

INTRODUCTION
Juvenile dermatomyositis (JDM) is a rare heterogeneous immune-medicated condition with characteristic skin rashes, muscle weakness, vasculopathy, and other organ involvement. Myositis-specific autoantibodies (MSAs) help define clinical associations within subgroups of JDM.1,2 The authors present key updates from the past 10 years on the classification, pathogenesis, MSA groups, assessment, and treatment of JDM.3
CLASSIFICATION
Historically, classification criteria for JDM have been limited to those of Bohan and Peter from 1975.4 These were not originally intended to be used, however, for JDM. Lundberg and colleagues5 recently conducted a large, international, consensus-based effort to develop new classification criteria for inflammatory myositis in both adults and children. The criteria assign scores to a variety of characteristics, such as presence of classic rash, age, or biopsy features, giving a total score, which can be used to determine the likelihood that a patient has an idiopathic inflammatory myopathy. Additional validation of the criteria in JDM suggest excellent sensitivity but specificity that is lower than previous criteria.6 The role of the new criteria for clinical research or diagnosis remains to be determined.
SEROLOGIC CLASSIFICATION OF JUVENILE DERMATOMYOSITIS
MSAs are found in 50% to 70% of juvenile myositis and have multiple clinical associations.1,2 MSAs are present exclusively in patients with myositis, as summarized in Table 1. Myositis-associated autoantibodies (MAAs) are present in patients with myositis as well as in patients with other autoimmune diseases but have clinical associations in myositis.
Table 1.
Myositis-specific autoantibodies in juvenile myositis
| MSA Group | Alias(es) | Frequency (%) | Cohort Sizes | Associations | References |
|---|---|---|---|---|---|
| Anti-TIF1 | p155/140, TIF1-gamma | 17.9–35.0 | 21–380 | Associated with photosensitivity, Gottron papules, malar rash, V-, and shawl-sign rash, cuticular overgrowth, cutaneous ulceration, and chronic or polycyclic disease course | 1,2,9,12,14 |
| Anti-NXP2 | MJ | 15.1–23.8 | 21–380 | Associated with muscle cramps, more weakness, dysphonia, calcinosis, hospitalization, younger age of onset, and less remision at 2 years | 1,2,11,14 |
| Anti-MDA5 | CADM-140 | 6.1–23.8 | 13–453 | Associated with oral and skin ulceration, amyopathic disease, ILD (sometimes RP-ILD), arthralgia/arthritis, fever, weight loss, adenopathy, and older age of diagnosis | 2,7,10,12,14,15 |
| Anti-synthetase | Jo-1, PL-7, PL-12, other aminoacyl-tRNA synthetases | 0.0–5.1 | 21–380 | Associated with ILD, arthralgia, less weakness, mechanic’s hands, lipoatrophy, and older age of onset | 1,2,12–14 |
| Anti-Mi-2 | NuRD | 0.0–3.9 | 21–380 | Associated with malar rash, more muscle weakness including pharyngeal weakness, edema, less ILD, lower mortality, and more common in Hispanic patients | 1,2,12–14 |
| Anti-SRP | 0.0–1.8 | 22–380 | Associated with polymyositis (without rash), severe onset, more weakness including distal weakness, falling episodes, necrosis on muscle biopsy, Raynaud phenomenon, cardiac involvement, cutaneous ulceration, high CK, frequent hospitalization, wheelchair use, poor response to treatment, chronic disease course, and more common in African American patients. | 1,2,12,13 | |
| Anti-HMGCR | 1.1 | 380–440 | Associated with severe proximal and distal weakness, necrosis on muscle biopsy, muscle atrophy, joint contractures, arthralgias, dysphagia, high CK, HLA DRB1*0701, poor response to medication, and chronic course. No previous statin medication exposure. | 2,8 | |
| Anti-SAE | 0.3–9.1 | 11–380 | Too few cases with in juvenile myositis for any clear clinical associations | 1,2,19 |
Abbreviations: CK, creatine kinase; NuRD, nucleosome remodeling deacetylase complex; RP-ILD, rapidly progressive ILD.
Myositis-specific Autoantibodies
In the past decade, several studies assessing clinical associations of MSAs have shown generally consistent findings. Anti-transcriptional intermediary factor 1 (TIF1) is the most common MSA, distinct from adult myositis in which anti-synthetase autoantibody is most common. The largest cohorts are based in North America (n = 365–454)1,7–9 and the United Kingdom (n = 285–380),2,10,11 with smaller cohorts from India12,13 and Japan.14,15 MSA-related clinical phenotype subgroups have been defined based on not only clinical features but also demographics and disease course (Table 1). Other distinctions are highlighted.
Environmental associations were also identified. Anti-p155/140 or TIF1 has an association with higher UV indices,16 consistent with the clinical association of anti-TIF1 with photosensitivity.1,2,9,14 Higher UV index also was associated with lower odds of antinuclear matrix protein 2 (NXP2), also known as anti-MJ.16
In one study, the IFN score of anti-TIF1 autoantibody-positive JDM patients had higher correlation with skin activity than in JDM generally or other disease activity measures, even though anti-TIF1 patients did not have significantly higher skin disease activity.17 This suggests that biomarkers may have distinct associations by MSA group, which may point to differences in pathogenic mechanisms, but this needs further evaluation.
JDM patients with anti-Mi2 were less likely to remain on treatment despite more severe muscle biopsy findings, indicating they may be more responsive to treatment.18 Thus, although muscle biopsy general indicates disease severity, MSA group also should be considered when considering disease course and treatment.
For the anti–melanoma differentiation-associated gene 5 (MDA5) MSA group, there are notable differences in cohorts based on geography. In general, the anti-MDA5 MSA group has less weakness with more joint symptoms (arthritis and arthralgia), skin ulcerations, and systemic features (eg, fever). In Japanese JDM cohorts, however, anti-MDA5 antibody was more frequent (23.8%–38.5%)14,15 and associated with rapidly progressive interstitial lung disease (ILD).15 In other cohorts, anti-MDA5 was less common (6.1%–7.7%), associated with ILD, but not associated with rapidly progressive ILD.2,7,10,12
Necrotizing myopathy includes anti–signal recognition particle (SRP), which is associated with juvenile polymyositis or JPM (without skin findings) and the recently described anti–3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR). Anti-HMGCR recently was detected in 1.1% of JDM, JPM, and necrotizing myositis of both North American and UK-based cohorts.2,8 Distinct from adults, it has not been associated with previous statin exposure and has a different HLA-type association (DRB1*0701) compared with adult myositis.8
Anti–small ubiquitin-like modifier activating enzyme (anti-SAE) is another rare MSA in juvenile myositis. There is limited information on the few cases from 3 cohorts (1/374 North American cohort, 3/380 UK cohort, and 1/11 Japanese cohort), which is too limited to establish clear clinical correlations.1,2,19
There can be differences in testing sensitivity and specificity by MSA testing method, so care should be taken when interpreting results.20 This is an important area of continued study.
Myositis-associated Autoantibodies Updates
Anti-NT5C1A, a more recently identified MAA, was identified in adults with inclusion body myositis. Of 380 US-based juvenile myositis patients, 27% were found to have anti-NT5C1A with greater pulmonary symptoms at diagnosis, more frequent hospitalizations, and required more medication.21 Also, anti-Ro52 MAA was assessed in juvenile myositis (14% in North American cohort of 698) to be more frequent with anti-synthetase and anti-MDA5 MSA groups. In general, it was associated with ILD, chronic disease course, less remission, and more medications.22
ASSESSMENT
Assessment of children with JDM for the purposes of clinical care or research is challenging due to the complexity of the illness. Consideration needs to be given to various aspects of muscle involvement, a wide range of skin manifestations, and involvement of multiple organ systems as well as other aspects of disease impact, including quality of life (QoL). Measurement tools in JDM are reviewed by Rider and colleagues.23 New or updated/modified assessments discussed in this article are listed in Table 2. A key development in the understanding of the assessment of JDM was the advent of core sets, consensus-derived groups of outcome measures, which attempt to assess the full spectrum of disease manifestations. The core sets were developed independently by the International Myositis Assessment and Clinical Studies Group (IMACS)24 and Pediatric Rheumatology International Trials Organization (PRINTO) groups25 and are summarized in Table 3. Although this approach acknowledged the multidimensional nature of JDM, completion of all recommended core set measures is a time-consuming, challenging process that is difficult to use in a clinical context.
Table 2.
New and updated/modified juvenile dermatomyositis assessment tools discussed in this article
| Measure | What is Assessed? | Author (Reference) |
|---|---|---|
| IMACS core set | Disease Activity | Miller et al,24 2001 |
| PRINTO core set | Disease Activity | Ruperto et al,25 2003 |
| Consensus core set | Comprehensive assessment | McCann et al,26 2014; McCann et al,27 2015; McCann et al,28 2014 |
| Hybrid MMT-8/CMAS | Muscle strength/function | Varnier et al,29 2018 |
| JDMAI | Muscle strength/function | Rosina et al,30 2019 |
| Abbreviated MMT-8 | Muscle strength | Rosina et al,31 2020 |
| Whole-body MRI score | MRI muscle inflammation | Malattia et al,33 2014 |
| Pelvic girdle MRI score | MRI muscle inflammation | Thyoka et al,34 2018 |
| Muscle biopsy score | Muscle biopsy assessment | Wedderburn et al,36 2007; Varsani et al,37 2015 |
| CAT | Skin disease activity | Huber et al,38 2008 |
| CDASI | Skin disease activity | Klein et al,39 2008 |
| DSSI | Skin disease activity | Klein et al,39 2008 |
| DAS | Skin disease activity | Bode et al,40 2003 |
| Inactive disease | Inactive disease | Lazarevic et al,47 2013; Almeida et al,48 2015 |
| Definition of improvement | Response/im provement | Ruperto et al,49 2010 |
| Response to therapy | Response/im provement | Rider et al,50–52 2017 |
Table 3.
Juvenile myositis disease activity core set measures
| IMACS Core Set variables59 | PRINTO Core Set variables60 |
|---|---|
| Physician global disease activity (VAS or Likert) | Physician global disease activity (VAS or Likert) |
| Parent/Patient Global Disease Activity (VAS or Likert) | Parent/patient assessment of overall well-being (VAS or Likert) |
| CHAQ and CMAS | CHAQ |
| MMT | CMAS and MMT |
| Muscle enzymes (at least 2) | Muscle enzymes |
| Extraskeletal muscle disease activity (to be developed) | Global JDM disease activity tool |
Consensus Core Data Set in Juvenile Dermatomyositis
To facilitate collaboration and standardization of data collection across international centers, McCann and colleagues26 developed a consensus core data set. They developed a preliminary minimal data set using consensus methods. Subsequently, this data set was reviewed and refined through an international email-based Delphi survey, including both medical specialists and patients/parents, followed by a smaller consensus meeting of JDM experts using nominal group technique.27 The resulting data set included 123 items and was renamed a “core” rather than “minimal” data set.28 Several organizations, including the Childhood Arthritis and Rheumatology Research Alliance (CARRA), have adopted the core data set. Collecting a common data set in both clinical and research contexts will facilitate future collaboration between sites and research networks.
Changes to Core Set Tools
Recent work seeks to streamline the assessment process. Varnier and colleagues29 published preliminary validation work of a hybrid tool, which combines the manual muscle test 8 (MMT 8) with 3 items from the Childhood Myositis Assessment Scale (CMAS) to give a score from 0 to 100. Their work supported face and content validity, construct validity, reliability, internal consistency, responsiveness and discriminant validity, while reducing completion time. Rosina and colleagues30 reported on the development of a composite measure of disease activity in JDM, called the Juvenile DermatoMyositis Activity Index (JDMAI). The JDMAI incorporates physician and parent/patient global scales, an assessment of muscle strength and an assessment of skin disease activity. All 6 versions of the JDMAI demonstrated face and content validity, construct validity, internal consistency, responsiveness, and the ability to discriminate between disease states and parental satisfaction. Future validation work will allow selection of a final version. Rosina and colleagues’ group also has reported on the measurement properties of shortened versions of the MMT-8.31 Rather than testing 8 muscle groups, they studied versions using 6 and 4 muscle groups, documenting similar construct validity, internal consistency, discriminant validity and somewhat better responsiveness to the original tool. Together, these publications demonstrate ongoing attempts to improve assessment of children with JDM, maintaining validity while facilitating use in clinical settings.
Magnetic Resonance Imaging
The use of magnetic resonance imaging (MRI) to assess and document muscle involvement in children with JDM has become common, even though MRI is not included in any diagnostic criteria. For example, in the CARRA Legacy Registry, which included 483 children with JDM, 90% had MRI as part of their evaluation.32 Malattia and colleagues33 reported on the use of whole-body MRI in 41 JDM patients and controls. They developed and validated a whole-body MRI score, demonstrating reliability, construct validity, and responsiveness, and showed the presence of muscle inflammation in muscles not clinically affected. More recently, Thyoka and colleagues refined a previous MRI scoring tool for children with JDM,34 using pelvic girdle MRI,35 demonstrating good inter-rater and intra-rater reliability. Unfortunately, MRI results from this study were not linked to weakness or disease activity, limiting further assessment of the clinical utility of the score.
Muscle Biopsy
The use of muscle biopsy in the assessment of JDM has declined, largely due to its invasiveness and replacement by other methods that document muscle involvement. Many experts, however, continue to advocate for its routine use. Wedderburn and colleagues36 described a consensus-derived scoring tool for comprehensive assessment of muscle biopsies. A subsequent update and validation showed good intra-rater and inter-rater reliability and strong correlations with clinical measures of disease activity.37 Its use allows standardization of muscle biopsy assessment in clinical and research contexts. Deakin and colleagues18 showed that muscle biopsy combined with myositis specific autoantibodies predicted clinically relevant outcomes, including future treatment status, suggesting that muscle biopsy could be part of an assessment to tailor therapy for individual patients.
Assessment of Skin Disease in Juvenile Dermatomyositis
The assessment of skin disease in JDM is complicated by the use of multiple tools, including the Cutaneous Assessment Tool (CAT),38 the Cutaneous Dermatomyositis Disease Activity and Severity Index (CDASI),39 the Dermatomyositis Skin Severity Index (DSSI),39 and the Disease Activity Score (DAS),40 with little consensus about which tool is optimal. Given the number of patients with persistent skin disease long after resolution of muscle disease, the lack of consensus on assessment negatively affects the study of effective therapies. Several recent studies have attempted to address this lack of consensus.
Tiao and colleagues41 compared the CDASI and the CAT when used by pediatric dermatologists, rheumatologists, and neurologists in children with JDM. They found that both tools had good inter-rater reliability and correlations with other measures of activity and damage when used by dermatologists and rheumatologists, although performance was worse when used by neurologists. Both tools also were compared in adults with dermatomyositis (DM), with demonstration of good inter-rater and intra-rater reliability, construct validity, and responsiveness.42 In the adult study, including 10 dermatologists, the CDASI was preferred. A review of the CDASI, primarily in adults with DM, recently has been published.43 Determination of the optimal tool to assess skin disease in children remains an area in need of consensus.
Quality of Life and Other Measures
There is increased recognition of the impact of JDM beyond physical manifestations like skin and muscle disease. Apaz and colleagues44 reported on QoL, as assessed by the Child Health Questionnaire, in an international cohort of children with JDM. They found that QoL was impaired in children with JDM, particularly in the physical domain, compared with healthy controls, with baseline disability and disease duration predicting lower scores 6 months later. Tory and colleagues45 surveyed health care providers and families of children with JDM to identify the most important components of high-quality care. They found that although patients and families generally gave higher scores than physicians, there was consensus on the 5 most important items: QoL, timely diagnosis, access to rheumatology care, normalization of function/strength, and ability for self-care. In another publication, Tory and colleagues46 studied discordance between physician and patient/family global visual analog scales (VASs). They found that nearly 40% of the global VAS scores in a large registry were discordant, defined as a difference of greater than or equal to 2 points on a 10-point scale. More work is needed to understand the reasons for discordance between physician and patient/family global assessments and the impact on patient care.
Inactive Disease
Achieving inactive disease, either on or off medications, is an important goal in the management of JDM. Lazarevic and colleagues47 used a large, international cohort of 275 children with JDM to develop and validate a data-driven definition (meeting at least 3 of 4 criteria: creatine phosphokinase (CPK) less than or equal to 150, CMAS greater than or equal to 48, MMT8 greater than or equal to 78, and physician global VAS less than or equal to 0.2). The definition, however, may overemphasize the importance of muscle disease, because a patient with marked persistent skin involvement or other active extramuscular disease would meet the definition of inactive disease if they had normal strength and normal muscle enzymes.48 Almeida and colleagues48 proposed an amendment whereby the physician global VAS is an essential criterion. Further work is needed to develop a more acceptable definition.
Definition of Improvement/Clinical Response in Juvenile Dermatomyositis
Defining improvement or response to therapy is critical to conducting clinical trials. Ruperto and colleagues49 initially reported provisional response criteria for JDM. Using previously established core set variables for JDM, the criteria defined permitted percentages of improvement and worsening for a patient to be “improved.” More recently, an international effort developed new criteria for response to therapy in JDM.50–52 The criteria generate a continuous total improvement score, based on points assigned for the absolute percent change of each of the core set variables. This approach allows for the definition of minimal, moderate, and major improvement, permitting quantification of the degree of response. The response calculator can be found online at https://www.niehs.nih.gov/research/resources/imacs/response_criteria/index.cfm.
PATHOGENESIS
Genetics
Although the etiology of JDM is not fully understood, genetic and environmental risk factors are key contributors. One study found that approximately half of JDM families had at least 1 other family member with autoimmune disease, most commonly type 1 diabetes mellitus or systemic lupus erythematosus.53 Genome-wide association studies of single-nucleotide polymorphisms (SNPs) comparing adult DM and JDM to controls found multiple major histocompatibility complex (MHC) region signals, strongest at HLA 8.1 ancestral haplotype, with HLA DRB1*03:01 strongest in adult and JDM. Three SNPs also were more common from PLCL1, BLK, and CCL21, as has been seen in other autoimmune diseases.54,55 Lower C4a gene copy number (deficiency) is more common in JDM than controls, particularly among those with an HLA DR3-positive background.56 Genetic changes have been validated as an important component in JDM, identifying variants associated with immune response, particularly MHC class II, B cell signaling, and C4.
Environment
Because photosensitivity is a common manifestation of disease and flares often occur after sun exposure, UV radiation is considered an environmental trigger of JDM.57 Higher UV index was associated more strongly with JDM and more prominently among those positive for anti-TIF1 MSA compared with juvenile myositis without skin involvement (JPM).16 Higher UV index in the month prior to symptom onset was associated with increased odds of developing calcinosis in non–African Americans with JDM.58 When matched to healthy control mothers, maternal exposures to school chalk dust, gasoline vapor, smoking, and carbon monoxide during the third trimester were significant risk factors for having a child with JDM.59 Direct UV exposure and maternal toxic exposures (chalk dust, gasoline vapor, smoking, and carbon monoxide) are environmental triggers associated with JDM.
Muscle Pathology
Perifascicular atrophy (PFA) is a characteristic finding on muscle biopsy in DM, with or without clinical myopathy.60 In early DM without PFA, there already is focal capillary depletion found on biopsy.61 In chronic DM, biopsies show a combination of chronic ischemia with replacement of normal capillaries by abnormal lumens and thickened walls as well as neoangiogenesis.60 RIG-I, an IFN-response gene part of innate immune system response, is overexpressed in areas of PFA. With hypoxia, RIG-I expression and type I IFN increase, indicating a connection between hypoxia, IFN, and PFA.62 When compared with adult myositis biopsies, JDM biopsies show predominantly hypoxia-driven pathology in perifascicular areas with atrophy, regional loss of capillaries, and less IFN-inducible gene expression.63 Decreased vascular density, ischemia, cell death, neoangiogenesis, and IFN expression all contribute to PFA, a characteristic finding of JDM muscle biopsies.
Interferon
An increased interferon (IFN) regulated signature is present in JDM.64 Human myogenic precursor cells from JDM patients’ muscle biopsies show an angiogenic gene expression signature recapitulated with type I IFN treatment, indicating JDM’s proangiogenic features may be type I IFN driven.65 Galectin-9 and CXCL10, IFN-stimulated proteins, were validated as reliable biomarkers for disease activity in multiple JDM cohorts, showing elevated or rising levels prior to flare in longitudinal analysis.66 Weakness and joint disease activity were the best predictors of a high IFN score from blood in JDM.17 Untreated JDM muscle biopsies had higher type I and type II IFN scores, both associated with muscle biopsy pathology and higher disease activity, whereas only type II IFN scores correlated with longer time to clinically inactive disease.67 A UK group found MxA (myxovirus-resistance protein A) staining of JDM muscle biopsies by immunohistochemistry correlated inversely with muscle strength measurements.68 Type I and type II IFN signatures are high in untreated JDM blood and muscle, correlating with disease activity and angiogenic changes.
Endothelial Markers
Endothelial dysfunction and vasculopathy are prominent in JDM. Low soluble intercellular adhesion molecule 1 (ICAM) and high endoglin levels are associated with increased vasculopathy assessed by nailfold capillary end-row loop scoring.69 Endothelial injury markers (von Willebrand factor (vWF) antigen and circulating endothelial cells) correlate with extramuscular activity, whereas circulating endothelial progenitor cells negatively correlate with muscle activity.70 Endothelial activation with elevated vascular cell adhesion molecule (VCAM)-1 was higher with shorter duration of untreated JDM (early in disease) in JDM muscle biopsy.71 Peripherally, endothelial activation (VCAM-1) and inhibition of angiogenesis (angiopoietin-2, soluble vascular endothelial growth factor receptor 1 (VEGFR-1)) also relate to JDM disease activity.72 As described previously, vasculopathy is a prominent feature of JDM with multiple endothelial markers that correlate to disease activity.
Immune Cells
Immature transitional B cells are expanded in pretreatment baseline JDM peripheral blood correlating with disease activity.73 Genes related to B-cell survival, including BAFF, correlate with disease activity.74 Increased plasmablasts correlate with some T-helper subsets and disease activity in JDM.75 Although peripheral T-regulatory cells were found in the same proportion as controls, those cells from active JDM patients were found to be functionally impaired.76 There are multiple immune cell subset changes in JDM, including B-cell and T-cell differences in level or function, which correlate with disease activity.
TREATMENT
There are few clinical trials to inform optimal treatment of JDM; therefore, treatment is largely based on empiric and expert opinion. Consensus recommendations and key clinical trial results are summarized.
Consensus Recommendations
Over the past decade, CARRA,77–79 Single Hub and Access point for paediatric Rheumatology in Europe (SHARE),80 the JDM working group of the Society for Pediatric Rheumatology81 and others have published standardized treatment approaches. Initial therapy for the average patient presenting with JDM typically includes a combination of steroids and methotrexate, with escalation or modification of therapy for resistant disease. See Figs. 1 and 2 for summaries of the CARRA and SHARE treatment recommendations.
Fig. 1.
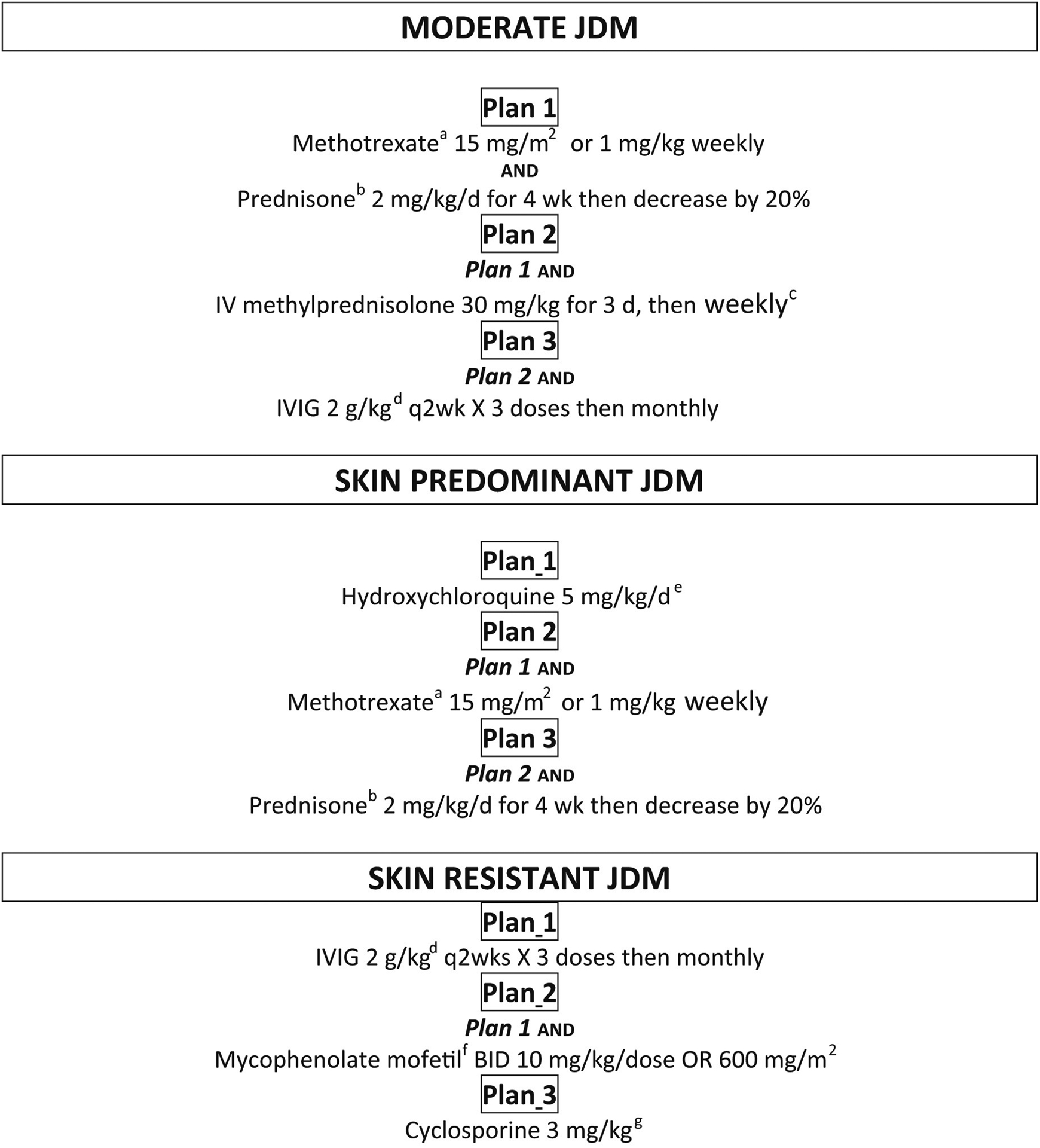
Recommended consensus treatment plans from CARRA, for JDM patients with moderate, skin-predominant, and skin-resistant disease. IV: intravenous, IG: immunoglobulin. a: Lesser of 15 mg/m2 or 1 mg/kg (max 40 mg), b: max 60mg, c: optional, d: max 70 grams, e: max 400mg, f: max 1500mg bid, g: higher doses based on toxicity, efficacy
Fig. 2.

Recommendations from SHARE, for treatment of JDM, ranging from mild to severe disease. * such as major organ involvement / extensive ulcerative skin disease; ** improvement based on clinical opinion, DMARD: disease modifying antirheumatic drug, MMF: mycophenolate mofetil, MTX: methotrexate
Clinical Trials
The Rituximab in Myositis trial was a randomized, double blind, placebo-phase trial82 for treatment of refractory myositis, which included 48 children. Although the study did not meet the primary outcome and definition of improvement, post hoc analyses suggest predictors of rituximab response included anti–Jo-1 and anti–Mi-2 or other anti-bodies compared with autoantibody negative patients.83 The findings suggest a role for B-cell–depleting therapy for subsets of patients with JDM.
The PRINTO Network completed an international, multicenter, randomized, open-label superiority trial using steroids with or without methotrexate or cyclosporine,84 enrolling 139 newly diagnosed JDM patients from 54 centers and 22 countries. The study confirmed the combination treatment with steroids and either methotrexate or cyclosporine to be superior to steroid monotherapy and methotrexate was more effective with less adverse events compared with cyclosporine.
Other Immunomodulatory Therapies
Intravenous immunoglobulin (IVIG) commonly is used as adjunctive treatment of JDM, but its efficacy is not well studied. A retrospective inception cohort of 78 Canadian JDM patients was assessed85 to determine whether IVIG-treated patients had less disease activity and achieved inactive disease sooner than patients not treated with IVIG. Marginal structural modeling showed IVIG-treated patients started with greater disease activity compared with patients not treated with IVIG. After controlling for confounding, IVIG-treated patients maintained similar or lower disease activity compared with controls.
Mycophenolate mofetil is used to treat JDM patients with resistant and refractory disease. A small case series of 8 patients published in 201286 suggested mycophenolate mofetil was safe and efficacious with improvement in strength and the ability to wean steroids.
Cyclophosphamide typically is reserved for severe or resistant disease, with limited published efficacy data. Cyclophosphamide was used to treat 19 steroid-resistant JDM patients from a single Japanese hospital87 and considered effective, including 4 patients whose interstitial pneumonitis resolved with treatment. However, 8 patients with calcinosis progressed without improvement. 56 patients from a UK JDM cohort received cyclophosphamide and showed improvement in global, skin, and muscle disease activity, with the greatest benefit in skin disease and global disease activity at 12 months.88
The use of biologic agents to treat refractory JDM is evolving. Two small studies suggested that etanercept was not beneficial and could cause worsening of disease.89,90 Other anti-TNF therapies, however, show promise for resistant disease. A retrospective analysis of 60 JDM patients treated with adalimumab or infliximab in the United Kingdom demonstrated benefit,91 including improvement of calcinosis. Smaller case reports and series also suggest anti-TNF therapies may be useful in resistant cases.92,93
Single case reports of the benefit of abatacept in the treatment of JDM with associated calcinosis have been reported44,94 and a larger pilot trial is under way (NCT02594735).
The use of Janus kinase inhibitors (JAKi) to treat JDM is increasing.95–98 In a 2020 case series, of 25 refractory JDM patients treated with JAKi, 96% (24/25) reported improvement and 66.7% (16/24) had complete resolution of rash.99,100 Kim and colleagues95 reported 4 patients with chronically active JDM in a compassionate use study of baricitinib, which also supported JAKi efficacy, according to physician, patient, and extramuscular VAS, as well as skin scoring measures, biomarker response, and MRI.
Exercise
JDM patients may have long-term issues with exercise intolerance, muscle weakness, and fatigue, despite aggressive treatment and apparent clinical remission.101,102 A growing body of evidence suggests a role for exercise training as an important non-pharmacologic adjunct treatment of JDM,103–107 without causing flares in disease.
LONG-TERM OUTCOMES
Long-term outcomes reported from several international JDM cohorts108–116 over the past decade continue to show suboptimal outcomes and long-term morbidity. These patient cohorts, followed for 3.7 to nearly 17 years, have chronic or polycyclic disease course in up to two-thirds of patients, suggesting that monocyclic disease course and remission are uncommon.
Although 60% of PRINTO’s 490 patient cohort had no impairment (Childhood Health Assessment Questionnaire [CHAQ] score = 0) after a mean disease duration of 7.7 years (range 2–25.2 y), approximately 7% had severe functional impairment (CHAQ score >1.5).108 Adult JDM patients from 2 US adult referral subspecialty centers (n = 49)109 had mild to moderate disability (median American College of Rheumatology [ACR] functional class 2, Health Assessment Questionnaire 0.4). More than half of JDM patients have reduced endurance and ongoing weakness,108,113 with up to 10% reporting severe impairment113 at long-term follow-up, after a median of at least 6 years disease duration (range 2–25.2 y)108,113 in 2 cohorts. These findings suggest that although some JDM patients have good functional outcomes, a subset of patients continue to have significant impairment due to JDM.
Cumulative damage at long-term follow-up, after a median of at least 6 years (range 2–25.2 years) disease duration,108,113 is common, reported in up to 69% of patients.108,113 Cutaneous damage also is frequent in these published JDM cohorts, including calcinosis and lipodystrophy, reported in up to 2% to 60% and 10% to 33% of patient, respectively.108,109,113,114 Long-term musculoskeletal damage based on MRI findings is present in up to 52% of patients110 and joint contractures in 18% to 63%.108,109 There also is long-term growth delay in 8% to 40% of long-term cohorts followed for a median of at least 6 years (range 2–25 years).108,109,114,115 In summary, the long-term damage described in JDM patients likely is related to many factors, including disease activity as well as medication toxicity from corticosteroids. The high frequency of damage in these cohorts point to the urgent need for better JDM treatments.
Although mortality rates for JDM have improved since the 1960s, when it was reported in one-third of patients,117 current rates remain unacceptably high. The JDM specific mortality rate from the National Institutes of Health natural history questionnaire–based protocols administered between 1989 and 2011 was estimated at 2.4%, most commonly attributed to ILD.118 In other cohorts, death was attributed to severe JDM manifestations, including cutaneous ulceration with infections, gastrointestinal vasculitis, and lung disease, including ILD and respiratory failure.114–116,118 The dramatic differences in mortality ranging from 2118 to 11%,114–116 suggest not only differences in patient populations, but disparities in access to care and treatment.
RISK FACTORS FOR WORSE OUTCOMES
The trajectory of disease is variable in JDM and it is challenging to predict long-term patient outcomes. Predicting treatment resistance or poor outcomes earlier in the course of disease could help guide treatment decisions to improve long-term prognosis.
There are inconsistent reports of age-based risk for worse outcomes in several cohorts that suggest possible regional or environmental differences contributing to worse outcome, and additional study is necessary.109,112,118,119
Longer disease duration, including chronic or continuous disease course, predicted long-term damage and worse outcomes, including functional impairment and death, in several cohorts.108,109,112,113,118 Similarly, calcinosis is reported to be less common in patients with a monocyclic course.115 High disease activity, according to muscle and total DAS scores, 1 year after diagnosis predicted poor outcomes in a Norwegian cohort.110 Worse baseline ACR functional class and baseline skin disease and severity (including erythroderma, heliotrope rash, and shawl sign) also were found to predict damage and risk for mortality in JDM.109,118 These findings suggest that patients with delayed or severe disease at diagnosis or persistent disease activity at 1 year may benefit from proactive, aggressive treatment to achieve rapid disease control and remission that may help limit unsatisfactory outcomes.
Ravelli and colleagues108 reported female patients had more long-term functional impairment and worse health-related QoL. The JDM CARRA Registry found that higher mean UV radiation exposure 1 month prior to disease onset increased calcinosis risk and varied by race.58 Decreased end row loops (ERLs) in nail fold capillaries at JDM diagnosis was associated with longer untreated disease and more active skin disease compared with children with normal ERLs.120 The UK Juvenile Dermatomyositis Cohort and Biomarker Study found differences in muscle biopsy scores when comparing MSA subgroups and increased severity of the histopathologic features predictive of longer treatment duration.18 Weight loss was found an early illness feature associated with mortality.118 These findings suggest that the clinical history, physical examination at onset, and diagnostic work-up may be useful to inform treatment recommendations and help to improve future long-term outcomes.
Earlier prognostication with modifications in therapy tailored to each patients’ risk for worse outcome may be possible if the current wide-ranging predictors are validated in future studies. Additional research is needed to identify more effective treatment approaches targeted toward these patients at risk for worse outcomes.
SUMMARY
JDM is a heterogeneous disease with new classification criteria and updates in MSA and MAA groups, helping define clinical subgroups. There are many validated assessment tools for assessing disease activity in JDM. Future studies will optimize the tools, improving feasibility of use in clinical and research contexts. Genetic and environmental risk factors, mechanisms of muscle pathology, role of IFN, vascular markers, and changes in immune cells provide insights to JDM pathogenesis. Outcomes have improved, but chronic disease activity, damage, and mortality highlight the need for improved predictors of outcome and treatment efficacy. Increased international collaboration of patients, clinicians, scientists, and other key stakeholders in patient registries and biorepositories as well as clinical trials may overcome current barriers that exist for improving research, treatment, and outcomes for this rare condition.
KEY POINTS.
New classification criteria for juvenile dermatomyositis (JDM) have been developed, but their role remains unclear. New serologic classifications that define distinct subgroups within juvenile myositis may be more useful in practice.
There are many validated assessment tools available to assess disease activity in JDM. Future studies will optimize these tools and improve feasibility in clinical and research contexts.
Genetic and environmental risk factors, causes of perifascicular atrophy in muscle, and vascular dysfunction provide new insights into the pathogenesis of JDM. Biomarkers related to endothelial function, interferon, and immune cells also have shown correlation to disease activity.
Outcomes of children with JDM have improved with immunomodulatory therapies but many patients have persistent disease sequelae.
An expanding repertoire of medications is helpful in subsets of JDM patients, but additional investigation to identify optimal individualized treatment regimens is essential to improve future outcomes in JDM
Clinical Care Points.
Validated tools should be used for the clinical assessment of children with JDM
Identification of specific JDM phenotypes using MSA and clinical features may help to predict course and outcome and influence treatment decisions
Prompt diagnosis and treatment are needed to improve long-term outcomes in JDM
Guidelines and recommendations developed through expert opinion and consensus should be used to treat JDM patients
DISCLOSURE
This research was support in part by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Arthritis and Musculoskeletal and Skin diseases (NIAMS). H.K. has been part of a clinical study at NIAMS that received grant support for under a government CRADA from Eli Lilly. The authors have no other disclosures.
REFERENCES
- 1.Rider LG, Shah M, Mamyrova G, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 2013; 92(4):223–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tansley SL, Simou S, Shaddick G, et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun 2017;84:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rider LG, Katz JD, Jones OY. Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am 2013;39(4):877–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975;292(8):403–7. [DOI] [PubMed] [Google Scholar]
- 5.Lundberg IE, Tjarnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017;76(12):1955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sag E, Demir S, Bilginer Y, Talim B, Haliloglu G, Ozen S. Validation of the EULAR/ACR 2017 idiopathic inflammatory myopathy classification criteria in juvenile dermatomyositis patients. Clin Exp Rheumatol. 2021. May-Jun;39(3):688–694. [PubMed] [Google Scholar]
- 7.Mamyrova G, Kishi T, Shi M, Targoff IN, Huber AM, Curiel RV, Miller FW, Rider LG; Childhood Myositis Heterogeneity Collaborative Study Group. Anti-MDA5 autoantibodies associated with juvenile dermatomyositis constitute a distinct phenotype in North America. Rheumatology (Oxford). 2021. Apr 6;60(4):1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kishi T, Rider LG, Pak K, et al. Association of Anti-3-Hydroxy-3-Methylglutary-lcoenzyme a reductase autoantibodies with DRB1*07:01 and severe myositis in juvenile myositis patients. Arthritis Care Res (Hoboken) 2017;69(7):1088–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habers GE, Huber AM, Mamyrova G, et al. Brief report: association of myositis autoantibodies, clinical features, and environmental exposures at illness onset with disease course in juvenile myositis. Arthritis Rheumatol 2016;68(3):761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tansley SL, Betteridge ZE, Gunawardena H, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther 2014;16(4):R138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tansley SL, Betteridge ZE, Shaddick G, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford) 2014;53(12):2204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain A, Rawat A, Jindal AK, et al. Autoantibodies in children with juvenile dermatomyositis: a single centre experience from North-West India. Rheumatol Int 2017;37(5):807–12. [DOI] [PubMed] [Google Scholar]
- 13.Srivastava P, Dwivedi S, Misra R. Myositis-specific and myositis-associated autoantibodies in Indian patients with inflammatory myositis. Rheumatol Int 2016; 36(7):935–43. [DOI] [PubMed] [Google Scholar]
- 14.Iwata N, Nakaseko H, Kohagura T, et al. Clinical subsets of juvenile dermatomyositis classified by myositis-specific autoantibodies: experience at a single center in Japan. Mod Rheumatol 2019;29(5):802–7. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi I, Okura Y, Yamada M, et al. Anti-melanoma differentiation-associated gene 5 antibody is a diagnostic and predictive marker for interstitial lung diseases associated with juvenile dermatomyositis. J Pediatr 2011;158(4): 675–7. [DOI] [PubMed] [Google Scholar]
- 16.Shah M, Targoff IN, Rice MM, et al. Childhood Myositis Heterogeneity Collaborative Study G. Brief report: ultraviolet radiation exposure is associated with clinical and autoantibody phenotypes in juvenile myositis. Arthritis Rheum 2013; 65(7):1934–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim H, Gunter-Rahman F, McGrath JA, et al. Expression of interferon-regulated genes in juvenile dermatomyositis versus Mendelian autoinflammatory interferonopathies. Arthritis Res Ther 2020;22(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deakin CT, Yasin SA, Simou S, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol 2016;68(11):2806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimoto M, Matsushita T, Hamaguchi Y, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis 2013;72(1):151–3. [DOI] [PubMed] [Google Scholar]
- 20.Tansley SL, Li D, Betteridge ZE, et al. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology (Oxford) 2020;59(8):2109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeker RM, Pinal-Fernandez I, Kishi T, et al. Anti-NT5C1A autoantibodies are associated with more severe disease in patients with juvenile myositis. Ann Rheum Dis 2018;77(5):714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabbagh S, Pinal-Fernandez I, Kishi T, et al. Anti-Ro52 autoantibodies are associated with interstitial lung disease and more severe disease in patients with juvenile myositis. Ann Rheum Dis 2019;78(7):988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rider LG, Werth VP, Huber AM, et al. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis: physician and Patient/Parent Global Activity, Manual Muscle Testing (MMT), Health Assessment Questionnaire (HAQ)/Childhood Health Assessment Questionnaire (C-HAQ), Childhood Myositis Assessment Scale (CMAS), Myositis Disease Activity Assessment Tool (MDAAT), Disease Activity Score (DAS), Short Form 36 (SF-36), Child Health Questionnaire (CHQ), physician global damage, Myositis Damage Index (MDI), Quantitative Muscle Testing (QMT), Myositis Functional Index-2 (FI-2), Myositis Activities Profile (MAP), Inclusion Body Myositis Functional Rating Scale (IBMFRS), Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), Cutaneous Assessment Tool (CAT), Dermatomyositis Skin Severity Index (DSSI), Skindex, and Dermatology Life Quality Index (DLQI). Arthritis Care Res (Hoboken) 2011;63(Suppl 11):S118–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller FW, Rider LG, Chung YL, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2001;40(11):1262–73. [DOI] [PubMed] [Google Scholar]
- 25.Ruperto N, Ravelli A, Murray KJ, et al. Preliminary core sets of measures for disease activity and damage assessment in juvenile systemic lupus erythematosus and juvenile dermatomyositis. Rheumatology (Oxford) 2003;42(12):1452–9. [DOI] [PubMed] [Google Scholar]
- 26.McCann LJ, Arnold K, Pilkington CA, et al. Developing a provisional, international minimal dataset for Juvenile Dermatomyositis: for use in clinical practice to inform research. Pediatr Rheumatol Online J 2014;12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCann LJ, Kirkham JJ, Wedderburn LR, et al. Development of an internationally agreed minimal dataset for juvenile dermatomyositis (JDM) for clinical and research use. Trials 2015;16:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCann LJ, Pilkington CA, Huber AM, et al. Development of a consensus core dataset in juvenile dermatomyositis for clinical use to inform research. Ann Rheum Dis 2018;77(2):241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varnier GC, Rosina S, Ferrari C, et al. Development and testing of a hybrid measure of muscle strength in juvenile dermatomyositis for use in routine care. Arthritis Care Res (Hoboken) 2018;70(9):1312–9. [DOI] [PubMed] [Google Scholar]
- 30.Rosina S, Consolaro A, van Dijkhuizen P, et al. Development and validation of a composite disease activity score for measurement of muscle and skin involvement in juvenile dermatomyositis. Rheumatology (Oxford) 2019;58(7):1196–205. [DOI] [PubMed] [Google Scholar]
- 31.Rosina S, Varnier GC, Pistorio A, Pilkington C, Maillard S, Civino A, Tsitsami E, Bracaglia C, Jelusic M, Cespedes-Cruz A, Espada G, Cimaz R, Couillault G, Joos R, Quartier P, Rao AP, Malattia C, Ruperto N, Consolaro A, Ravelli A; Pediatric Rheumatology International Trials Organization (PRINTO). Development and Testing of Reduced Versions of the Manual Muscle Test-8 in Juvenile Dermatomyositis. J Rheumatol. 2021. Jun;48(6):898–906. [DOI] [PubMed] [Google Scholar]
- 32.Robinson AB, Hoeltzel MF, Wahezi DM, et al. Clinical characteristics of children with juvenile dermatomyositis: the Childhood Arthritis and Rheumatology Research Alliance Registry. Arthritis Care Res (Hoboken) 2014;66(3):404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malattia C, Damasio MB, Madeo A, et al. Whole-body MRI in the assessment of disease activity in juvenile dermatomyositis. Ann Rheum Dis 2014;73(6): 1083–90. [DOI] [PubMed] [Google Scholar]
- 34.Thyoka M, Adekunle O, Pilkington C, et al. Introduction of a novel magnetic resonance imaging-based scoring system for assessing disease activity in children with juvenile dermatomyositis. Rheumatology (Oxford) 2018;57(9):1661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis WR, Halls JE, Offiah AC, et al. Assessment of active inflammation in juvenile dermatomyositis: a novel magnetic resonance imaging-based scoring system. Rheumatology (Oxford) 2011;50(12):2237–44. [DOI] [PubMed] [Google Scholar]
- 36.Wedderburn LR, Varsani H, Li CK, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthritis Rheum 2007;57(7): 1192–201. [DOI] [PubMed] [Google Scholar]
- 37.Varsani H, Charman SC, Li CK, et al. Validation of a score tool for measurement of histological severity in juvenile dermatomyositis and association with clinical severity of disease. Ann Rheum Dis 2015;74(1):204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber AM, Dugan EM, Lachenbruch PA, et al. Preliminary validation and clinical meaning of the Cutaneous Assessment Tool in juvenile dermatomyositis. Arthritis Rheum 2008;59(2):214–21. [DOI] [PubMed] [Google Scholar]
- 39.Klein RQ, Bangert CA, Costner M, et al. Comparison of the reliability and validity of outcome instruments for cutaneous dermatomyositis. Br J Dermatol 2008; 159(4):887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bode RK, Klein-Gitelman MS, Miller ML, et al. Disease activity score for children with juvenile dermatomyositis: reliability and validity evidence. Arthritis Rheum 2003;49(1):7–15. [DOI] [PubMed] [Google Scholar]
- 41.Tiao J, Feng R, Berger EM, et al. Evaluation of the reliability of the Cutaneous Dermatomyositis Disease Area and Severity Index and the Cutaneous Assessment Tool-Binary Method in juvenile dermatomyositis among paediatric dermatologists, rheumatologists and neurologists. Br J Dermatol 2017;177(4): 1086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goreshi R, Okawa J, Rose M, et al. Evaluation of reliability, validity, and responsiveness of the CDASI and the CAT-BM. J Invest Dermatol 2012;132(4):1117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed S, Chen KL, Werth VP. The validity and utility of the Cutaneous Disease Area and Severity Index (CDASI) as a clinical outcome instrument in dermatomyositis: a comprehensive review. Semin Arthritis Rheum 2020;50(3):458–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Apaz MT, Saad-Magalhaes C, Pistorio A, et al. Health-related quality of life of patients with juvenile dermatomyositis: results from the Pediatric Rheumatology International Trials Organisation multinational quality of life cohort study. Arthritis Rheum. 2009;61(4):509–517. [DOI] [PubMed] [Google Scholar]
- 45.Tory HO, Carrasco R, Griffin T, et al. Comparing the importance of quality measurement themes in juvenile idiopathic inflammatory myositis between patients and families and healthcare professionals. Pediatr Rheumatol Online J 2018; 16(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tory H, Zurakowski D, Kim S, et al. Patient and physician discordance of global disease assessment in juvenile dermatomyositis: findings from the Childhood Arthritis & Rheumatology Research Alliance Legacy Registry. Pediatr Rheumatol Online J 2020;18(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazarevic D, Pistorio A, Palmisani E, et al. The PRINTO criteria for clinically inactive disease in juvenile dermatomyositis. Ann Rheum Dis 2013;72(5):686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Almeida B, Campanilho-Marques R, Arnold K, et al. Analysis of published criteria for clinically inactive disease in a large juvenile dermatomyositis cohort shows that skin disease is underestimated. Arthritis Rheumatol 2015;67(9): 2495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruperto N, Pistorio A, Ravelli A, et al. The Paediatric Rheumatology International Trials Organisation provisional criteria for the evaluation of response to therapy in juvenile dermatomyositis. Arthritis Care Res (Hoboken) 2010;62(11):1533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rider LG, Aggarwal R, Pistorio A, et al. 2016 American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol 2017; 69(5):911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rider LG, Aggarwal R, Pistorio A, et al. 2016 American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis 2017;76(5): 782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rider LG, Ruperto N, Pistorio A, et al. 2016 ACR-EULAR adult dermatomyositis and polymyositis and juvenile dermatomyositis response criteria-methodological aspects. Rheumatology (Oxford) 2017;56(11):1884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niewold TB, Wu SC, Smith M, et al. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics 2011;127(5):e1239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller FW, Chen W, O’Hanlon TP, et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun 2015;16(7):470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller FW, Cooper RG, Vencovsky J, et al. Genome-wide association study of dermatomyositis reveals genetic overlap with other autoimmune disorders. Arthritis Rheum 2013;65(12):3239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lintner KE, Patwardhan A, Rider LG, et al. Gene copy-number variations (CNVs) of complement C4 and C4A deficiency in genetic risk and pathogenesis of juvenile dermatomyositis. Ann Rheum Dis 2016;75(9):1599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mamyrova G, Rider LG, Ehrlich A, et al. Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatology (Oxford) 2017;56(8):1342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neely J, Long CS, Sturrock H, et al. Association of short-term ultraviolet radiation exposure and disease severity in juvenile dermatomyositis: results from the childhood arthritis and rheumatology research alliance legacy registry. Arthritis Care Res 2019;71(12):1600–5. [DOI] [PubMed] [Google Scholar]
- 59.Orione MA, Silva CA, Sallum AM, et al. Risk factors for juvenile dermatomyositis: exposure to tobacco and air pollutants during pregnancy. Arthritis Care Res (Hoboken) 2014;66(10):1571–5. [DOI] [PubMed] [Google Scholar]
- 60.Gitiaux C, Kostallari E, Lafuste P, et al. Whole microvascular unit deletions in dermatomyositis. Ann Rheum Dis 2013;72(3):445–52. [DOI] [PubMed] [Google Scholar]
- 61.Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol 1990;27(4):343–56. [DOI] [PubMed] [Google Scholar]
- 62.De Luna N, Suarez-Calvet X, Lleixa C, et al. Hypoxia triggers IFN-I production in muscle: Implications in dermatomyositis. Sci Rep 2017;7(1):8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preusse C, Allenbach Y, Hoffmann O, et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta Neuropathol Commun 2016;4(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reed AM, Peterson E, Bilgic H, et al. Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course. Arthritis Rheum 2012;64(12):4078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gitiaux C, Latroche C, Weiss-Gayet M, et al. Myogenic progenitor cells exhibit Type I interferon-driven proangiogenic properties and molecular signature during juvenile dermatomyositis. Arthritis Rheumatol 2018;70(1):134–45. [DOI] [PubMed] [Google Scholar]
- 66.Wienke J, Bellutti Enders F, Lim J, et al. Galectin-9 and CXCL10 as biomarkers for disease activity in juvenile dermatomyositis: a longitudinal cohort study and multicohort validation. Arthritis Rheumatol 2019;71(8):1377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moneta GM, Pires Marafon D, Marasco E, et al. Muscle expression of Type I and Type II interferons is increased in juvenile dermatomyositis and related to clinical and histologic features. Arthritis Rheumatol 2019;71(6):1011–21. [DOI] [PubMed] [Google Scholar]
- 68.Soponkanaporn S, Deakin CT, Schutz PW, et al. Expression of myxovirus-resistance protein A: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol Appl Neurobiol 2019;45(4):410–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wienke J, Pachman LM, Morgan GA, et al. Endothelial and inflammation biomarker profiles at diagnosis reflecting clinical heterogeneity and serving as a prognostic tool for treatment response in two independent cohorts of patients with juvenile dermatomyositis. Arthritis Rheumatol 2020;72(7):1214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kishi T, Chipman J, Evereklian M, et al. Endothelial activation markers as disease activity and damage measures in juvenile dermatomyositis. J Rheumatol 2020;47(7):1011–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim E, Cook-Mills J, Morgan G, et al. Increased expression of vascular cell adhesion molecule 1 in muscle biopsy samples from juvenile dermatomyositis patients with short duration of untreated disease is regulated by miR-126. Arthritis Rheum 2012;64(11):3809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wienke J, Mertens JS, Garcia S, Lim J, Wijngaarde CA, Yeo JG, Meyer A, van den Hoogen LL, Tekstra J, Hoogendijk JE, Otten HG, Fritsch-Stork RDE, de Jager W, Seyger MMB, Thurlings RM, de Jong EMGJ, van der Kooi AJ, van der Pol WL; Dutch Juvenile Myositis Consortium, Arkachaisri T, Radstake TRDJ, van Royen-Kerkhof A, van Wijk F. Biomarker profiles of endothelial activation and dysfunction in rare systemic autoimmune diseases: implications for cardiovascular risk. Rheumatology (Oxford). 2021. Feb 1;60(2):785–801. [DOI] [PubMed] [Google Scholar]
- 73.Piper CJM, Wilkinson MGL, Deakin CT, et al. CD19(+)CD24(hi)CD38(hi) B cells are expanded in juvenile dermatomyositis and exhibit a pro-inflammatory phenotype after activation through toll-like receptor 7 and interferon-alpha. Front Immunol 2018;9:1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lopez De Padilla CM, McNallan KT, Crowson CS, et al. BAFF expression correlates with idiopathic inflammatory myopathy disease activity measures and autoantibodies. J Rheumatol 2013;40(3):294–302. [DOI] [PubMed] [Google Scholar]
- 75.Morita R, Schmitt N, Bentebibel SE, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011;34(1):108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vercoulen Y, Bellutti Enders F, Meerding J, et al. Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood. PLoS One 2014;9(8):e105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huber AM, Kim S, Reed AM, et al. Childhood arthritis and rheumatology research alliance consensus clinical treatment plans for juvenile dermatomyositis with persistent skin rash. J Rheumatol 2017;44(1):110–6. [DOI] [PubMed] [Google Scholar]
- 78.Kim S, Kahn P, Robinson AB, et al. Childhood Arthritis and Rheumatology Research Alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatr Rheumatol Online J 2017;15(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huber AM, Giannini EH, Bowyer SL, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res (Hoboken) 2010;62(2):219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bellutti Enders F, Bader-Meunier B, Baildam E, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017;76(2):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hinze CH, Oommen PT, Dressler F, et al. Development of practice and consensus-based strategies including a treat-to-target approach for the management of moderate and severe juvenile dermatomyositis in Germany and Austria. Pediatr Rheumatol Online J 2018;16(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 2013;65(2):314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aggarwal R, Bandos A, Reed AM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile Dermatomyositis and adult polymyositis. Arthritis Rheumatol 2014;66(3):740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ruperto N, Pistorio A, Oliveira S, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. The Lancet 2016;387(10019):671–8. [DOI] [PubMed] [Google Scholar]
- 85.Lam CG, Manlhiot C, Pullenayegum EM, et al. Efficacy of intravenous Ig therapy in juvenile dermatomyositis. Ann Rheum Dis 2011;70(12):2089–94. [DOI] [PubMed] [Google Scholar]
- 86.Dagher R, Desjonquères M, Duquesne A, et al. Mycophenolate mofetil in juvenile dermatomyositis: a case series. Rheumatol Int 2012;32(3):711–6. [DOI] [PubMed] [Google Scholar]
- 87.Kishi T, Miyamae T, Hara R, et al. Clinical analysis of 50 children with juvenile dermatomyositis. Mod Rheumatol 2013;23(2):311–7. [DOI] [PubMed] [Google Scholar]
- 88.Deakin C, Campanilho-Marques R, Simou S, et al. Efficacy and safety of cyclophosphamide treatment in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis Rheumatol 2018;70(5):785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2014. May;66(5):783–7. [DOI] [PubMed] [Google Scholar]
- 90.Liu SW, Velez NF, Lam C, Femia A, Granter SR, Townsend HB, Vleugels RA. Dermatomyositis induced by anti-tumor necrosis factor in a patient with juvenile idiopathic arthritis. JAMA Dermatol. 2013. Oct;149(10):1204–8. [DOI] [PubMed] [Google Scholar]
- 91.Campanilho-Marques R, Deakin CT, Simou S, et al. Retrospective analysis of infliximab and adalimumab treatment in a large cohort of juvenile dermatomyositis patients. Arthritis Res Ther 2020;22(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Boulter E, Beard L, Ryder C et al. Effectiveness of anti-TNF-α agents in the treatment of refractory juvenile dermatomyositis. Pediatr Rheumatol 9, O29 (2011) [Google Scholar]
- 93.Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford). 2008. Jun;47(6):877–80. [DOI] [PubMed] [Google Scholar]
- 94.Sukumaran S, Vijayan V. Abatacept in the Treatment of juvenile dermatomyositis-associated calcifications in a 16-year-old girl. Case Rep Rheumatol 2020; 2020:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim H, Dill S, O’Brien M, et al. Janus kinase (JAK) inhibition with baricitinib in refractory juvenile dermatomyositis Annals of the Rheumatic Diseases 2021;80:406–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Papadopoulou C, Hong Y, Omoyinmi E, Brogan PA, Eleftheriou D. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain. 2019. Mar 1;142(3):e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aeschlimann FA, Frémond ML, Duffy D, Rice GI, Charuel JL, Bondet V, Saire E, Neven B, Bodemer C, Balu L, Gitiaux C, Crow YJ, Bader-Meunier B. A child with severe juvenile dermatomyositis treated with ruxolitinib. Brain. 2018. Nov 1;141(11):e80. [DOI] [PubMed] [Google Scholar]
- 98.Yu Z, Wang L, Quan M, Zhang T, Song H. Successful management with Janus kinase inhibitor tofacitinib in refractory juvenile dermatomyositis: a pilot study and literature review. Rheumatology (Oxford). 2021. Apr 6;60(4):1700–1707. [DOI] [PubMed] [Google Scholar]
- 99.Ding Y, Huang B, Wang Y, Hou J, Chi Y, Zhou Z, Li J. Janus kinase inhibitor significantly improved rash and muscle strength in juvenile dermatomyositis. Ann Rheum Dis. 2020. Oct 28;80(4):543–5. doi: 10.1136/annrheumdis-2020-218582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levy DM, Bingham CA, Kahn PJ, et al. Favorable outcome of juvenile dermatomyositis treated without systemic corticosteroids. J Pediatr 2010;156(2):302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mathiesen PR, Ørngreen MC, Vissing J, Andersen LB, Herlin T, Nielsen S. Aerobic fitness after JDM--a long-term follow-up study. Rheumatology (Oxford). 2013. Feb;52(2):287–95. [DOI] [PubMed] [Google Scholar]
- 102.Berntsen KS, Tollisen A, Schwartz T, et al. Submaximal exercise capacity in juvenile dermatomyositis after longterm disease: the contribution of muscle, lung, and heart involvement. J Rheumatol 2017;44(6):827–34. [DOI] [PubMed] [Google Scholar]
- 103.Omori C, Prado DML, Gualano B, et al. Responsiveness to exercise training in juvenile dermatomyositis: a twin case study. BMC Musculoskelet Disord 2010; 11:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Omori CH, Silva CA, Sallum AM, Rodrigues Pereira RM, Lúciade Sá Pinto A, Roschel H, Gualano B. Exercise training in juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2012. Aug;64(8):1186–94. [DOI] [PubMed] [Google Scholar]
- 105.Riisager M, Mathiesen PR, Vissing J, Preisler N, Ørngreen MC. Aerobic training in persons who have recovered from juvenile dermatomyositis. Neuromuscul Disord. 2013. Dec;23(12):962–8. [DOI] [PubMed] [Google Scholar]
- 106.Samhan A, Mohamed N, Elnaggar R, et al. Assessment of the clinical effects of aquatic-based exercises in the treatment of children with juvenile dermatomyositis: a 2×2 controlled-crossover trial. Arch Rheumatol 2020;35(1):97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Habers EA, Van Brussel M, Langbroek-Amersfoort AC, et al. Design of the muscles in motion study: a randomized controlled trial to evaluate the efficacy and feasibility of an individually tailored home-based exercise training program for children and adolescents with juvenile dermatomyositis. BMC Musculoskelet Disord 2012;13:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ravelli A, Trail L, Ferrari C, et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res 2010;62(1):63–72. [DOI] [PubMed] [Google Scholar]
- 109.Tsaltskan V, Aldous A, Serafi S, et al. Long-term outcomes in Juvenile Myositis patients. Semin Arthritis Rheum 2020;50(1):149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanner H, Kirkhus E, Merckoll E, et al. Long-term muscular outcome and predisposing and prognostic factors in juvenile dermatomyositis: a case-control study. Arthritis Care Res 2010;62(8):1103–11. [DOI] [PubMed] [Google Scholar]
- 111.Sanner H, Sjaastad I, Flatø B. Disease activity and prognostic factors in juvenile dermatomyositis: a long-term follow-up study applying the Paediatric Rheumatology International Trials Organization criteria for inactive disease and the myositis disease activity assessment tool. Rheumatology (United Kingdom) 2014;53(9):1578–85. [DOI] [PubMed] [Google Scholar]
- 112.Mathiesen PR, Zak M, Herlin T, et al. Clinical features and outcome in a Danish cohort of juvenile dermatomyositis patients. Clin Exp Rheumatol 2010;28(5): 782–9. [PubMed] [Google Scholar]
- 113.Sharma A, Gupta A, Rawat A, et al. Long-term outcome in children with juvenile dermatomyositis: a single-center study from north India. Int J Rheum Dis 2020; 23(3):392–6. [DOI] [PubMed] [Google Scholar]
- 114.Okong’o LO, Esser M, Wilmshurst J, et al. Characteristics and outcome of children with juvenile dermatomyositis in Cape Town: a cross-sectional study. Pediatr Rheumatol 2016;14(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Malek A, Raeeskarami SR, Ziaee V, et al. Clinical course and outcomes of Iranian children with juvenile dermatomyositis and polymyositis. Clin Rheumatol 2014;33(8):1113–8. [DOI] [PubMed] [Google Scholar]
- 116.Singh S, Suri D, Aulakh R, et al. Mortality in children with juvenile dermatomyositis: two decades of experience from a single tertiary care centre in North India. Clin Rheumatol 2014;33(11):1675–9. [DOI] [PubMed] [Google Scholar]
- 117.Bitnum S, Daeschner CW Jr, Travis LB, et al. Dermatomyositis. J Pediatr 1964; 64:101–31. [DOI] [PubMed] [Google Scholar]
- 118.Huber AM, Mamyrova G, Lachenbruch PA, et al. Early illness features associated with mortality in the juvenile idiopathic inflammatory myopathies. Arthritis Care Res (Hoboken) 2014;66(5):732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Patwardhan A, Rennebohm R, Dvorchik I, et al. Is juvenile dermatomyositis a different disease in children up to three years of age at onset than in children above three years at onset? A retrospective review of 23 years of a single center’s experience. Pediatr Rheumatol 2012;10(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ostrowski RA, Sullivan CL, Seshadri R, et al. Association of normal nailfold end row loop numbers with a shorter duration of untreated disease in children with juvenile dermatomyositis. Arthritis Rheum 2010;62(5):1533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]