRNA manufacturer's protocol. 16‐h post‐mRNA transfection, one plate of DDX60 E890A and 5′ cap‐driven Fluc reporter mRNA transfected cells and one plate of DDX60 E890A and EMCV IRES‐driven Fluc reporter mRNA transfected cells were treated with 200 μM (~ 0.1 μg/ml) of puromycin for 20 min at 37°C by replacing the media on the cells with puromycin containing DMEM supplemented with 1.5% FBS, penicillin and streptomycin, and nonessential amino acids. Cells were then harvested. To harvest, all cells were first treated with 15 ml of complete growth media + cycloheximide (Sigma‐Aldrich C7698) at 100 μg/ml for 15 min at 37°C by replacing the media on the cells with cycloheximide containing DMEM supplemented with 10% FBS, penicillin and streptomycin, and nonessential amino acids. Cells were then washed once with 3 ml PBS + calcium and magnesium + cycloheximide at 100 μg/ml. Cells were then trypsinized with 3 ml trypsin +100 μg/ml cycloheximide. When cells lifted off, 10 ml PBS + 100 μg/ml cycloheximide supplemented protease and phosphatase inhibitors (Thermo Scientific 87786 and Thermo Scientific 78420) were added and cells transferred to 15‐ml tubes. Cells were pelleted by centrifugation at 150 g for 4 min at 4°C. Supernatant was aspirated and cells resuspended in 10 ml of PBS + cycloheximide supplemented protease and phosphatase inhibitors. 1 ml of cell suspension was collected in 1.5‐ml tubes for input. Cells in both 15‐ml and 1.5‐ml tubes were pelleted by centrifugation at 1,250 rpm for 4 min at 4°C. Supernatants were again discarded, and all cell pellets frozen using liquid nitrogen before storing at −80°C.
The day before running cell lysates through sucrose gradient, sucrose gradients were prepared by pipetting 5.5 ml of 50% sucrose solution (28.5 ml 1× LSB (20 mM Tris pH 7.4–7.5, 10 mM NaCl, 3 mM MgCl2), 21.5 g sucrose, 40 U/ml Riboblock (Thermo Scientific EO0382), 100 μg/ml cycloheximide) in a Beckman 14 × 89 mm tube (Beckman Coulter 331372) and adding 5.5 ml of 15% sucrose solution (42.5 ml 1× LSB [20 mM Tris pH 7.4–7.5, 10 mM NaCl, 3 mM MgCl2], 8 g sucrose, 40 U/ml Riboblock, 100 μg/ml cycloheximide) on top. Enough gradients were prepared for the number of samples being processed. The tubes with the gradients were then sealed with parafilm and laid horizontally at 4°C overnight. The next day, cell pellets were thawed on ice. Once pellets were thawed, one to two samples were processed at a time. 750 μl of 1× LSB + 100 μg/ml cycloheximide +40 U/ml riboblock was added to the cell pellet, and the pellet was incubated in a dounce homogenizer on ice for 3 min. Next, 250 μl of Triton X‐100 detergent buffer (1.2% Triton X‐100, 0.2 M sucrose (342 mg), 4.4 ml 1× LSB, 100 μg/ml cycloheximide, 40 U/ml riboblock) was added and cells were lysed using a dounce homogenizer. Sample was then transferred to a 1.5‐ml tube on ice. Once all pellets were lysed, samples were centrifuged at maximum speed for 5 min at 4°C. The supernatants were then transferred to 1.5‐ml tubes containing 100 μl of heparin solution (10 mg/ml heparin, 1.5 M NaCl, 1.4 ml 1× LSB, 100 μg/ml cycloheximide, 40 U/ml riboblock). RNA concentration in the tubes was then measured using a nanodrop. The sample volume being loaded into the sucrose gradients were removed from the gradients first and ~ 1 mg of RNA was pipetted onto the gradients. Gradient tubes were balanced and ultracentrifuged at 160,000 g for 2 h at 4°C using a SW 41 Ti rotor. Fractions from gradient tubes were then collected using a BR‐188 Density Gradient Fractionation System (Brandel) into 1.5‐ml tubes filled with 25 μl of 0.5 M EDTA (Ambion AM9260G). All fractions were frozen at −80°C.
RNA was isolated from each collected fraction per sample using an acid phenol chloroform extraction. Briefly, an equal volume of acid phenol chloroform was added, samples mixed 3–5 times by inverting tube up and down, and then vortexed to assure complete mix of phenol with sample. Samples were then centrifuged at maximum speed for 5 min at 4°C. The upper aqueous phase was transferred to a new 2‐ml tube. An equal volume of RNase‐free water was then added to the remaining phenol, samples mixed 3–5 times by inverting tube up and down, vortexed, and centrifuge at maximum speed for 5 min at 4°C. The aqueous phase from this spin was combined with the previously collected aqueous phase. To precipitate RNA, 1/10 volume of 3 M sodium acetate pH 5.1, 1.5 volume of isopropanol, and 1 μl of Glycoblue was added to each tube before mixing and incubating the tubes at −80°C overnight. The next day, all tubes were centrifuged at maximum speed for 30 min at 4°C and supernatant discarded. RNA pellet was washed twice with 1 ml of 70% ethanol, centrifuging at maximum speed for 15 min at 4°C in between washes. Supernatants were then discarded and pellets air dried. Pellets were resuspended in 20 μl of Rnase‐free water. Total RNA was analyzed on the Agilent Bioanalyzer 6000 using the pico chip kit according to the manufacturer's instructions (Agilent RNA 6000 Pico Kit Quick Start Guide). An equal volume of RNA from each fraction was used for cDNA synthesis (procedure described in Interferon‐ß treatment time course experiments above) and qPCR analysis was carried out as described in (Panda et al, 2017). Data were plotted as mean ± SEM. We performed ROUT outlier analysis (Q = 1%) on every individual fraction. We removed individual outliers as follows: one outlier from fraction 5 of DDX60 WT EMCV‐IRES Fluc mRNA, one from fraction 7 of DDX60 LOF cap‐Fluc mRNA, one from fraction 8 of DDX60 WT EMCV‐IRES Fluc mRNA.
Polysome profiling experiments with poliovirus and chimeric poliovirus
Three independent vials of H1 HeLa cells constitutively expressing wild‐type DDX60 were plated on 3 × 15‐cm dishes at a density of 1.5E4 cells/cm2. 48‐h postplating, cells were split 1:2 onto 6 × 15‐cm plates. 48 h post second plating, 3 × 15‐cm plate of cells was infected with parental poliovirus I (Mahoney) at an MOI of 1, and 3 × 15‐cm plate of cells was infected with chimeric poliovirus (EMCV‐IRES‐PV) at an MOI of 1. 6 hpi, cells were harvested. To harvest, all cells were first treated with 15 ml of complete growth media + cycloheximide at 100 μg/ml for 15 min on ice by replacing the media on the cells with cycloheximide containing MEM supplemented with 10% FBS and penicillin and streptomycin. Cells were then washed once with 3 ml of cold PBS + calcium and magnesium + cycloheximide at 100 μg/ml. Cells were then trypsinized with 3 ml trypsin + 100 μg/ml cycloheximide. When cells lifted off, 10 ml of cold PBS + 100 μg/ml cycloheximide supplemented protease and phosphatase inhibitors were added and cells transferred to 15‐ml tubes. Cells were pelleted by centrifugation at 1,250 rpm for 4 min at 4°C. Supernatant was aspirated and cells resuspended in 10 ml of PBS + cycloheximide supplemented protease and phosphatase inhibitors. 1 ml of cell suspension was collected in 1.5‐ml tubes for input. Cells in both 15 ml and 1.5 ml tubes were pelleted by centrifugation at 1,250 rpm for 4 min at 4°C. Supernatants were again discarded, and all cell pellets frozen using liquid nitrogen before storing at −80°C.
The day before running cell lysates through sucrose gradient, sucrose gradients were prepared by pipetting 5.5 ml of 50% sucrose solution (28.5 ml 1× LSB [20 mM Tris pH 7.4–7.5, 10 mM NaCl, 3 mM MgCl2], 21.5 g sucrose, 40 U/ml Riboblock, 100 μg/ml cycloheximide) in a Beckman 14 × 89 mm tube and adding 5.5 ml of 15% sucrose solution (42.5 ml 1× LSB [20 mM Tris pH 7.4–7.5, 10 mM NaCl, 3 mM MgCl2], 8 g sucrose, 40 U/ml Riboblock, 100 μg/ml cycloheximide) on top. Enough gradients were prepared for the number of samples being processed. The tubes with the gradients were then sealed with parafilm and laid horizontally at 4°C overnight. The next day, cell pellets were thawed on ice. Once pellets were thawed, one to two samples were processed at a time. 750 μl of 1× LSB + 100 μg/ml cycloheximide + 40 U/ml riboblock was added to the cell pellet, and the pellet was incubated in a dounce homogenizer on ice for 3 min. Next, 250 μl of Triton X‐100 detergent buffer (1.2% Triton X‐100, 0.2 M sucrose [342 mg], 4.4 ml 1× LSB, 100 μg/ml cycloheximide, 40 U/ml riboblock) was added, and cells were lysed using a dounce homogenizer. Sample was then transferred to a 1.5‐ml tube on ice. Once all pellets were lysed, samples were centrifuged at maximum speed for 5 min at 4°C. The supernatants were then transferred to 1.5‐ml tubes containing 100 μl of heparin solution (10 mg/ml heparin, 1.5 M NaCl, 1.4 ml 1× LSB, 100 μg/ml cycloheximide, 40 U/ml riboblock). RNA concentration in the tubes was then measured using a nanodrop. The sample volume being loaded into the sucrose gradients were removed from the gradients first and ~ 270 μg of RNA was pipetted onto the gradients. Gradient tubes were balanced and ultracentrifuged at 36,000 rpm for 2 h at 4°C using a SW 41 Ti rotor. Fractions from gradient tubes were then collected using a BR‐188 Density Gradient Fractionation System (Brandel) into 1.5‐ml tubes filled with 25 μl of 0.5 M EDTA. All fractions were frozen at −80°C.
RNA was isolated from each collected fraction per sample using an acid phenol chloroform extraction. Briefly, an equal volume of acid phenol chloroform was added, samples mixed 3–5 times by inverting tube up and down, and then vortexed to assure complete mix of phenol with sample. Samples were then centrifuged at maximum speed for 5 min at 4°C. The upper aqueous phase was transferred to a new 2‐ml tube. An equal volume of RNase‐free water was then added to the remaining phenol, samples mixed 3–5 times by inverting tube up and down, vortexed, and centrifuge at maximum speed for 5 min at 4°C. The aqueous phase from this spin was combined with the previously collected aqueous phase. To precipitate RNA, 1/10 volume of 3 M sodium acetate pH 5.1, 1.5 volume of isopropanol, and 1 μl of Glycoblue was added to each tube before mixing and incubating the tubes at −80°C overnight. The next day, all tubes were centrifuged at maximum speed for 30 min at 4°C and supernatant discarded. RNA pellet was washed once with 1 ml of 70% ethanol, centrifuging at maximum speed for 15 min at 4°C after wash. Supernatants were then discarded and pellets air dried. Pellets were resuspended in 20 μl of Rnase‐free water. Total RNA was analyzed on the Agilent Bioanalyzer 6000 using the pico chip kit according to the manufacturer's instructions (Agilent RNA 6000 Pico Kit Quick Start Guide). An equal volume of RNA from each fraction was used for cDNA synthesis (procedure described in Interferon‐ß treatment time course experiments above) and qPCR analysis was carried out as described in (Panda et al, 2017). We performed ROUT outlier analysis (Q = 1%) on every individual fraction and removed the following outliers: One from fraction 7 of EMCV‐IRES‐PV VP4, one from fraction 8 of EMCV‐IRES‐PV GAPDH.
Quantification and statistical analysis
All replicates of in vitro experiments are from biologically independent experiments unless directly stated. Statistical analyses were performed in Prism (GraphPad Prism Version 9.3.1 (350)). The statistical tests used, and number of biological replicates are indicated in each figure legend. Statistical significance was defined as a P value of 0.05.
Author contributions
Mohammad Sadic: Conceptualization; data curation; formal analysis; validation; investigation; methodology; writing – original draft. William M Schneider: Conceptualization; investigation; methodology; writing – review and editing. Olga Katsara: Conceptualization; data curation; investigation; methodology; writing – review and editing. Gisselle N Medina: Formal analysis; investigation; methodology; writing – review and editing. Ashley Fisher: Formal analysis; investigation; writing – review and editing. Aishwarya Mogulothu: Formal analysis; investigation; methodology; writing – review and editing. Yingpu Yu: Conceptualization; investigation; methodology; writing – review and editing. Meigang Gu: Investigation; methodology; writing – review and editing. Teresa de los Santos: Conceptualization; supervision; funding acquisition; investigation; methodology; writing – review and editing. Robert J Schneider: Supervision; funding acquisition; methodology; writing – review and editing. Meike Dittmann: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; methodology; writing – original draft; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 4
PDF+
Acknowledgments
We would like to thank Charles M. Rice for mentorship and support to initiate this project, bicistronic and monocistronic HCV reporters, and constructs used in (Schoggins et al, 2011) screen; Vincent Racaniello and Eckard Wimmer for strategizing the cloning and generation of chimeric poliovirus (EMCV‐IRES‐PV); Ann Palmenberg for pEC9 plasmid and pPVM‐2A‐144‐poliovirus‐GFP plasmid; Aaron Briley, Adil Mohamed, Maren de Vries, Ana M. Valero‐Jimenez, Keaton Crosse, Rachel Prescott, Austin Schinlever, and Ralf Duerr for helpful discussions; Aaron Briley, Sarah Ballentine, Chloe Adrienna Talana, Ashley Fisher, and Paige Loose for technical assistance. This work was funded by NIH grants: R01AI091707 (to CMR), R01AI143639 (to MD), R21AI139374 (to MD), 5R01CA248397 (to RJS), F32DK095666 (to WMS), T32AI007647, T32GM007308, and T32GM136573, USDA, ARS, CRIS Project No1940‐32000‐061‐00D (to GNM, AM, TDLS), The Rockefeller University Women & Science fellowship (to M.D.), and postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (to M.D.).
EMBO reports (2022) 23: e55218
Data availability
No large primary datasets have been generated and deposited.
References
- Abdullah SW, Wu J, Zhang Y, Bai M, Guan J, Liu X, Sun S, Guo H (2021) DDX21, a host restriction factor of FMDV IRES‐dependent translation and replication. Viruses 13: 1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander L, Lu HH, Wimmer E (1994) Polioviruses containing picornavirus type 1 and/or type 2 internal ribosomal entry site elements: genetic hybrids and the expression of a foreign gene. Proc Natl Acad Sci USA 91: 1406–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JS, Parker RP (1998) The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J 17: 1497–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev DE, Fernandez‐Miragall O, Ramajo J, Dmitriev SE, Terenin IM, Martinez‐Salas E, Shatsky IN (2007) Differential factor requirement to assemble translation initiation complexes at the alternative start codons of foot‐and‐mouth disease virus RNA. RNA 13: 1366–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev DE, Hirnet J, Terenin IM, Dmitriev SE, Niepmann M, Shatsky IN (2012) Glycyl‐tRNA synthetase specifically binds to the poliovirus IRES to activate translation initiation. Nucleic Acids Res 40: 5602–5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhab Y, Bulakhov AG, Pestova TV, Hellen CUT (2020) Dissemination of internal ribosomal entry sites (IRES) between viruses by horizontal gene transfer. Viruses 12: 612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzino BC, Fuchs G, Fraser CS (2017) Cellular cap‐binding protein, eIF4E, promotes picornavirus genome restructuring and translation. Proc Natl Acad Sci USA 114: 9611–9616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachboard DC, Horner SM (2016) Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol 32: 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales LP, Holzenburg A, Rowlands DJ (2003) Viral internal ribosome entry site structures segregate into two distinct morphologies. J Virol 77: 6574–6579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham GJ (1992) Dual initiation sites of protein synthesis on foot‐and‐mouth disease virus RNA are selected following internal entry and scanning of ribosomes in vivo. EMBO J 11: 1105–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM (2002) Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76: 13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochler A, Querido JB, Prilepskaja T, Soufari H, Simonetti A, Del Cistia ML, Kuhn L, Ribeiro AR, Valášek LS, Hashem Y (2020) Structural differences in translation initiation between pathogenic Trypanosomatids and their mammalian hosts. Cell Rep 33: 108534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Moon SL, Matheny T, Parker R (2019) RNase L reprograms translation by widespread mRNA turnover escaped by antiviral mRNAs. Mol Cell 75: 1203–1217.e1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrill CP, Strings VR, Andino R (2013) Poliovirus: Generation, quantification, propagation, purification, and storage. Curr Protoc Microbiol chapter 15: Unit 15H.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnero‐Montoro E, Barturen G, Povedano E, Kerick M, Martinez‐Bueno M, Ballestar E, Martin J, Teruel M, Alarcón‐Riquelme ME (2019) Epigenome‐wide comparative study reveals key differences between mixed connective tissue disease and related systemic autoimmune diseases. Front Immunol 10: 1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JR, Fraser TS, Fraser MJ (2008) Examining the relative activity of several dicistrovirus intergenic internal ribosome entry site elements in uninfected insect and mammalian cell lines. J Gen Virol 89: 3150–3155 [DOI] [PubMed] [Google Scholar]
- Cesaro T, Michiels T (2021) Inhibition of PKR by viruses. Front Microbiol 12: 757238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date T, Kato T, Miyamoto M, Zhao Z, Yasui K, Mizokami M, Wakita T (2004) Genotype 2a hepatitis C virus subgenomic replicon can replicate in HepG2 and IMY‐N9 cells. J Biol Chem 279: 22371–22376 [DOI] [PubMed] [Google Scholar]
- Decroly E, Ferron F, Lescar J, Canard B (2012) Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol 10: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Farzan M (2013) The broad‐spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol 13: 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann M, Hoffmann HH, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, Wilson SJ, Crotta S, Yu Y, Flatley B et al (2015) A serpin shapes the extracellular environment to prevent influenza a virus maturation. Cell 160: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Samulski RJ, Elsworth JD, Kaplitt MG, Leone P, Xiao X, Li J, Freese A, Taylor JR, Roth RH et al (1998) In vivo expression of therapeutic human genes for dopamine production in the caudates of MPTP‐treated monkeys using an AAV vector. Gene Ther 5: 820–827 [DOI] [PubMed] [Google Scholar]
- Feng Q, Hato Stanleyson V, Langereis Martijn A, Zoll J, Virgen‐Slane R, Peisley A, Hur S, Semler Bert L, van Rij Ronald P, van Kuppeveld Frank JM (2012) MDA5 detects the double‐stranded RNA replicative form in picornavirus‐infected cells. Cell Rep 2: 1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D (2021) Interferon regulatory factor 1 (IRF1) and anti‐pathogen innate immune responses. PLoS Pathog 17: e1009220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarelli M, Neil SJD, Swanson CM (2021) Targeted restriction of viral gene expression and replication by the ZAP antiviral system. Annu Rev Virol 8: 265–283 [DOI] [PubMed] [Google Scholar]
- Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, Nadjsombati MS, Hendricks MR, So L, Green R, Roy CN et al (2019) Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and type III interferons. Immunity 51: 451–464.e456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Doudna JA (2007) Structural and mechanistic insights into hepatitis C viral translation initiation. Nat Rev Microbiol 5: 29–38 [DOI] [PubMed] [Google Scholar]
- Freundt EC, Drappier M, Michiels T (2018) Innate immune detection of Cardioviruses and viral disruption of interferon signaling. Front Microbiol 9: 2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu TY, Wu CN, Sie HC, Cheng JT, Lin YS, Liou HH, Tseng YK, Shu CW, Tsai KW, Yen LM et al (2016) Subsite‐specific association of DEAD box RNA helicase DDX60 with the development and prognosis of oral squamous cell carcinoma. Oncotarget 7: 85097–85108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghattas IR, Sanes JR, Majors JE (1991) The encephalomyocarditis virus internal ribosome entry site allows efficient coexpression of two genes from a recombinant provirus in cultured cells and in embryos. Mol Cell Biol 11: 5848–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow I, Chaudhry Y, Gioldasi I, Gerondopoulos A, Natoni A, Labrie L, Laliberté J‐F, Roberts L (2005) Calicivirus translation initiation requires an interaction between VPg and eIF4E. EMBO Rep 6: 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, van der Veen AG, Chakravarty P, Lin R, Rogers N, Rehwinkel J, Deddouche S, Rosewell I, Hiscott J, Reis e Sousa C (2015) Mouse superkiller‐2‐like helicase DDX60 is dispensable for type I IFN induction and immunity to multiple viruses. Eur J Immunol 45: 3386–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Rice CM (2009) Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc Natl Acad Sci USA 107: 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M et al (2010) Transcriptome‐wide identification of RNA‐binding protein and microRNA target sites by PAR‐CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbach F, Rode M, Conti E (2012) The crystal structure of S. cerevisiae Ski2, a DExH helicase associated with the cytoplasmic functions of the exosome. RNA 18: 124–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Guo X, Zhang T, Wang J, Ye K (2022) Development of an RNA‐protein crosslinker to capture protein interactions with diverse RNA structures in cells. RNA 28: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Wimmer E (1995) Translation of encephalomyocarditis virus RNA by internal ribosomal entry. Curr Top Microbiol Immunol 203: 31–63 [DOI] [PubMed] [Google Scholar]
- Hernández G, Altmann M, Lasko P (2010) Origins and evolution of the mechanisms regulating translation initiation in eukaryotes. Trends Biochem Sci 35: 63–73 [DOI] [PubMed] [Google Scholar]
- Hoffmann HH, Schneider WM, Rice CM (2015) Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol 36: 124–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Kaneko S, Matsushita E, Kobayashi K, Abell GA, Lemon SM (2000) Cell cycle regulation of hepatitis C virus internal ribosomal entry site–directed translation. Gastroenterology 118: 152–162 [DOI] [PubMed] [Google Scholar]
- Hopfner K‐P, Hornung V (2020) Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol 21: 501–521 [DOI] [PubMed] [Google Scholar]
- Horwitz JA, Dorner M, Friling T, Donovan BM, Vogt A, Loureiro J, Oh T, Rice CM, Ploss A (2013) Expression of heterologous proteins flanked by NS3‐4A cleavage sites within the hepatitis C virus polyprotein. Virology 439: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL et al (2018) Recognition of RNA N6‐methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20: 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C‐T, Kung Y‐A, Li M‐L, Brewer G, Lee K‐M, Liu S‐T, Shih S‐R (2016) Additive promotion of viral internal ribosome entry site‐mediated translation by far upstream element‐binding protein 1 and an enterovirus 71‐induced cleavage product. PLoS Pathog 12: e1005959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SL, Kaminski A, Jackson RJ (1993) The influence of viral coding sequences on the efficiency of internal initiation of translation of cardiovirus RNAs. Virology 197: 801–807 [DOI] [PubMed] [Google Scholar]
- Jackson RJ (2013) The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb Perspect Biol: 5, a011569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan E, Sarnow P (2002) Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J Mol Biol 324: 889–902 [DOI] [PubMed] [Google Scholar]
- Jang SK, Kräusslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E (1988) A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol 62: 2636–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E (2011) RNA helicases at work: binding and rearranging. Trends Biochem Sci 36: 19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YM, Pardoe IU, Burness AT, Michalak TI (1994) Identification and characterization of the cell surface 70‐kilodalton sialoglycoprotein(s) as a candidate receptor for encephalomyocarditis virus on human nucleated cells. J Virol 68: 7308–7319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SJ, Jackson RN (2013) Ski2‐like RNA helicase structures: common themes and complex assemblies. RNA Biol 10: 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM (2007) Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81: 8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN et al (2010) Real‐time imaging of hepatitis C virus infection using a fluorescent cell‐based reporter system. Nat Biotechnol 28: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski A, Howell MT, Jackson RJ (1990) Initiation of encephalomyocarditis virus RNA translation: the authentic initiation site is not selected by a scanning mechanism. EMBO J 9: 3753–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa T, Sato R, Imaizumi T, Hashimoto S, Fujita M, Aizawa T, Tsugawa K, Kawaguchi S, Seya K, Terui K et al (2022) Glomerular endothelial expression of type I IFN‐stimulated gene, DExD/H‐box helicase 60 via toll‐like receptor 3 signaling: possible involvement in the pathogenesis of lupus nephritis. Ren Fail 44: 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Jang SK (1999) La protein is required for efficient translation driven by encephalomyocarditis virus internal ribosomal entry site. J Gen Virol 80: 3159–3166 [DOI] [PubMed] [Google Scholar]
- Kolupaeva VG, Lomakin IB, Pestova TV, Hellen CU (2003) Eukaryotic initiation factors 4G and 4A mediate conformational changes downstream of the initiation codon of the encephalomyocarditis virus internal ribosomal entry site. Mol Cell Biol 23: 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M (2011) Cellular IRES‐mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10: 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla M, Karginov FV (2016) Measuring mRNA translation by polysome profiling. Methods Mol Biol 1421: 127–135 [DOI] [PubMed] [Google Scholar]
- Lawrence P, Schafer EA, Rieder E (2012) The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 425: 40–52 [DOI] [PubMed] [Google Scholar]
- Lee KM, Chen CJ, Shih SR (2017) Regulation mechanisms of viral IRES‐driven translation. Trends Microbiol 25: 546–561 [DOI] [PubMed] [Google Scholar]
- Li D, Wu M (2021) Pattern recognition receptors in health and diseases. Signal Transduct Target Ther 6: 291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA et al (2005) Complete replication of hepatitis C virus in cell culture. Science 309: 623–626 [DOI] [PubMed] [Google Scholar]
- Linder P (2006) Dead‐box proteins: a family affair‐‐active and passive players in RNP‐remodeling. Nucleic Acids Res 34: 4168–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju M, Johnsen D, Schlundt A, Langer LM, Basquin J, Sattler M, Heick Jensen T, Falk S, Conti E (2019) The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch‐interacting motifs. Nat Commun 10: 3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yang D, Sun C, Wang H, Zhao B, Zhou G, Yu L, Pfeiffer JK (2020) hnRNP K is a novel internal ribosomal entry site‐transacting factor that negatively regulates foot‐and‐mouth disease virus translation and replication and is antagonized by viral 3C protease. J Virol 94: e00803‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Lozano G, Martínez‐Salas E (2015) Structural insights into viral IRES‐dependent translation mechanisms. Curr Opin Virol 12: 113–120 [DOI] [PubMed] [Google Scholar]
- Ma H‐W, Ye W, Chen H‐S, Nie T‐J, Cheng L‐F, Zhang L, Han P‐J, Wu X‐A, Xu Z‐K, Lei Y‐F et al (2017) In‐cell Western assays to evaluate Hantaan virus replication as a novel approach to screen antiviral molecules and detect neutralizing antibody titers. Front Cell Infect Microbiol 7: 269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Salas E, Francisco‐Velilla R, Fernandez‐Chamorro J, Embarek AM (2018) Insights into structural and mechanistic features of viral IRES elements. Front Microbiol 8: 2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn CL, Wimmer E, Racaniello VR (1989) Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56: 855–865 [DOI] [PubMed] [Google Scholar]
- Merrick WC, Pavitt GD (2018) Protein synthesis initiation in eukaryotic cells. Cold Spring Harb Perspect Biol 10: a033092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita M, Oshiumi H, Matsumoto M, Seya T (2011) DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG‐I‐like receptor‐mediated signaling. Mol Cell Biol 31: 3802–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat GN, Chapman WG (1962) Growth of foot‐and‐mouth disease virus in a fibroblastic cell line derived from hamster kidneys. Nature 194: 253–255 [DOI] [PubMed] [Google Scholar]
- Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J (1982) Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42: 3858–3863 [PubMed] [Google Scholar]
- Ohlmann T, Jackson RJ (1999) The properties of chimeric picornavirus IRESes show that discrimination between internal translation initiation sites is influenced by the identity of the IRES and not just the context of the AUG codon. RNA 5: 764–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, Matsumoto M, Seya T (2015) DDX60 is involved in RIG‐I‐dependent and independent antiviral responses, and its function is attenuated by virus‐induced EGFR activation. Cell Rep 11: 1193–1207 [DOI] [PubMed] [Google Scholar]
- Pacheco A, López de Quinto S, Ramajo J, Fernández N, Martínez‐Salas E (2009) A novel role for Gemin5 in mRNA translation. Nucleic Acids Res 37: 582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan PK, Ferreira GR, Zghidi‐Abouzid O, Oliveira C, Dumas C, Mariz Filipe C, Papadopoulou B (2021) Genetic depletion of the RNA helicase DDX3 leads to impaired elongation of translating ribosomes triggering co‐translational quality control of newly synthesized polypeptides. Nucleic Acids Res 49: 9459–9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Martindale JL, Gorospe M (2017) Polysome fractionation to analyze mRNA distribution profiles. Bio Protoc 7: e2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Sonenberg N (1992) Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF‐4A. EMBO J 11: 2643–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N (1988) Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334: 320–325 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Hellen CU, Shatsky IN (1996a) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol 16: 6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Shatsky IN, Hellen CU (1996b) Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol Cell Biol 16: 6870–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU (1998) A prokaryotic‐like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon during internal translation initiation of hepatitis C and classical swine fever virus RNAs. Genes Dev 12: 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM (1981) A unique cap(m7GpppXm)‐dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23: 847–858 [DOI] [PubMed] [Google Scholar]
- Pyle AM (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37: 317–336 [DOI] [PubMed] [Google Scholar]
- Ranji A, Boris‐Lawrie K (2010) RNA helicases: Emerging roles in viral replication and the host innate response. RNA Biol 7: 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB (2011) ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol 85: 12881–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyimu A, Chen Y, Song X, Zhou W, Dai J, Jiang F (2021) Identification of latent biomarkers in connection with progression and prognosis in oral cancer by comprehensive bioinformatics analysis. World J Surg Oncol 19: 240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder E, Bunch T, Brown F, Mason PW (1993) Genetically engineered foot‐and‐mouth disease viruses with poly(C) tracts of two nucleotides are virulent in mice. J Virol 67: 5139–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos‐Romero M, Cedro‐Tanda A, Peña‐Luna M, Mancera‐Rodríguez MA, Hidalgo‐Pérez L, Cisneros‐Villanueva M, Beltrán‐Anaya FO, Arellano‐Llamas R, Jiménez‐Morales S, Alfaro‐Ruíz LA et al (2020) FAM83H‐AS1 is a potential modulator of cancer driver genes across different tumors and a prognostic marker for ER/PR + BRCA patients. Sci Rep 10: 14145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Sáenz J, Goez Y, Tabares W, López‐Herrera A (2009) Cellular receptors for foot and mouth disease virus. Intervirology 52: 201–212 [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW (2019) Interferon‐stimulated genes: what do they all do? Annu Rev Virol 6: 567–584 [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz P, Karlberg T, van den Berg S, Collins R, Lehtiö L, Högbom M, Holmberg‐Schiavone L, Tempel W, Park H‐W, Hammarström M et al (2010) Comparative structural analysis of human DEAD‐box RNA helicases. PLoS ONE 5: e12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Meszaros T (2000) RNA helicase dynamics in pre‐mRNA splicing. EMBO J 19: 6582–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S (2006) Structural basis for RNA unwinding by the DEAD‐box protein drosophila vasa. Cell 125: 287–300 [DOI] [PubMed] [Google Scholar]
- Shen L, Pelletier J (2020) General and target‐specific DExD/H RNA helicases in eukaryotic translation initiation. Int J Mol Sci 21: 4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Bohnsack MT (2018) Unravelling the mechanisms of RNA helicase regulation. Trends Biochem Sci 43: 237–250 [DOI] [PubMed] [Google Scholar]
- Steimer L, Klostermeier D (2012) RNA helicases in infection and disease. RNA Biol 9: 751–771 [DOI] [PubMed] [Google Scholar]
- Stern‐Ginossar N, Thompson SR, Mathews MB, Mohr I (2019) Translational control in virus‐infected cells. Cold Spring Harb Perspect Biol 11: a033001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney TR, Dhote V, Yu Y, Hellen CU (2012) A distinct class of internal ribosomal entry site in members of the Kobuvirus and proposed Salivirus and Paraturdivirus genera of the Picornaviridae. J Virol 86: 1468–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney TR, Abaeva IS, Pestova TV, Hellen CUT (2014) The mechanism of translation initiation on type 1 picornavirus IRESs. EMBO J 33: 76–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SR, Sarnow P (2003) Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 315: 259–266 [DOI] [PubMed] [Google Scholar]
- Thoms M, Thomson E, Baßler J, Gnädig M, Griesel S, Hurt E (2015) The exosome is recruited to RNA substrates through specific adaptor proteins. Cell 162: 1029–1038 [DOI] [PubMed] [Google Scholar]
- Umate P, Tuteja N, Tuteja R (2011) Genome‐wide comprehensive analysis of human helicases. Commun Integr Biol 4: 118–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdaneta EC, Vieira‐Vieira CH, Hick T, Wessels H‐H, Figini D, Moschall R, Medenbach J, Ohler U, Granneman S, Selbach M et al (2019) Purification of cross‐linked RNA‐protein complexes by phenol‐toluol extraction. Nat Commun 10: 990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter BL, Nguyen JH, Ehrenfeld E, Semler BL (1999) Differential utilization of poly(rC) binding protein 2 in translation directed by picornavirus IRES elements. RNA 5: 1570–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lewis MS, Johnson AW (2005) Domain interactions within the Ski2/3/8 complex and between the ski complex and Ski7p. RNA 11: 1291–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widner WR, Wickner RB (1993) Evidence that the SKI antiviral system of Saccharomyces cerevisiae acts by blocking expression of viral mRNA. Mol Cell Biol 13: 4331–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JE, Pestova TV, Hellen CUT, Sarnow P (2000a) Initiation of protein synthesis from the a site of the ribosome. Cell 102: 511–520 [DOI] [PubMed] [Google Scholar]
- Wilson JE, Powell MJ, Hoover SE, Sarnow P (2000b) Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol 20: 4990–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin D, Liu J, Gu J, Ji Y, Jin J, Sun L, Tai Q, Cao J, Tian Y, Qin H et al (2020) Low expression of DDX60 gene might associate with the radiosensitivity for patients with breast cancer. J Oncol 2020: 8309492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Unbehaun A, Spahn CMT (2017) Ribosomal chamber music: toward an understanding of IRES mechanisms. Trends Biochem Sci 42: 655–668 [DOI] [PubMed] [Google Scholar]
- Yu Y, Sweeney TR, Kafasla P, Jackson RJ, Pestova TV, Hellen CU (2011) The mechanism of translation initiation on Aichivirus RNA mediated by a novel type of picornavirus IRES. EMBO J 30: 4423–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fu M, Zhang M, Zhang J, Du Z, Zhang H, Hua W, Mao Y (2021) DDX60 is associated with glioma malignancy and serves as a potential immunotherapy biomarker. Front Oncol 11: 665360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, He C (2017) Post‐transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinoviev A, Ayupov RK, Abaeva IS, Hellen CUT, Pestova TV (2020) Extraction of mRNA from stalled ribosomes by the ski complex. Mol Cell 77: 1340–1349.e1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Xiao X, Shen B, Jiang Q, Wang H, Lu Z, Wang F, Jin M, Min J, Wang F et al (2021) The N6‐methyladenosine RNA‐binding protein YTHDF1 modulates the translation of TRAF6 to mediate the intestinal immune response. Nucleic Acids Res 49: 5537–5552 [DOI] [PMC free article] [PubMed] [Google Scholar]