

Abstract
Associations between genetic variation and traits are often in non-coding regions with strong linkage disequilibrium (LD) where a single causal variant is assumed to underlie the association. We applied a massively parallel reporter assay (MPRA) to functionally evaluate genetic variants in high, local linkage disequilibrium for independent cis-expression quantitative trait loci (eQTL). We found that 17.7% of eQTLs exhibit more than one significant allelic effect in tight LD. The detected regulatory variants were highly and specifically enriched for activating chromatin structures and allelic transcription factor binding. Integration of MPRA profiles with eQTL/complex trait colocalizations across 114 human traits and diseases identified causal variant sets demonstrating how genetic association signals can manifest through multiple, tightly-linked causal variants.
INTRODUCTION
Genome-wide association studies (GWAS) have emerged as an important tool to assess the effect of individual genetic variants on phenotypes ranging from gene expression to complex traits and diseases (1, 2). However, due to linkage disequilibrium (LD), it is challenging to identify a single causal variant among multiple correlated variants. To address this challenge, statistical and functional fine-mapping approaches have been developed to identify credible sets of variants containing the causal variant (3). However, these approaches often cannot distinguish between proximal or highly-linked variants and lack systematic prior information on the number of causal variants underlying association signals.
One approach to systematically identify causal variants while controlling for LD is applying massively parallel reporter assays (MPRAs). MPRAs measure the effects of synthetic DNA libraries on the expression of a reporter gene, typically luciferase or GFP, containing a 3’ UTR barcode (4). Such assays have screened potential regulatory elements in diverse cellular contexts, and also have applications in saturation mutagenesis or tiling along regulatory regions of interest (5–7).
Beyond tests of regulatory function, MPRAs have also been applied to assay the differential regulatory effects of genetic variants (8–10). However, existing studies have either targeted variants with the strongest trait associations and/or applied extensive prior filtering limiting resolution of linked causal variants (8, 9, 11, 12). In the yeast, Saccharomyces cerevisiae, quantitative trait loci (QTL) mapping has identified loci containing multiple causal variants in tight LD, suggesting that the same genetic architecture may also underlie many human traits(13, 14).
RESULTS
Functional fine-mapping of eQTL reproducibly identifies regulatory and allelic hits
We applied an MPRA to systematically characterize causal variants underneath multiple expression QTL (eQTL) and GWAS loci. We selected independent, common, and top-ranked eQTL across 744 eGenes identified in the CEU cohort (comprising Utah residents of Northern and Western European ancestry). Each eQTL had a median of 6 lead associated variants (range 1–472) in perfect LD. For each lead variant, we identified all additional variants with r2 >= 0.85 that were associated with the same gene, as well as a set of variants (N=2,114 non-eQTLs) which were not associated with any gene’s expression. Our final library included 30,893 variants, with a median of 50 variants per eQTL (range 2–2824) (Fig. 1A).
Figure 1 -. Design and implementation of a variant-based massively parallel reporter assay.
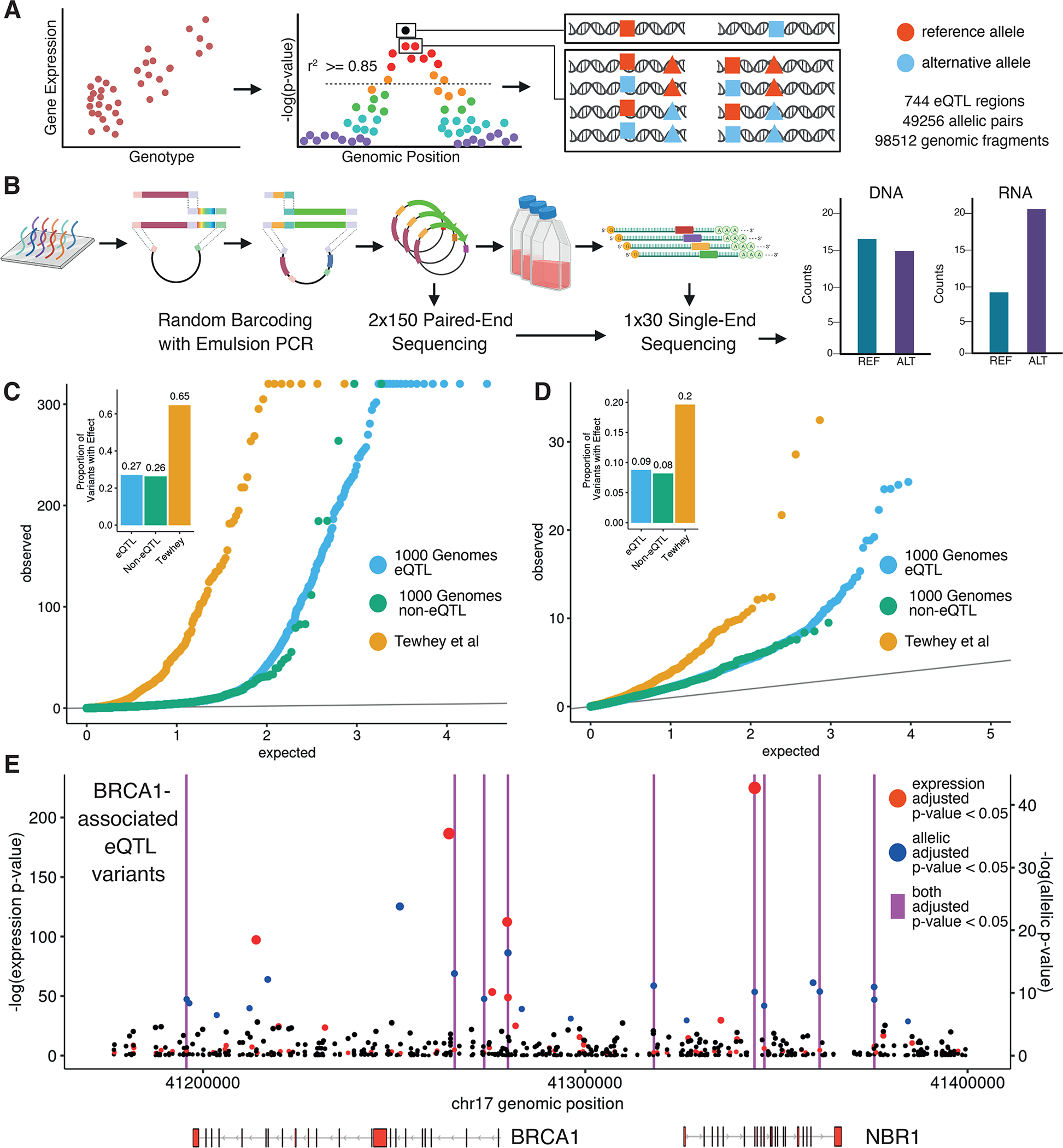
(A) Variant selection and oligonucleotide sequence design. (B) Random barcoding, sequencing and expression of the MPRA library. (C) Distribution of eQTLs (orange) and non-eQTLs (blue) from the 1000 Genomes Project compared to Tewhey et al. (green) (8) variant expression p-values (negative binomial regression) and relative effect proportions. Inset shows proportion of tested variants that are significant MPRA hits. (D) Same as in (C) but with allelic p-values (negative binomial regression) (E) Genomic position and unadjusted p-values for all tested BRCA1-associated variants with colors indicating Benjamini-Hochberg (BH) adjusted p-value <= 0.05. Vertical magenta lines indicate positions of variants that are both expression and allelic MPRA hits.
For each variant, we identified 150 bp sequences (centered on the variant) and generated a MPRA library by random barcoding (Fig. 1B). For allelic pairs, the fragment lengths and surrounding sequence were held constant to allow measurement of allele-specific effects. For indels, fragment lengths between allelic pairs differed by less than 9bp. Furthermore, in sequences with multiple variants, distinct oligonucleotides (oligos) were designed for each possible haplotype resulting in an average of 3.19 oligos per variant. Overall, this resulted in an assay of 49,256 total allelic pairs. After reporter gene insertion, the library was transfected into lymphoblastoid cell lines (LCLs) in triplicate, sequenced and then quantified for each oligo.
To measure regulatory effects from oligo counts, we used negative binomial regression. For each variant, we computed the allele-independent regulatory effects of an oligo (“expression” effects) and the difference in regulatory effects between reference and alternative allele-containing oligos (“allelic” effects). We detected 8,502 expression effects and 1,264 allelic effects across all tested variants.
We observed a modest increase in the total number of MPRA hits in eQTLs relative to non-eQTLs (27% vs 26% for expression hits and 9% vs 8% for allelic hits), reflecting the low proportion of eQTL variants overall that are expected to be causal (Fig. 1C, D). We observed a larger increase in allelic effect sizes among hits which are also eQTL vs non-eQTL (fig. S1D). This was the case when comparing MPRA hits between eQTL and non-eQTL for both expression effects (Kolmogorov–Smirnov, K-S p-value = 1.704e-4) and allelic effects (K-S p-value = 0.0116). Taken together, we obtained for each eQTL gene (eGene), a profile of allele-independent and -dependent effects across all highly-associated proximal variants (Fig. 1E).
By design, a subset of tested variants (N=782) were previously identified as expression-modulating variants in (8). This overlapping subset was highly enriched for expression and allelic effects (Fig. 1C, D). Further, we observed that 89.6% of allelic MPRA hits in both datasets were directionally concordant (fig. S2A). From these results, we constructed a concordant, high-confidence “MPRA positive” variant set containing 250 variants with expression effects and 120 with allelic effects (fig. S2B, C).
Diverse transcription factor programs contribute at eQTL
The large number of MPRA expression effects enabled identification of transcription factors (TFs) impacting gene expression within eQTLs. We observed widespread positive enrichment of ChIP-seq peaks for multiple TFsin MPRA expression effects (N=160 total TFs). Moreover, applying a more stringent filter, (adjusted p-value <= 5e-10) increases these enrichments in most TFs (Fig. 2A and table S5). While enrichments vary across a broad range (1.2- to 17-fold), many enriched TFs are members of the same family and exhibit highly correlated genome-wide binding profiles. This demonstrates the wide range of regulatory element effects captured in our assay and pinpoints specific TFs driving the regulatory effects of genetic variation.
Figure 2 -. General and allele-specific functional properties of regulatory variants.
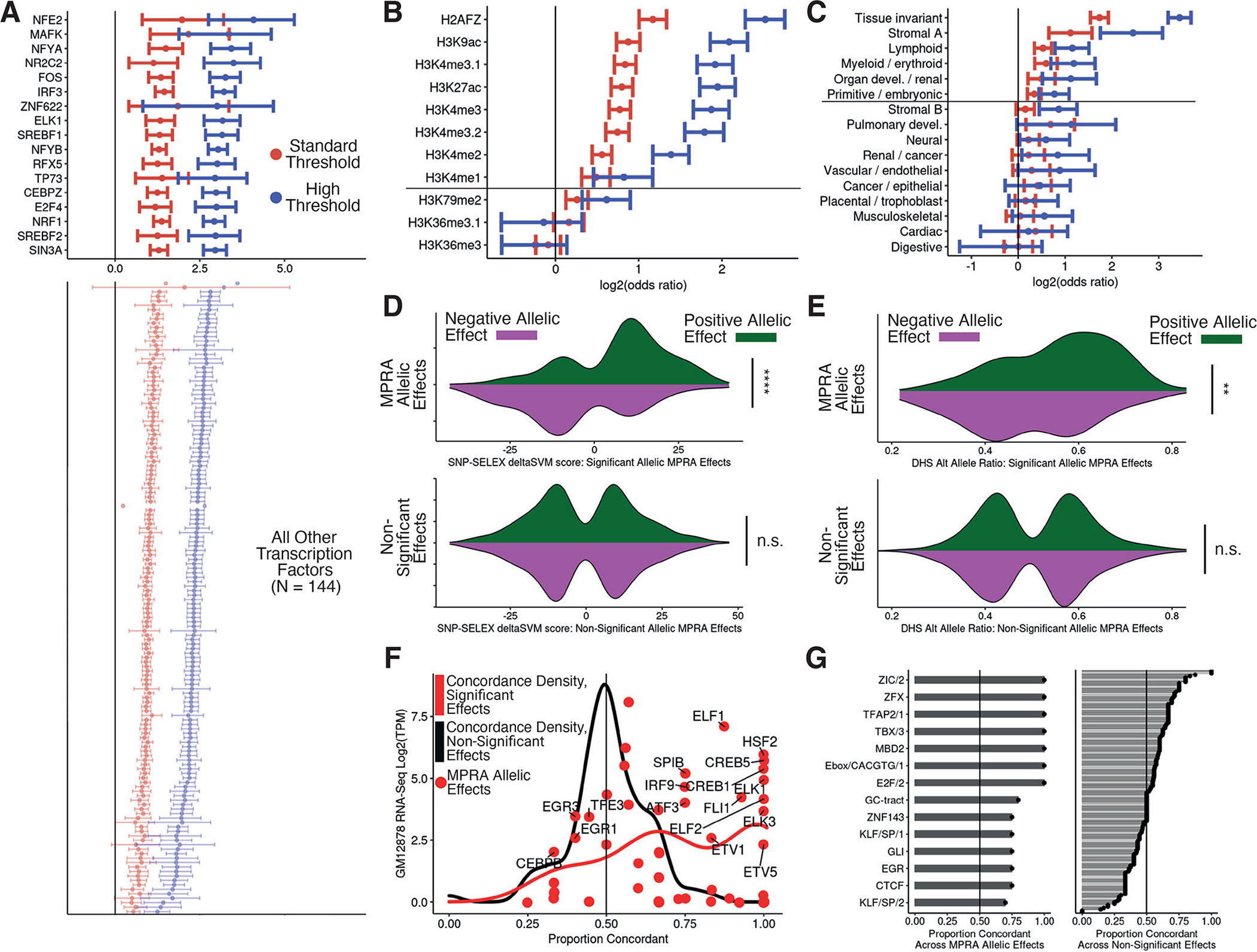
(A) Odds ratios and 95% confidence intervals for enrichment of peaks from 160 ENCODE ChIP-seq datasets within expression MPRA hitss. Standard and high thresholds required an expression BH-adjusted p-value of < 5e-2 or < 5e-10, respectively. Only TFtranscription factors with an enrichment adjusted p-value < 0.005 are shown, and listed TFtranscription factors have BH-adjusted enrichment p-value < 0.05 and odds ratio > 5 (Fisher’s exact test). (B) Same as in (A) but for histone modifications. Marks above the horizontal line have a BH-adjusted p-value < 0.05 at both thresholds. (C) Same as in (A) but for clustered chromatin accessibility regions in fragments with expression effects. (D) Distribution of SNP-SELEX deltaSVM scores at allele-specific binding variants stratified by MPRA allelic hit direction (color) and significance category (top and bottom); MPRA allelic hits have BH-adjusted expression and allelic p-values <= 0.05 while non-significant variants have p-values > 0.75 (negative binomial regression). (E) Same as in (D) but for allelic imbalance in chromatin accessibility from ENCODE. (F) For all TFs evaluated in (D), comparison of the concordance proportion across MPRA variants with the expression of each included TF in GM12878 cells; points indicate significant effect concordances. (G) Comparison of directional concordances within accessible chromatin motifs for significant (left) and non-significant (right) MPRA effects. Significance values for C and D were calculated with Fisher’s exact test and p-values are denoted as follows: * <0.05, **<0.005, ***<0.0005, ****<5e-5.
We next evaluated histone modifications and observed enrichments for activating histone modifications but not for repressive marks like H3K36me3 (Fig. 2B). We also observed the strongest enrichments in chromatin accessibility regions that were tissue invariant or specific to the Stromal A (representing JDP2 and other AP-1 TF families), Lymphoid, and Erythroid/Myeloid tissue clusters demonstrating detection of cell-type information encoded in accessible chromatin (Fig. 2C).
Identifying regulatory variants by allelic transcription factor binding and chromatin accessibility
To identify specific TFs affected by regulatory variation, we first characterized whether the direction of allelic MPRA hits was concordant with SNP-SELEX scores, a set of allele-specific binding models created from in vitro TF binding affinities. Here, we observed global concordance (Fisher’s exact p-value = 3.43e-15) that was absent in other tested sites (Fisher’s exact p-value = 0.63, Fig. 2D).
Next, we computed the concordance proportion (i.e. how often a SNP-SELEX score for a specific TF was concordant with the MPRA allelic effect) for all TFs overlapping at least three tested variants (Fig. 2E). The mean concordance proportion across TFs (N=59) was 0.733 when using allelic MPRA hits and 0.505 when using other tested sites (N=91) (binomial logistic generalized linear model, GLM p-value = 8.46e-12). Although allelic MPRA hits were enriched in SNP-SELEX variants, only 13.5% of SNP-SELEX variants had an MPRA effect. This suggests that many allelic effects can be explained by altered TF binding but altered binding itself does not typically affect transcription.
A similar pattern emerged when comparing allelic imbalance in accessible chromatin with MPRA allelic hits. We observed significant concordance between allelic imbalance and MPRA allelic effect directions for allelic MPRA hits but not other variants (Fisher’s exact p-value = 7.33e-3 and p-value = 0.839 respectively; Fig. 2F). Separation by functional footprints found within accessible chromatin regions revealed that several motifs, including Gli and a canonical E-box, were concordant across all allelic MPRA hits (Fig. 2G).
To further assess the relationship between regulatory variants and chromatin accessibility, we integrated chromatin accessible QTL (caQTL) data to identify variant annotations which increased MPRA signals. Using ENCODE allelic imbalance data, we separated all variants by whether they were inside or outside an associated peak. MPRA allelic hits were strongly concordant with allelic imbalance when inside their peaks, but not when adjacent to them (Fisher’s exact p-value = 3.2e-5 and p-value = 0.055, respectively; fig. S3A). Separately, in a set of caQTLs assessed across ten population groups, variants that were caQTLs in multiple populations were more enriched in MPRA allelic hits than caQTLs shared in only a few populations (fig. S3B). Taken together, MPRA allelic hits were significantly concordant with in vitro and in vivo measures of allelic regulatory activity while other tested sites were directionally random.
MPRAs inform non-coding variant effect prediction
An ongoing challenge is to summarize and predict the regulatory effect of non-coding variants using sequence and annotation alone. We evaluated whether genome-wide variant effect predictors could identify allelic MPRA hits. Using principal component scores from Enformer, a neural network that predicts variant effects by incorporating sequence information, we observed significant enrichment of allelic MPRA hits in the top percentiles of Enformer scores (K-S test, Fig. 3A inset, 3B) (15). We next assessed all tested variants with their annotation principal components (aPCs) from FAVOR, an integrated variant effect prediction tool (16). We again observed enrichment of allelic MPRA hits for multiple aPCs. These enrichments were strongest for the TF and epigenetics-based aPCs, while others like distance from TSS/TES were similarly enriched in both allelic MPRA hits and all other tested sites (K-S test, Fig. 3C inset, 3D).
Figure 3 -. Integrative non-coding variant effect prediction.
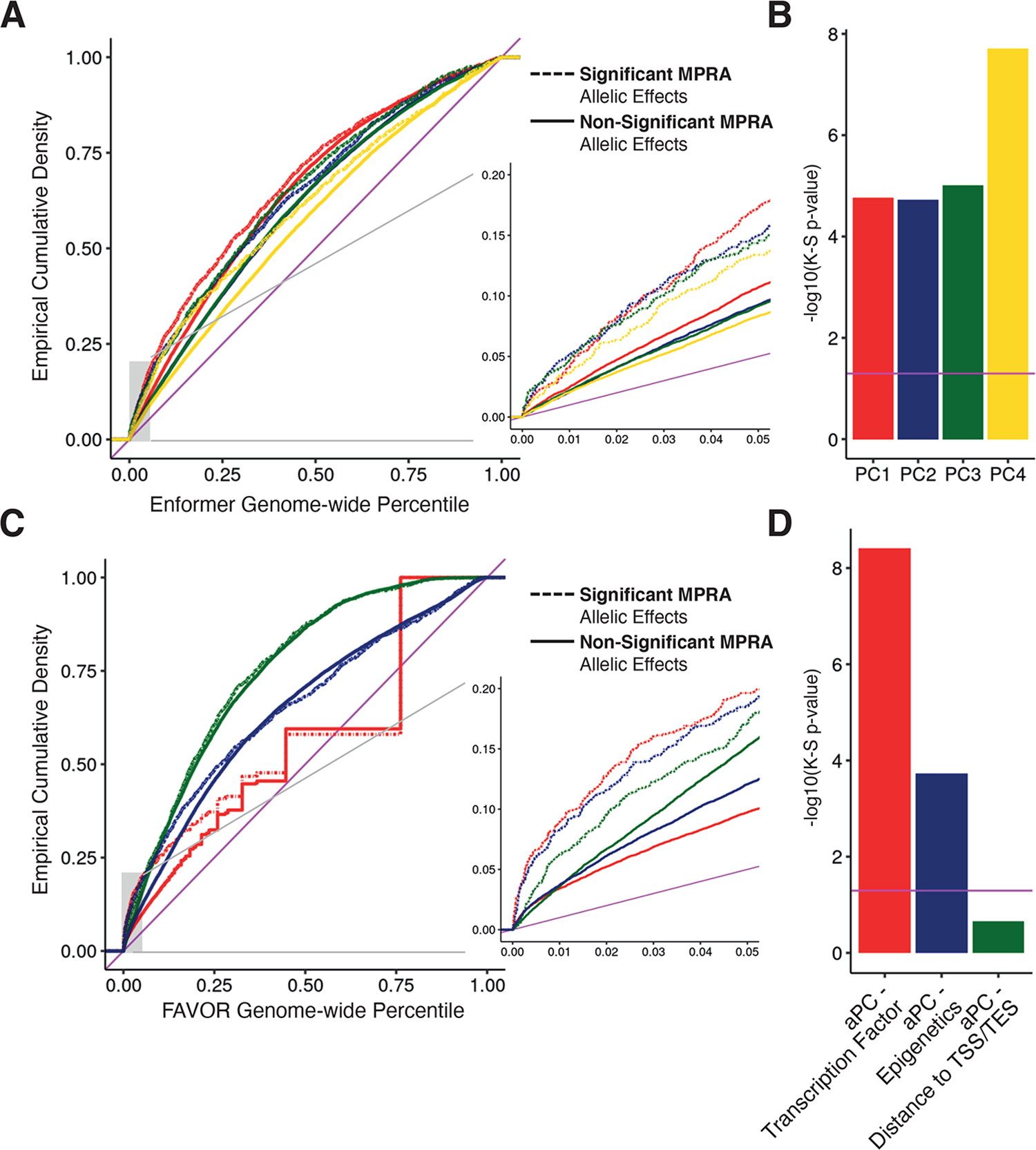
(A) Empirical cumulative probability distribution of the first through fourth principal component (PC) scores from Enformer for allelic MPRA hits and other tested variants significant and non-significant MPRA allelic hits; genome-wide percentiles computed across all common variants in 1000 Genomes Phase 3. Inset shows a blow up of lower genome-wide percentile curves (B) Significance of a Kolgomorov-Smirnov (K-S) test comparing the empirical distributions of Enformer scores for significant and non-significant allelic MPRA hits v; magenta horizontal line indicates significance by K-S test (p-value < 0.05). (C) Same as in (A) except showing annotation principle components from FAVOR; genome-wide percentiles computed across all variants in TOPMed Freeze5. (D) Same as in B, except testing FAVOR aPCs.
Both predictors could distinguish eQTL regions from genomic background; we also observed a positive enrichment in allelic MPRA hits up to the 50th percentile of these scores, with increasing enrichment at very high percentiles (Fig. 3A, C). Despite this overlap, when comparing allelic MPRA hits to other tested variants the distributions of all Enformer PCs and FAVOR aPCs except for Distance-to-TSS/TES were significantly different (K-S test, Fig. 3B, D). This suggests why functional fine-mapping approaches have not always benefitted from non-coding variant effect predictions, while also showing that the highest genome-wide percentiles of these scores identify variants enriched for MPRA allelic effects.
Multiple causal regulatory variants in high linkage disequilibrium underlie eQTL
In order to fine-map regulatory variants, we assessed MPRA hits within eQTLs. Across all loci, 76.7% (571/744) and 45.6% (339/744) had at least one expression or allelic MPRA hit, respectively (Fig. 4A, 4B). 17.7% (132/744) had more than one allelic MPRA hit, indicating that an appreciable number of genetic associations contain multiple regulatory variants in high LD (Fig. 4B). Notably, 69% of allelic hits were in perfect LD in Europeans from the 1000 Genomes Project limiting the use of statistical approaches (Fig. 4C). Even when additionally requiring a strong MPRA expression effect (|log2 effect size| > 1.4), 6.3% of all eQTL contained multiple regulatory variants.
Figure 4 -. Decomposition of allelic heterogeneity within regulatory loci.
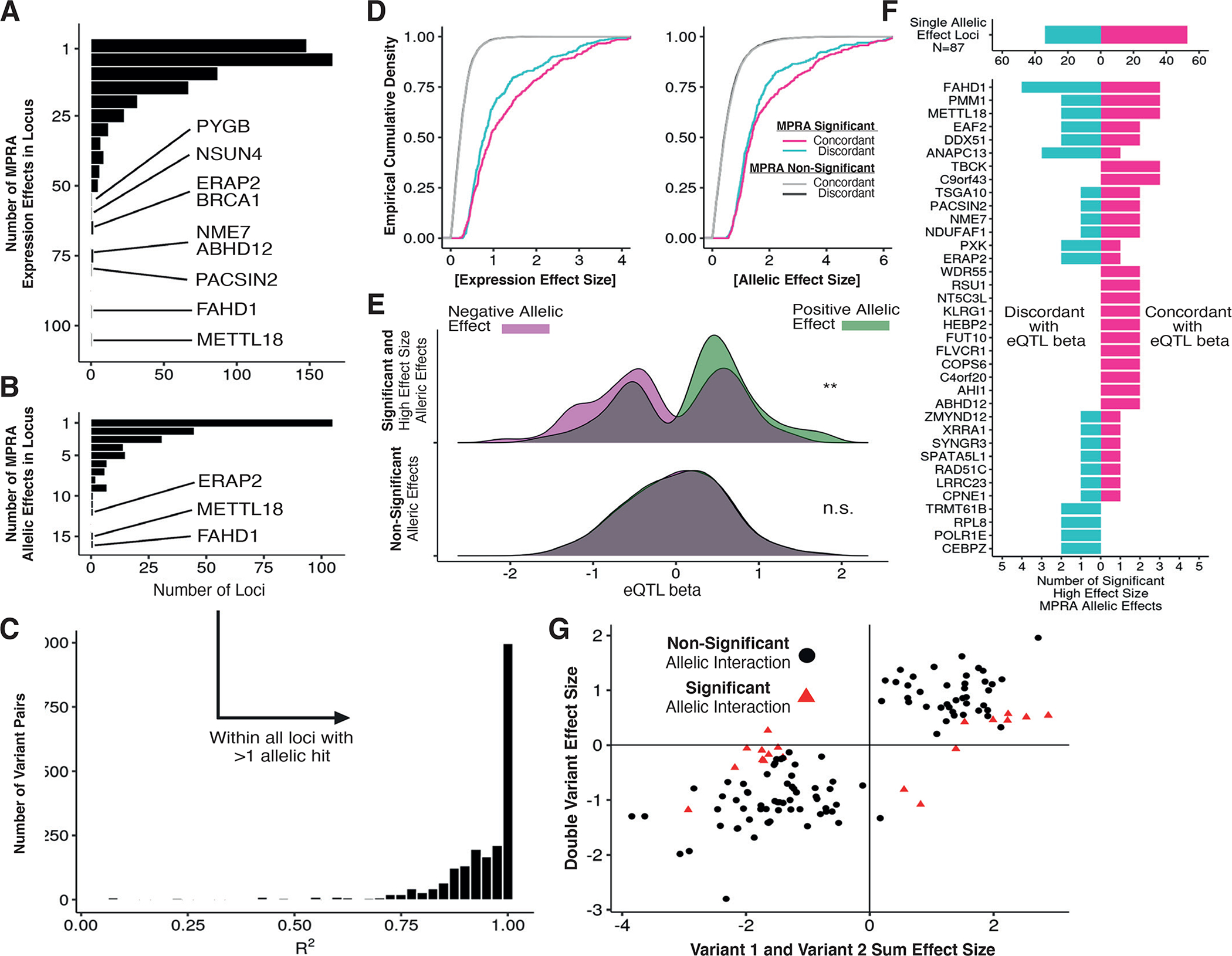
(A) Histograms of the number of expression MPRA hits per locus with BH-adjusted p-value <= 0.05 (negative binomial regression). (B) Same as in (A) but requiring BH-adjusted p-value <= 0.05 for allelic MPRA hits. (C) Distribution of linkage disequilibrium R2 values between all pairs of allelic MPRA hits within genes with multiple hits. (D) Cumulative distribution of effect sizes stratified by concordance; concordance is defined as the sign of the allelic effect size matching the sign of eQTL beta. (E) Distribution of eQTL betas measured in GTEx v8 LCLs for strong MPRA hits (log expression effect size >= 1.4), stratified by MPRA allelic effect direction and significance from negative binomial regression. (F) Using the same variants as (E), counts of directionally concordant and discordant allelic MPRA hits across all loci. (G) Comparison of haplotype regression coefficients for variants tested individually or jointly; red points indicate allelic interaction BH-adjusted p-value <= 0.05 (negative binomial regression). The x-axis displays the sum of effect sizes associated with oligos containing each variant individually, and the y-axis displays the effect size associated with the oligo containing both variants.
The degree to which eQTL are composite products of multiple causal variants is unknown due to high LD. We assessed whether allelic MPRA hits found within eQTL were more likely to be concordant with eQTL effect direction than other tested sites. We found that expression and allelic MPRA effect sizes were larger for concordant variants compared to discordant variants (Fig. 4D). Across strong allelic MPRA hits (|log2 effect size| > 1.4), we observed significant concordance with eQTL effect direction (Fisher’s exact p-value = 4.75e-3; Fig. 4E) and the strongest examples of allelic heterogeneity (Fig. 4F).
To rule out study-specific effects, we verified that eQTL effect sizes were consistent across multiple studies (fig. S4A) (17, 18). We found consistent patterns of concordance (fig. S4B). Additionally, to ensure that concordance patterns were not driven by individual eQTL with many concordant MPRA hits, we applied binomial count logistic regression to test whether concordance proportions were significantly shifted between allelic MPRA hits and other tested sites. We found that allelic MPRA hits, but not other sites, were significantly concordant (p-value = 2.85e-3; fig. S4C). We further found that concordance persists through the top four ranked variants per eQTL, with the set of third-strongest MPRA hits across all eQTL having a concordance rate of 0.67 (fig S4D). Altogether, these results indicate that several eQTL regions contain multiple, concordant allelic MPRA hits.
Haplotype decomposition identifies allelic regulation that is unlikely to be observed by population sampling
A major advantage of synthetic library design is separation of extremely proximal variants that are unlikely to be naturally separated by recombination. Our library included 2,097 pairs of eVariants within 75 bp. For these variants, we extended our statistical model to account for four haplotypes at each pair of variants and computed summary statistics for each of the three non-reference haplotypes (Fig. S5A). We then selected all variants included in at least one haplotype allelic MPRA hit. Combined, we identified 120 variant pairs (6.15% of all tests) with at least one haplotype allelic MPRA hit relative to all-reference sequence (negative binomial adjusted p-value < 0.05).
Most of the haplotype effects appeared additive, with a small number displaying non-additivity (Fig. 4G). Our linear contrast test allowed us to identify these non-additive interactions between allelic hits(fig. S5B–D and table S7). Of the variant pairs with at least one haplotype hit, 19 pairs also had a significant haplotype interaction effect (negative binomial adjusted p-value < 0.05; 14.7% of all significant haplotype effects and 0.91% of all tested variant pairs). Significant interactions were weaker than additive effects (Fig. S5E) and rarely reversed the direction of individual allelic effects. These results support other studies that have identified non-additive regulatory effects (14, 19–21) and find that 14.7% of significant haplotype effects (only 0.91% of all tests) have evidence of non-additivity.
Experimental fine-mapping of complex trait associations
In order to identify loci with shared genetic architecture between eQTL and human traits, we retrieved all genes tested in our dataset that had both an allelic MPRA hit and at least one LCL eQTL/GWAS colocalization (22). Out of 744 eGenes, 5.51% colocalized with at least one trait and contained at least one allelic MPRA hit. Notably, most colocalizations contained more than one allelic MPRA hit (71.9% of colocalizations and 82.9% of eGenes), with some loci containing as many as 13 (Fig. 5A). This suggests that the default assumption of one causal variant, often used in fine-mapping or GWAS colocalization, does not reflect causal variant biology at many regulatory regions. Traits with high-confidence co-localization were diverse, including blood-cell traits like ZC2HC1A/Lymphocyte Count or PACSIN2/Platelet Count, and highly polygenic traits like GNA12/Height (Fig. S6).
Figure 5 -. Resolving complex trait associations with multiple causal variants.
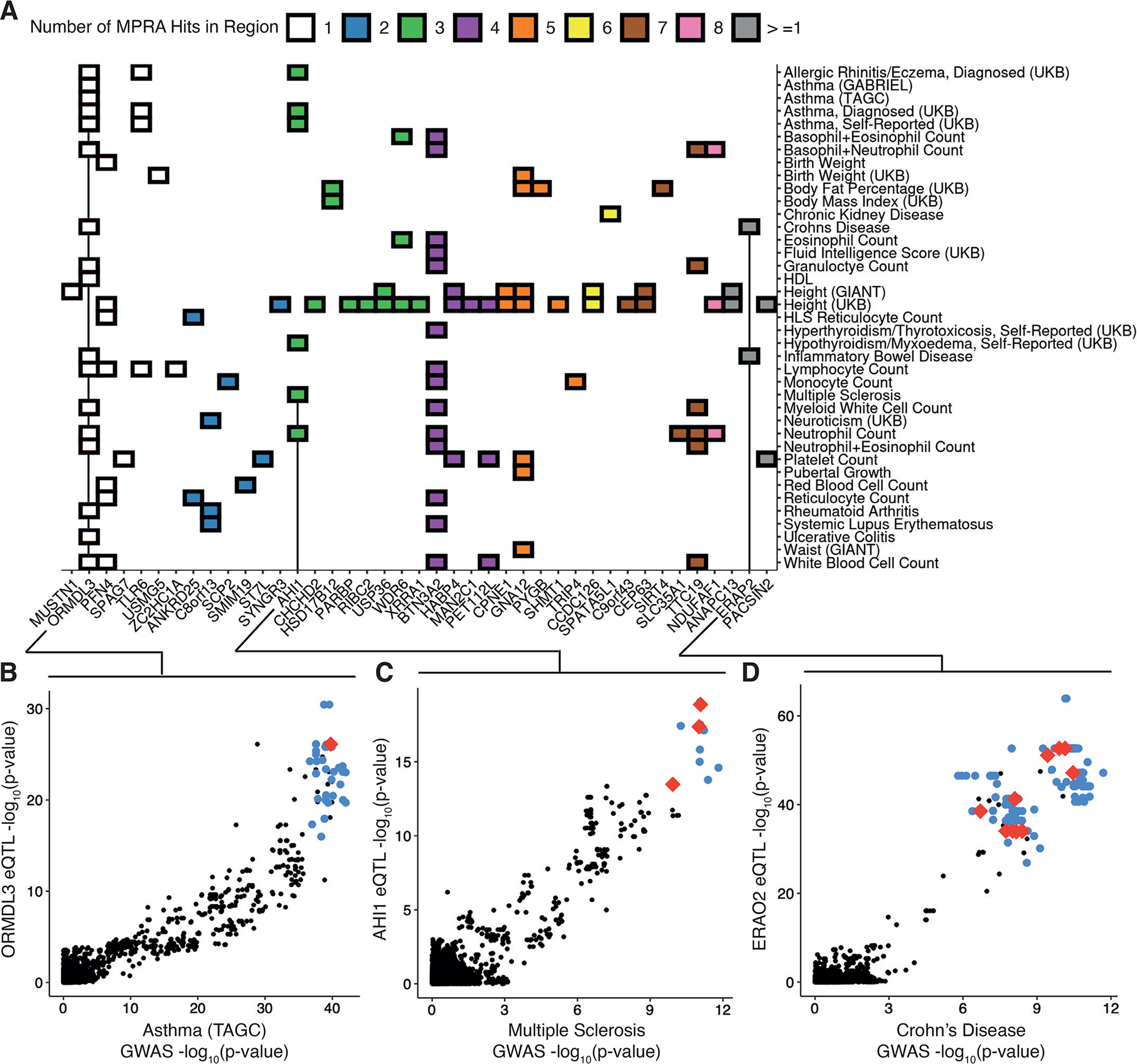
(A) Heatmap of significant colocalizations between eQTL loci and selected GWAS; color indicates the number of allelic MPRA hits within the colocalized regions. (B) Comparison of genetic associations for Asthma and ORMDL3 expression in GTEx v8 LCLs; red and blue points indicate allelic MPRA hits and other tested variants significant and non-significant allelic MPRA hits, respectively, black points indicate untested variants not included in our library. (C) Same as in (B) for associations with Multiple Sclerosis and AHI1 expression. (D) Same as in (B) and (C) for associations with Crohn’s Disease and ERAP2 expression which was also colocalized with Inflammatory Bowel Disease (fig. S5A). All GWAS and eQTL colocalizations are retrieved from (22), and lead variants were required to be genome-wide significant (reported GWAS p-value <= 5e-8 and reported eQTL p-value <= 5e-5) even if colocalization probability was high.
The 17q21 locus contains the most extensively replicated genetic association with Asthma which co-localizes with ORMDL3 eQTLs (Fig. 5B). This region contains a haplotype block with dozens of linked variants, flanked by two variants (rs4065275 and rs12936231) which induce loss and gain of CTCF binding, respectively. Further, other variants located between these two variants display allele-specific chromatin accessibility, histone modification, and CpG methylation (23, 24). Altogether, the risk haplotype results in increased ORMDL3 expression, which in turn negatively regulates interleukin-2 production in CD4+ T-cells. We identified a single allelic MPRA hit, rs12950743, that is linked to and located between the two CTCF variants (Fig. 5B). When tested by luciferase assay, this variant displayed a nominally significant but weak effect in the same direction as the MPRA (luciferase unpaired t-test p-value = 0.035, fig. S7A). Taken together, this suggests that two variants on the risk haplotype alter CTCF binding leading to distinct regulatory contacts with their own allelic specificity.
In contrast, a different colocalization that included three active variants was AHI1, a well-characterized gene strongly associated with Multiple Sclerosis (MS) (Fig. 5C) (25). This region contains a strong eQTL and colocalization signal in LCLs; however, its causal variant(s) are unknown. We identified rs6908428, rs9399148, and rs761357 as allelic MPRA hits. We validated the allelic effects of these variants via luciferase assay and found that rs6908428 (luciferase unpaired t-test p-value = 5.1e-6) and rs761357 (unpaired t-test p-value = 7.6e-3) showed allelic differences consistent with the MPRA, while rs9399148 (unpaired t-test p-value = 0.18) did not (fig. S7B). The first two of these variants have been highlighted by annotation overlap in prior studies of the role of AHI1 in MS pathology, particularly interferon gamma production and CD4+ T-cell differentiation, but were severely limited by linkage across the risk haplotype (25). When screened against known TF binding motifs, we found that rs6908428 and rs761357 overlapped predicted binding motifs for SMAD3/4 and HNF1A, respectively. Unlike HNF1A, SMAD3/4 are expressed in LCLs suggesting that rs6908428 may function to create a SMAD3/4 binding site (fig. S8A).
A complex multi-variant colocalization was identified at ERAP2, an aminopeptidase functionally implicated in both Inflammatory Bowel Disease and Crohn’s Disease (26). We detected thirteen active variants which span a strongly linked haplotype. While both eQTL and GWAS suggest a single top SNP, that top SNP differs between eQTL and GWAS and neither are MPRA hits (Fig. 5D and fig. S6A). Prior work has shown that a common splice variant in ERAP2 results in nonsense-mediated decay (NMD) and allele-specific expression, which can cause an eQTL signal (27). However, the haplotype with this variant contains hundreds of other linked variants and harbors a second conditional ERAP2 eQTL in GTEx LCLs. We evaluated eight of the thirteen active variants from our MPRA by luciferase assay and found significant allelic differences at four of the eight loci (luciferase unpaired t-test p-value < 0.05; rs1757538970, rs2549785, rs27298, and rs7713127; fig. S7C). This suggests ERAP2 is regulated by a complex allelic structure which operates via gene expression and splicing.
Another colocalization was PACSIN2 which contained thirteen variants and whose eQTL co-localized with Platelet Count. PACSIN2 is an F-BAR domain protein involved in vascular and platelet homeostasis (28). We evaluated eight of the thirteen allelic variants by luciferase assay and found significant effects at six of the eight loci (luciferase unpaired t-test p-value < 0.05; fig. S7D). Interestingly, two of the variants (rs5751402 and rs9607970) were predicted to disrupt known TF binding motifs in directions consistent with their luciferase assay result (fig. S8B). The two TF were PAX5 and NFKB1, both of which are very highly expressed in LCLs, suggesting that rs5751402 and rs9607970 may function through disruption of NFKB1 and PAX5 binding sites.
DISCUSSION
Linkage disequilibrium is a major barrier to identifying causal variants in genetic association studies. Furthermore, functional genomic annotations can be useful to prioritize likely causal variants but many annotations are also inconclusive, unattainable, or unknown (2). In this study, we demonstrate that MPRAs provide a scalable platform to separate and map the regulatory activities of expression and complex trait-associated natural genetic variants and highlight the limitations of existing approaches to variant interpretation and computational fine-mapping. Across positional annotations and variant scores, we observed that both allelic MPRA hits and other tested variants were shifted relative to the corresponding genome-wide distributions. This demonstrates how functional predictions may readily distinguish eQTL regions from the genomic background while struggling to discriminate regulatory activity between highly linked allelic MPRA hits within the same region.
We found that multiple, tightly-linked causal variants could be found under eQTL and GWAS loci. We identified that at least 17.7% of eQTL had more than one allelic hit. We further observed that most haplotype combinations exhibited additive effects, with 0.91% exhibiting non-additivity. Using these data, we demonstrate the power of MPRA-based experimental fine-mapping and report likely causal variants underlying hundreds of molecular and complex trait phenotypes, including a single variant underlying ORMDL3/Asthma, three variants underlying AHI1/Multiple Sclerosis, and up to thirteen variants each underlying PACSIN2/Platelet Count and ERAP2/Crohn’s Disease/Inflammatory Bowel Disease.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Montgomery lab for general guidance and feedback on this work and members of the M. Bassik laboratory for experimental advice. We also thank R. Tewhey and M. Love for experimental and statistical modeling advice, respectively and N. Cyr for assistance with figures.
Funding:
NSA is supported by the Stanford Department of Genetics T32 training grant and the Joint Institute for Metrology in Biology (JIMB) training program. EG is funded by the National Science Foundation Graduate Research Fellowship Program grant DGE-1656518. SBM is supported by National Institutes of Health grants R01AG066490, R01MH125244, U01HG009431 (ENCODE), R01HL142015 (TOPMed) and R01HG008150 (NoVa). This work in-part used supercomputing resources provided by the Stanford Genetics Bioinformatics Service Center, supported by National Institutes of Health S10 Instrumentation Grant S10OD023452.
Footnotes
Competing Interests: SBM has consulting agreements with MyOme, Biomarin and Tenaya Therapeutics. All other authors report no competing interests.
SUPPLEMENTAL MATERIALS LIST
Data and Materials Availability:
Sequencing data are available through the Gene Expression Omnibus under accession GSE174534. All code and supplementary tables are available through Zenodo (29).
REFERENCES
- 1.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H, The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D, Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Schaid DJ, Chen W, Larson NB, From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melnikov A, Zhang X, Rogov P, Wang L, Mikkelsen TS, Massively parallel reporter assays in cultured mammalian cells. J. Vis. Exp. (2014), doi: 10.3791/51719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen TS, Kellis M, Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat. Biotechnol. 34, 1180–1190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, Ahituv N, Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 10, 3583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patwardhan RP, Lee C, Litvin O, Young DL, Pe’er D, Shendure J, High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 27, 1173–1175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, Sabeti PC, Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 165, 1519–1529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, Sankaran VG, Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell. 165, 1530–1545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss CV, Harshman L, Inoue F, Fraser HB, Petrov DA, Ahituv N, Gokhman D, The cis-regulatory effects of modern human-specific variants. Elife. 10 (2021), doi: 10.7554/eLife.63713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi J, Zhang T, Vu A, Ablain J, Makowski MM, Colli LM, Xu M, Hennessey RC, Yin J, Rothschild H, Gräwe C, Kovacs MA, Funderburk KM, Brossard M, Taylor J, Pasaniuc B, Chari R, Chanock SJ, Hoggart CJ, Demenais F, Barrett JH, Law MH, Iles MM, Yu K, Vermeulen M, Zon LI, Brown KM, Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 11, 2718 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein JC, Keith A, Rice SJ, Shepherd C, Agarwal V, Loughlin J, Shendure J, Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.She R, Jarosz DF, Mapping Causal Variants with Single-Nucleotide Resolution Reveals Biochemical Drivers of Phenotypic Change. Cell. 172, 478–490.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renganaath K, Cheung R, Day L, Kosuri S, Kruglyak L, Albert FW, Systematic identification of cis-regulatory variants that cause gene expression differences in a yeast cross. Elife. 9 (2020), doi: 10.7554/eLife.62669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, Kelley DR, Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods. 18, 1196–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, Sun R, Dey R, Arnett DK, Aslibekyan S, Ballantyne CM, Bielak LF, Blangero J, Boerwinkle E, Bowden DW, Broome JG, Conomos MP, Correa A, Cupples LA, Curran JE, Freedman BI, Guo X, Hindy G, Irvin MR, Kardia SLR, Kathiresan S, Khan AT, Kooperberg CL, Laurie CC, Liu XS, Mahaney MC, Manichaikul AW, Martin LW, Mathias RA, McGarvey ST, Mitchell BD, Montasser ME, Moore JE, Morrison AC, O’Connell JR, Palmer ND, Pampana A, Peralta JM, Peyser PA, Psaty BM, Redline S, Rice KM, Rich SS, Smith JA, Tiwari HK, Tsai MY, Vasan RS, Wang FF, Weeks DE, Weng Z, Wilson JG, Yanek LR, NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, TOPMed Lipids Working Group, Neale BM, Sunyaev SR, Abecasis GR, Rotter JI, Willer CJ, Peloso GM, Natarajan P, Lin X, Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat. Genet. 52, 969–983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lappalainen T, Sammeth M, Friedländer MR, ’t Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlöf J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HPJ, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Pirinen M, Montgomery SB, Donnelly P, McCarthy MI, Flicek P, Strom TM, Geuvadis Consortium, Lehrach H, Schreiber S, Sudbrak R, Carracedo A, Antonarakis SE, Häsler R, Syvänen A-C, van Ommen G-J, Brazma A, Meitinger T, Rosenstiel P, Guigó R, Gut IG, Estivill X, Dermitzakis ET, Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA, Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. U. S. A. 109, 19498–19503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powell JE, Henders AK, McRae AF, Kim J, Hemani G, Martin NG, Dermitzakis ET, Gibson G, Montgomery GW, Visscher PM, Congruence of additive and non-additive effects on gene expression estimated from pedigree and SNP data. PLoS Genet. 9, e1003502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hivert V, Sidorenko J, Rohart F, Goddard ME, Yang J, Wray NR, Yengo L, Visscher PM, Estimation of non-additive genetic variance in human complex traits from a large sample of unrelated individuals. Am. J. Hum. Genet. (2021), doi: 10.1016/j.ajhg.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbeira AN, Bonazzola R, Gamazon ER, Liang Y, Park Y, Kim-Hellmuth S, Wang G, Jiang Z, Zhou D, Hormozdiari F, Liu B, Rao A, Hamel AR, Pividori MD, Aguet F, GTEx GWAS Working Group, Bastarache L, Jordan DM, Verbanck M, Do R, GTEx Consortium, Stephens M, Ardlie K, McCarthy M, Montgomery SB, Segrè AV, Brown CD, Lappalainen T, Wen X, Im HK, Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 22, 49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verlaan DJ, Berlivet S, Hunninghake GM, Madore A-M, Larivière M, Moussette S, Grundberg E, Kwan T, Ouimet M, Ge B, Hoberman R, Swiatek M, Dias J, Lam KCL, Koka V, Harmsen E, Soto-Quiros M, Avila L, Celedón JC, Weiss ST, Dewar K, Sinnett D, Laprise C, Raby BA, Pastinen T, Naumova AK, Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am. J. Hum. Genet. 85, 377–393 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rathod A, Duan J, Zhang H, Holloway JW, Ewart S, Arshad SH, Karmaus W, Interweaving Between Genetic and Epigenetic Studies on Childhood Asthma. Epigenet Insights. 13, 2516865720923395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaskow BJ, Buttrick TS, Klein H-U, White C, Bourgeois JR, Ferland RJ, Patsopoulos N, Bradshaw EM, De Jager PL, Elyaman W, MS AHI1 genetic risk promotes IFNγ+ CD4+ T cells. Neurol Neuroimmunol Neuroinflamm. 5, e414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christodoulou K, Wiskin AE, Gibson J, Tapper W, Willis C, Afzal NA, Upstill-Goddard R, Holloway JW, Simpson MA, Beattie RM, Collins A, Ennis S, Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut. 62, 977–984 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrés AM, Dennis MY, Kretzschmar WW, Cannons JL, Lee-Lin S-Q, Hurle B, NISC Comparative Sequencing Program, Schwartzberg PL, Williamson SH, Bustamante CD, Nielsen R, Clark AG, Green ED, Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet. 6, e1001157 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Begonja AJ, Pluthero FG, Suphamungmee W, Giannini S, Christensen H, Leung R, Lo RW, Nakamura F, Lehman W, Plomann M, Hoffmeister KM, Kahr WHA, Hartwig JH, Falet H, FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood. 126, 80–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abell N, nsabell/mpra-v2: FineMapMPRA (Zenodo, 2022; https://zenodo.org/record/5921041). [Google Scholar]
- 30.Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, Price A, Raj T, Nisbett J, Nica AC, Beazley C, Durbin R, Deloukas P, Dermitzakis ET, Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8, e1002639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magoč T, Salzberg SL, FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao L, Liu Z, Levy SF, Wu S, Bartender: a fast and accurate clustering algorithm to count barcode reads. Bioinformatics. 34, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Using RNA-seq DE methods to detect allele-specific expression (2017), (available at https://rpubs.com/mikelove/ase).
- 36.Chèneby J, Ménétrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, Bergon A, Lopez F, Ballester B, ReMap 2020: a database of regulatory regions from an integrative analysis of Human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 48, D180–D188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature. 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, A global reference for human genetic variation. Nature. 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan J, Qiu Y, Ribeiro Dos Santos AM, Yin Y, Li YE, Vinckier N, Nariai N, Benaglio P, Raman A, Li X, Fan S, Chiou J, Chen F, Frazer KA, Gaulton KJ, Sander M, Taipale J, Ren B, Systematic analysis of binding of transcription factors to noncoding variants. Nature. 591, 147–151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Teodosiadis A, Reynolds A, Haugen E, Nelson J, Johnson A, Frerker M, Buckley M, Sandstrom R, Vierstra J, Kaul R, Stamatoyannopoulos J, Index and biological spectrum of human DNase I hypersensitive sites. Nature. 584, 244–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Haugen E, Rynes E, Reynolds A, Nelson J, Johnson A, Frerker M, Buckley M, Kaul R, Meuleman W, Stamatoyannopoulos JA, Global reference mapping of human transcription factor footprints. Nature. 583, 729–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tehranchi A, Hie B, Dacre M, Kaplow I, Pettie K, Combs P, Fraser HB, Fine-mapping cis-regulatory variants in diverse human populations. Elife. 8 (2019), doi: 10.7554/eLife.39595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, Kelley DR, Effective gene expression prediction from sequence by integrating long-range interactions. bioRxiv (2021), p. 2021.04.07.438649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V, Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen X, Pique-Regi R, Luca F, Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 13, e1006646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coetzee SG, Coetzee GA, Hazelett DJ, motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 31, 3847–3849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, Makeev VJ, HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available through the Gene Expression Omnibus under accession GSE174534. All code and supplementary tables are available through Zenodo (29).