

Abstract
L.P. Li, J.W. Liang and H.J. Fu. An update on the association between traumatic brain injury and Alzheimer’s disease : focus on tau pathology and synaptic dysfunction. NEUROSCI BIOBEHAV REVXXX-XXX,2020.-Traumatic brain injury (TBI) and Alzheimer’s disease (AD) are devastating conditions that have long-term consequences on individual’s cognitive functions. Although TBI has been considered a risk factor for the development of AD, the link between TBI and AD is still in debate. Aggregation of hyperphosphorylated tau and intercorrelated synaptic dysfunction, two key pathological elements in both TBI and AD, play a pivotal role in mediating neurodegeneration and cognitive deficits, providing a mechanistic link between these two diseases. In the first part of this review, we analyze the experimental literatures on tau pathology in various TBI models and review the distribution, biological features and mechanisms of tau pathology following TBI with implications in AD pathogenesis. In the second part, we review evidences of TBI-mediated structural and functional impairments in synapses, with a focus on the overlapped mechanisms underlying synaptic abnormalities in both TBI and AD. Finally, future perspectives are proposed for uncovering the complex relationship between TBI and neurodegeneration, and developing potential therapeutic avenues for alleviating cognitive deficits after TBI.
Keywords: TRAUMATIC BRAIN INJURY, ALZHEIMER’S DISEASE, ANIMAL MODELS, TAU PATHOLOGY, SYNAPTIC DYSFUNCTION
1. Introduction
Traumatic brain injury (TBI) is a serious public health concern that affects an estimated 2.8 million people each year in the United States and represents a leading cause of death and disability worldwide (Peterson et al., 2019). Previous epidemiological studies have demonstrated an increased risk for long-term dementia following TBI, including Alzheimer’s disease (AD), the most frequent type of dementia in the elderly (Barnes et al., 2018; Fann et al., 2018; Plassman and Grafman, 2015). The link between TBI and AD has been supported by the identification of AD-like pathologies such as abnormal tau aggregates and Aβaccumulation following injuries in animal models and TBI survivors. However, recent data has brought challenges for reaching a general consensus on this link (Sugarman et al., 2019; Weiner et al., 2017), highlighting the need for a better understanding of the mechanistic underpinnings following TBI and how they related to the development of neurodegenerative process.
Several mechanisms have been proposed to link a history of TBI to the development of AD later in life, such as oxidative stress (Abdul-Muneer et al., 2015), cerebrovascular damage (Ramos-Cejudo et al., 2018) and persistent neuroinflammation (Kokiko-Cochran and Godbout, 2018). Recently, the pathophysiological link that has received increasing attention is the accumulation and spreading of tau pathology following TBI. Despite a wealth of data reporting tau changes following TBI, the type of tau pathology induced by TBI at different stages following the injury and how it is involved in the development of AD have not been systemically reviewed or addressed. In the first part of this critical review, we comprehensively analyze the available experimental literature on the tau pathology following TBI and present the evidence of molecular mechanisms linking TBI to AD. Long-term deficits in cognitive function is an overarching consequence of TBI that may be attributed to the impairments of synaptic function, which is also a neuropathological hallmark in AD (Forner et al., 2017). It remains unclear whether synaptic dysfunction in these two diseases share common features and/or are mediated by common mechanisms. In the second part, we review the current knowledge of TBI-induced synaptic dysfunction and provide evidence of the underlying molecular mechanisms. In particular, we discuss how the molecular pathways of TBI-induced synaptic failure overlap with those of AD. Finally, we proposed several future perspectives on uncovering the complex relationship between TBI and AD by taking advantage of cutting-edge technologies and newly developed TBI models. Currently, there are few pharmacological treatment options to prevent or alleviate cognitive impairments following TBI in humans (Janowitz and Menon, 2010). Although several pre-clinical treatment approaches, including anti-neuroinflammation, targeting neuromodulatory transmitter systems, targeting cell death pathways, stem cells transplantation and rehabilitative strategies, have shown beneficial effects on cognitive functions in animal models of TBI, translation of these preclinical studies to clinical practice has failed (Jarrahi et al., 2020; Kline et al., 2016). Collective evidence provided in this review will not only shed lights on the link between TBI and AD and/or AD-related dementia but also provide insights in developing new therapeutic strategies for the alleviation and/or reversion of cognitive impairments following TBI, or even in AD itself.
2. Tau pathology following TBI: experimental evidence, molecular mechanisms and implications in AD
Following TBI, tau undergoes several post-translational modifications, including cleavage and phosphorylation. Glutamate-mediated excitotoxicity and the resulted intracellular Ca2+ overload triggers a number of important downstream pathways, including mitochondrial oxidative stress and activation of proteases and kinases, which are thought to be key initiators of tau pathology following TBI (Walker and Tesco, 2013). Small fragments of tau (cleaved tau) and hyperphosphorylated tau are involved in the subsequent cascades of tau processing that eventually lead to tau aggregates and tangles. In this section, we review the TBI-induced molecular and conformational changes of tau in preclinical studies with various TBI models (summarized in Table 1), and discuss how these changes may be related to the tau pathology and neurodegeneration in AD.
Table 1.
Animal models of traumatic brain injury (TBI) and tau pathology
| TBI Model | Duration | Animal Model | Age/Weight | Tau Pathology | References |
|---|---|---|---|---|---|
| CCI (S) | Acute | Mouse (WT) | N/A | Increased cleaved tau | (Wu et al., 2020) |
| Mouse (C57BL/6J) | 2-3 mo | Increased t-tau, p-tau (S202/T205/T231/S416/T181) and oligomeric tau (T22) | (Wang and Han, 2018a; Wang et al., 2017a) | ||
| Mouse (C57BL/6J)) | 2-3 mo | Increased p-tau (S404) but not p-tau (T205/S262) | (Zhao et al., 2017c) | ||
| Mouse (Tau P301S) | 3 mo | Increased insoluble tau and p-tau (S202/T205) | (Edwards III et al., 2020) | ||
| Mouse (Tau P301L) | 6 mo | Increased t-tau and p-tau (S396/404/199) | (Tran et al., 2011b) | ||
| Mouse (3xTg-AD) | 6 mo | Increased t-tau | (Tran et al., 2011b) | ||
| Mouse (3xTg-AD) | 5-7 mo | Increased p-tau (S199/S422/S202/T205/T231/T212/S214/S396/S404) | (Tran et al., 2011a) | ||
| Rat (Sprague Dawley) | 230-300 g | Increased cleaved tau and p-tau (S199/202) | (Begum et al., 2014; Gabbita et al., 2005; Glushakova et al., 2018; Liu et al., 2010) | ||
|
|
|||||
| Subacute | Rat (Sprague Dawley) | 230-300 g | Increased cleaved tau and p-tau (S199/202) | (Begum et al., 2014; Glushakova et al., 2018) | |
| Rat (Sprague Dawley) | 280-300 g | No difference detected in cleaved tau | (Liu et al., 2010) | ||
|
|
|||||
| Chronic | Mouse (WT) | N/A | Increased cleaved Tau (N368), t-tau, p-tau (S202/T205), and NFTs (ThS/AT8) | (Wu et al., 2020) | |
| Mouse (WT) | 2 mo | No p-tau (S202/T205) detected | (Edwards III et al., 2020) | ||
| Mouse (C57BL/6J) | 8-9 wks | Increased t-tau and p-tau (S202/T205/S396/S404/T231) | (Zanier et al., 2018) | ||
| Mouse (C57BL/6J) | 3 mo | No difference detected in t-tau, p-tau (S202/T231/S396/S404), or conformational changes in tau (MC1) | (Mouzon et al., 2014) | ||
| Mouse (Tau P301S) | 3 mo | Increased p-tau (S202/T205) and NFT-like structure (AT8) | (Edwards III et al., 2020) | ||
| Mouse (3xTg-AD) | N/A | Increased cleaved Tau (N368), p-tau (S202/T205/T212/S214), and NFTs (ThS/AT8) | (Wu et al., 2020) | ||
| Rat (Sprague Dawley) | 230-300 g | Increased cleaved tau | (Glushakova et al., 2018) | ||
| Rat (Sprague Dawley) | 2 mo | Increased p-tau (S202/T205) and oligomeric tau (T22) | (Acosta et al., 2017) | ||
|
| |||||
| WD (S) | Acute | Mouse (C57BL/6J) | 2-3 mo | Increased cis p-tau, no p-tau (S202/T205/T212/S214) or oligomeric tau (T22) detected | (Albayram et al., 2017; Kondo et al., 2015) |
| Rat (Sprague Dawley) | 280-400 g | Increased p-tau (T231/S396), the ratios of p-tau (S396) : t-tau and p-tau (T231) : t-tau, and oligomeric tau (T22) | (Arun et al., 2015; Collins-Praino et al., 2018; Lv et al., 2014; McAteer et al., 2016) | ||
| Rat (Sprague Dawley) | 250-280 g | No difference detected in p-tau (S199/202/396/404) | (Wang et al., 2017b) | ||
|
|
|||||
| Subacute | Mouse (C57BL/6J) | 2-3 mo | Increased cis p-tau, no p-tau (S202/T205/T212/S214) or oligomeric tau (T22) detected | (Albayram et al., 2017; Kondo et al., 2015) | |
| Rat (Sprague Dawley) | 350-400 g | Increased ratio of p-tau (T231) : t-tau | (Collins-Praino et al., 2018) | ||
|
|
|||||
| Chronic | Mouse (C57BL/6J) | 2-3 mo | Increased cis p-tau, p-tau (S202/T205/T212/S214), oligomeric tau (T22) and NFTs (ThS) | (Albayram et al., 2017; Kondo et al., 2015) | |
| Rat (Sprague Dawley) | 350-400 g | Increased p-tau (T231) and the ratio of p-tau (T231) : t-tau | (Collins-Praino et al., 2018; McAteer et al., 2016) | ||
|
| |||||
| WD (R) | Acute | Mouse (C57BL/6J) | 2-3 mo | Increased cis p-tau | (Kondo et al., 2015) |
| Mouse (C57BL/6J) | 5-6 wks | No changes in t-tau or p-tau (S202/T205/S396/S404/S422/S202) | (Xu et al., 2016) | ||
|
|
|||||
| Rat (Sprague Dawley) | 300-400 g | Increased t-tau and p-tau (T231); increased ratio of p-tau (T231) : t-tau | (Collins-Praino et al., 2018; McAteer et al., 2016; Siahaan et al., 2018) | ||
|
|
|||||
| Subacute | Mouse (BALB/c) | 5-6 wks | Increased t-tau | (Nogueira et al., 2018) | |
|
|
|||||
| Chronic | Mouse (C57BL/6J) | 2-3 mo | Increased cis p-tau, p-tau (S202/T205/T212/S214/S396/404), oligomeric tau (T22) and NFTs (ThS) | (Albayram et al., 2017; Kondo et al., 2015) | |
| Mouse (C57BL/6J) | 3 mo | No changes in t-tau or p-tau (S202/T205/S396/S404/T181) | (Mannix et al., 2013) | ||
| Mouse (C57BL/6J) | 5-6 wks | No changes in t-tau or p-tau (S202/T205/S422) | (Xu et al., 2016) | ||
| Mouse (hTau) | N/A | Increased p-tau (S422) | (Xu et al., 2015) | ||
| Mouse (Tau P301S) | 5 wks | Increased p-tau (S422) | (Xu et al., 2015) | ||
| Rat (Sprague Dawley) | 350-400 g | Increased ratio of p-tau (T231) : t-tau and p-tau (T231) | (Collins-Praino et al., 2018; McAteer et al., 2016) | ||
|
| |||||
| Blast (S) | Acute | Mouse (C57BL/6J) | 2-3 mo | Decreased t-tau; increased cis p-tau and p-tau (T181) | (Chen et al., 2018b; Kondo et al., 2015) |
| Rat (Sprague Dawley) | 300-500 g | Increased oligomeric tau (T22) and p-tau (S396) | (Arun et al., 2015; Gerson et al., 2016) | ||
|
|
|||||
| Subacute | Mouse (C57BL/6J) | 2-5 mo | Increased p-tau (S202/S205/T181); increased cis p-tau; increased oligomeric tau (T22 and TOMA1); Increased ratios of pS202/T181/S396/S212/T214/cleaved tau : t-tau | (Bittar et al., 2019; Goldstein et al., 2012; Huber et al., 2013; Kondo et al., 2015) | |
| Mouse (C57BL/6J) | 2-3 mo | No changes in t-tau and decrease in p-tau (T181) | (Chen et al., 2018b) | ||
|
|
|||||
| Chronic | Mouse (C57BL/6J) | 2-3 mo | No changes in t-tau and p-tau (T181); increased cis p-tau | (Chen et al., 2018b; Kondo et al., 2015) | |
|
| |||||
| Blast (R) | Acute | Rat (Long-Evans) | 360-400 g | Increased t-tau, p-tau (S202/T205) and oligomeric tau (T22) | (Du et al., 2016) |
|
|
|||||
| Subacute | Mouse (C57BL/6J) | 5 mo | Increased oligomeric tau (T22 and TOMA1) | (Bittar et al., 2019) | |
| Rat (Long-Evans) | 360-400 g | Increased t-tau and oligomeric tau (T22); no difference in p-tau (S202/T205) | (Du et al., 2016) | ||
|
| |||||
| CHI* (S) | Subacute | Mouse (hTau) | 18 mo | No changes in p-tau (S202/S396/S404/T231) | (Ojo et al., 2013) |
|
| |||||
| CHI* (R) | Acute | Mouse (WT) | 3.5 mo | Increased p-tau (S202) | (Rubenstein et al., 2017) |
| Mouse (C57BL/6J) | 2-3 mo | No changes in p-tau (S396/S404) | (Bolton and Saatman, 2014) | ||
| Mouse (hTau/PS1) | 3-3.5 mo | Increased t-tau and p-tau (T231/S202) | (Rubenstein et al., 2019) | ||
| Mouse (hTau) | 3/12-13 mo | Increased t-tau and p-tau (T231) | (Mouzon et al., 2018; Ojo et al., 2016) | ||
| Mouse (3xTg-AD) | 4-6 mo | No changes in t-tau; increased p-tau (S396/S404/T205/S262); no changes in p-tau (S199/T212) | (Hu et al., 2018) | ||
| Mouse (3xTg-AD) | 18 mo | No changes in p-tau (T181/S202/T205/S199/T231/S422/S396/S404) or t-tau | (Winston et al., 2016) | ||
|
|
|||||
| Subacute | Mouse (hTau) | 18 mo | Increased p-tau (S202/S396/S404/T231) and NFTs (Gallyas silver) | (Ojoetal., 2013) | |
| Mouse (WT) | 3.5 mo | Increased p-tau (S202); no changes in t-tau | (Rubenstein et al.. 2017) | ||
| Mouse (C57BL/6J) | 2-3 mo | No changes in p-tau (S396/S404) | (Bolton and Saatman, 2014) | ||
| Mouse (3xTg-AD) | 18 mo | No changes in p-tau (T181/S202/T205/S199/T231/S422/S396/S404) or t-tau | (Winston et al., 2016) | ||
| Mouse (hTau/PS1) | 3-3.5 mo | Increased t-tau and p-tau (T231/S202) | (Rubenstein et al., 2019) | ||
|
|
|||||
| Chronic | Mouse (hTau/PS1) | 3-3.5 mo | Increased t-tau and p-tau (T231/S202) | (Rubenstein et al., 2019) | |
| Mouse (hTau) | 3 mo | Increased t-tau, p-tau (T231) and oligomeric tau (TOC1); no changes in p-tau (S396/S404/S202) or conformational tau (MC1/TNT) | (Ojo et al., 2016) | ||
| Mouse (WT) | 12 mo | No increases in p-tau (S396/S404/S262/T231) or NFTs | (Yoshiyama et al., 2005) | ||
| Mouse (T44 Tau) | 12 mo | Only one mouse showed increased p-tau (S396/S404/S262/T231) and extensive NFTs (Gallyas silver/ThS) | (Yoshiyama et al., 2005) | ||
|
| |||||
| FPI (S) | Acute | Rat (Sprague Dawley) | 400-500 g | Increased oligomeric tau (T22) and p-tau (T231/S202/T205); no changes in t-tau | (Gerson et al., 2016; Hawkins et al., 2013) |
| Rat (Long-Evans) | 250-300 g | Increased ratios of p-tau (S198) : t-tau and p-tau (S262): t-tau | (Shultz et al., 2015; Tan et al., 2016) | ||
|
|
|||||
| Subacute | Rat (Long-Evans) | 250-300 g | No changes in ratios of p-tau (S198) : t-tau or p-tau (S262) : t-tau | (Tan et al., 2016) | |
| Rat (Sprague Dawley) | 400-500 g | Increased oligomeric tau (T22) and p-tau (S202/T205) | (Hawkins et al., 2013) | ||
|
|
|||||
| Chronic | Rat (Long-Evans) | 250-300 g | Increased ratio of p-tau (S198) : t-tau but not of p-tau (S262) : t-tau | (Shultz et al., 2015) | |
| Rat (Sprague Dawley) | 3 mo | Increased p-tau (S202/T205) | (Hoshino et al., 1998) | ||
|
| |||||
| FPI (R) | Acute | Rat (Long-Evans) | 250-300 g | Increased ratios of p-tau (S198) : t-tau and p-tau (S262) : t-tau | (Tan et al., 2016) |
|
|
|||||
| Subacute | Rat (Long-Evans) | 250-300 g | Increased ratio of p-tau (S198) : t-tau but not of p-tau (S262) : t-tau | (Tan et al., 2016) | |
|
| |||||
| PBBI (S) | Acute | Rat (Sprague Dawley) | N/A | Decreased full-length tau and increased cleaved tau | (Cartagena et al., 2016) |
| Epidural compression (S) | Acute | Rat (Wistar) | 6-7 wks | Increased p-tau (T231/S262) | (Chen et al., 2010) |
| Compressed Gas CCI (S) | Acute | Mouse (Tg2576) | N/A | Increased ration of p-tau (T181) : t-tau | (Sawmiller et al., 2014) |
| “Hit & Run” (S) | Subacute | Mouse (C57BL/6J) | 8-12 wks | Increased p-tau (S396/T205/T231/S202/T205) | (Iliff et al., 2014) |
| CHIMERA (R) | Acute | Mouse (C57BL/6J) | 4 mo | Increased p-tau (S202/S396/S404/T231); no changes in t-tau | (Namjoshi et al., 2014) |
Note: Acute: 0 - 7 days, Subacute: 8 days - 1 month, Chronic: over 1 month; CCI, Controlled Cortical Impact; WD, Weight Drop; CHI, Closed Head Injury;
CHI was inflicted via an impact tip;
FPI, Fluid Percussion Injury; PBBI, Penetrating ballistic-like brain injury; CHIMERA, Closed Head Injury Model of Engineered Rotational Acceleration; S, Single; R, Repetitive; WT, wild-type littermate control mice; Tg, Transgenic; 3xTg-AD, Alzheimer’s disease (AD) model mice carrying SweAPP, MAPT P301L and PSEN1 M146V transgenes; Tau P301S, mice expressing the P301S mutant human tau gene; Tau P301L, mice expressing the P301L mutant human tau gene; hTau, mice expressing human wild-type tau on a null mouse tau background; hTau/PS1, mice expressing both wild-type human tau and the presenilin-1 (PS1) M146L human mutation on a null mouse tau background; T44 Tau, mice expressing the shortest form of human wild-type tau isoform; Tg2576, mice carrying a mutant form of amyloid precursor protein (APP), SweAPP; mo, months; wks, weeks; N/A, not applicable; t-tau, total tau; p-tau, phosphorylated tau; NFTs, neurofibrillary tangles; ThS: Thioflavin S.
2.1. Tau cleavage
Previous studies have shown that caspase-3-cleaved form of tau, TauC3, is exhibited in the brains of AD patients at the early stage of disease (Rissman et al., 2004). TauC3 can promote the oligomerization and fibrillization of tau in vitro and in vivo (Chung et al., 2001; de Calignon et al., 2010; Lin et al., 2011), accelerating neurodegeneration and cognitive deficits in rodents (Kim et al., 2016). Tau cleavage at N368 mediated by asparaginyl endopeptidase (AEP), a lysosomal cysteine protease, has also been detected in the human AD brains (Zhang et al., 2014), and the fragmented tau has been shown to be neurotoxic and possess the ability to form paired helical filaments (PHFs) (Zhang et al., 2014). AEP knockout can prevent tau cleavage and ameliorate tau pathology in P301S tau transgenic mice (Zhang et al., 2014). In addition, a more recent study has identified a role for novel tau fragments mediated by calpain in the disease progression of AD (Chen et al., 2018a). More interestingly, evidence has shown that cleaved tau may actively participate in tau misfolding and function similarly to prion protein to accelerate the formation of neurofibrillary tangles (NFTs) (Zilka et al., 2012). These findings collectively highlight the important role of tau cleavage in mediating AD-associated tau pathology.
Previous clinical studies have found an acute and significant increase of cleaved tau in TBI patients compared to healthy controls, and the level of cleaved tau in cerebrospinal fluid (CSF) and/or serum correlates with the severity of injury and predicts long-term clinical outcomes (Shaw et al., 2002; Zemlan et al., 2002). In consistency with these findings, altered tau cleavage has also been observed in rodents following various TBI models. A study using controlled cortical impact (CCI) model of different severities (mild, moderate and severe) in rats showed a severity-dependent increase of cleaved tau in the cortex and the hippocampus 3 days after injury compared with sham controls (Gabbita et al., 2005). The increase of cleaved tau was displayed as early as 6 h and was manifested in a time-dependent manner with the peak level observed 7 days following TBI (Gabbita et al., 2005). A later study found that severe CCI-induced increase of cleaved tau in the rat cortex was mediated by the activation of two major proteases, calpain and caspase-3, as determined by 35 kDa- and 45 kDa-tau breakdown product, respectively (Liu et al., 2010). A treatment of calpain-specific inhibitor SNJ-1945 significantly inhibited the formation of 35 kDa-tau fragment (Liu et al., 2010). In support of the caspase-3-mediated tau cleavage following TBI, a recent study detected a chronic activation of caspase-3 following CCI injury, which was accompanied by increase of caspase-3-cleaved tau observed up to 3 months after CCI (Glushakova et al., 2018). Utilizing a rat model of penetrating ballistic-like brain injury (PBBI), Amadoro et al. demonstrated that the full-length tau was substantially decreased 3 days and 7 days post-PBBI. But a 22-kDa tau fragment (tau22), a similarly sized tau fragment as that seen in the brains of AD patients (Amadoro et al., 2010), was increased early after injury and remains increased over 7 days post-PBBI (Cartagena et al., 2016). Interestingly, the 20-22 kDa NH2-trunicated tau fragments are enriched in mitochondria, suggesting their potential role in regulating mitochondrial function. Consistently, a recent study has found that caspase-mediated tau fragment impaired mitochondrial dynamics, which were also seen in AD (Perez et al., 2018). A recent study conducted by Wu and colleagues showed that AEP-mediated cleavage of tau at N368 was enhanced acutely post-CCI and was still evident months following injury in both wild-type (WT) and 3xTg AD mice, accompanied by hyperphosphorylation of tau and formation of NFTs, both of which are robustly diminished when AEP is depleted (Wu et al., 2020). These data indicates a critical role of AEP-mediated tau cleavage in the development of tau pathology, providing a potential link between TBI exposure and AD pathogenesis.
2.2. Tau hyperphosphorylation (P-tau)
As an axonal microtubule (MT)-associated protein, tau has a central role in regulating MT dynamics and stability (Hanger et al., 2009). Aberrant P-tau promotes the detachment of tau from the MTs and thus destabilizes MTs, causing deficits in axonal transport and neuronal functions (Hanger et al., 2009). When detached from MTs, hyperphosphorylated tau is prone to self-polymerize into tau oligomers, which may further aggregate into PHFs (Haase et al., 2004; Maeda et al., 2007). Assembly of PHFs gives rise to NFTs (Rankin et al., 2008), which is a characteristic AD brain pathology existing in the somas and dendrites of affected neurons. Accumulation of abnormally hyperphosphorylated tau is thus considered as one of the early and triggering steps in the process of tangle formation.
TBI has been shown to result in activation and accumulation of a series of kinases, such as c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases (ERK), glycogen synthase kinase 3-β(GSK3-β, and cycline-dependent kinase 5 (CDK5). These kinases cause hyperphosphorylation of tau at various sites, with Thr205/Thr212/Thr231 and Ser199/Ser202/Ser262/Ser396/Ser422 being the most reported phosphorylated sites following TBI (discussed below). Previous studies have demonstrated that Thr212/Thr231/Ser262 are the main sites that prevent tau binding to MTs, and abnormal P-tau at these sites can trigger caspase-3 mediated neuronal death (Alonso et al., 2010). P-tau at sites Ser202/Thr205 are recognized by AT8 antibody, and AT8-positive staining is commonly used as a marker of aberrant P-tau in the brains of AD patients (Augustinack et al., 2002; Goedert et al., 1995). Different stages of NFTs featured with differential hyperphosphorylation sites have been revealed in a previous study using immunostaining assay. In human AD brains, pretangles are observed with pSer262, pThr231 and pThr153 tau antibodies. While intraneuronal NFTs are especially stained with 12E8 (pSer262/pSer356), pThr175/181, pSer214 and pSer422 tau antibodies, extracellular NFTs are prominently recognized by AT8, AT100 (pThr212/pSer214) and PHF1 (pSer396/pSer404) tau antibodies, which also stain intracellular NFTs (Augustinack et al., 2002).
2.2.1. P-tau following single moderate-to-severe TBI (sTBI)
In preclinical studies with CCI model, increased P-tau at various sites have been reported as early as hours and through months following TBI. For example, it has been shown that P-tau at Ser404 was increased 24 hours after CCI and remained elevated up to 4 weeks post-CCI in various brain regions, including parietal cortex, prefrontal cortex, hippocampus and thalamus (Zhao et al., 2017a; Zhao et al., 2017b; Zhao et al., 2017c). P-tau at Ser404 has been shown to facilitate the exposure of microtubule-binding domain (MBD) sites to several kinases, therefore inducing tau detachment from MTs and promoting tau self-aggregation (Bibow et al., 2011; Steinhilb et al., 2007). P-tau at Ser404 also associates with PHFs in the AD brains (Otvos Jr et al., 1994). Increased P-tau at Ser404 is mediated by upregulation of adenosine A2A receptors (A2AR) (Zhao et al., 2017a). Inhibition of A2AR attenuated P-tau at Ser404, probably through 1) decreasing the activity of tau kinases, GSK-3γand protein kinase A (PKA) (Zhao et al., 2017a) and 2) restoring aquaporin-4 (AQP4) polarity (Zhao et al., 2017b), which is critical for the normal functioning of ‘glymphatic system’ that is used for brain-wide clearance of interstitial solutes (Iliff et al., 2014). In agreement of this finding, a study using the Hit & Run model, a modified CCI which only needs a very short period of anesthesia and minimizes the effects of isoflurane on tau phosphorylation, showed a great loss of perivascular AQP4 polarization following TBI. Furthermore, TBI-induced P-tau was exacerbated when Aqp4 gene was deleted (Iliff et al., 2014). P-tau at other sites as detected by AT8, AT180 and PHF1 have also been reported in the frontal cortex of rodents following CCI (Begum et al., 2014; Laskowitz et al., 2010; Zanier et al., 2018). Interestingly, accumulation of P-tau at Ser199/Ser202 following CCI was significantly reduced when TBI-induced endoplasmic reticulum (ER) stress was inhibited via a chronic post-TBI treatment of docosahexaenoic acid (DHA) (Begum et al., 2014), suggesting that ER stress may play a role in mediating TBI-induced tau phosphorylation.
Abnormal P-tau has also been observed in blast-induced TBI model (bTBI). A single blast significantly increased P-tau (Ser202/Thr205/Thr181) in various brain regions during the acute and subacute stages (Goldstein et al., 2012; Perez-Polo et al., 2015). Increased P-tau at Thr181 was detected as early as 3 hours post-bTBI (Chen et al., 2018b). A persistent increase of aberrant tau species (pSer396, AT8 and AT180) for at least 30 days was observed in the mouse hippocampus after single bTBI (Huber et al., 2013). Previous studies using fluid percussion injury (FPI) have revealed an important role of protein phosphatase 2A (PP2A) in the regulation of TBI-induced P-tau (Shultz et al., 2015; Tan et al., 2016). PP2A heterotrimers consist of PR55 regulatory B-subunit and represent the major tau phosphatase in the brain (Liu et al., 2005). An increase of P-tau (Ser198/Ser262) was observed in the rat cortex following FPI and was preceded by decreased expression and activity of PP2A. Post-FPI treatment of sodium selenate, a potent PR55 activator, increased PP2A activity and reduced the increased P-tau level (Shultz et al., 2015). Importantly, similar effects on PP2A and P-tau have been found in the brain cortex from human TBI patients (Shultz et al., 2015). In addition, selenate has been used in clinical trials for AD and a subtle but significant improvement in Mini-Mental Status Examination (MMSE) score was associated with the level of selenium in the cerebrospinal fluid (Cardoso et al., 2019; Malpas et al., 2016). Another protein phosphatase, tissue-nonspecific alkaline phosphatase (TNAP), also accounts for P-tau (Diaz-Hernandez et al., 2010). In both blast- and weight drop (WD)-induced TBI models, there was a significant decrease in TNAP expression and activity, accompanied by increased P-tau (Ser396) (Arun et al., 2015). Inhibited activity of TNAP may work in concert with enhanced activation of GSK-3βto increase P-tau (Ser396) following WD-induced injury (Lv et al., 2014).
To further investigate the potential causal relationship between TBI and AD-associated tau pathology, TBI-induced P-tau has been studied in AD-like mice. Evidence has shown that CCI causes an injury-severity-dependent increase in P-tau (notably at Ser199, Thr205, Thr231, Ser396, Ser422) in the ipsilateral fimbria/amygdala and contralateral CA1 regions of 3xTg AD mice (carrying SweAPP, MAPT P301L and PSEN1 M146V transgenes) compared with sham 3xTg AD mice (Tran et al., 2011a), which can be attenuated by a treatment of D-JNKil, a peptide inhibitor of JNK (Tran et al., 2012). Using a compressed gas CCI model, an increased level of P-tau (Thr181) was observed in the brain of mice carrying SweAPP mutation (Tg2567 mice) 3 days following TBI (Sawmiller et al., 2014). P-tau detected by PHF1 and pSer199 antibodies is increased acutely following moderate CCI in P301L tau transgenic mice compared with sham P301L tau mice (Tran et al., 2011b). Moderate CCI also induced a subacute increase of P-tau (Ser199, PHF1 and 12E8) and a chronic increase of P-tau (AT8) in hTau mice (express human wild-type tau) (Zhang et al., 2019c). Together, these findings suggest that TBI promotes the development of tau pathology in the presence of AD-associated mutations or overexpression of human WT tau.
2.2.2. P-tau following repetitive mild TBI (rmTBI)
Recent focus has been given to repetitive mild TBI (rmTBI) models due to the growing clinical evidence showing that aberrant P-tau has a close relationship with a history of repeated concussion (McKee and Daneshvar, 2015; McKee et al., 2013). Most rmTBI models employ closed head injury (CHI) where a tip or a weight impacts an intact skull. Recently, an improved CHI model, called CHIMERA (Closed Head Impact Model of Engineered Rotational Acceleration) has been developed to enable a better control of impact delivery. CHIMERA has been shown to be a useful model of investigating pathological and neurobehavioral consequences following rmTBI (Haar et al., 2019; Namjoshi et al., 2014). A series of preclinical studies have demonstrated an abnormal P-tau following rmTBI (Collins-Praino et al., 2018; Du et al., 2016; Haar et al., 2019; Kane et al., 2012; Luo et al., 2014; McAteer et al., 2016; Mei et al., 2018; Namjoshi et al., 2014; Petraglia et al., 2014b). For example, repeated CHI (3x, 24 h apart or 6x per day with 2 h interval last for 7 days) and blast injury (3x blasts with 1.5 min interval) significantly increased P-tau (AT8) throughout the brain regions such as cortex, hippocampus, amygdala and corpus callosum during early (7 days) and late (6 months) stages following insults (Du et al., 2016; Luo et al., 2014; Petraglia et al., 2014b). While P-tau (AT180) was significantly increased in the cortex acutely (24 h) after WD-induced single moderate severe injury or repeated mild injuries (3x, 5 days apart), a chronic increase of P-tau was only detected in mice received repeated insults (Collins-Praino et al., 2018). However, increased P-tau was not detected in other studies using the same type of injury, such as repetitive CHI (Bolton and Saatman, 2014; Mannix et al., 2013; Mouzon et al., 2014; Xu et al., 2016) and repetitive blast injury (Sosa et al., 2014). Previous studies have suggested that axonal injury plays a critical role in P-tau following TBI (McKee et al., 2014). A lack of traumatic axonal injury reported in the Mannix et al. study may explain why increased P-tau is not detected. However, this cannot explain for the absence of increased P-tau in other studies where axonal injury was observed diffusely distributed across the brain (Bolton and Saatman, 2014; Mannix et al., 2013; Mouzon et al., 2014; Sosa et al., 2014; Xu et al., 2016). Further studies are warranted to understand why P-tau is not changed following TBI where traumatic axonal injury is evident.
The repetitive CHI has also been performed in several AD-like mouse models. Using young and old hTau mice, the authors have shown an increased P-tau (Thr231, Ser202, PHF1) in the cortex and hippocampus following rmTBI, with a larger extend of increase in old hTau mice than young ones (Mouzon et al., 2018; Ojo et al., 2013). Increased P-tau at Ser202 and Thr231 has also been reported in the serum and cortex of hTau/PS1 mice following rmTBI (Rubenstein et al., 2019). However, increased P-tau was not found in the cortex of P301S tau transgenic mice following rmTBI compared with sham controls (Xu et al., 2015). This absence of difference in P-tau may be due in part to a lack of traumatic axonal injury. Studies with 3xTg AD mice has yield inconsistent results. A study using highly repetitive TBI model showed no change of P-tau at multiple sites (Thr205, Ser199, AT270, CP13, AT8, AT180 and PHF1) in the cortex of 3xTg AD mice (Winston et al., 2016), whereas another study using a more severe model found P-tau at Thr205, Ser262 and PHF1 sites in the forebrain and hippocampus of 3xTg AD mice (Hu et al., 2018). Differences in the severity of injury and the potential resultant different degrees of axonal damage may give rise to this discrepancy. Mechanistic studies showed that rmTBI-induced P-tau in 3xTg AD mice was associated with AEP activation, which indirectly inhibits PP2A by cleaving I2PP2A (inhibitor 2 of PP2A) and thus promoted P-tau (Hu et al., 2018). A recent study has replicated rbTBI in ApoE4 mice and found a significant increase of P-tau (AT8) in the hippocampus following insults (Cao et al., 2017). Down-regulation of synaptojanin 1 (synj1), a degrading enzyme of phosphoinositol phosphatase (PIP2), attenuated the increased P-tau in ApoE4 mice following rbTBI (Cao et al., 2017). It is worthy of note that the increased P-tau was only observed in the hippocampus, but not in the cortex, striatum and cerebellum in this study, suggesting a specific vulnerability of hippocampal neurons following TBI (Cao et al., 2017).
2.2.3. Conformational change of P-tau
P-tau at Thr231 following TBI is of particular interest as it triggers a trans-cis conformational change of P-tau. The trans P-tau is physiological and can promote MTs assembly, while cis P-tau is pathogenic and contributes to AD-related tau pathology (Nakamura et al., 2012). Both single TBI induced by WD or blast and rmTBI caused a robust and persistent increase of cis, but not trans P-tau starting at 12-24 hours following injury (Kondo et al., 2015). The cis P-tau is neurotoxic and can spread in the brain, causing the widespread of tau pathology (Kondo et al., 2015). A treatment of cis P-tau specific antibody reduced the cis P-tau-induced toxicity and prevented the development of tau pathology in TBI mice (Albayram et al., 2017; Kondo et al., 2015). These data suggest that cis P-tau may act as an early driver of disease after TBI and leads to overt tau pathology and neurodegeneration in AD.
2.3. Tau oligomers
Compelling evidence has shown that tau oligomers, a pre-filament form of tau is toxic and causes impairments in axonal transport, mitochondrial function and synaptic plasticity, resulting in chronic neuronal loss and memory deficits in AD mice (Fa et al., 2016; Lasagna-Reeves et al., 2011; Lasagna-Reeves et al., 2012). Tau oligomers have been reported in a variety of TBI models, including FPI, CCI, WD, blast and repetitive CHI. FPI triggered a rapid accumulation of tau oligomers recognized by T22 antibody as early as 4 hours following TBI (Hawkins et al., 2013), and a persistent increase of T22-positive signal in the cortex and hippocampus was observed 6 months following severe WD injury and CCI (Acosta et al., 2017; Albayram et al., 2017). The bTBI has also been reported to cause a marked accumulation of T22 immunoreactivity in the hippocampus 24 hours (Gerson et al., 2016) and 7 days (Du et al., 2016) after insults. Similarly, increased oligomerization of tau in the cortex was detected following a single moderate/severe WD injury (Collins-Praino et al., 2018). TBI-induced augmented oligomerization of tau appeared to be triggered by an elevated level of tau tyrosine phosphorylation, which was mediated by activation of tyrosine kinase, abelson murine leukemia viral oncogene homolog 1 (c-Abl) (Wang et al., 2017a). Following CCI, c-Abl was activated upon an inhibition of tyrosine-protein phosphatase non-receptor type 13 (PTPN13), a phosphatase susceptible to calpain 2 cleavage (Wang et al., 2017a). Post-injury treatment of a calpain 2 inhibitor (indirectly activates PTPN13) or a c-Abl inhibitor significantly attenuated tyrosine phosphorylated tau and the formation of tau oligomers (Wang et al., 2017a). Recently, the impact of single TBI-derived tau oligomers on neurotoxicity and long-term deficits has been investigated. It has been shown that single blast-derived tau oligomers significantly decreased cell viability in vitro (Gerson et al., 2016), and a single intra-hippocampal injection of FPI-induced tau oligomers accelerated the onset of cognitive deficits in hTau mice (Gerson et al., 2016).
TBI-induced tau oligomers has also been studied in several rmTBI models (Bittar et al., 2019; Ojo et al., 2016). For example, a study with repetitive mild CHI has demonstrated a robust increase in tau oligomer (TOC1-positive) level in hTau mice (Ojo et al., 2016). Repetitive mild WD injuries with 3 hits and 5 days apart failed to cause changes in the level of tau oligomers (Collins-Praino et al., 2018), 7 injuries in 9 days, however, induced robust tau oligomerization (Albayram et al., 2017). This indicates that the number of injury and the interval between injuries are important parameters in determining tau oligomerization following TBI. More interestingly, tau oligomers derived from rmTBI appeared to have distinct characteristics from those derived from sTBI. A recent study investigating tau oligomers derived from repetitive and single blast injury has found that these different sources of tau oligomers display differential neuronal toxicity, functional impairment and seeding profiles, providing a potential mechanism underlying the different risks for neurodegeneration following TBIs of different frequencies (Bittar et al., 2019).
2.4. Neurofibrillary tangles (NFTs)
NFTs are aggregates of hyperphosphorylated tau protein that are commonly known as a pathological hallmark of AD. A recent study using Thioflavin S/AT8 co-staining has found a deposition of NFTs in different brain regions, including cortex, hippocampus and striatum of WT mice at 12 months following CCI (Wu et al., 2020). Furthermore, tau aggregates were significantly attenuated in animals with AEP knockout, indicating a pivotal role of AEP in mediating TBI-induced AD-like pathology (Wu et al., 2020). A study using T44 mice (expressed with the shortest form of human WT tau isoform) and repetitive mild CHI has shown an increased NFT-like inclusions in the hippocampus, entorhinal cortex and inferolateral surface of the brain 9 months following injuries (Yoshiyama et al., 2005). In this study, animals are subjected to a total of 16 injuries, with 4 injuries a day and repeated every week for 4 weeks. NTF-like tau pathology was recognized by Gallyas and Thioflavin-S staining in combination of P-tau immunoreactivity with PHF1, PHF6 (pThr231) and 12E8 tau antibodies (Yoshiyama et al., 2005). The distribution pattern of NTF-like tau in the hippocampus and the globose-shaped, Pick body-like tau positive inclusions in the superficial layer of cortex are similar to those NFTs found in human AD (Yoshiyama et al., 2005). It is worthy of note that in this study only one out of eighteen animals was observed with mrTBI-induced NFTs (Yoshiyama et al., 2005). The reasons why others were resistant to TBI-induced formation of NFTs are remained to be elucidated. The expression of the shortest form of human tau instead of all six isoforms in these animals may be one of the possible reasons. In support of this possibility, a later study using hTau mice who express the complete human tau gene profile has demonstrated an augmented NFT-like signals in the cortex and hippocampus 3 weeks following rmTBI, which has an even lower frequency of injury (5 injuries in total during 9 day period with 48 hours interval) (Ojo et al., 2013).
2.5. Spreading of tau pathology
The propagation and spreading of tau protein is supposed to play an important role in the pathogenesis of AD (Jucker and Walker, 2018). Previous studies with different TBI models have found that TBI-induced tau pathology is not restricted to the ipsilateral impact region, but spreads to other related regions and the non-directly impacted contralateral areas hours or weeks following TBI, indicating a spatial spreading of abnormal tau species over time after insults (Acosta et al., 2017; Edwards III et al., 2020; Kondo et al., 2015; Ojo et al., 2013; Zanier et al., 2018; Zhao et al., 2017c). For example, a CCI study in WT mice demonstrated that increased P-tau at Ser404 was observed in the contralateral hippocampus, prefrontal cortex and thalamus as early as 24 hours after injury (Zhao et al., 2017c). Increased accumulation of AT8 reactivity was still evident in the contralateral cortex and hippocampus 6 months after CCI (Acosta et al., 2017). Increasing evidence has shown that tau pathology is able to spread along brain regions through synaptic connections (de Calignon et al., 2012; Liu et al., 2012). Therefore, the augmented phosphorylated tau in the contralateral brain areas may be transmitted from the ipsilateral regions. In supporting of this idea, a recent study has found that pathological tau (AT8) seemed to disseminate between synaptically connected brain regions after CCI, with the most affected brain regions being the ones projecting to or from the initial impact region, such as entorhinal cortex, hippocampus, amygdala and brainstem (Edwards III et al., 2020). However, P-tau did not increase over time in the brain regions in proximity to the impact area (Edwards III et al., 2020). A previous study showed that increased neuronal activity can promote the spreading of tau in vitro (Wu et al., 2016). It will be of great interest to investigate whether the spreading of tau pathology in TBI is also attributed to the sustained hyperactivity caused by massive glutamate release following TBI.
Recent studies have demonstrated that tau aggregates may acquire the seeding ability to propagate the transmission of tau pathology from one neuron to another in a prion-like manner (Clavaguera et al., 2013; Sanders et al., 2014). Whether the spreading of tau pathology following TBI is mediated in a prion-like manner has received increasing attention. In two recent studies, brain homogenates or tau oligomers from mice exposed to CCI or FPI are prepared and inoculated into the hippocampus of TBI-naïve animals. Immunochemistry assay in these animals demonstrates that the levels of abnormal tau species are increased in the injection site as well as the thalamus and cerebellum, providing evidence of a prion-like spreading of tau pathology after TBI (Gerson et al., 2016; Zanier et al., 2018).
3. Synaptic dysfunction following TBI
Synaptic dysfunction is not only a neuropathological hallmark of AD but also a frequent observation following TBI. Although by no means identical, long-lasting deficits in cognitive and memory function observed in AD and following TBI appear to have common roots at the synaptic level. In this section, we review the current knowledge of TBI-induced synaptic dysfunction from perspectives of synapse loss, impaired synaptic plasticity and imbalanced synaptic transmission, and discuss the potential common mechanisms underlying synaptic dysfunction post-TBI and in AD (summarized in Figure 1).
Figure 1. Common mechanisms underlying synaptic dysfunction in both TBI and AD.
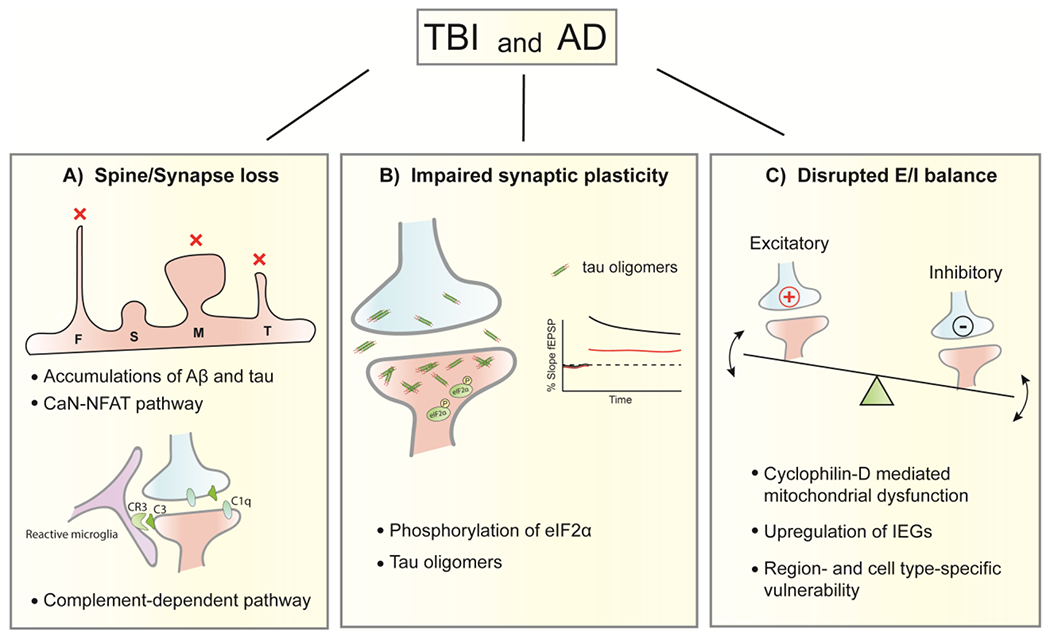
Synapse/spine loss, impaired synaptic plasticity and disrupted E/I balance are common synaptic features found post-TBI and in AD. A) Accumulations of Aβand tau and activation of CaN-NFAT pathway are thought to contribute to the dendritic spine loss after TBI and in AD. Following TBI, mature mushroom-like spines and immature filopodia/thin-shaped spines are more vulnerable to loss (indicated by red x marks) than transitional stubby-shaped spines. Synaptic loss mediated by complements (C1q and C3) and their receptor CR3 in microglia is found in both diseases. B) Phosphorylation of eIF2αcontributes to the deficits of long-term synaptic plasticity (e.g. LTP) in the hippocampus of brains from both TBI and AD. Tau oligomers derived from TBI, similar to those from AD, impair LTP. TBI-derived tau oligomers also impair short-term plasticity by reducing paired pulse facilitation. C) Hyperexcitation of cortical and hippocampal neuronal network is observed following TBI and in AD, indicated by an upregulation of IEGs. Cyclophilin-D mediated mitochondrial dysfunction is a common mechanism underlying this hyperexcitation. Region- and cell-type specific vulnerability to disrupted E/I balance, a featured characteristic of AD, is also displayed following TBI. F, Filopodia; S, Stubby; M, Mushroom; T, Thin; E/I, Excitation/Inhibition; CaN, calcineurin; NFAT, nuclear factor of activated T cells; eIF2α, α-subunit of eukaryotic translation initiation factor 2; LTP, long-term potentiation; IEGs, immediate early genes.
3.1. Degeneration of dendritic spines and synapses
3.1.1. Evidence for dendritic and synaptic disruption following TBI
Using Golgi staining or membrane-incorporating lipophilic dyes that mark the whole cell contour, a wealth of studies have shown an extensive dendritic degeneration and a decreased spine density in the cortex and hippocampus after TBI (Campbell et al., 2012a; Gao and Chen, 2011; Gao et al., 2011; Pijet et al., 2019; Sun et al., 2020; Winston et al., 2013; Winston et al., 2016; Zhang et al., 2019b; Zhao et al., 2016). CCI-induced acute reduction of dendritic spines was not only found in the ipsilateral (Gao and Chen, 2011; Gao et al., 2011; Pijet et al., 2019; Zhao et al., 2016), but also the contralateral cortex and hippocampus (Winston et al., 2013). In particular, the acute loss of dendritic spines at 1 or 3 days post-CCI was mainly found in mature mushroom-like spines and immature filopodia-shaped spines, with the number of transitional stubby-shaped spines being unchanged (Gao and Chen, 2011; Gao et al., 2011; Sun et al., 2020). These observations indicate that CCI preferably reduces the density of functional synapses and impairs the capacity of neurons to produce new synapses during the acute stage. Interestingly, at a later time point (e.g. 7 d) post-injury, more immature filopodia-type and thin-type spines were observed (Pijet et al., 2019; Sun et al., 2020), indicating a compensation for the loss of synaptic connections through synaptogenesis.
In accordance with reduced density of dendritic spines, loss of synapses has been consistently reported in a variety of TBI models, determined by reduced immunoactivity of synaptic protein markers such as synapsin 1, synaptophysin and PSD95 in the hippocampus and cortex at different time points following injury ranging from hours to weeks (Ansari et al., 2008a, b; Campbell et al., 2012a; Furman et al., 2016; Rachmany et al., 2017; Rehman et al., 2018; Sen et al., 2017; Wu et al., 2003; Zhang et al., 2019c). In support of the findings from immunochemistry studies, transmission electron microscopy (TEM) experiments also showed a significant loss of synapses in the CA1 area of hippocampus after CCI (Perez et al., 2016; Scheff et al., 2005). Although the synaptogenesis was observed 10 days after injury, the total number of synapses by 60 days post-injury was still significantly lower than pre-injury level (Scheff et al., 2005), indicating a sustained loss of synapses that may underlie the persistent cognitive dysfunction after injury. TBI-induced synapse loss in the hippocampus has been further confirmed in a more recent study using super-resolution imaging technique, known as SEQUIN (Synaptic Evaluation and Quantification by Imaging of Nanostructure) (Sauerbeck et al., 2020). By performing SEQUIN on brain sections derived from animals exposed to CHIMERA model of TBI, it has been shown that the number of synaptic loci was substantially reduced by 7 days after injury and remained decreased at least 30 days post-TBI (Sauerbeck et al., 2020).
3.1.2. Common mechanisms underlying the loss of synapses/spines following TBI and in AD
Accumulations of amyloid-β(Aβ) and tau, particularly oligomeric form of Aβand tau have pivotal roles in mediating synaptic loss in AD-like neurodegeneration (Forner et al., 2017). Although there is a rapid and sustained accumulation of Aβ(Johnson et al., 2010) and tau (discussed in section 2) following TBI in animal models, few studies have investigated whether these accumulations, similar to those found in AD, contribute to the synaptic loss following TBI. A previous study indicated that TBI-induced synaptic loss might not be attributable to the concurrent Aβaccumulation (Winston et al., 2013). Peripheral treatment of the γ-secretase inhibitor LY450139, although successfully attenuated Aβaccumulation, did not prevent synaptic loss post-CCI (Winston et al., 2013). However, it should be noted that the effect of Aβon synaptic density has only been assessed acutely (i.e. 24 h post-injury) in this study, whether TBI-induced subacute and/or chronic synaptic loss is mediated by injury-associated Aβdeposition remains unknown. In a more recent study, the role of Aβand tau accumulation in the regulation of synaptic density was investigated 6 months following TBI (Wu et al., 2020). The CCI-induced AEP activation led to an increased deposition of Aβand P-tau, which were accompanied by a significant decrease in synapse and dendritic spine density in CA1 area (Wu et al., 2020). However, the reduction of synapse/spine density was partially ameliorated in animals whose Aβand tau pathology were attenuated by AEP KO (Wu et al., 2020). These findings suggest that TBI-induced deposition of Aβand tau does play a role in mediating synapse/spine loss following TBI. Additional studies are needed to confirm this role in other TBI models and to further investigate whether oligomeric Aβ and/or tau is the toxic species executing this role following TBI.
Calcineurin (CaN) is a Ca2+-dependent phosphatase that has been shown to be activated following TBI (Furman et al., 2016; Kurz et al., 2005) and in AD (Wu et al., 2010) and contribute to the synaptic loss in these two diseases. It has been found that FPI caused an increased activity of CaN, which led to a cofilin-dependent disruption of the cytoskeleton of spines, resulting in the degeneration and eventual loss of dendritic spines (Campbell et al., 2012a). FPI-induced dendritic spine loss can be prevented by a post-injury treatment of CaN inhibitor, FK506 (Campbell et al., 2012b). In the context of AD, oligomeric Aβinduced the activation of CaN, which in turn activated the transcriptional factor, nuclear factor of activated T cells (NFAT), by translocating it into the nuclei (Wu et al., 2010). Inhibition of CaN activity or CaN-mediated NFAT activation can abolish Aβ-induced morphological abnormalities of spines and rescue spine loss in mutant APP-overexpressing mouse model of AD (Hudry et al., 2012; Rozkalne et al., 2011; Wu et al., 2010). A recent study showed that CaN activation and its associated spine loss can also be mediated by tau accumulation (Yin et al., 2016). In AD brains, CaN-NFAT signaling pathway has also been shown to be robustly activated in the astrocytes (Abdul et al., 2009; Norris et al., 2005). Astrocyte-specific blockade of CaN-NFAT signaling pathway was able to ameliorate cognitive and synaptic dysfunction in APP/PS1 mice (Furman et al., 2012). Recently, the role of astrocytic CaN-NFAT signaling pathway in modulating synaptic function has been revealed in a TBI model. Furman et al. showed that astrocytic NFAT activity within the hippocampus was significantly increased following CCI (Furman et al., 2016). Blocking astrocytic CaN-NFAT activation prevented the loss of several synaptic marker proteins including PSD95, synapsin 1 and GluR1 (Furman et al., 2016).
The complement pathway, of which C1q and C3 are the key molecular players, has a critical role in microglia-mediated synaptic loss in AD (Fonseca et al., 2004; Hong et al., 2016; Shi et al., 2017). Oligomeric Aβ-induced synaptic loss was associated with increased expression of C3 and C1q as well as enhanced microglial phagocytic activity (Hong et al., 2016). Genetic deletion of C3 has been shown to protect against synaptic loss in APP/PS1 mice (Shi et al., 2017). Both genetic deletion of C1q and pharmacological treatment of blocking antibody against C1q prevented oligomeric Aβ-induced synaptic loss in the hippocampus (Hong et al., 2016). Similar to oligomeric Aβ tau pathology has also been shown to induce microglia-mediated engulfment of synapses in a C1q-dependent manner (Dejanovic et al., 2018). Interestingly, a recent study has shown that the complement cascades may also plays a role in mediating TBI-induced synaptic loss (Krukowski et al., 2018). In aged mice brain exposed to CCI, chronic synaptic loss determined by reduced synaptic marker proteins was associated with an increase in synaptic expression of C1q and phagocytosis of synaptosomes by microglia. Additionally, both genetic deletion of C3 and pharmacological treatment of C1q-inhibiting antibody prevented memory deficits in aged mice after TBI (Krukowski et al., 2018).
3.2. Impaired synaptic plasticity
Temporary or long-term memory loss is commonly seen in patients with head injury (Rabinowitz and Levin, 2014) and experimental animals exposed to TBI (Xiong et al., 2013). Synaptic plasticity measured by long-term potentiation (LTP) is widely considered as the cellular mechanism underlying memory formation. A wealth of studies have examined synaptic plasticity in hippocampal slices from animals subjected to injury inflicted by CCI (Marshall et al., 2017; Perez et al., 2016; Wang and Han, 2018b), FPI (Chen et al., 2018c; Franklin et al., 2019), blast (Goldstein et al., 2012) or CHI (White et al., 2017; Zhang et al., 2019a). There are consistent reports of impairments of LTP at CA3-CA1 synapses, accompanied by memory deficits found in several hippocampal-dependent tasks, including Morris water maze, novel object recognition and contextual fear conditioning. Manipulations of NMDA receptors (Yaka et al., 2007) and its interacting EphB3 receptor (Perez et al., 2016) as well as its downstream molecular cascades such as PSD95-TrkB-BDNF pathway (Marshall et al., 2017) and cAMP-CREB pathway (Titus et al., 2013; Titus et al., 2016; Wilson et al., 2016) have been shown to attenuate TBI-induced hippocampal LTP deficits and improve neurobehavioral outcomes. TBI-induced deficits in CA1-LTP were observed after even a single mild CHI and these deficits were aggravated when animals express human mutant P301L tau protein, indicating that tau-related genetic predisposition promotes TBI-induced deficits in synaptic plasticity (Marschner et al., 2016). Despite of the consistent findings above, another study found that CA1-LTP was equally induced and maintained in both ipsilateral and contralateral hippocampal slices from rats following CCI (Norris and Scheff, 2009). Differences in the experimental settings (e.g. sacrifice time, LTP induction protocol), animal species (rats vs mice) and/or injury characteristics (e.g. impact speed and dwelling time) may partially explain this discrepancy. Similar to a single moderate FPI, rmFPI also rendered high frequency stimulation unable to potentiate excitatory postsynaptic potentials (EPSPs) in CA1 synapses (Aungst et al., 2014). In addition, rmTBI via CHI has been shown to produce LTP impairment in the cortex associated with glial activation and P-tau, which can be rescued by the treatment of memantine, an uncompetitive NMDA receptor antagonist (Mei et al., 2018).
TBI-induced impairments in LTP also extended to the dentate gyrus (DG) area of hippocampus. It has been reported that granule cells in the DG region ipsilateral to the injuries modeled by WD or FPI display a significant reduction in LTP post TBI (White et al., 2017; Yamashita et al., 2011). Neurotropins are a group of proteins playing pivotal roles in mediating synaptic plasticity (Gómez-Palacio-Schjetnan and Escobar, 2013). A recent single-cell RNA-seq study has found significant changes in the gene expressions of neurotrophins in DG granule cells following FPI, including brain-derived neurotrophic factor (Bdnf) and neurotrophin 3 (Ntf3) (Arneson et al., 2018). It is of great interest to investigate whether granule cell-specific manipulations of Bdnf and/or Ntf3 levels could restore synaptic plasticity and improve functionality in the DG following TBI.
Maintaining synaptic plasticity and long-lasting memory requires protein synthesis, which can be suppressed by phosphorylation of the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) (Costa-Mattioli et al., 2007). eIF2αphosphorylation is a central step of integrated stress responses (ISR) where various cellular stress signals (e.g. ER stress) converge on (Pakos-Zebrucka et al., 2016). Aberrant activation of eIF2α kinases and the resultant phosphorylation of eIF2αunder stress conditions represent one of the most important mechanisms mediating long-term deficits in synaptic plasticity and memory in AD (Ohno, 2014). Previous studies have shown that CCI induces acute and persistent phosphorylation of eIF2αin the cortex and hippocampus (Begum et al., 2014; Chou et al., 2017; Sen et al., 2017). A systemic treatment of ISRIB (ISR inhibitor), a small molecule blunting the effects of eIF2αphosphorylation on translation initiation, was found to reverse TBI-induced hippocampal LTP impairment and rescue TBI-induced behavioral deficits in various hippocampal-dependent memory tasks (Chou et al., 2017). Inhibition of PERK (double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase), a major kinase of eIF2α has also been shown to restore synaptic plasticity and memory deficits in both AD-like (APP/PS1) mice and mice subjected to CCI (Ma et al., 2013; Sen et al., 2017).
Tau pathology evolved following TBI has been shown to contribute to TBI-induced deficits in LTP and behavioral performance (Kondo et al., 2015). Treating animals exposed to severe CHI with an antibody specifically blocking cis-P-tau accumulation has been shown to prevent the development of tau pathology, reverse cortical LTP deficits and improve performance in the cortex-dependent anxiety/risk-taking paradigm (Kondo et al., 2015). A large body of research has suggested that the deleterious effects of tau protein on synaptic function during the process of AD is dependent on its oligomeric aggregates (Forner et al., 2017). Recent studies have found that tau oligomers derived from TBI models also display synaptic toxicity and produce long-lasting memory deficits when inoculated into the brain of TBI-naïve animals (Bittar et al., 2019; Gerson et al., 2016). Furthermore, tau oligomers derived from different TBI models (single blast vs repetitive blasts) decreased synaptic protein markers and impaired hippocampal LTP to different extents, with repetitive blast-derived tau oligomers being more toxic (Bittar et al., 2019). TBI-derived tau oligomers also reduced the probability of vesicular release (measured by paired pulse ratio), therefore impairing the short-term plasticity (Bittar et al., 2019). These findings suggest that TBI and AD result in similar biological changes in tau protein that increase its toxicity and contribute to the impairments of synaptic plasticity and long-term memory.
3.3. Imbalanced synaptic transmission
Proper functioning of neural circuits is ensured by a balance between the excitatory and inhibitory control of synaptic transmission (Hensch and Fagiolini, 2005). Aβ-induced disruption of the excitation/inhibition balance (E/I balance) in cortical and hippocampal neurons has been considered as one of the earliest synaptic dysfunction in the pathogenesis of AD (Busche and Konnerth, 2016). Recent studies support altered E/I balance in the cortex as a circuit mechanism underlying TBI-induced cognitive deficits (Bashir et al., 2012; Ding et al., 2011). It has been shown that lateral FPI induces an enduring deficit in working memory accompanied by shifts in both excitatory and inhibitory synaptic transmission in the layer 2/3 neurons of the prefrontal cortex (PFC) (Smith et al., 2015). The frequencies of spontaneous and miniature EPSCs (sEPSCs and mEPSCs) in layer 2/3 neurons were significantly increased, while the spontaneous and miniature inhibitory postsynaptic potentials (sIPSCs and mIPSCs) were much smaller following injury (Smith et al., 2015). Enhanced EPSCs and intrinsic excitability were also recorded in the layer 5 neurons of somatosensory cortex (SSC) following central FPI (Hanell et al., 2015). However, FPI-induced hyperexcitability in SSC-layer 5 neurons can be prevented in mice with a genetic deletion of cyclophilin-D (CypD) (Sun and Jacobs, 2016). CypD plays a critical role in cellular dysfunction by inducing the opening of mitochondrial permeability transition pore (mPTP), which leads to ATP depletion, synaptic deficits and eventual cell death (Baines et al., 2005; Du et al., 2008). Previous studies have showed that CypD deficiency restored altered synaptic transmission (Du et al., 2014) and significantly improved learning and memory in AD mice (Du et al., 2008). These findings together posit mitochondrial dysfunction as a common mechanism modulating synaptic abnormalities following TBI and in AD.
The elevated activity in cortical neurons following TBI is mirrored by increased expression of immediate early genes (IEGs), which has been widely used as an indicator of neuronal activity. In a recent study, Rodriques et al. have developed a novel technique, named Slide-Seq for measuring gene expression in situ with high spatial resolution (Rodriques et al., 2019). Using this technique, the authors showed that following cortical injury inflicted by intracranial injection, IEGs, such as Fos, Arc, Junb and the neuron-specific gene Npas4 have been upregulated in a large area around the injury site (Rodriques et al., 2019). IEGs upregulation has also been shown to associate with hyperexcitation of hippocampal neuronal network in TAU58/2 mouse, a tau P301S mouse model of AD. Furthermore, co-immunostaining study showed that IEG marker protein Arc was largely confined to tau pathology-bearing hippocampal neurons (Przybyla et al., 2020). These results suggest that upregulation of IEGs may disrupt the E/I balance in animal models of TBI and AD. Hyperexcitability induced by tau abnormalities appears to play an important role in the epileptogenesis in AD (Vossel et al., 2017). Interestingly, tau pathology has also been shown to be associated with epilepsy developed years after TBI (Zheng et al., 2014). Reducing the level of tau may suppress aberrant network activity and represent a promising therapy for TBI- and AD-associated epilepsy. Indeed, reducing phosphorylation of tau by sodium selenate has been shown to have anti-epileptogenic effects in TBI- and AD-related acquired epilepsy models (Liu et al., 2016).
TBI-induced hyperexcitability can also be linked to a loss of synaptic inhibition. Previous studies have reported a loss of GABAergic interneurons and impaired GABAergic activity in the cortex (Brizuela et al., 2017; Cantu et al., 2015) as well as the hippocampus (Almeida-Suhett et al., 2015; Gupta et al., 2012; Santhakumar et al., 2000) following various types of injury. Emerging evidence has showed that interneurons in the cortex and hippocampus do not respond to injures in the same way. Indeed, regional and/or cell-type specific vulnerability are observed. For example, in a recent study, Frankowski et al. comprehensively analyzed hippocampal interneurons following CCI and found a preferential loss of PV (parvalbumin)-and SST (somatostatin)-positive interneurons in the principal cell and polymorph layers, whereas nNOS (neuronal nitric oxide synthase)- and Reelin-positive interneurons occupying the molecular layer appeared to be well preserved (Frankowski et al., 2019). As to the cortical interneurons, a previous study using cell-type specific Cre-dependent juvenile mice has found a specific loss of PV- but not SST-expressing interneurons following CCI (Nichols et al., 2018). Cell-specific vulnerability to TBI has also been observed in the pyramidal neurons. It has been reported that subcortically-projecting layer 5 pyramidal neurons (type A) in the mPFC display an increase in the frequency of EPSCs following rmTBI, however, no effects were observed in the frequency or amplitude of EPSCs in the type B layer 5 neurons, which primarily project to other cortical regions (Krukowski et al., 2020). Furthermore, TBI-induced increased frequency of synaptic input in type A neurons and altered behavior in mPFC-dependent task (risk-taking) can be restored by a treatment of ISRIB (Krukowski et al., 2020). Taken together, these findings suggest that cell type-specific vulnerability, a featured characteristic in AD, is also displayed following TBI. ISR may serve as a potential target for preserving normal cortical synaptic transmission and high-order executive behavior following TBI.
4. Concluding Remarks and Future Perspectives
Although the association of TBI and increased risk for dementia, such as AD, is well described, the exact mechanisms underlying this association remain poorly understood, and are extremely complex. Abnormal accumulation of hyperphosphorylated tau aggerates and intercorrelated synaptic dysfunction, which are thought to play a pivotal role in the development of AD, are also observed following TBI. In this review, we discuss the distribution, molecular features and mechanisms of development of tau pathology in a variety of TBI models, with a particular focus on its implication in AD. TBI-induced tau pathologic changes in Chronic traumatic encephalopathy (CTE) is another hot topic and has been recently reviewed by Katsumoto et al. (Katsumoto et al., 2019). Emerging evidence suggests that rmTBI increases the risk of developing CTE, while a single moderate to severe TBI increases the risk of late-onset AD. It should be noted, however, that CTE pathology might not be unique to rmTBI as it has also been found in individuals who had no known exposure to repetitive neurotrauma (Iverson et al., 2019). Whether a single moderate to severe TBI can cause CTE is not well determined (Iverson et al., 2019). Clinical pathological studies reveal that the distribution and progression pattern of tau pathology in the brains of AD patients are quite different from that of professional sports players with CTE (Katsumoto et al., 2019). Therefore, it is of great importance to elucidate the molecular mechanisms underlying tau pathology following different types of TBI, e.g. sTBI vs rmTBI. Unfortunately, animal models of TBI that recapitulate the distinct features of tau pathology observed in AD and CTE are lacking, which thus significantly hinders a better understanding of TBI-induced tau pathology in humans. The newly developed CHIMERA model has shown to be more pathologically and biomechanically relevant to clinical TBI cases (Haar et al., 2019; Namjoshi et al., 2014). Using CHIMERA model on WT mice and/or human WT tau knock-in mice thus shows promise in facilitating a better understanding of tau pathology post TBI and shed insights on how tau pathology contributes to the development of different neurodegenerative diseases following TBI, such as AD and CTE. In addition, it is worthy of note that most TBI models utilize anesthesia during the injury delivery, which may confound the pathophysiology post-TBI. For example, isoflurane, a commonly used anesthetic agent in experimental TBI has been shown to increase tau phosphorylation (Dong et al., 2012) and have neuroprotective effects (Statler et al., 2006). Non-anesthetized models developed in both mice (Petraglia et al., 2014a) and rats (Meconi et al., 2018; Pham et al., 2019) avoid the effects of anesthesia and are valuable tools for pre-clinical studies of TBI.
Long-term cognitive deficits, particularly in high-order executive function and learning and memory resulted from TBI is a major health concern that may cause significant functional disability. TBI-induced impairments in synaptic function and network activity are thought to underlie these cognitive deficits. We review the experimental evidences of TBI-induced impairments in both structure and function at synaptic level, with a focus on the common mechanisms underlying synaptic dysfunction in both TBI and AD. Interestingly, tau aggregates derived from TBI models display biological similarities to those found in AD, leading to impaired synaptic plasticity and long-term memory. Given that tau pathology correlates better with cognitive impairments than does Aβpathology and the fact that most Aβ-based clinical trials failed, targeting of tau is rising to be a promising therapeutic, especially in AD cases whose clinical symptoms are evident. Suppression of transgenic tau by switching off its expression can stop the progression of tau pathology and reverse cognitive deficits in several mouse models (Xu et al., 2014). In cell and animal models, small interfering RNA (siRNA) has also been found to reduce tau pathology and associated functional impairments (Congdon and Sigurdsson, 2018). Reducing tau pathology with siRNA, antisense oligonucleotides (ASOs) or immunotherapy may restore synaptic function and represent a potential therapeutic avenue for mitigating cognitive deficits following TBI.
Region- and cell-type specific vulnerability to synaptic loss or dysfunction observed in AD is becoming increasingly evident in TBI. With the development of techniques such as single-cell or single-nucleus RNA-Seq and spatial transcriptomics like the Slide-Seq and 10x Genomics Visium Spatial Gene Expression Solution, future studies can be performed on different TBI models and postmortem brain tissues from TBI patients to investigate cell-type- and region-specific changes in gene signatures at different time points following TBI. Results from these investigations will address critical knowledge gaps in the pathogenesis of TBI, for example, which molecular pathways are affected by TBI in each cell type and how do they give rise to or involved in the development of different neurodegenerative diseases such as AD and CTE? Which cell types and brain regions are vulnerable to TBI at different disease stages? How neuronglia interactions following TBI contribute to the chronic progression of TBI pathology and neuroinflammation? Identifying the gene signatures enriched in the vulnerable regions and cell types can yield novel biomarkers and therapeutic targets that may alleviate and/or reverse the cognitive deficits and neurodegeneration following TBI.
Disruption of E/I balance observed in the cortex and hippocampus following TBI suggest potential intervention strategies for the amelioration of cognitive deficits. The pattern of excitation and inhibition in affected regions and neurons can be monitored and manipulated with optogenetic tools and Designer Receptors Exclusively Activated by Designer Drugs (DREADD)-based chemogenetic tools. By utilizing genetically encoded light-sensitive channels and engineered GPCRs that are selectively activated by light and a biologically inert compound, clozapine-N-oxide (CNO), respectively, optogenetics and DREADDs are able to activate or inhibit neuronal subpopulations with high spatiotemporal resolution (Delaney et al., 2020). These novel tools have recently been shown to monitor and modulate activities in specific types of neurons and promote neuronal function and survival in animal models of TBI (Adams et al., 2018; Chandrasekar et al., 2019; Zhao et al., 2018). The use of optogenetics and DREADDs will greatly improve our understanding of circuit-level pathology in TBI and ultimately help develop precise and selective neuromodulation methods that can promote the recovery of cognitive function following TBI (Delaney et al., 2020).
Highlights.
Tau pathology and synaptic dysfunction are common features in TBI and AD.
Tau pathology in animal models of TBI is analyzed, focusing on implications in AD.
Evidence of TBI-mediated synaptic impairments is reviewed.
Overlapped mechanisms underlying synaptic dysfunction in TBI and AD are discussed.
We propose future perspectives on uncovering the link between TBI and AD.
Acknowledgements
This work was supported by the Department of Defense [grant number, W81XWH1910309]; the National Institutes of Health [grant number, AG056673]; the Alzheimer’s Association [grant number, AARF-17-505009]; and the Neuroscience Research Institute Pilot Award and the Chronic Brain Injury Pilot Award from The Ohio State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Completing Interest
The authors have no conflict of interest to declare.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig T, Murphy MP, LeVine H 3rd, Kraner SD, Norris CM, 2009. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci 29, 12957–12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Muneer PM, Chandra N, Haorah J, 2015. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol 51, 966–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, Sanberg PR, Kaneko Y, Borlongan CV, 2017. Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. Journal of cellular physiology 232, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C, Bazzigaluppi P, Beckett TL, Bishay J, Weisspapir I, Dorr A, Mester JR, Steinman J, Hirschler L, Warnking JM, 2018. Neurogliovascular dysfunction in a model of repeated traumatic brain injury. Theranostics 8, 4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayram O, Kondo A, Mannix R, Smith C, Tsai C-Y, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J, 2017. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nature communications 8, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida-Suhett CP, Prager EM, Pidoplichko V, Figueiredo TH, Marini AM, Li Z, Eiden LE, Braga MF, 2015. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp Neurol 273, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AD, Di Clerico J, Li B, Corbo CP, Alaniz ME, Grundke-Iqbal I, Iqbal K, 2010. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. Journal of Biological Chemistry 285, 30851–30860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Stringaro A, Colone M, D’Aguanno S, Meli G, Ciotti M, Sancesario G, Cattaneo A, Bussani R, 2010. A NH 2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. Journal of Alzheimer’s Disease 21, 445–470. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW, 2008a. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med 45, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW, 2008b. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma 25, 513–526. [DOI] [PubMed] [Google Scholar]
- Arneson D, Zhang G, Ying Z, Zhuang Y, Byun HR, Ahn IS, Gomez-Pinilla F, Yang X, 2018. Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat Commun 9, 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Oguntayo S, Albert SV, Gist I, Wang Y, Nambiar MP, Long JB, 2015. Acute decrease in alkaline phosphatase after brain injury: A potential mechanism for tauopathy. Neurosci Lett 609, 152–158. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT, 2002. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103, 26–35. [DOI] [PubMed] [Google Scholar]
- Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI, 2014. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab 34, 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K, 2018. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA neurology 75, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir S, Vernet M, Yoo WK, Mizrahi L, Theoret H, Pascual-Leone A, 2012. Changes in cortical plasticity after mild traumatic brain injury. Restor Neurol Neurosci 30, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D, 2014. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 34, 3743–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibow S, Ozenne V, Biemat J, Blackledge M, Mandelkow E, Zweckstetter M, 2011. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. Journal of the American Chemical Society 133, 15842–15845. [DOI] [PubMed] [Google Scholar]
- Bittar A, Bhatt N, Hasan TF, Montalbano M, Puangmalai N, McAllen S, Ellsworth A, Carretero Murillo M, Taglialatela G, Lucke-Wold B, Logsdon A, Rosen C, Turner RC, Kayed R, 2019. Neurotoxic tau oligomers after single versus repetitive mild traumatic brain injury. Brain Commun 1, fcz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton AN, Saatman KE, 2014. Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. Journal of Neuropathology & Experimental Neurology 73, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizuela M, Blizzard CA, Chuckowree JA, Pitman KA, Young KM, Dickson T, 2017. Mild Traumatic Brain Injury Leads to Decreased Inhibition and a Differential Response of Calretinin Positive Interneurons in the Injured Cortex. J Neurotrauma 34, 2504–2517. [DOI] [PubMed] [Google Scholar]
- Busche MA, Konnerth A, 2016. Impairments of neural circuit function in Alzheimer’s disease. Philos Trans R Soc Lond B Biol Sci 371, 20150429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Low B, Kurz JE, Patel SS, Young MT, Churn SB, 2012a. Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury. J Neurotrauma 29, 218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Register D, Churn SB, 2012b. Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J Neurotrauma 29, 201–217. [DOI] [PubMed] [Google Scholar]
- Cantu D, Walker K, Andresen L, Taylor-Weiner A, Hampton D, Tesco G, Dulla CG, 2015. Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb Cortex 25, 2306–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Gaamouch FE, Meabon JS, Meeker KD, Zhu L, Zhong MB, Bendik J, Elder G, Jing P, Xia J, Luo W, Cook DG, Cai D, 2017. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyperphosphorylation after traumatic brain injury. Sci Rep 7, 11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso BR, Roberts BR, Malpas CB, Vivash L, Genc S, Saling MM, Desmond P, Steward C, Hicks RJ, Callahan J, 2019. Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: Results from a Randomized Controlled Pilot Trial in Alzheimer’s Disease. Neurotherapeutics 16, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartagena CM, Mountney A, Hwang H, Swiercz A, Rammelkamp Z, Boutte AM, Shear DA, Tortella FC, Schmid KE, 2016. Subacute Changes in Cleavage Processing of Amyloid Precursor Protein and Tau following Penetrating Traumatic Brain Injury. PLoS One 11, e0158576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar A, Heuvel FO, Tar L, Hagenston AM, Palmer A, Linkus B, Ludolph AC, Huber-Lang M, Boeckers T, Bading H, 2019. Parvalbumin interneurons shape neuronal vulnerability in blunt tbi. Cerebral Cortex 29, 2701–2715. [DOI] [PubMed] [Google Scholar]
- Chen H-H, Liu P, Auger P, Lee S-H, Adolfsson O, Rey-Bellet L, Lafrance-Vanasse J, Friedman BA, Pihlgren M, Muhs A, 2018a. Calpain-mediated tau fragmentation is altered in Alzheimer’s disease progression. Scientific reports 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LJ, Wang YJ, Tseng GF, 2010. Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons. J Neurotrauma 27, 1657–1669. [DOI] [PubMed] [Google Scholar]
- Chen M, Song H, Cui J, Johnson CE, Hubler GK, DePalma RG, Gu Z, Xia W, 2018b. Proteomic Profiling of Mouse Brains Exposed to Blast-Induced Mild Traumatic Brain Injury Reveals Changes in Axonal Proteins and Phosphorylated Tau. J Alzheimers Dis 66, 751–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Kuo T-T, Huang EY-K, Hoffer BJ, Chou Y-C, Chiang Y-H, Ma H-T, Miller JP, 2018c. Profound deficits in hippocampal synaptic plasticity after traumatic brain injury and seizure is ameliorated by prophylactic levetiracetam. Oncotarget 9, 11515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A, Krukowski K, Jopson T, Zhu PJ, Costa-Mattioli M, Walter P, Rosi S, 2017. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A 114, E6420–E6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK, 2001. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 8, 162–172. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Lavenir T, Falcon B, Frank S, Goedert M, Tolnay M, 2013. “Prion-like” templated misfolding in tauopathies. Brain Pathol 23, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-Praino L, Gutschmidt D, Sharkey J, Arulsamy A, Corrigan F, 2018. Temporal Changes in Tau Phosphorylation and Related Kinase and Phosphatases Following Two Models of Traumatic Brain Injury. Journal of Neuroinflammation and Neurodegenerative Diseases 2, 1–13. [Google Scholar]
- Congdon EE, Sigurdsson EM, 2018. Tau-targeting therapies for Alzheimer disease. Nature Reviews Neurology 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjevic K, Lacaille JC, Nader K, Sonenberg N, 2007. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT, 2010. Caspase activation precedes and leads to tangles. Nature 464, 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT, 2012. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, 2018. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100, 1322–1336. e1327. [DOI] [PubMed] [Google Scholar]
- Delaney SL, Gendreau JL, D’Souza M, Feng AY, Ho AL, 2020. Optogenetic Modulation for the Treatment of Traumatic Brain Injury. Stem Cells and Development 29, 187–197. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Gomez-Ramos A, Rubio A, Gomez-Villafuertes R, Naranjo JR, Miras-Portugal MT, Avila J, 2010. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem 285, 32539–32548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding MC, Wang Q, Lo EH, Stanley GB, 2011. Cortical excitation and inhibition following focal traumatic brain injury. J Neurosci 31, 14085–14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Wu X, Xu Z, Zhang Y, Xie Z, 2012. Anesthetic isoflurane increases phosphorylated tau levels mediated by caspase activation and Aβgeneration. PloS one 7, e39386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD, 2008. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Wu X, Sosunov AA, McKhann GM, Chen JX, Yan SS, 2014. Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta 1842, 2517–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, West MB, Cheng W, Ewert DL, Li W, Saunders D, Towner RA, Floyd RA, Kopke RD, 2016. Ameliorative Effects of Antioxidants on the Hippocampal Accumulation of Pathologic Tau in a Rat Model of Blast-Induced Traumatic Brain Injury. Oxid Med Cell Longev 2016, 4159357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards III G, Zhao J, Dash PK, Soto C, Moreno-Gonzalez I, 2020. Traumatic brain injury induces tau aggregation and spreading. Journal of Neurotrauma 37, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fa M, Puzzo D, Piacentini R, Staniszewski A, Zhang H, Baltrons MA, Li Puma DD, Chatterjee I, Li J, Saeed F, Berman HL, Ripoli C, Gulisano W, Gonzalez J, Tian H, Costa JA, Lopez P, Davidowitz E, Yu WH, Haroutunian V, Brown LM, Palmeri A, Sigurdsson EM, Duff KE, Teich AF, Honig LS, Sierks M, Moe JG, D’Adamio L, Grassi C, Kanaan NM, Fraser PE, Arancio O, 2016. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci Rep 6, 19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Grøn M, Christensen J, Benros ME, Vestergaard M, 2018. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. The Lancet Psychiatry 5, 424–431. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Zhou J, Botto M, Tenner AJ, 2004. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 24, 6457–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM, 2017. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci 40, 347–357. [DOI] [PubMed] [Google Scholar]
- Franklin W, Krishnan B, Taglialatela G, 2019. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci Rep 9, 8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowski JC, Kim YJ, Hunt RF, 2019. Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol Dis 129, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM, 2012. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci 32, 16129–16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Sompol P, Kraner SD, Pleiss MM, Putman EJ, Dunkerson J, Mohmmad Abdul H, Roberts KN, Scheff SW, Norris CM, 2016. Blockade of Astrocytic Calcineurin/NFAT Signaling Helps to Normalize Hippocampal Synaptic Function and Plasticity in a Rat Model of Traumatic Brain Injury. J Neurosci 36, 1502–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Scheff SW, Menard RM, Roberts K, Fugaccia I, Zemlan FP, 2005. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma 22, 83–94. [DOI] [PubMed] [Google Scholar]
- Gao X, Chen J, 2011. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J Neuropathol Exp Neurol 70, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Deng P, Xu ZC, Chen J, 2011. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One 6, e24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS, Hawkins BE, Kayed R, 2016. Tau Oligomers Derived from Traumatic Brain Injury Cause Cognitive Impairment and Accelerate Onset of Pathology in Htau Mice. J Neurotrauma 33, 2034–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakova OY, Glushakov AO, Borlongan CV, Valadka AB, Hayes RL, Glushakov AV, 2018. Role of Caspase-3-Mediated Apoptosis in Chronic Caspase-3-Cleaved Tau Accumulation and Blood-Brain Barrier Damage in the Corpus Callosum after Traumatic Brain Injury in Rats. J Neurotrauma 35, 157–173. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E, 1995. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neuroscience letters 189, 167–170. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC, 2012. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Palacio-Schjetnan A, Escobar ML, 2013. Neurotrophins and synaptic plasticity, Neurogenesis and neural plasticity. Springer, pp. 117–136. [DOI] [PubMed] [Google Scholar]
- Gupta A, Elgammal FS, Proddutur A, Shah S, Santhakumar V, 2012. Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J Neurosci 32, 2523–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haar CV, Martens KM, Bashir A, McInnes KA, Cheng WH, Cheung H, Stukas S, Barron C, Ladner T, Welch KA, 2019. Repetitive closed-head impact model of engineered rotational acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic innervation in the olfactory tubercle and generates white matter inflammation, tau phosphorylation and degeneration. Experimental neurology 317, 87–99. [DOI] [PubMed] [Google Scholar]
- Haase C, Stieler JT, Arendt T, Holzer M, 2004. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J Neurochem 88, 1509–1520. [DOI] [PubMed] [Google Scholar]
- Hanell A, Greer JE, Jacobs KM, 2015. Increased Network Excitability Due to Altered Synaptic Inputs to Neocortical Layer V Intact and Axotomized Pyramidal Neurons after Mild Traumatic Brain Injury. J Neurotrauma 32, 1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W, 2009. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in molecular medicine 15, 112–119. [DOI] [PubMed] [Google Scholar]
- Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR, DeWitt DS, Kayed R, 2013. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury possible link between traumatic brain injury and sporadic tauopathies. Journal of Biological Chemistry 288, 17042–17050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, 2005. Excitatory-inhibitory balance and critical period plasticity in developing visual cortex. Prog Brain Res 147, 115–124. [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B, 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Tamaoka A, Takahashi M, Kobayashi S, Furukawa T, Oaki Y, Mori O, Matsuno S, Inomata M, Teramoto A, 1998. Emergence of immunoreactivities for phosphorylated tau and amyloid-βprotein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9, 1879–1883. [DOI] [PubMed] [Google Scholar]
- Hu W, Tung YC, Zhang Y, Liu F, Iqbal K, 2018. Involvement of Activation of Asparaginyl Endopeptidase in Tau Hyperphosphorylation in Repetitive Mild Traumatic Brain Injury. J Alzheimers Dis 64, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER, Mayer C, Mehic E, Peskind ER, Cook DG, 2013. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis 37, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT, 2012. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J Neurosci 32, 3176–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh L, Deane R, Nedergaard M, 2014. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34, 16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson GL, Gardner AJ, Shultz SR, Solomon GS, McCrory P, Zafonte R, Perry G, Hazrati L-N, Keene CD, Castellani RJ, 2019. Chronic traumatic encephalopathy neuropathology might not be inexorably progressive or unique to repetitive neurotrauma. Brain 142, 3672–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowitz T, Menon D, 2010. Exploring new routes for neuroprotective drug development in traumatic brain injury. Science translational medicine 2, 27rv21–27rv21. [DOI] [PubMed] [Google Scholar]
- Jarrahi A, Braun M, Ahluwalia M, Gupta RV, Wilson M, Munie S, Ahluwalia P, Vender JR, Vale FL, Dhandapani KM, 2020. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 8, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2010. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nature Reviews Neuroscience 11, 361 –370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC, 2018. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MJ, Angoa-Perez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM, 2012. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods 203, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumoto A, Takeuchi H, Tanaka F, 2019. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front Neurol 10, 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Choi H, Lee W, Park H, Kam TI, Hong SH, Nah J, Jung S, Shin B, Lee H, Choi TY, Choo H, Kim KK, Choi SY, Kayed R, Jung YK, 2016. Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis 87, 19–28. [DOI] [PubMed] [Google Scholar]
- Kline AE, Leary JB, Radabaugh HL, Cheng JP, Bondi CO, 2016. Combination therapies for neurobehavioral and cognitive recovery after experimental traumatic brain injury: is more better? Progress in neurobiology 142, 45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokiko-Cochran ON, Godbout JP, 2018. The Inflammatory Continuum of Traumatic Brain Injury and Alzheimer’s Disease. Front Immunol 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, Wei S, Luo ML, Albayram O, Huang P, Rotenberg A, Ryo A, Goldstein LE, Pascual-Leone A, McKee AC, Meehan W, Zhou XZ, Lu KP, 2015. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukowski K, Chou A, Feng X, Tiret B, Paladini MS, Riparip LK, Chaumeil MM, Lemere C, Rosi S, 2018. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukowski K, Nolan A, Frias ES, Grue K, Becker M, Ureta G, Delgado L, Bernales S, Sohal VS, Walter P, Rosi S, 2020. Integrated Stress Response Inhibitor Reverses Sex-Dependent Behavioral and Cell-Specific Deficits after Mild Repetitive Head Trauma. J Neurotrauma 37, 1370–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JE, Parsons JT, Rana A, Gibson CJ, Hamm RJ, Churn SB, 2005. A significant increase in both basal and maximal calcineurin activity following fluid percussion injury in the rat. Journal of neurotrauma 22, 476–490. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R, 2011. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R, 2012. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, Dawson HN, 2010. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma 27, 1983–1995. [DOI] [PubMed] [Google Scholar]
- Lin WL, Dickson DW, Sahara N, 2011. Immunoelectron microscopic and biochemical studies of caspase-cleaved tau in a mouse model of tauopathy. J Neuropathol Exp Neurol 70, 779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX, 2005. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22, 1942–1950. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K, 2012. Trans-synaptic spread of tau pathology in vivo. PLoS One 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK, 2010. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN neuro 3, AN20100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-j., Zheng P, Wright DK, Dezsi G, Braine E, Nguyen T, Corcoran NM, Johnston LA, Hovens CM, Mayo JN, 2016. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139, 1919–1938. [DOI] [PubMed] [Google Scholar]
- Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, Bieri G, Castellano JM, Beaupre GS, Wyss-Coray T, 2014. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Q, Lan W, Sun W, Ye R, Fan X, Ma M, Yin Q, Jiang Y, Xu G, Dai J, Guo R, Liu X, 2014. Intranasal nerve growth factor attenuates tau phosphorylation in brain after traumatic brain injury in rats. J Neurol Sci 345, 48–55. [DOI] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E, 2013. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 16, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A, 2007. Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
- Malpas CB, Vivash L, Genc S, Saling MM, Desmond P, Steward C, Hicks RJ, Callahan J, Brodtmann A, Collins S, 2016. A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. Journal of Alzheimer’s Disease 54, 223–232. [DOI] [PubMed] [Google Scholar]
- Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, Peters NV, Lee C, Tien LW, Kaplan DL, Feany M, Whalen M, 2013. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol 74, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschner L, Ahmed T, Schreurs A, Lechat B, Leuven FV, Mogensen J, Balschun D, 2016. Mild Traumatic Brain Injury of Tau.P301L Mice Results in an Impairment of Neural Plasticity. Archives of Neuroscience 3. [Google Scholar]
- Marshall J, Szmydynger-Chodobska J, Rioult-Pedotti MS, Lau K, Chin AT, Kotla SKR, Tiwari RK, Parang K, Threlkeld SW, Chodobski A, 2017. TrkB-enhancer facilitates functional recovery after traumatic brain injury. Sci Rep 7, 10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAteer KM, Corrigan F, Thornton E, Turner RJ, Vink R, 2016. Short and Long Term Behavioral and Pathological Changes in a Novel Rodent Model of Repetitive Mild Traumatic Brain Injury. PLoS One 11, e0160220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Daneshvar DH, 2015. The neuropathology of traumatic brain injury, Handbook of clinical neurology. Elsevier, pp. 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Daneshvar DH, Alvarez VE, Stein TD, 2014. The neuropathology of sport. Acta Neuropathol 127, 29–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC, 2013. The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meconi A, Wortman RC, Wright DK, Neale KJ, Clarkson M, Shultz SR, Christie BR, 2018. Repeated mild traumatic brain injury can cause acute neurologic impairment without overt structural damage in juvenile rats. PloS one 13, e0197187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Z, Qiu J, Alcon S, Hashim J, Rotenberg A, Sun Y, Meehan WP 3rd, Mannix R, 2018. Memantine improves outcomes after repetitive traumatic brain injury. Behav Brain Res 340, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon B, Saltiel N, Ferguson S, Ojo J, Lungmus C, Lynch C, Algamal M, Morin A, Carper B, Bieler G, Mufson EJ, Stewart W, Mullan M, Crawford F, 2018. Impact of age on acute post-TBI neuropathology in mice expressing humanized tau: a Chronic Effects of Neurotrauma Consortium Study. Brain Inj 32, 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F, 2014. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 75, 241–254. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP, 2012. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 149, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA, 2014. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Molecular neurodegeneration 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Bjorklund GR, Newbern J, Anderson T, 2018. Parvalbumin fast-spiking interneurons are selectively altered by paediatric traumatic brain injury. J Physiol 596, 1277–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira ML, Hamraz M, Abolhassani M, Bigan E, Lafitte O, Steyaert J-M, Dubois B, Schwartz L, 2018. Mechanical stress increases brain amyloid β, tau, and α-synuclein concentrations in wild-type mice. Alzheimer’s & Dementia 14, 444–453. [DOI] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen K-C, Thibault V, Porter NM, Landfield PW, Kraner SD, 2005. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. Journal of Neuroscience 25, 4649–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Scheff SW, 2009. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma 26, 2269–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, 2014. Roles of eIF2alpha kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, Evans J, Mullan M, Bachmeier C, Stewart W, Crawford F, 2016. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J Neuropathol Exp Neurol 75, 636–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Greenberg MB, Bachmeier C, Mullan M, Crawford F, 2013. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol 72, 137–151. [DOI] [PubMed] [Google Scholar]
- Otvos L Jr, Feiner L, Lang E, Szendrei G, Goedert M, Lee VMY, 1994. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. Journal of neuroscience research 39, 669–673. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM, 2016. The integrated stress response. EMBO Rep 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EJ, Cepero ML, Perez SU, Coyle JT, Sick TJ, Liebl DJ, 2016. EphB3 signaling propagates synaptic dysfunction in the traumatic injured brain. Neurobiol Dis 94, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA, 2018. Caspase-Cleaved Tau Impairs Mitochondrial Dynamics in Alzheimer’s Disease. Mol Neurobiol 55, 1004–1018. [DOI] [PubMed] [Google Scholar]
- Perez-Polo JR, Rea HC, Johnson KM, Parsley MA, Unabia GC, Xu GY, Prough D, DeWitt DS, Spratt H, Hulsebosch CE, 2015. A rodent model of mild traumatic brain blast injury. J Neurosci Res 93, 549–561. [DOI] [PubMed] [Google Scholar]
- Peterson AB, Xu L, Daugherty J, Breiding MJ, 2019. Surveillance report of traumatic brain injury-related emergency department visits, hospitalizations, and deaths, United States, 2014. [DOI] [PMC free article] [PubMed]
- Petraglia AL, Plog BA, Dayawansa S, Chen M, Dashnaw ML, Czerniecka K, Walker CT, Viterise T, Hyrien O, Iliff JJ, 2014a. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. Journal of neurotrauma 31, 1211–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, Chen M, Hyrien O, Iliff JJ, Deane R, Huang JH, Nedergaard M, 2014b. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int 5, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham L, Shultz SR, Kim HA, Brady RD, Wortman RC, Genders SG, Hale MW, O’Shea RD, Djouma E, van den Buuse M, 2019. Mild closed-head injury in conscious rats causes transient neurobehavioral and glial disturbances: a novel experimental model of concussion. Journal of neurotrauma 36, 2260–2271. [DOI] [PubMed] [Google Scholar]
- Pijet B, Stefaniuk M, Kaczmarek L, 2019. MMP-9 Contributes to Dendritic Spine Remodeling Following Traumatic Brain Injury. Neural Plast 2019, 3259295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Grafman J, 2015. Traumatic brain injury and late-life dementia, Handbook of clinical neurology. Elsevier, pp. 711–722. [DOI] [PubMed] [Google Scholar]
- Przybyla M, Van Eersel J, van Hummel A, van der Hoven J, Sabale M, Harasta A, Müller J, Gajwani M, Prikas E, Mueller T, 2020. Onset of hippocampal network aberration and memory deficits in P301S tau mice are associated with an early gene signature. Brain. [DOI] [PubMed] [Google Scholar]
- Rabinowitz AR, Levin HS, 2014. Cognitive sequelae of traumatic brain injury. Psychiatr Clin North Am 37, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Li Y, Holloway HW, Kim DS, Ratliff WA, Saykally JN, Citron BA, Hoffer BJ, Greig NH, Pick CG, 2017. Exendin-4 attenuates blast traumatic brain injury induced cognitive impairments, losses of synaptophysin and in vitro TBI-induced hippocampal cellular degeneration. Sci Rep 7, 3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S, 2018. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 28, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin CA, Sun Q, Gamblin TC, 2008. Pre-assembled tau filaments phosphorylated by GSK-3b form large tangle-like structures. Neurobiol Dis 31, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman SU, Ahmad A, Yoon GH, Khan M, Abid MN, Kim MO, 2018. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb Cortex 28, 2854–2872. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW, 2004. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. The Journal of clinical investigation 114, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, Macosko EZ, 2019. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozkalne A, Hyman BT, Spires-Jones TL, 2011. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis 41, 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R, Chang B, Grinkina N, Drummond E, Davies P, Ruditzky M, Sharma D, Wang K, Wisniewski T, 2017. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta neuropathologica communications 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R, Sharma DR, Chang B, Oumata N, Cam M, Vaucelle L, Lindberg MF, Chiu A, Wisniewski T, Wang KKW, Meijer L, 2019. Novel Mouse Tauopathy Model for Repetitive Mild Traumatic Brain Injury: Evaluation of Long-Term Effects on Cognition and Biomarker Levels After Therapeutic Inhibition of Tau Phosphorylation. Front Neurol 10, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI, 2014. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Bender R, Frotscher M, Ross ST, Hollrigel GS, Toth Z, Soltesz I, 2000. Granule cell hyperexcitability in the early post-traumatic rat dentate gyrus: the ‘irritable mossy cell’hypothesis. The Journal of physiology 524, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbeck AD, Gangolli M, Reitz SJ, Salyards MH, Kim SH, Hemingway C, Gratuze M, Makkapati T, Kerschensteiner M, Holtzman DM, Brody DL, Kummer TT, 2020. SEQUIN Multiscale Imaging of Mammalian Central Synapses Reveals Loss of Synaptic Connectivity Resulting from Diffuse Traumatic Brain Injury. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawmiller D, Li S, Shahaduzzaman M, Smith AJ, Obregon D, Giunta B, Borlongan CV, Sanberg PR, Tan J, 2014. Luteolin reduces Alzheimer’s disease pathologies induced by traumatic brain injury. Int J Mol Sci 15, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C, 2005. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma 22, 719–732. [DOI] [PubMed] [Google Scholar]
- Sen T, Gupta R, Kaiser H, Sen N, 2017. Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J Neurosci 37, 5900–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GJ, Jauch EC, Zemlan FP, 2002. Serum cleaved tau protein levels and clinical outcome in adult patients with closed head injury. Ann Emerg Med 39, 254–257. [DOI] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA, 2017. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med 9, eaaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz SR, Wright DK, Zheng P, Stuchbery R, Liu SJ, Sashindranath M, Medcalf RL, Johnston LA, Hovens CM, Jones NC, O’Brien TJ, 2015. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138, 1297–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siahaan AMP, Japardi I, Rambe AS, Indharty RS, Ichwan M, 2018. Turmeric Extract Supplementation Reduces Tau Protein Level in Repetitive Traumatic Brain Injury Model. Open access Macedonian journal of medical sciences 6, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Xiong G, Elkind JA, Putnam B, Cohen AS, 2015. Brain Injury Impairs Working Memory and Prefrontal Circuit Function. Front Neurol 6, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MAG, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, Paulino AJ, Wicinski B, Shaughness MC, Maudlin-Jeronimo E, 2014. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta neuropathologica communications 2, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statler KD, Alexander H, Vagni V, Holubkov R, Dixon CE, Clark RS, Jenkins L, Kochanek PM, 2006. Isoflurane exerts neuroprotective actions at or near the time of severe traumatic brain injury. Brain research 1076, 216–224. [DOI] [PubMed] [Google Scholar]
- Steinhilb ML, Dias-Santagata D, Fulga TA, Felch DL, Feany MB, 2007. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol Biol Cell 18, 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman MA, McKee AC, Stein TD, Tripodis Y, Besser LM, Martin B, Palmisano JN, Steinberg EG, O’Connor MK, Au R, 2019. Failure to detect an association between self-reported traumatic brain injury and Alzheimer’s disease neuropathology and dementia. Alzheimer’s & Dementia 15, 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Jacobs KM, 2016. Knockout of Cyclophilin-D Provides Partial Amelioration of Intrinsic and Synaptic Properties Altered by Mild Traumatic Brain Injury. Front Syst Neurosci 10, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YY, Zhu L, Sun ZL, Feng DF, 2020. CRMP2 improves memory deficits by enhancing the maturation of neuronal dendritic spines after traumatic brain injury. Exp Neurol 328, 113253. [DOI] [PubMed] [Google Scholar]
- Tan XL, Wright DK, Liu S, Hovens C, O’Brien TJ, Shultz SR, 2016. Sodium selenate, a protein phosphatase 2A activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology 108, 382–393. [DOI] [PubMed] [Google Scholar]
- Titus DJ, Sakurai A, Kang Y, Furones C, Jergova S, Santos R, Sick TJ, Atkins CM, 2013. Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J Neurosci 33, 5216–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Wilson NM, Freund JE, Carballosa MM, Sikah KE, Furones C, Dietrich WD, Gurney ME, Atkins CM, 2016. Chronic Cognitive Dysfunction after Traumatic Brain Injury Is Improved with a Phosphodiesterase 4B Inhibitor. J Neurosci 36, 7095–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, LaFerla FM, Holtzman DM, Brody DL, 2011a. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31, 9513–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Sanchez L, Brody DL, 2012. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol 71, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Sanchez L, Esparza TJ, Brody DL, 2011b. Distinct temporal and anatomical distributions of amyloid-beta and tau abnormalities following controlled cortical impact in transgenic mice. PLoS One 6, e25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL, 2017. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. The Lancet Neurology 16, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KR, Tesco G, 2013. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Frontiers in aging neuroscience 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Han S, 2018a. Exosome-associated tau exacerbates brain functional impairments induced by traumatic brain injury in mice. Molecular and Cellular Neuroscience 88, 158–166. [DOI] [PubMed] [Google Scholar]
- Wang B, Han S, 2018b. Inhibition of Inducible Nitric Oxide Synthase Attenuates Deficits in Synaptic Plasticity and Brain Functions Following Traumatic Brain Injury. Cerebellum 17, 477–484. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hall RA, Lee M, Kamgar-Parsi A, Bi X, Baudry M, 2017a. The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci Rep 7, 11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z-F, Pan Z-Y, Xu C-S, Li Z-Q, 2017b. Activation of G-protein coupled estrogen receptor 1 improves early-onset cognitive impairment via PI3K/Akt pathway in rats with traumatic brain injury. Biochemical and biophysical research communications 482, 948–953. [DOI] [PubMed] [Google Scholar]
- Weiner MW, Crane PK, Montine TJ, Bennett DA, Veitch DP, 2017. Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology 89, 1923–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White ER, Pinar C, Bostrom CA, Meconi A, Christie BR, 2017. Mild Traumatic Brain Injury Produces Long-Lasting Deficits in Synaptic Plasticity in the Female Juvenile Hippocampus. J Neurotrauma 34, 1111–1123. [DOI] [PubMed] [Google Scholar]
- Wilson NM, Titus DJ, Oliva AA Jr., Furones C, Atkins CM, 2016. Traumatic Brain Injury Upregulates Phosphodiesterase Expression in the Hippocampus. Front Syst Neurosci 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP, 2013. Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J Neurotrauma 30, 1966–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Noel A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, Wilkins TE, Alikhani AD, Zapple DN, Villapol S, Planel E, Burns MP, 2016. Dendritic Spine Loss and Chronic White Matter Inflammation in a Mouse Model of Highly Repetitive Head Trauma. Am J Pathol 186, 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Molteni R, Ying Z, Gomez-Pinilla F, 2003. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience 119, 365–375. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT, 2010. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci 30, 2636–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nature neuroscience 19, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Wang ZH, Liu X, Zhang Z, Gu X, Yu SP, Keene CD, Cheng L, Ye K, 2020. Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer’s disease pathology. Prog Neurobiol 185, 101730. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M, 2013. Animal models of traumatic brain injury. Nat Rev Neurosci 14, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, W Rosler T, Carlsson T, de Andrade A, Fiala O, Hollerhage M, H Oertel W, Goedert M, Aigner A, U Hoglinger G, 2014. Tau silencing by siRNA in the P301S mouse model of tauopathy. Current gene therapy 14, 343–351. [DOI] [PubMed] [Google Scholar]
- Xu L, Nguyen JV, Lehar M, Menon A, Rha E, Arena J, Ryu J, Marsh-Armstrong N, Marmarou CR, Koliatsos VE, 2016. Repetitive mild traumatic brain injury with impact acceleration in the mouse: Multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system. Exp Neurol 275 Pt 3, 436–449. [DOI] [PubMed] [Google Scholar]
- Xu L, Ryu J, Nguyen JV, Arena J, Rha E, Vranis P, Hitt D, Marsh-Armstrong N, Koliatsos VE, 2015. Evidence for accelerated tauopathy in the retina of transgenic P301S tau mice exposed to repetitive mild traumatic brain injury. Exp Neurol 273, 168–176. [DOI] [PubMed] [Google Scholar]
- Yaka R, Biegon A, Grigoriadis N, Simeonidou C, Grigoriadis S, Alexandrovich AG, Matzner H, Schumann J, Trembovler V, Tsenter J, Shohami E, 2007. D-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J 21, 2033–2041. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Hasuo H, Tokutomi T, Shigemori M, Akasu T, 2011. Edaravone attenuates impairment of synaptic plasticity in granule cell layer of the dentate gyrus following traumatic brain injury. Kurume Med J 58, 47–58. [DOI] [PubMed] [Google Scholar]
- Yin Y, Gao D, Wang Y, Wang Z-H, Wang X, Ye J, Wu D, Fang L, Pi G, Yang Y, 2016. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proceedings of the National Academy of Sciences 113, E3773–E3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, Fujimoto S, McIntosh T, Lee VM, Trojanowski JQ, 2005. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma 22, 1134–1141. [DOI] [PubMed] [Google Scholar]
- Zanier ER, Bertani I, Sammali E, Pischiutta F, Chiaravalloti MA, Vegliante G, Masone A, Corbelli A, Smith DH, Menon DK, Stocchetti N, Fiordaliso F, De Simoni MG, Stewart W, Chiesa R, 2018. Induction of a transmissible tau pathology by traumatic brain injury. Brain 141, 2685–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlan FP, Jauch EC, Mulchahey JJ, Gabbita SP, Rosenberg WS, Speciale SG, Zuccarello M, 2002. C-tau biomarker of neuronal damage in severe brain injured patients: association with elevated intracranial pressure and clinical outcome. Brain Res 947, 131–139. [DOI] [PubMed] [Google Scholar]
- Zhang D, Xiao M, Wang L, Jia W, 2019a. Blood-Based Glutamate Scavengers Reverse Traumatic Brain Injury-Induced Synaptic Plasticity Disruption by Decreasing Glutamate Level in Hippocampus Interstitial Fluid, but Not Cerebral Spinal Fluid, In Vivo. Neurotox Res 35, 360–372. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chopp M, Rex CS, Simmon VF, Sarraf ST, Zhang ZG, Mahmood A, Xiong Y, 2019b. A Small Molecule Spinogenic Compound Enhances Functional Outcome and Dendritic Spine Plasticity in a Rat Model of Traumatic Brain Injury. J Neurotrauma 36, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wu F, Iqbal K, Gong CX, Hu W, Liu F, 2019c. Subacute to chronic Alzheimer-like alterations after controlled cortical impact in human tau transgenic mice. Sci Rep 9, 3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang JZ, Cheng L, Sun YE, Yu SP, Levey AI, Ye K, 2014. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20, 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M-L, Chen S-J, Li X-H, Wang L-N, Chen F, Zhong S-J, Yang C, Sun S-K, Li J-J, Dong H-J, 2018. Optical depolarization of DCX-expressing cells promoted cognitive recovery and maturation of newborn neurons via the Wnt/β-Catenin pathway. Journal of Alzheimer’s Disease 63, 303–318. [DOI] [PubMed] [Google Scholar]
- Zhao S, Gao X, Dong W, Chen J, 2016. The Role of 7,8-Dihydroxyflavone in Preventing Dendrite Degeneration in Cortex After Moderate Traumatic Brain Injury. Mol Neurobiol 53, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhao Y, Ning Y, Yang N, Peng Y, Li P, Chen X, Liu D, Wang H, Chen X, 2017a. Adenosine A 2A receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Translational psychiatry 7, e1123–e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z-A, Li P, Ye S-Y, Ning Y-L, Wang H, Peng Y, Yang N, Zhao Y, Zhang Z-H, Chen J-F, 2017b. Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A 2A receptor inactivation. Scientific reports 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZA, Ning YL, Li P, Yang N, Peng Y, Xiong RP, Zhao Y, Liu D, Zeng XJ, Chen JF, Zhou YG, 2017c. Widespread hyperphosphorylated tau in the working memory circuit early after cortical impact injury of brain (Original study). Behav Brain Res 323, 146–153. [DOI] [PubMed] [Google Scholar]
- Zheng P, Shultz SR, Hovens CM, Velakoulis D, Jones NC, O’Brien TJ, 2014. Hyperphosphorylated tau is implicated in acquired epilepsy and neuropsychiatric comorbidities. Molecular neurobiology 49, 1532–1539. [DOI] [PubMed] [Google Scholar]
- Zilka N, Kovacech B, Barath P, Kontsekova E, Novák M, 2012. The self-perpetuating tau truncation circle. Portland Press Ltd. [DOI] [PubMed] [Google Scholar]