

Abstract
The discovery, only a decade ago, of the genome editing power of clustered regularly interspaced short palindromic repeats (CRISPR)-associated nucleases is already reinventing the therapeutic process, from how new drugs are discovered to novel ways to treat diseases. CRISPR-based screens can aid therapeutic development by quickly identifying a drug’s mechanism of action and escape mutants. Additionally, CRISPR-Cas has advanced emerging ex vivo therapeutics, such as cell replacement therapies. However, Cas9 is limited as an in vivo therapeutic due to ineffective delivery, unwanted immune responses, off-target effects, unpredictable repair outcomes, and cellular stress. To address these limitations, controls that inhibit or degrade Cas9, biomolecule–Cas9 conjugates, and base editors have been developed. Herein, we discuss CRISPR-Cas systems that advance both conventional and emerging therapeutics.
Introduction
The genome editing power of CRISPR-Cas nucleases has propelled the development of both conventional and emerging therapeutic modalities. These nucleases (such as Streptococcus pyogenes Cas9, herein Cas9) are directed by a guide RNA (gRNA; see Glossary) to recognize a complementary target DNA sequence next to a protospacer adjacent motif (PAM) sequence. After a double-strand break (DSB) is introduced by the nuclease, the target DNA undergoes repair through the error-prone non-homologous end joining (NHEJ) pathway or the more precise homology directed repair (HDR) pathway. The ability of Cas9 to (i) recognize nucleic acid sequences with high affinities and (ii) precisely cut a target gene to trigger a repair mechanism to insert the desired sequence has led to the development of several biological tools and translational applications. The CRISPR-Cas field has seen unprecedented growth in under a decade owing to its ease of use, modular and versatile nature, and robust activity across myriad organisms [1,2]. Here, we describe how CRISPR-based tools are propelling the development of conventional therapeutic modalities (e.g., small molecules) and emerging therapeutic modalities (e.g., cell replacement therapies). Finally, we discuss the challenges in developing CRISPR-Cas systems as a therapeutic and diagnostic.
Target discovery and (in)validation
High-throughput CRISPR-based screens can rapidly identify genetic contributors to disease and, therefore, new drug targets and drivers of drug resistance. CRISPR screens directly connect genetic perturbations to their phenotype and are executed through standard techniques including CRISPR knockout (CRISPRko), CRISPR activation (CRISPRa), CRISPR interference (CRISPRi), or base editing (Figure 1A). CRISPR mutagenesis screens, which mutate a single target protein or subset of proteins (Figure 1B), have also been used to interrogate ligand–target interactions and escape mutants. The wide application of CRISPR in drug target discovery is evidenced by more than 1000 CRISPR screens conducted by independent groupsi.
Figure 1. (A) CRISPR screening workflows begin with the selection of the screening system.
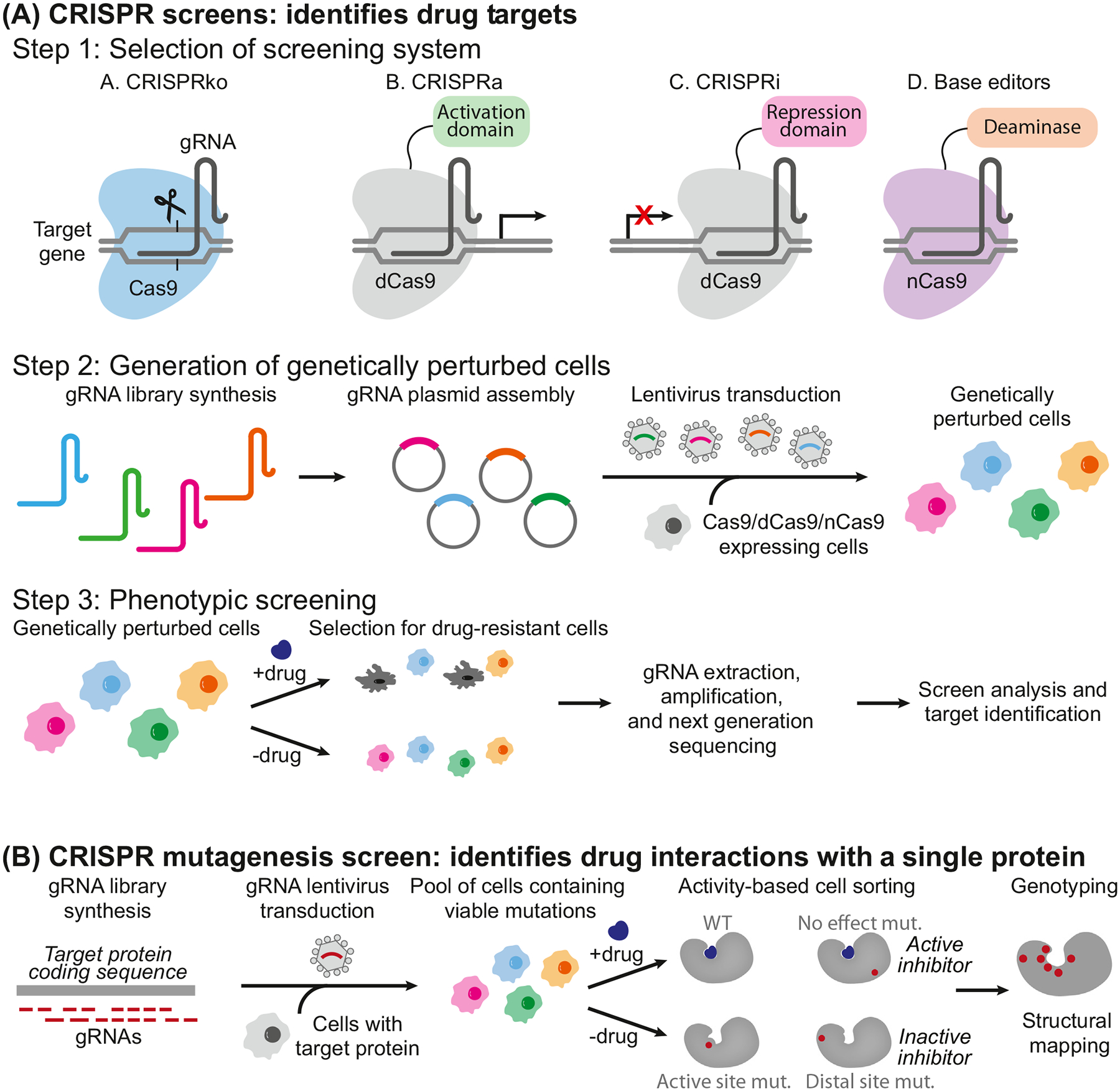
CRISPRko: Cas9 knocks out to generate premature stop codons or frameshifts; CRISPRa: activation domains (e.g., VPR, VP64) tethered to dCas9 increase transcription of target genes; CRISPRi: repression domains (e.g., KRAB) tethered to dCas9 decrease transcription of target genes; base editing screen tethers a base editor (e.g., cytosine deaminase or adenine deaminase) with or without uracil DNA glycosylase inhibitor (not shown) to create mutations without double-strand breaks. Next, the gRNA library is transduced to cells to create a genetically perturbed population of cells that are treated with drugs to select for drug-resistant populations. gRNAs are then extracted from the cells for PCR amplification, and next-generation sequencing identifies the targets. (B) CRISPR mutagenesis screening starts with a gRNA library to create in-frame mutations in the target protein coding sequence. After cells are transduced with the gRNA library, viable cells harboring protein variants are then treated with and without the drug. Activity-based cell sorting enriches cells harboring mutations that render the drug inactive (e.g., drug-resistant cells). Finally, enriched cells are genotyped by deep sequencing for structural mapping and to identify escape mutants. Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; CRISPRa, CRISPR activation; CRISPRi, CRISPR interference; CRISPRko, CRISPR knockout; dCas9, dead Cas9; gRNA, guide RNA; KRAB, Krüppel-associated box; nCas9, Cas9 nickase.
Furthermore, the Cancer Dependency Map (DepMap) consortium aims to use CRISPR-Cas9 screens to catalog essential genes in over 2000 cancer cell linesii. For example, CRISPR screening across multiple cell lines and cancers identified drivers of tumorigenesis and thus viable therapeutic targets [3]. Here, we discuss how these screening approaches are contributing to drug discovery and validation.
CRISPRko/a/i or base editor screens for target identification and mechanism-of-action studies
CRISPR screens are classified by type of screening system (Figure 1A, step 1). CRISPRko screens use active Cas9 to create premature stop codons or frameshifts in open reading frames to abolish the production of functional proteins. CRISPRa screens fuse transcriptional activation domains, such as VPR or VP64, to a catalytically dead Cas9 (dCas9) to increase the transcription of target genes, while CRISPRi screens fuse repression domains, such as Krüppel-associated box (KRAB), to dCas9 to decrease target transcription [4–6]. As a more recent strategy, base editors fuse an adenine or cytosine deaminase with or without a DNA glycosylase inhibitor to either dCas9 or catalytically impaired nuclease Cas9 nickase (nCas9) to create mutations without DSBs [7,8]. Cytosine base editors (CBEs) convert a cytosine•guanine (C•G) into a thymine•adenine (T•A) base pair [7], and adenine base editors (ABEs) convert an A•T into a G•C base pair [8]. Collectively, CBEs and ABEs can mediate all four possible transition mutations (C to T, A to G, T to C, and G to A).
To perturb the expression of thousands of genes in parallel, typical genome-wide CRISPR screens start with the high-throughput synthesis of gRNA libraries (Figure 1A, step 2), which are packaged into lentiviruses to transduce cells stably expressing Cas9, dCas9, or nCas9. Edited cells are put under selective pressure, such as treatment with a drug of interest, to enrich drug-resistant populations of cells. To identify genes or gRNAs responsible for driving drug resistance or sensitivity, the gRNAs are then extracted for PCR amplification and next-generation sequencing (Figure 1A, step 3). Compared with traditional phenotypic screening, where researchers may not know the drug target that produces a biological event, these CRISPR screens directly connect a phenotypic readout to the genes responsible for the phenotype.
Before CRISPR, target discovery screens were commonly performed by RNA interference (RNAi), which uses short hairpin RNAs (shRNAs) or siRNAs to knockdown RNA transcripts by hijacking mammalian machinery [9]. RNAi suffers from incomplete knockdowns (leading to false negatives) and high off-target effects (leading to false positives), which results in high levels of noise and inconsistent results [10]. Early side-by-side comparisons of RNAi and CRISPR screens demonstrated that CRISPR-based screening outperformed, providing higher signal-to-noise ratio, less off-target effects, and higher reproducibility [10–12]. While RNAi only interferes at the RNA level, CRISPRkos perturb both genomic (DNA) and transcriptomic (RNA) levels, which may identify a different set of drug-related targets. Performing both CRISPRko and RNAi screens can provide a complete picture of mechanism-of-action studies. For example, Bassik and coworkers performed parallel shRNA knockdown and Cas9 knockout to elucidate the previously unexplained toxicity of the antiviral GSK983 [13]. Not surprisingly, hits from the two screens did not overlap entirely, presumably because RNAi and CRISPR screens operate via different mechanisms; in some cases, the RNA knockdown was insufficient to affect the phenotype, and in CRISPRko screens, targeting essential genes can be lethal [14]. Nonetheless, the complementary screens allowed for a complete picture of the mechanism of action and identified dihydroorotate dehydrogenase as the target [13].
CRISPR screens have successfully provided hypothesis-free evaluations of drug–target interactions to elucidate a drug’s mechanism of action. Several complementary genome-wide screens have been performed to investigate drug interactions in BRAFV600E melanoma cell lines. For example, the Zhang group used a CRISPRko screen to identify novel negative regulators of BRAFV600E melanoma resistance to vemurafenib, such as NF1, MED12, and CUL3 [15]. CRISPRa screens have also been used to identify proteins mediating resistance to the BRAF inhibitor PLX-4720 in these BRAFV600E melanoma cell lines, identifying BCAR3 and EGFR, whose activation was previously implicated in driving drug resistance in several cancers [6]. In addition, Li et al. also investigated mediators of resistance to PLX-4720 in using BRAFV600E cells, but instead of an activation or repression domain, fused a kinase to dCas9 to perturb gene expression [16]. Furthermore, genome-wide screens using dCas9 fused with p300 or KRAB as epigenetic modulators have been employed to identify noncoding regulatory elements of β-globin and human epidermal growth factor receptor 2 (HER2), which may shed light on the influence of the noncoding genome on drugs modulating proteins of interest [17]. Moreover, CRISPR screens have also been used to provide information on target invalidation. For example, Jost et al. used a dual CRISPRa/i screen to show that rigosertib, whose mechanism of action was controversial, is a microtubule-destabilizing agent [19]. More recently, Lin et al. hypothesized that a mischaracterization of drug targets caused 97% of compounds in trials to never progress to United States Food and Drug Administration (FDA) approval [20]. They used CRISPR screening to investigate the off-target toxicity of compounds undergoing clinical trials, showing that the tested drugs were surprisingly unaffected by the loss of their identified target as they often kill cells via off-target effects. This suggested that comprehensive analyses, like those performed in their study, could decrease the failure rate for FDA approval [18]. This handful of examples shows that CRISPR screens are valuable tools for identifying drug on- and off-targets, even if their mechanism is thought to be largely understood.
Despite the apparent successes of CRISPR screens, each has its limitations. CRISPRko screens occasionally suffer from low cutting efficiencies and rely on DNA repair pathways to create heterogeneous populations of cells [21]. Furthermore, DSBs created by nucleases can cause toxicity and cell death [22,23], particularly in cancer cells with highly amplified genomic regions, which can lead to false positives [24]. Second-generation CRISPR screens have improved on this limitation through base editors, which convert a single base without creating DSBs [25]. For example, the Kim group recently used base editors to tile in stop codons to create a knockout screen for BRCA1 variants [26], and Doench and colleagues optimized CBEs for pooled screening to identify loss-of-function mutations in BRCA1 and BRCA2 [27]. However, the Joung group demonstrated that DNA base editors could induce transcriptome-wide off-targets, which should be accounted for when using base editors for genetic screening [28]. Efforts to reduce off-target RNA editing by base editors using different deaminase variants or manipulating the steric structural relationship between Cas9 and base editors have recently been described [29].
Additionally, CRISPRa and CRISPRi can be limited by high off-target effects, particularly in overlapping promoter regions (i.e., bidirectional promoters), since dCas9–VP64 or dCas9–KRAB binding can interfere with the expression of off-target neighboring genes [30]. This is a significant criticism for CRISPRa and CRISPRi, since observed effects may not perturb the gene of interest. Therefore, CRISPR screens that use the RNA-cutting nuclease Cas13, which does not cut DNA or interfere with promoters, may overcome these limitations and are often preferred [31]. Remaining challenges include adequately designing gRNAs to minimize off-target editing with high on-target efficiency and developing robust readout assays to ensure reproducible results with minimal false positives, which should be accounted for in every screen. Much progress has been made in this latter area with the development of CRISPR screening algorithms to better identify essential genes and minimize false positives. These algorithms, such as CERES and MAGeCK, can account for gene copy number, which can confound screening results due to unwanted DNA damage effects caused by CRISPR-induced DSBs at amplified genomic regions [32–35].
Recent advances combining CRISPRko screens with T cell killing assays may provide new approaches for developing drugs that modulate the immune response for cancer immunotherapy [36]. Likewise, recent developments of CRISPR screens for cancer 3D spheroids/organoids [37], which recapitulate the patient drug response, may propel the development of more CRISPR screens on organoids to evaluate drug action and resistance mechanisms. Further CRISPR screening studies on pathogen (e.g., virus and bacteria) interactions with host cells may shed light on the mechanism of infection and identify new targets for drug development to tackle future pandemics. For instance, a recent genome-wide CRISPR screening study identified cathepsin L, SMARCA4, and SMAD3 as novel host genes required for SARS-CoV-2 pathogenesis [38]. Small-molecule inhibitors of the three genes protected from virus-induced cell death and reduced viral infection frequency. Please see [9] for further reviews on CRISPR screening.
CRISPR mutagenesis screening
Unlike CRISPRko/a/i screens that sample a large portion of the genome, CRISPR mutagenesisor suppressor-scanning screens sample a particular protein, a small set of proteins, or a genomic region (e.g., enhancer) of interest using target-specific gRNAs in a pooled format (Figure 1B). Here, in-frame mutations produced by Cas9 during treatment can identify drug-resistant escape mutants and map the drug binding site, making this screen an effective hypothesis generator [39]. In addition to using this technology to interrogate functional protein domains [40] or genomic regulatory elements [41], CRISPR mutagenesis screening has immense potential for identifying novel drug mechanisms. For example, dCas9 recruits a hyperactive version of the activation-inducing cytidine deaminase (AID), termed CRISPR-X, and was used for a CRISPR mutagenesis screen employing 143 gRNAs tiling all coding exons of the PSMB5 protein to identify mutations that confer resistance to bortezomib, a proteasome inhibitor [42]. The hyperactive AID can create mutations at C or G bases of the tiled regions. The screen identified five mutations that strongly protect against bortezomib killing, including three novel mutations that likely disrupt the bortezomib-binding pocket and one new mutation in the protein domain distal to the binding pocket. Similarly, dCas9 fused to AID was used for a CRISPR mutagenesis screen targeting exon 6 of the ABL gene in the chronic myeloid leukemia cell line K562. This screen identified six new missense mutations that give rise to imatinib-resistant clones in addition to known imatinib-resistance mutations [43]. nCas9 was also fused with a fidelity-reduced variant of Escherichia coli DNA polymerase I for CRISPR mutagenesis screening, termed EvolvR [44]. Although the approach was limited to E. coli, the method could create all single-base substitutions (not restricted to C or G bases) and identified novel mutations in the ribosomal gene, rpsE, that confer spectinomycin resistance by disrupting the spectinomycin-binding pocket of the 30S ribosome [44].
In addition to dCas9 and nCas9, CRISPR mutagenesis screening was recently performed using Cas9 to generate in-frame mutations in the lysine-specific histone demethylase 1 (LSD1) gene to identify those that confer resistance to LSD1 inhibitors in acute myeloid leukemia (AML). Here, Liau and coworkers found that the inhibitors block AML proliferation not by disrupting LSD1 demethylase activity as expected, but by disrupting a complex of LSD1 with growth factor independent 1B transcriptional repressor (GFI1B) on chromatin, affecting GFI1B’s ability to repress the activity of the hematopoietic transcription factor PU.1, a cancer driver in AML [39].
A general challenge for CRISPR suppressor scanning includes adequately covering the protein of interest since the binding sites of Cas9 are restricted to NGG PAM sequences and can be difficult for larger targets. Combining Cas9 with Cas12, a more compact version of Cas9 recognizing a different PAM site (i.e., TTTV), may improve gRNA coverage of CRISPR mutagenesis scanning. An evolved version of Cas12, recognizing PAM sites beyond TTTV, employed for CRISPR mutagenesis screens uncovered new drug resistance mutations against inhibitors of NAMPT and KIF11 [45]. Evolved variants of Cas9 and Cas12 with broadened PAM compatibility may also address the coverage issue [46,47]. Another major drawback for CRISPR suppressor scanning is that Cas9-based screens can add random insertions and deletions due to NHEJ-mediated repair that result in out-of-frame mutations, abolishing the expression of proteins of interest instead of creating mutants. This issue can potentially be subverted using base or prime editors to tile in the gRNAs across the gene target to introduce the mutations (in-frame mutations). For additional reviews comparing CRISPR systems, multiplexing CRISPR technologies, and designing CRISPR screens, please see [48–50], respectively.
Emerging therapeutic modalities
Despite a growing CRISPR toolbox and the existence of over 800 cell and gene therapy programs, only a few CRISPR-based tools have progressed past preclinical trials [51,52]. Gene editing in clinical settings, particularly through CRISPR predecessors transcription activator-like effector nucleases (TALENs) and zinc finger nucleases (ZFNs), has been reviewed elsewhere [53]. Here, we discuss how CRISPR-Cas systems are propelling the development of ex vivo cell replacement therapies (Figure 2, left) and how they are being used for in vivo therapeutics (Figure 2, right) [72].
Figure 2. CRISPR-based cell-editing strategies in patients.
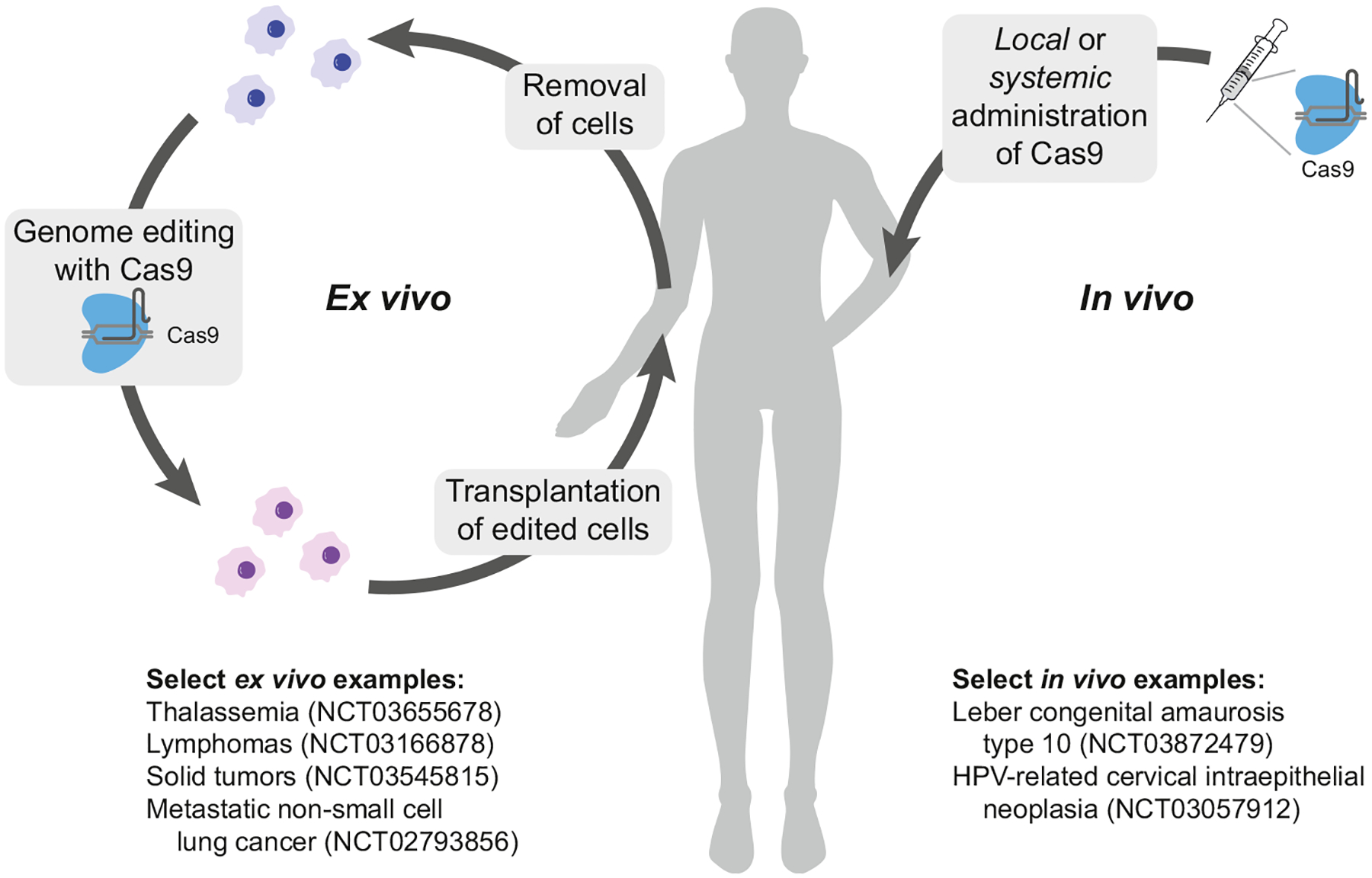
(Left) Ex vivo applications of CRISPR-Cas9 editing begin with the isolation of cells. Cells are typically expanded and then edited and filtered before being transplanted. (Right) For in vivo editing, CRISPR-Cas9 (or dCas9, not shown) is administered locally or systemically with the help of viral packaging or nanoparticles. Select clinical trials are highlighted in the figure. Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; dCas9, dead Cas9; HPV, human papillomavirus.
Cell replacement therapies
Cancer immunotherapy
To potentiate and customize adoptive cell therapies, immune cells should be engineered through gene knockout or transgene expression. For example, immune checkpoint receptors, such as programmed cell death protein 1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), have been safely and feasibly knocked out using Cas9 in T cells to improve their antitumor activity [54,55].
Chimeric antigen receptor (CAR)-T cells, which are generated by the introduction of CAR transgene into T cells, greatly advanced adoptive cell therapies through efficient cancer targeting and the stimulation of T cell activation signaling. Traditional use of viral vectors for transgene expression in CAR-T cells poses safety issues, such as immunogenicity to viral factors and potential oncogenesis arising from viral sequence integration. In addition, variable CAR expression complicates the quality control of cell therapeutics. To solve these, CRISPR-Cas can precisely knock in CAR transgenes at a defined genomic locus. For example, a CD19-specific CAR was inserted into the endogenous T cell receptor α constant (TRAC) locus, ensuring the safety of CAR-T cells while enhancing T cell potency [56].
Most clinical trials in this area have used autologous CAR-T cells. However, these cell therapies are not feasible in many patients due to the low quantity and quality of T cells, advanced disease states that require immediate treatment, or the high cost of manufacturing individual cell therapies [57,58]. The development of universal allogeneic CAR-T cells will broaden adoptive cell therapies by circumventing the aforementioned limitations [59]. Multiplex gene editing by CRISPR-Cas is becoming a key tool for generating safe and effective allogeneic CAR-T cells [60]. Additionally, CRISPR-Cas has been used to generate other types of therapeutic cells, such as CAR-natural killer cells and CAR-macrophages, which would overcome the limitations of T cell-based therapies, such as insufficient targeting of solid tumors [61,62].
Therapies for hemoglobinopathies and hematopoietic diseases
Hemoglobinopathies, such as sickle cell disease and β-thalassemia, arise from mutations in the β-globin gene cluster – thus representing an attractive area for genome editing. Studies have shown that CRISPR-Cas9 can successfully delete a large fragment at the β-globin locus, reactivating fetal γ-globin expression in adult erythroblasts and reversing sickle cell disease and β-thalassemia [63,64]. Similarly, knock in of the glucocerebrosidase gene within hematopoietic stem cells restores enzyme function in a Gaucher disease model [65]. CRISPR-Cas9 therapy was also useful in X-linked chronic granulomatous disease (X-CGD), in which correction of the CYBB mutations within immune cells produced functional human myeloid and lymphoid cells in a mouse model [66].
Endocrine cells for diabetes
Transplanting insulin-producing β cells is an effective and ultimate therapy for treating type 1 diabetes (T1D); though supply of primary β cells is extremely limited, methods to generate β cells from pluripotent stem cells are being extensively studied [67,68]. Here, pluripotent stem cells can be engineered by CRISPR-Cas to correct diabetes-causing mutations, ultimately giving functional stem cell-derived β cells (SC-β cells). For example, mutations at the insulin gene (INS) or mutations at the Wolfram syndrome (WSF1) gene that lead to misfolding, endoplasmic reticulum (ER) stress, and eventual β-cell death were corrected in T1D patient-derived iPSCs using CRISPR-Cas9. The corrected iPSCs were then differentiated into SC-β cells that displayed normal insulin folding and function, suggesting clinical utility [69,70]. In the future, CRISPR-Cas could be used to engineer β cells to circumvent inherent limitations of SC-β cells, including insufficient glucose-stimulated insulin secretion and vulnerability to immune attack after transplantation. For example, we engineered a β cell line to secrete interleukin-10, an anti-inflammatory factor that can protect β cells from the immune system [71]. Based on this example, we envision that regenerative medicine will benefit from ex vivo CRISPR editing, and we will examine the efficacy of this strategy in future in vivo animal models.
Therapies based on in vivo editing
The aforementioned cell therapies were demonstrated ex vivo, which involves cell extraction, gene editing, and transplantation. In contrast, in vivo systems edit directly within the body, and as a result, far fewer clinical trials with CRISPR have been performed in vivo (Figure 2, right) [72]. Several of the earliest preclinical efforts in this area used the enzyme to reverse a point mutation causing muscular dystrophy [73–75]. Successful in vivo use of CRISPR-Cas was also demonstrated in certain liver diseases and cancers [72]. Additionally, gene editing is particularly advantageous for genetic conditions related to blindness, as the eye is immune privileged, thereby reducing off-target risks [53]. For example, CRISPR was successfully applied in vivo in a mouse model of Leber congenital amaurosis, where it cured blindness derived from a mutation in the CEP290 gene [76]. Recently, a clinical trial (NCT04601051) established the safety and therapeutic efficiency of lipid nanoparticle (LNP) CRISPR-Cas9-mediated knockout of transthyretin in a small group of patients with hereditary transthyretin amyloidosis [77].
Despite the early successes of in vivo CRISPR-Cas therapies, some technical limitations remain, including in vivo delivery challenges and immune activation. Reports have shown pre-existing immunity to Cas in vivo; however, these data are limited and conflicting depending on the investigated population – one report measured pre-existing immunity to Cas9 at 58%, while another report recorded 2.5%. However, both reports agree there is higher pre-existing immunity to SaCas9 over SpCas9 [78,79]. Next, tissue-specific delivery of Cas9 is required for safe genome editing, which is currently being addressed through the development of modified LNPs for delivering Cas9 RNP into muscle, brain, liver, and lung tissue [80] and for the delivery of Cas9 mRNA into the liver, spleen, and lung [81]. Though these examples have demonstrated the possibility of tissue-specific editing, there was still some degree of unintended editing in non-targeted tissues, thus requiring more elaborate systems for tissue-specific editing. Additionally, methods for specific genome editing of other tissues should also be developed.
In addition to immunogenicity and tissue delivery, several other critical questions remain. First, although CRISPR off-targeting occurs at a slower rate than on-targeting [82], precise controls such as inhibitors and degraders are necessary to reduce off-target editing by constitutively active Cas9 [83]. Second, several groups, including ours, have explored conjugation chemistries to enhance CRISPR for a variety of purposes including biasing the desired editing outcome. Third, the DNA damage response after a Cas9-induced DSB can cause low editing efficiencies and cell death in certain cell types [22,23], as well as an innate immune activation response following the DSB [84]. In the next section, we highlight how these therapeutic challenges are being addressed by basic research to further improve CRISPR-based therapeutics.
Base and prime editors
In addition to using ABEs and CBEs in the aforementioned CRISPR screens, they can also be used to correct point mutations, the largest class of known human pathogenic mutations [85]. Additionally, there are new classes of base editors that can perform concurrent adenine and cytosine base editing [86]. Base editors are beneficial because they have fewer cytotoxic effects than Cas9, as their nickase activity produces clean deaminated products, unlike the DSBs from Cas9 that produce a mixture of insertion and deletion outcomes [85]. The non-disruptive editing capability of base editors was demonstrated by the Liu group, who used a CBE-based system to edit human mitochondrial DNA (mtDNA) with high target specificity and product purity [87]. Furthermore, base editing technologies showed promising preclinical results in devastating genetic disorders like Hutchinson-Gilford progeria, where base editors corrected a critical C-to-T point mutation in the gene encoding lamin A, improving the life span of the mice by 78% [88]. Recently, Musunuru et al. reported the therapeutic benefits of LNP-based delivery of ABEs in the treatment of proprotein convertase subtilisin/kexin type 9 serine protease (PCSK9)-mediated cardiovascular disorder in primates. They also reported no substantial off-target editing or serious long-term immune complications in primate models, encouraging clinical trials [89]. Therapeutic applications of base editors have been extensively reviewed elsewhere [90,91].
Despite multiple successes in preclinical models across several diseases, there are several limitations of base editors. First, specificity remains a major challenge as base editors are prone to off-target and transcriptome-wide deamination [28,92,93]. Reductions in off-target rates have been achieved using deaminase enzymes from different organisms, enzymes developed through directed evolution, or context-dependent base editors with narrow editing windows [28,85,94–96]. Moreover, current base editors cannot perform all 12 possible types of DNA base modifications. This limitation prompted the Liu group to pioneer ‘prime editing’ technology. Prime editing shows higher or similar efficiency with fewer byproducts than HDR and has similar strengths and weaknesses to base editing. Prime editing also has lower rates of off-target editing than Cas9 and offers clean DNA modification without DNA and RNA off-targets [97,98]. Current prime editors cannot perform insertion of long nucleotide sequences. We anticipate that optimizing the individual components of prime editors such as Cas9, reverse transcriptase and prime editing gRNA together with its delivery system could usher prime editor technology to therapeutic applications.
Concluding remarks and future perspectives
Over the past decade, substantial progress has been made using CRISPR as both a tool and a therapeutic. As discussed, CRISPR-Cas as a screening tool has become very effective, although it will benefit from better coverage of the genome, more refined gRNA designs, and analyses that can distinguish multigene transcripts. Clinically, as we have described, CRISPR-based editing has proven capable of advancing cell therapies for both ex and in vivo applications. Despite clear applications of CRISPR-based editing, several challenges remain (see Outstanding questions). These existing challenges (e.g., immune responses, delivery, off-targeting effects, DNA damage, etc.) exist mainly because nature has not optimized CRISPR nucleases for precise genome editing, but rather to function as a bacterial defense system, meaning much work must be done to make these systems amenable to precision editing in humans. Furthermore, there are large genomic heterogeneities in the human population, which complicate therapeutic development. In order to optimize on-target activity, and decrease off-target effects, tailor-made gRNAs based on a patient’s genetic makeup may be required [99–101]. In addition, to propel forward the development of CRISPR-based therapeutics, several methods are being developed for precision control of Cas9-based systems [83,102], and new editors are being developed (e.g., base and prime editors). Moreover, we are seeing a rise in translational dCas9-based technologies, which allow for dose and temporal control of a therapeutic target without DSBs, thereby averting existing challenges with Cas9 [53]. Especially considering how far CRISPR technologies have advanced since their discovery only a decade ago, we anticipate further engineering strategies over the next decade will better minimize and optimize the system, allowing for the development of transformational in vivo applications.
Outstanding questions.
Which nuclease is least immunogenic? Systematic studies have produced conflicting results, and larger population studies should be performed to answer this question.
Though an improvement over RNAi screens, off-target effects still remain the dominant confounding factor in high-throughput CRISPR screens, partly due to false positives caused by DSBs. Is it possible to improve upon existing algorithms for gRNA library design (e.g., predict gRNAs with less off-target and better on-target activity) and the analysis workflow to increase the signal-to-noise ratio? Can CRISPR nucleases that do not cut DNA like base/prime editors and the RNA-cutting nuclease, Cas13, generate less off-targets in high-throughput screens?
Can base editors be used to further sample the amino acid space in combination with CRISPR suppressor scanning? Alternatively, could we use PAM-less enzymes to cover more space?
Cas9-based technologies often involve genetic fusion of domains to endow Cas9 with new functions, but such fusions increase the size. Alternatively, can these domains be directed grafted into Cas9 to minimize the system?
Highlights.
CRISPR screens can advance conventional drug development by identifying key targets and escape mutants.
CRISPR-based therapies are rapidly advancing, with applications involving cell replacement therapies, gene drives, and diagnostics.
The use of CRISPR both ex vivo and in vivo is hindered by the immunogenicity of CRISPR components, limited delivery, low specificity, off-target effects, DNA damage, and repair pathways.
CRISPR limitations are being addressed through advances in controllers (degraders/inhibitors) of Cas9, conjugation strategies to endow Cas9 with new functions, and base and primer editors that do not produce double-strand breaks.
Acknowledgments
This work was supported by the Defense Advanced Research Projects Agency (N66001-17-2-4055) and National Institutes of Health (R01GM132825, R01GM137606).
Glossary
- Adenine base editors (ABEs)
an engineered system for A→G base conversion
- Cas9 nickase (nCas9)
created by mutating a catalytic residue (D10A or H840A) in Cas9 and is used to introduce a single-strand break
- Chimeric antigen receptor (CAR)-T cells
genetically engineered cells with an artificial chimeric T cell receptor that recognizes the antigens on the target cells
- CRISPR activation (CRISPRa)
expression of target genes using dCas9 fused to a transcriptional activator domain
- CRISPR interference (CRISPRi)
repression of the target genes using dCas9 fused to a transcriptional repressor domain
- CRISPR knockout (CRISPRko)
a gene knockout as a result of a Cas9-mediated double-strand break
- Cytosine base editors (CBEs)
an engineered system for C→T base conversion
- Dead Cas9 (dCas9)
catalytically inactive Cas9 variant that can bind to DNA but cannot catalyze strand breaks
- Guide RNA (gRNA)
an RNA component of the CRISPR-Cas9 system that allows the enzyme to identify the target DNA by base pairing
- Homology directed repair (HDR)
a mechanism by which cells precisely repair double-strand breaks using an exogenous DNA template
- Krüppel associated box (KRAB)
a DNA-binding transcriptional repressor domain
- Non-homologous end joining (NHEJ)
an error-prone repair pathway that results in insertions or deletions following double-strand break
- Protospacer adjacent motif (PAM)
a stretch of DNA used by Cas9 for target recognition
- VPR
VP64 fused with p65 and Rta transcriptional activators; together, they activate gene expression
Footnotes
Declaration of interests
Broad Institute has filed several applications for CRISPR inventions reviewed in this article. A.C. is a named inventor on several patent and patent applications filed from the Broad Institute, including U.S. Application Publication nos. 2021/0139872, 2021/0222164, and 2020/0354701, as well as International Patent Publication nos. WO 2020/041384 and WO 2020/041380.
References
- 1.Mali P et al. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong L et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behan FM et al. (2019) Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 [DOI] [PubMed] [Google Scholar]
- 4.Chavez A et al. (2015) Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbert LA et al. (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konermann S et al. (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudelli NM et al. (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boettcher M and McManus MT (2015) Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol. Cell 58, 575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Housden BE and Perrimon N (2016) Comparing CRISPR and RNAi-based screening technologies. Nat. Biotechnol 34, 621–623 [DOI] [PubMed] [Google Scholar]
- 11.Evers B et al. (2016) CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol 34, 631–633 [DOI] [PubMed] [Google Scholar]
- 12.Morgens DW et al. (2016) Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol 34, 634–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deans RM et al. (2016) Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol 12, 361–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jost M and Weissman JS (2018) CRISPR approaches to small molecule target identification. ACS Chem. Biol 13, 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shalem O et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J et al. (2021) Programmable human histone phosphorylation and gene activation using a CRISPR/Cas9-based chromatin kinase. Nat. Commun 12, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klann TS et al. (2017) CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol 35, 561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong CH et al. (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20, 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jost M et al. (2017) Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol. Cell 68, 210–223.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin A et al. (2019) Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med 11, eaaw8412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuster A et al. (2019) RNAi/CRISPR screens: from a pool to a valid hit. Trends Biotechnol. 37, 38–55 [DOI] [PubMed] [Google Scholar]
- 22.Ihry RJ et al. (2018) p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med 24, 939–946 [DOI] [PubMed] [Google Scholar]
- 23.Haapaniemi E et al. (2018) CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med 24, 927–930 [DOI] [PubMed] [Google Scholar]
- 24.Munoz DM et al. (2016) CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 6, 900–913 [DOI] [PubMed] [Google Scholar]
- 25.Kuscu C et al. (2017) CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods 14, 710–712 [DOI] [PubMed] [Google Scholar]
- 26.Kweon J et al. (2020) A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene 39, 30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanna RE et al. (2021) Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080.e20 [DOI] [PubMed] [Google Scholar]
- 28.Grunewald J et al. (2019) Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen Tran MT et al. (2020) Engineering domain-inlaid SaCas9 adenine base editors with reduced RNA off-targets and increased on-target DNA editing. Nat. Commun 11, 4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbluh J et al. (2017) Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat. Commun 8, 15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y et al. (2021) Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol. 22, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodapati S et al. (2020) A benchmark of algorithms for the analysis of pooled CRISPR screens. Genome Biol. 21, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B et al. (2019) Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc 14, 756–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyers RM et al. (2017) Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li W et al. (2014) MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawson KA et al. (2020) Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han K et al. (2020) CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei J et al. (2021) Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 184, 76–91.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vinyard ME et al. (2019) CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol 15, 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi J et al. (2015) Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Canver MC et al. (2015) BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess GT et al. (2016) Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma Y et al. (2016) Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 13, 1029–1035 [DOI] [PubMed] [Google Scholar]
- 44.Halperin SO et al. (2018) CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 [DOI] [PubMed] [Google Scholar]
- 45.Neggers JE et al. (2021) enAsCas12a enables CRISPR-directed evolution to screen for functional drug resistance mutations in sequences inaccessible to SpCas9. Mol. Ther 29, 208–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu JH et al. (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kleinstiver BP et al. (2019) Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol 37, 276–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pickar-Oliver A and Gersbach CA (2019) The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol 20, 490–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarty NS et al. (2020) Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun 11, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanna RE and Doench JG (2020) Design and analysis of CRISPR-Cas experiments. Nat. Biotechnol 38, 813–823 [DOI] [PubMed] [Google Scholar]
- 51.High KA and Roncarolo MG (2019) Gene therapy. N. Engl. J. Med 381, 455–464 [DOI] [PubMed] [Google Scholar]
- 52.Wu S-S et al. (2020) Advances in CRISPR/Cas-based gene therapy in human genetic diseases. Theranostics 10, 4374–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullard A (2020) Gene-editing pipeline takes off. Nat. Rev. Drug Discov 19, 367–372 [DOI] [PubMed] [Google Scholar]
- 54.Lu Y et al. (2020) Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med 26, 732–740 [DOI] [PubMed] [Google Scholar]
- 55.Shi L et al. (2017) CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 636, 36–41 [DOI] [PubMed] [Google Scholar]
- 56.Eyquem J et al. (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J et al. (2019) Building potent chimeric antigen receptor T cells with CRISPR genome editing. Front. Immunol 10, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacLeod DT et al. (2017) Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther 25, 949–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Depil S et al. (2020) ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov 19, 185–199 [DOI] [PubMed] [Google Scholar]
- 60.Li C et al. (2020) Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct. Genomics 19, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie G et al. (2020) CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine 59, 102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klichinsky M et al. (2020) Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol 38, 947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye L et al. (2016) Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and beta-thalassemia. Proc. Natl. Acad. Sci. U. S. A 113, 10661–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antoniani C et al. (2018) Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human beta-globin locus. Blood 131, 1960–1973 [DOI] [PubMed] [Google Scholar]
- 65.Scharenberg SG et al. (2020) Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun 11, 3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Ravin SS et al. (2017) CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med 9, eaah3480 [DOI] [PubMed] [Google Scholar]
- 67.Velazco-Cruz L et al. (2020) Advances toward engineering functionally mature human pluripotent stem cell-derived β cells. Front. Bioeng. Biotechnol 8, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hogrebe NJ et al. (2020) Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol 38, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma S et al. (2018) β cell replacement after gene editing of a neonatal diabetes-causing mutation at the insulin locus. Stem Cell Rep. 11, 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maxwell KG et al. (2020) Gene-edited human stem cell-derived beta cells from a patient with monogenic diabetes reverse pre-existing diabetes in mice. Sci. Transl. Med 12, eaax9106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim D et al. (2020) Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun 11, 4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li B et al. (2020) Strategies for the CRISPR-based therapeutics. Trends Pharmacol. Sci 41, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Long C et al. (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nelson CE et al. (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabebordbar M et al. (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maeder ML et al. (2019) Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med 25, 229–233 [DOI] [PubMed] [Google Scholar]
- 77.Gillmore JD et al. (2021) CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med 385, 493–502 [DOI] [PubMed] [Google Scholar]
- 78.Simhadri VL et al. (2018) Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev 10, 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Charlesworth CT et al. (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med 25, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei T et al. (2020) Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun 11, 3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng Q et al. (2020) Selective organ targeting (SORT) nano-particles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol 15, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho SW et al. (2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gangopadhyay SA et al. (2019) Precision control of CRISPR-Cas9 using small molecules and light. Biochemistry 58, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bednarski JJ and Sleckman BP (2019) At the intersection of DNA damage and immune responses. Nat. Rev. Immunol 19, 231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rees HA and Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 770–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grunewald J et al. (2020) A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol 38, 861–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mok BY et al. (2020) A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koblan LW et al. (2021) In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature 589, 608–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Musunuru K et al. (2021) In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 593, 429–434 [DOI] [PubMed] [Google Scholar]
- 90.Newby GA and Liu DR (2021) In vivo somatic cell base editing and prime editing. Mol. Ther 29, 3107–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Porto EM et al. (2020) Base editing: advances and therapeutic opportunities. Nat. Rev. Drug Discov 19, 839–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zuo E et al. (2019) Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grünewald J et al. (2019) CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol 37, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rees HA et al. (2019) Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv 5, eaax5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y et al. (2020) Very fast CRISPR on demand. Science 368, 1265–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee S et al. (2020) Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci. Adv 6, eaba1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anzalone AV et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hsu JY et al. (2021) PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun 12, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schmid-Burgk JL et al. (2020) Highly parallel profiling of Cas9 variant specificity. Mol. Cell 78, 794–800.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clement K et al. (2020) Technologies and computational analysis strategies for CRISPR applications. Mol. Cell 79, 11–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsai SQ and Joung JK (2016) Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet 17, 300–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Modell AE et al. (2021) Chemical and optical control of CRISPR-associated nucleases. Curr. Opin. Chem. Biol 60, 113–121 [DOI] [PubMed] [Google Scholar]